Embed Size (px)
Citation preview

Modeling the 3DStructure of GPCRsfrom Sequence
Sharon Shacham, Maya Topf,� Noa Avisar, Fabian Glaser, Yael Marantz, Shay Bar-Haim,Silvia Noiman, Zvi Naor,y Oren M. Becker
Bio IT (Bio Information Technologies) Ltd., 3 Hayetzira St., Ramat Gan, Israel
!
Abstract: G-protein-coupled receptors (GPCRs) are a large and functionally diverse protein
superfamily, which form a seven transmembrane (TM) helices bundle with alternating extra-
cellular and intracellular loops. GPCRs are considered to be one of the most important groups of
drug targets because they are involved in a broad range of body functions and processes and are
related to major diseases. In this paper we present a new technology, named PREDICT, for
modeling the 3D structure of any GPCR from its amino acid sequence. This approach takes into
account both internal protein properties (i.e., the amino acid sequence) and the properties of the
membrane environment. Unlike competing approaches, the new technology does not rely on the
single known structure of rhodopsin, and is thus capable of predicting novel GPCR conformations.
We demonstrate the capabilities of PREDICT in reproducing the known experimental structure of
rhodopsin. In principle, PREDICT-generated models offer new opportunities for structure-based
drug discovery towards GPCR targets. ß 2001 John Wiley & Sons, Inc. Med Res Rev, 21, No. 5, 472±483,
2001
Key words: GPCR; modeling; structure based drug discovery
1 . I N T R O D U C T I O N
G-protein-coupled receptors (GPCRs) are membrane embedded proteins, involved in communica-
tion between the cell and its environment by passing chemical signals across the cell membrane.
These proteins form a large and functionally diverse superfamily which consists of a single
polypeptide chain of variable length that traverses the lipid bilayer seven times, forming
characteristic transmembrane helices (TM) and alternating extracellular (ECL) and intracellular
(ICL) loops.1±2 Many and diverse ligands (e.g., ions, biogenic amines, nucleosides, lipids, peptides,
proteins, and even light) use this class of receptors to convert external and internal stimuli into
intracellular responses.
GPCRs activate one or more members of the guanine-nucleotide-binding signal transducing
proteins (G-proteins) that carry the information received by the receptor to cellular effectors such as
472
*Currentaddress:Departmentof Chemistry,NewChemistry Laboratory, South Parks Road,Oxford,OX13QT,United Kingdom.yCurrentaddress: Departmentof Biochemistry,Wise Facultyof Life Sciences,Tel Aviv University,Ramat Aviv,Tel Aviv 69978, Israel.Correspondence to: Oren M. Becker, Bio IT (Bio InformationTechnologies) Ltd., S.A.P. Building (11th floor), 3 Hayetzira St., RamatGan 52521, Israel.E-mail: [email protected]
Medical Research Reviews, Vol. 21, No. 5, 472^483, 2001ß 2001 John Wiley & Sons, Inc.

enzymes and ion channels.3±5 These effectors in¯uence levels of second messengers that regulate a
wide variety of cellular processes including cell growth and differentiation.6±7 Each G-protein
consists of three subunits, commonly denoted as a, b, and g. Sixteen distinct mammalian G-protein
a-subunits have been molecularly cloned. Similarly, 11 G-protein b-subunits and ®ve G-protein g-
subunits have been identi®ed. Thus, GPCRs are likely to represent the most diverse signal
transduction systems in eukaryotic cells. Furthermore, GPCRs may also couple to other proteins, for
example, those containing PDZ domains.8 The regulation of receptor-G-protein signal selectivity
and speci®city is highly complex and involves the activation of a network of mechanisms and
pathways that eventually lead to biological responses.
Based on nucleotide and amino acid sequence similarity, the superfamily of GPCRs can be
subdivided into six families of receptors whose protein sequences share signi®cant similarity.9
The main family (family A) is of the rhodopsin/adrenergic receptors, which consists of the majority
of G-protein-coupled receptors identi®ed to date. This family is the best studied from both the
structural and functional points of view.1,9±11 Receptors belonging to this family are activated by a
variety of stimuli including photons, odorants, hormones, and neurotransmitters with molecular
structures ranging from small biogenic amines (e.g., catecholamines and histamine) to peptides
(e.g., gonadotropin-releasing hormone (GnRH), thyrotropin-releasing hormone (TRH)), and
complex glycoproteins, such as luteinizing hormone (LH), follicle-stimulating hormone (FSH),
and thyroid-stimulating hormone (TSH).12±15 The other subfamilies are the secretin/vasointestinal
peptide (VIP) family (family B), which binds several neuropeptides and peptide hormones, the
metabotropic glutamate receptor family (family C), which comprises at least six closely related
subtypes of receptors that bind glutamate, the major excitatory neurotransmitter in the central
nervous system. Three additional GPCR families are the fungal pheromone P and a-factor (STE2/
MAM2) family (family D), the fungal pheromone A and M-factor (STE3/MAP3) receptors (family
E), and the cyclic adenosine monophosphate (cAMP) receptors of Dictyostelium (family F).
A number of new putative GPCR families have been discovered with varying degrees of similarity
to the established families, including frizzled, smoothened, basal vomeronasal receptors, and bride
of sevenless (BOSS) of Drosophila and mammals, latrophilin, several plant GPCRs, another yeast
GPCR, GPR1, as well as other mammalian sequences, p40 and pm1.8,16±19 Figure 1 depicts the
distribution of the 508 human GPCRs identi®ed so far, grouped according to the type of ligand.
Approximately half of these GPCRs are still orphans, indicating that their function is yet unknown.
It is expected that by the completion of the analysis of the human genome project even more
GPCRs, that are potential new drug targets, will be discovered.
The mechanisms controlling ligand binding, activation, and signal transduction of the GPCRs/
G-protein system as well as the mechanisms required for de®ning the speci®city of receptor-G
protein-effector interaction and the ef®ciency and regulation of signal transduction are highly
complex and multifactorial. Knowledge and mapping of the structural determinants and require-
ments for optimal receptor function are of paramount importance for understanding the molecular
basis of ligand action and receptor function in normal and abnormal conditions. Deciphering
structure-function relationships in GPCRs will promote computer-aided drug discovery by
studying the binding mode of known ligands into their receptor binding-sites and identify the
pharmacophores involved.
2 . D R U G D I S C O V E R Y A P P R O A C H E S
GPCRs are considered as one of the most important groups of drug targets. This is because GPCRs
are involved in a very wide range of body functions and processes, including cardiovascular,
nervous, endocrine, and immune systems; and are related to major diseases such as hypertension,
cardiac dysfunction, depression, eating disorders (obesity), certain types of cancer, pain,
MODELIN G-PROTEIN-COUPLED RECEPTORS * 473
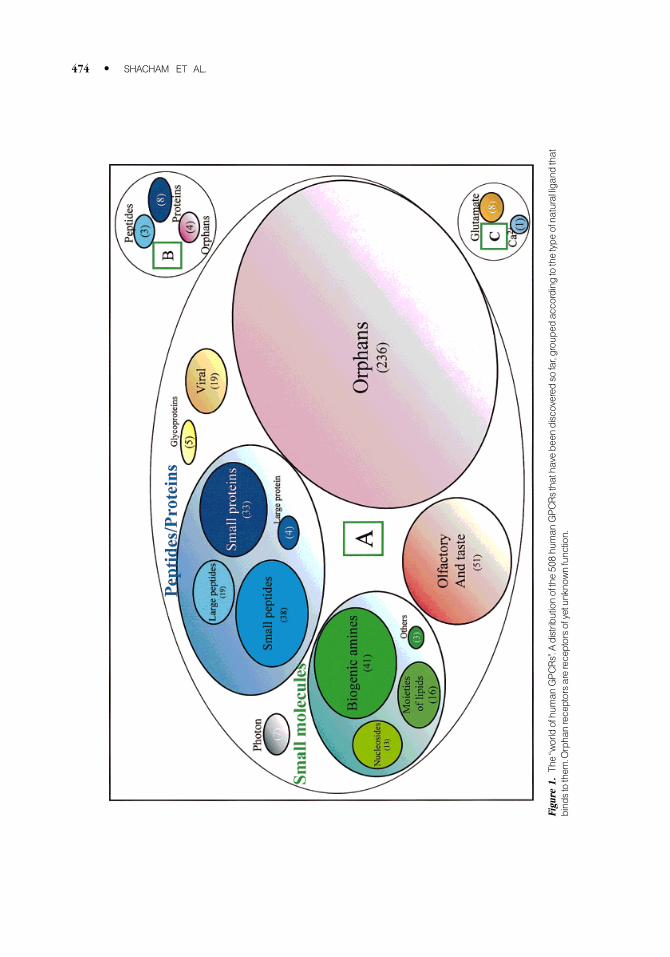
Figu
re1.
The`̀worldofhu
man
GPCRs''.A
distributionofthe50
8hu
man
GPCRsthatha
vebe
endiscovered
sofar,grou
ped
accordingtothetypeofna
turalliga
ndthat
bind
stothem
.Orpha
nreceptorsarereceptorsofyetunkno
wnfunction.
474 * SHACHAM ET AL.

schizophrenia, and viral infection. Thus, while GPCRs are only a small subset of the human genome
(2%±3% of the human genome) they constitute about 50% of the drug targets that are of interest to
the pharmaceutical industry.
Conventional drug discovery often involves combinatorial chemistry techniques to create (often
randomly) very large numbers of molecules that are subsequently screened for bioactivity using
high throughput screening tools. These methods suffer from drawbacks such as the consumption of
signi®cant resources for creating hundreds of thousands of molecules, only a small fraction of which
are active, and the fact that even very large molecular libraries can explore only a small portion of
the chemical of drug-like molecules.
Structure-based drug discovery is an alternative approach to conventional drug discovery,
which relies on the fact that interactions of molecules within the human body take place in three-
dimensions. Drug molecules compete with natural ligands by inserting themselves into the
functional site of the target protein and inducing (agonists) or inhibiting (antagonists) its activity.
The af®nity of these drugs to their respective target proteins is due to structural and chemical
complementarily, and can be explored by computational methods. This allows for using relatively
cheap and ef®cient computational screening technology instead of the expensive and low yield
experimental high-throughput screening (HTS) techniques. Furthermore, structure based drug
discovery can identify possible binding modes for ligands within the receptor cavity, typically
through the identi®cation of pharmacophoric centers complementary in character to the centers
found on the surface of the receptor.
3 . G P C R S T R U C T U R E
As indicated by the name, structure-based drug discovery requires knowledge of the targets'
structure, in this case GPCRs. Members of the GPCR family are characterized by seven regions,
each 20±25 amino acid sequences in length, that are believed to represent the TM hydrophobic
regions of the proteins. The seven TM domains are thought to form a barrel shape, oriented roughly
perpendicular to the plane of the membrane in a counterclockwise manner. Each receptor is believed
to have an extracellular N-terminal region that varies in length from less than 10 amino acids
(e.g., adenosine receptors) to several hundred amino acids (e.g., metabotropic glutamate receptors)
and an intracellular C-terminal region. The majority of intracellular and extracellular loops are
thought to be 10±40 amino acids long, although the third intracellular loop and the C-terminal
sequence may have more than 150 residues. The overall size of GPCRs varies signi®cantly from less
than 300 amino acids in the case of adrenocorticotrophin hormone receptor to more than the 1100
amino acids for the metabotropic glutamate receptors.20
Most of the primary sequence homology among the different groups of GPCRs is contained
within the TM domains. The most conserved residues among the GPCR superfamily are located
within the TM domains and apparently represent essential structural determinants of receptor
structure and function.
Due to technical dif®culties, which complicate experimental X-ray crystallography and NMR
structure determination of GPCRs, the 3D structure of most GPCRs is still unknown. The only
known GPCR structure, a 2.8 AÊ resolution structure of rhodopsin, was published only recently by
Palczewski et al.21 This structure sheds light onto the mechanism of receptor activation and on
speci®c ligand receptor interactions. Until the publication of this work the only structural
information that existed about any GPCR was the low-resolution structure of rhodopsin that was
solved by cryoelectron microscopy.22 To this one should add the 2.5 AÊ resolution X-ray structure of
bacteriorhodopsin, obtained from micro crystals grown in lipid cubic phases, that was determined a
few years ago,23 even though bacteriorhodopsin is not a GPCR. The projection maps of bacte-
riorhodopsin and rhodopsin clearly showed the presence of seven TMs and con®rmed the basic
MODELIN G-PROTEIN-COUPLED RECEPTORS * 475

seven-helix bundle structure. However, the spatial organization of the TMs in rhodopsin is different
from that in bacteriorhodopsin. To date, rhodopsin is still the only GPCR of known 3D structure.
A known fact is that the location of the ligand binding-site differs from one receptor to another,
depending on the type of GPCR. Mutagenesis and biophysical studies of several GPCRs have
indicated that small-molecule agonists and antagonists bind to a hydrophilic pocket buried in the
transmembrane core of the receptor.24 For example, binding of biogenic amines to their corre-
sponding receptors is characterized by a complex network of interactions involving several
transmembrane domains in which key residues in TM3, TM5, and TM6 are essential for forming the
binding pocket, with speci®city for agonist recognition.25 It is believed that in these receptors, the
ligand's amine group pairs with a carboxyl group from an Asp residue located in TM3, whereas its
catechol ring interacts with residues in TM5 and TM6. Interactions of the ligand with TM3 through
its amine group are important for binding, while interactions with TM5 and TM6 are more important
for receptor activation.26 On the other hand, peptide ligands bind to both extracellular and
transmembrane domains.11 For example, in the NK1 receptor (which binds substance P, an 11 amino
acids peptide), three residues in the ®rst extracellular segment (Asn23, Gln24, and Phe25) and two
in the second (Asp96 and His108) are particularly required for ligand binding.27 Several residues in
TM2 and TM7 domains of this receptor (Asn85, Asn89, Tyr92, and Asn96 in TM2, and Tyr287 in
TM7) are, however, also important in determining the af®nity of the receptor for its ligand.28±29 For
moderate-sized peptides, binding usually occurs in both the extracellular loops and the N-terminal
segment and for larger ligands such as the glycoprotein hormone receptors, the binding site usually
resides solely within the extracellular N-terminus.30 Receptors that bind large ligands are often
characterized by long N-termini.31 For example, in the parathyroid hormone/calcitonin receptor
subfamily, an approximately 100-residue extracellular N-terminus contains regions shown to be
critical for ligand binding speci®city.32 The binding sites of agonists and antagonists of small
peptides are different, whereas the binding sites of larger peptide hormones and endothelin overlap
for both agonists and antagonists.20,33
4 . T H E M O D E L I N G A P P R O A C H
Due to the lack of experimental three-dimensional structures of GPCR membrane protein receptors,
structural insights must be inferred with the aid of three-dimensional computer models. As
discussed above, the structures of only two heptahelical membrane proteins were determined to date
in high resolution, rhodopsin and bacteriorhodopsin (the latter is not a GPCR).21,23,34 Therefore,
rhodopsin (and before it bacteriorhodopsin) is widely used as a template for modeling the backbone
structures of the TM domains of many GPCRs using homology-modeling techniques. Homology
modeling describes an extended collection of techniques with the goal of modeling the 3D structure
of a protein with an unknown structure, based on the known structures of related proteins. The
accuracy of the prediction relies heavily on the number of structures that serve as a template and on
their homology to the protein of interest (typically it requires more than 35% homology).35 This
method has proven very successful in modeling certain types of globular proteins. However,
applying homology modeling to GPCRs is hampered by the low sequence homology between most
GPCRs and rhodopsin (or bacteriorhodopsin). Furthermore, the great diversity of ligands that bind
to GPCRs and the known diversity in binding sites (discussed above) suggest that ligands may
interact with the receptor in different and diverse ways. Since the main purpose of GPCR models is
to describe the ligand binding sites, the homology modeling approaches are clearly limited in their
ability to predict novel binding pocket structures for the vast space of GPCR's ligands.
To overcome these problems we developed a new and novel modeling technology, named
PREDICT,36 which is able to predict the 3D structure of any GPCR. This approach requires as input
only the protein's amino acid sequence and is not based on the limited known structural information
476 * SHACHAM ET AL.

from rhodopsin or bacteriorhodopsin. While this method is close in its concept to ab initio protein
folding approaches, it is speci®cally directed towards structure prediction of membrane-embedded
polyhelical proteins. In particular the new modeling technology stands on two pedestals: protein±
protein interactions encoded in the protein's amino acid sequence (primary structure) and protein±
membrane interactions that highlight the role of the unique environment in which these receptors
are embedded. Figure 2 schematically depicts the concepts underlying the PREDICT modeling
approach.
The role of the membrane in this context cannot be overstressed, since it determines to a large
extent the folded structure of the protein. As depicted in Figure 3, the membrane, which is a bilayer
formed by phospholipids, is a complex environment with three spatially and chemically distinct
regions: a hydrophobic core formed by the phospholipid hydrocarbon tails, polar (or charged)
interfaces on both sides formed by the phospholipid head groups, and regions of ordered water.37 It
should be noted, that while the overall structure of the membrane is determined by the lipid
components, the interactions of lipids with the surrounding water molecules and with membrane-
bound proteins are responsible for much of its diversity and function. In particular, the complexity of
the membrane environment indicates that transmembrane helices should exhibit different properties
when interacting with different regions of the membrane. These considerations are taken into
account in the new modeling approach.
The main driving force for GPCR folding, like that of any other protein, is hydrophobicity.38
This driving force has two general consequences in terms of the present modeling procedure. First, it
is reasonable to assume that the TM helices form a closed structure of some sort; especially since
most TM helices in multihelical membrane embedded proteins are amphipathic.39 This general
assumption is corroborated by, but does not depend on, the observed closed packing arrangements
of the seven TM helices in both bacteriorhodopsin22 and rhodopsin.40±42 Second, the fact that
packing is hydrophobically driven can be used for optimizing the conformation of the folded
protein.
The new technology is speci®cally tailored for GPCRs since it uses some GPCR-speci®c
assumptions. In particular it is assumed that the TM helices are arranged in a sequential manner, so
that the TM order along the sequence is also their order in the folded structure. This is based
on Baldwin's sequence analysis of minimal lengths of interhelical loops,43 performed across the
whole GPCR superfamily, which strongly supports the simple topology. We also assume, in
agreement with known residue contacts in various GPCRs, that the TM helices are arranged in a
counterclockwise manner when viewed from the extracellular side.43±44 These assumptions are
introduced mainly for computational ef®ciency, in principle the same modeling technology
can be adapted to additional protein groups also characterized by membrane embedded helical
bundles.
Figure 2. Aschematic representationof the concepts underlying the PREDICTmodelingapproach.
MODELIN G-PROTEIN-COUPLED RECEPTORS * 477

A signi®cant obstacle facing GPCR modeling is that while it is fairly easy to roughly identify
the TM sequences, it is very hard to pinpoint the exact `̀ ends'' of the TM helices, namely, the
location of the boundaries of the transmembrane regions. Our modeling approach is unique in the
careful way it treats this uncertainty. Most computational studies rely on hydropathy pro®les to
predict the putative TM domains of the receptor. However, secondary structure prediction tools,
such as the PredictProtein program,45 are not accurate enough to determine the exact location of the
boundaries of TM helices (or any secondary structure element for that matter) and the corrugated
character of the membrane surface itself37,46 makes the precise de®nition of such a boundary
inappropriate. Indeed, in the new structure of rhodopsin TM1, 2, 3, and 6 are signi®cantly longer
than those predicted from hydropathy plots (30, 30, 33, and 31 compared with 25, 25, 20, and 24,
respectively). This critical issue, where the TMs cross the membrane surface, is carefully treated by
our technology at several different stages of the modeling process, each time optimizing this
property rather than assuming it is known.
Since the folded structure of proteins is characterized by their lowest free energy, the modeling
procedure gets to this low-energy conformation by optimizing the model for a large number of
properties, including helical-packing geometry, multihelical tilts, helix orientations, sidechain
Figure 3. An atomistic picture of a phospholipid bilayer. Depicted is a snapshot from a molecular dynamics simulations of a 1,2-dipalmitoyl-3-sn-phosphatidylcholine, DPPC (details in Refs. 37, 46).The three regions of the membrane are color coded: (a) Thehydrophobic core formed by the phospholipid hydrocarbon tails (red), (b) Polar (or charged) interfaces on both sides formed bythe phospholipid head groups (yellow), and (c) Regions of ordered water (blue). Overlaid is a schematic representation of a trans-membranehelix.Different helix properties characterize its interactionswith the differentmembrane regions.
Table I. PREDICT Optimization
1. Helical-packinggeometry2. Multihelical tilts3. Helixorientations4. Sidechain rotamers5. Helixmembrane-surface crossing6. Helical kinks
478 * SHACHAM ET AL.
rotamers, helix membrane-surface crossing, and helical kinks (Table I). The new modeling
procedure deals with the huge size of the protein conformation space through a unique hierarchical
design, starting with a coarse representation and gradually increasing the complexity of the
representation until reaching a full atomistic model. This algori thmic design also has a bene®t of
being computationally very ef®cient.
5 . R E S U L T S
The new PREDICT technology was already used for generating 3D models of many family-A
GPCRs. As expected, the resulting models span a wide range of conformations, some of them
signi®cantly different from the structure of rhodopsin. This is not surprising, since, for example,
many peptide receptors have signi®cantly different binding-site characteristics than in rhodopsin.
These models also agree well with a broad range of biological mutagenesis data highlighting 3D
binding sites that can be used for structure-based drug discovery.
In most cases the PREDICT technology is able to distinguish between favorable folded
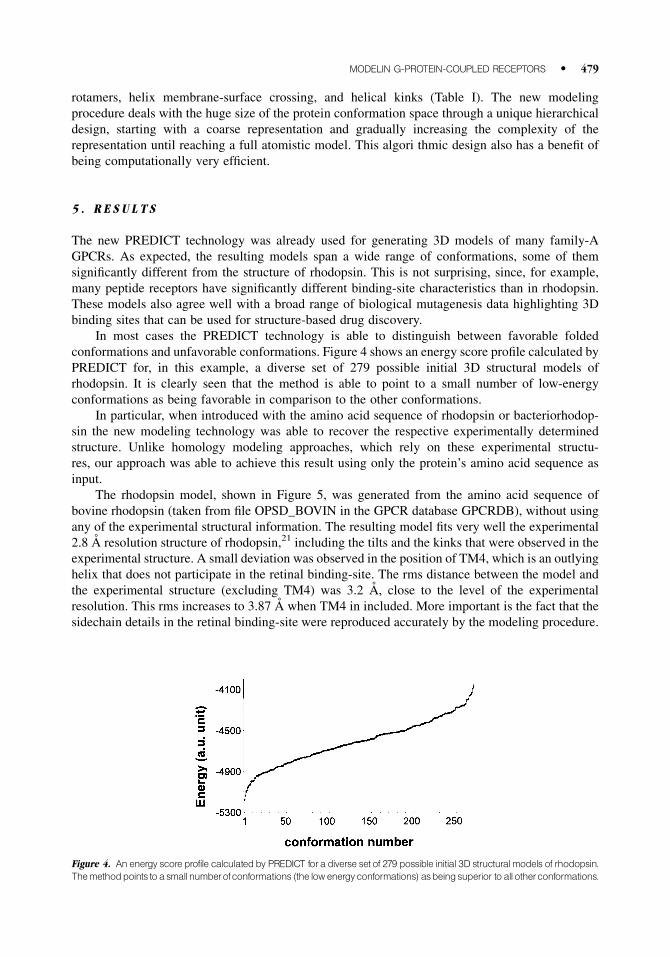
conformations and unfavorable conformations. Figure 4 shows an energy score pro®le calculated by
PREDICT for, in this example, a diverse set of 279 possible initial 3D structural models of
rhodopsin. It is clearly seen that the method is able to point to a small number of low-energy
conformations as being favorable in comparison to the other conformations.
In particular, when introduced with the amino acid sequence of rhodopsin or bacteriorhodop-
sin the new modeling technology was able to recover the respective experimentally determined
structure. Unlike homology modeling approaches, which rely on these experimental structu-
res, our approach was able to achieve this result using only the protein's amino acid sequence as
input.
The rhodopsin model, shown in Figure 5, was generated from the amino acid sequence of
bovine rhodopsin (taken from ®le OPSD_BOVIN in the GPCR database GPCRDB), without using
any of the experimental structural information. The resulting model ®ts very well the experimental
2.8 AÊ resolution structure of rhodopsin,21 including the tilts and the kinks that were observed in the
experimental structure. A small deviation was observed in the position of TM4, which is an outlying
helix that does not participate in the retinal binding-site. The rms distance between the model and
the experimental structure (excluding TM4) was 3.2 AÊ , close to the level of the experimental
resolution. This rms increases to 3.87 AÊ when TM4 in included. More important is the fact that the
sidechain details in the retinal binding-site were reproduced accurately by the modeling procedure.
Figure 4. An energy score profile calculatedby PREDICT fora diverse set of 279 possible initial 3D structural models of rhodopsin.Themethodpoints toasmallnumberofconformations (the low energyconformations) asbeingsuperior toall otherconformations.
MODELIN G-PROTEIN-COUPLED RECEPTORS * 479
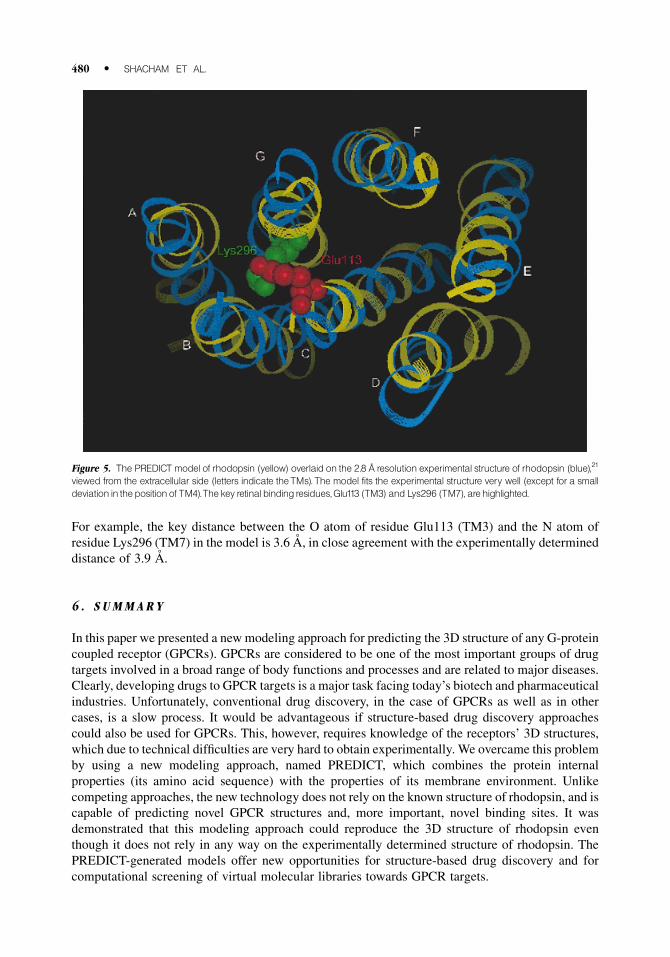
For example, the key distance between the O atom of residue Glu113 (TM3) and the N atom of
residue Lys296 (TM7) in the model is 3.6 AÊ , in close agreement with the experimentally determined
distance of 3.9 AÊ .
6 . S U M M A R Y
In this paper we presented a new modeling approach for predicting the 3D structure of any G-protein
coupled receptor (GPCRs). GPCRs are considered to be one of the most important groups of drug
targets involved in a broad range of body functions and processes and are related to major diseases.
Clearly, developing drugs to GPCR targets is a major task facing today's biotech and pharmaceutical
industries. Unfortunately, conventional drug discovery, in the case of GPCRs as well as in other
cases, is a slow process. It would be advantageous if structure-based drug discovery approaches
could also be used for GPCRs. This, however, requires knowledge of the receptors' 3D structures,
which due to technical dif®culties are very hard to obtain experimentally. We overcame this problem
by using a new modeling approach, named PREDICT, which combines the protein internal
properties (its amino acid sequence) with the properties of its membrane environment. Unlike
competing approaches, the new technology does not rely on the known structure of rhodopsin, and is
capable of predicting novel GPCR structures and, more important, novel binding sites. It was
demonstrated that this modeling approach could reproduce the 3D structure of rhodopsin even
though it does not rely in any way on the experimentally determined structure of rhodopsin. The
PREDICT-generated models offer new opportunities for structure-based drug discovery and for
computational screening of virtual molecular libraries towards GPCR targets.
Figure 5. The PREDICT model of rhodopsin (yellow) overlaid on the 2.8 Ð resolution experimental structure of rhodopsin (blue),21
viewed from the extracellular side (letters indicate theTMs).The model fits the experimental structure very well (except for a smalldeviation in thepositionof TM4).Thekey retinal bindingresidues,Glu113 (TM3) and Lys296 (TM7), arehighlighted.
480 * SHACHAM ET AL.

R E F E R E N C E S
1. Baldwin JM. Structure and function of receptors coupled to G proteins. Curr Opin Cell Biol 1994;6:
180±190.
2. Strader CD, Fong TM, Tota MR, Underwood D, Dixon RA. Structure and function of G protein-coupled
receptors. Annu Rev Biochem 1994;63:101±132.
3. Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell
functions. Nature 1990;348:125±132.
4. Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular
mechanism. Nature 1991;349:117±127.
5. Sprang SR. G protein mechanisms: insights from structural analysis. Annu Rev Biochem 1997;66:639±
678.
6. Dhanasekaran N, Heasley LE, Johnson GL. G protein-coupled receptor systems involved in cell growth
and oncogenesis. Endocr Rev 1995;16:259±270.
7. van Biesen T, Luttrell LM, Hawes BE, Lefkowitz RJ. Mitogenic signaling via G protein-coupled
receptors. Endocr Rev 1996;17:698±714.
8. Bockaert J, Pin JP. Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J
1999;18:1723±1729.
9. Probst WC, Snyder LA, Schuster DI, Brosius J, Sealfon SC. Sequence alignment of the G-protein coupled
receptor superfamily. DNA Cell Biol 1992;11:1±20.
10. Dalman HM, Neubig RR. Two peptides from the alpha 2A-adrenergic receptor alter receptor G protein
coupling by distinct mechanisms. J Biol Chem 1991;266:11025±11029.
11. Strader CD, Fong TM, Graziano MP, Tota MR. The family of G-protein-coupled receptors. FASEB J
1995;9:745±754.
12. Loosfelt H, Misrahi M, Atger M, Salesse R, Thi VH-L, Jolivet A, Guiochon-Mantel A, Sar S, Jallal B,
Garnier J. Cloning and sequencing of porcine LH-hCG receptor cDNA: variants lacking transmembrane
domain. Science 1989;245:525±528.
13. Heckert LL, Daley IJ, Griswold MD. Structural organization of the follicle-stimulating hormone receptor
gene. Mol Endocrinol 1992;6:70±80.
14. Kaiser UB, Zhao D, Cardona GR, Chin WW. Isolation and characterization of cDNAs encoding the rat
pituitary gonadotropin-releasing hormone receptor. Biochem Biophys Res Commun 1992;189:1645±
1652.
15. Kakar SS, Musgrove LC, Devor DC, Sellers JC, Neill JD. Cloning, sequencing, and expression of
human gonadotropin releasing hormone (GnRH) receptor. Biochem Biophys Res Commun 1992;189:
289±295.
16. Bargmann CI. Olfactory receptors, vomeronasal receptors, and the organization of olfactory information.
Cell 1997;90:585±587.
17. Slusarski DC, Corces VG, Moon RT. Interaction of Wnt and a Frizzled homologue triggers G-protein-
linked phosphatidylinositol signalling. Nature 1997;390:410±413.
18. Barnes MR, Duckworth DM, Beeley LJ. Frizzled proteins constitute a novel family of G protein-coupled
receptors, most closely related to the secretin family. Trends Pharmacol Sci 1998;19:399±400.
19. Sugita S, Ichtchenko K, Khvotchev M, Sudhof TC. Alpha-latrotoxin receptor CIRL/latrophilin 1 (CL1)
de®nes an unusual family of ubiquitous G-protein-linked receptors. G-protein coupling not required for
triggering exocytosis. J Biol Chem 1998;273:32715±32724.
20. Beck-Sickinger AG. Structural characterization and binding sites of G protein-coupled receptors. Drug
Discov Today 1996;1:502±513.
21. Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le TI, Teller DC, Okada T,
Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor.
Science 2000;289:739±745.
22. Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the stru-
cture of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol 1990;213:
899±929.
23. Pebay-Peyroula E, Rummel G, Rosenbusch JP, Landau EM. X-ray structure of bacteriorhodopsin at 2.5
angstroms from microcrystals grown in lipidic cubic phases. Science 1997;277:1676±1681.
MODELIN G-PROTEIN-COUPLED RECEPTORS * 481

24. Kim HK. Building a Bridge between G-protein-coupled receptor modelling, protein crystallography and
3D QSAR studies for ligand design. Pers Drug Discov Design 1998;12/13/14:233±255.
25. Dixon RA, Sigal IS, Rands E, Register RB, Candelore MR, Blake AD, Strader CD. Ligand binding to the
beta-adrenergic receptor involves its rhodopsin-like core. Nature 1987;326:73±77.
26. Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RA. Identi®cation of two serine residues involved
in agonist activation of the beta-adrenergic receptor. J Biol Chem 1989;264:13572±13578.
27. Fong TM, Yu H, Huang RR, Strader CD. The extracellular domain of the neurokinin-1 receptor is
required for high-af®nity binding of peptides. Biochemistry 1992;31:11806±11811.
28. Yokota Y, Akazawa C, Ohkubo H, Nakanishi S. Delineation of structural domains involved in the subtype
speci®city of tachykinin receptors through chimeric formation of substance P/substance K receptors.
EMBO J 1992;11:3585±3591.
29. Fong TM, Huang RR, Yu H, Strader CD. Mapping the ligand binding site of the NK-1 receptor. Regul
Pept 1993;46:43±48.
30. Ulloa-Aguirre A, Timossi C. Structure-function relationship of follicle-stimulating hormone and its
receptor. Hum Reprod Update 1998;4:260±283.
31. Ulloa-Aguirre A, Conn, PM. G protein-coupled receptors and the G protein family. In: P.M. Conn, editor.
Handbook of physiology-endocrinology. New York: Oxford University Press, 1998. p 87.
32. Juppner H, Schipani E, Bringhurst FR, McClure I, Keutmann HT, Potts JTJ, Kronenberg HM, Abou-
Samra AB, Segre GV, Gardella TJ. The extracellular amino-terminal region of the parathyroid hormone
(PTH)/PTH-related peptide receptor determines the binding af®nity for carboxyl-terminal fragments of
PTH-(1-34). Endocrinology 1994;134:879±884.
33. Gether U, Johansen TE, Snider RM, Lowe JA, Nakanishi S, Schwartz TW. Different binding epitopes on
the NK1 receptor for substance P and non-peptide antagonist. Nature 1993;362:345±348.
34. Kimura Y, Vassylyev DG, Miyazawa A, Kidera A, Matsushima M, Mitsuoka K, Murata K, Hirai T,
Fujiyoshi Y. Surface of bacteriorhodopsin revealed by high-resolution electron crystallography. Nature
1997;389:206±211.
35. Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling. In: Becker OM, MacKerell
AD, Jr., Roux B, Watanabe M, editors. Computational biochemistry and biophysics. New York: Marcel
Dekker; 2001. p 275±312.
36. PREDICT is a proprietary technology of Bio IT (Bio Information Technologies) Ltd., Israel.
37. Bachar M, Becker OM. Melittin at a membrane/water interface: effects on water orientation and water
penetration. J Chem Phys 1999;111:8672±8685.
38. Chothia C. Principles that determine the structure of proteins. Annu Rev Biochem 1984;53:537±572.
39. Eisenberg D, Weiss RM, Terwilliger TC. The hydrophobic moment detects periodicity in protein
hydrophobicity. Proc Natl Acad Sci USA 1984;81:140±144.
40. Unger VM, Schertler GF. Low resolution structure of bovine rhodopsin determined by electron cryo-
microscopy. Biophys J 1995;68:1776±1786.
41. Schertler GF, Villa C, Henderson R. Projection structure of rhodopsin. Nature 1993;362:770±772.
42. Schertler GF. Structure of rhodopsin. Eye 1998;12:504±510.
43. Baldwin JM, Schertler GF, Unger VM. An alpha-carbon template for the transmembrane helices in the
rhodopsin family of G-protein-coupled receptors. J Mol Biol 1997;272:144±164.
44. Du P, Salon JA, Tamm JA, Hou C, Cui W, Walker MW, Adham N, Dhanoa DS, Islam I, Vaysse PJ,
Dowling B, Shifman Y, Boyle N, Rueger H, Schmidlin T, Yamaguchi Y, Branchek TA, Weinshank RL,
Gluchowski C. Modeling the G-protein-coupled neuropeptide Y Y1 receptor agonist and antagonist
binding sites. Protein Eng 1997;10:109±117.
45. Rost B, Casadio R, Fariselli P, Sander C. Transmembrane helices predicted at 95% accuracy. Proteins
1995;13:59.
46. Bachar M, Becker OM. Protein induced membrane disorder: a molecular dynamics study of melittin in a
dipalmitoylphosphatidylcholine. Bilayer Biophys J 2000;78:1359±1375.
Dr. Sharon Shacham is head of development at Bio IT Ltd. She received a B.Sc. degree in chemistry, MBA
degree, and a Ph.D. degree in biochemistry and computational biology from Tel Aviv University. She has
academic and industry experience in information systems and bioinformatics programming.
482 * SHACHAM ET AL.

Maya Topf is a D.Phil student in the Department of Physical and Theoretical Chemistry, Oxford University,
UK. She received a B.Sc. degree and a M.Sc. degree in Chemistry from Tel Aviv University, Israel. She has been
working on protein design and on applications of QM/MM methods to model enzymatic reactions.
Noa Avisar is a research scientist at Bio IT Ltd. She received a B.Sc. degree in biology, a M.Sc. in
neuroendocrinolgy, and a Ph.D. degree in biochemistry and molecular biology from Tel Aviv University, Israel.
She has industry experience in bioinformatics and structural biology.
Fabian Glaser is a research scientist at Bio IT Ltd. and a Ph.D. student in the Department of Biochemistry, Tel
Aviv University, Israel. He rceived a B.Sc. degree and an M.Sc in Medicinal Chemistry from the Hebrew
University of Jerusalem, Israel. He has academic and industrial experience in analysis of protein surface
properties and in information technology.
Dr. Yael Marantz is a research scientist and project manager at Bio IT, Ltd. She received a B.Sc. degree in
chemistry, a M.Sc. degree in biochemistry and endocrinology, and a Ph.D. in biophysics and structural biology
from Tel Aviv University, Israel. She has industry experience in bioinformatics and computational drug
discovery.
Shay Bar-Haim is a research scientist in the development group at Bio IT, Ltd. He received a B.Sc. in chemistry
from Bar-Ilan University, Israel, and a M.Sc. in biology from the Weizmann Institute of Science, Israel. He has
industry experience in computer programming and bioinformatics.
Dr. Silvia Noiman is a cofounder and Chief Operations Of®cer of Bio IT. She is the author or coauthor of
numerous publications in molecular biology. She received a M.Sc. degree in population genetics, a MBA
degree, and a Ph.D. degree in molecular biology from Tel Aviv University, Israel. She formerly held an
academic position at the Weizmann Institute of Science, Israel, was the coordinator of the Israel National
Biotechnology Committee and founded and managed a diagnostic laboratory for genetic diseases in Tel-
Hashomer Hospital, Israel.
Prof. Zvi Naor is a Professor of biochemistry at Tel Aviv University, Israel. He formerly held an academic
position at the Weizmann Institute of Science, Israel and was a visiting scientist at the NIH and at Kobe Medical
School, Japan. He is the author or coauthor of numerous publications in endocrinology. He received a B.Sc.
degree in chemistry and M.Sc. in biochemistry from Bar-Ilan University, Israel. He received a Ph.D. degree in
biochemistry from the Weizmann Institute of Science, Israel and was a Postdoctoral Fellow at the University of
Texas, Health Scince Center. He is the recipient of the Chaim Weizmann Phostdoctoral Fellowship (1976), the
Charles H. Revson Career Development Chair (1980), the Juludan Prize (1989), and the Israel Fertility
Association Prize (1991).
Prof. Oren M. Becker is a cofounder, Chief Scientist, and Chief Technology Of®cer of Bio IT Ltd, formerly an
Assistant Professor at Tel Aviv University, Israel was a visiting Assistant Professor at Harvard University,
Cambridge, Massachusetts. He is the author or coauthor of numerous publication on protein modeling and
simulation. He is a coeditor of the textbook `̀ Computational Biochemistry and Biophysics'' (Marcel Dekker:
NY 2001). He received a B.A. degree in philosophy, a B.Sc. degree in chemistry and physics and a Ph.D. degree
in theoretical chemistry from the Hebrew University of Jerusalem, Israel, and was a Postdoctoral Fellow at
Harvard University, Cambridge, Massachusetts. He is the recipient of the Rothschild Postdoctoral Fellowship
(1991), the Fulbright Postdoctoral Fellowship (1991), and the Yig'al Alon Fellowship for Outstanding Young
Scientists (1994).
MODELIN G-PROTEIN-COUPLED RECEPTORS * 483





![Acquisition of 3D Data - Research Unit of Computer Graphics · 3D Data Animation, film: a sequence of images with time as the third coordinate: [x,y,t] Volume data: a sequence of](https://img.pdfslide.us/doc/110x75/6121c20d533c5226f370096b/acquisition-of-3d-data-research-unit-of-computer-graphics-3d-data-animation-film.jpg)























