Embed Size (px)
Citation preview

Mild Thiamine Deficiency and Chronic Ethanol ConsumptionModulate Acetylcholinesterase Activity Change and SpatialMemory Performance in a Water Maze Task
Ieda de Fátima Oliveira-Silva & Silvia R. Castanheira Pereira &
Paula A. Fernandes & Andrea F. Ribeiro & Rita G. W. Pires &Angela Maria Ribeiro
Received: 5 February 2014 /Accepted: 8 April 2014# Springer Science+Business Media New York 2014
Abstract Chronic thiamine deficiency may be responsiblefor pathologic changes in the brains of alcoholics, and sub-clinical episodes of this vitamin deficiency may cause cumu-lative brain damage. In the present work, the chronic effects ofethanol and its association to a mild thiamine deficiencyepisode (subclinical model) on neocortical and hippocampalacetylcholinesterase activity were assessed along with theirpossible association to spatial cognitive dysfunction. Theresults indicate that in the beginning of the neurodegenerativeprocess, before the appearance of brain lesions, chronic etha-nol consumption reverses the effects of mild thiamine defi-ciency on both spatial cognitive performance and acetylcho-linesterase activity without having significant effects on anymorphometric parameter.
Keywords Mild thiamine deficiency . Chronic ethanolconsumption . Acetylcholinesterase . Spatial learning andmemory . Rat
Introduction
Chronic alcoholism can result in thiamine deficiency (TD)through several mechanisms including inadequate food in-take, reduced absorption of thiamine, or a decrease in the rateof the vitamin’s conversion to its active form, thiamine pyro-phosphate (Hoyumpa 1980; Alexander-Kaufman and Harper2009). One hypothesis proposes that chronic TD may beresponsible for neurobiological changes in the brains of alco-holics and that subclinical episodes of this vitamin deficiencymay cause cumulative brain damage (Bowden 1990). In fact,the very existence of primary alcoholic dementia as a neuro-pathological entity has been questioned, and it has been pro-posed instead that the majority of cases of alcoholic dementiaare in fact Wernicke-Korsakoff syndrome (WKS) (Victor1994). Currently, the widely held view is that the cognitivedeficits of varying types and severities found in alcoholicsarise as a result of co-existing TD (Pitel et al. 2011).
WKS has been experimentally induced by using an asso-ciation of thiamine-deficient diet and IP administration ofpyrithiamine in a rat model (Carvalho et al. 2006;Homewood et al. 1997). This drug inhibits the enzyme thatphosphorylates thiamine to its metabolically active form(Peterson et al. 1975). Compared to animals treated with adeficient diet alone, animals treated with such a combinationshowmore severe clinical signs of WKS in a shorter period oftime (Chen et al. 1997).
Various authors have observed alterations of biochemicalparameters, e.g., in the biosynthesis and turnover of several
I. de Fátima Oliveira-Silva : P. A. FernandesDepartamento de Análises Clínicas e Toxicológicas - Faculdade deFarmácia, Universidade Federal de Minas Gerais, BeloHorizonte 31270-010, Brazil
S. R. C. Pereira :A. F. Ribeiro :A. M. Ribeiro (*)Programa de Pós-graduação emNeurociências, Universidade Federalde Minas Gerais, Belo Horizonte 31270-010, Brazile-mail: [email protected]
R. G. W. PiresDepartamento de Ciências Fisiológicas/CentroBiomédico-Laboratório de Neurobiologia Molecular eComportamental, Universidade Federal do Espírito Santo,Vitória 29043-910, Brazil
A. M. RibeiroDepartamento de Bioquímica e Imunologia, Laboratório deNeurociências Comportamental e Molecular, LaNeC, Faculdade deFilosofia e Ciências Humanas, FaFiCH, Universidade Federal deMinas Gerais, Belo Horizonte 31270-010, Brazil
J Mol NeurosciDOI 10.1007/s12031-014-0306-7

neurotransmitters such as glutamate, acetylcholine, and sero-tonin (Savage et al. 2012). Previous studies have observedneocortical and hippocampal cholinergic parameter changesin thiamine-deficient rats (Pires et al. 2007). These alterations,correlated to certain cognitive impairments, mainly deficits inaspects of learning and memory, were observed by severalauthors in both humans (Caulo et al. 2005; Noël et al. 2001)and animal models (Langlais et al. 1992; Bizon-Zygmańskaet al. 2011). However, the neurodegenerative mechanismsresponsible for the lesions caused by TD are not known.
In experimental animal models, chronic consumption ofethanol alone can result in cognitive deficit (Fehr et al. 1976;Pereira et al. 1998; Pitel et al. 2011) associated with choliner-gic hypofunction (Arendt et al. 1989; Casamenti et al. 1993).After abstinence, the cholinergic hypofunction and cognitivealteration induced by ethanol in rodents are partially or totallyreversible, depending on the length of the treatment and theperiod of abstinence (Arendt et al. 1989; Casamenti et al.1993). However, Fadda et al. (1999), working with alcohol-preferring rats detected neither cognitive deficits nor cholin-ergic hypofunction.
Studies of the interaction between chronic ethanol con-sumption and TD effects have yielded contradictory results.The effects of this association on morphological (Bâ et al.1999), biochemical (Pires et al. 2001), and behavioral param-eters (Ciccia and Langlais 2000; Homewood et al. 1997; Pireset al. 2001) can be synergistic or not. Thus, the relativecontribution of TD and ethanol consumption in determiningthe cerebral damage and behavioral deficits remainsunknown.
In this study, the association of the chronic effects ofethanol with a mild TD episode on aspects of spatial cognitionwas assessed by a water maze task. Moreover, acetylcholin-esterase (AChE) activity was evaluated in the neocortex andhippocampus areas, both important to spatial learning andmemory processing (Redish 2001; Takashima et al. 2007),permitting the analysis of a possible association betweenbehavioral and neurochemical parameters.
Materials and Methods
Animals and Treatments
Thirty-ninemaleWistar rats aged 3months at the beginning ofthe experiment were used. They were housed in single cagesand maintained on a regular 12:12 light/dark cycle. The careand use of animals in this study were in accordance with theguidelines set by the National Institutes of Health Guide forCare and Use of Laboratory Animals (National ResearchCouncil 1985). The animals were divided into two groups,control (W, n=19) and ethanol (E, n=20), according to thefollowing treatments, respectively: (i) free access to tap water
and (ii) free access to 20 %v/v ethanol solution as the solesources of fluid. Initial ethanol concentration was 5% and wasincreased progressively by 5 % every 2 days, until a 20 %concentration was reached. Initially, all rats had free access tocommercial chow. After 6 months, the two groups, W and E,were further divided into two subgroups, respectively: WS(n=9) and ES (n=10) whose animals were treated with astandard diet and groups WD (n=10) and ED (n=10) whoseanimals were treated with a thiamine-deficient diet. Standardand thiamine-deficient chows were prepared in the laboratorysuch that the only difference between them was the presenceor absence of thiamine. The TD episode lasted until theanimals displayed some of the first clinical signs of deficien-cy: aphagia and adipsia (Hakim and Pappius 1983). Thesesigns occurred around 48 days after the start of the TDepisode. Following that, all rats had free access to commercialchow. The water or ethanol treatment lasted for five moremonths. Body weight, chow consumption, and fluid ingestionwere recorded weekly and daily, respectively. After 20 days ofethanol abstinence, the rats were submitted to behavioral tests.Following the behavioral studies, the subjects were decapitat-ed, and their neocortex and hippocampus (from both hemi-spheres) were dissected for biochemical and morphologicalstudies. The experimenter carrying out these analyses wasblind to the experimental group fromwhich the samples came.Seven animals died during the treatment, three from the WDgroup and two from the other two groups, ES and ED.
Behavioral Study
Spatial Learning and Memory Task Spatial learning andmemory were assessed using the Morris Water Maze(MWM) task (Morris et al. 1982).
Apparatus A 1.80-m diameter fiberglass pool was used. Itwas filled with water to a depth of 27.5 cm at a controlledtemperature of 26±2 °C. A 15-cm diameter transparent Plex-iglas round platform was placed 2 cm below the water surfacein the center of one of the pool quadrants (southeast, south-west, northeast, or northwest). In order to avoid visual locationof the platform, 60 g of powdered milk were dissolved in thewater, making it turbid. The pool was placed in a 3×3m room,with extra-maze cues (e.g., posters, TV, video equipment, anda table). All sessions were recorded with a wide-angle videocamera fixed to the ceiling.
Procedure
Training Each animal had one daily session for five consec-utive days, and each session consisted of four trials. The inter-trial interval was the same for all rats in every session(30 min). Each trial began with the rat being placed into thepool, facing the wall of one of the four quadrant edges. The
J Mol Neurosci

quadrants were alternated in a pseudo-random fashion. Theplatform was located in the southeast quadrant in all sessions.The subject was allowed to swim around the pool until itfound the platform. If the animal did not find the platformwithin 60 s, it was gently guided to it by the experimenter.Once on the platform, the animals were allowed to stay therefor 20 s. The latency for finding the platform was recorded.Recorded videos were analyzed to measure path length man-ually by using a distance-measuring apparatus (Run-Mate™
Club) on the television screen.
Probe Trial On the day following the last training session,each animal was left in the pool without the platform for 120 s.The total and partial (only in the target quadrant) path lengthswere recorded. For each rat, a preference ratio was calculatedby dividing the partial length by the total length.
Biochemical Study
Acetylcholinesterase (AChE) activity was measured separate-ly in the neocortex and hippocampus, using a spectrophoto-metric method adapted from Ellman et al. (1961). Aliquots ofapproximately 20 mg of tissue from each area (neocortex andhippocampus) were cut into 400 μm slices with a McIlwaintissue chopper, mixed with a spatula, transferred to Eppendorftubes containing 1 mL of borate buffer and frozen at −20 °Cuntil the day of the assay. The samples were homogenized in aPotter Elvehjem, and the total volume was divided into twoaliquots of about 500 μL each. Of triton X-100, 0.2 % wasadded to one of the aliquots, which was subsequently used forthe total fraction activity assay. The second aliquot was cen-trifuged for 10 min at 12,300 g, and the supernatant wasseparated for soluble fraction activity analysis. Both fractionswere kept on ice. One hundred thirty-five microliters of thesoluble or total fractions were then added to a cuvette con-taining 35 μL of 5 mM dithiobisnitrobenzoate (DTNB) and820μL of borate buffer (pH=8.2). The reaction was started byadding 10 μL of 1 mM acetylthiocholine, and its rate wasrecorded every 10 s for a total time of 60 s using a Varian®spectrophotometer, mod. Cary 50. Soluble and total AChEactivities were determined. Bound AChE activity was workedout subtracting the soluble from total AChE activity, and theresults were expressed as micromoles of acetylthiocholinehydrolysed/min/g of tissue.
Morphological Study
Histopathological evaluations were performed in the hippo-campus of 32 animals. The cerebral hemispheres, chosen atrandom for morphological analysis, were fixed in 10 % salineformaldehyde, processed to be embedded in paraffin, and cutin 7-μm thick coronal sections at 140-μm intervals.Hematoxylin-eosin staining was used for optical microscopic
evaluation. We were not able to detect the hippocampus areain sections from four animals, so they were not included in theanalysis. Two histological sections from the remaining 28animals taken at the level of the temporal septum, correspond-ing to plates 31–33 on the Paxinos and Watson (2007) stereo-taxic atlas, were used for hippocampal morphometric analysis.The pyramidal cells of the CA1 and the granular cells of thedentate gyrus from the hippocampal regions were quantifiedseparately. Pyramidal cells from the CA2 and CA3 regionswere quantified together. Cell counting was carried out in asemi-automatic image analysis system KS-400 (Zeiss, Ger-many). Only neurons with a well-defined nucleus outline anda clear-cut cytoplasmwere counted. The neurons weremarkedwith a pencil to prevent either double counting or errors ofomission. Results are expressed in neuronal density (numberof cells/mm2).
In the analysis of nuclear size and diameter, the nucleararea and the highest nuclear diameter were also measured in100 cells of the dentate gyrus, from the CA1 and CA3 fieldsfrom each animal using an image system program developedby Pinto (1996). Nuclear area and nuclear diameter areexpressed in μm2 and μm, respectively.
Statistical Analysis
Data were analyzed for distribution normality using theKolmogorov-Smirnov test and homogeneity of variance usingLilliefors’ test (ANOVA). The biochemical experiments wereperformed in duplicates, and the mean was used for statisticalanalysis comparisons. All values are expressed as mean±standard error (S.E.M.). The morphometric, biochemical pa-rameters, and Morris water maze probe trial data were ana-lyzed using a 2×2 (chronic ethanol×TD) ANOVA followedby the Newman-Keuls multiple range post hoc test. Bodyweight, food and fluid intake, and Morris water maze trainingdata were analyzed using 2×2×r (chronic ethanol×TD×time)repeated-measures ANOVA, followed by the Newman-Keulsmultiple range post hoc test in the last element (r). The ethanolconsumption during last week of treatment was analyzed forES and ED groups using t test. Pearson’s linear correlationanalysis was used in order to determine the degree to whichbehavioral, biochemical, and morphological parameters werelinearly correlated. The significance level was set at p<0.05.
Results
Experimental Model
During the TD episode, animals treated with a deficient diet(from the WD and ED groups), but not those treated with astandard diet (from the WS and ES groups), lost weight(F3,34=15, 93, p=0.00). In the last week, comparisons among
J Mol Neurosci
groups revealed lower weight in ED than other groups(WS, p=0.0002; WD, p=0.02; and ES, p=0.0001). The WDgroup also exhibited low weight compared with the WS andES groups (p=0.005).
At the start of behavioral tests, there was no significantdifference in weight among groups (F3,34=1.27, p=0.29), butafter the TD episode, animals from the WD and ED groupsgained significantly more weight (F6,68=4.46, p=0.0007).
However, there was no significant difference in ethanolconsumption between the two groups on the last week oftreatment (t=0.23, p=0.82). The TD episode induced an in-crease in water and ethanol consumption for the WD and EDgroups (F3,34=20.77, p=0.00). The ED and WD groups ex-hibited higher solution intake than the ES and WS groups,respectively (p=0.03 and p=0.0004).
Behavioral Performance
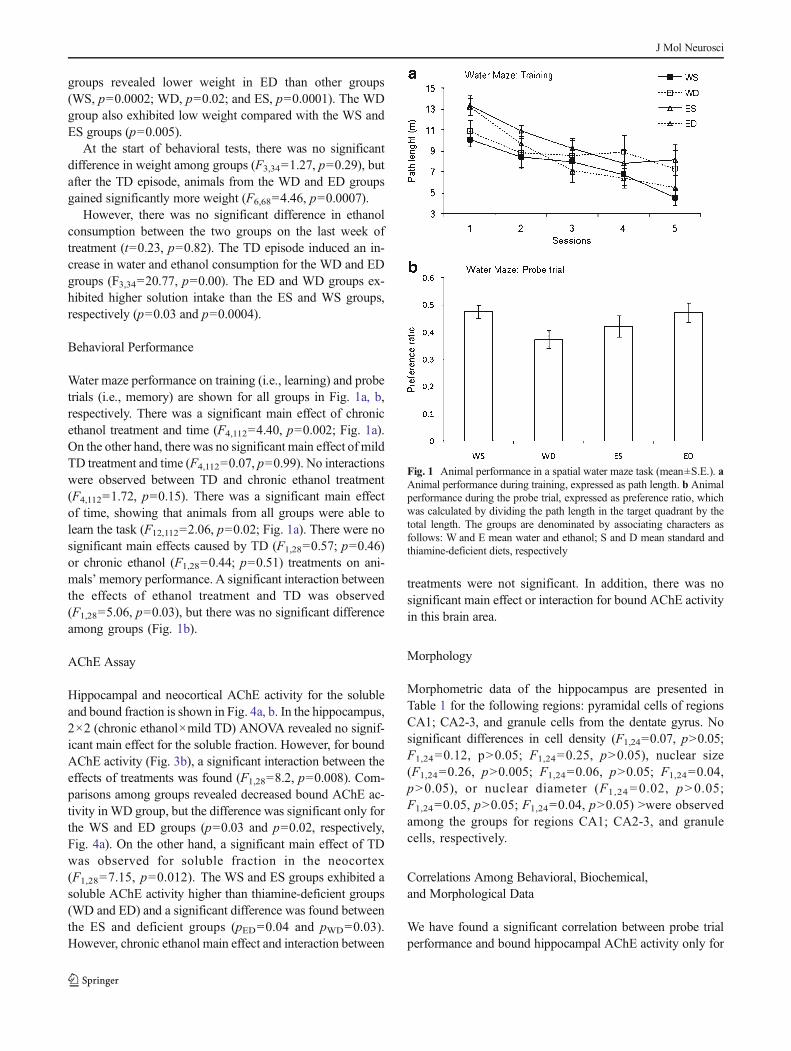
Water maze performance on training (i.e., learning) and probetrials (i.e., memory) are shown for all groups in Fig. 1a, b,respectively. There was a significant main effect of chronicethanol treatment and time (F4,112=4.40, p=0.002; Fig. 1a).On the other hand, there was no significant main effect of mildTD treatment and time (F4,112=0.07, p=0.99). No interactionswere observed between TD and chronic ethanol treatment(F4,112=1.72, p=0.15). There was a significant main effectof time, showing that animals from all groups were able tolearn the task (F12,112=2.06, p=0.02; Fig. 1a). There were nosignificant main effects caused by TD (F1,28=0.57; p=0.46)or chronic ethanol (F1,28=0.44; p=0.51) treatments on ani-mals’memory performance. A significant interaction betweenthe effects of ethanol treatment and TD was observed(F1,28=5.06, p=0.03), but there was no significant differenceamong groups (Fig. 1b).
AChE Assay
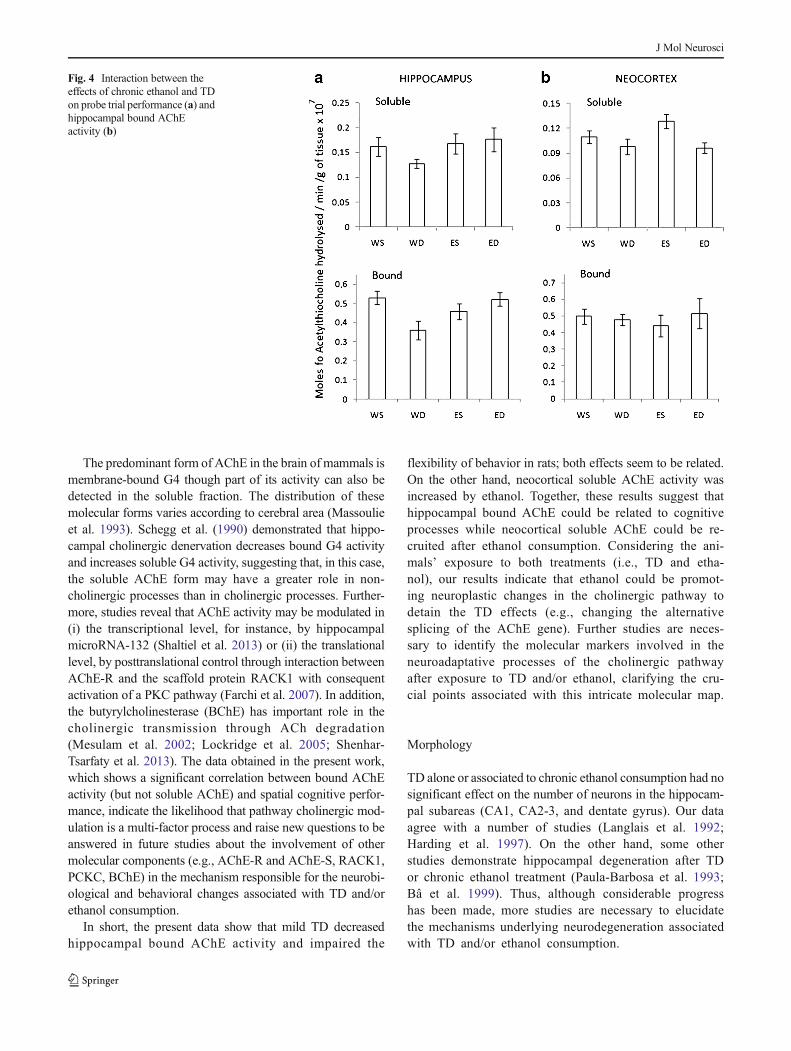
Hippocampal and neocortical AChE activity for the solubleand bound fraction is shown in Fig. 4a, b. In the hippocampus,2×2 (chronic ethanol×mild TD) ANOVA revealed no signif-icant main effect for the soluble fraction. However, for boundAChE activity (Fig. 3b), a significant interaction between theeffects of treatments was found (F1,28=8.2, p=0.008). Com-parisons among groups revealed decreased bound AChE ac-tivity inWD group, but the difference was significant only forthe WS and ED groups (p=0.03 and p=0.02, respectively,Fig. 4a). On the other hand, a significant main effect of TDwas observed for soluble fraction in the neocortex(F1,28=7.15, p=0.012). The WS and ES groups exhibited asoluble AChE activity higher than thiamine-deficient groups(WD and ED) and a significant difference was found betweenthe ES and deficient groups (pED=0.04 and pWD=0.03).However, chronic ethanol main effect and interaction between
treatments were not significant. In addition, there was nosignificant main effect or interaction for bound AChE activityin this brain area.
Morphology
Morphometric data of the hippocampus are presented inTable 1 for the following regions: pyramidal cells of regionsCA1; CA2-3, and granule cells from the dentate gyrus. Nosignificant differences in cell density (F1,24=0.07, p>0.05;F1,24=0.12, p>0.05; F1,24=0.25, p>0.05), nuclear size(F1,24=0.26, p>0.005; F1,24=0.06, p>0.05; F1,24=0.04,p>0.05), or nuclear diameter (F1,24 =0.02, p>0.05;F1,24=0.05, p>0.05; F1,24=0.04, p>0.05) >were observedamong the groups for regions CA1; CA2-3, and granulecells, respectively.
Correlations Among Behavioral, Biochemical,and Morphological Data
We have found a significant correlation between probe trialperformance and bound hippocampal AChE activity only for
Fig. 1 Animal performance in a spatial water maze task (mean±S.E.). aAnimal performance during training, expressed as path length. b Animalperformance during the probe trial, expressed as preference ratio, whichwas calculated by dividing the path length in the target quadrant by thetotal length. The groups are denominated by associating characters asfollows: W and E mean water and ethanol; S and D mean standard andthiamine-deficient diets, respectively
J Mol Neurosci
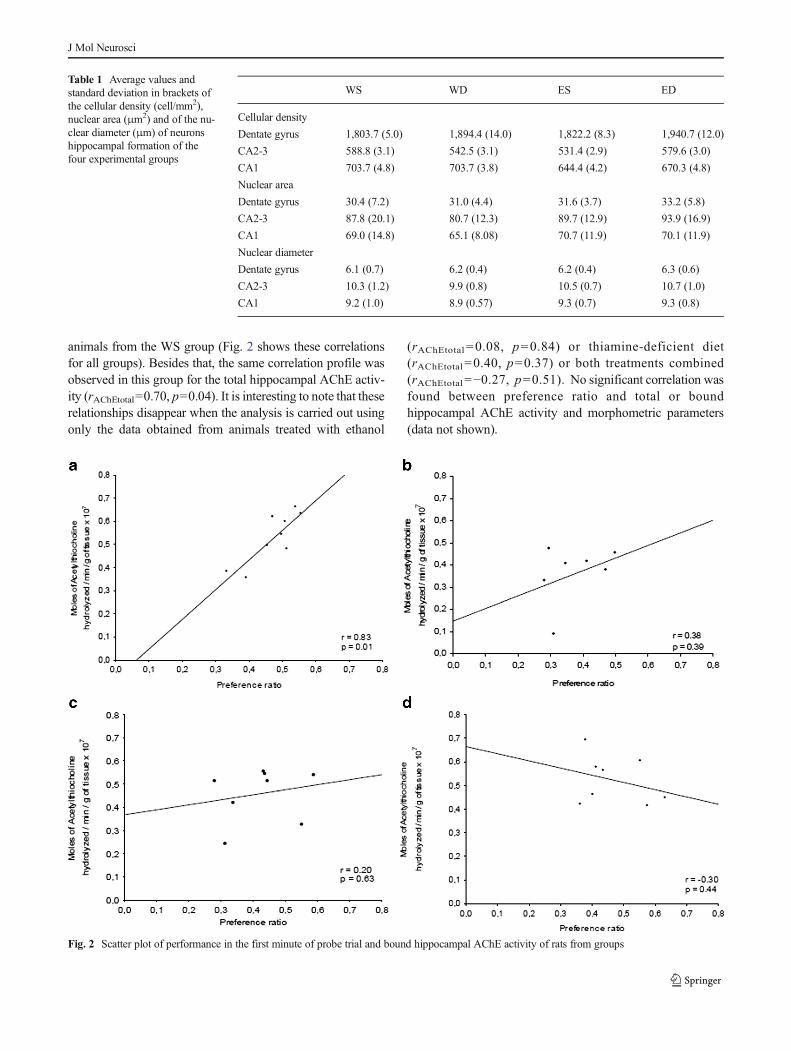
animals from the WS group (Fig. 2 shows these correlationsfor all groups). Besides that, the same correlation profile wasobserved in this group for the total hippocampal AChE activ-ity (rAChEtotal=0.70, p=0.04). It is interesting to note that theserelationships disappear when the analysis is carried out usingonly the data obtained from animals treated with ethanol
(rAChEtotal =0.08, p=0.84) or thiamine-deficient diet(rAChEtotal=0.40, p=0.37) or both treatments combined(rAChEtotal=−0.27, p=0.51). No significant correlation wasfound between preference ratio and total or boundhippocampal AChE activity and morphometric parameters(data not shown).
Table 1 Average values andstandard deviation in brackets ofthe cellular density (cell/mm2),nuclear area (μm2) and of the nu-clear diameter (μm) of neuronshippocampal formation of thefour experimental groups
WS WD ES ED
Cellular density
Dentate gyrus 1,803.7 (5.0) 1,894.4 (14.0) 1,822.2 (8.3) 1,940.7 (12.0)
CA2-3 588.8 (3.1) 542.5 (3.1) 531.4 (2.9) 579.6 (3.0)
CA1 703.7 (4.8) 703.7 (3.8) 644.4 (4.2) 670.3 (4.8)
Nuclear area
Dentate gyrus 30.4 (7.2) 31.0 (4.4) 31.6 (3.7) 33.2 (5.8)
CA2-3 87.8 (20.1) 80.7 (12.3) 89.7 (12.9) 93.9 (16.9)
CA1 69.0 (14.8) 65.1 (8.08) 70.7 (11.9) 70.1 (11.9)
Nuclear diameter
Dentate gyrus 6.1 (0.7) 6.2 (0.4) 6.2 (0.4) 6.3 (0.6)
CA2-3 10.3 (1.2) 9.9 (0.8) 10.5 (0.7) 10.7 (1.0)
CA1 9.2 (1.0) 8.9 (0.57) 9.3 (0.7) 9.3 (0.8)
Fig. 2 Scatter plot of performance in the first minute of probe trial and bound hippocampal AChE activity of rats from groups
J Mol Neurosci

Discussion
TD and chronic ethanol exert different effects on water- andethanol-consumer rats on spatial memory performance. More-over, the cholinergic system corresponds to a complex path-way in which molecular forms could contribute in distinctways to the cognitive and biochemical processes related tomaintenance of neuroadaptative mechanisms.
Experimental Animal Model
The experimental model used in the present work enabled usto study the effect of the association between mild TD andchronic ethanol consumption. A 48-day thiamine-restricteddiet succeeded in inducing aphasia and adipsia inWistar rats,consistent with a deficiency mild model. Gubler (1961) dem-onstrated that, by contrast, Sprague-Dawley rats exposed tothe TD diet, for at least 30 days, exhibited signs of severedeprivation (i.e., rapid loss of weight, anorexia, or convul-sions). In other studies, Sprague-Dawley rats submitted to athiamine-deficient diet for a period of 5 weeks showed anaccentuated decrease of thiamine levels in the brain after4 weeks of treatment (Molina et al. 1994). The differences inour results may be related to strains of rats, suggesting thatWistar rats show more resistance to developing severe defi-ciency. In addition, clinical and biochemical data from the TDanimal model in which dietary deprivation or central thiamineantagonist (e.g., pyrithiamine) was used reveal clear differ-ences (Butterworth 1986). This suggests that metabolic path-ways susceptible to thiamine effects may vary in differentstrains of rat, depending on the specific enzyme systemsimplicated in deficiency.
Pekkanen (1979) and Zimatkin (1996) found that after aTD episode, the ED group also consumed a significantlyhigher volume of ethanol solution compared with the ESgroup, which were not on a thiamine-deficient diet. Thus,even amild TD appears to trigger a behavioral risk with regardto the consumption of ethanol, which may predispose the ratto “abuse” this drug (i.e., to take in a higher volume ofethanol).
Cognitive deficits have been reported in rats experimental-ly submitted to severe TD (Langlais et al. 1992; Vigil et al.2010) or to chronic ethanol consumption (Fehr et al. 1976;Pereira et al. 1998). Although the association between ethanolconsumption and TD has not shown a synergistic effect(Ciccia and Langlais 2000; Palencia et al. 1994), some aspectsof memory seem to be more sensitive to the association ofethanol and TD, while others are more affected by individu-ally administered treatments (Ciccia and Langlais 2000;Zimitat et al. 1990). In addition, Homewood et al. (1997)did not observe memory deficits in rats submitted to chronicethanol consumption associated to TD. Likewise, impairedlearning performance was not found; however, the interaction
observed among the chronic ethanol and TD treatments on theanimals’ spatial memory performance (probe trial, Fig. 3a)indicates that the effect of mild TD varies according to liquidconsumption (i.e., water or ethanol).
Acetylcholinesterase Activity and Behavioral Performance
Evidence indicates that cholinergic hypoactivity impairs per-formance on a cognitive task (Everitt and Robbins 1997;Shaltiel et al. 2013). In addition, there is evidence that anincrease of acetylcholine release during the task performanceis associated with a better outcome (Roland et al. 2008; DeJaeger et al. 2013). However, in these studies, the rats were notin a physiological condition state (i.e., the rats were not fromcontrol group). Thus, in case of a decrease in AChE activity(e.g., nonphysiological hypoactive condition), an inhibition ofAChE would increase the level of ACh in the cleft, whichwould improve the animal’s performance, resulting in a neg-ative correlation between AChE activity and animal’s perfor-mance. On the other hand, interestingly, in a physiologicalcondition (i.e., in the control group), our results revealed apositive significant correlation between memory performanceand hippocampal-bound AChE activity for the WS group(Fig. 2a). Likewise, Segal et al. (1988) demonstrated that ratexposure to a water maze task exhibited a positive correlationbetween AChE activity and some aspects of learning (e.g.,“abandoning the old position” assessed by probe trial).Resultsobtained by Shaltiel et al. (2013), with which the present dataagree, show that the synaptic AChE (AChE-S) but not thesoluble read-through (AChE-R) isoform is related to animalperformance in spatial navigation tasks, suggesting the possi-ble importance of changes on the alternative splicing of theAChE gene to cognitive processes. Combined, these resultsindicate that high bound AChE activity might be related to ahigher cholinergic activity in a specific situation, which mayplay an important role in behavioral flexibility as displayed ina spatial memory task.
Both TD and chronic ethanol intake disrupted the linearassociation observed between spatial memory performanceand bound AChE activity under physiological conditions(Fig. 2). Nevertheless, our results demonstrate that the asso-ciation between the two treatments did not impair cognition(i.e., learning and memory, Fig. 1), even though during chron-ic exposure to ethanol both treatments had shown largereffects on cholinergic pathway (Pires et al. 2001). In spite ofthe fact that the animals from the ED group exhibited boundAChE activity similar to the ES and WS groups (Fig. 4a), asignificant linear correlation was not observed between thisparameter and memory performance as in the WS group(Fig. 2d). Moreover, neocortical soluble AChE activity ofES group was higher than WD and ED groups (Fig. 4b).Darreh-Shori et al. (2002) found an increase in the solubleAChE-R levels and improvement on cognitive abilities in
J Mol Neurosci

patients with Alzheimer disease (AD) after 12 months ofrivastigmine treatment. Although, in the present work, the ratsexhibited no significant changes on memory and learningbehaviors, we could consider the hypothesis that the ethanolpromoted a new “balance” between transcriptional isoformsof the AChE gene (i.e., an increase in AChE-R), contributingto the neural plasticity and consequently to the maintenance ofcognitive behavior performance.
An interaction between the ethanol consumption and thia-mine deficiency effects was observed for bound AChE activ-ity in the hippocampus (Fig. 3b). The WD group displayedreduced AChE activity in hippocampus (bound form, Fig. 4a)and neocortex (soluble form, Fig. 4b), compared with theother groups (WS, ES, and ED), without spatial learningdysfunction. Shaltiel et al. (2013) demonstrated that hippo-campal AChE can be suppressed by microRNA-132impairing cognitive performance. It is possible, then, thatTD could modulate the regulatory mechanism of the AChEgene and consequently could interfere with flexibility on a
spatial task. TD reduced brain AChE levels after 16 days ofdietary restriction (Vorhees et al. 1977), although the exactmechanism of this effect has not been determined. Heinrichet al. (1973) have not found AChE activity changes inrats treated with a thiamine-deficient diet, although theirmeasurement of the enzyme activity was derived fromwhole brain samples. Yet, the ACh level was notanalyzed in the present study; however, Pires et al.(2001) found similar results for AChE activity in thesame brain areas using a model approach to the thia-mine dietary restriction associated with ethanol intake.In addition, it was found that under stimulated condi-tions, the control group presented higher ACh releasethan all experimental groups, which had access to TDand/or ethanol (Pires et al. 2001). Also, chronic ethanolconsumption leads to TD, and the posttranslational mod-ifications in proteins related to acetyl-CoA synthesis(i.e., acetylation and deacetylation) are disturbed(Picklo 2008; for review Martin et al. 2003).
Fig. 3 Neocortical andhippocampal AChE activities(mean±S.E.). a and b show thesoluble and bound AChE activityobtained from hippocampus andneocortex, respectively. Thegroups are denominated byassociating characters as follows:Wand E mean water and ethanol;S and D mean standard andthiamine-deficient diets,respectively
J Mol Neurosci
The predominant form of AChE in the brain of mammals ismembrane-bound G4 though part of its activity can also bedetected in the soluble fraction. The distribution of thesemolecular forms varies according to cerebral area (Massoulieet al. 1993). Schegg et al. (1990) demonstrated that hippo-campal cholinergic denervation decreases bound G4 activityand increases soluble G4 activity, suggesting that, in this case,the soluble AChE form may have a greater role in non-cholinergic processes than in cholinergic processes. Further-more, studies reveal that AChE activity may be modulated in(i) the transcriptional level, for instance, by hippocampalmicroRNA-132 (Shaltiel et al. 2013) or (ii) the translationallevel, by posttranslational control through interaction betweenAChE-R and the scaffold protein RACK1 with consequentactivation of a PKC pathway (Farchi et al. 2007). In addition,the butyrylcholinesterase (BChE) has important role in thecholinergic transmission through ACh degradation(Mesulam et al. 2002; Lockridge et al. 2005; Shenhar-Tsarfaty et al. 2013). The data obtained in the present work,which shows a significant correlation between bound AChEactivity (but not soluble AChE) and spatial cognitive perfor-mance, indicate the likelihood that pathway cholinergic mod-ulation is a multi-factor process and raise new questions to beanswered in future studies about the involvement of othermolecular components (e.g., AChE-R and AChE-S, RACK1,PCKC, BChE) in the mechanism responsible for the neurobi-ological and behavioral changes associated with TD and/orethanol consumption.
In short, the present data show that mild TD decreasedhippocampal bound AChE activity and impaired the
flexibility of behavior in rats; both effects seem to be related.On the other hand, neocortical soluble AChE activity wasincreased by ethanol. Together, these results suggest thathippocampal bound AChE could be related to cognitiveprocesses while neocortical soluble AChE could be re-cruited after ethanol consumption. Considering the ani-mals’ exposure to both treatments (i.e., TD and etha-nol), our results indicate that ethanol could be promot-ing neuroplastic changes in the cholinergic pathway todetain the TD effects (e.g., changing the alternativesplicing of the AChE gene). Further studies are neces-sary to identify the molecular markers involved in theneuroadaptative processes of the cholinergic pathwayafter exposure to TD and/or ethanol, clarifying the cru-cial points associated with this intricate molecular map.
Morphology
TD alone or associated to chronic ethanol consumption had nosignificant effect on the number of neurons in the hippocam-pal subareas (CA1, CA2-3, and dentate gyrus). Our dataagree with a number of studies (Langlais et al. 1992;Harding et al. 1997). On the other hand, some otherstudies demonstrate hippocampal degeneration after TDor chronic ethanol treatment (Paula-Barbosa et al. 1993;Bâ et al. 1999). Thus, although considerable progresshas been made, more studies are necessary to elucidatethe mechanisms underlying neurodegeneration associatedwith TD and/or ethanol consumption.
Fig. 4 Interaction between theeffects of chronic ethanol and TDon probe trial performance (a) andhippocampal bound AChEactivity (b)
J Mol Neurosci

Conclusion
Our data support the notion that a mild TD episode, before theappearance of brain lesions, promotes perdurable neurochem-ical dysfunctions, and these alterations boost the ethanol in-take. Furthermore, hippocampal-bound AChE activity ap-pears to be relevant to behavioral flexibility assessed in aspatial navigation task. Moreover, neocortical soluble AChEactivity seems to be modulated by ethanol effects. Neverthe-less, further studies are necessary to clarify the connectionamong the common pathways related to TD and ethanoleffects, promoting a better understanding of the mechanismsassociated with cognitive and neurochemical processes.
Acknowledgments This work was supported by Fundação de Amparoà Pesquisa de Minas Gerais (FAPEMIG). The author thanks AparecidaGuerra de Jesus, received recipient of a FAPEMIG scholarship, for hertechnical assistance.
References
Alexander-Kaufman K, Harper C (2009) Transketolase: observations inalcohol-related brain damage research. Int J Biochem Cell Biol 41:717–720
Arendt T, Allen Y,Marchbanks RM, SchugensMM, Sinden J, Lantos PL,Gray JA (1989) Cholinergic system andmemory in the rat: effects ofchronic ethanol, embryonic basal forebrain brain transplants andexcitotoxic lesions of cholinergic basal forebrain projection system.Neuroscience 33:435–462
Bâ A, Seri BV, Aka KJ, Glin L, Tako A (1999) Comparative effects ofdevelopmental thiamine deficiencies and ethanol exposure on themorphometry of the CA3 pyramidal cells. Neurotoxicol Teratol 21:579–586
Bizon-Zygmańska D, Jankowska-KulawyA, Bielarczyk H, Pawełczyk T,Ronowska A, MarszałłM, Szutowicz A (2011) Acetyl-CoA metab-olism in amprolium-evoked thiamine pyrophosphate deficits in cho-linergic SN56 neuroblastoma cells. Neurochem Int 59:208–216
Bowden SC (1990) Separating cognitive impairment in neurologicallyasymptomatic alcoholism from Wernicke-Korsakoff-Syndrome: is the neuropsychological distinction justified?Psychol Bull 107:355–366
Butterworth RF (1986) Cerbral thiamine-dependent enzyme changes inexperimental Wernicke’s encephalopathy. Metab Brain Dis 1(3):165–175
Carvalho FM, Pereira SR, Pires RG, Ferraz VP, Romano-Silva MA,Oliveira-Silva IF, Ribeiro AM (2006) Thiamine deficiency de-creases glutamate uptake in the prefrontal cortex and impairs spatialmemory performance in a water maze test. Pharmacol BiochemBehav 83:481–489
Casamenti F, Scali C, Vannucchi MG, Bartolini L, Pepeu G (1993) Long-term ethanol-consumption by rats—effect on acetylcholine-releasein-vivo, choline-acetyltransferase activity, and behavior.Neuroscience 56:465–471
Caulo M, Van Hecke J, Toma L, Ferretti A, Tartaro A, Colosimo C,Romani GL, Uncini A (2005) Functional MRI study of diencephalicamnesia in Wernicke-Korsakoff syndrome. Brain 128:1584–1594
Chen Q, Okada S, Okeda R (1997) Causality of parenchymal andvascular changes in rats with experimental thiamine deficiencyencephalopathy. Pathol Int 47:748–756
Ciccia RM, Langlais PJ (2000) An examination of the synergistic inter-action of ethanol and thiamine deficiency in the development ofneurological signs and long-term cognitive and memory impair-ments. Alcohol Clin Exp Res 24:622–634
Darreh-Shori T, Almkvist O, Guan ZZ, Garlind A, Strandberg B,Svensson AL, Soreq H, Hellström-Lindahl E, Nordberg A (2002)Sustained cholinesterase inhibition in AD patients receivingrivastigmine for 12 months. Neurology 59:563–572
De Jaeger X, Cammarota M, Prado Marco AM, Izquierdo I, Prado VF,Pereira GS (2013) Decreased acetylcholine release delays the consol-idation of object recognition memory. Behav Brain Res 238:62–68
Everitt BJ, Robbins TW (1997) Central cholinergic systems and cogni-tion. Annu Rev Psychol 48:649–684
Fadda F, Cocco S, Stancampiano R, Rossetti ZL (1999) Long-termvoluntary ethanol consumption affects neither spatial nor passiveavoidance learning, nor hippocampal acetylcholine release inalcohol-preferring rats. Behav Brain Res 103:71–76
Farchi N, Shoham S, Hochner B, Soreq H (2007) Impaired hippocampalplasticity and erros in cognitive performance in mice withmaladaptative AChE splice site selection. Eur J Neurosci 25:87–98
Fehr KA, Kalant H, LeBlanc AE (1976) Residual learning deficit afterheavy exposure to cannabis or alcohol in rats. Science 192:1249–1251
Gubler CJ (1961) The effects of thiamine deficiency and thiamine antag-onists on the oxidation of α-keto acids by rat tissues. J Biol Chem236(12):3112–3120
Hakim AM, Pappius HM (1983) Sequence of metabolic, clinical, andhistological events in experimental thiamine-deficiency. AnnNeurol13:365–375
Harding AJ, Wong A, Svoboda M, Kril JJ, Halliday GM (1997) Chronicalcohol consumption does not cause hippocampal neuron loss inhumans. Hippocampus 7:78–87
Heinrich CP, Stadler H,Weiser H (1973) Effect of thiamine-deficiency onacetylcoenzyme-a and acetylcholine levels in rat-brain. JNeurochem 21:1273–1281
Homewood J, Bond NW, MacKenzie A (1997) The effects of single andrepeated episodes of thiamin deficiency on memory in alcohol-consuming rats. Alcohol 14:81–91
Hoyumpa AMJ (1980) Mechanisms of thiamin deficiency in chronicalcoholism. Am J Clin Nutr 33:2750–2761
Langlais PJ, Mandel RJ, Mair RG (1992) Diencephalic lesions, learningimpairments, and intact retrograde memory following acutethiamine-deficiency in the rat. Behav Brain Res 48:177–185
Lockridge O, Schopfer LM, Winger G, Woods JH (2005) Large scalepurification of butyrylcholinesterase from human plasma suitablefor infection into monkeys; a potential new therapeutic for protec-tion against cocaine and nerve agent toxicity. J Med Chem Biolradiol Def 3:nihms5095
Martin PR, Singleton CK, Hiller-Sturmhöfel S (2003) The role of thia-mine deficiency in alcoholic brain disease. Alcohol Res Health27(2):134–142
Massoulie J, Pezzementi L, Bon S, Krejci E, Vallette FM (1993) Molecularand cellular biology of cholinesterases. Prog Neurobiol 41:31–91
Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG, Lockridge O(2002) Acetylcholinesterase knockouts establish central cholinergicpathways and can use butyrylcholinesterase to hydrolyze acetylcho-line. Neuroscience 110(4):627–639
Molina PE, Myers N, Smith RM, Lang CH, Yousef KA, Tepper PG,Abumrad NN (1994) Nutritional and metabolic characterization of athiamine-deficient rat model. J Parenter Enter Nutr 18:104–111
Morris RG, Garrud P, Rawlins JN, O’Keefe J (1982) Place navigationimpaired in rats with hippocampal lesions. Nature 297:681–683
National Research Council (1985) Guide for the care and use of labora-tory animals: a report of the institute of laboratory animal resourcescommittee on care and use of laboratory animals
Noël X, Paternot J, Van der Linden M, Sferrazza R, Verhas M, Hanak C,Kornreich C, Martin P, De Mol J, Pelc I, Verbanck P (2001)
J Mol Neurosci

Correlation between inhibition, working memory and delimitedfrontal area blood flow measured by Tc-99 m-bicisate spect inalcohol-dependent patients. Alcohol Alcohol 36:556–563
Palencia G, Teixeira F, Ortiz A, Perez R, Rios C, Sotelo J (1994)Detrimental effects of malnutrition on the damage-induced by alco-holism - a study of animal-models that simulate chronic-alcoholismandmalnutrition of large human groups. J Stud Alcohol 55:113–120
Paula-Barbosa MM, Brandao F, Madeira MD, Cadete-Leite A (1993)Structural changes in the hippocampal formation after long-termalcohol consumption andwithdrawal in the rat. Addiction 88:237–247
Paxinos G and Watson C (2007) The rat brain in stereotaxic coordinates.6th,31-33
Pekkanen L (1979) Pyrithiamin shortens ethanol-induced narcosis andincreases voluntary ethanol drinking in rats. Int J VitamNutr Res 49:386–390
Pereira SRC, Menezes GA, Franco GC, Costa AEB, Ribeiro AM (1998)Chronic ethanol consumption impairs spatial remote memory in ratsbut does not affect cortical cholinergic parameters. PharmacolBiochem Behav 60:305–311
Peterson JW, Gubler CJ, Kuby SA (1975) Partial-purification and prop-erties of thiamine pyrophosphokinase from pig brain. BiochimBiophys Acta 397:377–394
Picklo MJ (2008) Ethanol intoxication increases hepatic N-lysyl proteinacetylation. Biochem Biophys Res Commun 376:615–619
Pinto LCM (1996) QUANTIKOV - Um analisador microestrutural para oambienteWindows. Doctorated thesis. IPEN - USP: São Paulo, p 167
Pires RG, Pereira SR, Carvalho FM, Oliveira-Silva IF, Ferraz VP, RibeiroAM (2007) Correlation between phosphorylation level of a hippo-campal 86 kDa protein and extinction of a behaviour in a model ofWernicke-Korsakoff syndrome. Behav Brain Res 180:102–106
Pires RGW, Pereira SRC, Pittella JE, Franco GC, Ferreira CL, FernandesPA, Ribeiro AM (2001) The contribution of mild thiamine deficien-cy and ethanol consumption to central cholinergic parameter dys-function and rats’ open-field performance impairment. PharmacolBiochem Behav 70:227–235
Pitel AL, Zahr NM, Jackson K, Sassoon SA, Rosenbloom MJ,Pfefferbaum A, Sullivan EV (2011) Signs of preclinicalWernicke’s encephalopathy and thiamine levels as predictors ofneuropsychological deficits in alcoholism without Korsakoff’s syn-drome. Neuropsychopharmacology 36:580–588
Redish AD (2001) The hippocampal debate: are we asking the rightquestions? Behav Brain Res 127:81–98
Roland JJ, Mark K, Vetreno RP, Savage LM (2008) Increasing hippo-campal acetylcholine levels enhance behavioral performance in ananimal model of diencephalic amnesia. Brain Res 1234:116–127
Savage LM, Hall JM, Resende LS (2012) Translational rodent models ofKorsakoff syndrome reveal the critical neuroanatomical substratesof memory dysfunction and recovery. Neuropsychol Rev 22:195–209
Schegg KM, Gillespie RP, Prym U, Peacock JH (1990) Changes inmembrane-bound and soluble molecular forms of acetylcholinester-ase in mouse hippocampus after cholinergic denervation. NeurosciLett 118:197–200
Segal M, Greenberger V, Israeli M, Biegon A (1988) A correlationbetween regional acetylcholinesterase activity rat brain and perfor-mance in a spatial task. Behav Brain Res 30:215–219
Shaltiel G, Hanan M, Wolf Y, Barbash S, Kovalev E, Shoham S, Soreq H(2013) Hippocampal microRNA-132 mediates stress-inducible cog-nitive deficits through its acetylcholinesterase target. Brain StructFunct 218:59–72
Shenhar-Tsarfaty S, Berliner S, Bornstein NM, Soreq H (2013)Cholinesterases as biomarkers for parasympathetic dysfunctionand inflammation-related disease. J Mol Neurosci 1559–1166
Takashima A, Nieuwenhuis ILC, Rijpkema M, Petersson KM, Jensen O,Fernandez G (2007) Memory trace stabilization leads to large-scalechanges in the retrieval network: a functional MRI study on asso-ciative memory. Learn Mem 14:472–479
Victor M (1994) Alcoholic dementia. Can J Neurol Sci 21:88–99Vigil FA, Oliveira-Silva IF, Ferreira LF, Pereira SR, Ribeiro AM (2010)
Spatial memory deficits and thalamic serotonergic metabolitechange in thiamine deficient rats. Behav Brain Res 210:140–142
Vorhees CV, Schmidt DE, Barrett RJ, Schenker S (1977) Effects ofthiamin deficiency on acetylcholine levels and utilization in vivoin rat brain. J Nutr 107:1902–1908
Zimatkin SM, Zimatkina TI (1996) Thiamine deficiency as predispositionto, and consequence of, increased alcohol consumption. AlcoholAlcohol 31:421–427
Zimitat C, Kril J, Harper CG, Nixon PF (1990) Progression of neurolog-ical disease in thiamin-deficient rats is enhanced by ethanol. Alcohol7:493–501
J Mol Neurosci





























