Embed Size (px)
Citation preview

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 1/52
---~- ---~
NGEE ANN POLYTECHNIC
Molecular Biology
Laboratory Manual
Student Name: _
Student Number: _
Module Group: _
ATBMS Ver 1.1 (Apr 06)

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 2/52

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 3/52
Practical Schedule for Molecular Biology:
Week PRACTICAL Page
12 Expt la: Isolation of bacterial chromosomal DNA 10
Expt Ib: Analysis of DNA by agarose gel electrophoresis 13
13 Expt 2: SDS-PAGE analysis of proteins 17
14 Expt 3: Transformation of E. coli competent cells using 21
pGLO plasmid DNA
15 Expt 4a: Detection of Alu repeats in human cheek cells using 30
Polymerase Chain Reaction
Expt 4b: DNA fingerprinting of crime scene DNA 33
16
2

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 4/52
INTRODUCTION TO THE MOLECULAR BIOLOGY LAB
Welcome to" the molecular biology laboratory course. We will
cover some common techniques employed in molecular genetics
laboratories. You will notice that many of the techniques involve bacteria
(e.g. Escherichia coli); this reflects their importance in molecular biologytoday. The rapid growth rate of bacteria makes them suitable for practicalclasses. '
Some of: the experiments here are adaptations from research
experimental protocols that would normally take a longer time than we
have available for each practical session: so the YIeld,and purity of DNA
we make may be compromised. Do not be surprised when you start
working, in a lab to find a longer or different version of a particular
technique; there is no one correct version. '
What is expected in the laboratory and lab reports:
PLEASE read lab protocol of the week thoroughly before the
practical class. Do ask questions to help yourself (as well as others)
understand the protocol.
Lab reports (10 marks) have to be submitted on A4 paper with
clear headings (e.g. aim, materials &methods, results & discussion). In
your lab manual, there is a set of questions at the end of each practicalsession that must be answered in your lab reports. Always draw large,
clear, and fully labeled diagrams when required. Make sure graphs have
labeled X and Y axis (with units), and headings. Where you are asked to
record data, try and imagine you are writing it for someone like yourself
who has to repeat the experiment. Add any helpful tips and hints that you
think may be necessary. You may be required to work in groups or
individually. If you are working in a group, try to make sure you are
, involved and know what is going on. PLEASE do not copy observations
and answers from your friends/lab partners as this is plagiarism and
students involved will receive nomarks for the practical.
3

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 5/52

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 6/52
10. Never remove material or equipment from the laboratory without the
permission of an academic member or lab staff.
11. Pipetting by mouth is EXPRESSIVELY FORBIDDEN. Use teats,
syringes or pumps provided.
12. All manipulations should be performed aseptically, using plugged
pipettes. After use, contaminated pipettes must be immediately sterilized
by total immersion in a suitable disinfectant.
13. Contaminated glassware, discarded or contaminated petri dishes etc.
must be placed in the appropriate receptacle provided for autoclaving;
then washed or disposed off in an appropriate way.
14. All microscope slides must be disposed off in the receptacles provided -
they contain sterilizing fluid.
15. Report all breakages to the academic member or lab staff-in-charge
immediately.
16. Report all personal accidents, including minor cuts and abrasions,spillages of culture fluid and reagents to the academic member or lab
staff-in-charge.
17. Before leaving workbench, swab it down with an appropriate
disinfectant fluid.
18. Before leaving the laboratory WASH YOUR HANDS with a suitable
germicidal soap and dry them with paper towels.
General suggestions for a safe and useful laboratory session.
1. Before each session read through the exercise. Try and understand the
principles and methods. This will decrease the likelihood of an accident
and increase the likelihood of a successful experiment. If any part of the
procedure is not clear, you must ask the academic member or lab staff-
in-charge.
2. Label clearly all materials, chemicals, media, culture. This is important
to avoid confusion, improper use or disposal.
5

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 7/52
STANDARD OPERATING PROCEDURE
Written by: Eric Tan Updated on 2 May 2006
Title: Standard Operation Procedure for use of Ethidium Bromide in
the Laboratories
1.1 This SOP outlines the correct procedure and safety precautions
required for the use of ethidium bromide in the laboratories.
1.0 INTRODUCTION:
1.2 It also outlines the de-contamination method and containment
procedure in times of spillage.
1.3 For further information with regards to the safety and chemical
properties of this chemical, please refer to the Material Safety
Data Sheet available on the LSCT Cabinet or Eric Tan.
2.0 PROCEDURE FOR CASTING OF AGAROSE GEL:
2.1 Wear safety goggles .:
2.2 Place all gel casting apparatus in a fumehood with benchkote .. 2.3 Set up the casting tray.
2.4 Place the comb about 1.5cm from the top end.
2.5 Weigh the appropriate amount of agarose powder into a glass
conical flask specially designated for use in casting of
agarose gel only.
2.6 Add the correct volume of 1x TBE buffer into the conical flask
and microwave until the agarose dissolves.
2.7 Cool to 60° C and add ethidium bromide (0.5flg/ml final
concentration). (NB: Wear gloves when handling ethidiumbromide).
2.8· Carefully pour agarose solution (with added ethidium
bromide) into the casting tray. The gel should cover about half
the height of the comb teeth. Make sure that there are no
bubbles.
2.9 After the agarose solidifies, carefully remove the comb and
tape.
6

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 8/52
2.10 Place the casting tray into the electrophoresis tank on a
benchkoted area specially assigned in the lab for gelelectrophoresis.
2.11 Add Ix TBE buffer into the electrophoresis tank
2.12 Switch on the power-pack and start the electrophoresis.
3.0 SAFETYPRECAUTIONS:
3.1 All used TBE buffer must be disposed into a specially
designated bin containing a CLP de-staining bag, in the
laboratory.
NB: Each bag will remove 99% of the dye from 2 liters of
a O.5p:g/mlsolutionafter overnight incubation.
After use, the destaining bag can be safely disposed off as
a chemical waste, usually destroyed by incineration by a
licensed hazardous waste collector, while the cleaned
solution is safe for normal disposal.
Please check the solution to ensure no fluorescence
using a handheld UV lamp before disposal.
3.2 All used agarose gels and tips are to be disposed into anotherdesignated bin labelled "For Solid Waste containing Ethidium
Bromide". When full, the bin will be collected by a licensed
hazardous waste collector for treatment and safe disposal.
(Reference: Technical Updatefor Continental Lab Products-
Ethidium Bromide Disposal, January 2002). .
3.3 Gloves must be worn in handling ethidiumbromide and agarose
gels containing ethidium bromide.
3.4 Casting and electrophoresis of agarose gels must be carried out
at a designated, benchkoted area specially for such procedures.
3.5 Ensure that the ethidium bromide is contained in a screw-
capped vial and open the cap slowlyand carefully.
7

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 9/52
3.6 Safety goggles must be worn.
3.7 Please use the fumehood for casting of agarose gels.
3.8 Only use the conical flasks specially designated for casting of
agarose gels.
3.9 Use of gel doc system: .
a) Remove gloves when using the keyboard/mouse and when
switching on/off the CPU.
b) Place the gel onto a gel handling sheet that is on the UVtransilluminator. .
3.10 Check with TSO if in doubt.
3.11 Follow the procedure below for spillage containment and de-
contamination.
i) Use eLP de-staining bags, orii) Use the method adapted from Quillardet and Hofnung
4.0 SPILLAGE CONTAINMENT AND DE-CONTAMINATION:
4.1 . In the event of any spillage of ethidium bromide, please use
one of the following de-contamination method:
4.2 CLP de-staining bag method:
i) Wearing gloves, soak up the spillage using a eLP de-
staining bag.
ii) Using a pair of tongs, remove the bag and place it into a
biohazard bin labeled "For used eLP de-staining bag ".
4.3 Quillardet and Hofnung method:
i) If necessary, add water to reduce the concentration of
ethidium bromide to less than O.Slng/ml.
ii) Add 1 volume of 0.05 M KMn04 and mix carefully
iii) Add 1volume of 0.25 N HCL and mix carefully
8

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 10/52
iv) Let the solution stand at room temperature for several
hours
.v) Add 1 volume 0.25N NaOH and mix carefully
vi) The de-contaminated area can then be wiped dry with
tissue paper.
vii) The tissue paper can be discarded into a regular trash.
4.4 If there is spillage onto a lab-coat, remove lab-coat
immediately, The lab-coat can be de-contaminated using the
Quillardet and Hofhung method (4.3i to v) before washing
thoroughly with detergent and plenty of water.
5.0 FIRST-AID MEASURES (recommended by Phannacia Biotech
MSDS)
5.1 Inhalation:
Remove from exposure. If discomfort persists, obtain medical
attention .
. 5.2 Eyes:
Wash thoroughly with water for 15 minutes. If discomfort
persists, obtain medical attention.
5.3 Skin:
Wash off thoroughly with water. If discomfort persists, seek
medical attention.
5.4 Ingestion:
Wash out mouth with water and if conscious give water to
drink. Seek medical attention.
Checked & Endorsed By:
Dr Sara Zaman
DrHedyGoh
Mr Chang Y.C. (Chairman, LSCT Safety Committee)
Dr Julia Gandhi
Mdm Huang Yan
9

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 11/52
EXPERIMENT 1A:ISOLATION OF BACTERIAL CHROMOSOMAL DNA
Introduction
Bacterial cells like Escherichia coli and Pseudomonas aeruginosa have
single chromosomes. In order to isolate the chromosomal DNA, the bacterial cell
membrane must be broken into. This can be achieved by using sodium dodecyl
sulfate (SDS, a detergent that ruptures the cell membrane). Proteins are
removed using phenol and chloroform and the chromosomal DNA is precipitated
using ethanol. The precipitated DNA is then dissolved in a buffer. DNA can be
degraded by enzymes known as nucleases secreted by bacteria and other
organisms. Therefore, it is, necessary (when working with DNA, and more so
when working with RNA) to use sterile solutions and techniques.
In this exercise, you will isolate chromosomal DNA from E. coli cells. RNA is
also present in the E. coli cells which can be degraded using the enzyme RNase
after cell lysis. However, due to time coristraints,RNA will not be removed for this
practical.
Materials
Overnight culture of E. coli
TE buffer (10mM Tris, 10mM EDTA, pH 8.0)
TES buffer (10mM Tris, 10mM EDTA, 2% SDS" pH 8.0)
Phenol:Chloroform (3:,1)saturated wi,th0.1M Tris-HCI pl+ 7.5 'Chloroform
4M Sodium Acetate pH 6.0
Ice cold absolute ethanol
Micropipettes, sterile yellow and bluetips
2 ml microcentrifuge tubes, microcentrifuge
68°C water bath
Method
1. Spin down 2 ml of E . coli cells in a 2 ml microcentrifuge tube at 10,000 rpm
for 2 min.
2. Using a micropipette, discard the supernatant.
3. Resuspend the E . coli pellet in 1 ml TE buffer.
4. Spin again (10,000 rpm, 2 min), and discard the supernatant.
5. Resuspend the pellet in 50 ) . 1 1 of TE buffer.
6. Add 450 ) . 1 1 of TES buffer to the resuspened E. coli pellet.
7. Incubate at 68°C Tor5 min.
10

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 12/52
8. Extract protein contaminants by adding 1 ml of phenol.chloroform (3:1).Vortex well and centrifuge at 14,000 rpm for 3 min. Remove upper aqueouslayer to a new microcentrifuge tube.
CAUTION: Phenol is very corrosive. Always wear gloves when handling
9. To the aqueous layer from step 7, add 1 ml of chloroform. Vortex well andcentrifuge at 14,000 rpm for 3 min. Remove upper aqueous layer to a fresh
microcentrifuge tube.
10. Add iii O th volume 4M Sodium Acetate pH 6.0 (i.e. 50 1 - 1 - 1 ) and 2 volumes of
absolute ethanol (i.e. 1 ml).
11. Gently invert the tube a few times to mix contents gently. Thin, white fibres
of DNA should appear.
12. Spin at 14,000 rpm for 2 min to pellet chromosomal DNA.
13. RemoVe as much alcohol as possible with a micropipettor and dry the DNA
pellet. .14. Redissolve DNA in 50 III of TE buffer. DO NOT VORTEX as DNA may
shear.
The DNA prepared may contain some RNA which can be removed using
RNase. However, for this practical, the RNA will not be removed.
15. Take 10 III of your DNA sample and add to it 990 III of sterile water. Transfer
your 1 ml sample to a quartz cuvette and measure the absorbance at260nm and 280nm respectively. 0
The following facts will enable you to quantify and indicate the quality of
your sample:
i) double-stranded DNA at 50 I lglml has an 0026000f1.
ii) pure DNA will have an 00260/280of between 1.65 and 1.85; lower ratiosindicate protein contamination and higher ratios indicate RNA
contamination (pure RNA has a ratio of 2.0).
iii) Glass and some plastics are not UV transparent (260nm and 280nm
are in the UV range) so quartz cuvettes are always used.
16. Add 20 III of loading buffer to your undiluted E. col i chromosomal DNA
preparation. Load 30 ilion a 1% agarose gel (see Experiment 1B, pg 12).
o 11

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 13/52
EXPERIMENT 1b
ANALYSIS OF DNA BY AGAROSE GEL ELECTROPHORESIS
Introduction
Agarose gel electrophoresis allows for the separation of different-
sized fragments of DNA by causing them to migrate under the influence of
an electric field. The DNA sample is mixed with a DNA loading buffer,
which contains a dye (such as bromophenol blue) and glycerol. The glycerol
gives the DNA sample added density allowing it to sink to the bottom of the
well during loading, while the dye provides a visual indication of the extent
of DNA migration. The agarose gel is totally immersed in a tank of buffer
(e.g. TBE) which is capable of conducting current. DNA is negatively
charged due to its phosphate groups, so DNA moves from negativelycharged pole (cathode) to positively charged pole (anode). Ethidium
bromide (which is usually incorporated into the agarose gel) is a fluorescent
dye that binds to the DNA molecule by intercalating between the base-pairs.
Thus, after electrophoresis, DNA bands can be visualized by placing the gel
on a transilluminator which emits UV light. DNA intercalated with ethidium
bromide will appear as orange bands.
The mobility of nucleic acids in agarose gels is influenced by:
i) agarose concentrationii) molecular size of the DNA
iii) molecular shape of the DNA
The key to separation is based on the matrix (pores) of the gel which
restricts migration of a larger molecule more than it restricts a smaller
molecule.: Ingeneral, the lower the agarose concentration in the gels, the
larger the DNA size that can be analyzed. For most routine analysis of
restriction fragments, agarose concentrations in the range of 0.7% to 1% are
most appropriate.
Nucleic acids migrate in an agarose medium at a rate that is inversely
proportional to their molecular weights (ie. the shorter the DNA fragment,
the faster it migrates through the gel). In fact, there is linear relationship
between DNA mobility and DNA molecular weight. A standard curve is
prepared by analyzing a restriction enzyme digest containing DNA
fragments of known molecular weight (ie. a marker), This is used as a
reference for estimating the molecular weights and sizes of DNA fragments
of interest.
·13

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 14/52
14

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 15/52
B) Loading of DNA san1ple onto agarose gel
1) Typically, 20 ).-Llof DNA is loaded into an individual "well" on an
agarose gel using a micropipettor. For example, 15 ul of reaction mixfrom a restriction enzyme digest is mixed with 5 ul of DNA loading
buffer. Remember to also load uncut DNA as a control.
2) Load the DNA molecular weight marker (1 kb DNA ladder or A HindIII)
into a sample well.
3) When all the samples have been loaded, connect the electrophoresis tank
up to the power supply. The black electrode is the negative pole and the
red electrode is the positive pole; since DNA is negatively charged it will
migrate to the red positive pole.
4) Run the gel at a constant voltage of 100V until the blue tracking dye hasreached the. bottom of the gel. [CAUTION: Do not touch the
electrophoresis tank or wires while it is running. Voltage may reach high
levels and electric shocks may be fataL] .
5) Remove the gel tray from electrophoresis tank USING GLOVES and
view it on an UV transilluminator. DNA fragments will appear as red-
orange fluorescent bands.
[CAUTION: Avoid looking into UV light. Wear a UV-absorbing face
shield or goggles when viewing the gel under UV light].
6) Take a photograph of the agarose gel.
Questions (cont'd from pg 12)
5. Analyze the photograph of your agarose gel and answer the following
questions:
a) What does your E. coli chromosomai DNA look like?b) What is the size of the fastest running band in each of the marker
lanes?
4. What is the net charge of DNA at pH 8.2?
16

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 16/52
Materials
Agarose (1.0% in IX TBE)
Ethidium bromide (10 mg/ml)DNA standards
DNA loading buffer (2.5 mg/ml bromophenol blue, 30% glycerol)
lOX TBE buffer (0.9M Tris, 25mM Na2EDTA, 0.9M Boric acid pH 8.2)
Horizontal electrophoresis equipment
Micro centrifuge
Polaroid camera and film
Transilluminator
Method
A) Casting of agarose gel
1. Tape both ends of the gel tray with masking tape. Place the comb about
1.5 em from the top end.
2. Prepare 1% slurry of agarose inIX TBE buffer.
3. Heat the slurry in the microwave oven until the agarose dissolves.
4. After the agarose solution has cooled to 60°C, add ethidium bromide
solution to give a final dye concentration of 0.5 ug/ml .
[CAUTION:. Ethidium bromide is a mutagen. Always wear gloves when
handling solutions or agarose gels containing ethidium bromide!]
5. Quickly pour the agarose solution into the gel tray (make sure there are
no bubbles).
6. Allow the gel to set for 30 min and carefully remove the comb and tape.
7. Place the gel into electrophoresis tank, containing TBE buffer.
8. Add IX TBE to electrophoresis tank to ensure gel is submerged in TBE.
15

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 17/52
EXPERIMENT 2
SDS-PAGE ANALYSIS OF PROTEINS
Introduction
Bacterial chromosomes specify several thousand polypeptides. A
common electrophoretic tec1mique called sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE) will be employed in this
exercise to separate and identify these bacterial proteins. This is an example
of discontinuous electrophoresis.
Polyacrylamide is made by cross-linking many acrylamide
monomers together, using bis-acrylamide, By varying the proportions ofacrylamide to bis-acrylamide, a variety of pore sizes can be created in the
resulting gel. These pores create a molecular sieving effect, which can be
used to separate proteins based on their molecular size, The SDS is an ionic
detergent, which readily. solubilizes proteins by binding to them. The
binding of SDS to a protein also overwhelms its native charge, giving the
different proteins equal negative-net charges. Thus, when subjected to SDS-
PAGE, these proteins are separated based solely on their molecular weights
and not their intrinsic charge. .
Proteins to be loaded on polyacrylamide gels must first be heated in
protein loading buffer to solubilize the proteins.
A protein gel is comprised of two parts:
a) the stacking gel (where proteins migrate without separation),
b) the resolving gel (where proteins separate according to their
molecular weight). The resolving gel is made with a higher pH than
the stacking gel.
In this practical, you are provided with 2 bacterial strains, E. coli andBacillus cereus. The following procedure will enable you to prepare total
cellular proteins from these cultures. The proteins will be separated by SDS-
polyacrylamide gel electrophoresis. Protein bands can be visualized after
staining with Coomassie Blue, followed by destaining in acetic
acid/methanol.
17
You will work in pairs, one student will process the E. coli sample
and the other student will process the B. cereus sample.

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 18/52
Materials
2X Protein loading buffer
0.062M Tris-HCl pH 6.810%glycerol
2%SDS
5% B-mercaptoethanol
0.00125% bromophenol blue
Bacterial Strains:
E . co l i
B. cereus
18

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 19/52
Method1. 50 m1 aliquots of nutrient broth have been inoculated with the above
strains and cultured overnight at 37°C with shaking.2. Transfer 1.5 ml of cells into microfuge tube and centrifuge cells at
13,000 rpm for 5min. Remove supernatant taking care not to lose the
cell pellet. .3. Resuspend cell pellet in 0.1 ml 2X protein loading buffer. Mix
thoroughly. . .
.4. Boil samples in a water bath for 5 min (to break open cells and denature
or "solubilize" the protein). .
5. Centrifuge in microfugefor 5 min at full speed (to pellet cell debris).
6. Transfer 10 ul of the supernatant (contains soluble protein) to a fresh
. tube and add 10 ul of 2X protein loading buffer to it.
7. Load 15ul aliquots onto a 3% stackingllO% resolving SDS:-·
polyacrylamide gel. Load 5 ul of protein molecular weight markers.
8.. Run gel at 100V until bromophenol blue marker reaches bottom of the
gel.
9. Disconnect power supply .. Remove one .of the glass plates from the gel .
and stain the gel with Coomassie blue for 30;.60min.
10. Pour Coomassie Blue into a beaker. Add about 200 ml of destain.· .
Destain overnight with several changes of destain. The background of ..
gel should be almost colourless. .-..11. Dry the gel for pennarient record.
NB: Ifpossible, load a protein marker for each set. .
19
8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 20/52
Questions
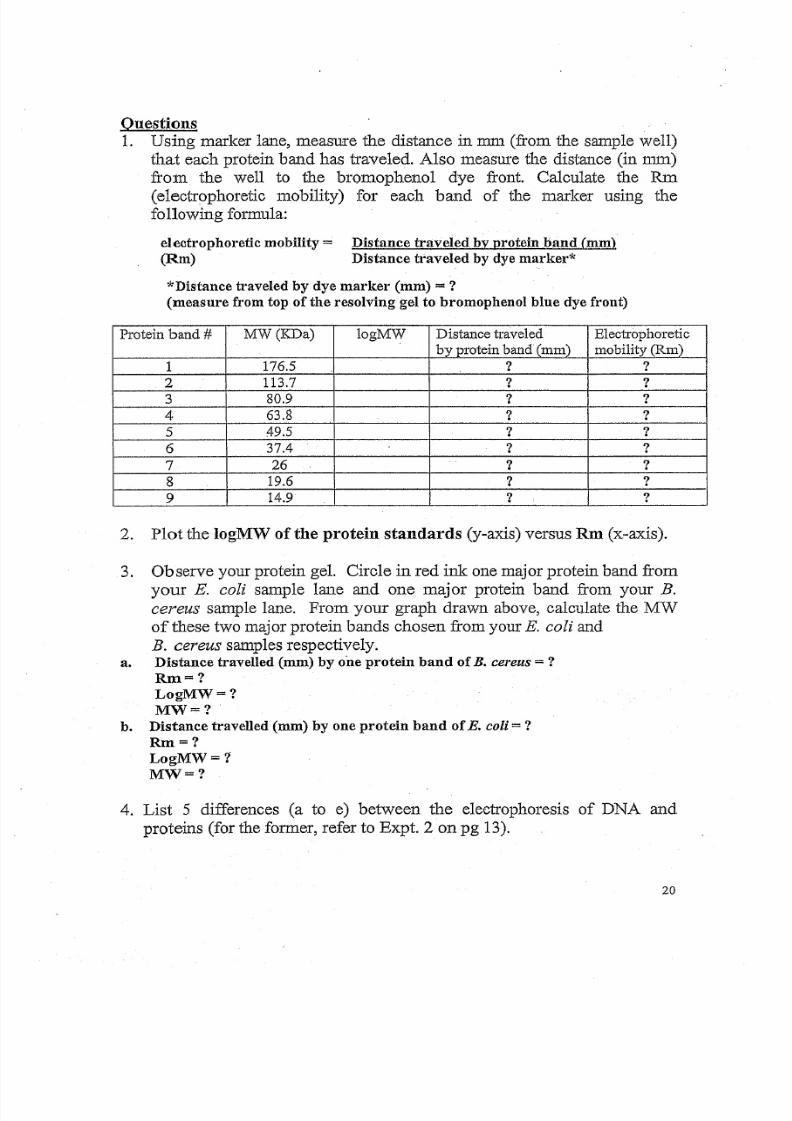
1. Using marker lane, measure the distance in mm (from the sample well)
that each protein band has traveled. Also measure the distance (in nun)
from the well to the bromophenol dye front. Calculate the RID
(electrophoretic mobility) for each band of the marker using the
following formula:
electrophoretic mobility =(Rm)
Distance traveled by protein band (mm)
Distance traveled by dye marker*
*Distance traveled by dye marker (mm) = ?
(measure from top of the resolving gel to bromophenol blue dye front)
P ro te in b and # MW(I<Da) logMW Distance traveled Electrophoretic
b yp ro te in b an d (mm) mobil i ty (Rm)
1 176.5 ? ?
2 113.7 ? ?
3 80.9 ? ?
4 63.8 ? ?
5 49.5 ? ?
6 37.4 ? ?
7 26 ? ?
8 19.6 ? ?
9 14.9 ? : ?
2. Plot the logMW of the protein standards (y-axis) versus Rm (x-axis).
3. Observe your protein gel. Circle in red ink one major protein band from
your E. coli sample lane and one major protein band from your B.
cereus sample lane. From your graph drawn above, calculate the MW
of these two major protein bands chosen from your E. coli and
B. cereus samples respectively.a. Distance travelled (mm) by one protein band of B. cereus = ?
Rm=?
LogMW=?
MW=?
b. Distance travelled (mm) by one protein band of E. col i = ?Rm=?
LogMW=?
MW=?
4. List 5 differences (a to e) between the electrophoresis of DNA and
proteins (for the former, refer to Expt. 2 on pg 13).
20

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 21/52
The pGLO System'
With the pGLO Transformation Kit, students use a simple procedure to transform bacte-
ria with a gene that codes for a Green Fluorescent Protein CGFe,),The real-life source of this
gene is the bioluminescent jellyfish Aequorea v i c tor i a, The gene codes for a Green Fluorescent
Protein which causes the jellyfish to fluoresce and glow in the dark. Following the transfor-
mation procedure, the bacteria express their newly acquired jellyfish gene and produce the flu-
crescent protein which causes them to glow a brilliant green color under ultraviolet light.
Inthis activity, students will learn about the process of moving genes from one organism
to another with the aid of a plasmid, In addition to one large chromosome, bacteria naturallycontain one or more small circular pieces of DNA called plasmids. Plasmid DNA usually
contains genes for one ormore traits that may be beneficial to bacterial survival . In nature: bac-
teria can transfer plasmids back and forth, allowing them to share these beneficial genes. This
natural mechanism allows bacteria to adapt to new environments. The recent occurrence of
bacterial resistance to antibiotics is due to the transmission of plasrnids.
Bio-.R.Cl.~'_:;.~~g.'::~.P " < ; ! ~ q p'~.~.~~_.~~~'?_~.~~~._~~~.~. fo.:_~~_g_r.I?!?_~,_F,!!-!:~~~~c_e.nt.~~.~~iIl
(Gr::P.2an~.~ _ ~ ~ E : ~ _or !esistance to_the~tibiotic, .~E£i l :~E::GLO also incorporates a spe-
cial gene regulation system which can be used to control expression of the fluorescent protein
in transformed cells. The gene for the Green Fluorescent Protein can be switched on in trans-
formed cells simply by adding the sugar arabinose to the cells nutrient medium. Selection for
cells that have been transformed with pGLO DNA is accomplished by growtli. on antibiotic
plates. Transformed cells will appear white (wild type phenotype) on plates not containing ara-binose, and fluorescent green when arabinose is included in the nutrient agar. The unique
construction of pGLO allows educators and students, for the very first time, to easily explore
mechanisms of gene regulation (Appendix D) and genetic selection. And, the entire process
is observable with an inexpensive long-wave UV lamp.
In order for your students to gain the most from this experiment they should know what
a gene is and understand the relationship between genes and proteins. For a more detailed
discussion of these and other basic molecular biology concepts and terms, refer to the review
provided in Appendix B.
. This pGLO Transformation Kit provides the opportunity for an additional experiment
involving purification of the recombinant fluorescent protein from transformed bacteria using
the GFP Purification Kit. (Bio-Rad catalog number l66-0005-EDU.)
EXPERIMENT 3TRANSFORMATION OF E. C<;lLI COMPETENT CELLS USING
pGLO PLAS,MID DNA
Introduction to Transformation
In this lab, your students will perform a procedure known as a genetic transformation.
Genetic transformation occurs when a cell takes up (takes inside) and expresses a new pieceof genetic material-DNA. This new genetic information often provides the organism with
a new trait which is identifiable after transformation ..Genetic transformation literally means
change caused by genes arid it involves the insertionofa genets) into an organism in orderto
change the organism 's traits. . 'J "
. Genetic transformation is used in many areas of biotechnology. In agriculture, genes cod-
ing for traits such as frost, pest, or drought resistance can be genetically transformed into
plants. ill bio-rernediation, bacteria can be genetically transformed with genes enabling them
to digest oil spills. In medicine, diseases caused by defective genes are beginning to be treat-
ed by gene therapy; that is, by genetically transforming a sick person's cells with healthy
copies of the gene involved in their disease.
Genes can be cut out of human, animal, or plant DNA and placed inside bacteria..For exam-
ple, a healthy human gene for the hormone insulin can be put into bacteria. Under the right con-ditions, these bacteria can make authentic human insulin. 'This insulin can then be used to treat
patients with the genetic disease, diabetes, whose insulin genes do not function normally.
21

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 22/52
Student Manual
pGlO Transformation,,'
Lesson 1 Introduction to Transformation
In this lab you will perform a procedure known as a genetic transformation. Remember
that a gene is a piece of DNA which provides the instructions for making (cod.ing for) a pro-
tein which gives an organism a particular trait. Genetic transformation literally means change
caused by genes and it involves the insertion of a gene(s) into an organism in order to change
the organism's trains). Genetic transformation is used in many areas of biotechnology. In
agriculture, genes coding for traits such as frost, pest, or spoilage resistance can be geneti-cally transformed into plants. In bio-remediation, bacteria can be genetically transformed
with genes enabling them to digest oil spills. In medicine, diseases caused by defective genes
are beginning to be treated by gene therapy; that is, by genetically transforming a sick person's
cells withhealthy copies of the gene involved in their disease.
You will use a procedure to transform bacteria with a gene that codes for a Green
Fluorescent Protein (OFP). The real-life source of this gene is the bioluminescent jellyfish
Aequorea victoria, The gene codes for a Green Fluorescent Protein which causes the jellyfish
to fluoresce and glow in the dark. Following the transformation procedure, the bacteria express
their newly acquired jellyfish gene and produce the fluorescent protein which causes them
to glow a brilliant green color under ultraviolet light.
In this activity, you wiIlleam about the process of moving genes from one organism to
another with the aid' of a plasmid, In addition to one large chromosome, bacteria naturally
contain one or more small circular pieces of DNA called plasmids. Plasmid DNA usually
contains genes for one or more traits that may be beneficial to bacterial survival. Innature,
bacteria can transfer plasmids back and forth allowing them to share these beneficial genes.
This natural mechanism allows bacteria to adapt to new environments. The recent occurrence
of bacterial resistance to antibiotics is due to the transmission of plasmids,
Bio-Rad's unique pOLO plasmid encodes the gene for the Green Fluorescent Protein
(OFP) and a gene for resistance to the antibiotic, ampicillin. pOLO also incorporates a spe-
cial gene regulation system which can be used to control expression of the fluorescent protein
in trans formed cells. The gene for the Green Fluorescent Protein can be switched on in trans-
formed cells by adding the sugar, arabinose, to the cells nutrient medium. Selection for cells
that have been transformed with pGLO DNA is accomplished by growth on antibiotic plates.
Transformed cells will appear white (wild type phenotype) on plates not containing arabi-
nose, and fluorescent green when arabinose is included in the nutrient agar.
You will be provided with the tools and a protocol for performing genetic transformation,
Your task will be:
1. To do the genetic transformation.
2, To determine the degree of success in your efforts to genetically alter an organism.
22

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 23/52
Consideration 3 The Genes
Genetic transformation involves the insertion of some new DNA into the E. coli cells.
In addition to one large chromosome, bacteria often contain one or more smaIl circular pieces
of DNA called plasrnids. Plasmid DNA usually contains genes for more than one trait.
Scientists can use a process called genetic engineering to insert genes coding for new traits into
a plasmid. Inthis case, the pGLO plasmid carries the gene (GFP) which produces the green
fluorescent protein and a gene (bla) that codes for a protein that. gives the bacteria resistance
to an antibiotic. The genetically engineered plasmid can-then be used to genetically trans-
form bacteria to give them this new trains). .. .
(l((ill('\ i·")'·:'C (S\:_j0r) C) (· ..: r (7:"\
I{' Cl( (~\bIn~;;:e 1$1)re.S0'll, I)
G f' P 1 : . . - • . ~ . t . il·\(--L,-," , 1 , . , \ 0:---" _~>- ..... _...... I ''I<.' . I I r
_ r I(.~::yaSC0J\- C1" 1 2 . rZII\ . J
I (' arYll ,.....' .."".' "c.. 0b~cZr)i.1: . . . ~ In._,.~~ .-", c..•, J
G·.f p. 'IS S:~-vH'C",~ 4'-:'>w i - . \ - r e - .
_'I ~ ) - lac·! (l;"(··\CI.~_~c..
C1 r' , F 'I c~II n
23

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 24/52
o oo
pGLO plasmid DNA
o 0
o o
o Beta lactamase
(antibiotic resIstance)Bacterial 0chromosomal
DNA·
o
Consideration 4 The Act of Transformation
This transformation procedure involves three main steps. These steps are intended to
in troduce the plasmid DNA into the E. coli cells and provide an environment for the cells to
express their newly acquired genes.
To move the plasmid DNA - pGLO through the cell membrane you will:
1. Use a transformation solution of CaC,," (calcium chloride)
2. Carry out a procedure referred to as heat shock
For transformed cells to grow in the presence of ampicillin you must:
3. Provide them with nutrients and a short incubation period to begin expressing their newlyacquired genes
24

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 25/52
Experimental Points
Practicing Techniques
Some educators like to dry run the procedures to explain sterile techniques, practice using
the pipettes and loops and streaking and spreading bacteria on the agar's surface. You willhave
to decide what is best for your students, based upon their lab experience and familiarity with,
these techniques. .
Transferring Bacterial Colonies from Agar Plates to Microtubes
The process of scraping a single colony off the starter plate leads to the temptation to get
more ceUsthan needed. A single colony 1nun in diameter contains' millions of bacterial cells ..
DNA Transfer
The transfer of plasmid DNA from it s stock tube to the transformation suspension is cru-
cial. Students must look carefuUy at the loop to see if there is a film of plasmid solution acrossthe ring. This is similar to seeing a soapy film across a wire ring for blowing soap bubbles.
Heat Shock
The procedure used to increase the bacterial uptake of foreign DNA is called heat shock.
It is important that students follow the directions regarding time. Also important is the rapid
temperature change and the duration of the heat shock. For optimal results, the tubes
containing the cell suspensions must be taken directly from ice, placed into the water bath
for 50 seconds and returned immediately to the ice. For example, the absence of the heat
shock will result in a 10 fold decrease in transformants while a 90 second heat shock wiIJ
give about half as many as 50 seconds. Either way the experiment will still work.
Spreading Transformants and Controls
Delivering more transformed culture to the plates with the disposable transfer pipette is
counter productive as the plates may not absorb the additional liqu id and spreading will be
uneven. Transferring bacterial suspensions from the microtubes to the Petri dishes requires
some care. The bacteria will settle to the bottom, so the students can hold the top of a closed
tube between the index finger and thumb of one hand and flick the bottom of the tube with the
index finger of the other hand. Be sure that students tap the tube with their finger or stir the
suspension with the pipette before drawing it up. Also, make sure that the students cover tile
Petri dishes with the li d immediately after pipetting in the transformation mixes and spread-
ing the cells.
Conceptual Points
Media
The liquid and solid media referred to as LB (named after Luria-Bertani) broth and agar
are made from an extract of yeast and an enzymatic digest of meat byproducts which pro- ,
vides a mixture of carbohydrates, amino acids, nucleotides, salts, and vitamins, aIJ of which
are nutrients for bacterial growth, Agar, which is derived from seaweed, melts when heated
and forms a solid gel when cooled (very analogous to Jell-O), and functions to provide a solid
support on which to culture bacteria.
25

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 26/52
Antibiotic Selection'
The paLO plasm id w hich contains th e G l 3 P gene alsq contains th e M . Q ~ J Q r . .. l :l ~ . t 9 , :, l i! . r; ; .w m g , s _ ~ ,
w hich provides resistance to th e antibiotic am picillin. T he beta-lactam ase protein is produced
a ~9 _~ ~~ ~t ::~ .I ?,~ l> ..Y Jl ac t~ !i ~h ic h c on ta in t he p !~ ~d , B e ta -Ia ct am a se i na ct iv at es t he ampi ci ll in
.~~nt in th e LB@ _gar, w hich allow s bacterial grow th . O nly transformed bacteria w hich con-
tain the plasm id, and express beta-lactam ase, can survive on th e plates w hich contain ampi-
cillin. O nly a very small percentage of th e cells take up the plasm id DNA and are transformed,
N on-transform ed cells can not grow o il t h e ampi ·c il J.l n' selection plates.
Transformation Solution
It is postulated th at th e C a2+ c ati on o f t he T ra nsfo rm ati on S ol uti on (50 mM C ae!." pH 7-.4)
neutralizes th e repulsive negative ch arges of th e phosphate backbone of the DNA and th e
pbosph olipids of the cell m embrane allow ing th e D NA to pass th rough the cell w all and enter
t he c el ls .
Heat Shock
The heat shock increases th e permeability of the cell membrane to DNA, and w hile the
mechanism is not know n, the time of the heat shock is critical and has been optim ized for the
cell line used and th e transform ation conditions em ployed,
Recovery
The 10 m inute incubation follow ing the addition of LB broth allow s th e cells to grow
and express th e am picillin resistance protein, beta-lactarnase, so th at th e transform ed cells
survive th e subsequent am picillin selection plates.
pGLO Gene Regulation
G ene expression in all organism s is carefully regulated to allow for adaptation to differ-
ing condi tions and to prevent w asteful overproduction of unneeded proteins. The genes
involved in the breakdow n of different food sources are good exam ples of h igh ly regulated
genes. For example the simple sugar arabinose is both a source of energy and a source of car-
bon for bacteria. Th e bacterial genes th at m ake digestive enzymes to breakdow n arabinose for
food are not expressed w hen arabinose is not in th e environm ent. B ut, w hen arabinose is pre-
sent, th ese genes are turned on. W hen th e arabinose runs out, the genes are turned off again.
A rabinose initiates transcription of th ese genes by prom oting the binding of RNA poly-
merase. In th e genetically engineered pO LO DNA, some of the genes in vol ved in the break-
down of arabinose have been replaced by th e jellyfish gene w hich codes for the G reen
Fluorescent Protein . ~~!!'Q ~£teria th at h ave been transform ed ;Hith pO LO DNA are growg.
in ilie presence of arabinose, th e O FP gene is turned on and the bacteria glow s brilliant greenw h en exp osed t o " U v lig~~."-·---·--..--.---.---------.--- .
Th is is an excellent example of th e central molecular framew ork of biology in action;
that is, DN A>R NA>PR OTEIN >TRA lT, W hen arabinose is absent from the grow t~_!TIcdia,
t h e GFP_g_~ne r ~. ~.~ !: !.~Y .: !0 .: ~9 .9 f.f ._ ~~.~~ .~010~_~~_~p .~~ .~ h .i te . A mo re d et ai le d d e sc ri pt io n
and analysis of gene regulation and the function of th e arabinose promoter can be found in
A ppendix A,
26
8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 27/52
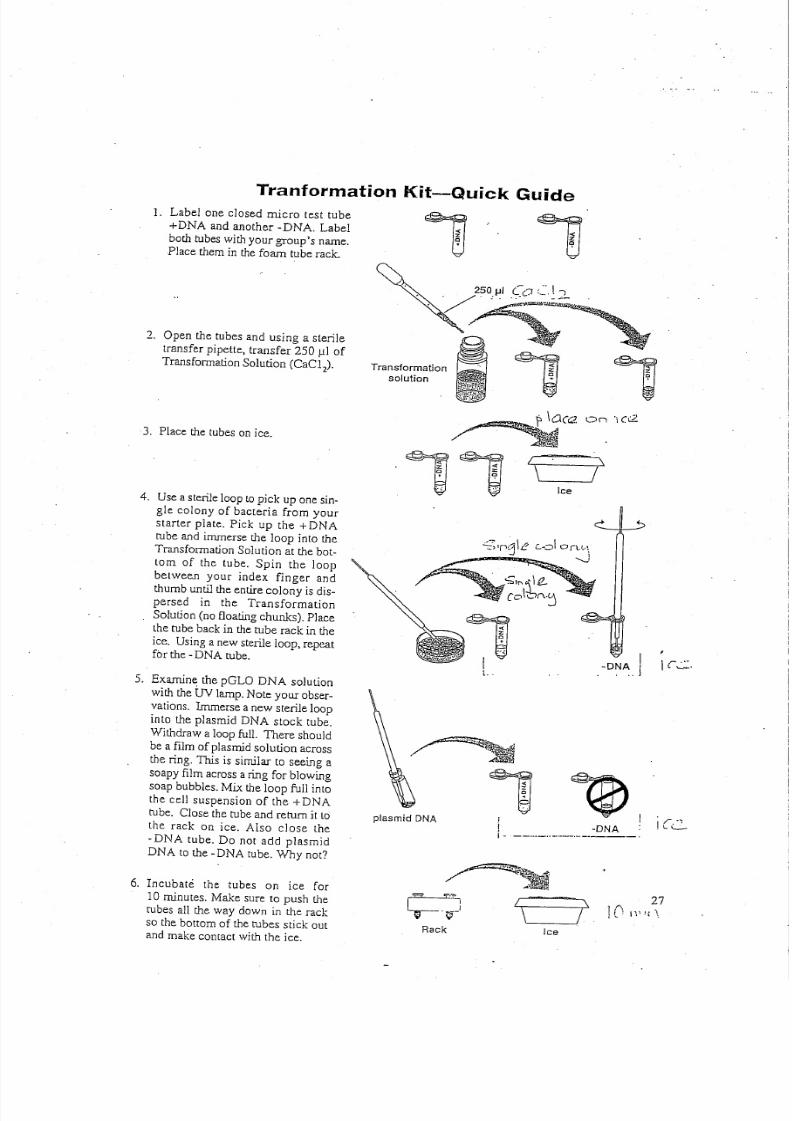
Tranformation Kit-Quick Guide
1. Label one closed m icro test tube
+DN A and anoth er -D NA . Label
b ot h tu be s w it h y ou r g ro up 's n am e.
Place th em in th e fo am tube rack .
2 . O pen th e tubes and using a steriletran sfer pipette, tran sfer 2 50 ul o f
T r an sf ormat ion So lu ti on (CaC 12),
3. P lace th e tubes o n ice.
4 . U se a sterile lo op to pick up o ne sin .
gle colony of bacteria from yourstarter plate. Pick up th e +DN A
tube and im merse th e loop into th e
T ra ns fo rm a tio n S ol ut io n a t t h e b ot -
tom of th e tube. Spin th e loop
betw een your index finger and
t humb u nt il t he e nt ir e co lony i s d is -
persed in th e Transform ation
So lu ti on ( no f lo a ti ng chunks) . P la ce
th e tube back in th e t ube rack in th e
ice . U sin g a n ew st erile loop , repeatfor th e . D NA tube.
5. Exam ine th e pGLO DNA solut ion
w ith t he UV lamp . N ote y our o bse r-v at io ns . Immer se a n ew s te ril e loop
into th e plasm id D NA stock tube.
W ith draw a lo op fu ll. T h er e sh ou ld
be a film o f p la sm id s olu tio n a cro ss
th e ring. T his is sim ilar to seein g a
s oa py f ilm a cr os s a r in g fo r b low in g
so ap b ub ble s. M ix t he lo op fu ll in to
th e cell suspension of the + DN A
tube. Close th e tube an d return it to
th e rack on ice. A lso close th e
·DN A tube. Do not add plasm id
DN A to th e -D NA tube. Why not?
6. Incubate the tubes on ice for
10 m inutes. M ake sure to push th e
rubes all th e w ay dow n in the rack
so th e bottom of th e tubes stick outan d m ak e co ntact w ith th e ice.
Transformation
solution ,On '\0:2.
./
Ice
O : = : " ' ; l r 1j\e c..d orv..
.. . _ ,
,zQ+.IL.
-DNA I...I
I r . . . , : : : .
plasmid DNA
,~,1 I
·DNAI . . -- . .. ..- . .. .. .. -- . ., -. ----.
Ice
8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 28/52
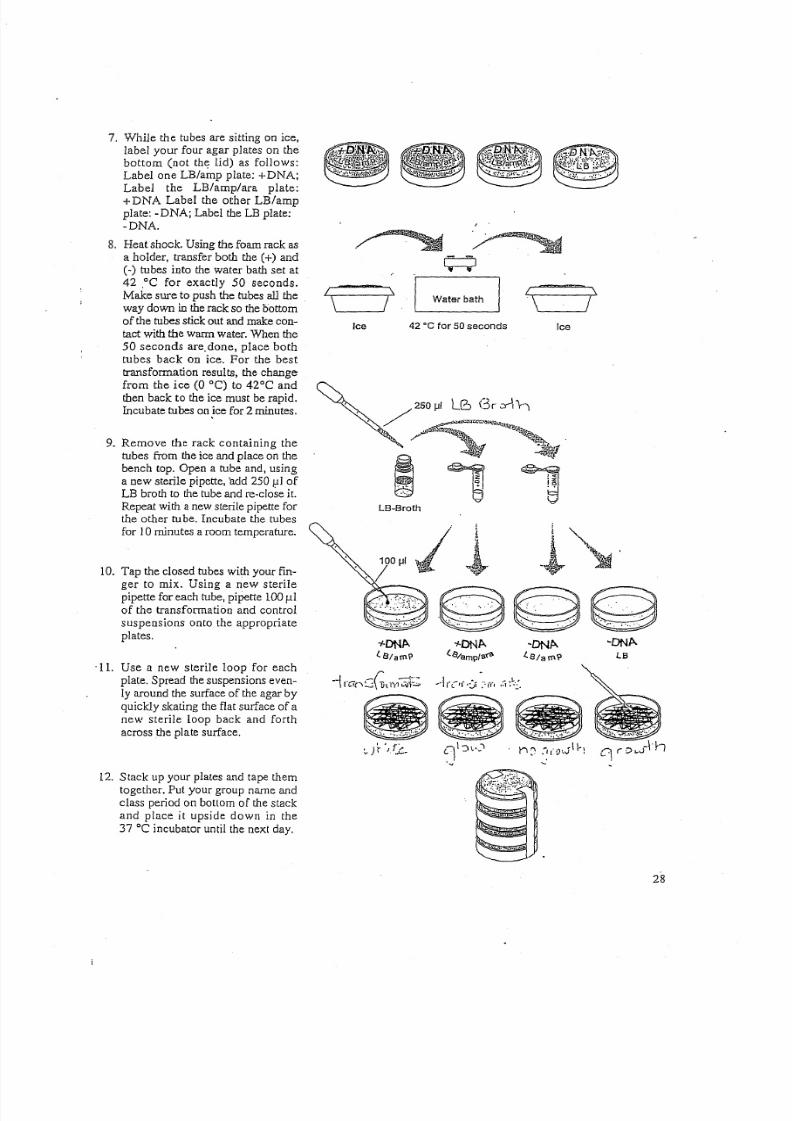
7. While the tubes are sitting on ice,
label your four agar plates on the
bottom (not the lid) as follows:
Label one LB/amp plate: +DNA;Label the LB/amp/ara plate:
+ DNA Label the other LB/amp
plate: -DNA; Label the LB plate:
-DNA.
8. Heat shock. Using the foam rack as
a holder, transfer both the (+) and
C - ) tubes into the water bath set at42 .oC for exactly 50 seconds.
Make sure to push the tubes all the
way down in the rack so the bottom
of the tubes stick out and make con-
tact with the warm water. When the
50 seconds are. done, place bothtubes back On ice. For the best
transformation results, the change
from the ice (0 DC) to 42°C and
then back to the ice must be rapid.
Incubate tubes onjce for 2 minutes.
9. Remove the rack containing the
tubes from the ice and place on the
bench top. Open a tube and, using
a new sterile pipette, 'add 250 1 : l 1 of
LB broth to the lube and re-close it.
Repeat with a new sterile pipette for
the other tube. Incubate the tubes
for 10minutes a room temperature.
10. Tap the closed tubes with your fin-
ger to mix. Using a new sterile
pipette for each tube, pipette 100 fl.1
of the transformation and control
suspensions onto the appropriate
plates.
'11. Use a new sterile loop for each
plate. Spread the suspensions even-
ly around the surface of the agar by
quickly skating the flat surface of anew sterile loop back and forth
across the plate surface.
12. Stack up your plates and tape them
together. Put your group name and
class period on bottom of the stack
and place it upside down in the
37°C incubator until the next day.
Water bathI \
\ ' - - - - - - 1 1Ice 42 ·cfor 50seconds Ice
;,"o. .. .,(.
LB·Broth
~~~
.~~~
~ D N A .
LB
~~~
~ . It · ',/.c- 0 ' ( : ) 1 . ' - ' " t ' l C ' :-~(O'JII'l
<
28

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 29/52
Lesson 3 Data Collection and Analysis
1. Observe and draw what you see on each of the four plates. Put your drawings in the data
table in the column on theright. Record your data to allow you to compare observations
of the "+ DNA" cells with those you record for the non-transformed E. coli. Write down
the following observations for each plate.
2. How much bacterial growth do you see on each, relatively speaking?
There should be multiple colonies on both the LB/amp and LB/amp/ara plates which
received the pGLO plasmid (-75-300 colonies). There should be no growth on the
LB/amp C o o ) DNA plate. There should be a lawn of bacteria on the LB (-) DNA plate.
3. What color are the bacteria?
The bacteria on the'{t) DNA LB/amp plate and the C - ) DNA LB plates should be
whitish in color. The bacteria on the C +) DNA LB/amp/ara plate should appear
whitish when exposed to normal, room lighting, but fluoresce green upon exposureto the UV light.
4. Count how many bacterial colonies there are on each plate (the spots you see).
There should be -75-300 bacterial colonies on the two C + ) DNA plates. The lawn of
bacteria on the LB plate contains an even spread of bacteria and individual colonies
can't be counted.
Plates Observations
+DNA, Lls/amp Many transformed colonies of bacteria (-75-300). Colonies
appear white.
+DNA,
LB/amp/ara
Many transformed colonies of bacteria (-75-300). Colonies
appear white when exposed to room light but fluoresce bright
green when exposed to UV light.
-D NA , LB /am p No bacterial growth present on this plate.
An even lawn of bacteria is present 011 this plate. The lawn
appears an off-white color.
-DNA, LB
_______ ....L • .. . _
29

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 30/52
EXPERIMENT 4a .
DETECTION OF ALU USING POLYMERASE CHAIN REACTION(Extracted from the DNA Learning Center, Cold Spring Harbour Laboratory)
In this experiment, polymerase chain reaction (PCR) is used to
amplify a nucleotide sequence from chromosome 8 to look for an insertion
of a short DNA sequence called Alu within the tissue plasminogen activator
(TPA) gene. Although the DNA from different individuals is more alike than
different, there are many regions of the human chromosomes that exhibit a
great deal of diversity. Such variable sequences are termed "polymorphic"
(meaning many forms) and provide the basis for genetic diagnosis, forensic
identification, and paternity testing.
The Alu family of short interspersed repeated DNA elements aredistributed throughout primate genome. Over the past 65 million years, the
Alu sequence has amplified via an RNA-mediated transposition process to a
copy number of about 500,000 - comprising an estimated 5% of the human
genome. Alu sequences are thought to be derived from the 7SL RNA gene
which encodes the RNA component of the signal recognition particle that
functions in protein synthesis. Alu elements are approximately 300 bp in
length and derive their name from a single recognition site for the
endonuclease Alu I located near the middle of the Alu sequence.
An estimated 500-2000 Alu elements are mostly restricted to the
human genome. A few of these have inserted recently, within the last one
million years, 'and are not fixed in the human species. One such element,
called TPA-25, is found within an intron of the tissue plasminogen activator
gene. This insertion is dimorphic, meaning that it is present in some
individuals and not in others. PCR can be used to screen individuals for the
presence (or absence) of the TPA-25 insertion.
In this experiment, oligonucleotide primers, flanking the insertion site,
are used to amplify a 400 bp fragment when TPA-25 is present and a 100 bpfragment when it is absent. Each of the three possible genotypes -
homozygotes for presence ofTPA-25 (400 bp fragment only), homozygous
for absence of TPA-25 (100 bp fragment only), and heterozygotes (400 bp
and 100 bp fragments) are distinguished following electrophoresis in agarose
gels.
The source of template DNA is a sample of several thousand cells
obtained by scraping with a sterile yellow tip. The cells are collected by
30

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 31/52
centrifugation and resuspended in a solution containing the resin "Chelex,"
which binds metal ions that inhibit the PCR reaction. The cells are lysed by
boiling and centrifuged to remove cell debris. A sample of the supernatantcontaining genomic DNA is mixed with Taq polymerase, oligonucleotide
primers, the four deoxynuc1eotides, and the cofactor magnesium chloride.
Temperature cycling is used to denature the target DNA, anneal the primers,
and extend a complementary DNA strand. The "upstream" primer -
(5' GTAAGAGTTCCGTAACAGGACAGCT 3') brackets on~ side of the
TPA locus, while the "downstream" primer -
(5' CCCCACCCTAGGAGAACTTCTCTTT 3') brackets the other side. The
size of the amplification product(s) depends on the presence or absence of
the Alu insertion at the TPA-25 locus on each copy of chromosome 8.
In order to co;rnpare the genotypes from a number of different
individuals, aliquots of the amplified sample and those of other
experimenters are loaded into wells of an agarose gel - along with the DNA
size markers and an unamplified control. Following electrophoresis and
staining, amplification products appear as distinct bands in the gel - the
distance moved from the well is inversely proportional to the presence or
absence of the TPA-25 insertion. One or two bands are visible in each lane,
indicating that an individual is either homozygous or heterozygous for the
Alu insertion.
31

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 32/52
Materials
10% Chelex beads
PCR reaction mix (from Promega) +25mM MgCb
PCR thermal cycler0.2 ml PCR tubes
1.5 ml Eppendorf tubes
Waterbath at 56°C & 100°C
100 bp DNA marker
1.5% agarose gel
Procedure
1. Use a sterile yellow tip to scrape some cheek cells from a volunteer.
2. Transfer cheek cells to 200 ul of 10% Chelex in an eppendorf tube, by
pippetting up and down using a micropipettor.3. Incubate sample tube in a 56°C water bath for 15 min, and then a 100
0ewaterbath for 6 min.
4. Spin tube for 5 min, 14;000 rpm"to pellet Chelex beads at bottom of tube.
5. During this 5 min, set up PCR reaction mix in a fresh eppendorf using:
PCR Master Mix
Upstream primer
Downstream primer
PCR reaction mix
6. Transfer supernatant (i.e. 15 J l I DNA sample from cheek cell) to 35 /-Llof
PCR reaction mix (avoid transferring any Chelex beads). The total
reaction volume should be 50 J . L 1 .
7. Amplify using the following program (30 cycles):
94°C for 3 min (initial denaturation step)
94°C for 1 min (denaturation step)
58°C for 1min (annealing step)
72°C for 1min (extension step)
72°C for 5 min (final extension step) and 4°C soak temperature
8. Electrophorese 20 J l l of the PCR reaction in a 1.5% agarose gel, together
with a 100 bp ladder.
Take a photograph of agarose gel and analyse your data.
32

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 33/52
EXPERIMENT 4bDNA FINGERPRINTING OF CRIME SCENE DNA
,"
I ,",,
Introduction
i
I i'jI:
i
! !
Technicians working in forensic labs are often asked to do DNA profiling or "finger-
printing" to analyze evidence in law enforcement cases and other applications. IThere are
two methods used to analyze the DNA: RFLp2 analysis and peR3 analysis. Itmay be irnpor-
tant for you to point out to your students that this laboratory exercise just models the RFLP
analysis technique. A step in this analysis requires the student (technician) to compare "band
patterns" produced by five DNA samples on a separation gel. The patterns are produced from
one sample of DNA taken at the crime scene and four obtained from suspectsin the case. We
will take this up again briefly.
Restriction Enzymes
Restriction enzymes sit on a DNA molecule and slide along the helix until they recognize
specific sequences of base pairs which signals the enzyme to stop sliding. The enzymes then
digest (chemically separate) the DNA molecule at that site-s-called a "restriction site"-act-
ing like molecular scissors, cutting DNA at a specific sequence of base pairs.
Ifa specific restriction site occurs in more than one location on. a DNA molecule, a restric-,
tion enzyme will make a cut at each of those sites, resulting in multiple fragments. Therefore,
if a given linear piece of DNA is cut with a restriction enzyme whose specific recognition
code is found at two different locations on the DNA molecule, the result will be three frag-
ments of different lengths. If the given piece of DNA is circular and is cut with a restriction
enzyme whose specific recognition code is found at two different locations on the DNA
molecule, the result will be two fragments of different lengths. The length of each fragment
will depend upon the location of restriction sites on the DNA molecule.
When restriction enzymes are used to cut a single strand of circular DNA, such as the
samples included in this kit, fragments of varying sizes are produced. DNA which has been
cut with restriction enzymes can 'be separated and observed using a process known asagarose
gel electrophoresis. The term electrophoresis means to carry with electricity.
Agarose Gel.Electrophoresis
Electrophoresis separates DNA fragments according to their relative size. DNA frag-
ments are loaded into an agarose gel slab, which is placed into a chamber filled with a con-
ductive liquid buffer solution. A direct current ispassed between wire electrodes at each endof the chamber. DNA fragments are negatively charged, and when placed in an electric field
will be drawn toward the positive pole and repelled by the negative pole. The matrix of the
agarose gel acts as a molecular sieve through which smaller DNA fragments can move more
easily than larger ones. Over a period of time smaller fragments will travel farther than larg-
er ones. Fragments of the same size stay together and migrate in single "b~ds" of DNA.
An analogy would be to equate this situation to your classroom in which all the desks
have been randomly pushed together. An individual student can wind his/her way through
the chair maze quickly and with little difficulty, whereas a string of four students would,
require more time and have difficulty working their way through the maze of chairs.
33 1 \
1

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 34/52
DNA Fingerprinting (RFLP analysis)
Each person h as sim ilarities and differences in th eir D NA sequences. T o sh ow th at a
p ie ce of DNA c on tain s a s pe cific n uc le otid e s eq ue nc e, a ra dio ac tiv e DNA p ro be c an b e m ad e
w h ic h w ill re co gn iz e a nd b in d th at se qu en ce . R ad io ac tiv e p ro be s allo w m ole cu lar b io lo gist s
t o l oc at e, id en ti fy , a nd c omp ar e t he DNA o f d iffe re nt in div id ua ls . T h is p ro be e an b e d es cr ib ed
as a "radioactive tag" th at w ill bind to a single stran,ded DNA fragm ent and produc e a band
ina gel or a band on a piece of nylon paper w hich is a replica of th e gel (also know n as a
S ou th ern b lo t). B ec au se o f it s spec ificity, th e radioactive probe c an be used to dem onstrate _
g en ot yp ic s im ila rit ie s b et w ee n in di vid ua ls . In DNA fin ge rp ri nt in g, t he r el at iv e p os it io ns o f
rad io lab ele d b an ds in a g el a re d ete rm in ed b y t he s iz e o f th e DNA fragme nts in e ac h b an d. T he '
siz e of t he fra gm en ts is re fle ct ed b y t he v ariat io ns in in div id uals' DNA.
W e a re ra pid ly g et tin g b eyo nd t he sc op e an d in te ntio n o f th is m an ua l. F or m ore de taile d
inform ation, w e recomm end a review of th e refe rences listed in A ppe ndix D .
T he e vid en ce n ee de d for DNA fin ge rp rin tin g c an b e o btain ed fro m a ny b io lo gic al m at e-
ria l th at c on tain s DNA: b od y tis su es, b od y fluid s (b lo od an d seme n), h air fo llic le s, e tc . T he
DNA analysis can even be done from drie d m aterial, such as blood stains or m umm ifie d tis-
sue. If a sam ple of D NA is too sm all it m ay be am plified using peR tec hniques. T he O NA is
th en tre at ed w ith re stric tio n e nz ym es th at c ut th e DNA in to fragme nt s o f v ario us le ng th ."
Restriction Digestion of DNA
B ec au se th ey c ut DNA, re stric tio n e nz ym es are t he " ch em ic al sc is so rs " o f t he m ole cu lar
biologist. W hen a particular r es tr ic tio n e nz ym e " re co gn iz es " a particular fo ur - or s ix -b ase
pair (bp) re cognitlon se quence on a segm ent of D NA , it cuts th e D NA m olecule at th at point.
T he re cognition sequence s for tw o comm only-use d enz ym es, Bam HI an d Hind III, are
sh ow n below . T he place on th e DNA backbones w he re th e D NA is ac tually cut is sh ow n w ith
a (~) symbol: "
For the enzyme: Hind III
G~~__.__~
CCTAG~
~
AlA: G C T~TTCGA
~
For the enzyme: Bam HI
34

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 35/52
Visualizing DNA Restriction Fragments
The DNA fragments in th e gel can't be seen be cause DNA is colorless, so a blue loading
dye is added to th e D NA solution. Th e loading dye does not stain the DNA . The dye front"m igrates" tow ard th e positive end of th e gel j ust ah ead of th e sm allest D NA fragm ents. T his
allo ws t he pro gre ss of t he DNA e le ct ro ph or es is ru n t o b e mo nit or ed .
Staining th e D NA pinpoints its location on th ~ gel. W hen th e gel is im me rsed in a d il ut e
solution of B io-Safe D NA stain, th e dye m olec ule s.attach to th e D NA m olecules trapped in
th e agarose gel. To enh ance contrast and to easily visualize th e D NA bands, excess back-
ground stain can be rem oved from the gel by destaining the gel w ith w ater. W hen the bands
are v is ible ,y our s tu de nts c an c om pare th e DNA re st ric tio n p atte rn s o f t he d iffe re nt sam ple s
of DNA .
T Ile gel below sh ow s th e DNA pattern th at w ill be obtained by your stude nts follow ing
electroph oresis. T he DNA from th e C rim e Scene h as be en labeled C S, th at from Suspec t # I,
Sl and so on. The DNA from the crim e scene is placed in lane 2; suspects' DN A is placed in
l an es 3 , 4,5, an d 6. Lane I contains H ind r u DNA s iz e s ta nd ar ds . [B y c on ve nt io n, t he la ne sare n um be re d fro m th e to p-Ie ft.) T he stu de nt's tas k is to lo ok at th e DNA b an din g' pat te rns an d
see if any of th e suspects bands m atch th ose of th e D NA found at th e crim e scene.~ ~ ,
> - 4 r I ~ . m : cs 51 S2 53 'Sit7' 1, 2 3 4 5 6
. . . : : . . : ~ : : . s . :
: : ' : ': ." .~: : ' : .'.
It's easy to see th at the D NA taken from the crim e scene and th e DNA [rom S3 is iden-
tical. Y ou m ay w ant to point out how "strong or w eak" th is evidence is in convicting a sus-
pe ct. T he DNA evidence m ay place th e suspec t at th e scene, but oth er evidence m ay be needed
to prove h im or h er guilty!5.6 F or exam ple, th e banding patterns of S2 and S3 m ay be sim ilar
e no ug h t o r eq uir e m o re c ar efu l 3 J1 8J ys is of a va ila ble e vi de nc e.
Y ou m ay point out [0 y ou r stu de nt s th at t his is a sim ulation . In ac tual DNA fing erp rint -
ing, tech nicians analyze m uch larger segm ents of D NA and m any m ore bands and Jane s are
produced. Th ese technicians are looking for a specific D NA segm ent, com mon to a given
po pu latio n, t hat w ill p rod uc e a un iq ue b nnd i n g p att ern for e ac h ind iv id ual org an ism .
Reliability of DNA Evidence
A majo r fa ct or a ffe ct in g t he r elia bili ty . o f DNA fi ng er pr in ti ng t ec h no lo gy 'i n f or en si cs i s
p op ulation ge ne tic s an d g e ne tic s tatis tic s. In h um ans th ere a re h un dre ds o f R FL P loc i o r DNA
s egm en ts t ha t c an b e s ele ct ed a nd u se d fo r fin ge rp rin ti ng a na ly si s. D e pe nd in g o n d emo gr ap hic
factors such as eth nicity or geograph ic isolation, som e segm ents w ill sh ow m ore variation
t h an o th e rs .
35
----------------

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 36/52
S om e p op ulatio ns sh ow m uc h less v aria tio n in p artic ular D NA segmen ts th an o th ers. T he
d eg re e o f v a riatio n w ill affec t th e statistic al o dd s o f m o re th an o ne in div id ual h av in g th e sam e
sequence. If 90% of a given population h as th e sam e frequency in its DN A fingerprintingpattern for a certain D NA segm ent, th en very little inform ation w ill be attained. B ut if th e
frequency of a D NA pattern turning up ina p op ul at io n f or a p art ic ul ar s egm en t i s e xtr em e ly
low , th en th is segm ent can serve as a pow erful tool to discrim inate betw een individuals in
th at p op ul at io n. D if fe re nt p op ul at io ns s how d iffe re nt p at te rn s i n t he ir g en ot yp es d ue to t he c on -
trib utio ns m ad e to th eir in div id ua l g en e p oo ls o ver tim e.
Th erefore, in analyzing h ow incrim inating th e D NA evidence is, one needs to ask th e
quest ion:
"S tatistically h ow m any people i.n a population m ay h ave th e sam e pattern as th at taken
from a crim e scene: 1 in 1,000,000? 1 in 1O ,000? Or" 1 in 10?"
References• "' •• ' 1
1. DNA Profil ing Fast BecomingAccepted. Tool For.Identificat1ob', 'Pamela Zurer, Chemical and.
Engineering News, Oct. 10, '1994.
2. RFLP means Restriction Fragment Length Polymorphisms ..."riff-l ips" in biotech jargon ...Pieces
of DNA are cut with restriction enzymes into fragments of various lengths. Individuals posses vari-
able restrict ion recognit ion sites 5. 0 that tw o pieces of DNA from separate sources may have different
fragment lengths when their DNA is cut by the same enzyme.
3. peR means Polymerase Chain Reaction: it is a technique used to amplify small am oun ts o f DNA
(in this case so that further analysis of the DNA can occur).
4. An excellent resource fer the classroom teacher is Genetic Fingerprinting, Pauline Lowrie and
Susan Wells, New Scientist, 16 November 1991.
5. Is DNA Fingerprinting ready fer the courts", Wm. C. Thompson and Simon Ford, New Scientist,
J [March 1990.
6. When Science Takes the Witness Stand, Peter Neufeld and Nevelle Coleman, Scientific Am erican ,
May 1990, Vol. 262:5.
36
8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 37/52
1)Lri
I \
\ 7ENZ Ice
l
Quick Guide for DNA Fingerprinting Kit
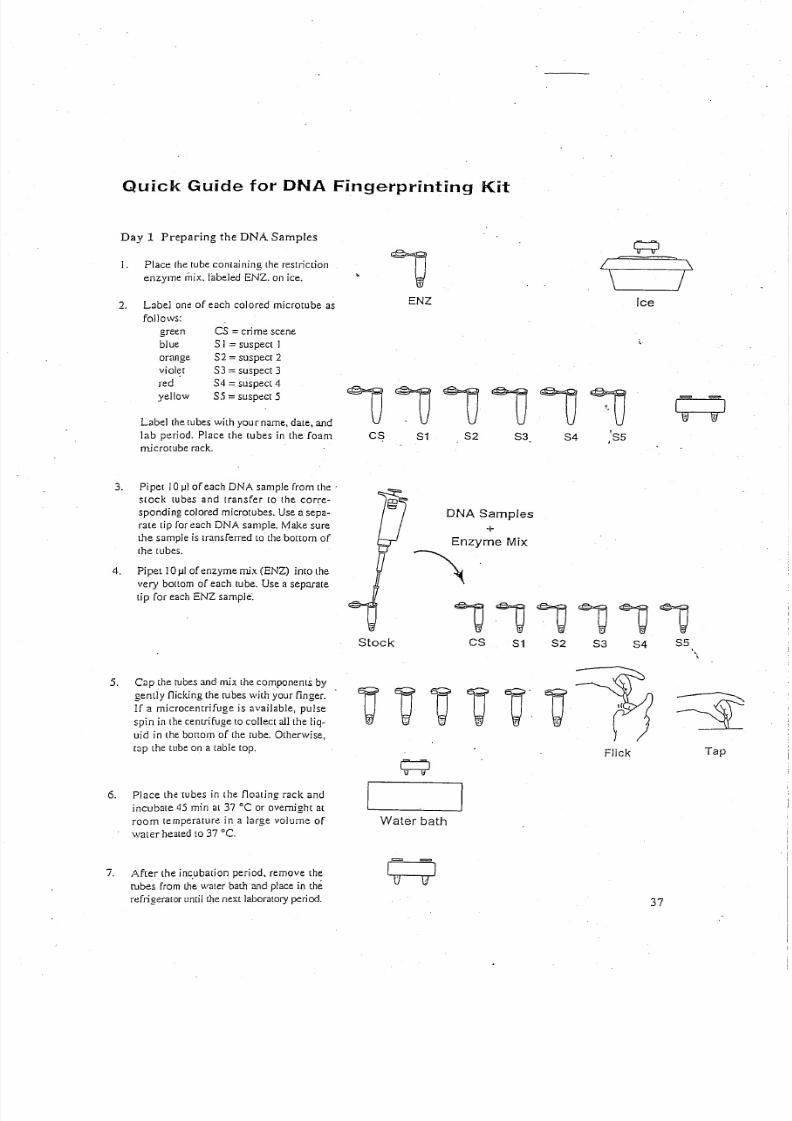
Day 1 Preparing the DNA Samples
I. Place the tube containing the restriction
enzym e m ix. labeled E N Z . on ice.
2. Label one of each colored rnicrotube as
follows:
green
blueorange
violet
red
yellow
CS = crim e scen e
S I = suspect IS 2 = suspect 2
S3 = suspect 3
S 4 = suspect 4
S5 =suspect 5
Label the tubes w ith you r nam e, date, and
lab period. Place the tubes in the foam
r ni cr ot ub e r ac k.
3. Pi pel 1 0 ) . 1 1 o f e ac h D N A sam ple from th e'
stock lubes and transfer to the corre-
s po nd in g c ol ore d rnicrotubes. U se a sepa-
rate lip for each D N A sam ple. M ake sure
the sample is transferred to t he b ot tom of
t he l ub es .
4 . Pipet 1 0 ) . 1 1 o f e nz yme mix ( E N Z ) into th e
very bot 10m of each tube. Use a separate
lip for each E N Z sample:
5. Cap the tubes and mix the components by
gently nicking the rubes w ith your finger.
If a microcenrrifuge is available, pulse
sp in in th e ce ntrifu ge [Q collect all th e liq-
uid in th e bortorn of the rube. O th erw ise,
rap the tube on a table lap.
6. Place the lubes in the floating rack and
incubate 45 min at 37°C or overnigh t at
room temperature in a luge volume of
w ater healed 1037 "C,
7. A fter the incubation period, remove the
rubes from Ih~ w ater bath and place in the
re fr ig er at or u nt il t he n ex t l ab or at or y p er io d.
111J111J'1JCS
Stock
S1 S2 ,'S53. S4
DNA Samples+
Enzyme Mix
~
v~~v,~~C8 84 S521 S3
Flick Tap
Water bath
37

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 38/52
D ay 2 G el Electroph oresis
I. Remove your digested DNA samples from the
refrigerator. If a centrifuge is available, pulse e r r i1 ' i1 n f - 1spin the tubes in the centrifuge to bring all of the . 1 . u J l d f J '§ ' t r Jliquid into the bottom of the tube.
Using a separate tip for each sample, add 5 j . llof
loading dye "LD" into each LU be. Cap th e tubes
and mix by gently nicking the tube w ith your
finger.
Place an agarose gel in the electrophoresis appa-
ratus. Fill the electroph oresis cham ber w ith I
TA B buffer to cover th e gel, using approxim ate-
ly 275 ml o f buf fe r.
Check [hat the w ells of the agarose gels are near
the black (- ) electrode and the base of the gel is
near th e red (+) electrode.
2.
3 .
Using a separate tip for each sample, load the
indicated volume of each sample into 7 w ells of
the gel in the follow ing order:
Lane 1: M, DNA size m arker, 10 j Jl
Lane 2: C S, green, 20 ]11
Lane 3: S I, blue, 20 ]11
Lane 4: S 2, o ra ng e, 20 jJl
Lane 5: S 3, v io let , 20 pi
Lane 6: S4, red, 20 jJl
Lane 7: S 5, y el low , 20 pi
Place the lid on (he electrophoresis chamber. Th e
lid w ili attach to the base i n o nl y o ne o ri en ta ti on .
Th e red and black jacks on the lid w ill matchw ith the red and black jacks on the base. Plug
th e electrodes into the.power supply.
\
. Turn on the power and electrophorese your
samples at 100 V fo r 3. 0 minutes.
When the electrophoresis is complete, turn off
the power and remove the top of the gel box.
Carefully remove the gel and tray from the gel
box. Be careful=-the gel is very slippery! Slide
th e gel into the staining tray.
. Add 60 ml of DNA stain [0 the tray. Cover the
tray w ith plastic w rap. Let the gel stain
overnigh t, w ith shaking for best results.
ay 3 Analysis of the Gel
Pour off the DNA stain into a bottle. Add 60 m l
or w ater to th e gel and let the gel destain IS min-
utes.
Pour off the w ater into a w aste beaker. Analyze
the results w ith the help of your teacher. 3.
Let the gel dry on gel support film or on y~ur lab
bench until completely dry. When the gel IS dry,
tape into your lab notebook for a permanent
record.
Centrifuge
P DNA Loadinq Dy'
~ \
~ V ~ I J V y
(+ )
o.
1.
38

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 39/52
Procedure for casting gels
Using Bio-Rad's Mirii Sub-CeIl® GT system, gels can be cast directly in the gel box by
using the casting gates with the gel tray. .
This section outlines the conventional tape-the-tray method for casting gels. Other meth-
ods are detailed in Bio-Rad's Sub-Cell GT instruction manual.
Step 1. Seal the ends of the gel tray securely with strips of standard laboratory tape. Press ..
the tape firmly to the edges of the gel tray to form a fluid-tight seal.
Step 2. Level the gel tray on a leveling table or workbench using the leveling bubble p r o -
vided with the instrument.
Step 3. Prepare the desired concentration and amount of agarose in lx TAB electrophore-
sis buffer.
Step 4. Cool the agarose to at least 60°C before pouring.
Step 5. While the agarose is cooling to 60°C, place the comb into the appropriate slot of the
gel tray. Gel Combs should be placed within 3/4 ofan inch of the end of the gel
casting tray (not in the middle of the gel).
Step 6.. Allow the gel to solidify at room temperature for 10 to 20 minutes-it will appear
cloudy, or opaque, when ready to use.
Step 7. Carefully remove the comb from the solidified gel.
Step 8. Remove the tape from the edges of the gel tray.
Step 9. Place the tray onto the leveled DNA electrophoresis cell so that the sample wells are
at the cathode (black) end of the base. DNA samples will migrate towards the anode
(red) end of the base during electrophoresis.
39

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 40/52
Lesson 4 Analyzing the DNA Patterns
Interpretation of Results
A ttac h a ph oto, X erox , or you r actu al drie d g el in th is s pace . Inc lic ate w h ic h sam ple is in
e ac h w e ll.
I. W hat are w e trying to determ ine? R e-state th e central question.
2. W hich of your DNA sam ples w ere fragmented? W hat w ould your gel look like if th e
DNA were not fragmented? "
3. W hat caused th e DN A to becom e fragm ented?
4 . W hat determ ines w here a restriction endonuclease w ill "cut" a D NA m olecule?
S. A r es tric tio n e nd on uc le as e " cu ts " two DNA m olecules at th e sam e location. W hat can
yo u assume is id en tical abou t th e m ole cule s at th at loc ation ?
6. D o any of your suspect sam ples appear to h ave BamH I or HilldIIT r ec og nit io n s it es a t t he
sam e location as th e DN A from th e crim e scene?
7. Based on th e above analysis, do any of the suspect sam ples of DN A seem to be from th e
sam e in divid ual as th e DNA fro m th e crim e sc ene ? D es cribe th e sc ien tific e vide nce th at
s up po ns you r con cl us io n.40

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 41/52
Appendix A
Alternative DNA Fingerprinting Scenarios (© 1996 Stanford University)
DNA typing, DNA profiling, and DNA fingerprinting are all names for the same pro-
cess, a process which uses DNA to show relatedness or identity of individual humans, plants,
or animals. DNA typing has become the subject of much debate and interest because of its uses
for forensics analysis in prominent criminal cases such' as' the O. J. Simpson case. The appli-
cations of DNA typing, however, are much broader than forensic science alone and are hav-
ing a profound impact on our society.
DNA typing is used in forensics, anthropology, and conservation biology not only to
determine the identity of individuals but also to determine relatedness. This process has been
used to free innocent suspects, reunite children with their relatives, identify stolen animals, and
prove that whale meat has been substituted for fish in sushi. It is used in times of war to help
identify the remains of soldiers killed in combat. It is also being used to find genetic linkages
to inherited diseases. In addition, scientists are learning a great deal about our evolutionary his-
tory from DNA analysis.
Each of the following paragraphs describes a scenario in which DNA has been used to
show how individuals are related to each other, or to show that a person is (or is not) the per-
petrator of a crime. These scenarios provide a context for using DNA typing for use in teach-
ing molecular biology, conservation biology, and biotechnology.
1. Food identification (endangered species identification).
The purity of ground beef (or irnpurityjhas been proven using DNA typing. Hamburger
has been 'shown to often be a mixture of pork, and other non-beef meats. Using portable
testing equipment, authorities have used DNA typing to determine that the fish served in
sushi was really meat from whales and dolphins. These are, many times, endangered
species that are protected by international law. (Angier, Natalie. "DNA Tests in Meat of
Endangered Whales for Sale in Japan." p. All, Sept. 13, 1994.)
2. Accused and convicted felons set free because of DNA typing.
A man imprisoned for 10 years was released when DNA testing, unavailable whim he
was convicted, was used to show that he could not have been the rapist. Statistics show
that about one-third of all sexual assault suspects are freed as a result of DNA test-
ing.("DNA Tests Free Man in Jail for Decade." New York Times, Oct. 22, 1994.)
3. Identifying of human remains.
Scientists have used DNA typing to confum that the body in the grave was (or was not)
the person that was supposed to be there. Bones found in Russia are believed to be those
of the Romanovs, Russia's last imperial family. Czar Nicholas ITand his family wereexecuted by the Bolsheviks in 1918. Experts from around the world have been studying
the bones to match skulls, teeth, and other features with photographs. DNA from the
bones will be compared to that of known descendants to determine whether the bones do
indeed belong to the Czar and his family. ("The Czar's Bones: Britons to Decide". New
York Times, Sept. 13, 1992.)

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 42/52
4. Determining relatedness of humans.
DNA typing has shown that the 5000 year old Ice Man found in a melting glacier is most
closely related to modern Europeans. ("Iceman Gets Real.tScience, Vol. 264:1669. 17
June 1994.) The DNA typing evidence also "removes all the suspicions that the body
was a fraud-that it had been placed on the ice" says Svante Paabo of the University of
, Mnnich, (Science, Vol'. 264:1775.17, June 1994).
5. Studying relatedness among ancient peoples.
DNA found at archeological sites in western Montana is being used to help determine
how many related groups of people (families) lived at a particular site. Morell, Virginia."PulIingHair from the Ground." (Science, Vol. 265:741-745 August 1994.)
6. DNA testing of families.
DNA testing of families has been used in Argentina and El Salvador to identify the chil-
dren of at least 9,000 citizens of these countries who disappeared between 1975 and 1983,
abducted by special units of the ruling military and police. Many of the children born to
the disappeared adults were kidnapped and adopted by military "parents" who claimed to
be their biological parents. After genetic testing of the extended family revealed the true
identity of a child, the child was placed in the home of its biological relatives. It was
feared that transferring a child from its military "parents" who were kidnappers, but who
had reared the child for years, would be agonizing. Inpractice, the transferred children
became integrated into their biological families with minimal trauma. (DNA Fingerprints:
I.M. Diamond "Abducted Orphans Identified by Grandpaternity Testing" 327:552-553.
1987).
7. Identifying organisms that cause disease.
Eva Harris, a UCSF scientist, is helping scientists in Nicaragua and Ecuador to learn to
use DNA technology to detect tuberculosis, and identify the dengue virus and various
strains of Leishmania. Other available tests cause waits of many weeks while disease
organisms are cultured and sent to foreign labs to be identified. (Marcia Barinaga, "A
Personal Technology Transfer Effort inDNA Diagnostics." Science, 266:1317-1318.
Nov. 25, 1994.)
8. Identifying birth parents (paternity testing).
Girls in Florida were discovered to have been switched at birth when one girl died of ahereditary disease. The disease was not in her family, but was known to be in the family
of another girl, born in the same hospital and about the same time she was born.
9. Proving paternity.
A woman, raped by her employer on Jan. 7, 1943, her l Sth birthday, became pregnant.
The child knew who her father was, but as long as he lived, he refused to admit being
her father. After the man died, DNA testing proved that she was his daughter and she
was granted a half of his estate. ("A Child of Rape Wins Award from Estate of her Father."
New York Times, July 10, 1994.)

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 43/52
r
10. Determining effectiveness of bone marrow transplants.
"DNA fingerprinting can help doctors to monitor bone marrow transplants. Leukemia isa cancer of the bone marrow and the diseased marro~ must be removed. The bone mar-
row makes new blood cells, so the leukemia sufferer will die without a transplant of
healthy marrow. Doctors can quickly tell whether the transplant has succeeded by DNA
typing of the patient and the donor. If the transplant has worked, a fingerprint from the
patient's blood shows the donor's bands. But if the cancerous bone marrow has not been
properly destroyed, then the cancerous cells multiply rapidly and the patient's own bands'
predominate." ("Our Ultimate Identity Card in Sickness and in Health," in "Inside
Science", New Scientist, Nov. 16, 1991.)
11. Proving relatedness of immigrants.
DNA fingerprinting has been used as proof of paternity for immigration purposes. In
1986, Britain's Home Office received 12,000 immigration applications from the wives and
children of Bangladeshi and Pakistani men residing in the United Kingdom. The burden
of proof is on the applicant, but establishing the family identity can be difficult because
of sketchy documentary evidence. Blood tests can also be inconclusive, but DNA f i n -
gerprinting results are accepted as proof of paternity by the Home Office. DNA finger-
prints (source unknown: Based on A. 1. Jeffreys, et al., "Positive Identification of an
Immigration Text-Case Using Human DNA Fingerprints." (Nature, 317:818-819,1985.)
12. Confirming relatedness among animals.
Scientists who extracted DNA from the hair of chimpanzees throughout Africa now have
evidence that there might be a third species of chimpanzee. At the same time, they and
have learned' things about chimp behavior and kinship patterns that would have' once
taken years to theorize. They discovered a group of chimps living in western Africa to be
genetically distinct from the chimps living in other parts of.Africa, suggesting that thegroup may be an endangered species. The have discovered that male chimps living in a
given area are often as closely 'related as half-brothers, and many so-called sub-species
may all be part of a single species. The male chimps' relatedness may explain why, unlike
other primates, the males are quite friendly to each other. ("Genetics Suggest Existence
of a Third Species of Chimp." The New York Times, National. Aug. 26, 1994, p. A9.)
13. DNA testing of plant material puts murderer at the scene.
Two small seed pods caught in the bed of his pick-up truck put an accused murderer at the
murder scene. Genetic testing showed that DNA in the seed pod exactly matched the
DNA of a plant found at the scene of the murder. The accused had admitted he had given
the victim a ride, but he denied ever having been near the crime scene. ("Genetic Finger
Printing, Seeds of Truth", Popular Science.)
I,
I

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 44/52
Appendix B Glossary of Terms
Agar Provides a solid matrix to support bacterial growth.Contains nutrient mixture of carbohydrates, amino acids,
nucleotides, salts, and vitamins.
Antibiotic Selection The plasmid used to move genes into bacteria also con-
tains the gene for beta-lactarnase which provides resis-
tance to the antibiotic ampicillin. The beta-Iactamase
protein is produced and secreted by bacteria which con-
tain the plasmid. The secreted beta-Iactamase inactivates
the ampicillin present in the LB/agar, which a l lows for
bacterial growth. Only bacteria which contain the plas-
mids, and express beta-lactamase can survive on the
plates which contain ampicillin. Only a very small per-
centage of the cells take up the plasmid DNA and aretransformed. Non-transformed cells, cells that do not
contain the plasmid, can not grow on the ampicillin
selection plates.
A carbohydrate, normally used as source of food by bac-
teria.
Arabinose
Beta-Lactamase Beta-lactamase is a protein which provides resistance to
the antibiotic ampicillin. The beta-lactarnase protein is
produced and secreted by bacteria which have been
transformed with a plasmid containing the gene for beta-
lactarnase. The secreted beta-lactamase inactivates the
ampicillin present in the LB/agar, which allows for bac-
terial growth and expression of newly acquired genes
also contained on the plasmid i.e. OFP.
Applying biology in the real world by the specific
manipulation of living organisms, especially at the
genetic level, to produce potentially beneficial products.
When a population of cells is prepared by growth from
a single cell, all the cells in the population will be genet-
ically identical. Such a populat ion is called clonal. The
process of creating a clonal population is called
"cloning". Identical copies of a specific DNA sequence,
or gene, can be accomplished following mitotic division
of a transformed host cell.
A clump of genetically identical bacterial cells growing
on an agar plate. Because all the cells in a single colony
are genetically identical they are called clones.
The liquid and solid media are referred to as LB (named
after Luria-Bertani) broth and agar are made from an
extract of yeast and an enzymatic digest of meat byprod-
ucts which provides a mixture of carbohydrates, amino
acids, nucleotides, salts, and vitamins, all of which are
nutrients for bacterial growth. Agar, which is from sea-
weed, polymerizes when heated to form a solid gel (very
analogous to Jell-O), and functions to provide a solidsupport on which to culture the bacteria.
Biotechnology
Cloning
Colony
Culture Media

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 45/52
Genetic Engineering
Gene Regulation
Green Fluorescent Protein
Plasmid
pGLO
Recombinant DNA Technology
Screening
Sterile Technique
Streaking
Vector
The manipulation of an organism's genetic material
(DNA) by introducing or eliminating specific genes.
Gene expression in all organisms is carefully regulated
to allow for differing conditions and to prevent wasteful
overproduction of unneeded proteins. The genes
invol ved in the transport and breakdo wn of food are
good examples of highly regulated genes. For example,
the simple sugar, arabinose, can be used as a source of
energy and carbon by bacteria. The bacterial enzymes
that are needed to breakdown or digest arabinose for
food are not expressed in the absence of arabinose but
are expressed when arabinose is present in the environ':
ment, In other words when arabinose is around the genes
for these digestive enzymes are turned on. When arabi-
nose runs out these genes are turned back off. See
Appendix D for a more detailed explanation of the role
that arabinose plays in the regulation and expression of
the green fluorescent protein gene.
Green Fluorescent Protein (GFP) was originally isolated
from the bioluminescent jellyfish, Aequorea victoria.
The gene for GFP has recently been cloned. The unique
three-dimensional conformation of GFP causes it to
resonate when exposed to ultraviolet and give off energy
in the form of visible green light,
A circular DNA molecule, capable of autonomous repli-
cation, carrying one or more genes for antibiotic resis-
tance proteins and a cloned foreign gene such as GFP.
Plasmid containing the GFP sequence and ampicillin
resistance gene' which codes for Beta-lactamase.
The process of cutting and recombining DNA fragments
as a means to isolate genes or to alter their structure and
funcLion.
Process of identifying wanted bacteria from a bacterial
library.
Minimizing the possibility of outside bacterial contam-
ination during an experiment through observance of
. cleanliness and using careful laboratory techniques.
Process of passing an inoculating loop with bacteria on
it across an agar plate
An autonomously replicating DNA molecule into which
foreign DNA fragments are inserted and then propagat-
ed in a host cell (i.e. plasmid).

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 46/52
Appendix C
Basic Molecular Biology Concepts and Terminology
A stu dy o f th e liv in g w o rld re ve als th at all liv in g o rg an ism s o rg an iz e th em se lv es in som e
u niq ue fa sh io n. A d eta ile d b lu ep rin t o f th is o rg an iz atio n is p ass ed o n to o ffsp rin g.
C e ll s a re t he smal le st f un ct io na l u ni ts c ap ab le o f i nd ep en de nt r ep ro du ct io n . Ma ny b ac te -
ria , fo r in sta nc e, c an s urv iv e as sin gle c ells . T h e c hem ic al m o le cu le s w ith in e ac h c ell are o rg a-
n ized to perform in concert. . .
Cells can be grown in culture and harvested
Ce ll s c an b e g at he re d f rom t he ir n at ur al i oc at io ns a nd g rown i ns id e l ab ora to ry c on ta in er s ..
A pp ro pria te fo od an d e nv iro nm en t m u st b e p ro vid ed fo r th e c ells to g row . B ac te ria an d y eas t
are very easy to gro w in culture . C ells taken from plants, insects and anim als can also be
gro wn , b ut are m ore d iffic ult to care fo r.
A fte r g row th is c om p le te , c ells in c ultu re c an b e h arv es te d an d s tu die d.
Cloning
Wh en a p op ulatio n o f c ells is p re pare d b y g row th from a sin gle c ell, all th e c ells in th e p op -
u la tio n w ill b e g en etic ally id en tic al. S uc h a p op ulatio n is c alle d c lo nal. T he p ro ce ss o f c re at-
ing a clonal population is called cloning . T he purpose o f streak ing bacteria on agar is to
g en era te s in gl e c ol on ie s, e ac h a ri si ng [ Toma s in gl e c el l.
Looking inside cells
T he m o le cu le s in sid e a c ell e ac h p erfo rm a g iv en fu nc tio n. F or in stan ce , DNA mo le cu le s
sto re in fo rm atio n (lik e th e h ard d riv e in a c om pu te r). P ro te in s a re th e w o rk ho rs es o f th e c ell.
T o study th ese m olecules w e prepare a clo nal population from a cell type of interest,b re ak o pe n th e c ells an d s ort th e c on te nts . F or in stan ce , it is fairly e asy to se parate a ll th e p ro -
te in s from all th e DNA mo le cu le s.
Purifying a single species of pro tein o ut of th e m ixture of proteins found inside a cell
ty pe is a lso p oss ib le . E ac h ty pe o f p ro te in h as u niq ue p hy sic al an d c hemic al p ro pe rtie s. T he se
p ro pe rtie s allo w th e se paratio n o f p ro te in s pe cie s b ase d o n siz e, c harg e o r h yd ro ph ob ic ity ,
fo r inst ance .
Special molecules, specialized functions
W e w ill take a close look at th ree very special kinds of m olecules found inside cells:
DN A, R NA and Pro teins. Each of th ese m olecules perform s a differen t function. D NA
m olecules are like file cab inets in w hich inform ation is stored. R NA h elps to retrieve and
e xe cu te th e in stru ctio ns w h ic h are s to re d in DNA. P ro te in s are d es ig ne d to p erfo rm c hemic alc ho re s in sid e (an d o fte n o uts id e) th e c ell.
DNA-The universal template for biological information'
T he m aste r s crip t fo r e ac h o rg an ism is e nc od ed w i th in it s d eo xy ri bo nu cl ei c a ci d (DNA ).
T he in fo rm atio n w ith in th e D NA m olec ule/s o f each c ell is suffic ien t to in itiate eve ry fun c-
t io n t ha t c el l w i ll p er fo rm .
D NA m olecules are very lon g ch ain s co mposed of repeatin g subunits. Eac h s ub un it
(" nu cle otid e" ) c on tain s o ne o f four p os si bl e b as es p ro tr ud in g f rom i ts s id e:
ADEN INE ("A" )
T HYM IN E ("T ")
CYTOS INE (" C" )
GUANINE ("GH
)

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 47/52
Since nucleotides are joined head-to-tail, a long strand of DNA essentially consists of a
chemical backbone with bases protruding along its side. The information carried by this
molecule is encoded in the sequence of As, Gs, Cs, and Ts along its length.
Some further points to note about DNA structure
1. Because the subunits of DNA chains are joined head-to-tail, the sequence is directional
"GTCAA". By-convention. wewrite DNA sequence from the free 5' end of the backbone
and work our way toward the other free end (3').
i .e. 5'".AACTG ...3'
2. The protruding bases along the chain are free form spontaneous bonds with available
bases on other DNA strands according to the following rules:
(i) "A" pairs with "T"
(ii) "C" pairs with "G"
Because of these rules, "A" and "T" are said to be complementary bases; "G" and "C"
are also complementary.
(iii) For two DNA strands to pair up, they must be complementary and run in
opposite directions.
i .e. (5'".AGGTC ...3') can pair with (5'..,GACCT ...3'). These two strands have comple-
rnentary sequences. The double-stranded pair is written as follows:
5' AGGTC 3'"
3' TCCAG 5'
The above molecule contains five base pairs. Indeed, in nature, DNA almost always
occurs in double-stranded form, the two strands containing complementary sequences.
3. DNA molecules are typically thousands, sometimes millions of base pairs long.
Sometimes the two ends of a DNA molecule are joined to form circular DNA.
4. Double-stranded DNA~ in its native form, occurs as a coiled spring, or helix. Because it
is two-stranded, it is often referred to as a double helix.
The architecture of DNA allows for a very simple strategy during reproduction: The two
strands of each DNA molecule unwind and "unzip"; then, each strand allows a new comple-mentary copy of itself to be made by an enzyme called DNA polymerase. This results in two
daughter molecules, each double-stranded, and each identical to the parent molecule.
Proteins and RNA are the workhorses of the cell
The biochemistry of life requires hundreds of very specific and efficient chemical inter-
actions, all happening simultaneously. The major players in these interactions are short-lived
protein and RNA molecules which can work together or independently to serve a variety of
functions. Like DNA, RNA and proteins are also long chains of repeating units .
RNA (ribonucleic acid), like DNA, consists of four types of building blocks strung togeth-
er in a chain. It differs from DNA in the following respects:
.RNA

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 48/52
The four bases in RNA are "A", "G", "C", and "U" (uracil); the pairing rules are the same
as for DNA except that "A" pairs with "U". Although RNA can pair with complementary
RNA or DNA, in cells RNA is usually single-stranded. The sugar in the RNA backbone
is ribose, not deoxyribose. RNA molecules are generally short, compared to DNAmolecules; this is because each RNA is itself a copy of a short segment from a DNA
molecule. The process of copying segments of DNA into RNA is called transcription,
and is performed by a protein called RNA polymerase.
Proteins
Proteins (more precisely, polypeptides) are also long, chain-like molecules but more struc-
turally diverse than either DNA or RNA. This isbecause the subunits of proteins called amino
acids, come in twenty different types. The exact sequence of amino acids along a polypeptide
chain determines how that chain will fold into a compact structure. The precise 3-dimensional
features of this structure, "intum, determine its function:
, .
What a protein will do depends on the exact sequence of its amino acids.
In most cases, a protein will perform a single function. Very diverse functions can be
performed by proteins: Some proteins, called enzymes, act as catalysts in chemical reactions;
some carry signals from one part of a cell to another-or, in the case of "hormones", [Tomone
cell to another; some proteins ("antibodies") have thetask of fighting intruders; many become
integra! pa r t s of the various physical structures inside cells; and still others (regulatory proteins)
police various activities within cells so as to keep them within legal limits.
Linear code, three-dimensional consequences
DNA is the primary depot for information in living systems. As mentioned, this infor-
mation is linear i.e, encoded in the sequence of "A", "G", "C", "T" building blocks along the
DNA molecule. This linear code can be passed on to offspring-because DNA can make
exact copies of itself.
Short segments of each DNA molecule are chosen for transcription at any given time.
These segments are called genes. The enzyme RNA polymerase copies the entire segment,
base by base, assembling an RNA molecule which contains a sequence of "A", "G", "C" and
"U'' exactly complementary to the DNA sequence of the transcribed gene.
In addition to providing a master template for copying RNAs, DNA also contains sequence
information which tells the RNA polymerase where to start transcribing a gene (promoter) and
where to stop; how many copies it should make and when; and it can even embed certain
information within the RNA sequence to determine the longevity and productivity of that
RNA .
There are three major classes of RNAs copied off DNA templates: messenger RNAs, or
mRNAs, which relay the sequence information required for assembling proteins; transfer
RNAs, or tRNAs, which work in the assembly line for proteins; and RNAs which perform
structural functions. For example, ribosomal RNAs, or rRNAs, help build the scaffolding for
ribosomes, the factories where proteins are assembled.
mRNAs carry the sequence information for making proteins. Ribosomes read this
. sequence of nucleotides, by a process called "translation" into a sequence of amino acids.
How is this accomplished? There are only four kinds of nucleotides, but twenty kinds of
amino acids"
During translation, the ribosome reads 3 nucleotides at a time and assigns an amino acid
to each successive triplet. Note: Triplets are often referred to as "codons", Each amino acid
is then attached to the end of the growing protein chain. There are 64 possible triplets, or
codons. Thus, the linear information residing in DNA is used to assemble a linear sequence

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 49/52
of amino acids in a protein. This sequence, in tum, will determine the way that protein will
fold into a precise shape with characteristic chemical properties.
In summary, the primary transfer of information.within cells follows the order:.
DNA »> RNA>>> PROTEIN»»TRAIT
Although the information itself is linear, the implications 'are 3-dimensionaL A funda-
me~tal assumption of recombinant DNA technology is that permanent and desirable changes
in the functioning of living cells can be accomplished by changing the linear sequence of
their DNA.
Genes are discrete files of DNA information
A gene is a segment within a DNA molecule singled out for copying into RNA. Directly
or indirectly, this RNA will perform a function. It is convenient to think of a gene, therefore,
as a unit of function.
Many traits, such as bacterial resistance to an antibiotic, are governed by single genes.
Most traits-such as the color of a rose, or the shape of a nose - are governed by several genes
acting in concert.
Genes can vary in length: Some are only a few hundred base pairs long; some can be tens
of thousands of base pairs long. A DNA molecule may carry from a handful to thousands of
genes. A cell, in tum, may contain one or several DNA molecules (chromosomes). Thus the
number of genes in a cell can vary ·greatly. Ecoli, a bacterium, contains one DNA molecule
with about five thousand genes on it. A human cell contains 46 DNA molecules carrying a total
of about 100,000 genes.
All genes in a given cell are not copied into RNA (i.e. "expressed") at the same time or
at the same rate. Thus, when speaking of gene function, one refers to its expression leveL'
This rate can be Controlled by the cell, according to predetermined rules which are themselves
written into the DNA.
An example: The cells in our bodies (all 100 trillion of them) each contain identical DNA
molecules. Yet liver cells, for example, express only those genes required for liver function,
whereas skin cells express a quite different subset of genes.
DNA can be cut into pieces with restriction enzymes
Restriction enzymes are proteins made by bacteria as a defense against foreign, invading
DNA (for example, viral DNA). Each restriction enzyme recognizes a unique sequence of
typically 4--6 base pairs, and will cut any DNA whenever that sequence occurs.
For example, the restriction enzyme BamH I recognizes the sequence ( S ' . .GGATCC. .3 ')
and cuts the DNA strand between the two G nuc1eotides in that sequence.
Restriction enzymes will cut DNA from any source, provided the recognition sequence
is present. It does not matter if the DNA is of bacterial, plant or human origin.
Pieces of DNA can bejoined by DNA ligase
DNA ligase is an enzyme that glues pieces of DNA together, provided the ends are
compatible.
Thus, a piece of human or frog or tomato DNA cut with BamH I can be easily joined to
. a piece of bacterial DNA also cut with BamHI . This allows the creation of recombinant DNAs
i.e. hybrids, created by joining pieces of DNA from two different sources.

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 50/52
Genes can be cut out of human DNA, or plant DNA, and-placed inside bacteria. For
example, the human gene for the hormone insulin can be put into bacteria. Under the right con-
ditions, these bacteria can make authentic human insulin.
Plasmids are small circular pieces ofDNA
Plasmids are small circular DNAs found inside some bacterial cells. They replicate their
own DNA by borrowing the cells' polymerases. Thus they can persist indefinitely inside cells
without doing very much work of their own. .: .
Because of their small size, plasmid DNAs are easy to extract and purify from bacterial
cells. When cut with a restriction enzyme, they can be joined to foreign DNAs - from any
source - which have been cut with the same enzyme.
The resulting hybrid DNAs can be re-introduced into bacterial cells by a procedure called
transformation. Now the hybrid plasmids can perpetuate themselves in the bacteria just as
before except that the foreign DNA which was joined to it is also being perpetuated. The for-eign DNA gets a free ride, so to speak.
Every hybrid plasmid now contains a perfect copy of the piece of foreign DNA original-
lyjoined to it. We say that foreign piece of DNA has been cloned; the plasmid which carried
the foreign DNA is called a cloning vehicle or vector.
In addition to their usefulness for cloning foreign genes, plasmids sometimes carry genes
of their own. Bacteria die when exposed to antibiotics. However, antibiotic-resistance genes
allow bacteria to grow in the presence of an antibiotic such as ampicillin. Such genes are
often found on plasrnids. When foreign PNA is inserted into such plasmids, and the hybrids
introduced into bacterial cells by transformation, it is easy to select those bacteria that have
received the plasmid - because they have acquired the ability to grow in the presence of the
antibiotic, whereas all other bacterial cells are killed.
DNA libraries
When DNA is extracted from a given cell type, it can be cut into pieces and the pieces can
be cloned en masse into a population of plasmids. This process produces a population of
hybrid (i.e. recombinant) DNAs. After introducing these hybrids back into celis, each trans-
formed cell will have received and propagated one unique hybrid. Every hybrid will contain
the same vector DNA but a different insert DNA.
If there are 1,000 different DNA molecules in the original mixture, 1,000 different hybrids
will be formed; 1,000 different transformant cells will be recovered, each carrying one of the
original 1,000 pieces of genetic information. Such a collection is called a DNA library. If the
original extract came from human cells, the library is a human library.
Individual DNAs of interest can be fished out of such a library by screening the librarywith an appropriate probe.

8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 51/52
Appendix 0
Gene Regulation, One Gene, One Protein
Our bodies contain thousands of different proteins which perform many different jobs.
Digestive enzymes are proteins; some of the hormone signals that run through our bodies and
the antibodies protecting us from disease are proteins. The information for assembling a
protein is carried in our DNA. The section of DNA which contains the code.for making a
protein is called a gene. There are over 100,000 genes in the human genome. Each gene codes
for a unique protein; one gene-one protein. The gene which makes a digestive enzyme in your
mouth is different from one which makes an antibody or the pigments that color your eyes.
Organisms regulate expression of their genes and ultimately the amounts and kinds of
proteins' present' within their cells for a myriad of reasons including developmental, cellular
specialization and adaptation to the environment. Gene regulation not only allows for adap-
. tation to differing conditions, but also prevents wasteful overproduction of unneeded proteins
which would put the organism at a competitive disadvantage. The genes involved in the trans-
port and breakdown (catabolism) of food are good examples of highly regulated genes. For
example, the sugar arabinose is both a source of energy and a source of carbon. E. coli bac-
teria produce three enzymes (proteins) needed to digest arabinose as a food source. The genes
which code for these enzymes are not expressed when arabinose is absent, but they are
expressed when arabinose is present in their environment. How is this so?
Regulation of the expression of proteins often occurs at the level of transcription from
DNA into RNA. This regulation takes place at a very specific location on the DNA template,
called a promoter, where RNA polymerase sits down on the DNA and begins transcription of
the gene. In bacteria, groups of related genes are often clustered together and transcribed into'
RNA from one promoter. These clusters of genes controlled by a single promoter are called
operons,
The three genes (a ra B, a ra A and araD) that code for three digestive enzymes involved
in the breakdown of arabinose are clustered together in what is known as the arabinose oper-
on.' These three proteins are dependent on initiation of transcription from a single promoter,
(PBAD) . Transcription of these three genes requires the simultaneous presence of the DNA
template (promoter and operon), RNA polymerase, a DNA binding protein called araC and
arabinose. araC binds to the DNA at the binding site for the RNA polymerase (the begin-
ning of the arabinose operon). When arabinose is present in the environment, bacteria take it
up. Once inside, the arabinose interacts directly with araC which is bound to the DNA. The
interaction causes araC to change its shape which in tum promotes (actually helps) the bind-
ing of RNA polymerase and the three genes B, A and D, are transcribed. Three enzymes are
produced, they do their job, and eventually the arabinose runs out. In the absence of arabinose
the araC returns to its original shape and transcription is shut off.
The DNA code of the pGLO plasmid has been engineered to incorporate aspects of the
arabinose operon. Both the promoter (PBAD) and the araC gene are present. However, the
genes which code for arabinose catabolism, araB, A and D, have been replaced by the single
gene which codes for the Green Fluorescent Protein (GFP). Therefore, in the presence of ara-
binose, araC protein promotes the binding of RNA polymerase and GFP is produced. Cells
fluoresce a brilliant green color as they produce more and more protein. In the absence of
arabinose, araC no longer facilitates the binding of RNA polymerase and the GFP gene is nottranscribed, ~llen the GFP protein is not made, bacteria colonies will appear to nave a wild
type (narural) phenotype - of white colonies with no florescence.
This is an excellent example of the central molecular framework of biology in action:
DNA>RNA>PROTEIN>TRAlT.
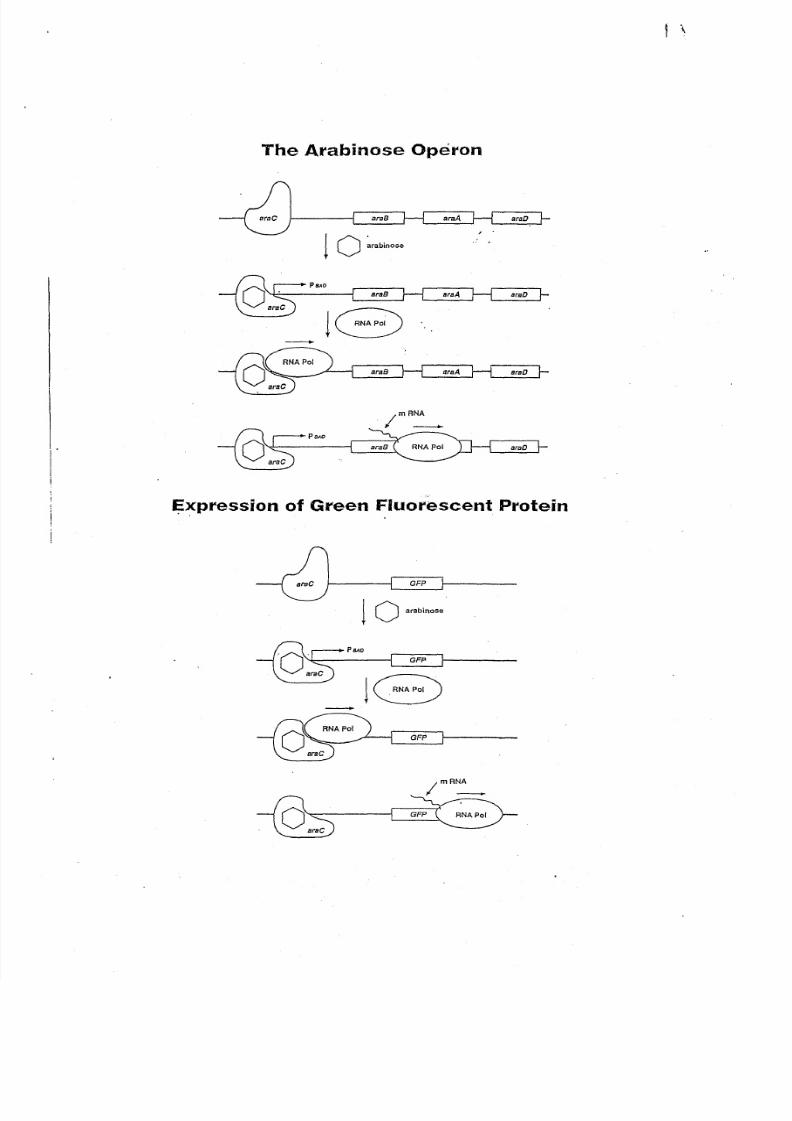
8/6/2019 Molecular Biology Practical Manual
http://slidepdf.com/reader/full/molecular-biology-practical-manual 52/52
\
The Arabinose Operon
araB
1 0~rabinos<>
, .
; m R : _ _ _PSAO
,?xpression of Green ~Iuotescent Protein
GFP
1 0 arabinose
- - A r = .. : :. . . : .. = : _ P B A O ~ _
~~
1~GFP
; m R ~



























