Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1986 by The American Society of Biological Chemists, Inc
Vol. 261, No .21, Issue of ‘July 25, pp. 9825-9831,1986 Printed in U. S. A.
Molecular Cloning and Nucleotide Sequence of a Complete Human Uroporphyrinogen Decarboxylase cDNA*
(Received for publication, August 1, 1985)
Paul-Henri Rom&$B, Natacha RaichS, Anne Dubart$, Denise Beaupain$, Marsha PryorV, James KushnerV, Michel Cohen-SolalS, and Michel GoossensS From the Slnstitut National de la Sante et de la Recherche Medicale U-91, H6pital Henri Mondor, 94010 Creteil, France and the TDepartment of Medicine, University of Utah, Salt Lake City, Utah 84132
We have cloned and sequenced a full-length cDNA coding for human uroporphyrinogen decarboxylase. The deduced 367-amino acid sequence is consistent with the molecular weight, the partial amino acid se- quence of cyanogen bromide peptides, and the total amino acid composition of the purified enzyme. South- ern analysis of human genomic DNA shows that its gene is present as a single copy in the human genome, and Northern analysis demonstrates the presence of a single size species of mRNA in erythroid and non- erythroid tissues and in several cultured cell lines. We have also demonstrated that the level of uroporphyrin- ogen decarboxylase mRNA is markedly increased in tissues or cell lines of erythroid origin and that this is due to a tissue-specific transcriptional activation of the uroporphyrinogen decarboxylase gene.
A coordinated expression of a number of proteins occurs during erythroid cell differentiation. Some proteins, like hemoglobin, are only expressed in these cells, and others, like the enzymes of the heme biosynthetic pathway, have an ubiquitous tissue expression but show increased activity in the red cell lineage (1). Moreover, there is a time-regulated coordination of the expression of different genes during eryth- roid differentiation, heme biosynthetic pathway enzymes dis- playing, for instance, an orchestrated increase of their activ- ities prior to accumulation of globin (2). Therefore, the genes coding for enzymes of the heme biosynthetic pathway may be viewed as a model of regulated house-keeping genes. A recent report by Grandchamp et al. (3) has shown that in mouse erythroleukemia cells, capable of undergoing in vitro erythroid differentiation, increases in the activity of two enzymes of the heme biosynthetic pathway are correlated with increases in the copy number of corresponding mRNAs and that this process precedes a- and &globin mRNAs accumulation. This increase could result either from a tissue-specific enhanced transcription of these genes, or from a post-transcriptional regulation as in the case of glyceraldehyde-3-phosphate de- hydrogenase (4).
Uroporphyrinogen decarboxylase (EC 4.1.1.37) is a cyto- solic enzyme involved in the biosynthesis of heme. It catalyzes the sequential removal of the four carboxyl groups of the
* This work was supported by the Institut National de la Sant6 et de la Recherche Mkdicale, by Ministere de la Recherche et de 1’In- dustrie Grant MRI 823140, and by National Institutes of Health Grant AM 20503. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisernent” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
8 To whom correspondence should be addressed.
carboxymethyl side chains of uroporphyrinogen to yield co- proporphyrinogen (5). Abnormal uroporphyrinogen decarbox- ylase activity has been associated with porphyria cutanea tarda (PCT’), the most common form of porphyria in man (6). It is characterized, in most cases, by a 50% decrease in the catalytic activity and immunoreactivity of uroporphyri- nogen decarboxylase in tissues tested, and this enzyme defect is inherited as an autosomal dominant trait ( 7 ) . The definition of the genetic locus coding for the enzyme and the knowledge of its primary structure will provide further basis for under- standing the structural alterations and the functional abnor- malities of uroporphyrinogen decarboxylase in patients with PCT.
This paper reports the cloning and analysis of a cDNA sequence complementary to uroporphyrinogen decarboxylase mRNA obtained from human spleen. Using this cDNA as hybridization probe, we show that uroporphyrinogen decar- boxylase is encoded by a single gene. We also report the nucleotide sequence of this cDNA, and we show that the predicted amino acid sequence of the enzyme agrees with the partial amino acid sequence that we have determined by direct protein sequencing. Analysis of the expression of this gene indicates that only one size species of mRNA is present in erythroid and non-erythroid tissues as well as in several cultured cell lines, and quantitative analysis shows that an accumulation of uroporphyrinogen decarboxylase mRNA oc- curs in tissues or cell lines having erythroid properties. We show, for the first time, that this accumulation results in part from an increase of transcription of uroporphyrinogen decar- boxylase gene in cell lines having erythroid properties.
MATERIALS AND METHODS
General Procedures-A previous communication (8) described the methods used for in vitro translation in a messenger-dependent rabbit reticulocyte cell-free system, uroporphyrinogen decarboxylase im- munoprecipitation from in vitro translation products, and SDS-pol- yacrylamide gel electrophoresis. RNA and DNA concentrations were determined by absorbance at 260 nm (1 A260 = 40 pg/ml for RNA and 1 AZw = 50 pg/ml for DNA).
Cell Culture-The K562, HEL, and HL60 cell lines (9-11) studied were a gift of Dr. U. Testa. Cells were grown in suspension culture in a modified RPMI 1640 medium containing 10% heat-inactivated newborn fetal calf serum plus 2% human serum in a 5% CO2 humi- dified atmosphere. All cell lines were harvested in their log phase having reached a density of lo6-LO6 cells/ml.
Plasmid Isolation, Znsert Purification, and Nick Translation-Plas- mids containing rat or human uroporphyrinogen decarboxylase cDNA inserts were prepared from chloramphenicol-treated bacteria by ther- mal denaturation and sedimentation of chromosomal DNA (12), followed by treatment with RNase and proteinase K and extraction
The abbreviations used are: PCT, porphyria cutanea tarda; SDS, sodium dodecyl sulfate; bp, base pair; Pipes, 1,4-piperazinedieth- anesulfonic acid.
9825

9826 cDNA for Human Uroporphyrinogen Decarboxylase
with bufferedphenol. The recombinant plasmids were further purified by the acid/phenol method (13). The cDNAs were excised from the recombinant plasmid, isolated by agarose gel electrophoresis, elec- troeluted, and nick-translated as described (14).
Preparation and Fractionation of Poly(A+) RNA-Total RNA was extracted by the LiCl method (15) from the spleen, removed for therapeutic reasons, of a child with 8-thalassemia major. Total RNA from the K562 cell line was prepared by extraction with the proteinase K/SDS procedure (16). Poly(A+) RNA was then selected by chro- matography on oligo(dT)-cellulose (type T3, Collaborative Research, Waltham, MA) (17). Messenger RNA obtained from the spleen was fractionated by preparative gel electrophoresis (la), and fractions containing mRNA enriched in uroporphyrinogen decarboxylase se- quences were pooled and ethanol-precipitated.
cDNA Cloning and Analysis-Synthesis of double-stranded cDNA complementary to enriched poly(A+) RNA was accomplished as de- scribed by Wickens et al. (19) or Land et al. (20). Double-stranded cDNAs longer than 800 bp were purified by polyacrylamide gel electrophoresis followed by electroelution. They were then inserted in the PstI site of pBR322 using the homopolymeric tailing and hybridization method (21). The resulting hybrid molecules were used to transform Escherichia coli strain MC1061 which was rendered competent for uptake of plasmids (22). Recombinant clones were stored frozen on nitrocellulose filters after high density plating (23).
Clones were screened with 32P-labeled cDNAs complementary to rat uroporphyrinogen decarboxylase mRNA using the colony hybrid- ization method of Grunstein and Hogness (24) as modified by Thayer (25). After hybridization and washing, positive colonies were visual- ized by exposing filters to Kodak AR5 x-ray films at -80 "C.
For further characterization, plasmids from positive colonies were isolated from 5-ml overnight cultures by the boiling method, digested with PstI, and electrophoresed in 1% agarose gel. The clone pUD3, which contains the largest insert, was further characterized and used as the labeled nucleic acid hybridization probe.
Primer Extension Analysis-pUD3 plasmid DNA was cut out with BamHI, 5' end-labeled with T4 polynucleotide kinase and [y"P] ATP, and cut again with either KpnI or PstI. The 66-bp KpnIIBamHI and 158-bp PstI/BamHI fragments were electroeluted and hybridized to 10 pg of mRNA isolated either from the K562 cell line or from a spleen of a patient with @-thalassemia. The hybridization was done in 70% formamide, 10 mM Pipes, pH 7.0, 1 mM EDTA, 0.4 M NaCl at 50 "C overnight. After ethanol precipitation, the pellet was redis- solved in 30 pl of 50 mM Tris-HCI, pH 8.3, 10 mM MgC12, 1 mM dXTP. 15 pl was incubated with 10 units of reverse transcriptase, 10 units of RNasin, and 100 pg/ml actinomycin D at 42 "C for 60 min, and 15 pl was incubated without reverse transcriptase. The mixture was phenol-extracted and precipitated with ethanol. The pellet was dissolved in formamide with marker dyes and electrophoresed on a 6% polyacrylamide gel containing 7 M urea.
Preparation and Blot Analysis of DNA-Human leucocyte DNA was isolated (26) and digested with different restriction enzymes (Boehringer Mannheim). Following digestion, the DNA was ethanol- precipitated, resuspended, and run on a 1% agarose gel. The gel was blotted onto a nitrocellulose filter and hybridized with the radioactive labeled probe at 42 "C in 50% formamide for 24 h. The filter was washed extensively and autoradiographed at -80 "C for 48 h with intensifying screens.
RNA Analysis-RNA denaturation, fractionation by electropho- resis in formaldehyde-agarose gels, and transfer to nitrocellulose filter were done as described (27). Hybridization and washing were per- formed according to Thomas (28), and autoradiograms were scanned with a densitometer.
In Vitro Transcription and Hybridization-Nuclei were extracted from cell lines (29), resuspended in 20 mM Tris, pH 7.9,75 mM NaC1, 0.5 mM EDTA, 0.85 mM dithiothreitol, 0.125 mM phenylmethylsul-
tion, isolation of 32P-labeled RNA, and hybridization to filter-bound fonyl fluoride, 50% glycerol, and stored at -70 "C. Nuclear transcrip-
DNA were done as described (30). To measure the relative rate of transcription, the following cloned DNAs were immobilized on Genescreen Plus (New England Nuclear): i) pUD3 cDNA which represents the DNA complementary to uroporphyrinogen decarbox- ylase mRNA (this cDNA recognizes a unique gene by Southern hybridization analysis), ii) pBR322, and iii) plasmid-containing se- quences coding for 28 S ribosomal RNA. Nonspecific binding to vector DNA was less than 50 cpm.
DNA Sequence Analysis-The chemical modification method of Maxam and Gilbert (31) was used for all the DNA sequence deter- minations. After digestion with the appropriate restriction endonu-
clease, the recombinant plasmid was labeled either at the 5' end with T4 polynucleotide kinase and [Y-~'P]ATP or at the 3' end with DNA polymerase I (Klenow fragment) and [cx-~'P]~ATP or terminal trans- ferase and [~~-~~P]dideoxy-ATP. After digestion with a second restric- tion enzyme, single end-labeled fragments were isolated from poly- acrylamide gels for DNA sequence analysis.
Purification of Uroporphyrinogen Decarboxylase, Preparation of Cyanogen Bromide Peptide Fragments, and Amino Acid Sequencing- Uroporphyrinogen decarboxylase was purified from 2.0 liters of nor- mal human erythrocytes by a modification of the methods of Straka and Kushner (32) and de Verneuil et al. (5). The erythrocyte lysate was rendered hemoglobin-free by ion exchange chromatography on DE52, followed by ammonium sulfate precipitation (20-45%) and chromatography on phenyl-Sepharose (Pharmacia Fine Chemicals). The eluate was dialyzed against stepwise decreasing concentrations of ethylene glycol in 50 mM potassium phosphate buffer, pH 6.8, until no ethylene glycol remained. Ultrafiltration concentrated the sample to a volume of approximately 20 ml. A substrate affinity column was prepared by covalently linking uroporphyrin I (Porphyrin Products, Logan, UT) to Affi-Gel102 (Bio-Rad) by a modification of the method of Spikes et al. (33).
The column was reduced in an anaerobic chamber with sodium amalgam, frequently adding small amounts of glacial acetic acid to maintain the pH near neutrality and finally equilibrated with 0.01 M dithiothreitol in 50 mM potassium phosphate buffer, pH 6.8. The sample was applied, and the column was extensively washed with the same buffer. Uroporphyrinogen decarboxylase was eluted with 0.5 M KC1 in 50 mM potassium phosphate buffer, pH 6.8.
Analytical SDS-polyacrylamide gel electrophoresis was performed (34) and stainedusing a silver stain (35). Enzyme activity was assayed by the method of Straka et al. (36).
The purified protein was carboxymethylated, reduced with thiog- lycolic acid, and cleaved with cyanogen bromide (37). The resulting peptide fragments were separated by reverse-phase high pressure liquid chromatoraphy using a Cs column (Beckman Instruments) with a linear gradient of propanol in 0.1% trifluoroacetic acid. The amino acid sequence of individual peptide fragments was determined using a Beckman 890D instrument (38). Amino acid analysis of the entire protein was performed after hydrolysis of the sample in 6 N HCI for 24 h.
RESULTS
Identification of Uroporphyrinogen Decarboxylase cDNA Clones-As uroporphyrinogen decarboxylase mRNA was found to be present in low amounts in all human tissues tested, we finally selected as a source of RNA the spleen removed from a patient with p-thalassemia major. This organ contained erythropoietic islands, and uroporphyrinogen de- carboxylase mRNA represented about 0.05% of the total mRNA. After fractionation, the uroporphyrinogen decarbox- ylase sequence was enriched to 0.5% and used to construct a cDNA library. Screening of this library with specific rat cDNA obtained previously (8) permitted us to isolate several human uroporphyrinogen decarboxylase clones. Plasmid DNA was then prepared, and the size of the inserts was examined by agarose gel electrophoresis. The largest uroporphyrinogen decarboxylase cDNA, pUD3, contains 1300 bp. Since the size of uroporphyrinogen decarboxylase mRNA is 1300 bp (see below), this clone was likely to correspond to most, if not all, of the coding sequence and was used for further analysis.
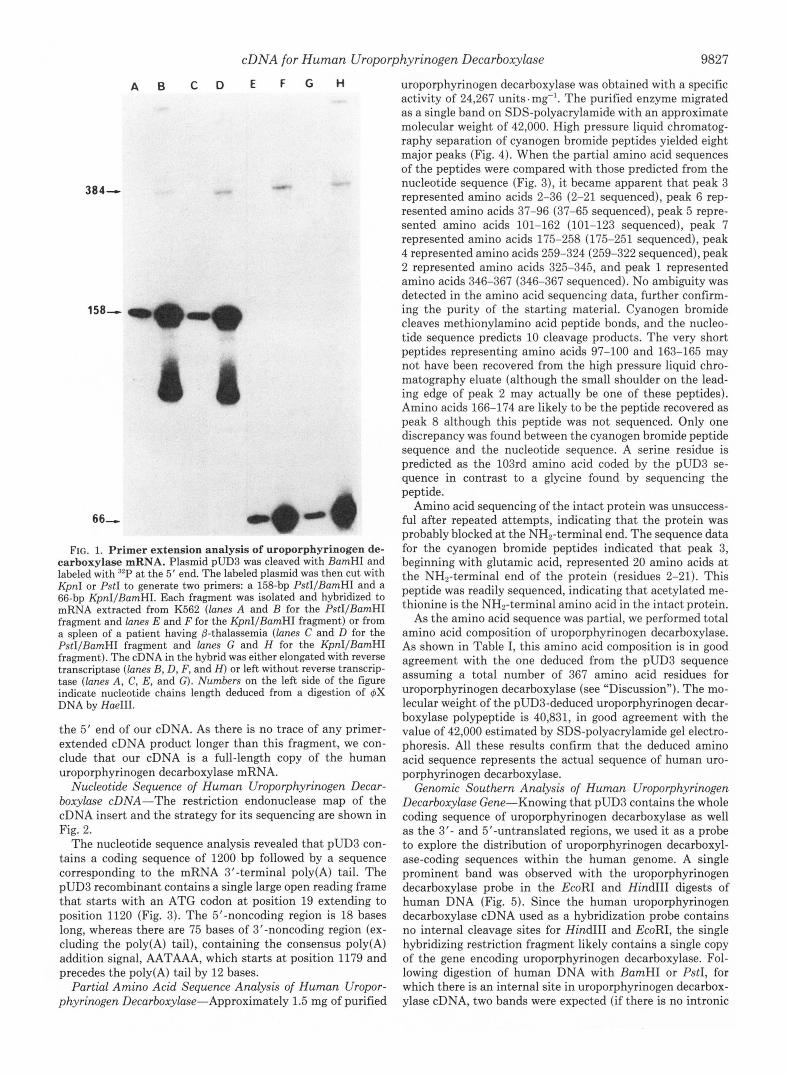
Mapping of the 5 ' End of the Message-To show that pUD3 extends to the 5' end of uroporphyrinogen decarboxylase mRNA, we performed a primer extension analysis. Two pUD3 restriction fragments of different length, having a common 5' BamHI end-labeled extremity, were hybridized to mRNA isolated from a cell line (K562) or a tissue (human erythroid spleen) and elongated with reverse transcriptase. The elon- gated cDNA products and the original end-labeled primers were then electrophoresed on a sequencing gel and autoradi- ographed. As shown in Fig. 1, the length of the elongated fragment was 384 bp whichever RNA was tested. This size represents the exact distance between the BamHI site and
cDNA for Human Uroporphyrinogen Decarboxylase 9827
A B
384-
66,
C D E F G H
FIG. 1. Primer extension analysis of uroporphyrinogen de- carboxylase mRNA. Plasmid pUD3 was cleaved with BamHI and labeled with n2P at the 5’ end. The labeled plasmid was then cut with KpnI or PstI to generate two primers: a 158-bp PstIIBamHI and a 66-bp KpnI/BamHI. Each fragment was isolated and hybridized to mRNA extracted from K562 (lanes A and B for the PstI/BamHI fragment and lanes E and F for the KpnIIBamHI fragment) or from a spleen of a patient having &thalassemia (lanes C and D for the PstIIBamHI fragment and lanes G and H for the KpnI/BamHI fragment). The cDNA in the hybrid was either elongated with reverse transcriptase (lanes B, D, F, and H) or left without reverse transcrip- tase (lanes A, C, E, and G). Numbers on the left side of the figure indicate nucleotide chains length deduced from a digestion of 4X DNAby HaeIII.
the 5’ end of our cDNA. As there is no trace of any primer- extended cDNA product longer than this fragment, we con- clude that our cDNA is a full-length copy of the human uroporphyrinogen decarboxylase mRNA.
Nucleotide Sequence of Human Uroporphyrinogen Decar- boxylase cDNA-The restriction endonuclease map of the cDNA insert and the strategy for its sequencing are shown in Fig. 2.
The nucleotide sequence analysis revealed that pUD3 con- tains a coding sequence of 1200 bp followed by a sequence corresponding to the mRNA 3‘-terminal poly(A) tail. The pUD3 recombinant contains a single large open reading frame that starts wit.h an ATG codon at position 19 extending to position 1120 (Fig. 3). The 5“noncoding region is 18 bases long, whereas there are 75 bases of 3”noncoding region (ex- cluding the poly(A) tail), containing the consensus poly(A) addition signal, AATAAA, which starts at position 1179 and precedes the poly(A) tail by 12 bases.
Partial Amino Acid Sequence Analysis of Human Uropor- phyrinogen Decarboxylase-Approximately 1.5 mg of purified
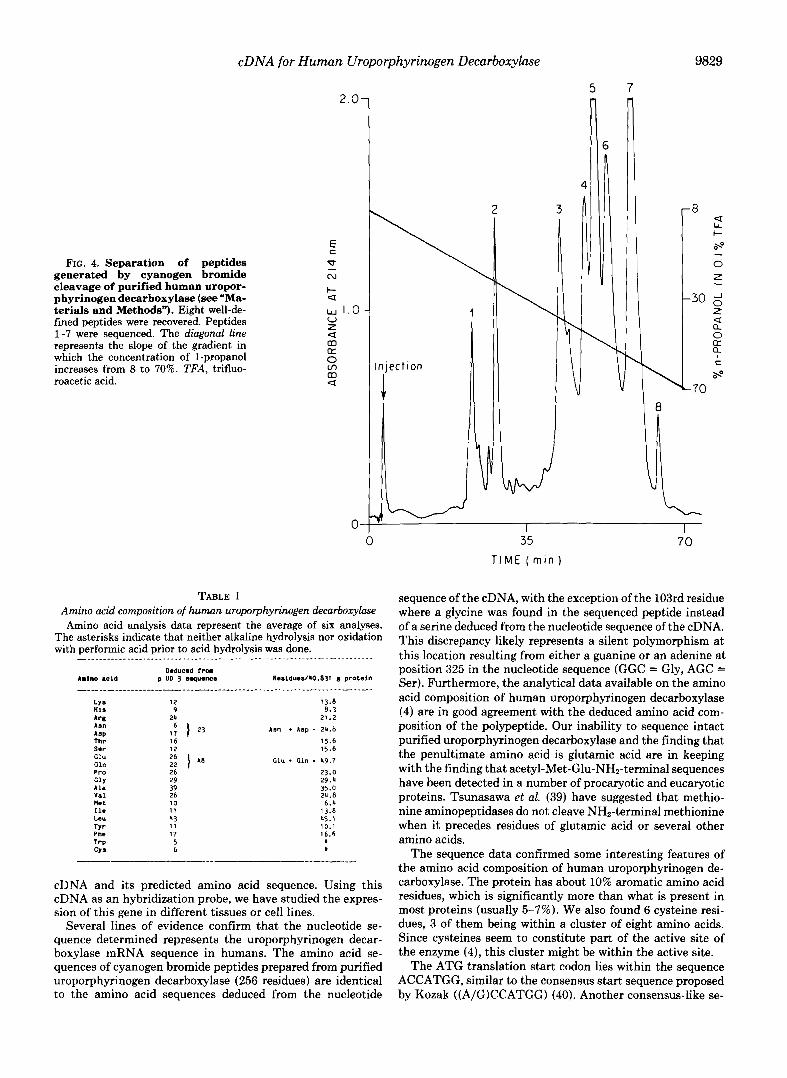
uroporphyrinogen decarboxylase was obtained with a specific activity of 24,267 units. mg”. The purified enzyme migrated as a single band on SDS-polyacrylamide with an approximate molecular weight of 42,000. High pressure liquid chromatog- raphy separation of cyanogen bromide peptides yielded eight major peaks (Fig. 4). When the partial amino acid sequences of the peptides were compared with those predicted from the nucleotide sequence (Fig. 3), it became apparent that peak 3 represented amino acids 2-36 (2-21 sequenced), peak 6 rep- resented amino acids 37-96 (37-65 sequenced), peak 5 repre- sented amino acids 101-162 (101-123 sequenced), peak 7 represented amino acids 175-258 (175-251 sequenced), peak 4 represented amino acids 259-324 (259-322 sequenced), peak 2 represented amino acids 325-345, and peak 1 represented amino acids 346-367 (346-367 sequenced). No ambiguity was detected in the amino acid sequencing data, further confirm- ing the purity of the starting material. Cyanogen bromide cleaves methionylamino acid peptide bonds, and the nucleo- tide sequence predicts 10 cleavage products. The very short peptides representing amino acids 97-100 and 163-165 may not have been recovered from the high pressure liquid chro- matography eluate (although the small shoulder on the lead- ing edge of peak 2 may actually be one of these peptides). Amino acids 166-174 are likely to be the peptide recovered as peak 8 although this peptide was not sequenced. Only one discrepancy was found between the cyanogen bromide peptide sequence and the nucleotide sequence. A serine residue is predicted as the 103rd amino acid coded by the pUD3 se- quence in contrast to a glycine found by sequencing the peptide.
Amino acid sequencing of the intact protein was unsuccess- ful after repeated attempts, indicating that the protein was probably blocked at the NH2-terminal end. The sequence data for the cyanogen bromide peptides indicated that peak 3, beginning with glutamic acid, represented 20 amino acids a t the NH2-terminal end of the protein (residues 2-21). This peptide was readily sequenced, indicating that acetylated me- thionine is the NHz-terminal amino acid in the intact protein.
As the amino acid sequence was partial, we performed total amino acid composition of uroporphyrinogen decarboxylase. As shown in Table I, this amino acid composition is in good agreement with the one deduced from the pUD3 sequence assuming a total number of 367 amino acid residues for uroporphyrinogen decarboxylase (see “Discussion”). The mo- lecular weight of the pUD3-deduced uroporphyrinogen decar- boxylase polypeptide is 40,831, in good agreement with the value of 42,000 estimated by SDS-polyacrylamide gel electro- phoresis. All these results confirm that the deduced amino acid sequence represents the actual sequence of human uro- porphyrinogen decarboxylase.
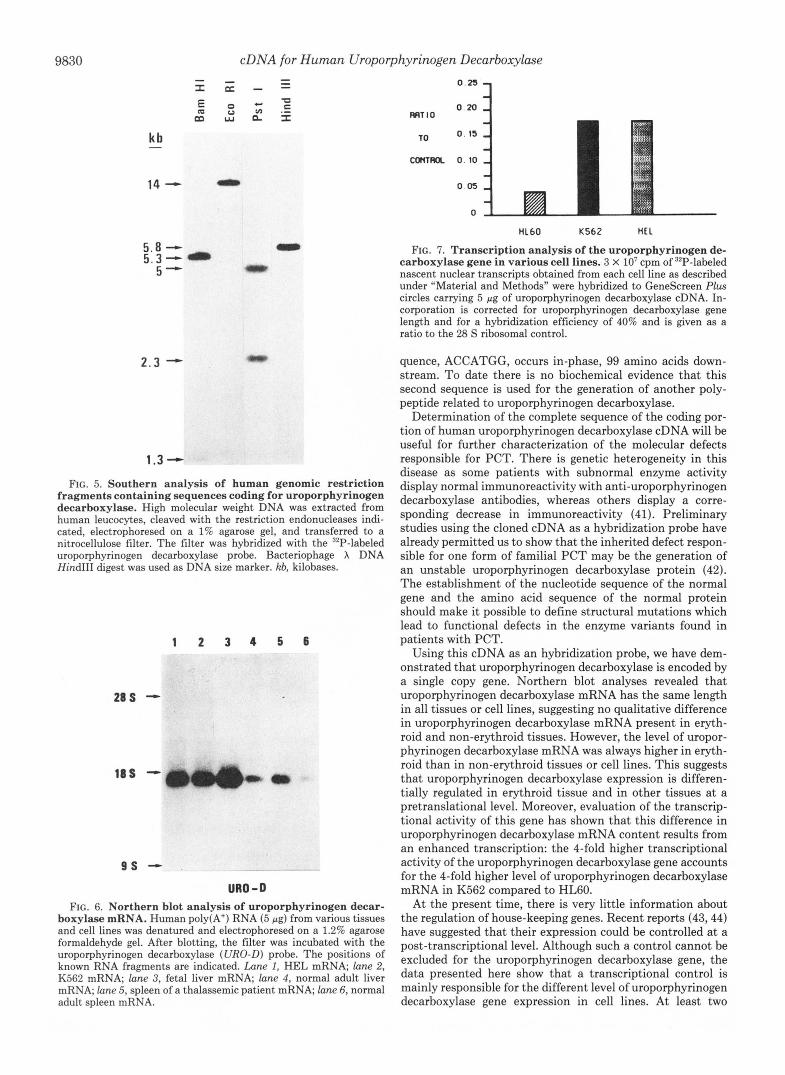
Genomic Southern Analysis of Human Uroporphyrinogen Decarboxylase Gene-Knowing that pUD3 contains the whole coding sequence of uroporphyrinogen decarboxylase as well as the 3‘- and 5”untranslated regions, we used it as a probe to explore the distribution of uroporphyrinogen decarboxyl- ase-coding sequences within the human genome. A single prominent band was observed with the uroporphyrinogen decarboxylase probe in the EcoRI and HindIII digests of human DNA (Fig. 5). Since the human uroporphyrinogen decarboxylase cDNA used as a hybridization probe contains no internal cleavage sites for HindIII and EcoRI, the single hybridizing restriction fragment likely contains a single copy of the gene encoding uroporphyrinogen decarboxylase. Fol- lowing digestion of human DNA with BamHI or PstI, for which there is an internal site in uroporphyrinogen decarbox- ylase cDNA, two bands were expected (if there is no intronic

9828 cDNA for Human Uroporphyrinogen Decarboxylase
- FIG. 2. Strategy for sequencing human uroporphyrinogen decarboxylase cDNA pUD3. The top line
is a restriction map for the entire cDNA sequence including the two PstI cloning sites. The map numbering proceeds in a 5' to 3' direction. The arrows below the map indicate the extent and direction of sequences determined. Each segment was sequenced at least two times.
FIG. 3. cDNA and primary amino acid sequence of human uropor- phyrinogen decarboxylase. The DNA sequence of the strand correspond- ing to the mRNA is displayed above the protein sequence. The poly(A) nucleo- tide tail is not shown. Experimentally determined amino acid sequences of pu- rified erythrocyte uroporphyrinogen de- carboxylase peptides produced from CNBr cleavage are indicated in capital letters. The poly(A) addition signal is indicated by an open box.
site) and obtained. All these results are consistent with the existence of a single uroporphyrinogen decarboxylase gene.
Northern Blot Analysis of Uroporphyrinogen Decarboxylase mRNA-Northern blot analysis of various erythroid or non- erythroid tissues and cultured cell lines was performed. Only one size species of mRNA was found in all cases, suggesting that these mRNAs sequences are not qualitatively different in erythroid and non-erythroid cells. The size of uroporphyr- inogen decarboxylase mRNA was estimated to be 1300 bases, based upon its electrophoretic mobility relative to known standards (Fig. 6).
The analysis of the relative intensity of the bands obtained after autoradiography showed that the amount of uropor- phyrinogen decarboxylase mRNA was always higher in eryth- roid than in non-erythroid tissues or cell lines. Indeed, when we compared mRNA quantities present in cell lines with or without some erythroid properties, we observed that K562 or HEL, erythroid human cell lines, contains 4 times more uroporphyrinogen decarboxylase mRNA than does HL60, a lymphoid human cell line (data not shown). This is in good agreement with the respective enzymatic activities measured in these cell lines and suggests a tissue-specific control of the
uroporphyrinogen decarboxylase gene expression at a pre- translational level.
Higher Rate of Uroporphyrinogen Decarboxylase Gene Tran- scription in Cell Lines with Erythroid Properties-To deter- mine whether the accumulation of uroporphyrinogen decar- boxylase mRNA in cell lines with erythroid properties is due to stabilization of the mRNA or is a consequence of an enhanced transcription of the uroporphyrinogen decarboxyl- ase gene, we measured the rate of transcription of this gene in isolated nuclei. Since reinitiation does not occur when isolated nuclei are transcribed in uitru, the amount of nucleo- tide incorporation into RNA in such an assay is an accurate indication of the rate of transcription initiation on a particular gene in vivo. As shown in Fig. 7 , we found that the relative rate of transcription of the uroporphyrinogen decarboxylase gene is &fold higher in the K562 or HEL cell lines than in HL60 cell lines. This precisely indicates a transcriptional enhancement of the uroporphyrinogen decarboxylase gene in cell lines having erythroid properties.
DISCUSSION
The results of the present study reveal the primary struc- ture of a full-length human uroporphyrinogen decarboxylase
cDNA for Human Uroporphyrinogen Decarboxylase
FIG. 4. Separation of peptides generated by cyanogen bromide cleavage of purified human uropor- phyrinogen decarboxylase ( s e e "Ma- terials and Methods"). Eight well-de- fined peptides were recovered. Peptides 1-7 were sequenced. The diagonal line represents the slope of the gradient in which the concentration of I-propanol increases from 8 to 70%. TFA, trifluo- roacetic acid.
2.0-
c E
x N
I- U
w 1.0- V z U
U 0 m U
m
m
0 0-
TABLE I
9829
5 7
Amino acid composition of human uroporphyrinogen decarboxylase Amino acid analysis data represent the average of six analyses.
The asterisks indicate that neither alkaline hydrolysis nor oxidation with performic acid prior to acid hydrolysis was done.
A m l n o acid 9 UD 3 abquence Reaidueal40,831 8 proteln Deaucea frm
- - - . - - " - - - - - - " " " - - - - - "- - - " - "_ - - - - -. " _" - - - - I - - - LYS His
12 13 .8 8.3
AS" 21.2
Thr
A m * Asp - 24.8
16 15.6 Ser 12 15.6 Cl" 2 6
ClU f Cl" - 49.7
PPO 26 23 .0 C l Y 29 A18 39
29.4
Val 35.0
wet 26 24 .8 10
IlC 6.4
I 1 Leu
13.8 43
Tyr 45.1
11 P ns 17
10.1 16.6
Trp 5 CYS 6 .
9 24 APg
ASP 1: I 23
Cl" 22 I
" "
cDNA and its predicted amino acid sequence. Using this cDNA as an hybridization probe, we have studied the expres- sion of this gene in different tissues or cell lines.
Several lines of evidence confirm that the nucleotide se- quence determined represents the uroporphyrinogen decar- boxylase mRNA sequence in humans. The amino acid se- quences of cyanogen bromide peptides prepared from purified uroporphyrinogen decarboxylase (256 residues) are identical to the amino acid sequences deduced from the nucleotide
:i. 35 70
T IME ( m i n )
sequence of the cDNA, with the exception of the 103rd residue where a glycine was found in the sequenced peptide instead of a serine deduced from the nucleotide sequence of the cDNA. This discrepancy likely represents a silent polymorphism at this location resulting from either a guanine or an adenine at position 325 in the nucleotide sequence (GGC = Gly, AGC = Ser). Furthermore, the analytical data available on the amino acid composition of human uroporphyrinogen decarboxylase (4) are in good agreement with the deduced amino acid com- position of the polypeptide. Our inability to sequence intact purified uroporphyrinogen decarboxylase and the finding that the penultimate amino acid is glutamic acid are in keeping with the finding that acetyl-Met-Glu-NH2-terminal sequences have been detected in a number of procaryotic and eucaryotic proteins. Tsunasawa et al. (39) have suggested that methio- nine aminopeptidases do not cleave NH2-terminal methionine when it precedes residues of glutamic acid or several other amino acids.
The sequence data confirmed some interesting features of the amino acid composition of human uroporphyrinogen de- carboxylase. The protein has about 10% aromatic amino acid residues, which is significantly more than what is present in most proteins (usually 5-7%). We also found 6 cysteine resi- dues, 3 of them being within a cluster of eight amino acids. Since cysteines seem to constitute part of the active site of the enzyme (4), this cluster might be within the active site.
The ATG translation start codon lies within the sequence ACCATGG, similar to the consensus start sequence proposed by Kozak ((A/G)CCATGG) (40). Another consensus-like se-
9830 cDNA for Human Uroporphyrinogen Decarboxylase ” - - x = - -
kb
14 - - 5.8 - 0 5.3” 5- 0
2.3 -
1.3 - FIG. 5. Southern analysis of human genomic restriction
fragments containing sequences coding for uroporphyrinogen decarboxylase. High molecular weight DNA was extracted from human leucocytes, cleaved with the restriction endonucleases indi- cated, electrophoresed on a 1% agarose gel, and transferred to a nitrocellulose filter. The filter was hybridized with the “P-labeled uroporphyrinogen decarboxylase probe. Bacteriophage X DNA Hind111 digest was used as DNA size marker. kb, kilobases.
1 2 3 4 5 6
28s -
16s -
9 s - URO - D
FIG. 6. Northern blot analysis of uroporphyrinogen decar- boxylase mRNA. Human poly(A+) RNA (5 pg) from various tissues and cell lines was denatured and electrophoresed on a 1.2% agarose formaldehyde gel. After blotting, the filter was incubated with the uroporphyrinogen decarboxylase (URO-D) probe. The positions of known RNA fragments are indicated. Lune I, HEL mRNA; lane 2, K562 mRNA; lane 3, fetal liver mRNA, lane 4, normal adult liver mRNA; lane 5, spleen of a thalassemic patient mRNA; lane 6, normal adult spleen mRNA.
WIT I O 0 2 0
0.15
mmL 0.10
0.05
0
HL60 K562 Hf L
FIG. 7. Transcription analysis of the uroporphyrinogen de- carboxylase gene in various cell lines. 3 X lo7 cpm of 32P-labeled nascent nuclear transcripts obtained from each cell line as described under “Material and Methods” were hybridized to GeneScreen Plus circles carrying 5 pg of uroporphyrinogen decarboxylase cDNA. In- corporation is corrected for uroporphyrinogen decarboxylase gene length and for a hybridization efficiency of 40% and is given as a ratio to the 28 S ribosomal control.
quence, ACCATGG, occurs in-phase, 99 amino acids down- stream. To date there is no biochemical evidence that this second sequence is used for the generation of another poly- peptide related to uroporphyrinogen decarboxylase.
Determination of the complete sequence of the coding por- tion of human uroporphyrinogen decarboxylase cDNA will be useful for further characterization of the molecular defects responsible for PCT. There is genetic heterogeneity in this disease as some patients with subnormal enzyme activity display normal immunoreactivity with anti-uroporphyrinogen decarboxylase antibodies, whereas others display a corre- sponding decrease in immunoreactivity (41). Preliminary studies using the cloned cDNA as a hybridization probe have already permitted us to show that the inherited defect respon- sible for one form of familial PCT may be the generation of an unstable uroporphyrinogen decarboxylase protein (42). The establishment of the nucleotide sequence of the normal gene and the amino acid sequence of the normal protein should make it possible to define structural mutations which lead to functional defects in the enzyme variants found in patients with PCT.
Using this cDNA as an hybridization probe, we have dem- onstrated that uroporphyrinogen decarboxylase is encoded by a single copy gene. Northern blot analyses revealed that uroporphyrinogen decarboxylase mRNA has the same length in all tissues or cell lines, suggesting no qualitative difference in uroporphyrinogen decarboxylase mRNA present in eryth- roid and non-erythroid tissues. However, the level of uropor- phyrinogen decarboxylase mRNA was always higher in eryth- roid than in non-erythroid tissues or cell lines. This suggests that uroporphyrinogen decarboxylase expression is differen- tially regulated in erythroid tissue and in other tissues at a pretranslational level. Moreover, evaluation of the transcrip- tional activity of this gene has shown that this difference in uroporphyrinogen decarboxylase mRNA content results from an enhanced transcription: the 4-fold higher transcriptional activity of the uroporphyrinogen decarboxylase gene accounts for the 4-fold higher level of uroporphyrinogen decarboxylase mRNA in K562 compared to HL60.
At the present time, there is very little information about the regulation of house-keeping genes. Recent reports (43,44) have suggested that their expression could be controlled a t a post-transcriptional level. Although such a control cannot be excluded for the uroporphyrinogen decarboxylase gene, the data presented here show that a transcriptional control is mainly responsible for the different level of uroporphyrinogen decarboxylase gene expression in cell lines. At least two

cDNA for Human Uroporphyrinogen Decarboxylase 9831
mechanisms could account for this regulation: one possibility being that the gene possesses a single promotor, the efficiency of which is enhanced in a tissue-specific manner; another being that this gene has two promotors, one used in all tissues and the other active only in erythroid tissue. Gene character- ization will clarified the promoter organization and elucidate some of the mechanisms involved in differential expression.
Acknowledgments-We thank Pr. J. Rosa for continuous support and encouragement, Y. Blouquit, B. Grandchamp, and A. Munnich for helpful discussions, M. Titeux for technical assistance in cell cultures, C. Valentin for technical assistance in nucleotide sequenc- ing, W. R. Gray (Department of Biology, University of Utah) for technical assistance with the amino acid sequencing, and A. M. Dulac and M. Tassier for assistance in the preparation of the manuscript.
1. 2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
REFERENCES Sassa, S. (1976) J. Exp. Med. 143, 305-315 Sassa, S. (1980) in In vivo and in vitro Erythropoiesis: The Friend
System (Rossi, G. B, ed) pp. 219-228, Elsevier/North-Holland, New York
Grandchamp, B., Beaumont, C., de Verneuil, H., and Nordman, Y. (1985) J. Biol. Chem. 260,9630-9635
Pierchaczyk, M., Blanchard, J. M., Marty, L., Dani, C., El Sa- boutv, S., Fort, Ph., and Jeanteur, P. (1984) Nucleic Acids Res. 12,6951-6963
. .
de Verneuil. H.. Sassa. H.. and KaDDaS. A. (1983) J. Biol. Chem. 258,2454-2460
Elder, G. H., Sheppard, D. M., DeSalamanca, R. E., and Olmos, A. (1980) Clin. Sci. (Lond. ) 58, 477-484
Kushner, J. P., Barbuto, A. J., and Lee, G. R. (1976) J. Clin. Invest. 299, 274-278
Romiio, P. H., Dubart, A., Grandchamp, B., de Verneuil, H., Rosa, J., Nordmann, Y., and Goossens, M. (1984) Proc. Natl. Acad. Sci. U. S. A. 81, 3346-3350
Anderson, L. C., Jokinen, M., and Gahmberg, C. G. (1979) Nature
Martin, P., and Papayannopoulou, T. (1982) Science 216,1233-
Collins, S. J., Gallo, R. C., and Gallagher, R. E. (1977) Nature
Holmes, D. S., and Quigley, M. (1981) Anal. Biochem. 114, 193-
Zasloff, A., Ginder, G. D., and Felsenfeld, G. (1978) Nucleic Acids
Durnam, D. M., and Palmiter, R. D. (1983) Anal. Biochem. 131,
Itoh, N., Nose, K., and Okamoto, H. (1979) Eur. J. Biochem. 97,
Ares, M. J., and Howell, S. H. (1982) Proc. Natl. Acad. Sci. U. S.
, , " I . .
278,364-365
1235
270,347-349
197
Res. 5, 1139-1151
385-393
1-9
A. 79,5577-5581
17. Aviv, H., and Leder, P. (1972) Proc. Natl. Acad. Sci. U. S. A. 69,
18. BxGlet, P., and Roskam, W. G. (1983) Gene (Amst.) 22,59-68 19. Wickens, M. P., Buell, G. N., and Schimke, R. T. (1978) J. Biol.
20. Land, H., Grez, M., Hauser, H., Lindenmaier, W., and Schutz,
21. Michelson, A. M., and Orkin, S. H. (1982) J. Biol. Chem. 257,
22. Hanahan, D. (1983) J. Mol. Biol. 166,557-580 23. Hanahan, D., and Meselson, M. (1980) Gene (Amst.) 10,63-67 24. Grunstein, M., and Hogness, D. S. (1975) Proc. Natl. Acad. Sci.
25. Thayer, R. E. (1979) A d . Biochem. 98,60-63 26. Goossens, M., and Kan, Y. W. (1980) Methods Enzymol. 76,805-
27. Hoffman, L. M., Fritsch, M. K., and Gorski, J. (1981) J. Biol.
28. Thomas, P. S. (1980) Proc. Natl. Acad. Sci. U. S. A. 77, 5201-
29. Weber, J., Jelinek, W., and Darnell, J. E., Jr. (1977) Cell 10,
30. Vaulont, S., Munnich, A., Marie, J., Reach, G., Pichard, A. L., Simon, A. P., Besmond, C., Barbry, P., and Kahn, A. (1984) Biochem. Biophys. Res. Commun. 125,135-141
31. Maxam, A. M., and Gilbert, W. (1980) Methods Enzymol. 65,
32. Straka, J. G., and Kushner, J. P. (1983) Biochemistry 22, 4664- 4672
33. Spikes, J. D., Burnham, F. B., and Bommer, J. C. (1984) in Porphyrins in Tumor Phototherapy (Andreoni, A., and CU- beddu, R., eds) pp. 61-65, Plenum Press, New York
34. Laernmli, U. K. (1970) Nature 227,680-685 35. Morrissey, J. H. (1981) A d . Biochem. 117,307-310 36. Straka, J. G., Kushner, J. P., and Pryor, M. A. (1982) Enzyme
(Basel) 28,170-185 37. Kasper, C. B. (1975) in Molecular Biochemistry and Biophysics
(Needleman, S. B., ed) pp. 114-124, Springer-Verlag, New York 38. Winge, D. R., Nielson, K. B., Zeikus, R. D., and Gray, W. R.
(1984) J. BWL Chern. 259, 11419-11425 39. Tsunasawa, S., Stewart, J. W., and Sherman, F. (1985) J. Biol.
Chem. 260,5382-5391 40. Kozak, M. (1984) Nucleic Acids Res 12,857-872 41. de Verneuil, H., Beaumont, C., Deybach, J. C., Nordman, Y.,
619 Sfar, Z., and Katally, R. (1984) Am. J. Hum. Genet. 36, 613-
42. de Verneuil, H., Grandchamp, B., Romeo, P. H., U c h , N., Beaumont, C., Goossens, M., Nicolas, H., and Nordman, y. (1986) J. Clin. Invest. 77,431-435
43. Groudine, M., and Casimir, C. (1984) Nucleic Acids Res. 12,
44. Cleveland, D. W., and Havercroft, J. C. (1983) J. Cell. Physiol.
1408-1412
Chem. 253,2483-2495
G. (1981) Nucleic Acids Res. 9,2251-2266
14773-14782
U. S. A. 72,3961-3966
807
Chem. 256,2597-2600
5205
611-616
499-560
1427-1446
97,919-924





























