Embed Size (px)
Citation preview

Modelling of Defects DFT and complementary methods
Tor Svendsen BjørheimPhD fellow, FERMiO, Department of Chemistry
University of Oslo
NorFERM-2008, 3rd -7th of October 2008

Outline Background / Introduction DFT
Theory DFT calculations in practice Case studies
Supplementary Methods Nudged Elastic Band Molecular Dynamics Monte Carlo approach
Summary

Introduction to defect modeling Experimental techniques
Time consuming Expensive Inconclusive results Supplement with computational studies
Condensed systems Mutually interacting particles Described by the full Hamiltonian:
Increasingly complex with larger number of electrons (3N variables) Need simplifications!
Hartree-Fock DFT

DFT -
Density Functional
Theory

Density Functional Theory Ab initio ground state theory
Basic variable electron density, n(r) n(r) depends only on the three spatial variables
Hohenberg-Kohn theorems (1964): For a system of interacting particles in an external potential, the external
potential and hence the total energy is a unique functional of n(r)
The ground state energy can be obtained variationally; the density that minimizes the total energy is the exact ground state density
Problem: kinetic energy of the electrons…
Practical use: Kohn-Sham approach (1965) Introduce a reference system of non-interacting particles (with the same n(r)) Kinetic energy easily determined:
Functional = Function of a function

Total energy:
Exc:
Need to determine the orbitals of the reference system Kohn-Sham equations:
Kohn-Sham potential:
Depends on the electron density Can not be solved directly Self consistent solutions using iterative schemes
Kohn-Sham approach In principle exact Do not know the form of EXC Approximate Modern DFT: new and improved EXC

Simplest approach: Local Density Approximations - LDA Assume Exc equal to Exc in a homogeneous electron gas:
Locally constant electron density Systems with slowly varying electron densities Inadequate for systems with quickly varying electron densities
Improvements: Generelized-Gradient Approximations (GGA) Include gradients of the electron density at each point
E.g. GGA-PW91 and GGA-PBE
Problems: Band gaps and Van der Waals forces…..
Exchange-Correlation Functionals

DFT calculations in practice Real solids ~1023 atoms
Huge number of wave functions.. Need further simplifications!
Popular approach: plane waves Periodically repeating unit cell Bloch’s theorem:
Finite number of wave functions over an infinite number of k-points in the 1. Brillouin Zone!
K-point sampling Electronic states at a finite number of k-points Finite number of wave functions at a finite number of k-points in the 1.
Brillouin Zone! K-mesh needs to be chosen carefully

Defects in solids
Solids with one or more defects Aperiodic systems Bloch’s theorem can not be applied
Can not use plane wave basis sets
Introduce the concept of a periodically repeating supercell
Supercell method Defects in a ’box’ consisting of n unit cells Define the supercell with the defect as the new unit cell Periodic boundaries 3D periodic ordering of defects Typical size: <500 atoms Surfaces/interfaces & molecules

Finite-Cluster approach Defects in finite atomic clusters No interaction with defects in neighboring unit cells Avoid surface effects: large clusters
Green’s Function Embedding Technique Purely mathematical Defect regions embedded in known DFT Green’s
function of bulk Ideal for studies of isolated defects (in theory) Numerically challenging…..
Alternative approaches
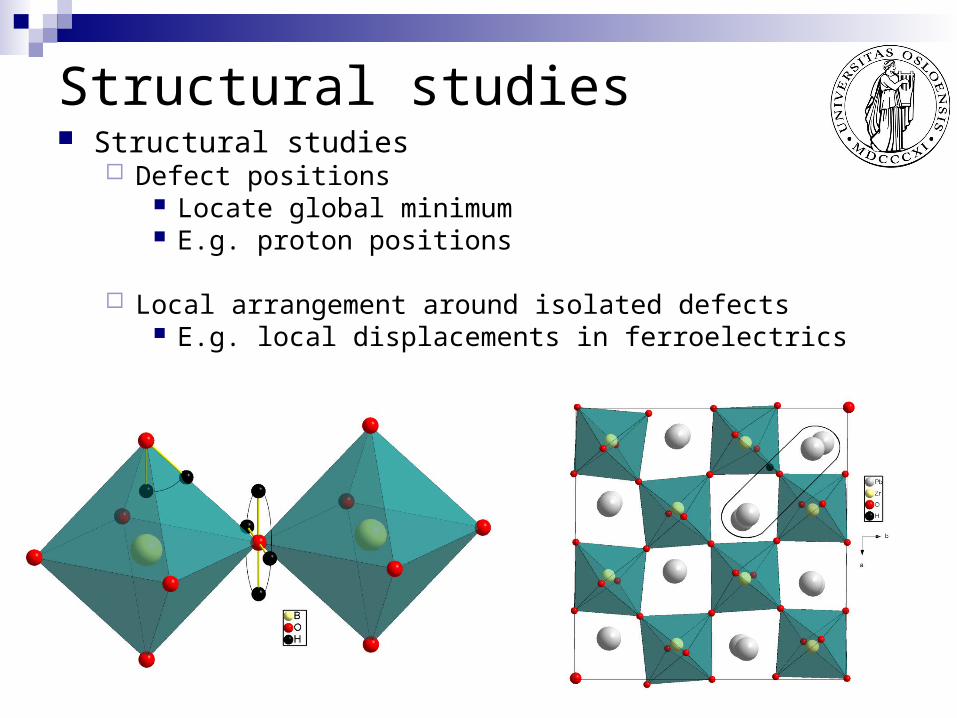
Structural studies Structural studies
Defect positions Locate global minimum E.g. proton positions
Local arrangement around isolated defects E.g. local displacements in ferroelectrics

Thermodynamics Formation energy of isolated point defects:
Defects that change the composition: Total energies and a set of chemical potentials
E.g. isolated protonic defects & oxygen vacancies: Formation:
Formation energy:
Effective defect concentration
Supercell size (unit cells)
Atmospheric conditions
- Thermodynamic tables
-
Fermi level:
CBMVBTe ε,εμ 2tot
H HEμ
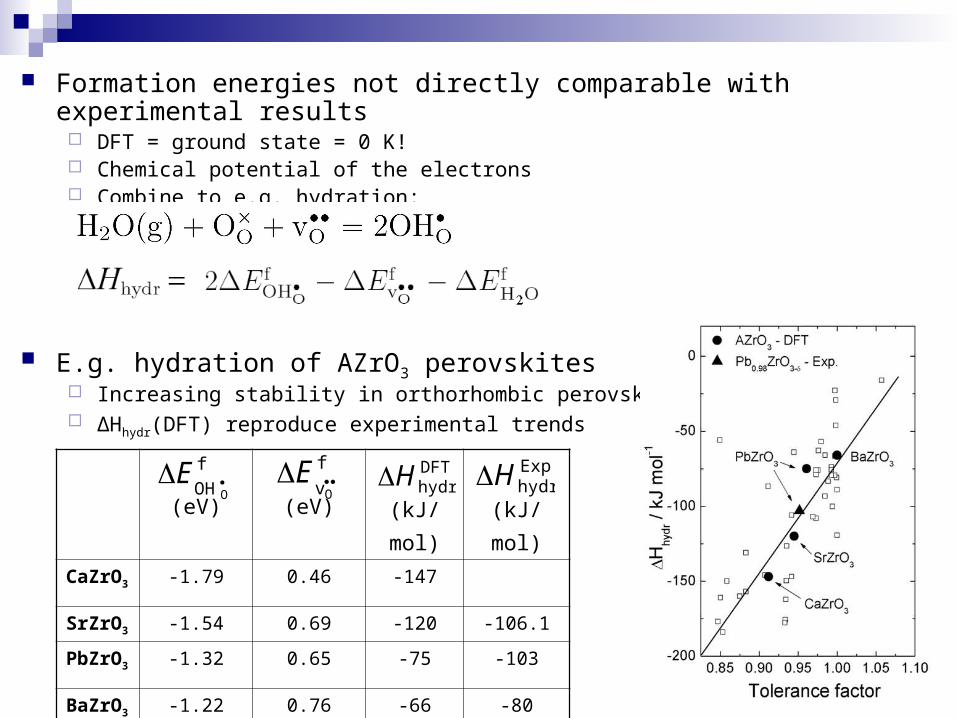
Formation energies not directly comparable with experimental results DFT = ground state = 0 K! Chemical potential of the electrons Combine to e.g. hydration:
E.g. hydration of AZrO3 perovskites Increasing stability in orthorhombic perovskites ∆Hhydr(DFT) reproduce experimental trends
f
vOEf
OHOE
(eV) (eV) (kJ/mol) (kJ/mol)
CaZrO3 -1.79 0.46 -147
SrZrO3 -1.54 0.69 -120 -106.1
PbZrO3 -1.32 0.65 -75 -103
BaZrO3 -1.22 0.76 -66 -80
DFThydrH Exp
hydrH

Defect levels Defect levels
Transition between charge states: Ef(q/q’) Experimentally: DLTS Determine the preferred charge state of defects
In supercell calculations: Determine ΔEf for all charge states ΔEf for all Fermi level positions Most stable: charge state with lowest ΔEf
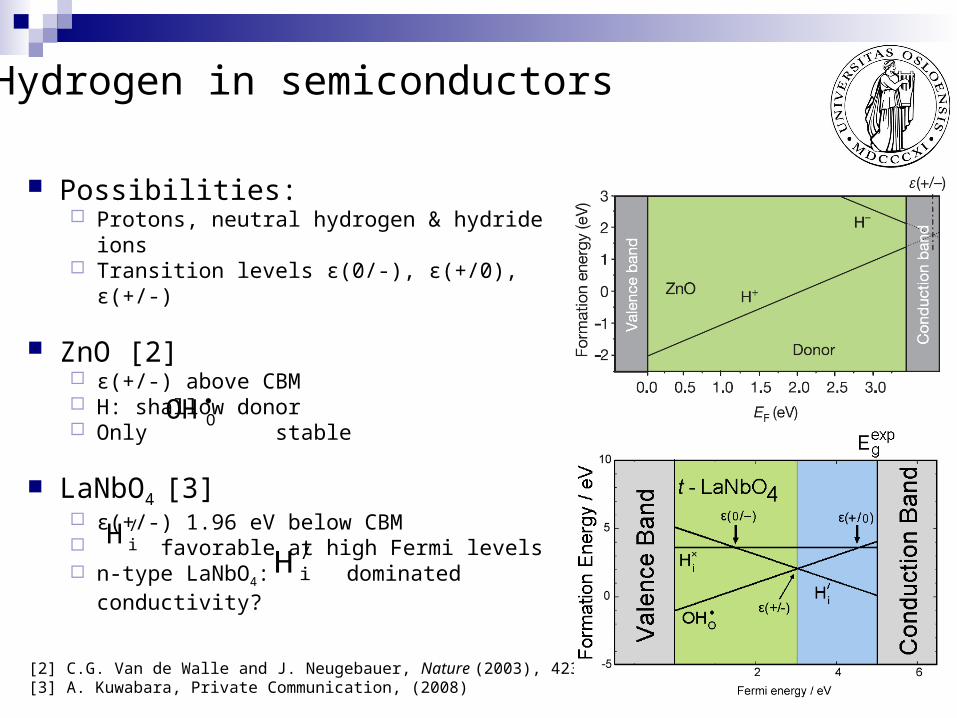
Possibilities: Protons, neutral hydrogen & hydride ions Transition levels ε(0/-), ε(+/0), ε(+/-)
ZnO [2] ε(+/-) above CBM H: shallow donor Only stable
LaNbO4 [3] ε(+/-) 1.96 eV below CBM favorable at high Fermi levels n-type LaNbO4: dominated
conductivity?
/iH
[2] C.G. Van de Walle and J. Neugebauer, Nature (2003), 423[3] A. Kuwabara, Private Communication, (2008)
OOH
/iH
Hydrogen in semiconductors
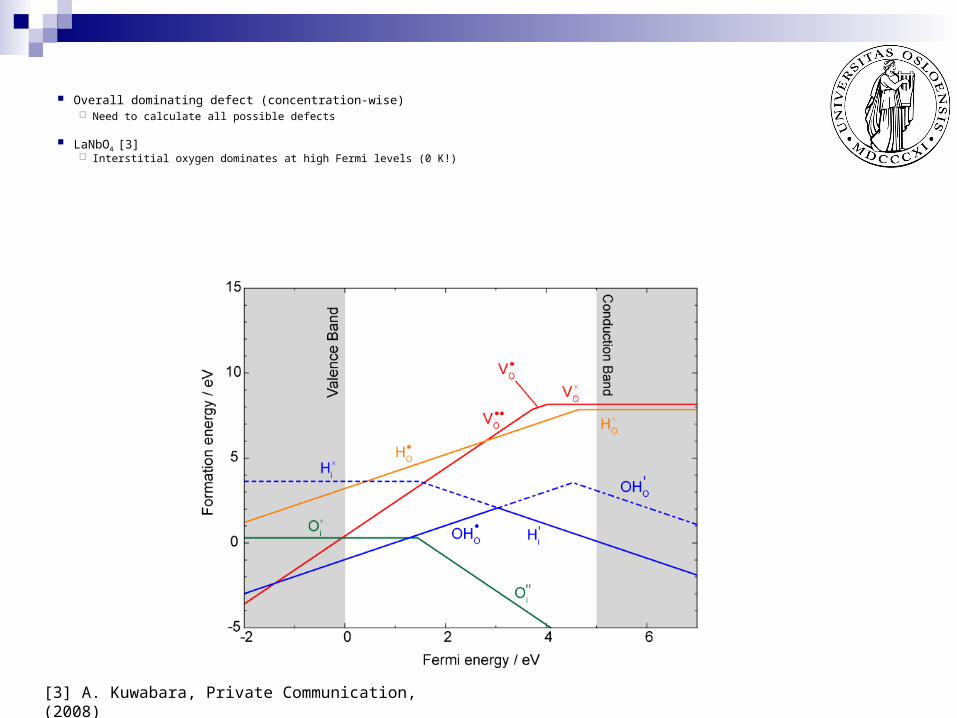
Overall dominating defect (concentration-wise) Need to calculate all possible defects
LaNbO4 [3] Interstitial oxygen dominates at high Fermi levels (0 K!)
[3] A. Kuwabara, Private Communication, (2008)

When DFT fails… In defect calculations: band gaps
Heavily underestimated Both LDA/GGA functionals Affects:
Defect levels Defect formation energies
Correction: scissor operation! Shift the conduction band Align Eg(DFT) and Eg(EXP)
Donor type defects follow CB Correct formation energies
Alternative: hybrid or semi-empirical functionals
/ eV / eV
BaZrO3 3.2 5.3
PbZrO3 3.0 3.7
ZnO 1.9 3.4
DFTgE
ExpgE

Complementary methods
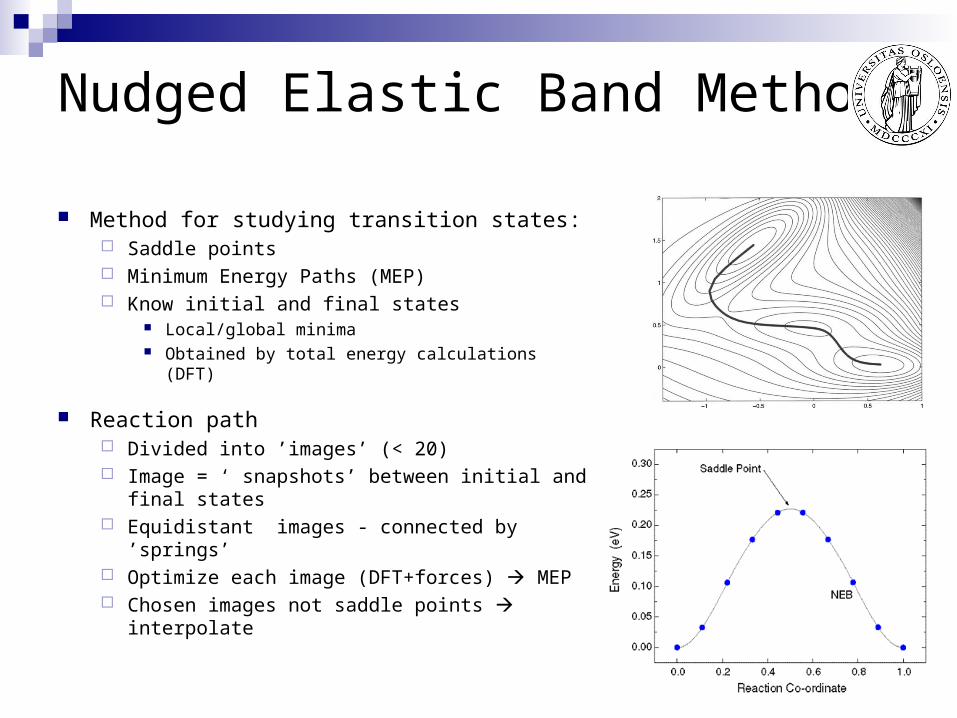
Nudged Elastic Band Method
Method for studying transition states: Saddle points Minimum Energy Paths (MEP) Know initial and final states
Local/global minima Obtained by total energy calculations (DFT)
Reaction path Divided into ’images’ (< 20) Image = ‘ snapshots’ between initial and final
states Equidistant images - connected by ’springs’ Optimize each image (DFT+forces) MEP Chosen images not saddle points interpolate
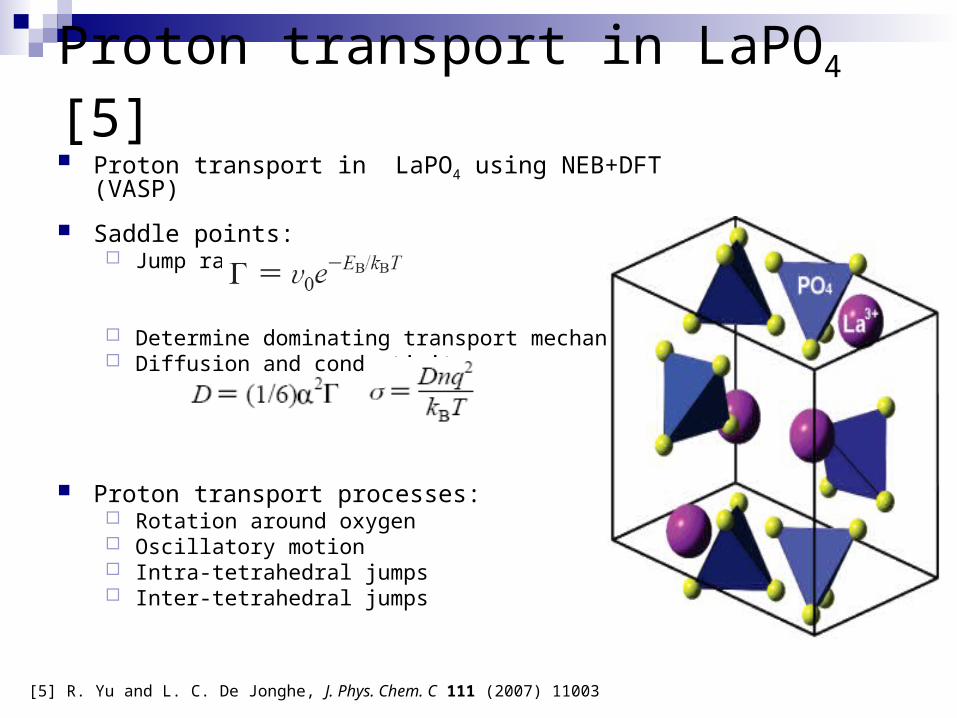
Proton transport in LaPO4 [5] Proton transport in LaPO4 using NEB+DFT (VASP)
Saddle points: Jump rates:
Determine dominating transport mechanism Diffusion and conductivity:
Proton transport processes: Rotation around oxygen Oscillatory motion Intra-tetrahedral jumps Inter-tetrahedral jumps
[5] R. Yu and L. C. De Jonghe, J. Phys. Chem. C 111 (2007) 11003
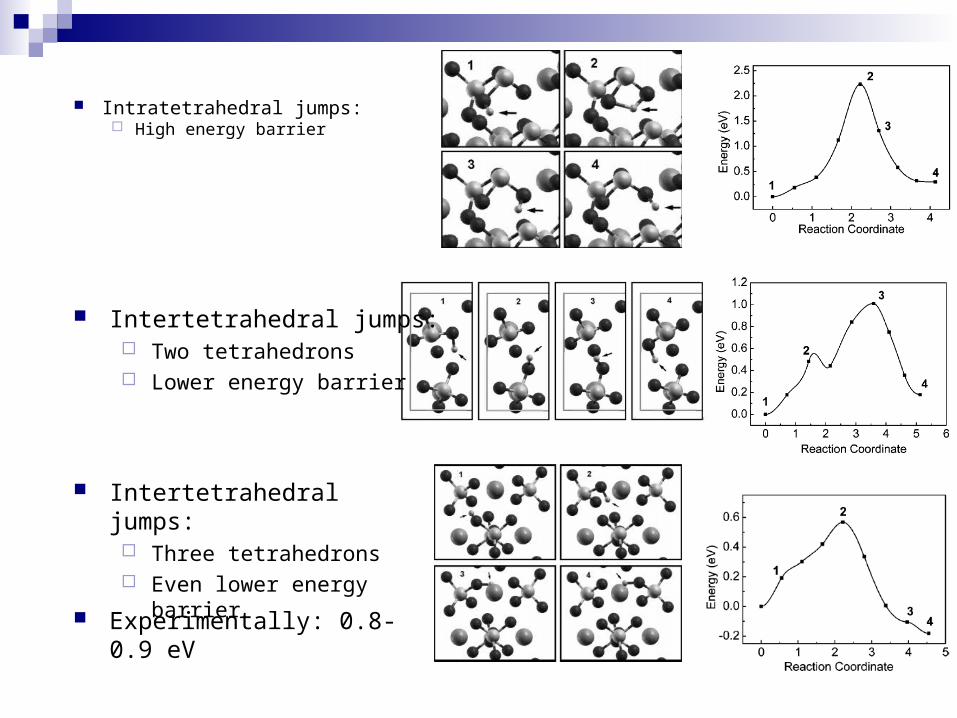
Intratetrahedral jumps: High energy barrier
Intertetrahedral jumps: Two tetrahedrons Lower energy barrier
Intertetrahedral jumps: Three tetrahedrons Even lower energy barrier
Experimentally: 0.8-0.9 eV
Molecular Dynamics Used to simulate time evolution of classical many-particle systems
Obey the laws of classical mechanics
Condensed systems: Classical particles moving under influence of an interaction potential, V(R1,…,RN)
Forces on the ions:
MD algorithms: Discretize equation of motion Trajectories: stepwise update positions and velocities
),...,,...,(V 1j NjjRRRF R

Interionic interactions: Model potential vs. first-principles
Model potential Parameterized to fit experimental or first principles data Advantages
Possible to treat large systems Long time evolution
Disadvantages Inaccurate potentials Poor force representation
First-principles First-principles electronic structure calculation (e.g. DFT) at each ionic step Advantages:
Accurate forces Realistic dynamic description
Disadvatages Computationally demanding Small systems (~100 ions) Short time periods (~ps)
Good statistical accuracy
Poor statistical accuracy..

Monte-Carlo approach Loosely described:
Statistical simulation methods
Conventional methods (MD): Discretize equations describing the physical process E.g. equations of motion
Monte-Carlo approach Simulate the physical process directly No need to solve the underlying equations Requirement: process described by probability distribution functions (PDF) Average results over the number of observations
Proton diffusion Jump frequency and PDF:
N
nmmP
1n/ TEii Bia0 k/exp

Summary
Many different computational approaches
Systematic trends
Fundamental processes
Predict defect properties of real materials
Combine different methods

Acknowledgements
Akihide Kuwabara
Espen Flage-Larsen
Svein Stølen
Truls Norby
Colleagues at the Solid State Electrochemistry group in Oslo
Thank You !!

Thank You!




























![[Adam D.M. Svendsen] Intelligence Cooperation and](https://img.pdfslide.us/doc/110x75/55cf8f89550346703b9d3a56/adam-dm-svendsen-intelligence-cooperation-and.jpg)
