-
8/13/2019 MicroRNAs in Organogenesis and Disease
1/13
698 Current Molecular Medicine 2008, 8, 698-710
1566-5240/08 $55.00+.00 2008 Benth am Science Publis hers Ltd
.
MicroRNAs in Organogenesis and Disease
Naisana S. Asli, Mara E. Pitulescu and Michael Kessel*
Research Group Developmental Biology, Department of Molecular
Cell Biology, Max-Planck-Institute forBiophysical Chemistry, 37077
Gttingen, Germany
Ab st rac t: Large numbers and quantities of different, small
RNA molecules are present in the cytoplasm ofanimal and plant
cells. One subclass of these molecules is represented by the
noncoding microRNAs. Sincetheir discovery in the 1990s a multitude
of basic information has accumulated, which has identified
theirfunction in post-transcriptional control, either via
degradation or translational inhibition of target mRNAs.
Thisfunction is in most of the cases a finetuning of gene
expression, working in parallel with transcriptionalregulatory
processes. MicroRNA expression profiles are highly dynamic during
embryonic development and inadulthood. Misexpression of microRNAs
can perturb embryogenesis, organogenesis, tissue homeostasis andthe
cell cycle. Evidence from gain- and loss-of function studies
indicates roles for microRNAs inpathophysiologic states including
cardiac hypertrophy, muscle dystrophy, hepatitis infection,
diabetes,Parkinson syndrome, hematological malignancies and other
types of cancer. In this review, we focus onstudies addressing the
role of various microRNAs in heart, muscle, liver, pancreas,
central nervous system,and hematopoiesis.
Keywords: microRNA, development, disease, cell cycle.
INTRODUCTIONMicroRNAs represent a previously overlooked
class
of abundant, relatively small molecules, which hasbecome a major
focus of research in the last years.From an exponentially growing
number of studies theyemerged as important factors in a
post-transcriptionalregulatory network, relevant in nearly all
processes oflife.
By using high-throughput sequencing of small RNAlibraries from
different organ systems and cell types,416 different human, 386
mouse and 325 rat microRNAgenes were identified [1]. These numbers
match veryclosely the entries in miRBase, the microRNA
registrydatabase (http://microrna.sanger.ac.uk/sequences).However,
predicted numbers based on bioinformaticsand genomic sequences are
often larger, withestimates as high as 1000 genes in the human
genome[2].
The microRNA nomenclature follows guidelines ofmiRBase, which
also decides on the acceptance ofnewly characterized microRNAs.
MicroRNAs (alsocalled miRNAs) are named using a miR-
prefix,followed by a unique identifying number.
OrthologousmicroRNAs with the same sequence, but from
differentspecies, receive the same numbers, and the species
name can be added as a prefix (e.g. hsa-miR-124).Identical
microRNAs from different genomic loci anddifferent precursors
within a given species areregarded as paralogous, and are
differentiated by anadditional numbering (e.g. hsa-miR-1-1).
ParalogousmicroRNAs which have a difference of one or two
*Address correspondence to this author at the Research
GroupDevelopmental Biology, Department of Molecular Cell Biology,
Max-Planck-Institute for Biophysical Chemistry, 37077
Gttingen,Germany; Tel: ++49-551-2011752; Fax:
++49-551-2011504;E-mail: [email protected]
bases, are differentiated by the addition of small letters(e.g.
hsa-miR-20a). MicroRNAs which originate fromdifferent arms of the
same stem loop structure, arefurther differentiated by the addition
of 3p or 5paccording to which arm (3 or 5 respectively) they
aredriven from (e.g. miR-17-5p) [3-6].
The expression of microRNAs involves initially themachinery used
for the transcription of proteinencoding genes, and subsequently a
highly conservedprocessing mechanism. RNA polymerase II
generateslarge primary transcripts known as pri-microRNA whichcan
have a length of more than 1kb (Fig. 1) [7-9].
These molecules pass through a first processing stepby the
nuclear RNAse Drosha, which results in theformation of a hairpin
molecule of around 70nucleotides, the pre-microRNA [9]. These RNAs
enterthe next processing step in the cytoplasm, where theDicer
RNAse cleaves the hairpin structure and yieldsa dsRNA of 21-24 base
pairs, with the typical feature ofa 2 nucleotides overhang at the 3
end [9]. From thedouble stranded Dicer product, the strand with
lesserthermodynamic stability at its 5 end is
selectivelystabilized, whereas the complementary strandbecomes
subsequently degraded. The stable, maturemicroRNA molecule of 21-24
nucleotide length is thenready to act as the guide strand in the
silencingprocess [9] (Fig. 1). Mature microRNAs exert theirfunction
within a ribonucleoprotein complex, themicroRNA-induced silencing
complex (miRISC). ThemiRISC complex includes the mature
microRNAtogether with a number of proteins of the Argonautefamily
[10]. Within the miRISC complex the binding of amicroRNA to a mRNA
occurs through base-pairingbetween the nucleotides 2-8 at the 5end
of themicroRNA (seed region) and the complementarysequence in the
3UTR of the target mRNA. Theconsequences of this binding depend to
a large extent
-
8/13/2019 MicroRNAs in Organogenesis and Disease
2/13
MicroRNAs in Organogenesis and Disease Current Molecular
Medicine, 2008, Vol. 8, No. 8 699
on the degree of complementation between the twoRNA
molecules.
The most common effect of the binding of miRNA tothe 3 UTR is to
significantly affect translational
efficiency, resulting from non-perfect base
pairing.MicroRNA-mediated inhibition of translation can occurvia
two mechanisms: 1) At the translation initiationstep, by impairing
the cap recognition process [10, 11]or 2) via premature termination
of translation or co-translational protein degradation [11]. In
addition totranslational repression of mRNAs by microRNA, thereis
also evidence for microRNA-mediated destabilizationof target
transcripts. This process involves thedeadenylation and subsequent
decapping of a mRNAmolecule which results in mRNA degradation in
specialcytoplasmic foci, known as P-bodies [11]. A RISC-
associated microRNA recognizes its target mRNA bybase pairing at
a target sequence on the mRNA. The
Argonaute protein then interacts with a P-body protein,and is
delivered to the P-bodies [11]. The target mRNAis then either
decapped and degraded, or just stored ina translationally static
state. Which mechanism is finallyadopted depends on an individual
microRNA-mRNApair, and the specific sequence context [11]. A
translationally-stalled mRNA, can be reshuffled intoactive
polysomes in special cases of stress or otherexternal stimuli
[10].
Although most of the microRNAs recognize theirtargets through
imperfect pairing, there are also someexamples of perfect
complementarity between themiRNA guide strand and the mRNA target.
Thisgenerates an A-form microRNA-mRNA double helix,which marks the
duplex for the siRNA pathway, andeventually results in the
degradation of the targetmRNA by the endonucleolytic capacity of
the siRISCthrough the cleavage of the mRNA in the central
part(positions 10 and 11) of the duplex [10].
The high conservation of microRNAs in differentspecies combined
with highly dynamic expressionpatterns suggested that they might
have regulatoryfunctions in gene expression [12, 13]. The
firstexperimental evidence for a significant function
duringembryonic development came from embryos, wherethe complete
microRNA supplies were depletedthrough an inactivation of the
pre-microRNA processingenzyme Dicer. Dicer knockout mice die at
very earlystages of development, before gastrulation [14].Deletion
of the zebrafish Dicer homologue also resultedin an early arrest of
embryonic development, thoughthe embryos survived longer than
murine dicermutants, possibly as a result of maternal
contributions
[15-17]. The Dicer RNase is also involved in theprocessing of
interfering RNA molecules other thanmicroRNAs. Therefore, to draw
far reachingconclusions from the absence of a central molecule
likeDicer should only be done with caution, since functionsin
development and homeostasis not related tomicroRNAs could also be
affected [14].
The study of microRNAs, involves a variety oftechniques from
high throughput screening methodslike small RNA library sequencings
and microarrayexpression profilings, to finer methods of
functionalanalysis. The latter, include loss of functionapproaches,
such as application of antisenseoligonucleotides against microRNA
sequences, orcomplete deletion of a microRNA coding sequence
byconventional knockout strategies. Additionalapproaches include
ectopic expression of themicroRNAs to further elucidate their
downstreameffects. In the following paragraphs we will
describeexemplary cases, where the function of microRNAswere
experimentally validated. We will review evidencefor the role of
microRNAs in development and disease,with a particular focus on
heart, musculature, liver,pancreas, the nervous system,
hematopoiesis, and thecell cycle.
Fig. (1). Mechanism of microRNA production and
function.MicroRNAs are transcribed by RNA polymerase II,processed
through two successive steps by RNAses Droshaand Dicer, and the
mature microRNA attacks the targetmRNA within the RISC, inhibiting
its translation, or leading toits degradation.
-
8/13/2019 MicroRNAs in Organogenesis and Disease
3/13
700 Current Molecular Medicine, 2008, Vol. 8, No. 8 Asli et
al.
MicroRNAs in Heart and Muscle Development andDisease
Somatic and cardiac muscles are derived frommesodermal cells
ingressing through the primitivestreak during gastrulation. These
progenitors developinto still dividing myoblasts, before they exit
from thecell cycle and fuse to form myotubes, which thenmature into
muscle fibres. Cardiac and skeletalmuscles represent principally
different tissues, whichdiffer in origin, migratory pathways,
inductiverequirements and molecular determinants.
The complete loss of all microRNAs in zebrafishembryos due to a
mutation of the Dicer gene ledamong other defects also to problems
in heartdevelopment [15]. However, it was the analysis ofmouse
embryos with a conditional inactivation of theDicer gene in the
developing heart which clearlydefined the essential role of
microRNAs incardiogenesis [18]. These mice died from cardiacfailure
on day 12.5, presenting an underdevelopedventricular myocardium and
pericardial edema.
Up- or downregulation of several microRNAsaccompanied
pathological cardiac remodeling inrodents and humans [19-24].
Several candidates,including miR-1, miR-21, miR-133, miR-181,
miR-195,miR-206 and miR-208 were more specifically studied ingain-
or loss-of-function experiments (Fig. 2A ). Thecardiac-specific
microRNA miR-208 is encoded by anintron of the alpha myosin heavy
chain (MHC) gene[25]. Mice carrying a miR-208 mutation demonstrate
itsinvolvement in cardiomyocyte hypertrophy, fibrosis,and
expression of beta MHC in response to stress andhypothyroidism.
miR-208 downregulates THRAP1, anegative regulator of beta MHC after
birth [25]. Thus,the alpha MHC gene, in addition to encoding a
majorcardiac contractile protein, regulates cardiac growthand gene
expression in response to stress andhormonal signaling through
miR-208.
Roles in cardiac growth were found for miR-195,miR-21, miR-133,
and miR-1. Moderate overexpressionof miR-195 in the developing
heart of mice inducedhypertrophy, and higher levels of expression
led tocardiomyopathy with ventricular dilatation and wallthinning,
finally resulting in heart failure [19]. MiR-21 isone of the
microRNAs found increased in hypertrophy-induced mouse hearts and
in angiotensin II orphenylephrine-induced cultured rat
neonatalhypertrophic cardiomyocytes [20]. However, while
Cheng et al . observed that knocking-down miR-21interfered with
the induction of hypertrophy in culturedcardiomyocytes [20], the
opposite was found in thestudy by Tatsuguchi et al . [22]. These
authorsdescribed that miR-21 over-expression reducedcardiomyocyte
hypertrophy, while its inhibition inducedmyocyte hypertrophy. The
reason for the observeddiscrepancies after functional modulation of
miR-21levels is currently unclear.
Another microRNA which functions as a negativeregulator of
cardiac hypertrophy is miR-133 [26]. The
miR-133 family consists of three members, encoded bytwo miR-133a
and one miR-133b genes, all beingexpressed in both cardiac and
skeletal muscle tissuesof adults [27, 28]. MiR-133 is
down-regulated in threemurine models of cardiac hypertrophy, and in
heartsfrom patients with hypertrophic cardiomyopathy andatrial
dilatation [26]. Overexpression of miR-133 in bothneonatal and
adult cultured cardiomyocytes reduced
hypertrophy, while suppression of endogenous miR-133 resulted in
marked hypertrophy in the absence ofany hypertrophic stimulus.
However, changes of miR-133 levels were not detected by expression
profiling inthree different types of human cardiac
disease,emphasizing the cautions necessary when concludingfrom one
model to the other [24].
From Drosophila to higher vertebrates, miR-1 isexpressed in
developing skeletal and heart muscle [18,29-36]. miR-1 and miR-133a
are derived from the samebicistronic RNA, and have identical mature
sequences[28]. Downregulation of miR-1 was observed in amouse model
of thoracic aortic constriction-inducedcardiac hypertrophy [21].
The authors concluded thatlow levels of miR-1 might be important
for the inductionof hypertrophy, releasing its growth-related
targets frominhibitory influence. Previous studies had shown
thatthe overexpression of miR-1 in the developing mouseheart lead
to a decreased proliferation of myocytes,with a developmental
arrest on day 13.5 [34]. Thisphenotype was due to a failure of
ventricularcardiomyocyte expansion, caused by a decrease of
themiR-1 target Hand2, a cardiac transcription factorexpressed
early in heart development and required forventricular growth [18,
34, 37]. A further study on theloss of miR-1-2 function in mice
confirmed its role inrepressing the proliferation of cardiomyocytes
[18].
Adult mice lacking miR-1-2 had hyperplas tic hearts withstill
proliferating cardiomyocytes, while a normal adultmouse heart has
only terminally differentiatedcardiomyocytes. In mice, miR-1
expression is regulatedby the transcription factors MyoD, Mef2 and
SRF [34].This reminds of the situation in Drosophila, where miR-1
expression in the presumptive and early mesoderm isregulated by the
transcription factors Twist, Mef2, andSRF [34-36]. Severely
affected Drosophila miR-1mutant embryos showed abnormal cardiac and
musclepatterning, their heart progenitor cells were in excessand
failed to differentiate into distinct lineages [36].
Anoverexpression of miR-1 in cardiac mesoderm ofDrosophila had an
opposite effect. Precursors
differentiated prematurely, so that fewer progenitorswere
observed [36]. Another loss of function miR-1study in Drosophila
showed that it is not necessary fornormal specification and
patterning of the somaticmuscle, but is essential for maintaining
thedevelopment and integrity of body wall muscle duringphases of
rapid muscle growth [35].
Besides their function in regulating cardiachypertrophy, miR-1
and miR-133 play roles inmodulating heart electrical conduction
[18, 38-40]. miR-1-2 mouse mutants had a decreased heart rate
andproblems in ventricular repolarization [18]. 50% of the
-
8/13/2019 MicroRNAs in Organogenesis and Disease
4/13
MicroRNAs in Organogenesis and Disease Current Molecular
Medicine, 2008, Vol. 8, No. 8 701
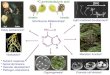
Fig. (2). Exemplary microRNAs and their field of function.
-
8/13/2019 MicroRNAs in Organogenesis and Disease
5/13
702 Current Molecular Medicine, 2008, Vol. 8, No. 8 Asli et
al.
miR-1-2 knockout embryos died during embryonicdevelopment due to
defects in their ventricular septum,and 15% died 2-3 months after
birth. The ones thatsurvived to adulthood died frequently by sudden
death.
After myocardial infarction, the surviving heartsbecame
hypertrophic and underwent an electricalremodeling process favoring
arrhythmia [38]. miR-1 levels were increased in rats with
experimental
myocardic infarcts, in rat myocardium that hadundergone ischemia
and reperfusion, and in individualsaffected by coronary artery
disease [39]. MiR-1 is aproarrhythmic and arrhythmogenic factor
directlytargeting the cardiac gap junction channel connexin 43,and
the K + channel subunit Kir2.1. Knocking-down miR-1 in myocardial
infarction normalized the expression ofthese targets and reduced
arrhythmias. The direct co-silencing of miR-1 with the two targets
inducedarrhythmias, indicating that the role of miR-1 inelectrical
remodeling and the proarrhythmic effect wereat least partially due
to downregulation of these ionchannels proteins. MiR-133 plays an
important role inQT prolongation, a disorder of the hearts
electrical
conduction system, and the associated arrhythmias indiabetic
hearts [40]. This syndrome is furtherassociated with episodic
ventricular arrhythmias andsudden death. MiR-133 targets a subunit
of a cardiacK+ channel that conducts one of the
cardiacrepolarization currents [40]. This protein was repressedin
rabbit hearts of a diabetes mellitus model wheremiR-133 and its
upstream regulator, the SerumResponse Factor (SRF), were increased.
Inhibition orsilencing of SRF decreased miR-133 levels andincreased
the cardiac repolarization current density inventricular
myocytes.
The multiple functions of miR-1 and miR-133 furtherinclude the
regulation of cardiac cell fate through themodulation of apoptosis,
possibly targeting the heatshock and caspase proteins [41]. Induced
by oxidativestress, miR-1 and miR-133 produced opposing effectson
apoptosis, with miR-1 being pro-apoptotic and miR-133 being
anti-apoptotic. Therefore, the miR-1/miR-133ratio is very important
for cardiac cell fate regulation.Several studies focused on the
role of microRNAs inthe acquisition of a myogenic fate. They
identified miR-1, miR-133, miR-181 and miR-206 as
functionalregulators of the fine balance between cell
proliferationand differentiation (Fig. 2B ). MiR-181 is
poorlyexpressed in terminally differentiated muscle, but isstrongly
upregulated in differentiated, regenerated
muscle fibers of a muscle injury mouse model. MiR-181directly
targets Hoxa11 mRNA, which encodes asuppressor of MyoD, a trigger
of the muscledifferentiation program. However, neither
upregulationof miR-181, nor downregulation of Hoxa11 wassufficient
to trigger terminal differentiation ofproliferating myoblasts [42].
The murine myoblast-derived cell line C2C12 is frequently used as a
modelsystem for myogenesis. The overexpression of theclosely
related microRNAs miR-1 or miR-206 in C2C12cells decreased
proliferation, and increasedmyogenesis through the regulation of
targets including
a histone deacetylase (HDAC4), a subunit of DNApolymerase
(Pola1), and of a gap-junction protein(Connexin43) [28, 43-45].
Even in HeLa cells, miR-1 iscapable of promoting myogenesis [46].
Although miR-133 derives from the same bicistron as miR-1, it
stillhas an opposite effect on myogenesis, promotingrather than
inhibiting myoblast proliferation, anddecreasing myogenesis [28].
Its effect was attributed to
a direct downregulation of the muscle transcriptionfactor SRF.
These findings indicate a negativeregulatory loop, where
tissue-specific miR-1 expressionis regulated by SRF, whose
expression is regulated bymiR-133 [28] (Fig. 3A ).
In conclusion, a full range of miroRNAs isexpressed in heart
and/or skeletal muscle, and theirmisregulation alone or in
association affects cell fate,regulates the balance between cell
proliferation anddifferentiation, and induces or influences heart
diseasestates, such as hypertrophy, fibrosis, electricalremodeling
and arrhythmia (Fig. 2A, B ).
MicroRNA Functio ns in the LiverThe formation of the foregut
pocket marks the
beginning of endodermal organogenesis, leading notonly to an
elaborated intestinal system, but also to thedevelopment of
adjacent endodermal organs, like lung,liver, and pancreas. The
hepatic primordium forms inthe cardiac region, and becomes obvious
by anincrease of proliferation and cell migration in a
cord-likefashion into the mesenchyme of the septumtransversum.
Hepatic ducts begin to form in thegrowing liver parenchyme, and
finally a common ductconnects the rapidly developing, distinct
organ to thegut tube. While many embryonic signals and
inductiveinteractions during liver development had
beencharacterized, an involvement of microRNAs has notbeen shown so
far, but appears most likely. Theavailable data rather focus on the
adult liver, and inparticular the role of miR-122, a
liver-specificmicroRNA which was cloned from mouse, rat andhuman
liver tissues, as well as from several hepatic celllines [29, 47].
The potential importance of miR-122 indiseases like hepatitis
fuelled the interest in thismicroRNA. Expression studies revealed
its relation tothe replication competence of the Hepatitis C
virus(HCV), and showed that the cellular abundance of miR-122 often
correlated with the replication ability of theHCV RNA [47]. A
sequence comparison of the HCV
genome and the miR-122 microRNA suggested thattwo potential
target sequences were located in the 5and 3 UTRs of the HCV coding
core [47]. Mutationalanalysis of the two putative targets showed,
that the5UTR, but not the 3UTR is an efficient targetsequence for
miR-122, necessary for the accumulationof viral RNA [47].
Furthermore, the analysis of HCVRNA in miR-122 transfected cells
revealed that theeffect on HCV is rather at the level of the
genomic RNAreplication, than of translation. Therefore, it
wasconcluded that miR-122 affects the replication of theHCV genome
upon binding to its 5UTR, and probably
-
8/13/2019 MicroRNAs in Organogenesis and Disease
6/13
MicroRNAs in Organogenesis and Disease Current Molecular
Medicine, 2008, Vol. 8, No. 8 703
changing the secondary structure of the viral genomeby making it
replication competent [47]. The sequenceof miR-122 is an integral
constituent of the non-codinghcr RNA, a 166 nucleotide molecule
formerly identifiedin liver tumors, different stages of embryonic
liver, andliver cell lines [48]. The computational analysis of
thehcr secondary structure predicted a stem loopconfiguration, with
the miR-122 sequence in one arm.
Together these data confirmed that the non-coding hcris the
unprocessed form of miR-122, which showsdifferential expression in
different types of hepatictumors [48]. Computational target
predictions for miR-122 predicted that the 3UTR of a mRNA encoding
thecationic amino acid transporter (CAT)-1 was a target.This was
further confirmed by reporter analysis. Furtherevidence was
obtained by following the expression ofmiR-122 and CAT-1 in
different stages of embryonicand adult livers, and in several cell
lines, showing aconverse correlation [48]. A systemic ablation of
miR-122 in mouse liver by intraperitoneal injection of the
2-O-methoxyethyl (2OME) phosphorothioate antisenseoligonucleotides,
showed a further role of miR-122 in
lipid metabolism [49]. Mice injected with the miR-122antisense
oligonucleotides, showed a normal plasmalevel of glucose, but a
reduction of cholesterol andtriglycerides, comparing to the control
animals [49].Further microarray analysis of liver gene expression
inthese mice, showed a high fluctuation of genesinvolved in lipid
metabolism, which was furtherconfirmed by ex-vivo studies showing a
reduction offatty acid synthesis and an increase of fatty
acidoxidation [49].
All in all, the results from these functional studieselucidate
the function of a microRNA in the replicationcompetence of the HCV
genome, and more generally,a role in the lipid metabolism of the
liver.
MicroRNAs and Pancreas Development
Pancreas development begins early in vertebrateembryogenesis
with dorsal and ventral evaginationsfrom the endodermal epithelium
of the foregut. The twopancreatic rudiments grow together to form a
singlemass of cells, which then becomes structured into
acharacteristic pattern of lobules, separated bymesenchyme and
drained by a duct system.Cytodifferentiation then gives rise to
exocrine andendocrine cell types. The latter develop in
smallclusters, the islets of Langerhans and secrete
hormones, most importantly insulin, glucagon
andsomatostatin.
The proper development and function of thepancreas is a highly
regulated and tuned process,which involves an extensive array of
transcriptionfactors and tight regulatory pathways.
MicroRNAexpression profiles show a considerable degree ofdynamics
from the early bud stages to the formation ofthe definitive
pancreas [50]. Among the manymicroRNAs which are suggested either
by high-throughput RNA profiling or computational methods tobe
involved in pancreas development or function, only
a few were functionally validated. The mainconclusions on the
role of microRNAs in pancreasmorphogenesis stem from
loss-of-function experimentsin zebrafish embryos, where
morpholinooligonucleotides against the precursor or the
maturemicroRNA sequence of interest were applied, whichimpaired the
microRNA function [51].
A morphlino knockdown approach was used tostudy the function of
miR-375, which was originallyreported as a microRNA specifically
expressed in thepancreatic islet and the pituitary gland [33], and
waslater isolated from pancreatic beta cells [52]. The lossof
miR-375 function in zebrafish embryos, did not resultin any defect
of the pituitary gland as demonstrated bythe expression of pit1 as
an early marker for thepituitary gland formation [51]. The
pancreatic islets,however, showed a scattered pattern of
islet-1expression which was also followed by the expressionof
further endocrine pancreatic markers, such asglucagon and
somatostatin [51]. The results suggestthat miR-375 is involved in
the maintenance ofpancreatic integrity, but the exact mechanism and
thedownstream pathway still remains to be understood.The study of
miR-375 in pancreatic beta cells furthershowed a direct effect on
insulin secretion in thesecells [52]. The overexpression of miR-375
in the murinepancreatic beta cell line MIN6 resulted in a decrease
ofglucose-induced insulin secretion. The functionalknockdown of
miR-375 on the other hand, using 2-O-methyl antisense
oligonucleotides, had a converseeffect, and increased the insulin
secretion uponinduction of these cells with glucose. The function
ofmiR-375 was not resulting from impaired signalingpathways, but
rather from a direct effect on the insulinexocytosis [52]. Among
the different targets of miR-375, myotrophin, an ankyrin
repeat-rich cytoplasmicprotein which promotes insulin secretion,
was validatedas a functional mediator of miR-375 in the
pancreaticbeta cells [52]. According to these
studies,overexpression of miR-375 in MIN-6 cells impaired
theglucose-induced insulin secretion by downregulatingmyotrophin
and subsequently resulting in a defect ininsulin exocytosis
[52-54].
The insulin secretory pathway is further regulatedby miR-9, a
microRNA expressed in neurons,pancreatic beta cells and the rat
pancreatic beta cellline INS-1E [55]. The overexpression of miR-9
in INS-1E cells resulted in a decrease of insulin exocytosis,which
was further proved to result from a defect in the
secretory signaling pathway [55]. In the subsequentcheck for
misregulated downstream effectorsconnected to the insulin secretion
signaling cascade,Granuphilin/Slp4, a Rab GTPase effector
associatedwith beta cell secretory granules, was shown to
bedramatically upregulated. As microRNAs are known todownregulate
their target mRNAs, the study wasfurther followed to find a
putative miR-9 target whichwould be acting upstream of the
Granuphilin/SLP gene,finally resulting in the identification of the
transcriptionfactor Onecut-2 which is associated with
theGranuphilin promoter, and represses its transcription.
-
8/13/2019 MicroRNAs in Organogenesis and Disease
7/13
-
8/13/2019 MicroRNAs in Organogenesis and Disease
8/13
MicroRNAs in Organogenesis and Disease Current Molecular
Medicine, 2008, Vol. 8, No. 8 705
markers. The genes encoding miR-124, miR-9 andmiR-132 are
targets of the transcriptional repressorREST in non-neural cells,
similar to many otherneuronal genes [65, 66]. When progenitors
mature intoneurons, REST relieves miR-124, which then
becomesinvolved in the inactivation of non-neural genes,including
REST itself and its cofactors MeCP2 andCoREST, which have binding
sites for miR-124 in their
3 UTRs. These findings indicate a double negativefeedback loop
at the transition from non-neural toneural gene expression (Fig. 3B
).
The first direct studies of microRNAs in the centralnervous
system of vertebrate embryos were performedby electroporation of
the neural tube in chick embryos.Cao et al . observed that neither
inhibition noroverexpression of miR-124 altered the acquisition
ofneuronal fate, indicating that miR-124 did not act as aprimary
determinant of neural differentiation [67]. Incontrast, Visvanathan
et al . claim to have observed astimulation of neuronal
differentiation by miR-124,which is antagonized by the target SCP1
[68]. Theprimary function of miR-124 seemed to be the inhibitionof
target genes such as SCP1, lamin, and integrin,which need to be
downregulated in order to proceedfrom a neural progenitor cell to a
differentiated neuron.
Among the many targets of miR-124 is also the globalrepressor of
differential splicing PTBP1 [69]. Byrepressing PBTP1, an
alternatively spliced mRNAencoding PTBP2 becomes favored. This
transition isimportant for the proceeding from a neural progenitor
toa differentiated neuron. Thus, the regulation by aneural specific
microRNA becomes connected to asuperimposed regulatory level, to
alternative splicing.
Similar to miR-124, miR-134 is confined to braincells [70]. It
was found localized in synaptic sites of
dendrites of cultured hippocampal neurons.Overexpression of
miR-134 decreased the dendriticspine volume, whereas a knockdown
increased thevolume slightly. As a molecular target of miR-134,
themRNA encoding LIM domain containing kinase Limk1was identified,
which inhibits Limk1 translation not inthe cell body, but locally
within dendrites. DendriticRNAs are normally transported to
synaptic sites,remain dormant, and are translated only
uponstimulation by factors such as BDNF. The findingssuggest a
miR-134 associated silencing complex,which needs to be inactivated
in order to allow Limk1translation, thereby contributing to
synaptic plasticity[70] .
The neuron enriched microRNA miR-132, wasidentified in a
genome-wide screen as a target of thecAMP-response element binding
protein (CREB) [71].Expression of miR-132 in cortical neurons
inducedneurite outgrowth. Conversely, inhibition of miR-132function
attenuated neuronal outgrowth. The studyestablished a neurogenetic
pathway from an extrinsicneurotrophin, to the transcription factor
CREB, to miR-132 and to the GTPase-activating protein p250GAP.
MiR-133b was identified in a subtractive screensearching for
microRNAs enriched in dopaminergic
neurons [72, 73]. It is enriched in normal adult humanand rodent
midbrain samples, and deficient in samplesstemming from Parkinson
patients, as well as inmidbrains from murine dopamine deficiency
models.The influence of miR-133b in midbrain neurons wasanalyzed in
embryonic stem cell derived culturesystems, or in primary midbrain
cells. Overexpressionusing a lentivirus vector did not affect early
markers of
dopaminergic neuron development. However, a latemarker
indicating mature dopaminergic neurons wassuppressed. Knocking down
miR-133b on the otherhand, induced markers for dopaminergic
neuronformation. A concrete target for miR-133b lies in the3UTR of
the Pitx3 gene, a necessary transcriptionfactor for the development
of dopaminergic neurons.These findings suggests a negative feedback
circuit:Pitx3 activates midbrain-specific neuronal geneexpression,
including miR-133b, which then in turnpost-transcriptionally
represses its inducer Pitx3 (Fig.3C ).
Taken together these examples demonstrate thatneural specific
microRNAs play significant roles in thelast step of differentiation
towards a mature neuron(Fig. 2D ). They are part of an environment,
whichrepresses non-neural molecules at a relatively late stepin
development. Effects on earlier steps are much lessobvious, and may
either not occur at all, or may turnout to be much more difficult
to measure. However, itseems conceivable that also for early
intermediates ofa developmental pathway specific
functionalmicroRNAs exist.
MicroRNAs in Hematopoiesis
During hematopoiesis pluripotent, self-renewingstem cells give
rise to the different blood cell lineages.These include the
erythroid cells, the lymphoid B and Tcells, and the myeloid cells,
including granulocytes,megakaryocytes and macrophages. Aberrations
ofregulatory pathways in hematopoiesis can result insevere disease
states, such as leukemias, lymphomasor myelomas. Recently, it was
demonstrated that alsomicroRNAs are misregulated in various
hematopoieticdiseases or relevant experimental model systems
[74,75]. As shown before for many protein encoding genes,also the
genomic loci encoding microRNAs were foundto be altered by
translocations or deletions [76].
Erythrocyte differentiation normally coincides withthe
downregulation of a number of microRNAs,
including miR-24, miR-221 and miR-222 [77]. Theforced expression
of miR-24 in K562 erythroleukemiccells impairs the activin-induced
maturation of erythroidcells, and represses the accumulation of
hemoglobin inthese cells [77]. This effect results from
thedownregulation of Activin type I receptor ALK4, whichis targeted
by miR-24 [77]. The differentiation of cordblood CD34+ cells
associates with an increase in thereceptor protein Kit, a critical
factor for erythropoiesis.The two microRNAs miR-221 and miR-222
target themRNA for Kit, and therefore a
pro-erythropoietic,undifferentiated state is maintained [78, 79].
The
-
8/13/2019 MicroRNAs in Organogenesis and Disease
9/13
706 Current Molecular Medicine, 2008, Vol. 8, No. 8 Asli et
al.
downregulation of the two microRNAs and theconsequent induction
of the Kit receptor is sufficient torelease the inhibition of
erythropoietic differentiation[79].
MiR-181 is expressed in thymus and bone marrowwith an expression
increasing during B-lymphocytematuration [76]. Ectopic expression
of miR-181 inhematopoietic progenitor cells, doubled the amount
ofB-lymphocytes, with little change in the number of T-cells,
indicating a specific role of this microRNA in B-lymphocyte
differentiation [76]. Also miR-142 and miR-223 have a preferential
expression in hematopoietictissues [76]. In contrast to miR-181,
however, theectopic expression of these two microRNAs,
drovehematopoietic differentiation towards T-lymphocyteformation,
indicating a preferential role of thesemicroRNAs in the maturation
of T-cells [76].
MicroRNAs are also shown to be involved in thedifferentiation of
hematopoietic progenitors to themyeloid cell lineages. When induced
towards themonocyte/macrophage fate, the level of miR-424
increases in cord blood CD34+ cells [80].Concomitantly, the
transcription factor PU.1, requiredfor the terminal maturation
steps of monocyte/macrophage cells, becomes upregulated. The
PU.1protein interacts physically with the promoter of themiR-424
gene, and enhances its transcription. MiR-424itself then targets
the mRNA of NFI-A, a factor whichneeds to be downregulated during
monocyte/macrophage maturation [80]. Through these
regulatoryinteractions a molecular circuitry becomes
establishedwith miR-424 as its central component, controlling
theterminal differentiation of the monocyte/macrophagelineage (Fig.
3D ) [80]. The level of miR-223 increasesduring granulocytic
differentiation [81]. Studies of the
miR-223 promoter revealed overlapping binding sites ofthe
transcription factors NFI-A and C/EBP . Inundifferentiated cells,
the NFI-A protein maintains a lowlevel of miR-223 expression by
binding to its promoter.During granulocyte differentiation however,
the level ofC/EBP as a mediator of differentiation
processesincreases, competes eventually for the binding of NFI-Ato
the miR-223 promoter, and acts as a strongeractivator of microRNA
transcription [81]. The presenceof a target site for miR-223 in the
3UTR of the NFI-AmRNA, enhances the effect of miR-223 on
granulocyticdifferentiation (Fig. 3E ) [81].
Expression profiling and the functionalcharacterizations of
microRNAs in differenthematopoietic lineages indicate their
relevance innormal as well as malignant hematopoiesis (Fig. 2E
).The available medical possibilities in the treatment ofleukemias
or lymphomas make it conceivable, thathematopoietic microRNAs
qualify as targets or agentsin therapy one day
MicroRNAs in t he Cell Cycle and Cancer
Nature has developed multiple mechanisms ofcoupling cell
proliferation with apoptosis in order to
Fig. (3). Feedback regulation between microRNAs
andtranscriptional regulators.
A . Promotion of muscle fate through a feedback loop,repressing
the translation of myoblast proteins afterdifferentiation [28, 34,
40 , 44, 45].B. Balancing between a non-neural and a neuronal
fatethrough a double negative feedback loop repressing inneuronal
cells the translation of the REST complexmembers, which themselves
would maintain a non-neuralfate [65, 66, 68].C. Double negative
feedback inhibition involved in thedifferentiation of a neural
progenitor to a mature neuron [73].
D. Differentiation of promyelocytes intomonocyte/macrophages
upon downregulation of NFI-A [80].E. The balance of NFI-A and
C/EBP- in the differentiation ofundifferentiated cord blood cells
into granulocytes. C/EBP-competes NFI-A in binding the microRNA
promoter andresults in a higher microRNA expression [81].F .
Feedback inhibition of E2F transcription factors bymicroRNAs after
depletion of c-myc from B-cells. ThemicroRNA genes are under
positive control in the presenceof c-myc [85, 86, 89, 91].For a
detailed discussion please see the main text.
-
8/13/2019 MicroRNAs in Organogenesis and Disease
10/13
MicroRNAs in Organogenesis and Disease Current Molecular
Medicine, 2008, Vol. 8, No. 8 707
control aberrant proliferation and to avoid cancerformation. One
cause for cancer formation andprogression is the misregulation of
this link. Obviously,the involvement of microRNAs in the regulation
of thecell cycle and apoptotic pathways, and their functionalroles
in tissue homeostasis is of considerable interest.In the last years
a number of microRNAs with highlydynamic and complex patterns in
cancer tissues were
identified by expression profiling, suggesting functionsin tumor
formation through the regulation of factorslinked to cell cycle
control and apoptosis [82-84].
A group of transcription factors related in manyrespects to cell
cycle regulation is the E2F family. E2Ftranscription factors are
regulated by severalmicroRNAs, including miR-17-5p, miR-20,
miR-34a,miR-221 and miR-222 [85-89]. MicroRNAs from themiR-17-92
cluster, namely miR-17-5p and miR-20a,influence the translation of
E2F1 mRNA [85], whilemiR-20a targets in addition E2F2 and E2F3
[86]. Thismodulation occurs through binding sites in the
3'-untranslated region of the respective mRNAs. Viceversa, E2F1-3
directly regulates the transcription of themiR-17-92 cluster,
suggesting an auto regulatoryfeedback loop between E2F factors and
miRNAs (Fig.3F ). A further component in this regulatory network
isc-Myc, a transcription factor known to regulate
cellproliferation, growth and apoptosis [90]. C-Myc
istranscriptionally induced by E2F1, and itself directlyactivates
the transcription of the miR-17-92 cluster [85,86, 91]. Since
miR-17-5p and miR-20 preventuncontrolled reciprocal activation of
c-Myc and E2F1products, they are involved in the control of
c-Mycmediated cellular proliferation. MicroRNAs from themiR-17-92
cluster have an oncogenic activity whenoverexpressed with c-myc in
a mouse model of humanB-cell lymphoma [92]. In addition,
miR-20aoverexpression decreased apoptosis in a prostatecancer cell
line, and its inhibition resulted in anincrease of cell death after
DNA damage cell treatment[86].
E2F3 is not only targeted by miR-20, but also bymiR-34a, a
microRNA which is downregulated inneuroblastoma derived cell lines
and primaryneuroblastoma tumors, a tumor type accounting for15% of
cancer related childhood deaths [87]. MiR-34aamounts increased in
parallel with decreased E2F3protein levels, when differentiation of
neuroblastomawas induced by all-trans retinoic acid. Proliferation
ofneuroblastoma cells became repressed upon
overexpression of miR-34 by inducing a caspase-dependent
apoptotic pathway.
Another well-known cell cycle regulator, the p27 Kip1 protein,
was found to be subject to control bymicroRNAs. Drosophila germline
stem cells with animpaired dicer-1 activity were delayed at the
transitionfrom G1 to S, due to an increase of Dacapo, a homologof
human p27 Kip1 [93]. Also in human glioblastoma cellsthe
downregulation of Dicer led to a decrease of cellproliferation
involving p27 Kip1 [89]. MiR-221 and miR-222, two homologous
microRNAs, were shown to
regulate p27 Kip1 [88, 89]. These two microRNAs arepresent at
high levels in tumor tissues, such as primaryglioblastomas,
papillary thyroid carcinoma andpancreas tumors [92, 94-96]. In many
glioblastomacancers low levels of p27 Kip1 were observed
[88].Suppression of miR-221 and miR-222 levels in severalcancer
cell lines with antagomirs led to a proliferationarrest and
increase of p27 Kip1 levels [88]. Thus, miR-
221 and miR-222 seem to function as oncogenes bycontrolling cell
cycle progression through inhibition ofp27 Kip1.
The occurrence of chronic lymphocytic leukemia(CLL) is often
correlated with the upregulation of theanti-apoptotic factor BCL2
[97]. On the other hand, thetwo microRNAs miR-15a and miR-16-1 are
significantlydownregulated in many CLL cells, often due to
analtered or deleted genomic locus [97]. This inversecorrelation
suggested BCL2 as a target of these twomicroRNAs. Experimental
testing proved that bothmicroRNAs were able to repress the
translation ofBCL2, resulting finally in the activation of the
APAF-1-caspase 9-PARP apoptotic pathway [97]. Comparativeanalysis
of microRNA pools in normal and p53-mutantembryonic fibroblasts of
mice identified a correlation ofthe expression levels of miR-34a,
miR-34b, and miR-34c with the p53 status [98]. Moreover,
theoverexpression of miR-34 could mimic the downstreamfunctions of
p53 and its canonical effector p21 inactivating the DNA damage
response pathway, inapoptosis, and in cell cycle arrest [98, 99].
Chromatinimmunoprecipitation showed a direct interaction of p53and
the miR-34 promoter, leading to the conclusionthat p53 is in fact
acting upstream of miR-34. A screenfor miR-34 targets revealed many
cell cycle regulationgenes thus further strengthening the evidence
for afunction of miR-34 in the regulation of the cell cycle asa
downstream effector of p53 [98, 99].
Together, these results suggest that microRNAs areimportant for
maintaining the balance between cellularproliferation and
apoptosis, and thus can function asboth tumor promoters or
suppressors, depending oncomplex regulatory networks (Fig. 2F
).
OUTLOOK
The studies discussed in this review support thesteadily growing
evidence that microRNAs function askey players of
post-transcriptional regulation. Mostphysiological processes seem
to involve this level ofregulation, and appear to require microRNAs
for theexact final decision on the states of gene expression.
Inparticular, microRNAs seem to increase therobustness, speed, and
stability in response to dynamicchanges. Fig. ( 2 ) lists the
microRNA studies which arediscussed in the current review. The body
of suchfunctional studies is continuously growing in a
realavalanche of publications. However, if the emergingpicture is
true that microRNAs are involved rather in thefine tuning of many
targets, than in very specific on oroff decisions, it may turn out
to become increasinglydifficult to obtain a systematic
understanding of
-
8/13/2019 MicroRNAs in Organogenesis and Disease
11/13
708 Current Molecular Medicine, 2008, Vol. 8, No. 8 Asli et
al.
microRNA functions. Already now, the importance ofup-to date
high-throughput methods and broadscreening approaches are the way
of choice to tacklesuch questions.
Currently, the field of microRNA research is mostlyfocused on
basic research in many aspects of the lifesciences, including
developmental and cell biology.However, the many disease models in
various animalsystems indicate already that the time is ripe to
transferthe knowledge into clinical research, and then possiblyalso
to clinical applications [100]. The possibility toaddress many
targets simultaneously within a givencontext may eventually turn
out to be an explicitadvantage, as compared to highly specific
techniques.MicroRNAs may finally represent ideal tools for a
so-called one-hit-multiple-target approach [100]. Thesmall size of
the active molecules will allow theintroduction of chemical
modifications, and thus themodulations of microRNA functions,
finally providingprecisely tailored, agonistic or antagonistic
moleculeswith defined cellular, and therefore also
medicalconsequences. The development of methods for asystemic
delivery or applications of microRNA willcertainly represent a
major hurdle to take beforepharmacological applications come into
reach [100].Numerous patents have been filed in recent years in
anattempt to ensure later commercial exploitations of thebasic
findings. A growing number of biotech companiesare being founded to
initiate the next steps inmicroRNA applications. Taken together,
there can beno doubt that the importance of microRNAs inresearch,
clinic, and business will increase in the nextyears. The
expectations are high.
ACKNOWL EDGEMENTS
We thank Dr. Samuel M. Young for critical readingof the
manuscript and Hartmut Sebesse for graphics.
AB BREVIA TIONS
CAT = Cationic Amino acid TransporterCLL = Chronic Lymphocytic
LeukemiaCREB protein = cAMP-response element binding
proteinHCV = Hepatitis C VirusMHC = Myosin Heavy ChainSRF =
Serum Response Factor
REFERENCES
[1] Landgraf, P., Rusu, M., Sheridan, R., Sewer, A., Iovino, N.,
Aravin, A., Pfeffer, S ., Rice, A., Kamphorst, A.O., La
ndthaler,M., Lin, C., Socci, N.D., Hermida, L., Fulci, V.,
Chiaretti, S.,Foa, R., Schliwka, J., Fuchs, U., Novosel, A.,
Muller, R.U.,Schermer, B., Bissels, U., Inman, J., Phan, Q., Chien,
M.,Weir, D.B., Choksi, R., De Vita, G., Frezzetti, D.,
Trompeter,H.I., Hornung, V., Teng, G., Hartmann, G., Palkovits, M.,
DiLauro, R., Wernet, P., Macino, G., Rogler, C.E., Nagle, J.W.,Ju,
J., Papavasiliou, F.N., Benzing, T., Lichter, P., Tam,
W.,Brownstein, M.J., Bosio, A., Borkhardt, A., Russo, J.J.,
Sander, C., Zavolan, M., and Tuschl, T. (2007) Cell ,
1291401-1414.
[2] Griffiths-Jones, S. (2007) Annu Rev. Genomics. Hum.Genet. ,
8 , 279-298.
[3] Ambros, V., Bartel, B., Bartel, D.P., Burge, C.B.,
Carrington,J.C., Chen, X., Dreyfuss, G., Eddy, S.R.,
Griffiths-Jones, S.,Marshall, M., Matzke, M., Ruvkun, G., and
Tuschl, T. (2003)RNA , 9 , 277-279.
[4] Griffiths-Jones, S. (2004) Nucleic Acids Res. , 32 ,
D109-111.[5] Griffiths-Jones, S. (2006) Methods. Mol. Biol. , 342 ,
129-138.[6] Griffiths-Jones, S., Grocock, R.J., van Dongen, S.,
Bateman,
A., and Enright, A.J. (2006) Nucleic Acids Res. , 34 ,
D140-144.
[7] Cai, X., Hagedorn, C.H., and Cullen, B.R. (2004) RNA ,
101957-1966.
[8] Lee, Y., Kim, M., Han, J., Yeom, K.H., Lee, S., Baek,
S.H.,and Kim, V.N. (2004) EMBO J. , 23 , 4051-4060.
[9] Zeng, Y. (2006) Oncogene , 25 , 6156-6162.[10] Pillai, R.S.,
Bhattacharyya, S.N., and Filipowicz, W. (2007)
Trends. Cell Biol. , 17 , 118-126.[11] Nilsen, T.W. (2007)
Trends Genet. , 23 , 243-249.[12] Ason, B., Darnell, D.K.,
Wittbrodt, B., Berezikov, E.,
Kloosterman, W.P., Wittbrodt, J., Antin, P.B., and Plasterk,R.H.
(2006) Proc. Natl. Acad. Sci. USA , 103 , 14385-14389.
[13] Kloosterman, W.P., and Plasterk, R.H. (2006) Dev. Cell ,
11441-450.
[14] Bernstein, E., Kim, S.Y., Carmell, M.A., Murchison, E.P.,
Alcorn, H., Li, M.Z., Mills, A.A., Elledge, S.J ., Anderson,
K.V.,and Hannon, G.J. (2003) Nat. Genet. , 35 , 215-217.
[15] Giraldez, A.J., Cinalli, R.M., Glasner, M.E., Enright,
A.J.,Thomson, J.M., Baskerville, S., Hammond, S.M., Bartel,D.P.,
and Schier, A.F. (2005) Science , 308 , 833-838.
[16] Wienholds, E., Koudijs, M.J., van Eeden, F.J., Cuppen,
E.,and Plasterk, R.H. (2003) Nat. Genet. , 35 , 217-218.
[17] Ying, S.Y., and Lin, S.L. (2005) Biochem. Biophys
Res.Commun. , 335 , 1-4.
[18] Zhao, Y., Ransom, J.F., Li, A., Vedantham, V., von
Drehle,M., Muth, A.N., Tsuchihashi, T., McManus, M.T.,
Schwartz,R.J., and Srivastava, D. (2007) Cell , 129 , 303-317.
[19] van Rooij, E., Sutherland, L.B., Liu, N., Williams,
A.H.,McAnally, J., Gerard, R.D., Richardson, J.A. and Olson,
E.N.(2006) Proc. Natl. Acad. Sci. USA , 103 , 18255-18260.
[20] Cheng, Y., Ji, R., Yue, J., Yang, J., Liu, X., Chen, H.,
Dean,
D.B., and Zhang, C. (2007) Am. J. Pathol. , 170 , 1831-1840.[21]
Sayed, D., Hong, C., Chen, I.Y., Lypowy, J., and Abdellatif,M.
(2007) Circ. Res. , 100 , 416-424.
[22] Tatsuguchi, M., Seok, H.Y., Callis, T.E., Thomson,
J.M.,Chen, J.F., Newman, M., Rojas, M., Hammond, S.M., andWang,
D.Z. (2007) J. Mol. Cell. Cardiol., 42 , 1137-1141.
[23] Thum, T., Galuppo, P., Wolf, C., Fiedler, J., Kneitz, S.,
vanLaake, L.W., Doevendans, P.A., Mummery, C.L., Borlak,
J.,Haverich, A., Gross, C., Engelhardt, S., Ertl, G.,
andBauersachs, J. (2007) Circulation , 116 , 258-267.
[24] Ikeda, S., Kong, S.W., Lu, J., Bisping, E., Zhang, H.,
Allen,P.D., Golub, T.R., Pieske, B., and Pu, W.T. (2007)
Physiol.Genomics. , 31 (3), 367-373.
[25] van Rooij, E., Sutherland , L.B., Qi, X., Richardson, J.A.,
Hill,J., and Olson, E.N. (2007) Science , 316 , 575-579.
[26] Care, A., Catalucci, D., Felicetti, F., Bonci, D., Addario,
A.,Gallo, P., Bang, M.L., Segnalini, P., Gu, Y., Dalton, N.D.,Elia,
L., Latronico, M.V., Hoydal, M., Autore, C., Russo, M.A.,Dorn,
G.W., 2nd, Ellingsen, O., Ruiz-Lozano, P., Peterson,K.L., Croce,
C.M., Peschle, C., and Condorelli, G. (2007)Nat. Med. , 13 ,
613-618.
[27] van Rooij, E. and Olson, E.N. (2007) J. Clin. Invest. ,
1172369-2376.
[28] Chen, J.F., Mandel, E.M., Thomson, J.M., Wu, Q.,
Callis,T.E., Hammond, S.M., Conlon, F.L., and Wang, D.Z. (2006)Nat.
Genet ., 38 , 228-233.
[29] Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer,
J.,Lendeckel, W., and Tuschl, T. (2002) Curr. Biol. , 12 ,
735-739.
[30] Lee, R.C., and Ambros, V. (2001) Science , 294 ,
862-864.
-
8/13/2019 MicroRNAs in Organogenesis and Disease
12/13
MicroRNAs in Organogenesis and Disease Current Molecular
Medicine, 2008, Vol. 8, No. 8 709
[31] Sempere, L.F., Freemantle, S., Pitha-Rowe, I., Moss,
E.,Dmitrovsky, E., and Ambros, V. (2004) Genome Biol. , 5 ,R13.
[32] Mansfield, J.H., Harfe, B.D., Nissen, R., Obenauer,
J.,Srineel, J., Chaudhuri, A., Farzan-Kashani, R., Zuker,
M.,Pasquinelli, A.E., Ruvkun, G., Sharp, P.A., Tabin, C.J.,
andMcManus, M.T. (2004) Nat. Genet ., 36 , 1079-1083.
[33] Wienholds, E., Kloosterman, W.P., Miska, E.,
Alvarez-Saavedra, E., Berezikov, E., de Bruijn, E., Horvitz,
H.R.,Kauppinen, S., and Plasterk, R.H. (2005) Science , 309 ,
310-311.
[34] Zhao, Y., Samal, E., and Srivastava, D. (2005) Nature , 436
,214-220.
[35] Sokol, N.S., and Ambros, V. (2005) Genes Dev. , 19 ,
2343-2354.
[36] Kwon, C., Han, Z., Olson, E.N., and Srivastav a, D.
(2005)Proc. Natl. Acad. Sci. USA , 102 , 18986-18991.
[37] Srivastava, D., Thomas, T., Lin, Q., Kirby, M.L., Brown,
D.,and Olson, E.N. (1997) Nat. Genet , 16 , 154-160.
[38] Anderson, M.E., and Mohler, P.J. (2007) Nat. Med. , 13 ,
410-411.
[39] Yang, B., Lin, H., Xiao, J., Lu, Y., Luo, X., Li, B.,
Zhang, Y.,Xu, C., Bai, Y., Wang, H., Chen, G., and Wang, Z.
(2007)Nat. Med. , 13 , 486-491.
[40] Xiao, J., Luo, X., Lin, H., Zhang, Y., Lu, Y., Wang, N.,
Yang,B., and Wang, Z. (2007) J. Biol. Chem. , 282 ,
12363-12367.
[41] Xu, C., Lu, Y., Pan, Z., Chu, W., Luo, X., Lin, H., Xiao,
J.,Shan, H., Wang, Z., and Yang, B. (2007) J. Cell. Sci., 120
,3045-3052.
[42] Naguibneva, I., Ameyar-Zazoua, M., Polesskaya, A., Ait-Si-
Ali, S., Groisman, R., Souidi, M., Cuvellier, S., and Harel-Bellan,
A. (2006) Nat. Cell. Biol. , 8 , 278-284.
[43] Lu, J., McKinsey, T.A., Zhang, C.L., and Olson, E.N.
(2000)Mol. Cell. , 6 , 233-244.
[44] Kim, H.K., Lee, Y.S., Sivaprasad, U., Malhotra, A., and
Dutta, A. (2006) J. Cell. Biol. , 174 , 677-687.
[45] Anderson, C., Catoe, H., and Werner, R. (2006) Nucleic
Acids Res. , 34 , 5863-5871.
[46] Lim, L.P., Lau, N.C., Garrett-Engele, P., Grimson,
A.,Schelter, J.M., Castle, J., Bartel, D.P., Linsley, P.S.,
andJohnson, J.M. (2005) Nature , 433 , 769-773.
[47] Jopling, C.L., Yi, M., Lancaster, A.M., Lemon, S.M.,
andSarnow, P. (2005) Science , 309 , 1577-1581.
[48] Chang, J., Nicolas, E., Marks, D., Sander, C., Lerro,
A.,Buendia, M.A., Xu, C., Mason, W.S., Moloshok, T., Bort,
R.,Zaret, K.S., and Taylor, J.M. (2004) RNA Biol. , 1 ,
106-113.
[49] Esau, C., Davis, S., Murray, S.F., Yu, X.X., Pandey,
S.K.,Pear, M., Watts, L., Booten, S.L., Graham, M., McKay,
R.,Subramaniam, A., Propp, S., Lollo, B.A., Freier, S.,
Bennett,C.F., Bhanot, S., and Monia, B.P. (2006) Cell Metab. , 3 ,
87-98.
[50] Joglekar, M.V., Parekh, V.S., and Hardikar, A.A.
(2007)Trends Endocrinol. Metab. , 18 , 393-400.
[51] Kloosterman, W.P., Lagendijk, A.K., Ketting, R.F.,
Moulton,J.D., and Plasterk, R.H. (2007) PLoS Biol. , 5 , e203.
[52] Poy, M.N., Eliasson, L., Krutzfeldt, J., Kuwajima, S., Ma,
X.,Macdonald, P.E., Pfeffer, S., Tuschl, T., Rajewsky, N.,Rorsman,
P., and Stoffel, M. (2004) Nature , 432 , 226-230.
[53] Mello, C.C., and Czech, M.P. (2004) Nat. Med. , 10 ,
1297-1298.
[54] Gauthier, B.R., and Wollheim, C.B. (2006) Nat. Med. , 12 ,
36-38.
[55] Plaisance, V., Abderrahmani, A., Perret-Menoud,
V.,Jacquemin, P., Lemaigre, F., and R egazzi, R. (2006) J.
Biol.Chem. , 281 , 26932-26942.
[56] Baroukh, N., Ravier, M.A., Loder, M.K., Hill, E.V.,
Bounacer, A., Scharfmann, R., Rutter, G.A., and Van Obberg hen,
E.(2007) J. Biol. Chem. , 282 , 19575-19588.
[57] Yatoh, S., Dodge, R., Akashi, T., Omer, A., Sharma,
A.,Weir, G.C., and Bonner-Weir, S. (2007) Diabetes , 56 ,
1802-1809.
[58] Joglekar, M.V., Parekh, V.S., Mehta, S., Bhonde, R.R.,
andHardikar, A.A. (2007) Dev. Biol. , 311 , 603-612.
[59] Cuellar, T.L., and McManus, M.T. (2005) J. Endocrinol. ,
187327-332.
[60] Kolfschoten, I.G., and Regazzi, R. (2007) Nat. Clin.
Pract.Endocrinol. Metab., 3 , 827-834.
[61] Johnston, R.J., and Hobert, O. (2003) Nature , 426 ,
845-849.[62] Johnston, R.J., Jr., Chang, S., Etchberger, J.F.,
Ortiz, C.O.,
and Hobert, O. (2005) Proc. Natl. Acad. Sci. USA ,
10212449-12454.
[63] Giraldez, A.J., Mishima, Y., Rihel, J., Grocock, R.J.,
VanDongen, S., Inoue, K., Enright, A.J., and Schier, A.F.
(2006)Science, 312 , 75-79.
[64] Krichevsky, A.M., Sonntag, K.C., Isacson, O., and
Kosik,K.S. (2006) Stem Cells , 24 , 857-864.
[65] Wu, J., and Xie, X. (2006) Genome Biol. , 7 , R85.[66]
Conaco, C., Otto, S., Han, J.J., and Mandel, G. (2006) Proc.
Natl. Acad. Sci. USA , 103 , 2422-2427.[67] Cao, X., Pfaff,
S.L., and Gage, F.H. (2007) Genes Dev. , 21
531-536.[68] Visvanathan, J., Lee, S., Lee, B., Lee, J.W., and
Lee, S.K.
(2007) Genes Dev. , 21 , 744-749.[69] Makeyev, E.V., Zhang, J.,
Carrasc o, M.A., and Maniatis, T.
(2007) Mol. Cell, 27 , 435-448.[70] Schratt, G.M., Tuebing, F.,
Nigh, E.A., Kane, C.G., Sabatini,
M.E., Kiebler, M., and Greenberg, M.E. (2006) Nature ,
439283-289.
[71] Vo, N., Klein, M.E., Varlamova, O., Keller, D.M.,
Yamamoto,T., Goodman, R.H., and Impey, S. (2005) Proc. Natl.
Acad.Sci. USA , 102 , 16426-16431.
[72] Hebert, S.S., and De Strooper, B. (2007) Science ,
3171179-1180.
[73] Kim, J., Inoue, K., Ishii, J., Vanti, W.B., Voronov,
S.V.,Murchison, E., Hannon, G., and Abeliovich, A. (2007)Science ,
317 , 1220-1224.
[74] Kluiver, J., Kroesen, B.J., Poppema, S., and van den Berg,
A. (2006) Leukemia , 20 , 1931-1936.
[75] Lawrie, C.H. (2007) Br. J. Haematol. , 137 , 503-512.[76]
Chen, C.Z., Li, L., Lodish, H.F., and Bartel, D.P. (2004)
Science , 303 , 83-86.[77] Wang, Q., Huang, Z., Xue, H., Jin,
C., Ju, X.L., Han, J.D.,
and Chen, Y.G. (2008) Blood , 111 , 588-595.[78] Georgantas,
R.W., 3rd, Hildreth, R., Morisot, S., Alder, J.,
Liu, C.G., Heimfeld, S., Calin, G.A., Croce, C.M., and
Civin,C.I. (2007) Proc. Natl. Acad. Sci. USA , 104 , 2750-2755.
[79] Felli, N., Fontana, L., Pelosi, E., Botta, R., Bonci,
D.,Facchiano, F., Liuzzi, F., Lulli, V., Morsilli, O., Santoro,
S.,Valtieri, M., Calin, G.A., Liu, C.G., Sorrentino, A.,
Croce,C.M., and Peschle, C. (2005) Proc. Natl. Acad. Sci. USA102 ,
18081-18086.
[80] Rosa, A., Ballarino, M., Sorrentino, A., Sthandier, O., De
Angelis, F.G., Marchioni, M., Masella, B., Guarini, A., Fatica, A.,
Pesch le, C., and Bozzoni, I. (2 007) Proc. Natl. Acad. Sci.USA ,
104 , 19849-19854.
[81] Fazi, F., Rosa, A., Fatica, A., Gelmetti, V., De Marchis,
M.L.,Nervi, C., and Bozzoni, I. ( 2005) Cell , 123 , 819-831.
[82] Chen, C.Z. (2005) N. Engl. J. Med. , 353 , 1768-1771.[83]
Garzon, R., Fabbri, M., Cimmino, A., Calin, G.A., and Croce,
C.M. (2006) Trends. Mol. Med. , 12 , 580-587.[84] Cho, W.C.
(2007) Mol. Cancer , 6 , 60.[85] O'Donnell, K.A., Wentzel, E.A.,
Zeller, K.I., Dang, C.V., and
Mendell, J.T. (2005) Nature , 435 , 839-843.[86] Sylvestre, Y.,
De Guire, V., Querido, E., Mukhopadhyay,
U.K., Bourdeau, V., Major, F., Ferbeyre, G., and Chartrand,P.
(2007) J. Biol. Chem. , 282 , 2135-2143.
[87] Welch, C., Chen, Y., and Stallings, R.L. (2007) Oncogene26
, 5017-5022.
[88] le Sage, C., Nagel, R., Egan, D.A., Schrier, M., Mesman,
E.,Mangiola, A., Anile, C., Maira, G., Mercatelli, N., Ciafre,
S.A.,Farace, M.G., and Agami, R. (2007) Embo. J. , 26 ,
3699-3708.
[89] Gillies, J.K., and Lorimer, I.A. (2007) Cell Cycle , 6 ,
2005-2009.
[90] Tsantoulis, P.K., and Gorgoulis, V.G. (2005) Eur. J. Cancer
41 , 2403-2414.
-
8/13/2019 MicroRNAs in Organogenesis and Disease
13/13
710 Current Molecular Medicine, 2008, Vol. 8, No. 8 Asli et
al.
[91] Matsumura, I., Tanaka, H., and Kanakura, Y. (2003)
CellCycle , 2 , 333-338.
[92] He, L., Thomson, J.M., Hemann, M.T., Hernando-Monge, E.,Mu,
D., Goodson, S., Powers, S., Cordon-Cardo, C., Lowe,S.W., Hannon,
G.J., and Hammond, S.M. (2005) Nature ,435 , 828-833.
[93] Hatfield, S.D., Shcherbata, H.R., Fischer, K.A., Nakahara,
K.,Carthew, R.W., and Ruohola-Baker, H. (2005) Nature , 435
,974-978.
[94] Ciafre, S.A., Galardi, S., Mangiola, A., Ferracin, M.,
Liu,C.G., Sabatino, G., Negrini, M., Maira, G., Croce, C.M.,
andFarace, M.G. (2005) Biochem. Biophys Res. Commun. , 334
,1351-1358.
[95] Pallante, P., Visone, R., Ferracin, M., Ferraro, A.,
Berlingieri ,M.T., Troncone, G., Chiappetta, G., Liu, C.G.,
Santoro, M.,Negrini, M., Croce, C.M., and Fusco, A. (2006)
Endocr.Relat. Cancer , 13 , 497-508.
[96] Lee, E.J., Gusev, Y., Jiang, J., Nuovo, G.J., Lerner,
M.R.,Frankel, W.L., Morgan, D.L., Postier, R.G., Brackett,
D.J.,
and Schmittgen, T.D. (2007) Expression profiling
identifiesmicroRNA signature in pancreatic cancer. Int. J. Cancer
120 , 1046-1054.
[97] Cimmino, A., Calin, G.A., Fabbri, M., Iorio, M.V.,
Ferracin, M.,Shimizu, M., Wojcik, S.E., Aqeilan, R.I., Zupo, S.,
Dono, M.,Rassenti, L., Alder, H., Volinia, S., Liu, C.G., Kipps,
T.J.,Negrini, M., and Croce, C.M. (2005) Proc. Natl. Acad. Sci.USA
, 102 , 13944-13949.
[98] He, L., He, X., Lim, L.P., de Stanchina, E., Xuan, Z.,
Liang,Y., Xue, W., Zender, L., Magnus, J., Ridzon, D., Jackson,
A.L., Linsley, P.S., Chen, C., Lowe, S.W., Cleary, M.A.,
andHannon, G.J. (2007) Nature , 447 , 1130-1134.
[99] Bommer, G.T., Gerin, I., Feng, Y., Kaczorowski, A.J.,
Kuick,R., Love, R.E., Zhai, Y., Giordano, T.J., Qin, Z.S.,
Moore,B.B., MacDougald, O.A., Cho, K.R., and Fearon, E.R.
(2007)Curr. Biol. , 17 , 1298-1307.
[100] Mack, G.S. (2007) Nat. Biotechnol. , 25 , 631-638.
Received: January 31, 2008 Revised: April 18, 2008 Accepted: May
01, 2008










![Direct Organogenesis from Cotyledonary Node Explants of ... · shoot organogenesis in C. peporeported [19] direct organogenesis in Cucumis sativus [20] and reported L. cy-lindrica](https://img.pdfslide.us/doc/110x75/5fac27dc76c37d66627b9b5d/direct-organogenesis-from-cotyledonary-node-explants-of-shoot-organogenesis.jpg)