Embed Size (px)
Citation preview

Chinese Pharmaceutical Association
Institute of Materia Medica, Chinese Academy of Medical Sciences
Acta Pharmaceutica Sinica B
Acta Pharmaceutica Sinica B 2012;2(6):535–548
2211-3835 & 2012 In
hosting by Elsevier B
http://dx.doi.org/10.1
nCorresponding au
E-mail address: e
Peer review under r
www.elsevier.com/locate/apsbwww.sciencedirect.com
REVIEW
Milestones in the discovery of antiviral agents: nucleosides
and nucleotides
Erik de Clercqn
Rega Institute for Medical Research, Katholieke Universiteit Leuven, Minderbroedersstraat 10, Leuven B-3000, Belgium
Received 20 June 2012; revised 18 August 2012; accepted 5 September 2012
KEY WORDS
Valacyclovir;
Brivudin;
FV-100;
Emtricitabine;
(S)-HPMPA;
(S)-HPMPC;
Cidofovir;
Adefovir;
Tenofovir;
Truvadas;
Phosphonoamidate
stitute of Materia M
.V. All rights rese
016/j.apsb.2012.10
thor. Tel.: þ32 16
rik.declercq@rega
esponsibility of Ins
Abstract In this review article, a number of milestones in the antiviral research field on
nucleosides and nucleotides are reviewed in which the author played a significant part, especially
in the initial stages of their development. Highlighted are the amino acyl esters of acyclovir,
particularly valacyclovir (VACV), brivudin (BVDU) and the valine ester of Cf1743 (FV-100), the
20,30-dideoxynucleosides (nucleoside reverse transcriptase inhibitors, NRTIs), the acyclic nucleoside
phosphonates (S)-HPMPA, (S)-HPMPC (cidofovir) and alkoxyalkyl esters thereof (HDP-, ODE-
CDV), adefovir and adefovir dipivoxil, tenofovir and tenofovir disoproxil fumarate (TDF),
combinations containing TDF and emtricitabine, i.e., Truvadas, Atriplas, Compleras/Evipleras
and the Quad pill, and the phosphonoamidate derivatives GS-7340, GS-9131, GS-9191 and
GS-9219.
& 2012 Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical
Association. Production and hosting by Elsevier B.V. All rights reserved.
edica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association. Production and
rved.
.001
337367; fax: þ32 16 337340.
.kuleuven.be (Erik de Clercq).
titute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.

Figure 1 Prodrugs of acyclovir, ganciclovir and penciclovir.
Erik de Clercq536
1. Introduction
The era of antiviral drug therapy started with idoxuridine
(IDU) and trifluridine (TFT). IDU was first synthesized as a
potential anticancer drug by Prusoff in 19591; it was first
shown in 1961 to possess activity against herpes simplex virus
(HSV) and vaccinia virus by Herrmann2, before it was
launched in the clinic, for the topical treatment of herpetic
keratitis by Kaufman3. Two years later, Kaufman and
heidelbrger4 also unleashed trifluridine (TFT) for the topical
treatment of herpetic keratitis. As of today, IDU and TFT are
still used in the topical treatment of herpetic eye infections.
Still in the 1960s, arabinosyladenine (ara-A), originally
synthesized as a potential anticancer agent by Lee et al.5 was
first shown by Privat de Garilhe and de Rudder6 to be active
against HSV and vaccinia virus before it was further described
as an antiviral agent by Schabel7, before it became the first
antiviral drug to be used systemically, i.e., by Whitley et al.8 in
1976, in the therapy of varicella-zoster virus (VZV) infections.
Ara-A is no longer used in the clinic, essentially for a number
of reasons: it has low aqueous solubility, is rapidly deaminated
to the inactive arabinosylhypoxanthine (ara-Hx), but primar-
ily, because it was superseded from the 1980s by acyclovir.
Meanwhile, in the early 1970s ribavirin (virazole) had been
described by Sidwell et al.9 as a broad-spectrum antiviral
agent. For circa 30 years, ribavirin was looking for a disease
against which it could be useful, until it found its niche,
together with pegylated interferon, in the treatment of chronic
hepatitis C virus (HCV) infections. The combination of
pegylated interferon-a with ribavirin has since the last 10
years been the standard of care (SOC) for the treatment of
HCV infections, but is likely to be, first complemented and
then replaced by direct-acting antiviral agents (DAAs).
Of crucial importance in the treatment of herpesvirus (i.e.,
HSV and VZV) infections was the discovery of acyclovir, the
first truly specific antiviral agent, by Elion et al.10 and
Schaeffer et al11. Now, 35 years after it was originally
described, acyclovir can still be considered as the ‘‘gold
standard’’ for the treatment of HSV and VZV infections12.
How the antiviral research field, that started with the
nucleoside analogs IDU, TFT, ara-A, ribavirin and acyclovir,
further evolved (and thrived) will be the subject of the present
review. This review will focus specifically on nucleoside
analogs such as amino acyl acyclovir esters, bromovinyldeox-
yuridine and 20,30-dideoxynucleoside analogs, and nucleotide
analogs (i.e., acyclic nucleoside phosphonates (ANPs)).
Twenty-five years ago, the era of the ANPs started with the
birth of (S)-HPMPA. It has now grown to a large family of
marketed drugs, including cidofovir, adefovir and tenofovir,
and various prodrugs and drug combinations derived thereof.
2. Antiviral agents
2.1. Valacyclovir (VACV)
Acyclovir suffers from some drawbacks in that it is relatively
insoluble in aqueous medium and poorly absorbed after oral
administration. To circumvent the first problem, amino acid
(i.e., glycine, alanine) esters of acyclovir (Fig. 1) were synthe-
sized13. An advantage of such amino acyl esters is that for
topical use, i.e., for the treatment of herpetic keratitis, they can
be administered as eye drops14, whereas acyclovir has to be
applied as an eye ointment. A second advantage of such amino
acyl esters is that for systemic use they could be injected
intramuscularly or subcutaneously, in small volumes, whereas
the parent compound, acyclovir, has to be administered
intravenously in large volumes. However, from a practical
viewpoint, parenteral injection of the amino acid esters of
acyclovir has never been pursued.
Of the various amino acid esters of acyclovir that were
subsequently studied, the valine ester, valacyclovir (Zelitrexs,
Valtrexs) (Fig. 1) appeared to be the most suitable for
increasing the oral bioavailability of acyclovir15, and valacyclo-
vir has now replaced acyclovir in the oral treatment of HSV and
VZV infections. Likewise, to increase the oral bioavailability,
Discovery of antiviral agents 537
valganciclovir, the valine ester of ganciclovir (Fig. 1), and
famciclovir, the prodrug of penciclovir (Fig. 1), were introduced
for the oral treatment of, respectively, cytomegalovirus (CMV)
infections and HSV and VZV infections15.
VACV is the only antiviral drug approved for once-daily
suppressive therapy for genital herpes16. VACV has been FDA-
approved for the treatment of HSV and VZV infections in
immunocompromised patients. It is more effective than acyclovir
in the treatment of herpes zoster, particularly with regard to the
duration of post-herpetic neuralgia (mean duration of pain: 40
days as compared to 60 days)17. VACV at a dose of 1 g twice
daily from month 4 to 24 after hematopoietic stem cell
transplantation was found effective in suppressing herpes zos-
ter18. Oral VACV could also be recommended for early treat-
ment of HSV encephalitis in resource-limited settings19.
Single-day famciclovir (1000 mg administered twice daily)
proved as effective as a 3-day VACV regimen (500 mg adminis-
tered twice daily) for recurrent genital herpes20. One-year
suppressive therapy with oral VACV (500 mg daily) was shown
to be as effective and as well tolerated as acyclovir (400 mg twice
daily) in reducing the rate of recurrent ocular HSV disease21.
Administration of VACV (500 mg twice daily) beginning at
36 weeks’ gestation until delivery to women with a history of
recurrent genital HSV reduced the number of subsequent
clinical HSV recurrences22. Exposure to acyclovir or VACV
in the first trimester of pregnancy was not associated with an
increased risk of major birth defects23.
Preemptive valganciclovir and VACV prophylaxis would
be equally effective in the prevention of CMV disease
after renal transplantation24. Valganciclovir and VACV were
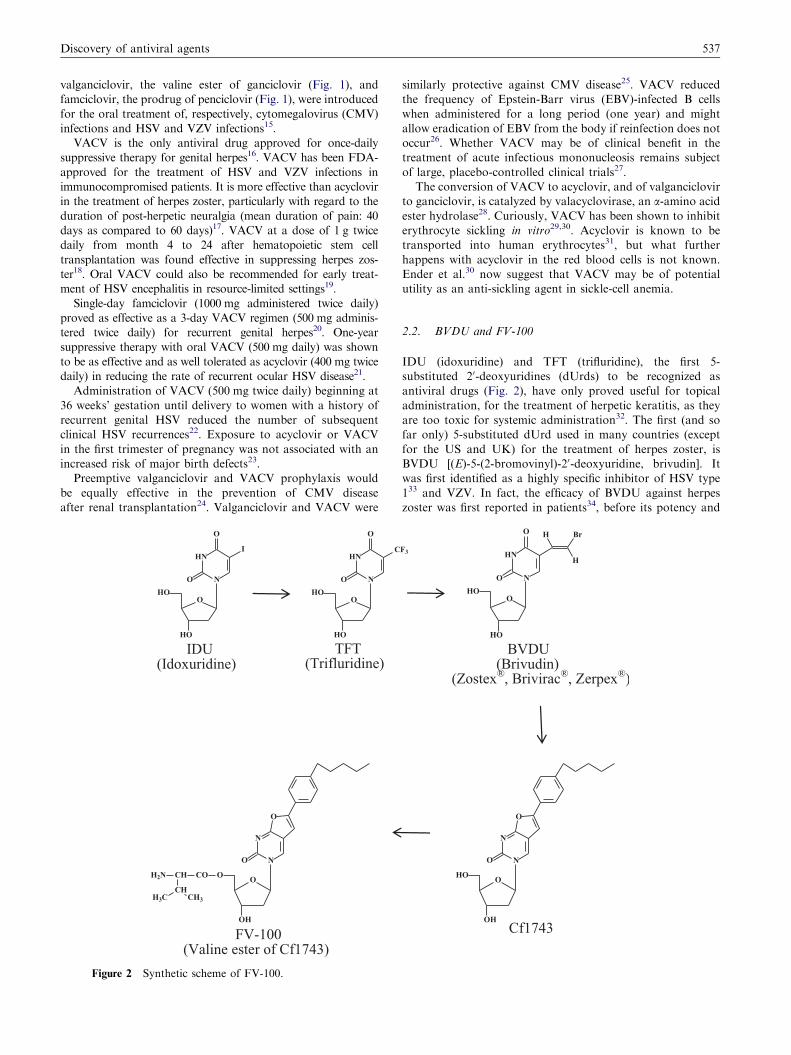
Figure 2 Synthetic scheme of FV-100.
similarly protective against CMV disease25. VACV reduced
the frequency of Epstein-Barr virus (EBV)-infected B cells
when administered for a long period (one year) and might
allow eradication of EBV from the body if reinfection does not
occur26. Whether VACV may be of clinical benefit in the
treatment of acute infectious mononucleosis remains subject
of large, placebo-controlled clinical trials27.
The conversion of VACV to acyclovir, and of valganciclovir
to ganciclovir, is catalyzed by valacyclovirase, an a-amino acid
ester hydrolase28. Curiously, VACV has been shown to inhibit
erythrocyte sickling in vitro29,30. Acyclovir is known to be
transported into human erythrocytes31, but what further
happens with acyclovir in the red blood cells is not known.
Ender et al.30 now suggest that VACV may be of potential
utility as an anti-sickling agent in sickle-cell anemia.
2.2. BVDU and FV-100
IDU (idoxuridine) and TFT (trifluridine), the first 5-
substituted 20-deoxyuridines (dUrds) to be recognized as
antiviral drugs (Fig. 2), have only proved useful for topical
administration, for the treatment of herpetic keratitis, as they
are too toxic for systemic administration32. The first (and so
far only) 5-substituted dUrd used in many countries (except
for the US and UK) for the treatment of herpes zoster, is
BVDU [(E)-5-(2-bromovinyl)-20-deoxyuridine, brivudin]. It
was first identified as a highly specific inhibitor of HSV type
133 and VZV. In fact, the efficacy of BVDU against herpes
zoster was first reported in patients34, before its potency and

Erik de Clercq538
selectivity as an anti-VZV agent in cell culture was demon-
strated by Shigeta et al35.
BVDU (Fig. 2) was first approved for clinical use (as
Helpins) in the former DDR (East Germany) in immunosup-
pressed patients, before it was licensed for clinical use in
immunocompetent persons for the treatment of herpes zoster.
It is now available in several countries, under different
Figure 3 Structures of ddN analogues.
trade names (Zostexs (Germany); Briviracs (Italy); Zerpexs
(Belgium)). While even more active than acyclovir against
both HSV-1 and VZV, BVDU is specifically used for the
treatment of VZV infections (herpes zoster)36. It owes its
selective anti-VZV activity to a specific phosphorylation by the
VZV-encoded thymidine kinase to the mono- and dipho-
sphate, which upon further phosphorylation by the (cellular)

Discovery of antiviral agents 539
nucleoside diphosphate (NDP) kinase to the triphosphate,
interferes with the viral DNA synthesis.
Recent review articles have pointed to FV-100 as an
emerging new candidate drug for the treatment of VZV
infections37–39. Migliore39 qualified FV-100 as the most potent
and selective anti-VZV agent reported to date.
Where did FV-100 originate from? The parent compound of
FV-100 (FV standing for ‘‘Fermavir’’) is Cf1743 (Cf standing for
‘‘Cardiff’’) (Fig. 2). Cf1743 belongs to the so-called BCNAs
(bicyclic furopyrimidine nucleoside analogs) originally described
by McGuigan et al40,41. Cf1743 is the most potent of a class of
compounds that are specifically active against VZV. The mechan-
ism of action of Cf1743 still needs to be elucidated but clearly
depends on a specific phosphorylation by the VZV-encoded
thymidine kinase42. Its potency against VZV exceeds that of all
other antiviral agents including BVDU43.
FV-100 is the 50-valine ester of Cf174344 (Fig. 2) and in this
sense related to the valine esters of acyclovir and ganciclovir
(Fig. 1). FV-100 has recently been the subject of 3 randomized,
double-blind, placebo-controlled clinical trials45. These studies
support further investigations with FV-100 (at a daily dosage
of 100 mg, 200 mg, 400 mg or 800 mg, for 7 days) for the
treatment of herpes zoster45. Herpes zoster patients can now
be monitored for VZV DNA in the saliva46. Such non-invasive
analysis would allow to follow the response of VZV infection
to anti-VZV agents such as VACV, BVDU and FV-100.
Figure 4 Structures of (S)-HPMPC and (S)-HPMPA.
2.3. 20,30-Dideoxynucleoside (ddN) analogs
The first 20,30-dideoxynucleoside (ddN) described, in 1985, for
its inhibitory effect on the infectivity and cytopathicity of HIV
(then called HTLV-III for human T-cell lymphotropic virus
type 3) was azidothymidine (AZT, retrovirs) (Fig. 3)47.
This compound was discovered serendipitously as an anti-
HIV agent, as it had been originally synthesized as a potential
anticancer agent by Horwitz et al48. In fact, in 1980 we had
described the anti-HSV activity of several ddN analogs,
including azidothymidine49. The mechanism of anti-HIV
action of azidothymidine was described by Furman et al50.
It resided in the inhibitory effect of its 50-triphosphate as an
HIV alternate substrate/competitive inhibitor on the reverse
transcriptase (RT).
Following azidothymidine, several other 20,30-dideoxynu-
cleoside analogs, including dideoxyinosine (ddI, didanosine,
Videxs, Videx ECs) and dideoxycytidine (ddC, zalcitabine,
Hivids) (Fig. 3) were found inhibitory to the infectivity and
cytopathicity of HIVal 51. Like AZT, ddI and ddC were found
to inhibit the HIV RT as chain terminators, ddI, following its
conversion to ddATP in competition with dATP, and ddC,
following its conversion to ddCTP in competition with dCTP,
both dATP and dCTP being regular substrates for the RT
reaction.
Following AZT, ddI and ddC, the fourth ddN analog to be
discovered was d4T (stavudine, Zerits), which was announced
successively by Baba et al52. Lin et al.53 and Hamamoto
et al54. The mode of action of d4T was resolved by Balzarini
et al55. It would become one of the most popular, world-wide
used anti-HIV drugs, later to be superseded only by tenofovir
(see infra).
Following stavudine (d4T), three more ddN analogs, lamivu-
dine (3TC), abacavir (ABC) and emtricitabine [(–)FTC] would
subsequently be licensed as anti-HIV drugs56,57. Lamivudine was
originally discovered by the late Belleau58 as a racemic mixture of
which the (–)enantiomer (3TC) was proven to be most active.
The anti-HIV activity of abacavir was originally described
by Daluge59, whereas the discovery of (–)FTC was credited to
Schinazi60. 3TC, ABC, and (–)FTC would later be commercia-
lized under the trade names of Epivirs, Ziagens and Emtrivas,
respectively.
Although many more ddNs than those reported above,
have been reported in the meantime, the number of approved
ddN analogs (now also referred to as nucleoside reverse
transcriptase inhibitors (NRTIs)) have been limited to the
seven ddN analogs shown in Fig. 3. In the past few years, no
more ddN analogs or NRTIs have been added to the current
anti-HIV drug armamentarium. Instead, further progress in
the field of anti-HIV therapy has been focused on the judicious
choice of combination of the appropriate NRTIs (such as
Emtrivas) with the appropriate NtRTIs (such as tenofovir
disoproxil fumarate) and NNRTIs (such as efavirenz and
rilpivirine) (see infra: Atriplas and Compleras/Evipleras).
2.4. (S)-HPMPA and (S)-HPMPC (Cidofovir)
The era of the acyclic nucleoside phosphonates (ANPs)61 started
in 1986 with the description of (S)-HPMPA [(S)-9-(3-hydroxy-2-
phosphonylmethoxypropyl)adenine] as a novel selective broad-
spectrum anti-DNA virus agent62. One year later followed the
description of (S)-HPMPC [(S)-1-(3-hydroxy-2-phosphonyl-
methoxypropyl)cytosine], the cytosine counterpart (cidofovir) of
(S)-HPMPA (Fig. 4)63. (S)-HPMPC showed an activity spectrum
similar to that of (S)-HPMPA; nine years later, in 1996, it would
be formally approved as Vistides for the treatment, by intrave-
nous injection, of CMV retinitis in AIDS patients. This indication
has virtually disappeared with the efficient treatment of AIDS.
Yet, cidofovir is still used off label in the treatment of poxvirus
infections (i.e., molluscum contagiosum) and human papilloma-
virus (HPV) infections. It has proven more efficacious than
smallpox vaccination upon lethal monkeypox virus infection64.
Cidofovir bears an hydroxyl group that is equivalent to the
30-hydroxyl group of dCMP and permits its incorporation into
DNA, creating a significant impediment to trans-lesion DNA
synthesis in a manner resembling DNA damage65. This leads
for vaccinia virus to an inhibition of genome encapsidation
and virus assembly66.
Cidofovir has proven highly effective in different models of
poxvirus infection, whether the compound was administered
either intraperitoneally, intranasally or topically67, including

Erik de Clercq540
camelpox in athymic nude mice68 as well as an aerosol
rabbitpox model69. Also, the sea lion parapoxvirus has proven
susceptible to cidofovir70. When co-administered with the
smallpox vaccine, cidofovir effectively reduced vaccination
side effects71.
In earlier reports, both topical and systemic cidofovir were
shown to resolve recalcitrant molluscum contagiosum virus
lesions72–75. The efficacy of intravenous cidofovir in the
treatment of giant molluscum contagiosum was most drama-
tically shown recently in a patient with HIV infection76.
Intralesional cidofovir has proven successful in the treat-
ment of acyclovir-resistant HSV infection77, whereas intrave-
nous cidofovir was used successfully in the treatment of dual
infection with polyomavirus BK and acyclovir-resistant HSV
in a bone marrow transplant recipient78. Cidofovir may be a
potentially effective therapy for the treatment of BK virus-
associated hemorrhagic cystitis, a severe complication after
allogeneic hematopoietic stem cell transplantation (HSCT)79.
BK virus-associated nephopathy in a kidney transplant reci-
pient was successfully treated with cidofovir, the first case in
Japan80.
Cidofovir markedly reduced the growth of HPV-positive
cervical cancer cell xenografts in nude mice81. This result was
confirmed in a clinical setting where cidofovir proved effective
in the topical treatment of cervical intraepithelial neoplasia
(CIN) stage 282.
The principal off-label use of cidofovir, however, is cur-
rently for intralesional injection for recurrent respiratory
papillomatosis in both children83 and adults84, and, even more
so, the local administration of cidofovir for HPV-associated
skin lesions in transplant recipients85. Successful treatment of
cutaneous warts has been reported with intravenous cidofovir
in an 11-year old girl86, and several case reports have pointed
to the successful use of topical cidofovir at 1% in the
treatment of HPV warts in children87–89.
2.5. CMX001 (HDP-CDV)
CMX001 (HDP-CDV) (Fig. 5) corresponds to hexadecylox-
ypropyl cidofovir, an ether lipid conjugate of cidofovir, which,
in principle, should be active against the same DNA viruses
against which cidofovir itself is active, but, in comparison with
CDV, CMX001, should have increased oral bioavailability
and increased cellular uptake, facilitated by the lipid moiety of
the molecule90. The increased cellular uptake accounts for the
increased antiviral activity of HDP-CDV91. Its oral activity
Figure 5 Structures of HDP-CDV and ODE-CDV.
has been demonstrated particularly for the treatment of
smallpox92 and in a lethal mousepox model93.
Preclinical studies have demonstrated the efficacy of
CMX001 in the treatment of vaccinia and cowpox virus
infections in mice, which suggested that the compound could
be used in the event of a bioterror attack by poxviruses94. The
advantage of CMX001 over its parent compound, cidofovir, is
that it could be administered orally (as a tablet or liquid)
(presumably) without nephrotoxicity, which limits the dosing
of the parent compound95. It has proven efficacious as a post
exposure antiviral in rabbits infected with rabbitpox virus, a
model for orthopoxvirus infection of humans96.
In pregnant guinea pigs, CMX001 improved he outcome
of congenital CMV infection97, suggesting the potential of
CMX001 for the treatment of congenital CMV infection in
humans. CMX001 may also be effective in the treatment of
acyclovir-resistant HSV infection, and combination of
CMX001 with acyclovir led to a synergistic inhibition of
HSV infections98.
CMX001 has been shown to prevent adenovirus-induced
mortality in a permissive immunosuppressed animal model
(cyclophosphamide-treated hamsters)99, and in a pediatric
hematopoietic stem cell transplantation recipient CMX001
led to the eradication of disseminated adenovirus infection100.
CMX001 also seems effective in the treatment of human
polyomavirus replication in primary human renal tubular
epithelial cells101, polyoma JC virus replication in human fetal
brain SVG cell cultures102 and polyomavirus JC replication in
human brain progenitor-derived astrocytes103.
In addition to CMX001, various other prodrugs of cidofo-
vir have been reported, i.e., Ala–Ser and Val–Ser prodrugs104.
Similarly, tyrosine-based prodrugs of cidofovir have been
synthesized which may be of potential use in the treatment
of the same DNA virus infections that are susceptible to
cidofovir105. The advantage of these prodrugs, if any, over
that of the parent compound, cidofovir, remains to be
demonstrated.
2.6. HDP-(S)-HPMPA
Alkoxypropyl, i.e., hexadecyloxypropyl (HDP) and octadecy-
loxyethyl (ODE) esters have been prepared from both
(S)-HPMPC and (S)-HPMPA. (S)-HPMPA was the prototype
of the acyclic nucleoside phosphonates62, which was never
commercialized for clinical use. The reason why (S)-HPMPC
(cidofovir), PMEA (adefovir), and (R)-PMPA (tenofovir) were
successfully introduced in clinical medicine, whereas the

Discovery of antiviral agents 541
prototype compound (S)-HPMPA was not, even not for any
of the DNA virus infections for which it was more effective
than (S)-HPMPC, has remained an enigma.
HDP-(S)-HPMPA and ODE-(S)-HPMPA (Fig. 6) were
recently reported to be active against HIV replication106.
This was not surprising given the broad-spectrum anti-
DNA virus activity of the parent compound, (S)-HPMPA62.
More surprising was the activity reported for ODE- and
HDP-(S)-HPMPA against hepatitis C virus (HCV), which,
being an RNA virus, was certainly not expected to be
sensitive to the inhibitory action of acyclic nucleoside
phosphonates107.
Of the alkoxyalkyl esters, the most active anti-HCV agent was
ODE-(S)-3-methoxy-2-(phosphonylmethoxy)propyl-guanine with
an EC50o0.01 nM and a selectivity index of 44.4 million106.
(S)-HPMPA has been found active against parasites, i.e., the
growth of Plasmodium falciparum and P.berghei108,109. The HDP-
cyclic(S)-HPMPA was found to have antischistosomal activity110,
and this has given a new impetus to a long forgotten therapeutic
potential of (S)-HPMPA and acyclic nucleoside phosphonates,
Figure 7 Structures of adefovir, adefovir dipivoxil, tenofovir and TD
Figure 6 Structures of HDP-(S)-HPMPA and ODE-(S)-HPMPA.
their unexplored therapeutical potential in the treatment of
malaria and other parasitic infections.
ODE and HDP esters of HPMPA are potent inhibitors of
HIV111, while its parent compound has not been recognized as an
anti-HIV agent. Alkoxyalkyl esters of (S)-HPMPAmay, like those
of cidofovir, exhibit wide spectrum antiviral activity against pox-,
adeno-, herpes-, cytomegalovirus infections, with HDP-(S)-
HPMPA being at least as promising as HDP-(S)-HPMPC as
orally active against various DNA virus infections112.
2.7. Adefovir and tenofovir
The oral bioavailability of cidofovir (CDV) is increased by linking
CDV to an alkoxyalkyl ester, as in hexadecyloxypropyl (HDP)-
CDV (see supra, section 2.5). The oral prodrugs designed for
adefovir (first described by de Clercq et al.62) and tenofovir (first
described by Balzarini et al.113) have been adefovir dipivoxil (AD,
Hepseras) and tenofovir disoproxil fumarate (TDF, Vireads)
(Fig. 7). The design of AD and TDF has made the parent
F.

Erik de Clercq542
compounds adefovir and tenofovir rapidly available, through
cleavage of the ester bonds, after oral administration.
Adefovir dipivoxil [bis(pivaloyloxymethyl)-9-(2-phosphonyl-
methoxyethyl)adenine] has been described by Starrett et al.114,
Cundy et al.115,116 and Shaw et al117.
The pioneering clinical studies of Hadziyannis et al.118 and
Marcellin et al.119 helped the approval of Hepseras, in 2002,
for the treatment of chronic hepatitis B. These results were
nicely confirmed by a cross-over study upon long-term
treatment with Hepseras in HBeAg-negative chronic HBV
patients120.
Tenofovir disoproxil [bis(isopropyloxycarbonyloxy-
methyl)-9-(R)-(2-phosphonylmethoxy-propyl)adenine] was
described by Robbins et al.121 and Naesens et al.122 Teno-
fovir disoproxil fumarate (TDF) was approved for clinical
use for the treatment of HIV infection (AIDS) in 2001, and
for the treatment of chronic HBV infection in 2008. As to
the latter, the key observation was that TDF at a daily dose
of 300 mg had superior antiviral activity with a similar
safety profile as compared to adefovir dipivoxil at a daily
dose of 10 mg123.
Twenty years after its original discovery, tenofovir has acquired
a crucial position in the fight against HIV: it is not only efficacious
against AIDS but also hepatitis B; it can be used in combination
with emtricitabine, efavirenz, rilpivirine, elvitegravir, atazanavir, or
darunavir, as a single once-daily oral pill; and it can be used
in combination with emtricitabine as a once-daily oral pill,
Truvadas, in the prophylaxis of sexual HIV transmission124.
The increased oral bioavailability and increased cellular
uptake noted for the hexadecyloxypropyl derivative of cido-
fovir (see supra, section 2.5) also holds for tenofovir. Hex-
adecyloxypropyl tenofovir (CMX157) shows markedly
increased oral bioavailability and cellular uptake as compared
with the parent compound125,126.
Figure 8 Drug combination of Quad.
For the treatment of HBV infections, there are officially six
compounds available: interferon-a, lamivudine (3TC), adefo-
vir dipivoxil, entecavir, telbivudine and TDF. For HIV
infections, however, 25 years after the discovery of HIV, there
are now more than 25 compounds approved as anti-HIV
drugs56,57. Tenofovir, in its oral prodrug form, TDF, is the
cornerstone for the treatment of HIV infections127,128, appar-
ently due to the magic of the phosphonate bond129.
2.8. Truvada, Atripla, Complera, Quad (Stribild TM)
When first approved in 2001, Vireads could hardly be
anticipated to grow to a double-drug combination (Truvadas)
in 2004, a triple-drug combination (Atriplas) in 2006, another
triple-drug combination (Compleras in the US, Evipleras in
the EU) in 2011, and even a quadruple-drug combination
Quad (Stribild TM) in 2012 (Fig. 8). The single once-daily oral
pill containing three active ingredients (Truvadas and efavir-
enz) was Atriplas130. Then followed Compleras/Evipleras,
like Atripla, containing three active ingredients: Truvadas
and rilpivirine (Edurants) (Fig. 8)131.
Scheduled for 2012 is the first Quad pill, containing in
addition to Truvadas, the integrase inhibitor (INI) elvitegra-
vir and the pharmacoenhancer cobicistat (Fig. 8). Also
expected to be approved in 2012 is Truvadas for prophylactic
use to prevent sexual transmission of HIV infection.
In addition to the ‘‘real’’ Quad (Quad no. 1 Stribild TM)
containing elvitegravir, cobicistat, emtricitabine and TDF, several
other Quads are forthcoming131: Quad no. 2, consisting of
Truvadas, cobicistat and atazanavir; Quad no. 3, consisting of
GS-7340, emtricitabine ((–)FTC), cobicistat and darunavir; and
Quad no. 4, consisting of GS-7340, emtricitabine ((–)FTC),
cobicistat and elvitegravir131.

Figure 9 Structures of GS-7340, (R)-PMPA, GS-9131 and GS-9148.
Discovery of antiviral agents 543
2.9. GS-7340, GS-9131, GS-9191 and GS-9219
GS-7340 (Fig. 9) (see supra, section 2.8) is an isopropylalaninyl
monoamidate phenyl monoester prodrug of tenofovir; it was first
described by Lee et al132. Phosphonoamidate prodrugs of
adefovir and tenofovir had originally been described by Ballatore
et al133. A novel one-pot synthesis of bis-amidate prodrugs of
acyclic nucleoside phosphonates was reported by Jansa et al134.
GS-9131 (Fig. 9) is the orally bioavailable phosphonoami-
date prodrug of GS-9148, a cyclic nucleoside phosphonate
analogue135 with a low nephrotoxic potential136. The 20-fluoro
group fulfills a specific role in this compound, as it may cause
steric hindrance to the side chain of the Q151L mutation137.
Cathepsin A is the major hydrolase catalyzing the intracellular
hydrolysis of GS-7340 to tenofovir and of GS-9131 to GS-
9148138. Cathepsin is also held responsible for the intracellular
hydrolysis of GS-9191 to cPrPMEDAP139. GS-9191 can be
considered as a ‘‘double’’ prodrug, being converted to PMEG
via cPrPMEDAP as intermediate, thus requiring hydrolysis
followed by deamination to yield PMEG, which is then
converted to PMEG diphosphate as the final active metabolite.
GS-9191 has been pursued for the topical treatment of
human papilloma virus (HPV) infections140. cPrPMEDAP
could also be directly applied topically or transdermally141.
Like GS-9191, GS-9219 (Fig. 10) is a double prodrug of
PMEG via cPrPMEDAP: it has potent antineoplastic activity
in dogs with spontaneous non-Hodgkin’s lymphoma142,143.
As for GS-9191, the final active metabolite of GS-9219 is
assumed to be PMEG diphosphate142.
3. Conclusions
The principal conclusions reached from this overview are as
follows:
3.1.
Valacyclovir (VACV) has succeeded acyclovir as thegold standard for the oral treatment of both herpes
simplex virus (HSV) and varicella-zoster virus (VZV)
infections;
3.2.
BVDU (brivudin) and FV-100 (the valine ester of Cf1743)have remained the most potent inhibitors of VZV
infections; Cf1743 does not suffer from the liability of
potentiating the toxicity of 5-fluorouracil, as noted for
bromovinyluracil, the degradation product of BVDU;
3.3.
Most of the 20,30-dideoxynucleoside (ddN) analogs thathave been licensed for clinical use, are still used for the
treatment of human immunodeficiency virus (HIV)
infections, i.e. (–)FTC (emtricitabine) in combination
with tenofovir disoproxil fumarate (TDF);
3.4.
The acyclic nucleoside phosphonates (S)-HPMPA and(S)-HPMPC (cidofovir) possess broad-spectrum anti-
viral activity against DNA (pox, herpes, adeno, papil-
loma, polyoma) viruses;
3.5.
The alkoxyalkyl (HDP: hexadecyloxypropyl, ODE: octa-decyloxyethyl) esters of (S)-HPMPA and (S)-HPMPC
show increased oral bioavailability and increased cellular
uptake as compared to the parent compounds;
3.6.
Adefovir dipivoxil, the prodrug of PMEA, is widelyaccepted for the treatment of hepatitis B virus (HBV)
infections, whereas tenofovir disoproxil fumarate, the
prodrug of (R)-PMPA, is widely used for the treatment
of both HIV and HBV infections;
3.7.
Combinations containing TDF and (–)FTC (Truvadas),if extended with efavirenz to Atriplas, or with rilpivirine
to Compleras (or Evipleras) are available as single pills
for once-daily use in the treatment of HIV infections.
was recently appeared is the Quad pill containing TDF,
(–)FTC, elvitegravir and cobicistat Quad (Stribild TM).
3.8.
In development are phosphonoamidate prodrugs,i.e., GS-7340 and GS-9131, for the oral treatment of
HIV infections, GS-9191 for topical treatment of human
papilloma virus (HPV) infections, and GS-9219 for the
treatment of non-Hodgkin’s lymphoma (NHL) in dogs.

Figure. 10 Structures of prodrugs of PMEG.
Erik de Clercq544
Acknowledgments
The author thanks Mrs. Christiane Callebaut for her profi-
cient editorial assistance.
References
1. Prusoff WH. Synthesis and biological activities of iododeoxyur-
idine, an analog of thymidine. Biochim Biophys Acta 1959;32:295–6.
2. Herrmann EC Jr. Plaque inhibition test for detection of specific
inhibitors of DNA containing viruses. Proc Soc Exp Biol Med
1961;107:142–5.
3. Kaufman HE. Clinical cure of herpes simplex keratitis by
5-iodo-20-deoxyuridine. Proc Soc Exp Biol Med 1962;109:251–3.
4. Kaufman HE, Heidelberger C. Therapeutic antiviral action of
5-trifluoromethyl-20-deoxyuridine in herpes simplex keratitis.
Science 1964;145:585–6.
5. Lee WW, Benitez A, Goodman L, Baker BR. Potential antic-
ancer agents. XL. Synthesis of the b-anomer of 9-(b-D-arabino-furanosyl)-adenine. J Am Chem Soc 1960;82:2648–9.
6. Privat de GM, de Rudder J. Effet de deux nucleosides de
l’arabinose sur la multiplication des virus de l’herp�es et de la
vaccine en culture cellulaire. C R Acad Sc Paris 1964;259:2725–8.
7. Schabel FM Jr. The antiviral activity of 9-b-D-arabinofuranosy-ladenine (Ara-A). Chemotherapy 1968;13:321–38.
8. Whitley RJ, Ch’ien LT, Dolin R, Galasso GJ, Alford CA Jr.
Adenine arabinoside therapy of herpes zoster in the immuno-
suppressed. NIAID collaborative antiviral study. N Engl J Med
1976;294:1193–9.
9. Sidwell RW, Huffman JH, Khare GP, Allen LB, Witkowski JT,
Robins RK. Broad-spectrum antiviral activity of virazole: 1-beta-D-
ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 1972;177:705–6.
10. Elion GB, Furman PA, Fyfe JA, de Miranda P, Beauchamp L,
Schaeffer HJ. Selectivity of action of an antiherpetic agent,
9-(2-hydroxyethoxymethyl)guanine. Proc Natl Acad Sci USA
1977;74:5716–20.
11. Schaeffer HJ, Beauchamp L, de Miranda P, Elion GB, Bauer
DJ, Collins P. 9-(2-Hydroxyethoxymethyl) guanine activity
against viruses of the herpes group. Nature 1978;272:583–5.
12. Field HJ, de Clercq E. Antiviral drugs – a short history of their
discovery and development. Microbiol Today 2004;31:58–61.
13. Colla L, de Clercq E, Busson R, Vanderhaeghe H. Synthesis
and antiviral activity of water-soluble esters of acyclovir [9-
(2-hydroxyethoxymethyl)guanine]. J Med Chem 1983;26:
602–4.
14. Maudgal PC, de Clercq E, Descamps J, Missotten L. Topical
treatment of experimental herpes simplex keratouveitis with 20-
O-glycylacyclovir. A water-soluble ester of acyclovir. Arch
Ophthalmol 1984;102:140–2.
15. de Clercq E, Field HJ. Antiviral prodrugs – the development of
successful prodrug strategies for antiviral chemotherapy. Brit J
Pharmacol 2006;147:1–11.
16. Brantley JS, Hicks L, Sra K, Tyring ST. Valacyclovir for the
treatment of genital herpes. Expert Rev Anti Infect Ther
2006;4:367–76.
17. Rajalakshmi R, Kumari R, Thappa DM. Acyclovir versus
valacyclovir. Indian J Dermatol Venereol Leprol 2010;76:439–44.
18. Klein A, Miller KB, Sprague K, DesJardin JA, Snydman DR. A
randomized, double-blind, placebo-controlled trial of valacyclo-
vir prophylaxis to prevent zoster recurrence from months 4 to 24
after BMT. Bone Marrow Transplant 2011;46:294–9.
19. Pouplin T, Pouplin JN, Van Toi P, Lindegardh N, Rogier van
DH, Hien TT. Valacyclovir for herpes simplex encephalitis.
Antimicrob Agents Chemother 2011;55:3624–6.
20. Abudalu M, Tyring S, Koltun W, Bodsworth N, Hamed K.
Single-day patient-initiated famciclovir therapy versus 3-day

Discovery of antiviral agents 545
valacyclovir regimen for recurrent genital herpes: a randomized,
double-blind, comparative trial. Clin Infect Dis 2008;47:651–8.
21. Miserocchi E, Modorati G, Galli L, Rama P. Efficacy of
valacyclovir vs. acyclovir for the prevention of recurrent herpes
simplex virus eye disease: a pilot study. Am J Ophthalmol
2007;144:547–51.
22. Andrews WW, Kimberlin DF, Whitley R, Cliver S, Ramsey PS,
Deeter R. Valacyclovir therapy to reduce recurrent genital herpes in
pregnant women. Am J Obstet Gynecol 2006;194:774–81.
23. Pasternak B, Hviid A. Use of acyclovir, valacyclovir, and
famciclovir in the first trimester of pregnancy and the risk of
birth defects. J Am Med Assoc 2010;304:859–66.
24. Reischig T, Jindra P, Hes O, Svecova M, Klaboch J, Treska V.
Valacyclovir prophylaxis versus preemptive valganciclovir ther-
apy to prevent cytomegalovirus disease after renal transplanta-
tion. Am J Transplant 2008;8:69–77.
25. Leone F, Akl A, Giral M, Dantal J, Blancho G, Soulillou JP. Six
months anti-viral prophylaxis significantly decreased cytomega-
lovirus disease compared with no anti-viral prophylaxis follow-
ing renal transplantation. Transpl Int 2010;23:897–906.
26. Hoshino Y, Katano H, Zou P, Hohman P, Marques A, Tyring
SK. Long-term administration of valacyclovir reduces the
number of Epstein-Barr virus (EBV)-infected B cells but not
the number of EBV DNA copies per B cell in healthy volunteers.
J Virol 2009;83:11857–61.
27. Balfour HH Jr, Hokanson KM, Schacherer RM, Fietzer CM,
Schmeling DO, Holman CJ. A virologic pilot study of
valacyclovir in infectious mononucleosis. J Clin Virol
2007;39:16–21.
28. Lai L, Xu Z, Zhou J, Lee KD, Amidon GL. Molecular basis of
prodrug activation by human valacyclovirase, an alpha-amino
acid ester hydrolase. J Biol Chem 2008;283:9318–27.
29. de Bellis RH, Chen BX, Erlanger BF. Inhibition of sickling
in vitro by three purine-based antiviral agents: an approach to
the treatment of sickle cell disease. Blood Cells Mol Dis
2003;31:286–90.
30. Ender KL, de Bellis RH, Erlanger BF, Billote GB, Brittenham
GM. Safety of short-term valacyclovir as an anti-sickling agent
in sickle-cell anemia. Pediatr Blood Cancer 2011;56:843–5.
31. Mahony WB, Domin BA, McConnell RT, Zimmerman TP.
Acyclovir transport into human erythrocytes. Biol Chem
1988;263:9285–91.
32. Field HJ, de Clercq E. Antiviral chemistry and chemotherapy’s
current antiviral agents factfile (2nd edition): DNA viruses.
Antiviral Chem Chemother 2008;19:51–62.
33. de Clercq E, Descamps J, de Somer P, Barr PJ, Jones AS,
Walker RT. E-5-(2-Bromovinyl)-20-deoxyuridine: a potent and
selective antiherpes agent. Proc Natl Acad Sci USA 1979;76:
2947–51.
34. de Clercq E, Degreef H, Wildiers J, de Jonge G, Drochmans A,
Descamps J. Oral (E)-5-(2-bromovinyl)-20-deoxyuridine in severe
herpes zoster infections. Brit Med J 1980;281:1178.
35. Shigeta S, Yokota T, Iwabuchi T, Baba M, Konno K, Ogata M.
Comparative efficacy of antiherpes drugs against various strains
of varicella-zoster virus. J Infect Dis 1983;147:576–84.
36. de Clercq E. Discovery and development of BVDU (brivudin) as
a therapeutic for the treatment of herpes zoster. Biochem
Pharmacol 2004;68:2301–15.
37. Dropulic LK, Cohen JI. Update on new antivirals under
development for the treatment of double-stranded DNA virus
infections. Clin Pharmacol Ther 2010;88:610–9.
38. Andrei G, Snoeck R. Emerging drugs for varicella-zoster virus
infections. Expert Opin Emerg Drugs 2011;16:507–35.
39. Migliore M. FV-100: the most potent and selective anti-varicella
zoster virus agent reported to date. Antivir Chem Chemother
2010;20:107–15.
40. McGuigan C, Yarnold CJ, Jones G, Velazquez S, Barucki H,
Brancale A, et al. Potent and selective inhibition of varicella-
zoster virus (VZV) by nucleoside analogs with an unusual
bicyclic base. J Med Chem 1999;42:4479–84.
41. McGuigan C, Barucki H, Blewett S, Carangio A, Erichsen JT,
Andrei G, et al. Highly potent and selective inhibition of
varicella-zoster virus by bicyclic furopyrimidine nucleosides
bearing an aryl side chain. J Med Chem 2000;43:4993–7.
42. de Clercq E. Highly potent and selective inhibition of varicella-
zoster virus replication by bicyclic furo[2,3-d]pyrimidine nucleo-
side analogs. Med Res Rev 2003;23:253–74.
43. Andrei G, Sienaert R, McGuigan C, de Clercq E, Balzarini J, Snoeck
R. Susceptibilities of several clinical varicella-zoster virus (VZV)
isolates and drug-resistant VZV strains to bicyclic furano pyrimidine
nucleosides. Antimicrob Agents Chemother 2005;49:1081–6.
44. McGuigan C, Pathirana RN, Migliore M, Adak R, Luoni G,
Jones AT, et al. Preclinical development of bicyclic nucleoside
analogs as potent and selective inhibitors of varicella zoster
virus. J Antimicrob Chemother 2007;60:1316–30.
45. Pentikis HS, Matson M, Atiee G, Boehlecke B, Hutchins JT,
Patti JM, et al. Pharmacokinetics and safety of FV-100, a novel
oral anti-herpes zoster nucleoside analog, administered in single
and multiple doses to healthy young adult and elderly adult
volunteers. Antimicrob Agents Chemother 2011;55:2847–54.
46. Mehta SK, Tyring SK, Gilden DH, Cohrs RJ, Leal MJ, Castro
VA, et al. Varicella-zoster virus in the saliva of patients with
herpes zoster. J Infect Dis 2008;197:654–7.
47. Mitsuya H, Weinhold KJ, Furman PA, St Clair MH, Lehrman SN,
Gallo RC, et al. 30-Azido-30-deoxythymidine (BW A509U): an
antiviral agent that inhibits the infectivity and cytopathic effect of
human T-lymphotropic virus type III/lymphadenopathy-associated
virus in vitro. Proc Natl Acad Sci USA 1985;82:7096–100.
48. Horwitz JP, Chua J, Noel M. Nucleosides V. The monomesy-
lates of 1-(2-deoxy-beta-D-lyxofuranosyl) thymine. J Org Chem
1964;29:2076–8.
49. de Clercq E, Balzarini J, Descamps J, Eckstein F. Antiviral,
antimetabolic and antineoplastic activities of 20- or 30-amino or -
azido-substituted deoxyribonucleosides. Biochem Pharmacol
1980;29:1849–51.
50. Furman PA, Fyfe JA, St Clair MH, Weinhold K, Rideout JL,
Freeman GA, et al. Phosphorylation of 30-azido-30-deoxythymi-
dine and selective interaction of the 50-triphosphate with human
immunodeficiency virus reverse transcriptase. Proc Natl Acad Sci
USA 1986;83:8333–7.
51. Mitsuya H, Broder S. Inhibition of the in vitro infectivity and
cytopathic effect of human T-lymphotrophic virus type III/
lymphadenopathy-associated virus (HTLV-III/LAV) by 20,30-
dideoxynucleosides. Proc Natl Acad Sci USA 1986;83:1911–5.
52. Baba M, Pauwels R, Herdewijn P, de Clercq E, Desmyter J,
Vandeputte M. Both 20,30-dideoxythymidine and its 20,30-unsaturated
derivative (20,30-dideoxythymidinene) are potent and selective inhibi-
tors of human immunodeficiency virus replication in vitro. Biochem
Biophys Res Commun 1987;142:128–34.
53. Lin TS, Schinazi RF, Prusoff WH. Potent and selective in vitro
activity of 30-deoxythymidin-20-ene (30-deoxy-20,30-didehydrothy-
midine) against human immunodeficiency virus. Biochem Phar-
macol 1987;36:2713–27.
54. Hamamoto Y, Nakashima H, Matsui T, Matsuda A, Ueda T,
Yamamoto N. Inhibitory effect of 20,30-didehydro-20,30-dideox-
ynucleosides on infectivity, cytopathic effects, and replication of
human immunodeficiency virus. Antimicrob Agents Chemother
1987;31:907–10.
55. Balzarini J, Herdewijn P, de Clercq E. Differential patterns of
intracellular metabolism of 20,30-didehydro-20,30-dideoxythymi-
dine and 30-azido-20,30-dideoxythymidine, two potent anti-
human immunodeficiency virus compounds. J Biol Chem
1989;264:6127–33.
56. de Clercq E. Anti-HIV drugs: 25 compounds approved within 25
years after the discovery of HIV. Int J Antimicrob Agents
2009;33:307–20.

Erik de Clercq546
57. de Clercq E. The history of antiretrovirals: key discoveries over
the past 25 years. Rev Med Virol 2009;19:287–99.
58. Soudeyns H, Yao XI, Gao Q, Belleau B, Kraus JL, Nguyen-Ba
N, et al. Anti-human immunodeficiency virus type 1 activity and
in vitro toxicity of 20-deoxy-30-thiacytidine (BCH-189), a novel
heterocyclic nucleoside analog. Antimicrob Agents Chemother
1991;35:1386–90.
59. Daluge SM, Good SS, Faletto MB, Miller WH, St Clair MH,
Boone LR, et al. 1592U89, a novel carbocyclic nucleoside analog
with potent, selective antihuman immunodeficiency virus activ-
ity. Antimicrob Agents Chemother 1997;41:1082–93.
60. Schinazi RF, McMillan A, Cannon D, Mathis R, Lloyd RM,
Peck A, et al. Selective inhibition of human immunodeficiency
viruses by racemates and enantiomers of cis-5-fluoro-1-[2-(hydro-
xymethyl)-1,3-oxathiolan-5-yl]cytosine. Antimicrob Agents Chemother
1992;36:2423–31.
61. de Clercq E, Holy A. Acyclic nucleoside phosphonates: a key
class of antiviral drugs. Nat Rev Drug Discov 2005;4:928–40.
62. de Clercq E, Holy A, Rosenberg I, Sakuma T, Balzarini J,
Maudgal PC. A novel selective broad-spectrum anti-DNA virus
agent. Nature 1986;323:464–7.
63. de Clercq E, Sakuma T, Baba M, Pauwels R, Balzarini J, Rosenberg
I, et al. Antiviral activity of phosphonylmethoxyalkyl derivatives of
purine and pyrimidines. Antiviral Res 1987;8:261–72.
64. Stittelaar KJ, Neyts J, Naesens L, van Amerongen G, van
Lavieren RF, Holy A, et al. Antiviral treatment is more effective
than smallpox vaccination upon lethal monkeypox virus infec-
tion. Nature 2006;439:745–8.
65. Julien O, Beadle JR, Magee WC, Chatterjee S, Hostetler KY,
Evans DH, et al. Solution structure of a DNA duplex containing
the potent anti-poxvirus agent cidofovir. J Am Chem Soc
2011;133:2264–74.
66. Jesus DM, Costa LT, Gonc-alves DL, Achete CA, Attias M,
Moussatche N, et al. Cidofovir inhibits genome encapsidation
and affects morphogenesis during the replication of vaccinia
virus. J Virol 2009;83:11477–90.
67. Andrei G, Snoeck R. Cidofovir activity against poxvirus infec-
tions. Viruses 2010;2:2803–30.
68. Duraffour S, Matthys P, van den Oord JJ, de Schutter T, Mitera
T, Snoeck R, et al. Study of camelpox virus pathogenesis in
athymic nude mice. PLoS One 2011;6:e21561.
69. Verreault D, Sivasubramani SK, Talton JD, Doyle LA, Reddy
JD, Killeen SZ, et al. Evaluation of inhaled cidofovir
as postexposure prophylactic in an aerosol rabbitpox model.
Antiviral Res 2012;93:204–8.
70. Nollens HH, Gulland FM, Jacobson ER, Hernandez JA, Klein
PA, Walsh MT, et al. In vitro susceptibility of sea lion poxvirus
to cidofovir. Antiviral Res 2008;80:77–80.
71. Wei H, Huang D, Fortman J, Wang R, Shao L, Chen ZW.
Coadministration of cidofovir and smallpox vaccine reduced
vaccination side effects but interfered with vaccine-elicited immune
responses and immunity to monkeypox. J Virol 2009;83:1115–25.
72. Meadows KP, Tyring SK, Pavia AT, Rallis TM. Resolution of
recalcitrant molluscum contagiosum virus lesions in human
immunodeficiency virus-infected patients treated with cidofovir.
Arch Dermatol 1997;133:987–90.
73. Davies EG, Thrasher A, Lacey K, Harper J. Topical cidofovir
for severe molluscum contagiosum. Lancet 1999;353:2042.
74. Zabawski EJ Jr, Cockerell CJ. Topical cidofovir for molluscum
contagiosum in children. Pediatr Dermatol 1999;16:414–5.
75. Toro JR, Wood LV, Patel NK, Turner ML. Topical cidofovir: a
novel treatment for recalcitrant molluscum contagiosum in
children infected with human immunodeficiency virus 1. Arch
Dermatol 2000;136:983–5.
76. Erickson C, Driscoll M, Gaspari A. Efficacy of intravenous
cidofovir in the treatment of giant molluscum contagiosum in a
patient with human immunodeficiency virus. Arch Dermatol
2011;147:652–4.
77. Castelo-Soccio L, Bernardin R, Stern J, Goldstein SA, Kovarik C.
Successful treatment of acyclovir-resistant herpes simplex virus with
intralesional cidofovir. Arch Dermatol 2010;146:124–6.
78. Andrei G, Fiten P, Goubau P, van Landuyt H, Gordts B,
Selleslag D, et al. Dual infection with polyomavirus BK and
acyclovir-resistant herpes simplex virus successfully treated with
cidofovir in a bone marrow transplant recipient. Transpl Infect
Dis 2007;9:126–31.
79. Cesaro S, Hirsch HH, Faraci M, Owoc-Lempach J, Beltrame A,
Tendas A, et al. Cidofovir for BK virus-associated hemorrhagic
cystitis: a retrospective study. Clin Infect Dis 2009;49:233–40.
80. Akioka K, Okamoto M, Ushigome H, Nobori S, Kaihara S,
Yoshimura NBK. Virus-associated nephropathy in a kidney
transplant recipient successfully treated with cidofovir, the first
case in Japan. Int J Urol 2008;15:369–71.
81. Yang Y, Zhao X, Chen W, Gao Z, Liu A, Guo J, et al. Effects of
cidofovir on human papillomavirus-positive cervical cancer cells
xenografts in nude mice. Oncol Res 2010;18:519–27.
82. van Pachterbeke C, Bucella D, Rozenberg S, Manigart Y, Gilles
C, Larsimont D, et al. Topical treatment of CIN 2þ by cidofovir:
results of a phase II, double-blind, prospective, placebo-
controlled study. Gynecol Oncol 2009;115:69–74.
83. Bielecki I, Mniszek J, Cofa"a M. Intralesional injection of
cidofovir for recurrent respiratory papillomatosis in children.
Int J Pediatr Otorhinolaryngol 2009;73:681–4.
84. Tanna N, Sidell D, Joshi AS, Bielamowicz SA. Adult intralesional
cidofovir therapy for laryngeal papilloma: a 10-year perspective. Arch
Otolaryngol Head Neck Surg 2008;134:497–500.
85. Bonatti H, Aigner F, de Clercq E, Boesmueller C, Widschwend-
ner A, Larcher C, et al. Local administration of cidofovir for
human papilloma virus associated skin lesions in transplant
recipients. Transpl Int 2007;20:238–46.
86. Cusack C, Fitzgerald D, Clayton TM, Irvine AD. Successful
treatment of florid cutaneous warts with intravenous cidofovir in
an 11-year-old girl. Pediatr Dermatol 2008;25:387–9.
87. Field S, Irvine AD, Kirby B. The treatment of viral warts with
topical cidofovir 1%: our experience of seven paediatric patients.
Br J Dermatol 2009;160:223–4.
88. Fernandez-Morano T, del Boz J, Gonzalez-Carrascosa M,
Tortajada B, de Troya M. Topical cidofovir for viral warts in
children. J Eur Acad Dermatol Venereol 2011;25:1487–9.
89. Kralund HH, Broesby-Olsen S, Bistrup C, Lorentzen HF.
Substantial effect of topical cidofovir 1% on recalcitrant warts
in a renal-transplanted adolescent: a case report. Transplantation
2011;91:52–4.
90. Hostetler KY. Synthesis and early development of hexadecylox-
ypropylcidofovir: an oral antipoxvirus nucleoside phosphonate.
Viruses 2010;2:2213–25.
91. Aldern KA, Ciesla SL, Winegarden KL, Hostetler KY.
Increased antiviral activity of 1-O-hexadecyloxypropyl-
[2-14C]cidofovir in MRC-5 human lung fibroblasts is explained
by unique cellular uptake and metabolism. Mol Pharmacol
2003;63:678–81.
92. Huggins JW, Baker RO, Beadle JR, Hostetler KY. Orally active
ether lipid prodrugs of cidofovir for the treatment of smallpox.
Antiviral Res 2002;53:104.
93. Parker S, Touchette E, Oberle C, Almond M, Robertson A,
Trost LC, et al. Efficacy of therapeutic intervention with an oral
ether-lipid analog of cidofovir (CMX001) in a lethal mousepox
model. Antiviral Res 2008;77:39–49.
94. Quenelle DC, Kern ER. Treatment of vaccinia and cowpox virus
infections in mice with CMX001 and ST-246. Viruses
2010;2:2681–95.
95. Lanier R, Trost L, Tippin T, Lampert B, Robertson A, Foster S,
et al. Development of CMX001 for the treatment of poxvirus
infections. Viruses 2010;2:2740–52.
96. Rice AD, Adams MM, Wallace G, Burrage AM, Lindsey SF,
Smith AJ, et al. Efficacy of CMX001 as a post exposure antiviral in

Discovery of antiviral agents 547
New Zealand white rabbits infected with rabbitpox virus, a model
for orthopoxvirus infections of humans. Viruses 2011;3:47–62.
97. Bravo FJ, Bernstein DI, Beadle JR, Hostetler KY, Cardin RD.
Oral hexadecyloxypropyl-cidofovir therapy in pregnant guinea
pigs improves outcome in the congenital model of cytomegalo-
virus infection. Antimicrob Agents Chemother 2011;55:35–41.
98. Prichard MN, Kern ER, Hartline CB, Lanier ER, Quenelle DC.
CMX001 potentiates the efficacy of acyclovir in herpes simplex
virus infections. Antimicrob Agents Chemother 2011;55:4728–34.
99. Toth K, Spencer JF, Dhar D, Sagartz JE, Buller RM, Painter
GR, et al. Hexadecyloxypropyl-cidofovir, CMX001, prevents
adenovirus-induced mortality in a permissive, immunosuppressed
animal model. Proc Natl Acad Sci USA 2008;105:7293–7.
100. Paolino K, Sande J, Perez E, Loechelt B, Jantausch B, Painter
W, et al. Eradication of disseminated adenovirus infection in a
pediatric hematopoietic stem cell transplantation recipient using
the novel antiviral agent CMX001. J Clin Virol 2011;50:167–70.
101. Rinaldo CH, Gosert R, Bernhoff E, Finstad S, Hirsch HH. 1-O-
hexadecyloxypropyl cidofovir (CMX001) effectively inhibits
polyomavirus BK replication in primary human renal tubular
epithelial cells. Antimicrob Agents Chemother 2010;54:4714–22.
102. Jiang ZG, Cohen J, Marshall LJ, Major EO.
Hexadecyloxypropyl-cidofovir (CMX001) suppresses JC virus
replication in human fetal brain SVG cell cultures. Antimicrob
Agents Chemother 2010;54:4723–32.
103. Gosert R, Rinaldo CH, Wernli M, Major EO, Hirsch HH.
CMX001 (1-O-hexadecyloxypropyl-cidofovir) inhibits polyoma-
virus JC replication in human brain progenitor-derived astro-
cytes. Antimicrob Agents Chemother 2011;55:2129–36.
104. Peterson LW, Kim JS, Kijek P, Mitchell S, Hilfinger J, Breitenbach J,
et al. Synthesis, transport and antiviral activity of Ala-Ser and Val-
Ser prodrugs of cidofovir. Bioorg Med Chem Lett 2011;21:4045–9.
105. Zakharova VM, Serpi M, Krylov IS, Peterson LW, Breitenbach
JM, Borysko KZ, et al. Tyrosine-based 1-(S)-[3-hydroxy-2-
(phosphonomethoxy)propyl]cytosine and -adenine ((S)-HPMPC
and (S)-HPMPA) prodrugs: synthesis, stability, antiviral activity,
and in vivo transport studies. J Med Chem 2011;54:5680–93.
106. Valiaeva N, Wyles DL, Schooley RT, Hwu JB, Beadle JR,
Prichard MN, et al. Synthesis and antiviral evaluation of 9-(S)-
[3-alkoxy-2-(phosphonomethoxy)propyl]nucleoside alkoxyalkyl
esters: inhibitors of hepatitis C virus and HIV-1 replication.
Bioorg Med Chem 2011;19:4616–25.
107. Wyles DL, Kaihara KA, Korba BE, Schooley RT, Beadle JR,
Hostetler KY. The octadecyloxyethyl ester of (S)-9-[3-hydroxy-2-
(phosphonomethoxy) propyl]adenine is a potent and selective
inhibitor of hepatitis C virus replication in genotype 1A, 1B, and
2A replicons. Antimicrob Agents Chemother 2009;53:2660–2.
108. de Vries E, Stam JG, Franssen FF, Nieuwenhuijs H, Chavalit-
shewinkoon P, de Clercq E, et al. Inhibition of the growth of
Plasmodium falciparum and Plasmodium berghei by the DNA
polymerase inhibitor HPMP.Mol Biochem Parasitol 1991;47:43–50.
109. Smeijsters LJ, Franssen FF, Naesens L, de Vries E, Holy A,
Balzarini J, et al. Inhibition of the in vitro growth of Plasmodium
falciparum by acyclic nucleoside phosphonates. Int J Antimicrob
Agents 1999;12:53–61.
110. Botros SS, William S, Beadle JR, Valiaeva N, Hostetler KY.
Antischistosomal activity of hexadecyloxypropyl cyclic 9-(S)-[3-
hydroxy-2-(phosphonomethoxy)propyl]adenine and other alkox-
yalkyl esters of acyclic nucleoside phosphonates assessed by
schistosome worm killing in vitro. Antimicrob Agents Chemother
2009;53:5284–7.
111. Magee WC, Valiaeva N, Beadle JR, Richman DD, Hostetler
KY, Evans DH. Inhibition of HIV-1 by octadecyloxyethyl esters
of (S)-[3-hydroxy-2-(phosphonomethoxy)propyl] nucleosides and
evaluation of their mechanism of action. Antimicrob Agents
Chemother 2011;55:5063–72.
112. Ruiz J, Beadle JR, Buller RM, Schreiwer J, Prichard MN, Keith
KA, et al. Synthesis, metabolic stability and antiviral evalua-
tion of various alkoxyalkyl esters of cidofovir and 9-(S)-
[3-hydroxy-2-(phosphonomethoxy)propyl]adenine. Bioorg Med
Chem 2011;19:2950–8.
113. Balzarini J, Holy A, Jindrich J, Naesens L, Snoeck R, Schols D,
et al. Differential antiherpesvirus and antiretrovirus effects of the
(S) and (R) enantiomers of acyclic nucleoside phosphonates:
potent and selective in vitro and in vivo antiretrovirus activities of
(R)-9-(2-phosphonomethoxypropyl)-2,6-diaminopurine. Antimi-
crob Agents Chemother 1993;37:332–8.
114. Starrett JE Jr, Tortolani DR, Hitchcock MJ, Martin JC,
Mansuri MM. Synthesis and in vitro evaluation of a phosphonate
prodrug: bis(pivaloyloxymethyl) 9-(2-phosphonylmethoxyethy-
l)adenine. Antiviral Res 1992;19:267–73.
115. Cundy KC, Fishback JA, Shaw JP, Lee ML, Soike KF, Visor
GC, et al. Oral bioavailability of the antiretroviral agent 9-(2-
phosphonylmethoxyethyl)adenine (PMEA) from three formula-
tions of the prodrug bis(pivaloyloxymethyl)-PMEA in fasted
male cynomolgus monkeys. Pharm Res 1994;11:839–43.
116. Cundy KC, Sue IL, Visor GC, Marshburn J, Nakamura C, Lee
WA, et al. Oral formulations of adefovir dipivoxil: in vitro
dissolution and in vivo bioavailability in dogs. J Pharm Sci
1997;86:1334–8.
117. Shaw JP, Louie MS, Krishnamurthy VV, Arimilli MN, Jones RJ,
Bidgood AM, et al. Pharmacokinetics and metabolism of selected
prodrugs of PMEA in rats. Drug Metab Dispos 1997;25:362–6.
118. Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT,
Kitis G, Rizzetto M, et al. Adefovir dipivoxil for the treatment of
hepatitis B e antigen-negative chronic hepatitis B. N Engl J Med
2003;348:800–7.
119. Marcellin P, Chang TT, Lim SG, Tong MJ, Sievert W, Shiffman
ML, et al. Adefovir dipivoxil for the treatment of hepatitis B e
antigen-positive chronic hepatitis B. N Engl J Med 2003;348:808–16.
120. Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT,
Kitis G, Rizzetto M, et al. Long-term therapy with adefovir
dipivoxil for HBe Ag-negative chronic hepatitis B. N Engl J Med
2005;352:2673–81.
121. Robbins BL, Srinivas RV, Kim C, Bischofberger N, Fridland A.
Anti-human immunodeficiency virus activity and cellular meta-
bolism of a potential prodrug of the acyclic nucleoside phospho-
nate 9-R-(2-phosphonomethoxypropyl)adenine (PMPA),
bis(isopropyloxymethylcarbonyl)-PMP. Antimicrob Agents Che-
mother 1998;42:612–7.
122. Naesens L, Bischofberger N, Augustijns P, Annaert P, van den
Mooter G, Arimilli MN, et al. Antiretroviral efficacy and
pharmacokinetics of oral bis(isopropyloxycarbonyloxymethyl)-
9-(2-phosphonylmethoxypropyl)adenine in mice. Antimicrob
Agents Chemother 1998;42:1568–73.
123. Marcellin P, Heathcote EJ, Buti M, Gane E, de Man RA,
Krastev Z, et al. Tenofovir disoproxil fumarate versus adefovir
dipivoxil for chronic hepatitis B. N Engl J Med 2008;359:
2442–55.
124. de Clercq E. Tenofovir: quo vadis anno 2012 (where is it going in
the year 2012)?Med Res Rev 2012;32:765–85.
125. Lanier ER, Ptak RG, Lampert BM, Keilholz L, Hartman T,
Buckheit RW Jr, et al. Opment of hexadecyloxypropyl tenofovir
(CMX157) for treatment of infection caused by wild-type and
nucleoside/nucleotide-resistant HIV. Antimicrob Agents Che-
mother 2010;54:2901–9.
126. Painter GR, Almond MR, Trost LC, Lampert BM, Neyts J, de
Clercq E, et al. Evaluation of hexadecyloxypropyl-9-R-[2-(Phos-
phonomethoxy)propyl]-adenine, CMX157, as a potential
treatment for human immunodeficiency virus type 1 and hepatitis
B virus infections. Antimicrob Agents Chemother 2007;51:
3505–9.
127. de Clercq E. Acyclic nucleoside phosphonates: past, present and
future. Bridging chemistry to HIV, HBV, HCV, HPV, adeno-,
herpes-, and poxvirus infections: the phosphonate bridge. Bio-
chem Pharmacol 2007;23:911–22.

Erik de Clercq548
128. de Clercq E. The acyclic nucleoside phosphonates from inception
to clinical use: historical perspective. Antiviral Res 2007;75:
1–13.
129. de Clercq E. The clinical potential of the acyclic (and cyclic)
nucleoside phosphonates: the magic of the phosphonate bond.
Biochem Pharmacol 2011;82:99–109.
130. de Clercq E. From adefovir to AtriplaTM via tenofovir, VireadTM
and TruvadaTM. Future Virol 2006;1:709–15.
131. de Clercq E. Where rilpivirine meets with tenofovir, the start of a
new anti-HIV drug combination era. Biochem Pharmacol
2012;84:241–8.
132. Lee WA, He GX, Eisenberg E, Cihlar T, Swaminathan S,
Mulato A, et al. Selective intracellular activation of a novel
prodrug of the human immunodeficiency virus reverse transcrip-
tase inhibitor tenofovir leads to preferential distribution and
accumulation in lymphatic tissue. Antimicrob Agents Chemother
2005;49:1898–906.
133. Ballatore C, McGuigan C, de Clercq E, Balzarini J. Synthesis
and evaluation of novel amidate prodrugs of PMEA and PMP.
Bioorg Med Chem Lett 2001;11:1053–6.
134. Jansa P, Baszczynski O, Dracınsky M, Votruba I, Zıdek Z,
Bahador G, et al. A novel and efficient one-pot synthesis of
symmetrical diamide (bis-amidate) prodrugs of acyclic nucleoside
phosphonates and evaluation of their biological activities. Eur J
Med Chem 2011;46:3748–54.
135. Cihlar T, Ray AS, Boojamra CG, Zhang L, Hui H, Laflamme G,
et al. Design and profiling of GS-9148, a novel nucleotide analog
active against nucleoside-resistant variants of human immuno-
deficiency virus type 1, and its orally bioavailable phosphonoa-
midate prodrug, GS-9131. Antimicrob Agents Chemother
2008;52:655–65.
136. Cihlar T, Laflamme G, Fisher R, Carey AC, Vela JE, Mackman
R, et al. Ovel nucleotide human immunodeficiency virus reverse
transcriptase inhibitor GS-9148 with a low nephrotoxic potential:
characterization of renal transport and accumulation. Antimicrob
Agents Chemother 2009;53:150–6.
137. Scarth BJ, White KL, Chen JM, Lansdon EB, Swaminathan S,
Miller MD, et al. Mechanism of resistance to GS-9148 conferred
by the Q151L mutation in HIV-1 reverse transcriptase. Anti-
microb Agents Chemother 2011;55:2662–9.
138. Birkus G, Wang R, Liu X, Kutty N, MacArthur H, Cihlar T,
et al. Cathepsin A is the major hydrolase catalyzing the
intracellular hydrolysis of the antiretroviral nucleotide phospho-
noamidate prodrugs GS-7340 and GS-9131. Antimicrob Agents
Chemother 2007;51:543–50.
139. Birkus G, Kutty N, Frey CR, Shribata R, Chou T, Wagner C,
et al. Role of cathepsin A and lysosomes in the intracellular
activation of novel antipapillomavirus agent GS-9191. Antimi-
crob Agents Chemother 2011;55:2166–73.
140. Wolfgang GH, Shibata R, Wang J, Ray AS, Wu S, Doerrfler E,
et al. GS-9191 is a novel topical prodrug of the nucleotide analog
9-(2-phosphonylmethoxyethyl)guanine with antiproliferative
activity and possible utility in the treatment of human papillo-
mavirus lesions. Antimicrob Agents Chemother 2009;53:
2777–84.
141. Vavrova K, Kovarıkova P, Skolova B, Lıbalova M, Roh J, Cap
R, et al. Enhanced topical and transdermal delivery of anti-
neoplastic and antiviral acyclic nucleoside phosphonate cPr-
PMEDAP. Pharm Res 2011;28:3105–15.
142. Reiser H, Wang J, Chong L, Watkins WJ, Ray AS, Shibata R,
et al. GS-9219–a novel acyclic nucleotide analog with potent
antineoplastic activity in dogs with spontaneous non-Hodgkin’s
lymphoma. Clin Cancer Res 2008;14:2824–32.
143. Vail DM, Thamm DH, Reiser H, Ray AS, Wolfgang GH,
Watkins WJ, et al. Assessment of GS-9219 in a pet dog
model of non-Hodgkin’s lymphoma. Clin Cancer Res 2009;15:
3503–10.





























