Embed Size (px)
Citation preview

protocol
408 | VOL.5 NO.3 | 2010 | nature protocols
p
uor
G g
n ih si l
bu
P eru ta
N 010 2©
nat
ure
pro
toco
ls/
moc. e r
ut an .
ww
w / /:pt t
h
IntroDuctIonChromatographic separations are extensively employed in the downstream processing of recombinant protein bioproducts in both laboratory and industrial scale operations1–6. However, in most cases, chromatographic methods development traditionally involves time-consuming trial and error approaches. Typically, chromatographic workstations are employed in order to carry out large numbers of labor-intensive experimental runs and fraction analyses to identify a successful purification method. This approach can take upto several days. As a result, there is great interest in the development of new methods that can accelerate the identification of chromatographic steps for protein purification, both for labora-tory-scale research applications and preparative-scale processes in the biotechnology industry.
High-throughput screening (HTS) and combinatorial methods have been used extensively for the discovery of drug leads in the pharmaceutical industry7–10 and for the identification of affin-ity ligands for proteins from epitope libraries11,12. Combinatorial methods have been recently employed in applications relating to protein purification. For example, combinatorial techniques were used to facilitate the identification of biospecific ligands to allow the purification of a wide range of proteins by affinity chromatography13–22. Methods based on microtiter-plate tech-niques have been employed for the miniaturized purification of histidine-tagged proteins23,24 using immobilized metal-affinity chromatography. Recently, we have employed parallel screening techniques to rapidly identify small molecules as high affinity and selective displacers for protein purification using displacement chromatography25–29.
Here we describe a miniaturized method that uses 96-well micro-titer plates for the high-throughput identification of chromato-graphic steps for obtaining pure protein from expression broths30. Protein mixtures present in cell-free expression broths are used as the feed for screening chromatographic purification steps using the miniaturized screen. Note that while the following protocol
is described for expression in Escherichia coli using a suitable promoter (e.g., T7 promoter), the screening procedure itself is inde-pendent of the expression methods used. Thus, the screening can be used with clarified broths of bacterial, yeast, insect, fungal and mammalian cultures. The microtiter plate based parallel screening approach successfully identifies chromatographic resins (or media; note that the terms chromatographic resins and chromatographic media are used interchangeably throughout the text), operating conditions and protein purity levels necessary to allow scaled-up purification, illustrating that the results are transferable across dif-ferent scales of operation. Extension of the current approach using automated liquid handling systems and sophisticated robotics can serve to enhance the screening throughput, resulting in further acceleration of methods development31.
In this method, feed mixture containing the protein of inter-est (bioproduct) is equilibrated with aliquots of commercially available chromatographic resins in membrane-bottomed 96-well plates placed on top of a vacuum manifold. A variety of chro-matographic modes, including, anion-exchange (AEx), cation-exchange (CEx), hydrophobic interaction chromatography (HIC) and hydrophobic charge-induction chromatography (HCIC) can be employed in the screen. Serial step elution is carried out using different concentrations of the eluent (e.g., increasing salt con-centrations in ion-exchange chromatography) in order to elute the bioproduct from chromatographic resins; negative pressure (vacuum) is applied in order to facilitate the elution. Fractions are collected in a 96-well collection plate at the bottom of the vacuum manifold after elution from the membrane-bottomed plate and their purity is analyzed using SDS-PAGE to identify the elution conditions and chromatographic media that result in high purities of the desired protein. Densitometry analysis using SDS-PAGE gels can be employed to generate semi-quantitative infor-mation on bioproduct purity in eluted fractions. Other analytical methods such as protein chips using the Agilent Bioanalyzer,
Miniaturized parallel screens to identify chromatographic steps required for recombinant protein purificationKaushal Rege1 & Meng Heng2
1Department of Chemical Engineering, ECG 202, Arizona State University, Tempe, Arizona, USA. 2Process Engineering and Technology, Genencor, A Danisco Division, Palo Alto, California, USA. Correspondence should be addressed to K.R. ([email protected]).
Published online 11 February 2010; doi:10.1038/nprot.2009.149
Methods development in chromatography is a time-consuming, trial-and-error process that requires laborious experimentation. We describe a high-throughput screening (Hts) protocol for the rapid identification of chromatographic steps for protein purification from cell-free expression broths. Broths containing the protein are loaded on different chromatographic resins aliquotted in membrane-bottomed microtiter plates. serial step elution of protein from resins results in fraction collection in 96-well plates. choice of the optimal chromatographic operating conditions is based on protein purity in eluted fractions, determined using sDs-paGe analysis or similar analytical techniques. the screening procedure is then repeated in order to identify the subsequent chromatographic steps, ultimately leading to high purities of the protein. the protocol takes ~24 h in order to determine the required sequence of chromatographic steps. the use of a miniaturized screen facilitates screening of a range of media and operating conditions (i.e., pH, salt concentration, and so on.) in parallel and is a novel approach to chromatographic methods development.

protocol
nature protocols | VOL.5 NO.3 | 2010 | 409
p
uor
G g
n ih si l
bu
P eru ta
N 010 2©
nat
ure
pro
toco
ls/
moc. e r
ut an .
ww
w / /:pt t
h
ELISAs and capillary electrophoresis assays, are also viable alter-natives to SDS-PAGE analysis. It is important to note that the throughput of the screening protocol is greatly influenced by the throughput of the protein analysis method employed.
The selection of appropriate chromatographic mode (i.e., ion exchange), media (i.e., type of chromatographic resin) and operat-ing conditions (i.e., pH and salt concentration) for the first chro-matographic purification step is based on the purity of protein in the eluted fractions. Considerations such as the relative purity of the bioproduct and number of contaminants are important in selecting appropriate chromatographic conditions, and this proto-col allows the selection of those chromatographic conditions that result in highest purities (and least number of contaminating pro-teins). Orthogonal chromatographic techniques can be employed subsequently to remove the remaining purities. An intermediate step involving a resin volume of 1 ml is then used to obtain enough protein as feed load for the second step, and the first chroma-tographic dimension that results in the greatest enrichment of the bioproduct is used to identify the next chromatographic step. Similar to the first dimension, fractions from the second chro-matographic dimension are analyzed to identify a set of chroma-tographic operating conditions that result in high purities of the bioproduct. This screening procedure can be repeated using mul-tiple chromatographic dimensions, within which varying operat-ing conditions and modes of chromatography can be tested until high purities (e.g., ‘single band’ purities using SDS-PAGE analysis)
of the protein of interest are achieved. The optimal sequence of chromatographic steps and operating conditions determined using the microtiter-plate screening are then recommended for purify-ing the bioproduct from expression broths using linear gradient column chromatography. While step elutions are employed in the microtiter screening, linear gradient chromatography is employed in the column chromatography-based protein purification because of the high resolving power of linear gradient chromatography. The overall high-throughput approach is shown in Figure 1.
Advantages and limitations of the HTS methodThe most significant advantage of this approach is that parallel screening of chromatographic modes and operating conditions can result in the rapid identification of the sequence of chroma-tographic steps required for purification of the bioproduct of interest. This is a considerable improvement over traditional methods involving column chromatographic runs that require days, if not weeks, for identifying the appropriate purification methods. In principle, the method allows for the identification of multiple sequences of chromatographic steps that can result in high purities of the desired protein. This, in turn, presents users with different options that they can use for obtaining a purified bioproduct.
The volatility of organic modifiers under vacuum makes this screening approach unsuitable for reversed phase chromatography (RPC). This is not a significant limitation in cases where functional proteins are required at the end of the purification steps, as organic solvents used in RPC (e.g., acetonitrile) often compromise protein structure and function32,33. An additional limitation of the present method is that size-exclusion chromatography (SEC) cannot be employed in the screen. SEC requires a large number of theoretical plates for the high-resolution separation of proteins by size. The small volumes of resin used in the screening protocol do not allow for a large number of theoretical plates. Another limitation of the microtiter screening approach is that it is not possible to gener-ate continuous gradients using a vacuum manifold as a result of which, serial step elutions are employed in the screening. Finally, although the screening of chromatographic modes and operating conditions is rapid and can be automated, the throughput of con-comitant methods for analyzing protein purity can limit the overall throughput of this approach.
Feed equlibration: equilibration of feed protein mixture on aliquots ofchromatographic media in 96-well membrane bottomed plates
Elution: serial step elution of feed; fraction collection in 96-well plates
Analytical: analysis of protein purity in eluted fractions using sds-page
Screen recommendation: selection of chromatographic media and operatingconditions based on protein purity
Protein purification: column chromatographic purificationbased on screening recommendations
Figure 1 | Flowsheet schematic of the screening approach employed for the high-throughput identification of chromatographic purification steps.
MaterIalsREAGENTS
Chromatographic media used in the screening are listed in Table 1. crItIcal The screen may be expanded to include different commercially available media. The media should be stored as recommended by the respective vendors.Acetic acid (Sigma-Aldrich, cat. no. A6283-500ML)Ammonium chloride (Sigma-Aldrich, cat. no. A4514-500G)Ammonium sulfate (Sigma-Aldrich, cat. no. A5132-500G)Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, cat. no. 276855-100ML)Halt protease inhibitor cocktail (Pierce Biotechnology, cat. no. 78425)Isopropyl β-D-1-thiogalactopyranoside (IPTG; Research Products International Corporation, cat. no. I56000)β-Mercaptoethanol (BME) (Sigma-Aldrich, cat. no. M6250-250ML)Sodium phosphate monobasic (Sigma-Aldrich, cat. no. S0751-500G)Sodium phosphate dibasic (Sigma-Aldrich, cat. no. S0876-500G)Sodium chloride (Sigma-Aldrich, cat. no. S9625-500G)Tris acid (Sigma-Aldrich, cat. no. T5941-500G)
•
••••••
•••••
Tris base (Sigma-Aldrich, cat. no. T1503-1KG)Sodium hydroxide (Sigma-Aldrich, cat. no. S5881-500G)Sodium acetate (Sigma-Aldrich, cat. no. S2889-1KG)Trifluoroacetic acid (TFA) (Sigma-Aldrich, cat. no. T62200-500G)SDS-PAGE gels (4–12% Bis-Tris, 1 mm) (Invitrogen, cat. no. NP0321BOX)4× LDS buffer (Invitrogen, cat. no. NP0007)20× MES and MOPS buffers (Invitrogen, cat. no. NP0002 and NP0001, respectively)Protein molecular weight markers (Invitrogen, cat. no. BP 3603-500 or BP 3602-500)Antioxidant solution (Invitrogen, cat. no. NP0005)Gel staining solution (Invitrogen, cat. no. 161-0786)Gel drying solution (Invitrogen, cat. no. LC4025)Gel-drying cellophane sheets (Invitrogen, cat. no. NC2200)Durapore membrane-bottomed (multiscreen) 96-well plates (Millipore, cat. no. MAHV N45 50)Flat-bottomed, polystyrene 96-well plates (Corning, cat. no. 3596)
•••••••
•
•••••
•

protocol
410 | VOL.5 NO.3 | 2010 | nature protocols
p
uor
G g
n ih si l
bu
P eru ta
N 010 2©
nat
ure
pro
toco
ls/
moc. e r
ut an .
ww
w / /:pt t
h
Eight-channel Finpipette (50–300 µl) pipetter (Eppendorf, cat. no. 21-377-247)5 ml Sephadex G10 desalting column (Sigma-Aldrich, cat. no. 54822 and /or G10120-10G)
EQUIPMENTPerkin–Elmer HTS 7000 plus plate reader with HTSoft 2 software (or equivalent system) (Perkin–Elmer)
••
•
taBle 1 | Chromatographic media used in the microtiter plate screening.
Mode of chromatographic separation Media Vendor cat. no.
Cation exchange chromatography POROS HS Applied Biosystems 1-3,359
EMD Fractogel SO3 − EMD 116,882
CM Ceramic HyperD Pall Corporation 20,050
S Ceramic HyperD Pall Corporation 20,038
Anion exchange chromatography POROS Q Applied Biosystems 1-2,559
TMAE Fractogel EMD 116,881
DEAE Ceramic HyperD Pall Corporation 20,067
Hydrophobic interaction chromatography (HIC)
Source Ether GE Healthcare 17-0146
Source Isoropyl GE Healthcare 17-0148
Source Phenyl GE Healthcare 17-0147
POROS HP2 Applied Biosystems 1-4,529
Hydrophobic charge induction chromatography (HCIC) MEP HyperCel Pall Corporation 12,035
Vacuum manifold for microtiter plates (MultiScreenHTS Vacuum Manifold; Millipore, cat. no. MSVMHTS00)Novex gel station (Invitrogen, cat. no. EI0002) crItIcal The above gel electrophoresis equipment and reagents are recommended; however, selection of actual equipment is left to the discretion of individual laboratories.
•
•
proceDureFeed preparation ● tIMInG ~ 24–36 h1| Prepare a glycerol stock of E. coli transformed with the expression plasmid for the bioproduct of interest, as previously described34. Add a swab of the glycerol stock of E. coli transformed with the expression plasmid for the bioproduct of interest to four, 250 ml baffled culture flasks, each containing 25 ml of LB broth plus the resistance antibiotic. Shake the flasks at 250 r.p.m. overnight at 37 °C.
2| Add the entire culture (25 ml) from each of the four individual flasks into four individual 2 liter baffled culture flasks each containing 1 liter LB broth; shake the culture at 250 r.p.m. at 37 °C until the optical density (OD) of the bacterial suspension in broth reaches 0.5–0.8. Measure the OD at 600 nm at different times following the inoculation, as follows: turn on a spectrophotometer and select 600 nm wavelength for analysis. Use LB broth media (without bacterial cells) in order to blank in the spectrophotometer and measure the OD of the bacterial culture relative to the LB broth blank at 600 nm wave-length. Typically, a volume of 1 ml LB broth and bacterial culture is used in these analyses; actual volumes will depend on the spectrophotometer and the cuvette used in individual laboratories.
3| Add 0.5 mM IPTG to each 2 liter culture flask when the OD at 600 nm is in the range of 0.5–0.8 to induce protein expression.
4| Carry out the expression for 12–18 h at 37 °C with shaking the culture at 250 r.p.m.
5| After expression, centrifuge the culture broth using a table-top centrifuge at 8,000g for 15 min at 4 °C.
6| Discard the supernatant and resuspend the cells in 50 ml of the same buffer that will be used to load the feed on a particular chromatographic dimension for screening (e.g., resuspend the feed in 50 ml sodium phosphate buffer pH 6.0 for CEx; see table 2). The four culture flasks are pooled at this stage such that cells from all four 2-liter flasks are suspended in a final volume of 50 ml.

protocol
nature protocols | VOL.5 NO.3 | 2010 | 411
p
uor
G g
n ih si l
bu
P eru ta
N 010 2©
nat
ure
pro
toco
ls/
moc. e r
ut an .
ww
w / /:pt t
h
7| Sonicate the cells on ice using a dismembrator; typical operation involves eight sonication cycles with each cycle involving the switching on of the sonication tip for 1 min and switching off for 1 minute.
8| Centrifuge the broth at 10,000–15,000g at 4 °C for 45 min following sonication.
9| Decant the bioproduct-containing supernatant (feed solution) into a 50 ml centrifuge tube on ice and dispose of the cell debris in an appropriate fashion. crItIcal step It is important to remove all cell debris before loading the broth on chromatographic media as presence of cell debris can clog chromatographic media leading to irreversible damage.
10| Add a broad specificity protease inhibitor in amounts recommended by the manufacturer to the cell-free broth in order to inhibit protease activity. (e.g., SIGMAFAST protease inhibitor tablets, Sigma-Aldrich, St. Louis, MO, USA; 10 ml of prepared stock (1 tablet in 100 ml de-ionized water) per 20 g of cell extracts.)
11| It is necessary to adjust the conditions (pH, salt concentration, and so on) of the feed solution to be loaded on differ-ent chromatographic media to ensure adequate binding of the feed to the resins. In order to do this, dialyze the feed solu-tion35 against 2–5 liter of the loading buffer appropriate for a particular chromatographic mode (see table 2) three times at 4 °C using a 1,000 molecular weight cut-off membrane; each dialysis step should be carried out for at least 4 h. crItIcal step All buffer solutions should be sterile filtered through a 0.22 µm membrane filter under vacuum.
12| In order to determine how to adjust the salt concentration of the feed solutions to be used in the chromatographic screening, firstly determine the conductivity of different concentrations sodium phosphate buffer (0–1.5 M in steps of 0.05 M (i.e., 50 mM); pH 6.0). Use a conductivity meter to determine the conductivity (mS cm − 1) of phosphate buffer containing different salt concentrations. Carry out measurements at ~25 °C. Plot the conductivity values displayed on the meter readout against salt concentration. Use this calibration to adjust the conductivity of buffers as described in the following steps (Steps 13 A–C). crItIcal step This calibration is a useful method for estimating and adjusting the salt concentration of feed solutions to be used in the chromatographic screening step, particularly for CEx and AEx chromatography. Care must be taken to ensure that the temperature of the buffers being measured is ~25 °C as that used in the calibration.
13| The salt concentration of the feed (clarified cell culture broth) can be adjusted using option A for AEx and CEx chromatography, option B for HIC and option C for HCIC.(a) adjustment of the salt concentration of the feed for aex and cex chromatography (i) Adjust the conductivity of the feed for AEx and CEx chromatography to an equivalent of 50 mM sodium phosphate
buffer by desalting through a Sephadex G10 column36. (ii) Measure the conductivity of the flow through as described above and repeat the desalting if required. (iii) Adjust the pH of the feed solution to 7.0 or 8.4 for AEx chromatography using sodium hydroxide (1M) or hydrochloric acid
(1N) to screen different pH conditions. Adjust the feed pH to 7.0, 6.0 or 5.0 for CEx chromatography in a similar fashion.(B) adjustment of salt concentration of feed for HIc (i) Weigh and add an appropriate amount so as to bring the cell-free culture broth (feed) to a final concentration of 4 M
sodium chloride or 1.5 M ammonium sulfate.(c) adjustment of salt concentration of feed for HcIc (i) Adjust the salt concentration by adding sodium chloride to increase the salt concentration or dialysis to reduce the
salt concentration. Ensure that the feed salt concentration is between 200 and 300 mM based on conductivity measurements.
(ii) Adjust the pH to 8.4 using either 1 M NaOH or 1N HCl as appropriate.
chromatographic media preparation ● tIMInG ~6 h14| Store the chromatographic media as indicated by individual vendors. In case the bulk media is stored at 4 °C, then let the media equilibrate to room temperature before using in the screening.
taBle 2 | Loading buffers used in the microtiter plate screening.
chromatograpic mode loading buffer
Cation exchange 50 mM sodium acetate buffer, pH 5
50 mM sodium phosphate buffer, pH 6
Anion exchange 50 mM Tris buffer, pH range 8.4–8.6
50 mM sodium phosphate buffer, pH range 7.0–7.4
Hydrophobic charge-induction chromatography (HCIC)
50 mM Tris buffer + 250 mM NaCl, pH 8.4
Hydrophobic interaction chromatography (HIC)
50 mM sodium phosphate buffer + 4 M NaCl or 1.5 M (NH4)2SO4

protocol
412 | VOL.5 NO.3 | 2010 | nature protocols
p
uor
G g
n ih si l
bu
P eru ta
N 010 2©
nat
ure
pro
toco
ls/
moc. e r
ut an .
ww
w / /:pt t
h
15| Agitate each bottle containing chromatographic media to obtain a uniform slurry.
16| As all experiments are to be carried out in triplicate, aliquot at least 750 µl of the media in a 1.5 ml microcentrifuge tube. crItIcal step This volume takes into account the excess liquid volume in the slurry and therefore will contain enough media for screening which requires 3 (triplicate) × 100 µl = 300 µl. Note that this volume is sufficient only for one screening condition (i.e., single pH and loading salt concentration). Scale up the media volume appropriately in case multiple operating conditions are to be used with the same media (e.g., different loading pHs, salt concentrations, and so on). Use a 15 ml centrifuge tube if necessary.
17| Wash media twice with de-ionized water (ratio of de-ionized water volume to media slurry volume = 3:1). Let the media settle under gravity (1 h for each wash). pause poInt The media may be stored overnight in the de-ionized water at the storage temperature recommended by the vendor.
18| Using the same volume ratio, wash the media with the loading buffer for the corresponding mode of chromatography (e.g., 50 mM sodium phosphate pH 6.0 for CEx chromatography, 50 mM tris buffer, pH 8.4 for AEx chromatography, and so on; see table 2 for appropriate buffers). Wash three times and allow the media to settle under gravity each time (1 h for each wash). pause poInt The media may be stored overnight in the loading buffer at the storage temperature recommended by the vendor.
19| After the final wash, pipette out the supernatant and add aliquots of the media (100 µl) to each well of the membrane-bottomed plate. Allow the media to settle under gravity (~30 min) and remove excess supernatant by applying negative pressure using the vacuum manifold. This results in the formation of a chromatographic ‘bed’ in the membrane- bottomed plate. Discard the collection plate containing the wash solution. crItIcal step It is important to aliquot equivalent amounts of media in each well of the membrane-bottomed plate. Using a repeater pipette can help minimize the error involved in distributing media aliquots.
parallel screening in microtiter plates ● tIMInG ~2 h20| Feed loading: Load the protein feed mixture (cell-free culture broth) onto the media aliquots in the wells of the 96-well membrane-bottomed plate from Step 19. Use a total load volume of 800 µl per well, loaded in four steps of 200 µl each with 10 min equilibration time in between loadings.? trouBlesHootInG
21| Apply vacuum to the manifold to collect the supernatant in the flat-bottomed 96-well plate below after each individual loading step. The flow through (200 µl) is collected in a unique 96-well plate for each step such that a total of 800 µl flow through is collected in four different 96-well plates. The negative pressure facilitates the movement of the liquid through the chromatographic media and the membrane into the collection plate below. Store plates containing the flow-through at 4 °C before SDS-PAGE analysis.? trouBlesHootInG
22| Wash: After loading, wash the resin twice with 200 µl of loading buffer employed for each step. Collect the wash under vacuum and save at 4 °C before SDS-PAGE analysis.? trouBlesHootInG
23| Elution of bound protein: Follow option A for CEx and AEx chromatography, option B for HIC and option C for HCIC.(a) elution of bound protein using cex and aex chromatography (i) For both, CEx and AEx media, carry out elution under vacuum in the manifold in a serial step format using
200 µl of the loading buffer containing the following final counter ion (i.e., sodium for CEx and chloride for AEx) concentrations at the corresponding pH: 100, 150, 200, 250, 300, 350, 400, 450, 500, 600, 700, 800 and 1,000 mM. crItIcal step These solutions should be prepared before carrying out the serial-step elution, preferably by diluting a stock solution of the most concentrated elution condition (i.e., 1,000 mM salt concentration in the buffer). In cases where narrower salt concentrations might be required, prepare buffer of buffered salt concentrations with step increases of 20 mM counter-ion concentration. crItIcal step Add elution solutions carefully to the chromatographic media in the membrane-bottomed plates so as to not disturb the chromatographic ‘bed’ formed in individual wells. crItIcal step Do not let the chromatographic media run dry in the membrane-bottomed plates upon application of negative pressure in the serial step elution steps.

protocol
nature protocols | VOL.5 NO.3 | 2010 | 413
p
uor
G g
n ih si l
bu
P eru ta
N 010 2©
nat
ure
pro
toco
ls/
moc. e r
ut an .
ww
w / /:pt t
h
(ii) Collect eluted fractions in flat-bottomed 96-well polystyrene collection plates and store at 4 °C unless SDS-PAGE analysis is to follow immediately. Use a fresh 96-well collection plate for each fraction collection step. crItIcal step Collection plates should not be stored at 4 °C for more than 12–18 h. ? trouBlesHootInG
(B) elution of bound protein using HIc (i) For HIC, the loading buffer should contain 1.5 M ammonium sulfate or 4 M sodium chloride. In the case of ammonium
sulfate loading buffer, carry out the elution with 200 µl buffer containing 1,400; 1,200; 1,000; 800; 600; 400; and 200 mM ammonium sulfate salt followed by two final MilliQ water elution steps. In the case of sodium chloride salt (4 M salt concentration loading), carry out elution using 200 µl buffer containing 3,600; 3,200; 2,800; 2,400; 2,000; 1,600; 1,200; 800; and 400 mM salt concentration. crItIcal step Narrower salt concentration steps (e.g., 200 mM) may also be employed in order to improve resolution. crItIcal step Add elution solutions carefully to the chromatographic media in the membrane-bottomed plates so as to not disturb the chromatographic ‘bed’ formed in individual wells. crItIcal step Do not let the chromatographic media run dry in the membrane-bottomed plates upon application of negative pressure in the serial step elution steps.
(ii) Collect eluted fractions in 96-well collection plates and store at 4 °C unless SDS-PAGE analysis is to follow immediately. Use a fresh collection plate for each fraction collection step. crItIcal step Collection plates should not be stored at 4 °C for more than 12–18 h. ? trouBlesHootInG
(c) elution of bound protein using HcIc (i) For HCIC, elute proteins adsorbed on the resin (loading buffer: 200–300 mM salt concentration, pH 8) using a serial
step reducing pH elution range from pH 8 to pH 2.5. Use appropriate buffers to prepare solutions of pH 8, pH 7.5, pH 7.0, pH 6.5, pH 6.0, pH 5.5, pH 5, pH 4.5 and two steps of pH 4.0. crItIcal step Add elution solutions carefully to the chromatographic media in the membrane-bottomed plates so as to not disturb the chromatographic ‘bed’ formed in individual wells. crItIcal step Do not let the chromatographic media run dry in the membrane-bottomed plates upon application of negative pressure in the serial step elution steps.
(ii) Collect eluted fractions in 96-well collection plates; use a fresh collection plate for each fraction collection step and store at 4 °C unless SDS-PAGE analysis is to follow immediately. crItIcal step Collection plates should not be stored at 4 °C for more than 12–18 h. ? trouBlesHootInG
analysis of individual fraction purity: sDs-paGe purity analysis ● tIMInG ~8–10 h24| Use either 10-lane or 12-lane 4–12% Bis-Tris 1 mm gels with a Novex gel station. Fill the gel station with 1× MES (or MOPS) buffer. Approximately 400–500 ml buffer is required for each run (two gels).
25| Dilute the eluted samples in appropriate gel running buffer and heat at 95 °C for 10 min, then allow the samples to equilibrate to room temperature (approximately 25 °C). Centrifuge samples (at 10,000g in 0.4 or 1.5 ml microcentrifuge tubes for 1 min at room temperature) before loading onto gels.
26| Load individual fractions (10–20 µl) in individual wells of the gel. Load appropriate molecular weight marker in the first well. If available, load a purified sample of the protein of interest as a standard.
27| The gels should be run at 100 V for 0.5–1 h depending on the migration of the proteins. Turn the power off when the buffer front is about 0.5 cm from the bottom of the gel.
28| Remove the gel from the station, separate the gel cassette using a gel-knife and gently add the gel to a tray containing 50–100 ml de-ionized water for 10 min. Following this wash, stain the gel with 50 ml Coomassie Blue (or other appropriate staining solutions) for 1 h. Destain with de-ionized water containing 5–10% glacial acetic acid overnight.
29| Scan gel using ImageScanner II (GE Healthcare, Piscataway, NJ, USA) or a similar scanning instrument.
30| Identify fractions with high protein purities (>60% purity) using image analysis software, ImageQuant TL (GE Healthcare). Identify the mode of chromatography (e.g., CEx) and operating conditions (chromatographic media, loading and elution pH and salt concentrations) that result in fractions containing high purities of the bioproduct. This is the screen recommendation for the first chromatographic purification step.
protein preparation for the second chromatographic screening step: intermediate ‘1 ml column’ step ● tIMInG 2 h31| Pipette the recommended chromatographic media into an empty 5 ml column such that the media volume is 1 ml. This step is tenfold higher than the screening step and is employed to obtain enough protein to facilitate screening and identification of the second chromatographic dimension.

protocol
414 | VOL.5 NO.3 | 2010 | nature protocols
p
uor
G g
n ih si l
bu
P eru ta
N 010 2©
nat
ure
pro
toco
ls/
moc. e r
ut an .
ww
w / /:pt t
h
32| Repeat the steps described in Steps 11–30 above for the recommended chromatographic conditions. All volumes in this step should be scaled up by a factor of ten compared with those used originally in the screening Steps 11–30. All loadings, washes and elutions are carried out under gravity, without the use of the vacuum manifold.
33| Pool the fractions containing the enriched bioproduct from Step 32.
34| Adjust the salt concentration by first measuring the conductivity of the fraction pool and either adding salt for increas-ing concentration or by desalting in Sephadex G10 columns for reducing salt concentration to the following equivalent concentrations: 50 mM for ion-exchange, 1.5 M for HIC and 200–300 mM for HCIC. As before, adjust pH using 1M NaOH or 1N HCl (see Steps 12 and 13, and table 2). pause poInt The fraction pool can be stored at 4 °C overnight at this point before resuming the second screening step.
screening for subsequent chromatographic dimensions ● tIMInG approximately the same required for steps 23–3435| The mode of chromatography identified (in Steps 20–30) as the first chromatographic step from screening can be eliminated from the second screening step. The screening procedure described for the first chromatographic step (Steps 23–34) should be repeated to identify appropriate conditions for the second chromatographic dimension. crItIcal step The above screening procedure can be repeated in a sequential manner to identify subsequent chromatographic steps (more than two as described here) for purifying the bioproduct. It is recommended that two to three chromatographic steps be used in the purification of the protein starting from cell-free culture broth. ? trouBlesHootInG
● tIMInGSteps 1–13, Feed preparation: 24–36 hSteps 14–19, Chromatographic media preparation: ~6 hSteps 20–23, Parallel screening in microtiter plates: ~2 hSteps 24–30, Analysis of individual fraction purity: SDS-PAGE purity analysis: ~8–10 hSteps 31–34, Protein preparation for the second chromatographic screening step: 2 hStep 35, Screening for subsequent chromatographic steps: as for Steps 20–30
? trouBlesHootInGTroubleshooting advice can be found in table 3.
taBle 3 | Troubleshooting table.
step problem possible reason solution
20–23 and 35 Well-to-well variability in the eluate volume during screening
Variability of vacuum in manifold
Ensure that the vacuum is steady across the entire plate during the course of the screening
Ensure that the top and bottom portions of the vacuum manifold are sealed appropriately
Accumulation of liquid on the bottom of the membrane-bottomed plates
Variability of vacuum in manifold Ensure that the vacuum is steady across the entire plate during the course of the screening
Ensure that the top and bottom portions of the vacuum manifold are sealed appropriately
Very little or no bioprod-uct is found in the elution fractions
Poor binding of the bioproduct protein on the chromatographic media
Expand the screen to select other media
High retention of bioproduct pro-tein on chromatographic media
Choose different operating conditions (pH, salt concentration, and so on)

protocol
nature protocols | VOL.5 NO.3 | 2010 | 415
p
uor
G g
n ih si l
bu
P eru ta
N 010 2©
nat
ure
pro
toco
ls/
moc. e r
ut an .
ww
w / /:pt t
h
antIcIpateD resultsAs an example of the results obtained using this protocol, we describe here the previous ‘purification of α-amylase from cell-free culture broth,’ which was carried out according to this protocol30. In this experiment, a recombinant mutant of a Bacillus licheniformis α-amylase (MW = 55.3 kDa; pI = 6.41), as described in patents WO 98/26078 and US 005736499A was used in this investigation. The protein was expressed in Bacillus subtilis and the fermentation broth was clarified and concen-trated using a 10 K molecular weight cut-off membrane. Four different chromatographic modes, HIC, CEx, AEx and HCIC, were screened for the first purification step using the above protocol. The resins employed in the screen are given in table 1. Four HIC resins, source ether, source isopropyl, source phenyl and POROS HP2 were used for HIC. Binding and elution of α-amylase was carried out in the presence of sodium chloride and ammonium sulfate. Two AEx resins, POROS HQ and TMAE Fractogel, and two pH conditions (pH 7.4 and 8.4) were screened. Four CEx resins, POROS SO3 − , EMD Fractogel SO3 − , S Ceramic HyperD (strong CExs) and CM Ceramic HyperD (weak CEx), and two pH conditions (pH 5.0 and 6.0) were used in the screen. HCIC, using MEP Hyper Cel resin, was also screened as a potential first step for α-amylase purification.
recommendation for the first chromatographic purification stepBased on the purity of α-amylase in eluted fractions (determined using SDS-PAGE; Fig. 2) obtained upon screening 11 resins (4 HIC, 6 ion-exchange and 1 HCIC resins), and 7 different loading and elution conditions, CEx using CM Ceramic HyperD resin was identified as the best first chromatographic purification step. The following recommendations were made for the first chromatographic step:
Chromatographic media: CM Ceramic HyperD.Loading condition: 50 mM acetate buffer, pH 5. Elution: Linear salt gradient between 50–1,000 mM sodium ion concentration; anticipated α-amylase elution between 300 and 500 mM sodium ion concentration.
screening for the second chromatographic stepThe presence of impurities in α-amylase fractions after the first purification step (CM Ceramic Hyper D) necessitated a second chromatographic step. Consequently, eluted fractions from the CM Ceramic HyperD resin were pooled and a second screen was set up to identify the next chromatographic step. The fraction pool containing α-amylase was adjusted for pH and salt concentration and used as feed load for the second screening step.
Cation exchange chromatography was eliminated from the second chromatographic screen since it was recommended as the first purification step. AEx chromatography was the first mode of chromatography chosen in the second screening step. Four AEx resins (two weak AExs, TMAE fractogel and DEAE HyperD; and two strong AEs, POROS HQ and Q Ceramic HyperD) were screened at pH 7.4 and pH 8.6. Stronger binding of α-amylase was observed on POROS HQ and TMAE Fractogel resins at pH 8.6 and the protein eluted between 100 and 150 mM salt concentration (chromato-grams not shown). SDS-PAGE analysis of these fractions
1
1 2
Feed CM ceramichyperDfractions
MEP hyperCellfractions
Poros HSfractions
Fractogel SO1–
fractions
3 4 5 6 7 8 9
a
b 10 1 2 3 4 5 6 7 8 9 10
2 3 4 5 6 7 8 9 10 11 12
Figure 2 | Analysis of fractions eluted during the miniaturized screening using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). (a) Fractions eluted from source isopropyl and POROS phenyl hydrophobic interaction chromatography (HIC) resins using decreasing concentrations of ammonium sulfate for elution. Lane 1: α-amylase standard. Lane 2: cell-free α-amylase culture broth. Lanes 3, 4, 5, 6 and 7: 1,200, 1,000, 800, 600 and 400 mM ammonium sulfate fractions from source isopropyl, respectively. Lanes 8, 9, 10, 11 and 12: 1,000, 800, 600, 400 and 200 mM ammonium sulfate fractions from POROS phenyl, respectively. (b) Fractions eluted from cation exchange (CM Ceramic HyperD) and hydrophobic charge-induction chromatography (MEP HyperCel) media in the first microtiter-screening step. Lane 1: cell-free α-amylase culture broth. Lanes 2, 3, 4 and 5: 400, 450, 500 and 600 mM sodium chloride fractions from CM Ceramic Hyper D, respectively. Lanes 6, 7, 8, 9 and 10: pH 5.0, 5.0, 4.5, 4.0 and 4.0 fractions from MEP HyperCel (hydrophobic charge-induction chromatography (HCIC)) resin, respectively. The line around lane 5 indicates the fraction which is most enriched in α-amylase. (c) Fractions eluted from Fractogel SO3− and POROS SO3−. Lanes 1, 2, 3, 4, 5 and 6: 150, 200, 250, 300, 350 and 400 mM sodium chloride fractions from Fractogel SO3−. Lanes 7, 8, 9 and 10: 200, 250, 300 and 350 mM sodium chloride fractions from POROS SO3−.
protocol
416 | VOL.5 NO.3 | 2010 | nature protocols
p
uor
G g
n ih si l
bu
P eru ta
N 010 2©
nat
ure
pro
toco
ls/
moc. e r
ut an .
ww
w / /:pt t
h
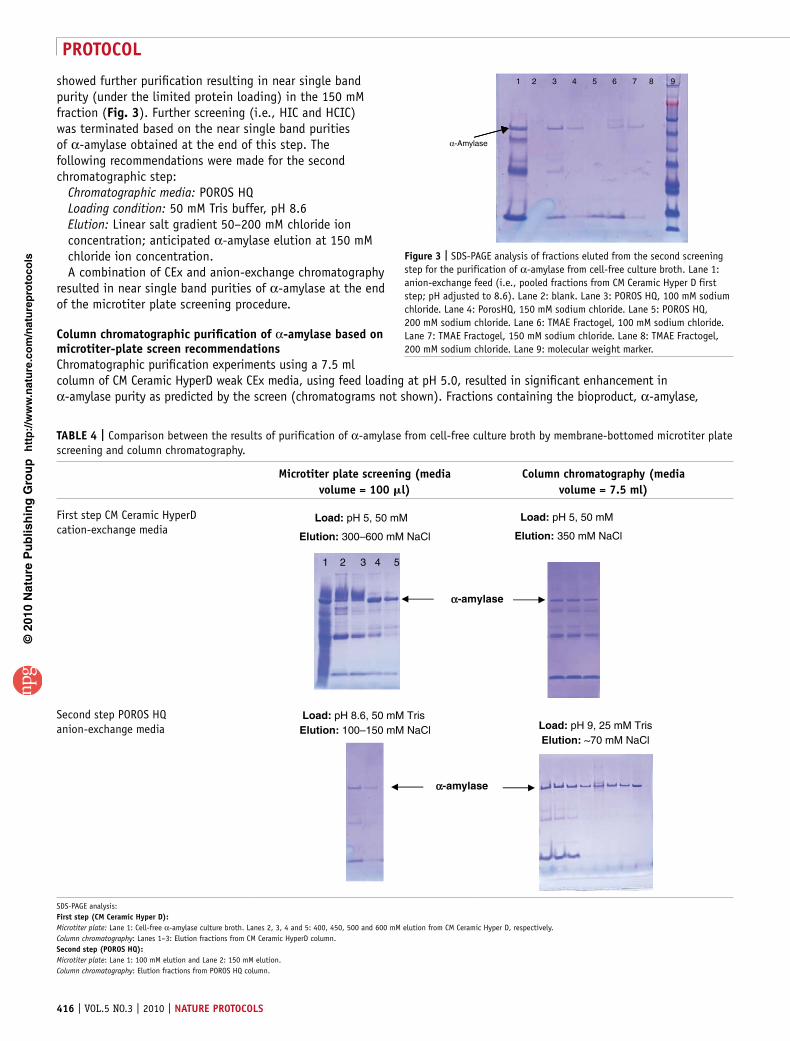
showed further purification resulting in near single band purity (under the limited protein loading) in the 150 mM fraction (Fig. 3). Further screening (i.e., HIC and HCIC) was terminated based on the near single band purities of α-amylase obtained at the end of this step. The following recommendations were made for the second chromatographic step:
Chromatographic media: POROS HQLoading condition: 50 mM Tris buffer, pH 8.6 Elution: Linear salt gradient 50–200 mM chloride ion concentration; anticipated α-amylase elution at 150 mM chloride ion concentration.A combination of CEx and anion-exchange chromatography
resulted in near single band purities of α-amylase at the end of the microtiter plate screening procedure.
column chromatographic purification of α-amylase based on microtiter-plate screen recommendationsChromatographic purification experiments using a 7.5 ml column of CM Ceramic HyperD weak CEx media, using feed loading at pH 5.0, resulted in significant enhancement in α-amylase purity as predicted by the screen (chromatograms not shown). Fractions containing the bioproduct, α-amylase,
Figure 3 | SDS-PAGE analysis of fractions eluted from the second screening step for the purification of α-amylase from cell-free culture broth. Lane 1: anion-exchange feed (i.e., pooled fractions from CM Ceramic Hyper D first step; pH adjusted to 8.6). Lane 2: blank. Lane 3: POROS HQ, 100 mM sodium chloride. Lane 4: PorosHQ, 150 mM sodium chloride. Lane 5: POROS HQ, 200 mM sodium chloride. Lane 6: TMAE Fractogel, 100 mM sodium chloride. Lane 7: TMAE Fractogel, 150 mM sodium chloride. Lane 8: TMAE Fractogel, 200 mM sodium chloride. Lane 9: molecular weight marker.
taBle 4 | Comparison between the results of purification of α-amylase from cell-free culture broth by membrane-bottomed microtiter plate screening and column chromatography.
Microtiter plate screening (media volume = 100 l)
column chromatography (media volume = 7.5 ml)
First step CM Ceramic HyperD cation-exchange media
Load: pH 5, 50 mM
Elution: 350 mM NaCl
Load: pH 5, 50 mM
Elution: 300–600 mM NaCl
α-amylase
1 2 3 4 5
Second step POROS HQ anion-exchange media Load: pH 9, 25 mM Tris
Elution: ~70 mM NaCl
Load: pH 8.6, 50 mM Tris Elution: 100–150 mM NaCl
α-amylase
SDS-PAGE analysis:First step (cM ceramic Hyper D):Microtiter plate: Lane 1: Cell-free α-amylase culture broth. Lanes 2, 3, 4 and 5: 400, 450, 500 and 600 mM elution from CM Ceramic Hyper D, respectively.Column chromatography: Lanes 1–3: Elution fractions from CM Ceramic HyperD column.second step (poros HQ):Microtiter plate: Lane 1: 100 mM elution and Lane 2: 150 mM elution.Column chromatography: Elution fractions from POROS HQ column.
1
α-Amylase
2 3 4 5 6 7 8 9

protocol
nature protocols | VOL.5 NO.3 | 2010 | 417
p
uor
G g
n ih si l
bu
P eru ta
N 010 2©
nat
ure
pro
toco
ls/
moc. e r
ut an .
ww
w / /:pt t
h
were then pooled, desalted and were used as feed for the second recommended step (AEx chromatography using POROS HQ). Single-band purities of the bioproduct were observed following this AEx step; however, the protein eluted at a lower salt concentration than predicted by the screen, indicating a narrower window of operation than expected (data not shown). table 4 compares the performance of the microtiter plate (100 µl resin volume) with that of the chromatographic column (7.5 ml resin volume). The protein (α-amylase) eluted between 300 and 600 mM sodium ion concentration from the first CEx step at the microtiter scale, while it eluted at ~350 mM from the chromatographic column at comparable purity levels. Importantly, the three main bands (including α-amylase) eluted from the resin at the microtiter scale were also present in the column eluate. For the second AEx step, α-amylase eluted from the resin in microtiter plates between 100 and 150 mM salt concentration. A slight modification in the loading conditions for the column chromatographic purification (25 mM salt and pH 9) compared to that recommended by the microtiter screen (50 mM salt and pH 8.6) resulted in the elution of α-amylase at 64 mM with similar (single-band) purities. To summarize, a two-step chromatographic sequence, derived from the recommendations of a microtiter plate screening protocol, resulted in the successful chromatographic purification of α-amylase from cell-free culture broth.
autHor contrIButIons K.R. and M.H. designed the work, K.R. carried out experiments, and K.R. and M.H. analyzed the data and wrote the manuscript.
Published online at http://www.natureprotocols.com/. Reprints and permissions information is available online at http://npg.nature.com/ reprintsandpermissions/.
1. Sunasara, K.M., Xia, F., Gronke, R.S. & Cramer, S.M. Application of hydrophobic interaction displacement chromatography for an industrial protein purification. Biotechnol. Bioeng. 82, 330–339 (2003).
2. Shepard, S.R. et al. Large-scale purification of recombinant human angiostatin. Protein Expr. Purif. 20, 216–227 (2000).
3. Prazeres, D.M., Schluep, T. & Cooney, C. Preparative purification of supercoiled plasmid DNA using anion-exchange chromatography. J. Chromatogr. A 806, 31–45 (1998).
4. Haldankar, R., Kopchick, J.J. & Ridgway, D. Stable production of a human growth hormone antagonist from CHO cells adapted to serum-free suspension culture. Biotechnol. Prog. 15, 336–346 (1999).
5. Shukla, A.A. et al. Preparative purification of a recombinant protein by hydrophobic interaction chromatography: modulation of selectivity by the use of chaotropic additives. Biotechnol. Prog. 18, 556–564 (2002).
6. Levison, P.R. Large-scale ion-exchange column chromatography of proteins. Comparison of different formats. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 790, 17–33 (2003).
7. Van Hijfte, L., Marciniak, G. & Froloff, N. Combinatorial chemistry, automation and molecular diversity: new trends in the pharmaceutical industry. J. Chromatogr. B Biomed. Sci. Appl. 725, 3–15 (1999).
8. Gallop, M.A., Barrett, R.W., Dower, W.J., Fodor, S.P. & Gordon, E.M. Applications of combinatorial technologies to drug discovery. 1. Background and peptide combinatorial libraries. J. Med. Chem. 37, 1233–1251 (1994).
9. Gordon, E.M., Barrett, R.W., Dower, W.J., Fodor, S.P. & Gallop, M.A. Applications of combinatorial technologies to drug discovery. 2. Combinatorial organic synthesis, library screening strategies, and future directions. J. Med. Chem. 37, 1385–1401 (1994).
10. Woodbury Jr., C.P. & Venton, D.L. Methods of screening combinatorial libraries using immobilized or restrained receptors. J. Chromatogr. B Biomed. Sci. Appl. 725, 113–137 (1999).
11. Scott, J.K. & Smith, G.P. Searching for peptide ligands with an epitope library. Science 249, 386–390 (1990).
12. Barrett, R.W. et al. Selective enrichment and characterization of high affinity ligands from collections of random peptides on filamentous phage. Anal. Biochem. 204, 357–364 (1992).
13. Mondorf, K., Kaufman, D.B. & Carbonell, R.G. Screening of combinatorial peptide libraries: identification of ligands for affinity purification of proteins using a radiological approach. J. Pept. Res. 52, 526–536 (1998).
14. Teng, S.F., Sproule, K., Hussain, A. & Lowe, C.R. A strategy for the generation of biomimetic ligands for affinity chromatography. Combinatorial synthesis and biological evaluation of an IgG binding ligand. J. Mol. Recognit. 12, 67–75 (1999).
15. Burton, N.P. & Lowe, C.R. Design of novel affinity adsorbents for the purification of trypsin-like proteases. J. Mol. Recognit. 5, 55–68 (1992).
16. Sproule, K. et al. New strategy for the design of ligands for the purification of pharmaceutical proteins by affinity chromatography. J. Chromatogr. B Biomed. Sci. Appl. 740, 17–33 (2000).
17. Lowe, C.R. Combinatorial approaches to affinity chromatography. Curr. Opin. Chem. Biol. 5, 248–256 (2001).
18. Huang, P.Y. et al. Affinity purification of von Willebrand factor using ligands derived from peptide libraries. Bioorg. Med. Chem. 4, 699–708 (1996).
19. Kaufman, D.B. et al. Affinity purification of fibrinogen using a ligand from a peptide library. Biotechnol. Bioeng. 77, 278–289 (2002).
20. Oldenburg, K.R., Loganathan, D., Goldstein, I.J., Schultz, P.G. & Gallop, M.A. Peptide ligands for a sugar-binding protein isolated from a random peptide library. Proc. Natl. Acad. Sci. USA 89, 5393–5397 (1992).
21. Clonis, Y.D. Affinity chromatography matures as bioinformatic and combinatorial tools develop. J. Chromatogr. A 1101, 1–24 (2006).
22. Saraswat, L.D. et al. Affinity ligand selection from a library of small molecules: assay development, screening, and application. Biotechnol. Prog. 21, 300–308 (2005).
23. Paborsky, L.R., Dunn, K.E., Gibbs, C.S. & Dougherty, J.P. A nickel chelate microtiter plate assay for six histidine-containing proteins. Anal. Biochem. 234, 60–65 (1996).
24. Draveling, C., Ren, L., Haney, P., Zeisse, D. & Qoronfleh, M.W. SwellGel: an affinity chromatography technology for high-capacity and high-throughput purification of recombinant-tagged proteins. Protein Expr. Purif. 22, 359–366 (2001).
25. Mazza, C.B. et al. High-throughput screening and quantitative structure-efficacy relationship models of potential displacer molecules for ion-exchange systems. Biotechnol. Bioeng. 80, 60–72 (2002).
26. Rege, K., Hu, S., Moore, J.A., Dordick, J.S. & Cramer, S.M. Chemo-enzymatic synthesis and high-throughput screening of an aminoglycoside-polyamine library: identification of high-affinity displacers and DNA-binding ligands. J. Am. Chem. Soc. 126, 12306–12315 (2004).
27. Rege, K., Ladiwala, A. & Cramer, S.M. Multidimensional high-throughput screening of displacers. Anal. Chem. 77, 6818–6827 (2005).
28. Rege, K., Ladiwala, A., Tugcu, N., Breneman, C.M. & Cramer, S.M. Parallel screening of selective and high-affinity displacers for proteins in ion-exchange systems. J. Chromatogr. A 1033, 19–28 (2004).
29. Rege, K., Tugcu, N. & Cramer, S.M. Predicting column performance in displacement chromatography from high throughput screening batch experiments. Sep. Sci. Technol. 38, 1499–1517 (2003).
30. Rege, K., Pepsin, M., Falcon, B., Steele, L. & Heng, M. High-throughput process development for recombinant protein purification. Biotechnol. Bioeng. 93, 618–630 (2006).
31. Coffman, J.L., Kramarczyk, J.F. & Kelley, B.D. High-throughput screening of chromatographic separations: I. Method development and column modeling. Biotechnol. Bioeng. 100, 605–618 (2008).
32. McNay, J.L. & Fernandez, E.J. Protein unfolding during reversed-phase chromatography: I. Effect of surface properties and duration of adsorption. Biotechnol. Bioeng. 76, 224–232 (2001).
33. McNay, J.L., O’Connell, J.P. & Fernandez, E.J. Protein unfolding during reversed-phase chromatography: II. Role of salt type and ionic strength. Biotechnol. Bioeng. 76, 233–240 (2001).
34. Bernard, A. & Payton, M. Selection of Escherichia coli expression systems. Curr. Protoc. Protein Sci. Ch. 5, Unit 5.2, 5.2.1–5.2.18 (2001).
35. Andrew, S.M., Titus, J.A. & Zumstein, L. Dialysis and concentration of protein solutions. Curr. Protoc. Cell Biol. Appendix 3, A.3C.1–A.3C.5 (2001).
36. Hagel, L. Gel-filtration chromatography. Curr. Protoc. Mol. Biol. Chapter 10, unit 10 19 (2001).





























