Embed Size (px)
Citation preview

React. Kinet. Catal. Lett., Vol. 18, No. 1 -2 , 13 -16 (1981)
MINIMUM ENERGY REACTION PATHS BY A MODIFIED PAULING RELATION
G. Harlschmarm
Zentralinstitut flit Isotopen- und Strahlenforschung, Akademie der Wissenschaften der DDR, DDR-7050 Leipzig, GDR
Received March 6, 1981 Accepted April 7, 1981
A simple procedure for adjustment of the constant in the Pauling relation between bond order and bond length to generate reaction coordinates is proposed by means of a proton transfer as an example. The magnitude of this constant reflects bond and reaction types.
1-IpoDJIo>KeH npocToi~ MeT0~ npHcIIOCO6IIeHHH HOCTO$IHI-I0iCl B COOTHOIIIeHHH I"Io/II,IHFa
Me~y IIopH~KOM H ~HHHO~ CBH3H ~qH yCTaI-IOBJIeHti,~ peaKL~HOHHblX K00p;DIHaT. I]pH 3TOM nepeHOC IIpOTOHa cIIy~'rlT HpnMepOM. BeHHqHHa 3T01~ HOCTO~IKHOi~ oTpa~aeT xapaKTep CBH3H r~ peaKttttH.
INTRODUCTION
Several years ago we showed the possibility of the combined use of semiempir- ical rules and quantum-chemical methods in the calculation of minimum energy paths of hydrogen atom abstraction reactions/I-3/. Considering a simple exchang~ reaction of the type
A + B C = A B + C (1)
we employed the following two relations to generate the reaction coordinate, namely the well-known Pauling relation/4/
R = R s - 0 . 6 1 o g n (2)
and the principle of bond-order conservation
nAB + nBr = 1 (3)
13

HANSCHMANN: PAULING RELATION
both of which are basically used in the BEBO method /5 -10 / (for other applica-
tions of Eqs (2) and (3), see Refs . /11-15/) . In Eq. (2) R denotes the covalent
radius (in units of 0.1 nm) of an atom which carries n ligands, whereas R s refers
to the case of a single ligand. Johnston and Par r /5 / applied Eq. (2) to reactions where n also covers the range
between one and zero. So the bond number is altered to a formal bond-order (nAB and nBc in Eq. (3) e.g.), but the same constant (0.6) as in Eq. (2) is accepted for
all possible bonds and reactions. This seems to be somewhat arbitrary, for we should expect the constant to be dependent on the bond type as well as on the
reaction mechanism. Some authors have mentioned such a correlat ion/! 6, 17/ and proposec~ a pos-
sible generalization of Eq. (2) /17/
R x y = (RxY)s - C log n x v (4)
where R xy and (Rxv)s designate the bond lengths of the partial and single bonds, respectively, and the constant C is treated as an adjustable parameter.
In this paper we suggest a simple procedure for the determination of that con-
stant.
Determination of constant C
When dealing with a thermo-neutral reaction, one can solve Eq. (4) for the con-
stant C, because in this case the value of the bond-order nxv is immediately known
for one special point on the reaction path, namely nxv = 0.5 in the saddle point
of the corresponding potential energy (PE) surface. So we write
C = Rxy - (RxY)s / log 2 (5)
The only unknown quantity in Eq, (5), Rxy , the bond length in the transition
state, can be obtained from a sufficiently accurate ab initio PE surface.
This is briefly demonstrated by the reaction
N'H] + NH3 = N'H3 + NH] (6)
for which precise ab initio - SCF calculations are known /18, 19/. The value of 1.2935 • 0.1 nm for the N ' -H 1 (or N - H 1) distance in the transition state of the reaction complex [ H 3 N ' . . . H 1 . . . NH3] § together with the ab initio equilibrium bond length of 1.016 • 0.1 nm/18/ , yield 0.92 for constant C in Eq. (5).
14
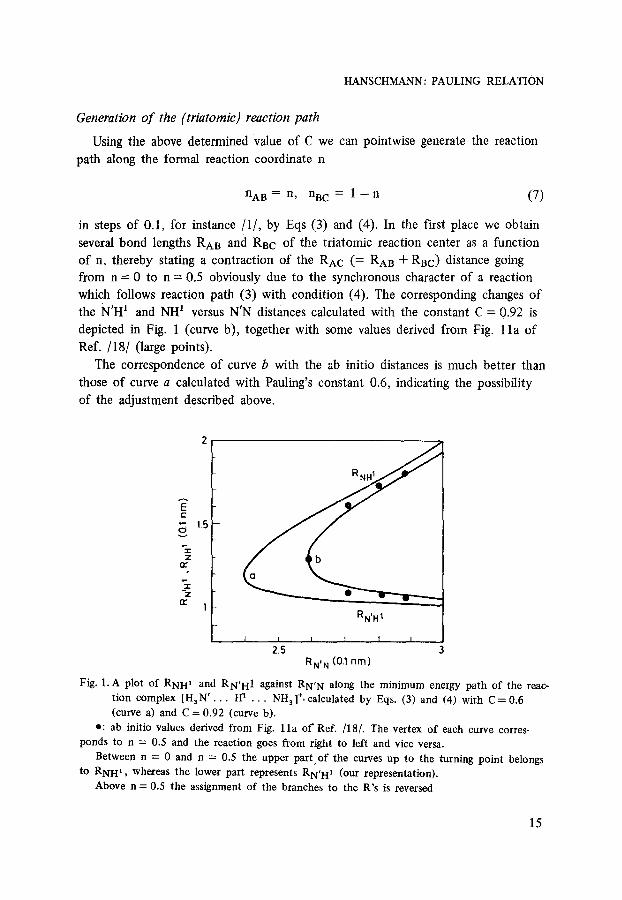
HANSCHMANN: PAULING RELATION
Generation of the (triatomic) reaction path
Using the above determined value of C we can pointwise generate the reaction
path along the formal reaction coordinate n
n A B = n, nBC = 1 - - n (7)
in steps o f 0.1, for instance /1/ , by Eqs (3) and (4). In the first place we obtain
several bond lengths RAB and R a c o f the t r iatomic reaction center as a function
of n, thereby stating a contract ion of the RAC (= RAB + RBC) distance going
from n = 0 to n = 0.5 obviously due to the synchronous character o f a reaction
which follows reaction pa th (3) with condit ion (4). The corresponding changes o f
the N'H 1 and NH 1 versus N'N distances calculated with the constant C = 0.92 is
depicted in Fig. 1 (curve b), together with some values derived from Fig. 1 l a o f
Ref. /18/ (large points).
The correspondence of curve b with the ab initio distances is much bet ter than
those o f curve a calculated with Pauling's constant 0.6, indicating the possibili ty
of the adjustment described above.
E r
v
15
-1-
Z t - r
"3"
Z
1 RN'H1
I I I I I I 2.5
R N , N (0.1 n m )
Fig. 1. A plot of RNH, and RN'H1 against RN' N along the minimum energy path of the reac- tion complex [Ha N ' . . . H a . . . NH 3]+, calculated by Eqs. (3) and (4) with C = 0.6 (curve a) and C = 0.92 (curve b).
r ab initio values derived from Fig. l l a of Ref. /18/. The vertex of each curve corres- ponds to n = 0.5 and the reaction goes from right to left and vice versa.
Between n = 0 and n = 0.5 the upper part of the curves up to the turning point belongs to RNH 1 , whereas the lower part represents RN, H, (our representation).
Above n = 0.5 the assignment of the branches to the R's is reversed
15

HANSCHMANN: PAULING RELATION
Changes of atomic coordinates aside from the triatomic reaction center one can
determine by further empirical structural correlations like that presented in Ref . /16/ .
DISCUSSION
Our aim was to show by an example a possibility to adjust the constant in the
Pauling relation for generation of the nearly true minimum energy reaction path.
The modified Pauling relation can then be used for the construction o f analytical
potentials (like BEBO) as well as in ab initio quantum-chemical PE calculations,
where a straightforward multidimensional geometry variation is not practicable.
The magnitude o f C reflects the farces which act in a reaction. A small value
of C (0.6 in H2 + H = H + H~ /20/) corresponds to a covalent character o f the
reaction with a strong contraction o f the reaction complex due to orbital overlap,
whereas a higher value o f C (0.92 in a proton transfer) counts for long-range ionic
forces and results in a less strong contraction of the system.
Theoretical foundation and practical extension of the proposed concept to other
(synchronous) reactions and cases with nonsymmetrical PE courses require further
investigation. However, we should apply the "symmetrically" determined constant
C rather than the single constant 0.6 as a first approximation for cases with similar
bonds and reaction mechanisms, too.
REFERENCES
1, G. 2. G. 3. G. 4. L. 5. H. 6. H. 7. R. 8. R. 9. C.
10. B. 11. T. 12. O. 13. A. 14. R.
Hanschmann, H.-J. K6hler: Z. Chem., 13, 470 (1973). Hanschmann, H.-J. K6hler: Z. Phys. Chem. (Leipzig), 255, 1192 (1974). Hanschmann, H.-J. K6hler: Z. Chem., 14, 455 (1974). Pauling: J. Amer. Chem. Soc., 69, 542 (1947). S. Johnston, C. Parr: J. Amer. Chem. Soc., 85, 2544 (1963). S. Johnston: Gas Phase Reaction Rate Theory. The Ronald Press Co., New York 1966. D. Gilliom: J. CheM. Phys., 65, 5027 (1976). D. Gilliom: J. Amer. Chem. Soc., 99, 8399 (1977). M. Marschoff, A. Jatem: Chem. Phys. Lett., 56, 35 (1978). C. Garrett, D. G. Truhlar: J. Amer. Chem. Soc., 101, 4534 (1979). B&ces, J. Dombi: React. Kinet. Catal. Lett., 5, 28t (1976). Kafri, M. J. Berry: Faraday Discuss. Chem. Sor 62, 127 (1977). R. Miller: J. Amer. Chem. Soc., 100, 1984 (1978). F. Nalewajski, T. S. Carlton: Acta Phys. Polon., A53, 321 (1978).
15. N. Agmon: J. C. S., Faraday Trans. II, 74, 388 (1978). 16. H.-B. Btirgi: Angew. Chem., 87, 461 (1975). 17. N. Agmon: Chem. Phys. Lett., 45, 343 (1977). 18. J.-J. Delpuech, G. Serratrice, A. Strich, A. Veillard: Mol. Phys,, 29, 849 (1975). 19. P. Merlet, S. D. Peyerimhoff, R. J. Buenker: J. Amer. Chem. Soc., 94, 8301 (1972). 20. D. G. Truhlar: J. Amer. Chem. Soc., 94, 7584 (1972).
16





























