Embed Size (px)
Citation preview

JOURNAL OF BACTERIOLOGY, Feb. 1978, p. 641-6490021-9193/78/0133-0641$02.00/0Copyright i 1978 American Society for Microbiology
Vol. 133, No. 2
Printed in U.S.A.
Microbial Oxidation of Methane and Methanol: Crystallizationof Methanol Dehydrogenase and Properties of Holo- and Apo-Methanol Dehydrogenase from Methylomonas methanica
RAESH N. PATEL,* CHING T. HOU, AND ANDRE FELIX
Corporate Research Laboratories, Exxon Research and Engineering Company, Linden, New Jersey 07036
Received for publication 11 August 1977
Procedures are described for the purification and crystallization of methanoldehydrogenase from the soluble fraction of the type I obligate methylotrophMethylomonas methanica strain Si. The crystallized enzyme is homogeneous asjudged by acrylamide gel electrophoresis and ultracentrifugation. The enzymehad a high pH optimum (9.5) and required ammonium salt as an activator. Inthe presence of phenazine methosulfate as an electron acceptor, the enzymecatalyzed the oxidation of primary alcohols and formaldehyde. Secondary, ter-tiary, and aromatic alcohols were not oxidized. The molecular weight as well assubunit size of methanol dehydrogenase was 60,000, indicating that it is mono-meric. The sedimentation constant (s2o,w) was 3.1S. The amino acid compositionof the crystallized enzyme is also presented. Antisera prepared against thecrystalline enzyme were nonspecific; they cross-reacted with and inhibited theisofunctional enzyme from other obligate methylotrophic bacteria. The crystallinemethanol dehydrogenase had an absorption peak at 350 nm in the visible regionand weak fluorescence peaks at 440 and 470 nm due to the presence of a pteridinederivative as the prosthetic group. A procedure was developed for the preparationofapo-methanol dehydrogenase. The molecular weights, sedimentation constants,electrophoretic mobilities, and immunological properties of apo- and holo-meth-anol dehydrogenases are identical. Apo-methanol dehydrogenase lacked the ab-sorption peak at 350 nm and the fluorescence peaks at 440 and 470 nm and wascatalytically inactive. All attempts to reconstitute an active enzyme from apo-methanol dehydrogenase, using various pteridine derivatives, were unsuccessful.
Methylotrophs are microorganisms that grownon-autotrophically on compounds containingone or more carbon atoms but no carbon-carbonbonds (3). There are both obligate and faculta-tive methylotrophs.The methane-utilizing bacteria are obligately
dependent on methane, methanol or dimethylether (obligate methylotrophs) as sole sourcesof carbon and energy for growth (7, 29). Re-cently, Patt et al. (19) isolated facultative meth-ane-ulizing bacteria that can also utilize morecomplex organic molecules as carbon and energysources. On the basis of the structural organiza-tion of their intracytoplasmic membrane andthe pathway of carbon assimilation, the meth-ane-utilizing bacteria are divided into two dis-tinct groups (5, 20, 22). Bacteria with the so-called type I membrane structure utilize a pen-tose phosphate pathway of formaldehyde fixa-tion for carbon assimilation and have an incom-plete tricarboxylic acid cycle. The type II mem-brane bacteria utilize a serine pathway for car-bon assimilation and have a complete tricarbox-
ylic acid cycle (4, 5, 20, 22).A soluble methanol dehydrogenase containing
a pteridine derivative as a prosthetic group wasfirst reported from a facultative methylotroph,Pseudomonas M27 (1). This methanol dehydro-genase uses only phenazine methosulfate (PMS)as its artificial electron acceptor and is activatedby ammonium ions (1). Subsequently, a similarsoluble methanol dehydrogenase was isolatedfrom a type I obligate methylotroph, Methylo-coccus capsulatus (15), and from various meth-anol-utliing bacteria (24). Recently, we crys-tallized methanol dehydrogenase from the typeII obligate methylotroph Methylosinus sporium(18).Wazinski and Ribbons (27) reported that M.
capsulatus contained particulate PMS-inde-pendent methanol oxidase, particulate PMS-de-pendent methanol dehydrogenase, and solublePMS-dependent methanol dehydrogenase activ-ities. The properties of the purified PMS-de-pendent methanol dehydrogenase activity in thesoluble fraction and that solubilized from partic-
641
on August 6, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

642 PATEL, HOU, AND FELIX
ulate fractions are similar, indicating that theymay be identical proteins (27).Methylomonas methanica strain Si is a type
I obligate methylotroph, containing particulatePMS-independent methanol oxidase as well as
soluble PMS-dependent methnol dehydrogen-ase activities (6). In this report, we describe thecUystallization and properties of the solublemethanol dehydrogenase from AM. methanica.We also report procedures for preparation andproperties of apo-methanol dehydrogenase.
MATEIALS AND METHODSBacterial trains. Methylomonas methnica
strains S1 and A4, Methylosuus sporium strain 5,Methylosinus trichosporium stains OB3b and PG,Methylococcus capsulatus strain TRMC, Methylo-bacter capsuatu strain Y, and Methylocystisparvusstrain OBBP were lindly provided by R. Whittenbury(School of Biological Sciences, Univeraity ofWarwick,Coventry, Warwickhe, Engla). The organ
were maintained on mineral salts (7) agar plates in adesiccator under an atmosphere of methane and air(1:1, vol/vol) at 30°C.Chemicals All alcohols were obtained from
Matheson Coleman and Bell Manufacting Co., Nor-wood, Ohio. Bio-Gel agarose A-1.5 was obtained from
Bio-Rad Laboratoriea Whatman microgranular DE-32 (diethylaminoethyl [DEAE])-cellulose was precy-cled according to the maufacturer's instructions priorto use. Acrylamide, bisacrylamide, and ammonium
persulfate were purchased from Canalco Inc., Rock-vile, Md. Aminoptern, phenazine methosulfate,pterin (2-amino-4-hydroxypteridine), and pterin--carboxylate (2-amino-4-hydroxypteridine-6-carboxylicacid) were obtained from Sigma Chemical Co., StLouis, Mo. Lumazine monohydrate (2,4-pteridinediol)and 2-amino-6,7-dimethyl-4-hydroxy-5,6,7,8-tetrahy-dropteridine hydrochloride were purchased from Ald-rich Chemical Co.Enzyme asays. Methanol dehydrogenase activity
was measured spectrophotometrically as well as polar-ographically as descibed previously (15). Specific ac-
tivities were expressed as nanomoles ofdichlorophenolindophenol reduced per minute per milligram of pro-tein in the spectrophotometric assays and as nano-
moles of oxygen consumed per minute per milligramof protein in polarographic anays Protein concentra-
J. BAcTERIOL
tions were determined by the method of Lowry et aL
(10).Growth of organisms. Small-scale cultures of
methane-utilizing were grown at 30°C in2.8-liter fls otainin 700 ml of mineral salts me-dium (7) with methane (mane and air, 1:1, vol/vol)or (0.4%, vol/vol) as sole carbon and energy
sources.
Large-scale cultures ofM. metwnica stain S1 weregrown with aeration at 30'C in a 100-liter New Bruns-wick Fermacell model CF 130 fermentor in a mineralsalts medium (7) containing methanol (0.4%, vol/vol)as the sole carbon source. Cells were harvested with
a refrigeratedSarplescenzifup andstoredat-20C.Purification of metanl dehydrogenase. AM
purification procedures were caTied out at 40C unlessotherwise stated. The cels (700 g, wet weight) were
suspended in 1 liter of 50 mM sodium phosphatebuffer, pH 7.0 (buffer A), and crude extracts were
prepared as descrbed previously (13). To the crudeeracts, 45 ml of protamine sulfate solution [2% so-
lution in 0.1 M tris(hydroymethyl) ne
(Tris) base] was added dropwise with continuous stir-
ing. After sanding for 30 min, the extracts werecentrifuged at 20,000 x g for 60 min. Tbe supernatantsolution (Table 1, step 2) was fractionated with solidammonium sulfate. Exracts were brought to 60% ofsaturation with respect to ammonium sulfate by ad-dit of 390 g of the salt per liter of extact Precipi-tated protein was removed by ceniuation, and 143g of ammonium sulfate was added per liter of thesupernatant liquid to bring it to 80% of saturation.Material pcipitating between 60 and 80% of satura-tion was collected by centigation and dissolved inbuffer A (Table 1, step 3). This preparation was di-alyzed overnight aginst buffer A, and the dialyzedmaterial was applied to a DEAE-ceulose column (5by 40 cm) that had been equilibrated with buffer A.The manol dehydrogenase activty that was elutedin the void volume (Table 1, step 4) wasdignated
DEAE-cellulose eluate. The DEAE-cellulose eluatewas concentrated by ammoniuim sulfate fractionation.Material precipitating between 60 and 80% of satura-tion with respect to ammonium sulfate was collectedby centrifugation and dissolved in buffer A. This prep-aration was dialyzed overnight against buffer A, and4-ml samples were passed through a Bio-Gel agaroseA-1.5 column (2.5 by 100 cm) that had been equili-brated with buffer A. The elution profile of methanol
TABLE 1. Purification ofmethanol dehydrogenaseafrom M. methanica
Volume Protein Sp ac YieldMaterial Vol e Units (U/mg Of )protein)
1. Crude extracts 2,250 17,000 340,000 20.0 1002. Protamine sulfate treatment 2,300 15,000 321,000 21.4 943. Ammonium sulfate fractionation (60-80% 180 4,000 304,000 76.0 89
saturation)4. DEAE-cellulose eluate 350 1,700 263,000 155 775. Bio-Gel chromatography 55 595 208,000 350 616. First crystallization 25 500 200,000 400 587. Second crystallization 20 450 180,900 402 53
a Methanol dehydrogenase actiVity was estimated spectrophotometrically.
on August 6, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
METHANOL DEHYDROGENASES FROM M. METHANICA
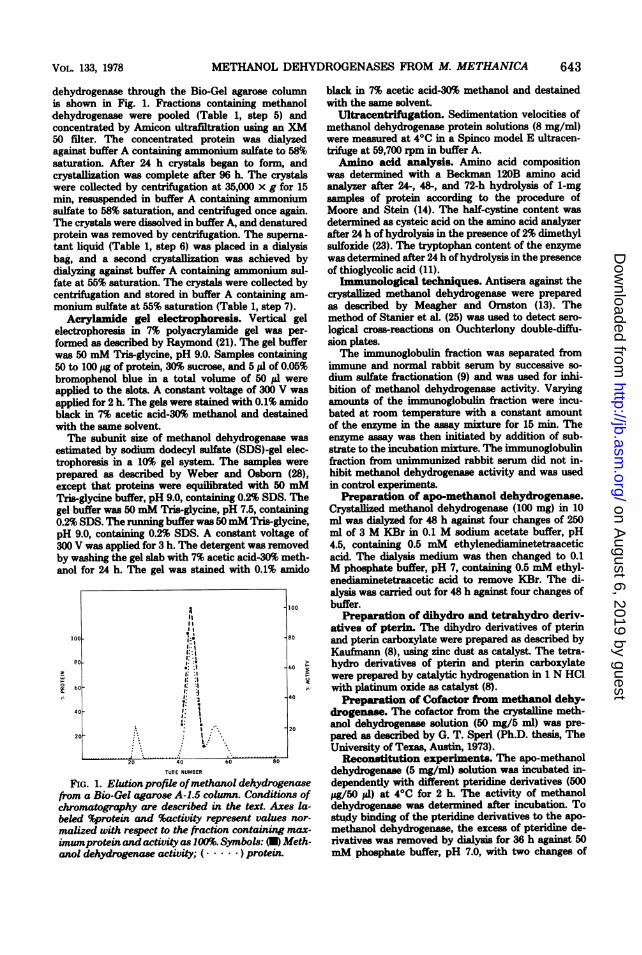
dehydrogenase through the Bio-Gel agarose columnis shown in Fig. 1. Fractions containing methanoldehydrogenase were pooled (Table 1, step 5) andconcentrated by Amicon ultrafiltration using an XM50 filter. The concentrated protein was dialyzedagainst buffer A containing ammonium sulfate to 58%msaturation. After 24 h crystals began to form, andcrystallization was complete after 96 h. The crystalswere collected by centrifugation at 35,000 x g for 15min, resuspended in buffer A containing ammomumsulfate to 58% saturation, and centrifuged once again.The crystals were dissolved in buffer A, and denaturedprotein was removed by centrifugation. The superna-
tant liquid (Table 1, step 6) was placed in a dialysisbag, and a second crystallization was achieved bydialyzing against buffer A containing ammonium sul-fate at 55% saturation. The crystals were collected bycentrifugation and stored in buffer A containing am-
monium sulfate at 55% saturation (Table 1, step 7).Acrylamide gel electrophoresis. Vertical gel
electrophoresis in 7% polyacrylamide gel was per-formed as described by Raymond (21). The gel bufferwas 50 mM Tris-glycine, pH 9.0. Samples containing50 to 100 ,ug of protein, 30% sucrose, and 5 pl of 0.05%bromophenol blue in a total volume of 50 I1 wereapplied to the slots. A constant voltage of 300 V was
applied for 2 h. The gels were stained with 0.1% amidoblack in 7% acetic acid-30% methanol and destainedwith the same solvent.The subunit size of methanol dehydrogenase was
estimated by sodium dodecyl sulfate (SDS)-gel elec-trophoresis in a 10% gel system. The samples were
prepared as described by Weber and Osborn (28),except that proteins were equilibrated with 50 mMTris-glycine buffer, pH 9.0, containing 0.2% SDS. Thegel buffer was 50 mM Tris-glycine, pH 7.5, containing0.2% SDS. The running buffer was 50mM Tris-glycine,pH 9.0, containing 0.2% SDS. A constant voltage of300 V was applied for 3 h. The detergent was removedby washing the gel slab with 7% acetic acid-30% meth-anol for 24 h. The gel was stained with 0.1% amido
loo1
w
a° 60
20
A11
1:,: . I*: :1,: :.1
.. I
~~~I
:: ~ ~~~:- *...~~~~~~~~
;t. ..~~~.
100
-80
20 40 60 00
TUBE NUMBER
FIG. 1. Elutionprofile ofmethanol dehydrogenasefrom a Bio-Gel agarose A-1.5 column. Conditions ofchromatography are described in the text. Axes la-beled %protein and %activity represent values nor-
malized with respect to the fraction containing max-imumprotein and activity as 100%. Symbols: (U) Meth-anol dehydrogenase activity; (. ) protein.
black in 7% acetic acid-30% methanol and destainedwith the same solvent.
UltracentrfUigation. Sedimentation velocities ofmethanol dehydrogenase protein solutions (8 mg/ml)were measured at 4°C in a Spinco model E ultracen-trifuge at 59,700 rpm in buffer A.Amino acid analysis. Amino acid composition
was determined with a Beckman 120B amino acidanalyzer after 24-, 48-, and 72-h hydrolysis of 1-mg
samples of protein according to the procedure ofMoore and Stein (14). The half-cystine content was
determined as cysteic acid on the amino acid analyzerafter 24 h of hydrolysis in the presence of 2% dimethylsulfoxide (23). The tryptophan content of the enzymewas determined after 24 h of hydrolysis in the presenceof thioglycolic acid (11).
Immunological techniques. Antisera against thecrystallized methanol dehydrogenase were preparedas described by Meagher and Ormston (13). Themethod of Stanier et al. (25) was used to detect sero-
logical cross-reactions on Ouchterlony double-diffu-sion plates.The immunoglobulin fraction was separated from
immune and normal rabbit serum by successive so-dium sulfate fractionation (9) and was used for inhi-bition of methanol dehydrogenase activity. Varyingamounts of the immunoglobulin fraction were incu-bated at room temperature with a constant amountof the enzyme in the assay mixture for 15 min. Theenzyme assay was then initiated by addition of sub-strate to the incubation mixture. The immunoglobulinfraction from unimmunized rabbit serum did not in-hibit methanol dehydrogenase activity and was usedin control experiments.
Preparation of apo-methanol dehydrogenase.Crystallized methanol dehydrogenase (100 mg) in 10ml was dialyzed for 48 h against four changes of 250ml of 3 M KBr in 0.1 M sodium acetate buffer, pH4.5, containing 0.5 mM ethyleneiaminetetraaceticacid. The dialysis medium was then changed to 0.1M phosphate buffer, pH 7, containing 0.5 mM ethyl-endfiaminetetraacetic acid to remove KBr. The di-alysis was carried out for 48 h against four changes ofbuffer.
Preparation of dihydro and tetrahydro deriv-atives of pterin The dihydro derivatives of pterinand pterin carboxylate were prepared as described byKaufmann (8), using zinc dust as catalyst. The tetra-hydro derivatives of pterin and pterin carboxylatewere prepared by catalytic hydrogenation in 1 N HCIwith platinum oxide as catalyst (8).Preparation of Cofactor from methanol dehy-
drogenase. The cofactor from the crystalline meth-anol dehydrogenase solution (50 mg/5 ml) was pre-pared as described by G. T. Sperl (Ph.D. thesis, TheUniversity of Texas, Austin, 1973).
Reconstitution experiments. The apo-methanoldehydrogenase (5 mg/ml) solution was incubated in-dependently with different pteridine derivatives (500jAg/50 id) at 40C for 2 h. The activity of methanoldehydrogenase was determined after incubation. Tostudy binding of the pteridine derivatives to the apo-
methanol dehydrogenase, the excess of pteridine de-rivatives was removed by dialysis for 36 h against 50mM phosphate buffer, pH 7.0, with two changes of
VOL. 133, 1978 643
on August 6, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

644 PATEL, HOU, AND FELIX
buffer. After dialyss, the absorption spectrum andmethanol dehydrogenase activity of the dialyzed so-lution were determined.
RESULTSPurification and properties of methanol
dehydrogenase fromM. methanic We havedeveloped procedures for the crystallization ofmethanol dehydrogenase from a soluble fractionof M. methanica (Table 1). During purification,methanol dehydrogenase was eluted in the voidvolume from a DEAE-cellulose column. Gel fil-tration on a Bio-Gel agarose A-1.5 column gavea major protein peak consisting of enzyme anda minor peak conng of a c-type cytochrome(Fig. 1). Similar results were obtained duringpurification of methanol dehydrogenase fromMethylococcus capsulatus (15) and Pseudomo-nas M27 (1), where cytochrome was separatedfrom methanol dehydrogenase in the final stepby gel filtration. A photomicrograph of the crys-talline methanol dehydrogenase showed that theenzyme crystallized in the shape of needles.The crystalline enzyme preparations migrated
as a single protein band when subjected to elec-trophoresis on polyacrylamide gel (Fig. 2). Byultracentrifugal analysis, the Schlieren profile ofthe methanol dehydrogenase revealed a singlesymmetrical peak, with a sedimentation con-stant (s2o,w) of 3.0S (Fig. 3).The purified methanol dehydrogenase re-
quired ammonium chloride as an activator. Nocatalytic activity was detected in {he absence ofammonium chloride. Activity of the enzyme in-creased as the anmonium chloride concentra-tion increasd up to 35 pamol per 3.0 ml of assaymixture. Further addition of ammonium chlo-ride neither increased nor inhibited activity.Ammonium chloride could be replaced by otherammonium salts as activator for methanol de-hydrogenase. The optimum pH for methanoloxidation was found to be 9.5, with 50 and 25%activity at pH 8.5 and 7.5, respectively. Amongvarious artificial electron acceptors tested, onlyphenazines (e.g., phenazine methosulfate, phen-azine ethosulfate) and phennium compounds(e.g., phenazinium ethosulfate, 5-methylphena-zinium methylsulfate) could act as a primaryhydrogen acceptor for the methanol dehydro-genase. Biological electron acceptors like nico-tinamide adenine dinucleotide, nicotinamide ad-enine dinucleotide phosphate, flavine adeninedinucleotide, and cytochrome c were not activeas electron carriers.The crystalline methanol dehydrogenase cat-
alyzed the oxidation of primary alcohols (Clthrough C10 tested), 3-chloro-l-propanoL 4-chloro-l-butanoL 6-chloro-1-hexanoL 2-phenox-yethanol, 2-methoxyethanoL 2-aminoethanoL
J. BACTERIOL.
.-J ..
U.:
*..I..*:
~~~Is~~~~~ ~~~~ss ilkillub.-llliilifM -16NNIO' 4-UNlNW Ii
FIG. 2. Vertical gel electrophoresis of apo- andholo-methanol dehydrogenases. Slots 1 and 2 re-ceived 50 pg and 100 pg of apoenzyme, respectively,and slots 3 and 4 received 50 pg and 100 pg ofholoenzyme, respectively. Slot 5 received 50 pg ofapoenzyme and 50 pg ofholoenzyme.
and formaldehyde. Propan-2-ol, butan-2-ol, oc-tan-2-ol, cyclohexanoL isobutanol, benzyl alco-hoL acetaldehyde, and propionaldehyde werenot oxidized (Table 2). Oxidation of formalde-hyde by methanol dehydrogenase presumablyis due to the fact that in aqueous solution form-aldehyde is more than 99% hydrated and ap-pears to the enzyme to be an analog of methanol(24). The methanol dehydrogenase from M.methanica was stable to acid treatment and didnot lose activity when the pH was lowered to4.0 with 0.1 N HCI. Activity was completely lostwhen the pH was lower than 3.3.The molecular weight of methanol dehydro-
genase was estimated by gel filtration to be60,000. The subunit size estimated by electro-phoresis on polyacrylamide in the presence ofSDS was 60,000. The amino acid content wasdetermined after 24-, 48-, and 72-h acid hydrol-
on August 6, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

METHANOL DEHYDROGENASES FROM M. METHANICA
ysis of the crstaline enzyme preparations. Thevalues are expressed as average number of resi-dues per molecule, assuming a molecular weightof 60,000 (Table 3).The visible and UV absorption spectra of the
purified enzyme preparations are shown in Fig.4. The UV spectrum represents a typical absorp-tion pattern of a protein solution. In the visiblerange, an absorption peak at 350 nrm was re-corded. The enzyme had weak fluorescencepeaks at 440 and 470 nm (Table 4). The emissionof the bound cofactor was different from thatof the cofactor in its free form. Studies on cofac-tor and circular dichroism of the methanol de-hydrogenase will be reported elsewhere.
Serological properties. The ability of anti-sera prepared against the crystafline methanoldehydrogenase from M. methanica strain Si tocross-react with isofunctional enzymes in thesoluble fraction from various obligate methylo-
FIG. 3. Ultracentrifugal pattern of the methanoldehydrogenase. Protein concentration was 6 mg/mlin 50mMphosphate buffer, pH 7.0. Photograph wastaken at 48 min after reaching 59,700 rpm at 4°C.Sedimentation was from right to left.
trophs was determined by immunodfffusiontechniques. Strong precipitinbandswere formed,and spur formation was not detected when themethanol dehydrogenases were derived from M.methanica strain S1 or A4 (Fig. 5). Thus, twostrains of M. methanica appear to form immu-nologically imilar enzymes. In contrast, meth-anol dehydrogenases from Methylosinus spor-ium strain 5, Methylosinus trichosporiumstrains OB3b and PG, Methylocystis parvusstrain OBBP, Methylobacter capsulatus strainY, and Methylococcus capsulatus strain TRMCformed weak precipitin bands, and methanoldehydrogenase from M. methanica strain S1formed a strong spur when it was placed in
TABLE 3. Amino acid composition of methanoldehydrogenase
Amino acid No. of residues/60,000daltonsa
Lysine ........ 45Histidine ........ 10Arginine ........ 12Aspartic acid .. ...... 56Threonine ........ ?4 bSerine .... ........... 12bGlutamic acid .. ...... 32Proline ........ 22Glycine ........ 50Alanine ........ 37Cysteic acid .. ...... 2Valine . .............. 26CMethionine .. ...... 8bIsoleucine .......... 15CLeucine........ 32cTyrosine ........ 10Phenylalanine ........ 14Tryptophan 6
a Average of 24, 48, and 72 h of hydrolysis.b Determined by extrapolation to zero-time hydrol-
ymB.c Average of 48 and 72 h of hydrolysis.
TABLE 2. Substrate specificity ofmethanol dehydrogenasea
Substrate Rate of oxidation Substrate Rate of oxidation
Methanol .......................... 100 2-Phenoxyethanol 79Ethanol ............................ 68 2-Methoxyethanol 74Propan-l-ol ......................... 65 2-Aminoethanol 0Butan-l-ol .......................... 63 Propan-2-ol ............. 0Pentan-l-ol ......................... 84 Butan-2-ol .............. 0Hexan-l-ol ......................... 84 Octan-2-ol ............... 0Heptan-l-ol ........................ 84 Isobutyl alcohol .......... 0Octan-l-ol .......................... 80 Cyclohexanol ............ 0Nonan-l-ol ......................... 84 Benzyl alcohol ........... 0Decan-l-ol ......................... 79 Formaldehyde ........... 683-Chloro-l-propanol ......... ........ 84 Acetaldehyde ............ 04-Chloro-l-butanol ......... ......... 74 Propionaldehyde ......... 06-Chloro-l-hexanol .......... ........ 63
aMethanol dehydrogenase was estimated spectrophotometrically. Reaction was started by addition of 20,umol of substrate and rates of dichlorophenol indophenol reduction were measured in the presence of PMS.
I
VOL. 133, 1978 645
on August 6, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
646 PAT11L, HOU, AND FELIX
z
oco< 0.5 -
200 300 400 500 600
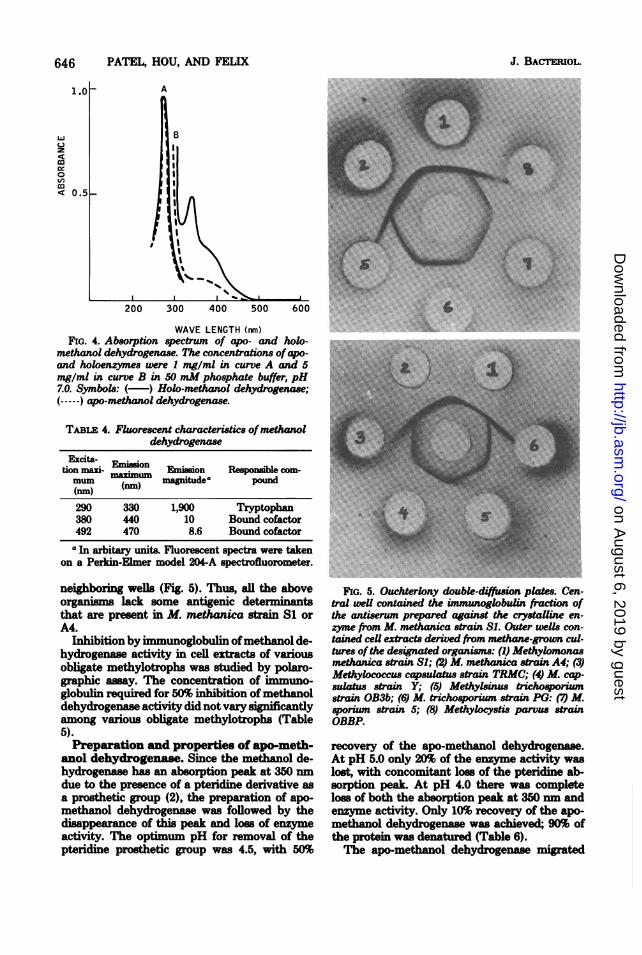
WAVE LENGTH (rnm)FIG. 4. Absorption spectrum of apo- and holo.
methanol dehydrogenase. The concentrations ofapo-and holoenzymes were 1 mg/ml in curve A and 5mg/ml in curve B in 50 mM phosphate buffer, pH7.0. Symbols: ( ) Holo-methanol dehydrogenase;(-----) apo-methanol dehydrogenase.
TABLE 4. Fluorescent characteristics ofmethanoldehydrogenase
Excita- Eisotion mai- Eimon Emisson Responsible com-mum m) magnitude* pound(nm) (m
290 330 1,900 Tryptophan380 440 10 Bound cofactor492 470 8.6 Bound cofactoraIn arbitary units. Fluorescent spectra were taken
on a Perkin-Elmer model 204-A spectrofluorometer.
neighboring wells (Fig. 5). Thus, all the aboveorganims lack some antigenic determinantsthat are present in M. methanica strain Si orA4.
Inhibition by immunoglobulin ofmethanol de-hydrogenase activity in cell extracts of variousoblUgate methylotrophs was studied by polaro-graphic assay. The concentration of immuno-globulin required for 50% inhibition ofmethanoldehydrogenase activity did not vary significantlyamong various obligate methylotrophs (Table5).Preparation and properties of apo-meth-
anol dehydrogenase. Since the methanol de-hydrogenase has an absorption peak at 350 nmdue to the presence of a pteridine derivative asa prosthetic group (2), the preparation of apo-methanol dehydrogenase was followed by thedisappearance of this peak and loss of enzymeactivity. The optimum pH for removal of thepteridine prosthetic group was 4.5, with 50%
FIG. 5. Ouchterlony double-ditsion plates. Cen-tral well contained the immunoglobulin fraction ofthe antiserum prepared against the crystalline en-zyme from M. methanica strain SI. Outer weUs con-tained ceU extracts derived from methane-grown cul-tures ofthe desinated organisms: (1) Methylomonasmethanica strain S; (2) M. methanica strain A4; (3)Methylococcus capsulatus strain TRMC; (4) M. cap-sulatus strain Y; (5) Methylsinus trichosporiumstrain OB3b; (6) M. trichosporium strain PG: (7) M.sporium strain 5; (8) Methylocystis parvus strainOBBP.
recovery of the apo-methnol dehydrogenase.At pH 5.0 only 20% of the enzyme activity waslost, with concomitant loss of the pteridine ab-sorption peak. At pH 4.0 there was completeloss of both the absorption peak at 350 nm andenzyme activity. Only 10% recovery of the apo-methanol dehydrogenase was achieved-; 90% ofthe protein was denatured (Table 6).The apo-methanol dehydrogenase migrated
J. BACTEP1OL.
C2
on August 6, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

METHANOL DEHYDROGENASES FROM M. METHANICA
TABLE 5. Inhibition ofmethanol dehydrogenase byimmunoglobulina
Concn of immuno-Obligate methylotroph globulin required for
50% inhibition (mg)Type IMethylomonas methanica 0.4
strain SiMethylomonas methanica 0.5
strain A4Methylobacter capsulatus 1.0
strain YMethylococcus capsulatus 1.2
strain TRMCType IIMethylosinus trichosporium 1.0
strain OB3bMethylosinus tricho8porium 1.5
strain PGMethylocystis parvus strain 1.2OBBP
a Methanol dehydrogenase was estimated spectro-photometrically.
TABLE 6. Effect ofpotassium bromide treatment onmethanol dehydrogenasea at variouspH values
Absorb- Methanol de- Apoen-pH ance at hydrogenase zyme re-
(mg) 350 nm activity (%) covery
7.0 10 0.98 1006.0 10 0.98 1005.0 10 0.76 714.5 10 0.0 0 504.0 10 0.0 0 10
a Crystallized methanol dehydrogenase (20 mg/2ml) was dialyzed for 48 h against four changes of 250ml of 3 M potassium bromide in 0.1 M sodium acetatebuffer containing 5 x 10' M ethylenediaminetetraace-tic acid at various pH values, as indicated. The dialysismedium was then changed to 0.1 M phosphate buffer,pH 7.0, containing 5 x 10-4 M ethylenediaminetetra-acetic acid to remove potassium bromide. After 48 hof dialysis, the dialysate was centrifuged and the su-pernatant solution was analyzed spectrophotometri-cally.
as a single protein band having electrophoreticmobility similar to that of holo-methanol dehy-drogenase when subjected to electrophoresis onpolyacrylamide gel (Fig. 2). By ultracentrifugalanalysis, the Schlieren profile of the apo-meth-anol dehydrogenase revealed a single peak witha sedimentation constant (s2o,,w) of 3.OS, similarto that of holo-methanol dehydrogenase. Theapo-methanol dehydrogenase peak was elutedin the same fractions as holo-methanol dehydro-genase when passed through a Bio-Gel agaroseA-1.5 column with a molecular weight of 60,000.Immunologically, the apo- and holo-methanol
dehydrogenases were found to be homologous,since they gave precipitin bands of completeidentity and no spur formation was detectedwhen examined by the immunodiffusion tech-mque on Ouchterlony double-diffusion plates(Fig. 6). The absorption spectrum of apo-meth-anol dehydrogenase is shown in Fig. 4. In thevisible region, the loss of the absorption peakat 350 nm indicates removal of the pteridineprosthetic group. The apo-methanol dehydro-genase did not have fluorescence peaks at 440and 470 nm.Attempts to reconstitute apo-methanol
dehydrogenase. AU attempts to reconstituteapo-methanol dehydrogenase using variouspteridine derivatives have been unsuccessfuLApo-methanol dehydrogenase (5 mg/ml) wasincubated for 2 h at 4°C with pterin, pterincarboxylate, dihydropterin, dihydropterin car-boxylate, tetrahydropterin, tetrahydropterincarboxylate, lumazine monohydrate, 2-amino-6,7-dimethyl-4-hydroxy-5,6,7,8-tetrahydropteri-dine hydrochloride (500 ug each), and cofactorextracted from the purified enzyme independ-ently in different tubes. No catalytic activity orabsorption at 350 nm was observed after dialyz-ing out the excess of pteridine derivative, indi-cating that no binding of the pteridine deriva-tives to the apoenzyme had occurred.
DISCUSSIONCrystalline methanol dehydrogenase from the
type I obligate methylotroph M. methanica re-quired ammonium chloride as an activator and
FIG. 6. Ouchterlony double-diffusion plates. Cen-tral well contained the imnumoglobulin fraction ofthe antiserum prepared against the crystalline en-zyme from M. methanica strain SI. Outer wells 1, 3,and 5 contained holo-methanol dehydrogenase, andwells 2, 4, and 6 contained apo-methanol dehydro-genase.
VOL. 133, 1978 647
on August 6, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

648 PATEL, HOU, AND FELIX
catalyzed the phenazine methosulfate-depend-ent oxidation of all primary alcohols tested. Therate of oxidation of primary alcohols varied from65 to 85% that of methanol. The purified enzymealso catalyzed the oxidation of substituted pri-mary alcohols and formaldehyde, but was unableto oxidize secondary, tertiary, and aromatic al-cohols.The crystalline enzyme has a molecular
weight of 60,000 and subunit size of 60,000, in-dicating that it is a monomer. Similar molecularweight and subunit size were estimated for meth-anol dehydrogenase from the type HI obligatemethylotroph Methylosinus sporium (16).Methanol dehydrogenases from Methylococcuscapsulatus (15), Pseudomonas M27 (1), andother methanol utilizers (24) have molecularweights of 120,000 and contain subunits of 60,000molecular weight. The sedimentation constant( s2o,w) of the purified enzyme from M. methanicaat pH 7.0 is 3.OS. It has been reported that thesedimentation constant (s2o,w) of methanol de-hydrogenases from M. capsulatus and Pseu-domonas M27 at pH 7.0 is 7.2S (15). The meth-anol dehydrogenase from M. methanica is anacid-stable protein; it does not lose activity atpH 4.0. The methanol dehydrogenases fromPseudomonas M27 (1) and M. capsulatus aresimilarly acid stable. In contrast, the purifiedmethanol dehydrogenase from M. sporium (16)and Pseudomonas Wi (24) lost all activity onlowering the pH to 5.0.The purified enzyme from M. methanica has
an absorption peak at 350 nm. A similar absorp-tion peak was recorded for methanol dehydro-genases from other methane- and methanol-uti-lizing organisms (1, 15, 24, 27). The purifiedenzyme exhibits fluorescence peaks at 440 and470 nm. It has been reported by Anthony andZatman (2) that the purified enzyme from Pseu-domonas M27 does not fluoresce, but green flu-orescent material diffusible on dialysis is pro-duced when the enzyme is treated with acid oralkali or when it is boiled. On the basis of thefluorescence and absorption spectra, the pros-thetic group of methanol dehydrogenase hasbeen characterized as a pteridine derivative (2).Two possible mechanisms have been proposedfor the involvement of a pteridine prostheticgroup. In the first, the pteridine acts directly asa hydrogen acceptor; in the second, the hydrogentransfer function of the pteridine derivative islinked to its role as a Cl carrier. Urushibara etal. (26) demonstrated that pteridine present inthe methanol dehydrogenase is not a folate de-rivative and isolated three pteridines from theM. capsulatus: 2-amino-4-hydroxy-6-carboxy-pteridine, 2-amino-4-hydroxy-6-methylpteri-dine, and L-threoneopterin 2',3'-phosphate. Re-
cently, Sperl et al. (24) presented evidence thatthe pteridine in methanol dehydrogenase fromM. capsulatus may be a 2,4-dihydroxypteridine(lulmaine).There has been no previous report on the
removal of the prosthetic group and the prepa-ration of apo-methanol dehydrogenase. We havefor the first time prepared apo-methanol dehy-drogenase fromM. methanica, using a techniqueanalogous to that used for the removal of flavinfrom flavoproteins (12). The physicochemicaland immunological properties of apo- and holo-methanol dehydrogenases are practically iden-tical, indicating that the apoenzyme is not de-natured or degraded protein. So far, however,all attempts to reconstitute the apoenzyme usingvarious pteridines have been unsuccessful. Itmay be that a slight change in structure of theenzyme on removal of the prosthetic group dis-turbs the binding site for the pteridine or thata change in structure of the prosthetic groupupon release from the holoenzyme makes it dif-ficult to reconstitute the apoenzyme.
Antisera prepared against the purified enzymefrom M. methanica cross-reacted and inhibitedisofunctional enzymes from both type I and typeII obligate methylotrophs. In contrast, antiseraprepared against the methanol dehydrogenasefrom another type I obligate methylotroph, M.capsulatus (15), cross-reacted only with en-zymes from type I obligate methylotrophs.
ACKNOWLEDGMENTEWe thank Allen I. Laskin for his continued encouragement
during this work and assistance in preparing this manuscript.We gratefully acknowledge R Whittenbury for providing uscultures of methane-utilizing bacteria and L. N. Onaton forallowing use of fermentor facilities.
LITERATURE CITED1. Anthony, C., and L J. Zatman. 1967. The microbial
oxidation of methanol. Purification and properties ofthe alcohol dehydrogenase of Pseudomonas M27. Bio-chem. J. 104:963-959.
2. Anthony, C., and L J. Zatman 1967. The microbialoxidation of methanol. The prosthetic group of thealcohol dehydrogenase of Pseudomonas M27: a newoXidoreductase prosthetic group. Biochem. J.104:960-969.
3. Colby, J., and L J. Zatman. 1972. Hexose phosphatesynthase and tricarboxylic acid cycle enzymes in bac-terium 4B6, and obligate methylotroph. Biochem. J.128:1373-1376.
4. Davey, J. F., R. Whittenbury, and J. F. Wilkinson.1972. The distribution in the methylobacteria of somekey enzyme concerned with intermediary metabolism.Arch. Mikrobiol. 87:359-66.
5. Davis, 8. L, and R. Whlttenbury. 1970. Fine strctureof methane and other hydrocarbon utilizing bacteria.J. Gen. Microbiol. 61:227-232.
6. Ferenci, T., T. Strom, and J. R. Quayle. 1975. Oxida-tion of carbon monoxide and methane by Pseudomonasmethanica. J. Gen. Microbiol. 91:79-91.
7. Foster, J. W., and R. IL Davis. 1966. A methane de-pendent coccus, with notes on classification and nomen-
J. BACTERIOL.
on August 6, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

METHANOL DEHYDROGENASES FROM M. METHANICA
clature of obligate methane-utilizing bacteria. J. Bac-terioL 91:1924-1931.
8. Kaufmann, S. 1967. Metabolism of the phenylalaninehydroxylation cofactor. J. Biol. Chem. 242:3934-3943.
9. Kekwick, R. A. 1940. The serum proteins in multiplemyelomatois. Biochem. J. 34:1248-1257.
10. Lowry, 0. IL, N. J. Rosebrough, A. L Farr, and R.J. Randall. 1951. Protein measurement with the Folinphenol reagent. J. Biol. Chem. 193:266-275.
11. MatMubara,M., and R. PL Sa8akl 1969. High recoveryof tryptophan from acid hydrolysis of proteins. Bio-chem. Biophys. Res. Commun. 35:175-181.
12. Mayhew, S. G. 1971. Studies on the binding of FMNand other flavin compounds to apo flavodoxin, p.185-209. In H. Kamin (ed.), Flavins and flavoproteins.University Park Press, Baltimore.
13. Meagher, R. B., and L N. Ornton. 1973. Relationshipamong enzymes of the 8-ketoadipate pathway. I. Prop-erties of cim, cisnmuconate lactonig enzyme and mu-conolactone isomerase from Pseudomonasputida. Bio-chemistry 12:3523-3530.
14. Moore, S., and W. IL Stein. 1963. Chromatographic# determiination of amino acids by the use of automatic
recording equipment. Methods Enzymol. 3:819.15. Patel, R. N., IL R. Bowe, W. J. Mandy, and D. S.
Hoare. 1972. Physiological studies on methane andmethanol-oxidizing bacteria: comparison of a primaryalcohol dehydrogenase from Methylococcu capsulatus(Texas strain) and Pseudomonas M27. J. Bacteriol.110:570-577.
16. Patel, R. N., and A. Felix. 1976. Microbial oxidation ofmethane and methanol: crystallization and propertiesof methanol dehydrogenase from Methylosinus spor-wun. J. Bacteriol. 118:413-424.
17. Patel, R. N., S. L Hoare, D. S. Hoare, and B. F.Taylor. 1975. Incomplete tricarboxylic acid cycle in atype I methylotroph, Methylococcus capsadatus. J. Bac-teriol. 123:382-384.
18. Patel, R. N., W. J. Mandy, and D. S. Hoare. 1973.Physiological studies of methane and methanol-oxidiz-
ing bacteria: immunological comparison of a primaryalcohol dehydrogenase from Methylococcus capsulatusand Pseudomonas M27. J. Bacteriol. 113:937-945.
19. Patt, T. E., G. C. Cole, J. Bland, and RI S. Hanson.1974. Isolation and characterization of bacteria thatgrow on methane and organic compounds as solesources ofcarbon and energy. J. Bacteriol. 120:955-964.
20. Quayle, J. R. 1972. The metabolism of C-1 compoundsby microorganisms. Adv. Microb. Physiol. 7:119-203.
21. Raymond, S. 1962. A convenient apparatus for verticalgel electrophoresis. Clin. Chem. 8:455-470.
22. Ribbons, D. W., J. E. Harrison, and A. M. Wadzinski.1970. Metabolism of single carbon compounds. Annu.Rev. Microbiol. 24:135-158.
23. Spencer, R. L, and F. Wold. 1969. New convenientmethod for estimation of total cystine and cysteine inprotein. Anal. Biochem. 32:185-190.
24. SperL G. T., IL S. Forrest, and D. T. Gibson. 1974.Substrate specificity of the purified primary alcoholdehydrogenases from methanol-oxidizing bacteria. J.Bacteriol. 118:541-550.
25. Stanier, R. Y., D. Wachter, C. Gasser, and A. C.Wilson. 1970. Comparative immunological studies oftwo Pseudomonas enzymes. J. Bacteriol. 102:351-362.
26. Urushibara, T., H. S. Forrest, D. S. Hoare, and R.N. PateL 1971. Pteridines produced by Methylococcuscapsudatus. Isolation and identification of a Neopterin2':3'-phosphate. Biochem. J. 125:141-146.
27. Wadznki, A. M., and D. W. Ribbons. 1975. Oxidationof C-1 compounds by particulate fraction from Methy-lococcus capsulatus properties of methanol oxidaseand methanol dehydrogenase. J. Bacteriol. 122:1364-1374.
28. Weber, K., and M. Osborn. 1969. The reliability ofmolecular weight determination by dodecyl sulfatepolyacrylamide gel electrophoresis. J. Biol. Chem.244:4406-4412.
29. Whittenbury, RI, K. C. Phillips, and J. F. Wilkinson.1970. Enrichment, isolation and properties of methaneutilizing bacteria. J. Gen. Microbiol. 61:205-218.
VOL. 133, 1978 649
on August 6, 2019 by guest
http://jb.asm.org/
Dow
nloaded from