Embed Size (px)
Citation preview

The Journal of Immunology
Mice Producing Less Reactive Oxygen Species Are RelativelyResistant to Collagen Glycopeptide Vaccination againstArthritis
Tsvetelina Batsalova, Balik Dzhambazov, Dorota Klaczkowska, and Rikard Holmdahl
The bottleneck for the induction of collagen-induced arthritis in mice is the recognition of immunodominant type II collagen (CII)
peptide (CII259-273) bound to the MHC class II molecule Aq. We have shown previously that the posttranslationally glycosylated
lysine at position 264 in this epitope is of great importance for T cell recognition and tolerance induction to CII as well as for
arthritis development. The Ncf1 gene, controlling oxidative burst, has been shown to play an important role for immune tolerance
to CII. To investigate the effect of oxidation on the efficiency of immune-specific vaccination with MHC class II/glycosylated–CII
peptide complexes, we used Ncf1 mutated mice. We demonstrate that normal reactive oxygen species (ROS) levels contribute to
the establishment of tolerance and arthritis protection, because only mice with a functional oxidative burst were completely
protected from arthritis after administration of the glycosylated CII259–273 peptide in complex with MHC class II. Transfer of
T cells from vaccinated mice with functional Ncf1 protein resulted in strong suppression of clinical signs of arthritis in B10.Q
mice, whereas the Ncf1 mutated mice as recipients had a weaker suppressive effect, suggesting that ROS modified the secondary
rather than the primary immune response. A milder but still significant effect was also observed in ROS deficient mice. During the
primary vaccination response, regulatory T cells, upregulation of negative costimulatory molecules, and increased production of
anti-inflammatory versus proinflammatory cytokines in both Ncf1 mutated and wild type B10.Q mice was observed, which could
explain the vaccination effect independent of ROS. The Journal of Immunology, 2010, 185: 2701–2709.
Rheumatoid arthritis (RA) is a chronic autoimmune diseasewith unknown etiology that affects ∼0.5% of the pop-ulation worldwide. It is characterized by inflammation of
peripheral joints and bone destruction due to synovial tissue in-filtration with immune cells, secretion of proinflammatory cyto-kines, and matrix metalloproteases (1). A specific prognostic markerfor RA development is the presence of Abs against citrullinatedproteins (ACPAs) and rheumatoid factors (RFs; anti-IgG Fc) (2). Ithas been shown that ACPAs together with certain MHC class II(MHC II) and PTPN22 alleles are strong predictive factors arguingfor a role of adaptive immunity in the disease pathogenesis (3). TheMHC II region, coding a shared amino acid motif in the peptide-binding pocket of the b-chain of HLA-DR4 and DR1 haplotypes(the shared epitope hypothesis) (4, 5) is the strongest genetic factor.This epitope includes amino acid residues 67, 70, 71, 74, which aresimilar in all RA-associated HLA-DR alleles and have crucial im-portance in determining the pocket structure as well as the T cellrepertoire and its ability to recognize self peptides (4).T cells are believed to play a central role in RA pathogenesis. Im-
portant arguments for this are the MHC II association, the presence
of T cell-dependent ACPA and RF IgG in the serum, and T cell-derived cytokines in the synovial fluid and serum of patients withRA. Moreover, T lymphocytes is a large fraction of the cellular infil-trate, could be∼40%, in the inflamed synovium (6). It has been shownthat the specificity of this milieu could be directed toward differentautoantigens—ubiquitously expressedmolecules, such as glucose-6–phospoisomerase, heterogenous nuclear ribonucleoprotein-A2, thestress protein BiP (7–9), citrullinated filaggrin and vimentin (10),or joint-specific proteins (collagen II, aggrecan, human cartilagegp39) (11–14). Despite decades of intensive research, a single dis-ease-causative autoantigen has not been identified yet. One of thesecandidate autoantigens, collagen type II (CII), is the major proteinin hyaline cartilage and is of particular interest because immuniza-tion with CII induces collagen induced arthritis (CIA), the mostcommonly used animal model for RA.Research based on CIA has been proved useful for studies of
pathologic mechanisms relevant to RA development and in vali-dating novel therapies, such as anti–TNF-a treatment (15).Importantly, an MHC II (Abeta) gene has been positioned that isa major factor for development of CIA in mice, and the mostsusceptible allele is Aq, which has a peptide-binding pocket similarto the shared epitope in humans (16–18). Autoreactive T cells inboth RA and CIA are directed toward the same immunodominantCII peptide 259–273 (14, 19, 20), which binds both Aq and sharedepitope human MHC II molecules. The lysine residue at positions264 and 270 in this epitope could be posttranslationally modified byhydroxylation and subsequent glycosylation with monosaccharidesor disaccharides. It has been demonstrated that glycosylation of thelysine side chains at position 264 is of particular importance forCIA development (21–23). In addition, we have previously shownthat CIA in Aq mice could be blocked by therapeutic administrationof soluble MHC II molecules in complex with the galactosylatedCII peptide 259–273 (24). Furthermore, we have recently demon-strated the important role of reactive oxygen species (ROS) in
Division of Medical Inflammation Research, Department of Medical Biochemistryand Biophysics, Karolinska Institute, Stockholm, Sweden
Received for publication February 4, 2010. Accepted for publication June 16, 2010.
This work was supported by grants from the King Gustaf V’s 80-Year Foundation,the Anna-Greta Craaford Foundation, and European Union Grants Autocure(LSHB-2006-018661) and MUGEN (LSHG-CT-2005-005203).
Address correspondence and reprint requests to Prof. Rikard Holmdahl, Division ofMedical Inflammation Research, Department of Medical Biochemistry and Biophys-ics, Karolinska Institute, SE-17177 Stockholm, Sweden. E-mail address: [email protected]
Abbreviations used in this paper: ACPA, Ab against citrullinated proteins; CII, col-lagen type II; CIA, collagen-induced arthritis; MHC II, MHC class II; Ncf1*/*, Ncflmutated; RA, rheumatoid arthritis; ROS, reactive oxygen species.
Copyright� 2010 by The American Association of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1000385
on August 19, 2010
ww
w.jim
munol.org
Dow
nloaded from

regulating autoimmune reactions and tolerance induction in animalmodels of autoimmunity (25–28). This finding came from the po-sitional cloning of the Ncf1 gene (coding p47phox subunit of theNADPH oxidase complex) and the identification of Ncf1 polymor-phism in the DA rat strain, resulting in increased susceptibility toarthritis because of low production of ROS. The discovery was laterconfirmed in mice, where a mutation in the same gene leads tosignificantly reduced levels of ROS, highly increased autoimmu-nity, impaired tolerance to CII, and more severe arthritis (27, 28).In this study, we investigated the importance of ROS for the
effectiveness of our vaccination therapy. We show that CIA in B10.Q mice with reduced ROS production cannot be ameliorated byMHC-II/CII–peptide treatment, which implicates a new importantaspect in vaccine development.
Materials and MethodsMice
Ncf1 mutated (Ncf1p/p) mice on the B10.Q background were used and de-scribed previously (28). A point mutation in exon 8 (29) of the Ncf1 gene ina B6 strain was introduced to the B10.Q/rhd strain by backcrossing for 13generations, resulting in a genetically clean strain as checked with 10 kilo-base single nucleotide polymorphism markers. B10.Q/rhd mice with a func-tional Ncf1 gene were used as a control in all experiments. B10.Q mice wereoriginally provided by J. Klein (Tubingen, Germany), but were kept in ouranimal house for .10 y. The animals were age- (8–12 wk old) and sex-matched; they were bred and kept at the animal facility of Medical Inflam-mation Research (Lund University, Karolinska Institute) under temperature-,light- and food-controlled conditions. The Lund-Malmo or Stockholmethical committee approved all animal experiments. All experiments wereblinded, and the mice of different experimental groups were mixed in cagesto avoid cage-dependent effects.
Ags
Rat collagen type II (CII) was prepared from Swarm chondrosarcoma vialimited pepsin digestion as described previously (30). The protein was dis-solved in 0.1 M acetic acid and stored at 4˚C. The following CII259–273peptides were used: K264 (unmodified rat CII259–273 with lysine at position264; GIAGFKGEQGPKGEP) and GalOK264, denoted also as Gal264(CII259–273 with b-D-galactopyranosyl-5-hydroxy-L-lysine at position 264).The synthesis and purification of these peptides were described previously(31, 32). The MOG79–96 peptide (mouse myelin oligodendrocytic glyco-protein; GKVTLRIQNVRFSDEGGY) was produced by Schafer-N (Copen-hagen, Denmark). All peptides were dissolved in PBS and kept at 4˚Cprior to use. C1 (CII359–369; ARGLTGRPGDA) and U1 (CII494–504;LVGPRGERGFP) peptides were synthesized by Schafer-N, dissolved in 0.1M acetic acid and stored at 4˚C. Con A was purchased from Sigma-Aldrich(St. Louis, MO), dissolved in PBS, sterile filtered, and kept at –20˚C until use.
Purification of soluble Aq molecules and preparation ofAq–peptide complexes
The design of the constructs for Aq a- and b-chains, their cotransfection inSL2Drosophila cells (CRL-1963; American Type Culture Collection, Man-assas, VA), and establishing a stably transfected cell line were describedpreviously (24). For large-scale expression of soluble Aq, the transfectedcells were expanded in serum-free Insect Express medium (PAA Laborato-ries, Pasching, Austria) and protein expression was induced using 0.7 mMCuSO4 for 4 d at 25˚C. The cell culture medium was then collected andcleared from cell debris by centrifugation and filtration. Soluble Aq mole-cules were purified from the derived supernatant by nickel–nitrilotriaceticacid affinity chromatography using the manufacturer’s protocol. The proteinfractions were concentrated with Amicon centrifugal filter devices (Milli-pore, Bedford, MA) and evaluated for recombinant protein content andquality by ELISA, SDS-PAGE, and Western blot analysis. The proteinwas then incubated with 5- to 10-fold molar excess of GalOK264, K264or MOG79–96 peptides for 4 d at 4˚C. MHC–peptide complexes were pu-rified by anion-exchange HPLC (ResourseQ column, purchased from GEHealthcare Bio-Sciences, Uppsala, Sweden) using AKTA explorer 100 Airsystem (Amersham Biosciences, Uppsala, Sweden) and UNICORN V4.00software. The separation was performed using increasing elution gradientup to 1 M NaCl in 100 mM Tris buffer. The positive protein fractionswere concentrated, dialyzed against PBS, and purified further with Superdex200 gel filtration column (Amersham Biosciences). Finally, the fractionswith correct protein mass were concentrated and stored at 4˚C until use.
Vaccination protocol
Intravenous injections were given through the tail vein. Mice received 100mg protein complex (Aq/GalOK264 or Aq/K264) in 200 ml PBS on days 20and 34 postimmunization. The control groups were injected with eitherPBS or 100 mg Aq/MOG in the same volume (200 ml) and time.
CII immunization and CIA evaluation
To induce CIA, all mice were injected in the skin at the base of the tail with100 ml emulgate, prepared by mixing 1:1 CFA (Difco/Beckton Dickinson,Sparks, MD) and 100mg CII. After 35 d, the animals received a 50-ml boosterinjection of 50-mg CII emulsified 1:1 with immunofluorescence assay (Difco/Beckton Dickinson). Clinical signs of arthritis were evaluated using a60 points system, giving a maximum score of 15 points per paw. Themice were examined two to three times per week for at least 70 d, and bloodwas collected at days 35 and 70 postimmunization.
Anti-CII Abs measurement
Serum was collected from all blood samples after centrifugation at 6500rpm for 20 min, diluted 10 times in PBS, and stored at220˚C. The levels ofanti-CII Abs were measured by quantitative ELISA. For this aim, 96-wellplates (Costar plates, Corning, Lowell, MA) were coated overnight with50 ml per well of rat CII (10 mg/ml solution in phosphate buffer, pH 9).The plates were then washed three times with PBS, containing 0.1%Tween 20, and nonspecific binding to the plastic was blocked using 100ml per well 2% PBS solution of nonfat milk for 1 h at room temperature.After washing, samples were added to the plate in serial dilutions rangingfrom 1:100 to 1.105 and incubated for 2 h at room temperature. To detectspecific anti-CII Abs 50 ml per well solution of 1 mg/ml goat anti-mouseIgG (H+L chain) HRP-conjugated Ab (Jackson ImmunoReseach Labora-tories, West Grove, PA) was added for 1 h at room temperature. The plateswere washed five times and developed using the ABTS system (RocheDiagnostic Systems, Somerville, NJ) and absorbance was measured atthe 405 nm wavelength with Wallac Victor 1420 multilabel counter (Per-kinElmer, Wellesley, MA). Pooled polyclonal serum from DBA1 mice withknown anti-CII Abs concentration was added in serial dilutions to all platesas positive control.
CII epitope-specific ELISAwas done using Costar plates coated for 2 h atroom temperature with either 4 mg/ml C1 peptide or U1 peptide. The block-ing and sample additions were performed as already described; 50 ml perwell rat anti-mouse IgG k biotinylated Ab (1 mg/ml concentration, clone187.1 from our Ab collection) was added to the plates after washing. Thepresence of anti-CII epitope specific Abs was revealed via the dissociation-enhanced lanthanide fluorescent immunoassay system using Eu3+-labeledstreptavidin (Wallac, Turku, Finland), and time-resolved fluorescence wasmeasured with Wallac Victor 1420 ELISA reader (PerkinElmer). Mono-clonal C1- or U1-specific Abs in serial dilutions were included to each plateas a positive control.
Cytokine ELISA and T cell assays
To determine the T cell response, mice were sacrificed 25 and 50 d afterimmunization, corresponding to day 5 after the first vaccination injectionand day 16 after the second vaccination injection, respectively. Lymph nodeand spleen cells (1 3 106 cells/well) were collected and restimulatedin vitro with 5 mg/ml ConA in DMEM (Life Technologies, Rockville, MD)supplemented with 10% heat-inactivated FCS, 100 IU/ml penicillin, and100 mg/ml streptomycin. After 96 h, cell-free supernatant was collectedand stored at –20˚C until use.
IL-10 and IL-17 cytokines levels were measured by sandwich ELISA.Ninety-six–well Costar plates were coated for 2 h at room temperaturewith 50 ml per well anti-mouse IL-10 or IL-17 mAbs in a concentrationof 2 mg/ml and 1 mg/ml, respectively. After blocking and washing, 50 mlsupernatant from ConA-stimulated lymph node and spleen cells were addedto the plates in duplicates or triplicates. The samples were incubated at roomtemperature for 2 h. After washing three times with PBS 0.1% Tween 20,anti-mouse biotinylated IL-10 or IL-17 Abs (1 mg/ml) were used for de-tection of the respective cytokines. The plates were developed with Eu3+-labeled streptavidin and fluorescence was measured as denoted in the pre-vious section. Recombinant mouse IL-10 and supernatant from ConA-stimulated splenocytes with known IL-17 concentration were added to eachplate as positive controls for the IL-10 and IL-17 ELISA, respectively.
T cell transfer
Two donor groups of B10.Qmicewere vaccinated i.v. (day 0) with 200mg ofeither Aq/GalOK264 or Aq/MOG in a total volume of 100 ml PBS withoutCa2+ and Mg2+ (Life Technologies). The animals were sacrificed 5 d later;spleens were harvested and pooled in two groups according to the type
2702 REACTIVE OXYGEN SPECIES INFLUENCE THE VACCINATION EFFECT
on August 19, 2010
ww
w.jim
munol.org
Dow
nloaded from

of treatment. The organs were passed through a 40-mm cell strainer (BDDiscovery, Labware, Sparks, MD), and erythrocytes were lysed using 0.84%NH4Cl. The cells were washed again with PBS without Ca2+ and Mg2+, andT lymphocytes were isolated via negative selection. Rat anti-mouse MHCII Abs (M5/114; BD Pharmingen) were added to the cell suspension andincubated for 20 min. The unbound Abs were then washed out and anti-ratIgG Dynabeads (Dynal Biotech, Oslo, Norway) were incubated withthe cells according to the manufacturers recommendations. After magneticsorting, the cells were washed and resuspended (5 3 105 cells/ml) in PBSwithout Ca2+ and Mg2+ (Life Technologies). The purity of the cell suspen-sion was evaluated by flow cytometry and was found to be 95% CD3expressing cells.
At the same time (day 0), four recipient groups (2 groups of B10.Q and2 groups of B10.Q Ncf1p/p mice) were immunized with CII in CFA andevaluated for arthritis development. On day 5 postimmunization, the animalsreceived an i.v. injection of 200 ml with 13 106 T cells: one group of B10.Qand one group of B10.Q Ncf1p/pmice got T cells from Aq/GalOK264-treatedanimals, the other two groups (B10.Q and B10.Q Ncf1p/p) received T cellsfrom Aq/MOG-treated mice. The mice were bled and evaluated for clinicalsigns of arthritis.
Flow cytometry analysis
The following Abs labeled with different fluorochromes or biotin were usedfor staining of surface epitopes: anti-mouse CD4 (clone L3T4, Southern Bio-technolgy Associates, Birmingham, AL), anti-mouse CD25 (clone PC61,eBioscience, Hatfield, U.K.), anti-mouse CD3 (clone 145-2C11; BD Phar-mingen), anti-mouse CD8 (clone 53-6.7; BD Pharmingen), anti-mouseMHC II (clone 7.16.17; BD Pharmingen), anti-mouse PD-1 (clone RMP1-30, BioLegend, San Diego, CA). Anti-FcRIIε Ab (clone 2.4.G2 from our Abcollection) was used prior to staining with the aim of blocking unspecificbinding to Fc receptors. Anti-Foxp3 (NRRF-30; eBioscience) and anti–CTLA-4 (UC10-4f10-11; BD Pharmingen) fluorochrome-conjugated Abswere used for intracellular staining.
Erythrocyte-free blood cells and lymph node cells pooled with erythrocyte-free spleen cells were analyzed by flow cytometry. The cells were washed,resuspended in PBS supplemented with 0.5% BSA (Sigma-Aldrich) and0.01% NaN3 (FACS buffer), and stained for various surface epitopes withmAbs for 15 min at room temperature. The samples were then washedwith FACS buffer and either analyzed immediately or used for additionalintracellular staining. The intracellular staining was performed using eBio-science Foxp3 Staining Buffer Set according to the manufacturer’s instruc-tions. The samples were analyzed on an LSRII flow cytometer (BectonDickinson) using Diva software (Becton Dickinson).
Statistics
The statistical significance of the data were calculated by Mann-Whitney Utest or Kruskal Wallis test using the StatView program (SAS Institute,Cary, NC). p , 0.05 was considered statistically significant.
ResultsNormal ROS levels are of importance for vaccination
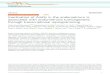
To investigate the effect of reduced ROS levels on peptide–MHC IItherapy, we induced CIA in B10.QNcf1p/pmice and then vaccinatedthem on days 20 and 34 after CII immunization. The mice weredivided in three groups: 1) injected with glycosylated CII peptide(GalOK264) in complex with Aq; 2) treated with unmodified CIIpeptide (K264)/Aq complex; and 3) PBS control group. Three moregroups of B10.Q wild type littermates, treated as indicated above,were included in this experiment to compare the result of the therapyand estimate the effects of low redox production on tolerance de-velopment. Consistent with our previous data (24) administration ofAq/GalOK264 dramatically and specifically reduced arthritis se-verity, incidence, and onset in normal (nonmutated) B10.Q mice(Fig. 1A, Table I). In contrast, B10.Q Ncf1p/p mice showed no de-crease in the incidence of disease (Table I), and all animals in thethree treated groups developed CIA. Evaluation of clinical signs ofarthritis indicated a slight tendency for reduced disease severity inthe Aq/GalOK264-treated group that was significant only at the endof the experiment (Fig. 1B). The animals, treated with glycosylatedCII peptide started to develop arthritis later than the other twogroups of Ncf1p/p mice (Table I). These data demonstrate that Aq/GalOK264 administration has a specific effect in B10.Q Ncf1p/p
mice like in normal B10.Q strain, but this effect is not pronouncedand is extremely mild, indicating that reduced ROS levels havea crucial effect on tolerance development and arthritis susceptibility.
MHC II/GalOK264 vaccination reduces anti-CII Ab levels
Anti-CII Ab levels in all treated animals were measured on days 35and 70 after CII immunization. Concordant with the arthritis data inB10.Q mice, we observed significantly decreased serum concentra-tion of anti-CII IgG (Fig. 1C, 1D), confirming that glycosylated CIIpeptide–MHC II treatment affects Ag-specific T cells and they failto stimulate pathogenic anti-CII Ab secreting plasma cells. Inter-estingly, the Aq/GalOK264-treated Ncf1p/pmice also exhibited sig-nificantly lower levels of anti-CII IgG (Fig. 1E, 1F). This result wassurprising for us considering the lack of strong arthritis suppressionin this strain. The data illustrate that the CII peptide vaccinationoperates in the same way in Ncf1p/p and normal littermates sup-pressing both T and B cell anti-CII responses. However, the sup-pression of CIA is not efficient in the absence of normal ROS levelsin mice with mutated Ncf1 gene.Possibly the fine specificity of the anti-CII Ab responsemight dif-
fer between the B10.Q Ncf1p/p and B10.Q mice, and we attemptedto find this difference by analyzing the response to certain CIIepitopes. It has been shown that Abs directed to C1 and U1 epitopesfrom CII are arthritogenic and correlate with chronic arthritis (33).The response against C1 and U1 epitopes is immunodominant inboth CIA and RA (34). In our vaccination experiment, as expected,the levels of anti-C1 and anti-U1 Abs were reduced in the Aq/GalOK264-treated groups (Fig. 2), confirming again the specificeffect of Aq/GalOK264 vaccination. However, a trend was observedthat the suppression of the response to these specific epitopes wasless pronounced in the B10.Q Ncf1p/p mice.
Aq/GalOK264 treatment leads to expansion of Tregs andelevated expression of PD-1 on CD4+ T cells
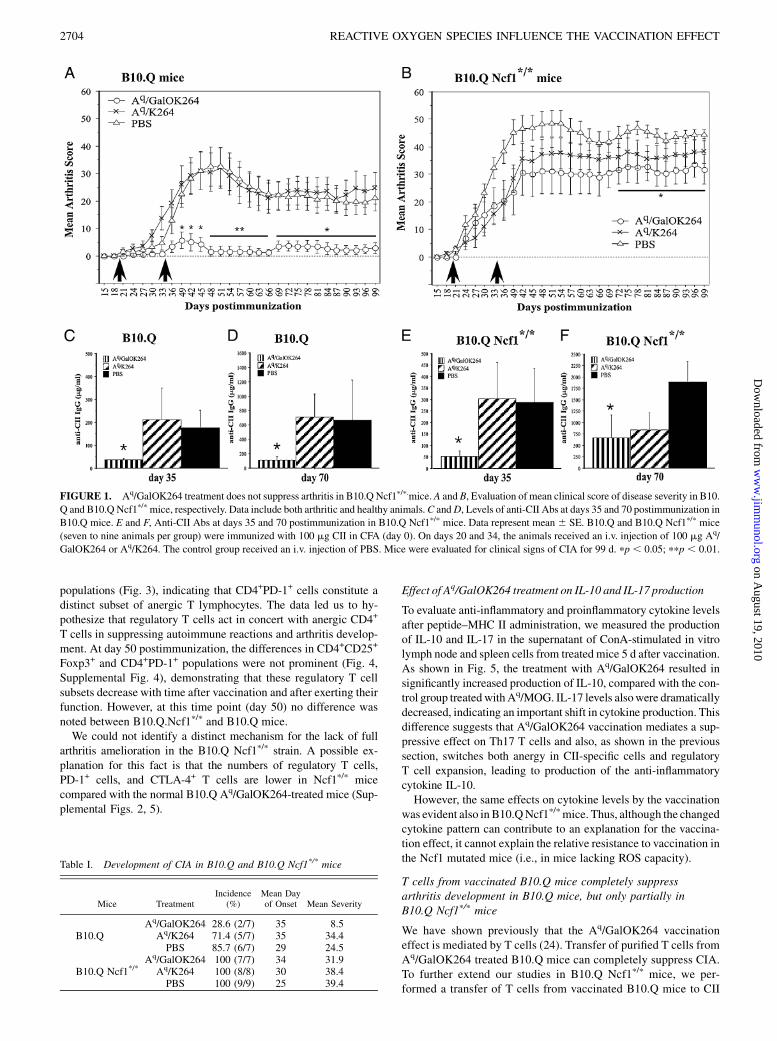
Nextwe analyzed the effect of vaccination ondifferent T cell subsets,hoping for a clue to the role of ROS production. B10.Q and B10.QNcf1p/p mice were immunized with CII as described in the previoussection. The animals from each strain were divided in two groupsand vaccinated on days 20 and 34 postimmunization with either Aq/GalOK264 or Aq/MOG79–96. The Aq/MOG79–96 complex sup-presses experimental autoimmune encephalomyelitis (B. Dzham-bazov, unpublished data), showing that it is functional in vivo, andthus an optimal negative control. Two different time points aftergiving the CII treatment were chosen to evaluate possible changes invarious T cell subgroups. Five days after the first vaccination (day 25postimmunization), and 16 d after the second vaccination (day 50postimmunization) blood was collected from each group and ana-lyzed by flow cytometry. These time points were chosen with theaim of studying the dynamics of the T cell populations during andafter arthritis onset. Erythrocyte-free cell suspensions were stainedfor the following T cell markers: CD4, CD8, CD25, Foxp3,PD-1 and CTLA-4. Figs. 3 and 4 indicate the differences observedamong the CD4+ T cell population. No differences were found in theCD8+ T cell subset (Supplemental Figs. 3, 6). Five days after Aq/GalOK264 treatment, we detected an increase in the number ofCD4+CD25+Foxp3+ T cells (Fig. 3, Supplemental Fig. 1), althoughit was not statistically significant (p = 0.08). This finding suggeststhat the MHCII–glycosylated CII peptide vaccine mediates its effectvia active suppression of the induction of regulatory T cells. At thesame time, we observed upregulation of inhibitory costimulatorymolecules PD-1 and CTLA-4 (Fig. 3). The majority of CD4+
CTLA-4+ T cells were also Foxp3+ (Fig. 3), which confirms theimportance of CTLA-4 as a specific marker for regulatory T cells.There was no significant difference in the Foxp3+PD-1+CD4+ T cell
The Journal of Immunology 2703
on August 19, 2010
ww
w.jim
munol.org
Dow
nloaded from
populations (Fig. 3), indicating that CD4+PD-1+ cells constitute adistinct subset of anergic T lymphocytes. The data led us to hy-pothesize that regulatory T cells act in concert with anergic CD4+
T cells in suppressing autoimmune reactions and arthritis develop-ment. At day 50 postimmunization, the differences in CD4+CD25+
Foxp3+ and CD4+PD-1+ populations were not prominent (Fig. 4,Supplemental Fig. 4), demonstrating that these regulatory T cellsubsets decrease with time after vaccination and after exerting theirfunction. However, at this time point (day 50) no difference wasnoted between B10.Q.Ncf1p/p and B10.Q mice.We could not identify a distinct mechanism for the lack of full
arthritis amelioration in the B10.Q Ncf1p/p strain. A possible ex-planation for this fact is that the numbers of regulatory T cells,PD-1+ cells, and CTLA-4+ T cells are lower in Ncf1p/p micecompared with the normal B10.Q Aq/GalOK264-treated mice (Sup-plemental Figs. 2, 5).
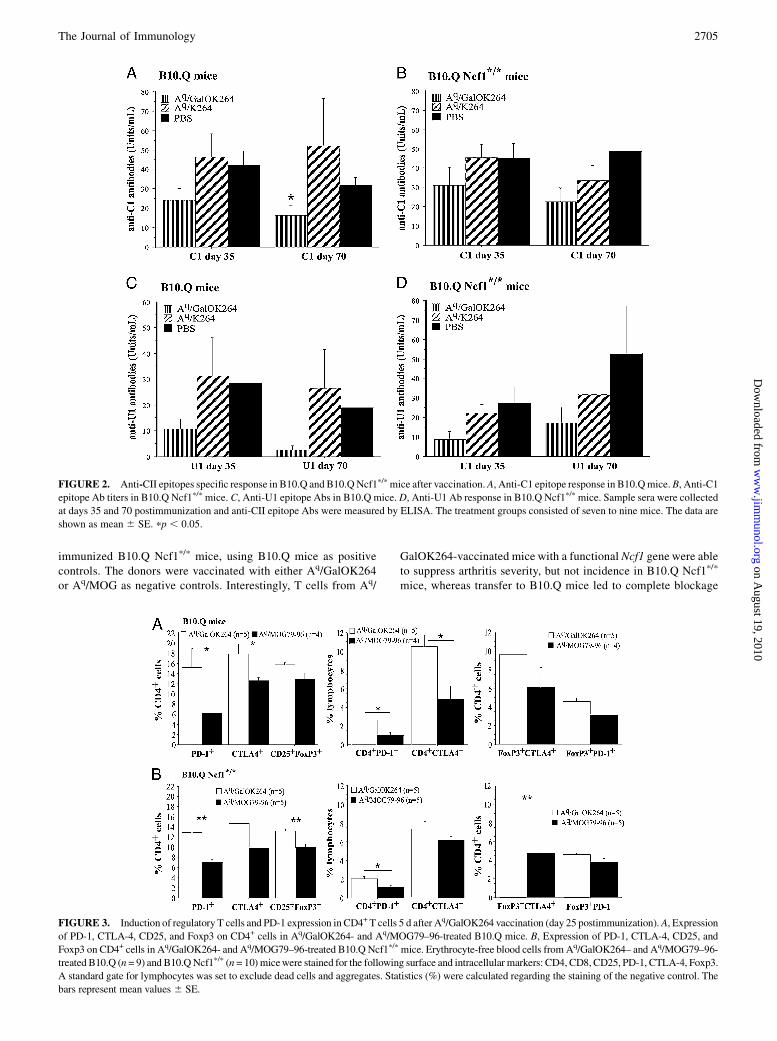
Effect of Aq/GalOK264 treatment on IL-10 and IL-17 production
To evaluate anti-inflammatory and proinflammatory cytokine levelsafter peptide–MHC II administration, we measured the productionof IL-10 and IL-17 in the supernatant of ConA-stimulated in vitrolymph node and spleen cells from treated mice 5 d after vaccination.As shown in Fig. 5, the treatment with Aq/GalOK264 resulted insignificantly increased production of IL-10, compared with the con-trol group treatedwithAq/MOG. IL-17 levels alsowere dramaticallydecreased, indicating an important shift in cytokine production. Thisdifference suggests that Aq/GalOK264 vaccination mediates a sup-pressive effect on Th17 T cells and also, as shown in the previoussection, switches both anergy in CII-specific cells and regulatoryT cell expansion, leading to production of the anti-inflammatorycytokine IL-10.However, the same effects on cytokine levels by the vaccination
was evident also in B10.QNcf1p/pmice. Thus, although the changedcytokine pattern can contribute to an explanation for the vaccina-tion effect, it cannot explain the relative resistance to vaccination inthe Ncf1 mutated mice (i.e., in mice lacking ROS capacity).
T cells from vaccinated B10.Q mice completely suppressarthritis development in B10.Q mice, but only partially inB10.Q Ncf1p/p mice
We have shown previously that the Aq/GalOK264 vaccinationeffect is mediated by T cells (24). Transfer of purified T cells fromAq/GalOK264 treated B10.Q mice can completely suppress CIA.To further extend our studies in B10.Q Ncf1p/p mice, we per-formed a transfer of T cells from vaccinated B10.Q mice to CII
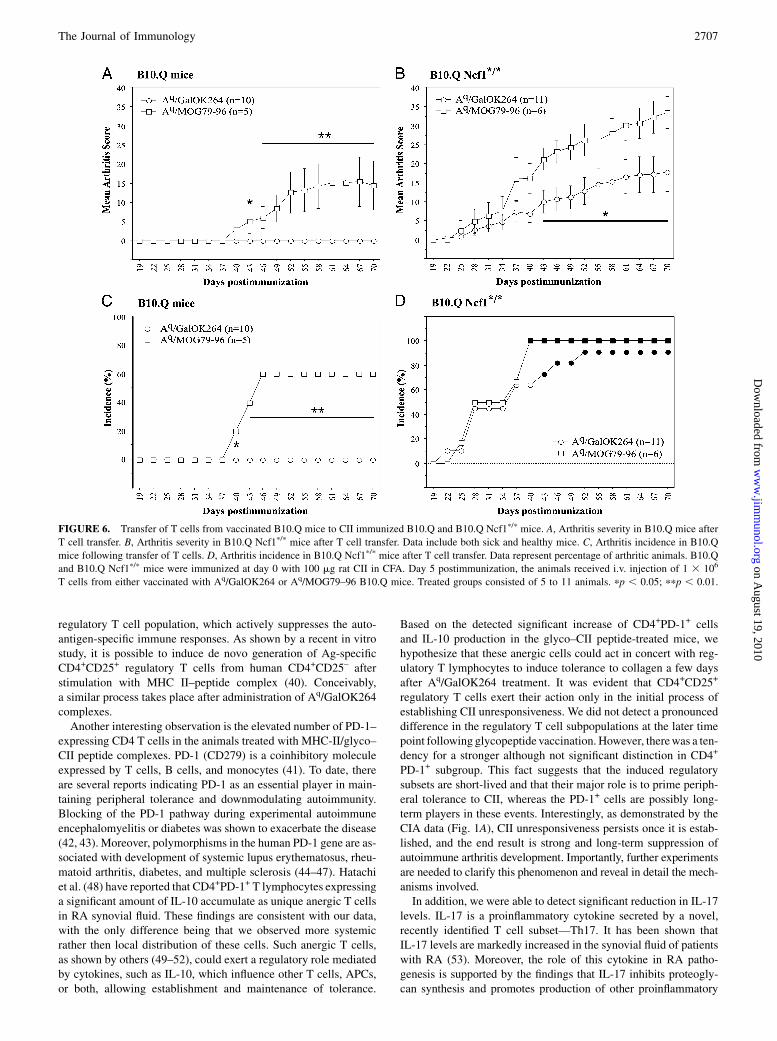
FIGURE 1. Aq/GalOK264 treatment does not suppress arthritis in B10.Q Ncf1p/p.mice. A and B, Evaluation of mean clinical score of disease severity in B10.
Q and B10.Q Ncf1p/pmice, respectively. Data include both arthritic and healthy animals.C andD, Levels of anti-CII Abs at days 35 and 70 postimmunization in
B10.Q mice. E and F, Anti-CII Abs at days 35 and 70 postimmunization in B10.Q Ncf1p/p mice. Data represent mean 6 SE. B10.Q and B10.Q Ncf1p/p mice
(seven to nine animals per group) were immunized with 100 mg CII in CFA (day 0). On days 20 and 34, the animals received an i.v. injection of 100 mg Aq/
GalOK264 or Aq/K264. The control group received an i.v. injection of PBS. Mice were evaluated for clinical signs of CIA for 99 d. pp , 0.05; ppp , 0.01.
Table I. Development of CIA in B10.Q and B10.Q Ncf1*/* mice
Mice TreatmentIncidence
(%)Mean Dayof Onset Mean Severity
Aq/GalOK264 28.6 (2/7) 35 8.5B10.Q Aq/K264 71.4 (5/7) 35 34.4
PBS 85.7 (6/7) 29 24.5Aq/GalOK264 100 (7/7) 34 31.9
B10.Q Ncf1*/* Aq/K264 100 (8/8) 30 38.4PBS 100 (9/9) 25 39.4
2704 REACTIVE OXYGEN SPECIES INFLUENCE THE VACCINATION EFFECT
on August 19, 2010
ww
w.jim
munol.org
Dow
nloaded from
immunized B10.Q Ncf1p/p mice, using B10.Q mice as positivecontrols. The donors were vaccinated with either Aq/GalOK264or Aq/MOG as negative controls. Interestingly, T cells from Aq/
GalOK264-vaccinated mice with a functional Ncf1 gene were ableto suppress arthritis severity, but not incidence in B10.Q Ncf1p/p
mice, whereas transfer to B10.Q mice led to complete blockage
FIGURE 3. Induction of regulatory T cells and PD-1 expression in CD4+ T cells 5 d after Aq/GalOK264 vaccination (day 25 postimmunization).A, Expression
of PD-1, CTLA-4, CD25, and Foxp3 on CD4+ cells in Aq/GalOK264- and Aq/MOG79–96-treated B10.Q mice. B, Expression of PD-1, CTLA-4, CD25, and
Foxp3 on CD4+ cells in Aq/GalOK264- and Aq/MOG79–96-treated B10.Q Ncf1p/pmice. Erythrocyte-free blood cells fromAq/GalOK264– and Aq/MOG79–96-
treated B10.Q (n = 9) and B10.QNcf1p/p (n = 10)micewere stained for the following surface and intracellular markers: CD4, CD8, CD25, PD-1, CTLA-4, Foxp3.
A standard gate for lymphocytes was set to exclude dead cells and aggregates. Statistics (%) were calculated regarding the staining of the negative control. The
bars represent mean values 6 SE.
FIGURE 2. Anti-CII epitopes specific response in B10.Q and B10.QNcf1p/pmice after vaccination.A, Anti-C1 epitope response in B10.Qmice. B, Anti-C1
epitope Ab titers in B10.Q Ncf1p/pmice. C, Anti-U1 epitope Abs in B10.Q mice.D, Anti-U1 Ab response in B10.Q Ncf1p/pmice. Sample sera were collected
at days 35 and 70 postimmunization and anti-CII epitope Abs were measured by ELISA. The treatment groups consisted of seven to nine mice. The data are
shown as mean 6 SE. pp , 0.05.
The Journal of Immunology 2705
on August 19, 2010
ww
w.jim
munol.org
Dow
nloaded from
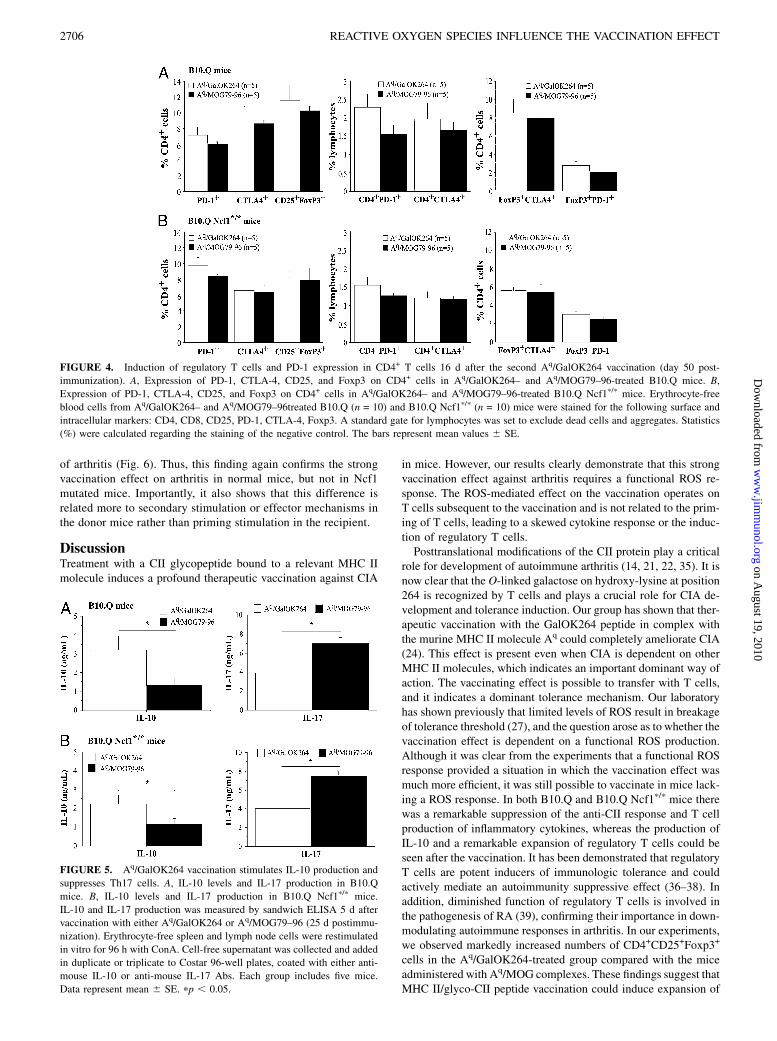
of arthritis (Fig. 6). Thus, this finding again confirms the strongvaccination effect on arthritis in normal mice, but not in Ncf1mutated mice. Importantly, it also shows that this difference isrelated more to secondary stimulation or effector mechanisms inthe donor mice rather than priming stimulation in the recipient.
DiscussionTreatment with a CII glycopeptide bound to a relevant MHC IImolecule induces a profound therapeutic vaccination against CIA
in mice. However, our results clearly demonstrate that this strongvaccination effect against arthritis requires a functional ROS re-sponse. The ROS-mediated effect on the vaccination operates onT cells subsequent to the vaccination and is not related to the prim-ing of T cells, leading to a skewed cytokine response or the induc-tion of regulatory T cells.Posttranslational modifications of the CII protein play a critical
role for development of autoimmune arthritis (14, 21, 22, 35). It isnow clear that the O-linked galactose on hydroxy-lysine at position264 is recognized by T cells and plays a crucial role for CIA de-velopment and tolerance induction. Our group has shown that ther-apeutic vaccination with the GalOK264 peptide in complex withthe murine MHC II molecule Aq could completely ameliorate CIA(24). This effect is present even when CIA is dependent on otherMHC II molecules, which indicates an important dominant way ofaction. The vaccinating effect is possible to transfer with T cells,and it indicates a dominant tolerance mechanism. Our laboratoryhas shown previously that limited levels of ROS result in breakageof tolerance threshold (27), and the question arose as to whether thevaccination effect is dependent on a functional ROS production.Although it was clear from the experiments that a functional ROSresponse provided a situation in which the vaccination effect wasmuch more efficient, it was still possible to vaccinate in mice lack-ing a ROS response. In both B10.Q and B10.Q Ncf1p/p mice therewas a remarkable suppression of the anti-CII response and T cellproduction of inflammatory cytokines, whereas the production ofIL-10 and a remarkable expansion of regulatory T cells could beseen after the vaccination. It has been demonstrated that regulatoryT cells are potent inducers of immunologic tolerance and couldactively mediate an autoimmunity suppressive effect (36–38). Inaddition, diminished function of regulatory T cells is involved inthe pathogenesis of RA (39), confirming their importance in down-modulating autoimmune responses in arthritis. In our experiments,we observed markedly increased numbers of CD4+CD25+Foxp3+
cells in the Aq/GalOK264-treated group compared with the miceadministered with Aq/MOG complexes. These findings suggest thatMHC II/glyco-CII peptide vaccination could induce expansion of
FIGURE 4. Induction of regulatory T cells and PD-1 expression in CD4+ T cells 16 d after the second Aq/GalOK264 vaccination (day 50 post-
immunization). A, Expression of PD-1, CTLA-4, CD25, and Foxp3 on CD4+ cells in Aq/GalOK264– and Aq/MOG79–96-treated B10.Q mice. B,
Expression of PD-1, CTLA-4, CD25, and Foxp3 on CD4+ cells in Aq/GalOK264– and Aq/MOG79–96-treated B10.Q Ncf1p/p mice. Erythrocyte-free
blood cells from Aq/GalOK264– and Aq/MOG79–96treated B10.Q (n = 10) and B10.Q Ncf1p/p (n = 10) mice were stained for the following surface and
intracellular markers: CD4, CD8, CD25, PD-1, CTLA-4, Foxp3. A standard gate for lymphocytes was set to exclude dead cells and aggregates. Statistics
(%) were calculated regarding the staining of the negative control. The bars represent mean values 6 SE.
FIGURE 5. Aq/GalOK264 vaccination stimulates IL-10 production and
suppresses Th17 cells. A, IL-10 levels and IL-17 production in B10.Q
mice. B, IL-10 levels and IL-17 production in B10.Q Ncf1p/p mice.
IL-10 and IL-17 production was measured by sandwich ELISA 5 d after
vaccination with either Aq/GalOK264 or Aq/MOG79–96 (25 d postimmu-
nization). Erythrocyte-free spleen and lymph node cells were restimulated
in vitro for 96 h with ConA. Cell-free supernatant was collected and added
in duplicate or triplicate to Costar 96-well plates, coated with either anti-
mouse IL-10 or anti-mouse IL-17 Abs. Each group includes five mice.
Data represent mean 6 SE. pp , 0.05.
2706 REACTIVE OXYGEN SPECIES INFLUENCE THE VACCINATION EFFECT
on August 19, 2010
ww
w.jim
munol.org
Dow
nloaded from
regulatory T cell population, which actively suppresses the auto-antigen-specific immune responses. As shown by a recent in vitrostudy, it is possible to induce de novo generation of Ag-specificCD4+CD25+ regulatory T cells from human CD4+CD25– afterstimulation with MHC II–peptide complex (40). Conceivably,a similar process takes place after administration of Aq/GalOK264complexes.Another interesting observation is the elevated number of PD-1–
expressing CD4 T cells in the animals treated with MHC-II/glyco–CII peptide complexes. PD-1 (CD279) is a coinhibitory moleculeexpressed by T cells, B cells, and monocytes (41). To date, thereare several reports indicating PD-1 as an essential player in main-taining peripheral tolerance and downmodulating autoimmunity.Blocking of the PD-1 pathway during experimental autoimmuneencephalomyelitis or diabetes was shown to exacerbate the disease(42, 43). Moreover, polymorphisms in the human PD-1 gene are as-sociated with development of systemic lupus erythematosus, rheu-matoid arthritis, diabetes, and multiple sclerosis (44–47). Hatachiet al. (48) have reported that CD4+PD-1+ T lymphocytes expressinga significant amount of IL-10 accumulate as unique anergic T cellsin RA synovial fluid. These findings are consistent with our data,with the only difference being that we observed more systemicrather then local distribution of these cells. Such anergic T cells,as shown by others (49–52), could exert a regulatory role mediatedby cytokines, such as IL-10, which influence other T cells, APCs,or both, allowing establishment and maintenance of tolerance.
Based on the detected significant increase of CD4+PD-1+ cellsand IL-10 production in the glyco–CII peptide-treated mice, wehypothesize that these anergic cells could act in concert with reg-ulatory T lymphocytes to induce tolerance to collagen a few daysafter Aq/GalOK264 treatment. It was evident that CD4+CD25+
regulatory T cells exert their action only in the initial process ofestablishing CII unresponsiveness. We did not detect a pronounceddifference in the regulatory T cell subpopulations at the later timepoint following glycopeptide vaccination. However, therewas a ten-dency for a stronger although not significant distinction in CD4+
PD-1+ subgroup. This fact suggests that the induced regulatorysubsets are short-lived and that their major role is to prime periph-eral tolerance to CII, whereas the PD-1+ cells are possibly long-term players in these events. Interestingly, as demonstrated by theCIA data (Fig. 1A), CII unresponsiveness persists once it is estab-lished, and the end result is strong and long-term suppression ofautoimmune arthritis development. Importantly, further experimentsare needed to clarify this phenomenon and reveal in detail the mech-anisms involved.In addition, we were able to detect significant reduction in IL-17
levels. IL-17 is a proinflammatory cytokine secreted by a novel,recently identified T cell subset—Th17. It has been shown thatIL-17 levels are markedly increased in the synovial fluid of patientswith RA (53). Moreover, the role of this cytokine in RA patho-genesis is supported by the findings that IL-17 inhibits proteogly-can synthesis and promotes production of other proinflammatory
FIGURE 6. Transfer of T cells from vaccinated B10.Q mice to CII immunized B10.Q and B10.Q Ncf1p/p mice. A, Arthritis severity in B10.Q mice after
T cell transfer. B, Arthritis severity in B10.Q Ncf1p/p mice after T cell transfer. Data include both sick and healthy mice. C, Arthritis incidence in B10.Q
mice following transfer of T cells. D, Arthritis incidence in B10.Q Ncf1p/p mice after T cell transfer. Data represent percentage of arthritic animals. B10.Q
and B10.Q Ncf1p/p mice were immunized at day 0 with 100 mg rat CII in CFA. Day 5 postimmunization, the animals received i.v. injection of 1 3 106
T cells from either vaccinated with Aq/GalOK264 or Aq/MOG79–96 B10.Q mice. Treated groups consisted of 5 to 11 animals. pp , 0.05; ppp , 0.01.
The Journal of Immunology 2707
on August 19, 2010
ww
w.jim
munol.org
Dow
nloaded from

cytokines like IL-1, TNF-a, IL-6, and IL-8 by synoviocytes, mono-cytes, and macrophages (54). IL-17–deficient mice have signifi-cantly reduced CIA severity and incidence (55). In our study, thereduced IL-17 levels after Aq/GalOK264 treatment support thepossibility of active suppression on pathogenic T and B cells.There are several mechanisms that could provide an explanation
for the vaccination effect. In addition, the pronounced effects on theinduction of regulatory T cells, induction of phenotypic tolerantT cells, and the cytokine response suggest that the glycosylated CIIpeptide evoked a broader response than activation of only CII-specific T cells. This finding could provide an explanation for thehighly pronounced vaccination effect with glyco CII peptides inCIA compared with the relatively mild vaccination of experimentalautoimmune encephalomyelitis using the MHC-II/MOG peptidecomplex (B. Dzhambazov, personal communication). Importantlyhowever, these mechanisms could not explain why the vaccinationwas not operating as efficiently in ROS-deficient mice. Importantly,the vaccination effect on arthritis was much stronger when T cellswere transferred to normal B10.Qmice than to B10.Qwith deficientROS production. Thus, the effects on the immune response duringthe induction of vaccination, such as a skewed cytokine response andinduction of regulatory T cells, operating in the donor B10.Q are notthe critical factors. Instead there are ROS dependent mechanisms inthe recipient mice that could involve a secondary activation of thetransferred T cells in the recipient mice. A corresponding effect wasobserved in rats treated with phytol, an inducer of the ROS response(56). In this case treatment of the recipient of arthritogenic T cellscould downregulate the development of arthritis.These findings also could have implications for human RA. The
glycosylatedCII peptidebinds to thehumanDR4/DR1MHC-IImol-ecules in a manner similar to Aq, extending its galactose side chainsto the TCR (4, 57, 58). Moreover, T cell specificity directed to thisposttranslationally modified epitope also has been shown in humanpatients with RA (14), which indicates that therapy with this pep-tide would lead to a promising result in humans.
AcknowledgmentsWe thank Carlos Palestro, Kristina Palestro, and Isabell Bohlin for taking
care of the animals.
DisclosuresThe authors have no financial conflicts of interest.
References1. Feldmann, M., F. M. Brennan, and R. N. Maini. 1996. Rheumatoid arthritis. Cell
85: 307–310.2. Rantapaa-Dahlqvist, S., B. A. de Jong, E. Berglin, G. Hallmans, G. Wadell,
H. Stenlund, U. Sundin, and W. J. van Venrooij. 2003. Antibodies againstcyclic citrullinated peptide and IgA rheumatoid factor predict the developmentof rheumatoid arthritis. Arthritis Rheum. 48: 2741–2749.
3. Johansson, M., L. Arlestig, G. Hallmans, and S. Rantapaa-Dahlqvist. 2006.PTPN22 polymorphism and anti-cyclic citrullinated peptide antibodies in com-bination strongly predicts future onset of rheumatoid arthritis and has a specific-ity of 100% for the disease. Arthritis Res. Ther. 8: R19.
4. Diab, B. Y., N. C. Lambert, F. E. L’Faqihi, P. Loubet-Lescoulie, C. de Preval,and H. Coppin. 1999. Human collagen II peptide 256-271 preferentially binds toHLA-DR molecules associated with susceptibility to rheumatoid arthritis. Im-munogenetics 49: 36–44.
5. Gregersen, P. K., J. Silver, and R. J. Winchester. 1987. The shared epitope hy-pothesis. An approach to understanding the molecular genetics of susceptibilityto rheumatoid arthritis. Arthritis Rheum. 30: 1205–1213.
6. Toh, M. L., and P. Miossec. 2007. The role of T cells in rheumatoid arthritis: newsubsets and new targets. Curr. Opin. Rheumatol. 19: 284–288.
7. Kamradt, T., and D. Schubert. 2005. The role and clinical implications of G6PIin experimental models of rheumatoid arthritis. Arthritis Res. Ther. 7: 20–28.
8. Fritsch, R., D. Eselbock, K. Skriner, B. Jahn-Schmid, C. Scheinecker, B. Bohle,M. Tohidast-Akrad, S. Hayer, J. Neumuller, S. Pinol-Roma, et al. 2002. Char-acterization of autoreactive T cells to the autoantigens heterogeneous nuclearribonucleoprotein A2 (RA33) and filaggrin in patients with rheumatoid arthritis.J. Immunol. 169: 1068–1076.
9. Corrigall, V.M.,M.D.Bodman-Smith,M. S. Fife, B. Canas, L.K.Myers, P.Wooley,C. Soh, N. A. Staines, D. J. Pappin, S. E. Berlo, et al. 2001. The human endoplasmicreticulum molecular chaperone BiP is an autoantigen for rheumatoid arthritis andprevents the induction of experimental arthritis. J. Immunol. 166: 1492–1498.
10. Mimori, T. 2005. Clinical significance of anti-CCP antibodies in rheumatoidarthritis. Intern. Med. 44: 1122–1126.
11. Cremer, M. A., E. F. Rosloniec, and A. H. Kang. 1998. The cartilage collagens:a review of their structure, organization, and role in the pathogenesis of exper-imental arthritis in animals and in human rheumatic disease. J. Mol. Med. 76:275–288.
12. Zou, J., Y. Zhang, A. Thiel, M. Rudwaleit, S. L. Shi, A. Radbruch, R. Poole,J. Braun, and J. Sieper. 2003. Predominant cellular immune response to thecartilage autoantigenic G1 aggrecan in ankylosing spondylitis and rheumatoidarthritis. Rheumatology (Oxford) 42: 846–855.
13. Verheijden, G. F., A. W. Rijnders, E. Bos, C. J. Coenen-de Roo, C. J. vanStaveren, A. M. Miltenburg, J. H. Meijerink, D. Elewaut, F. de Keyser,E. Veys, and A. M. Boots. 1997. Human cartilage glycoprotein-39 as a candidateautoantigen in rheumatoid arthritis. Arthritis Rheum. 40: 1115–1125.
14. Backlund, J., S. Carlsen, T. Hoger, B. Holm, L. Fugger, J. Kihlberg,H. Burkhardt, and R. Holmdahl. 2002. Predominant selection of T cells specificfor the glycosylated collagen type II epitope (263-270) in humanized transgenicmice and in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 99: 9960–9965.
15. Brand, D. D., A. H. Kang, and E. F. Rosloniec. 2003. Immunopathogenesis ofcollagen arthritis. Springer Semin. Immunopathol. 25: 3–18.
16. Brunsberg, U., K. Gustafsson, L. Jansson, E. Michaelsson, L. Ahrlund-Richter,S. Pettersson, R. Mattsson, and R. Holmdahl. 1994. Expression of a transgenicclass II Ab gene confers susceptibility to collagen-induced arthritis. Eur. J.Immunol. 24: 1698–1702.
17. Wooley, P. H., H. S. Luthra, J. M. Stuart, and C. S. David. 1981. Type II collagen-induced arthritis in mice. I. Major histocompatibility complex (I region) linkageand antibody correlates. J. Exp. Med. 154: 688–700.
18. Andersson, E. C., B. E. Hansen, H. Jacobsen, L. S. Madsen, C. B. Andersen,J. Engberg, J. B. Rothbard, G. S. McDevitt, V. Malmstrom, R. Holmdahl, et al.1998. Definition of MHC and T cell receptor contacts in the HLA-DR4restrictedimmunodominant epitope in type II collagen and characterization of collagen-induced arthritis in HLA-DR4 and human CD4 transgenic mice. Proc. Natl.Acad. Sci. USA 95: 7574–7579.
19. Michaelsson, E., M. Andersson, A. Engstrom, and R. Holmdahl. 1992. Identi-fication of an immunodominant type-II collagen peptide recognized by T cellsin H-2q mice: self tolerance at the level of determinant selection. Eur. J. Immu-nol. 22: 1819–1825.
20. Corthay, A., J. Backlund, and R. Holmdahl. 2001. Role of glycopeptide-specificT cells in collagen-induced arthritis: an example how post-translational modifica-tion of proteins may be involved in autoimmune disease. Ann. Med. 33: 456–465.
21. Michaelsson, E., V. Malmstrom, S. Reis, A. Engstrom, H. Burkhardt, andR. Holmdahl. 1994. T cell recognition of carbohydrates on type II collagen. J.Exp. Med. 180: 745–749.
22. Michaelsson, E., J. Broddefalk, A. Engstrom, J. Kihlberg, and R. Holmdahl.1996. Antigen processing and presentation of a naturally glycosylated proteinelicits major histocompatibility complex class II-restricted, carbohydrate-specific T cells. Eur. J. Immunol. 26: 1906–1910.
23. Corthay, A., J. Backlund, J. Broddefalk, E. Michaelsson, T. J. Goldschmidt,J. Kihlberg, and R. Holmdahl. 1998. Epitope glycosylation plays a critical rolefor T cell recognition of type II collagen in collagen-induced arthritis. Eur. J.Immunol. 28: 2580–2590.
24. Dzhambazov, B., K. S. Nandakumar, J. Kihlberg, L. Fugger, R. Holmdahl, andM. Vestberg. 2006. Therapeutic vaccination of active arthritis with a glycosy-lated collagen type II peptide in complex with MHC class II molecules. J.Immunol. 176: 1525–1533.
25. Olofsson, P., J. Holmberg, J. Tordsson, S. Lu, B. Akerstrom, and R. Holmdahl.2003. Positional identification of Ncf1 as a gene that regulates arthritis severityin rats. Nat. Genet. 33: 25–32.
26. Hultqvist, M., and R. Holmdahl. 2005. Ncf1 (p47phox) polymorphism deter-mines oxidative burst and the severity of arthritis in rats and mice. Cell.Immunol. 233: 97–101.
27. Hultqvist, M., J. Backlund, K. Bauer, K. A. Gelderman, and R. Holmdahl. 2007.Lack of reactive oxygen species breaks T cell tolerance to collagen type II andallows development of arthritis in mice. J. Immunol. 179: 1431–1437.
28. Hultqvist, M., P. Olofsson, J. Holmberg, B. T. Backstrom, J. Tordsson, andR. Holmdahl. 2004. Enhanced autoimmunity, arthritis, and encephalomyelitis inmice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc.Natl. Acad. Sci. USA 101: 12646–12651.
29. Huang, C. K., L. Zhan, M. O. Hannigan, Y. Ai, and T. L. Leto. 2000. P47(phox)-deficient NADPH oxidase defect in neutrophils of diabetic mouse strains,C57BL/6J-m db/db and db/+. J. Leukoc. Biol. 67: 210–215.
30. Andersson, M., and R. Holmdahl. 1990. Analysis of type II collagen-reactiveT cells in the mouse. I. Different regulation of autoreactive vs. non-autoreactiveanti-type II collagen T cells in the DBA/1 mouse. Eur. J. Immunol. 20: 1061–1066.
31. Holm, B., J. Backlund, M. A. Recio, R. Holmdahl, and J. Kihlberg. 2002.Glycopeptide specificity of helper T cells obtained in mouse models for rheu-matoid arthritis. ChemBioChem 3: 1209–1222.
32. Broddefalk, J., M. Forsgren, I. Sethson, and J. Kihlberg. 1999. Preparation ofa Diglycosylated Hydroxylysine Building Block Used in Solid-Phase Synthesisof a Glycopeptide from Type II Collagen. J. Org. Chem. 64: 8948–8953.
33. Bajtner, E., K. S. Nandakumar, A. Engstrom, and R. Holmdahl. 2005. Chronicdevelopment of collagen-induced arthritis is associated with arthritogenic
2708 REACTIVE OXYGEN SPECIES INFLUENCE THE VACCINATION EFFECT
on August 19, 2010
ww
w.jim
munol.org
Dow
nloaded from

antibodies against specific epitopes on type II collagen. Arthritis Res. Ther. 7:R1148–R1157.
34. Burkhardt, H., T. Koller, A. Engstrom, K. S. Nandakumar, J. Turnay, H. G. Kraetsch,J. R. Kalden, and R. Holmdahl. 2002. Epitope-specific recognition of type II colla-gen by rheumatoid arthritis antibodies is shared with recognition by antibodies thatare arthritogenic in collagen-induced arthritis in the mouse. Arthritis Rheum. 46:2339–2348.
35. Myers, L. K., J. Myllyharju, M. Nokelainen, D. D. Brand, M. A. Cremer,J. M. Stuart, M. Bodo, K. I. Kivirikko, and A. H. Kang. 2004. Relevance ofposttranslational modifications for the arthritogenicity of type II collagen. J.Immunol. 172: 2970–2975.
36. Sakaguchi, S., N. Sakaguchi, J. Shimizu, S. Yamazaki, T. Sakihama, M. Itoh,Y. Kuniyasu, T. Nomura, M. Toda, and T. Takahashi. 2001. Immunologic tol-erance maintained by CD25+ CD4+ regulatory T cells: their common role incontrolling autoimmunity, tumor immunity, and transplantation tolerance.Immunol. Rev. 182: 18–32.
37. Sakaguchi, S., M. Ono, R. Setoguchi, H. Yagi, S. Hori, Z. Fehervari, J. Shimizu,T. Takahashi, and T. Nomura. 2006. Foxp3+ CD25+ CD4+ natural regulatoryT cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 212:8–27.
38. Suri-Payer, E., A. Z. Amar, A. M. Thornton, and E. M. Shevach. 1998. CD4+CD25+ T cells inhibit both the induction and effector function of autoreactiveT cells and represent a unique lineage of immunoregulatory cells. J. Immunol.160: 1212–1218.
39. Ehrenstein, M. R., J. G. Evans, A. Singh, S. Moore, G. Warnes, D. A. Isenberg,and C. Mauri. 2004. Compromised function of regulatory T cells in rheumatoidarthritis and reversal by anti-TNFalpha therapy. J. Exp. Med. 200: 277–285.
40. Walker, M. R., B. D. Carson, G. T. Nepom, S. F. Ziegler, and J. H. Buckner.2005. De novo generation of antigen-specific CD4+CD25+ regulatory T cellsfrom human CD4+CD25- cells. Proc. Natl. Acad. Sci. USA 102: 4103–4108.
41. Fife, B. T., K. E. Pauken, T. N. Eagar, T. Obu, J. Wu, Q. Tang, M. Azuma,M. F. Krummel, and J. A. Bluestone. 2009. Interactions between PD-1 andPD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immu-nol. 10: 1185–1192.
42. Salama, A. D., T. Chitnis, J. Imitola, M. J. Ansari, H. Akiba, F. Tushima,M. Azuma, H. Yagita, M. H. Sayegh, and S. J. Khoury. 2003. Critical role of theprogrammed death-1 (PD-1) pathway in regulation of experimental autoimmuneencephalomyelitis. J. Exp. Med. 198: 71–78.
43. Ansari, M. J., A. D. Salama, T. Chitnis, R. N. Smith, H. Yagita, H. Akiba,T. Yamazaki, M. Azuma, H. Iwai, S. J. Khoury, et al. 2003. The programmeddeath-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic(NOD) mice. J. Exp. Med. 198: 63–69.
44. Prokunina, L., C. Castillejo-Lopez, F. Oberg, I. Gunnarsson, L. Berg,V. Magnusson, A. J. Brookes, D. Tentler, H. Kristjansdottir, G. Grondal, et al.2002. A regulatory polymorphism in PDCD1 is associated with susceptibility tosystemic lupus erythematosus in humans. Nat. Genet. 32: 666–669.
45. Nielsen, C., D. Hansen, S. Husby, B. B. Jacobsen, and S. T. Lillevang. 2003.Association of a putative regulatory polymorphism in the PD-1 gene with sus-ceptibility to type 1 diabetes. Tissue Antigens 62: 492–497.
46. Kroner, A., M. Mehling, B. Hemmer, P. Rieckmann, K. V. Toyka, M. Maurer,and H. Wiendl. 2005. A PD-1 polymorphism is associated with disease pro-gression in multiple sclerosis. Ann. Neurol. 58: 50–57.
47. Lin, S. C., J. H. Yen, J. J. Tsai, W. C. Tsai, T. T. Ou, H. W. Liu, and C. J. Chen.2004. Association of a programmed death 1 gene polymorphism with the de-velopment of rheumatoid arthritis, but not systemic lupus erythematosus. Ar-thritis Rheum. 50: 770–775.
48. Hatachi, S., Y. Iwai, S. Kawano, S. Morinobu, M. Kobayashi, M. Koshiba,R. Saura, M. Kurosaka, T. Honjo, and S. Kumagai. 2003. CD4+ PD-1+ T cellsaccumulate as unique anergic cells in rheumatoid arthritis synovial fluid. J.Rheumatol. 30: 1410–1419.
49. Jooss, K., B. Gjata, O. Danos, H. von Boehmer, and A. Sarukhan. 2001. Reg-ulatory function of in vivo anergized CD4(+) T cells. Proc. Natl. Acad. Sci. USA98: 8738–8743.
50. Buer, J., A. Lanoue, A. Franzke, C. Garcia, H. von Boehmer, and A. Sarukhan.1998. Interleukin 10 secretion and impaired effector function of major histo-compatibility complex class II-restricted T cells anergized in vivo. J. Exp. Med.187: 177–183.
51. Lechler, R., J. G. Chai, F. Marelli-Berg, and G. Lombardi. 2001. T-cell anergyand peripheral T-cell tolerance. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:625–637.
52. Gabrysova, L., K. S. Nicolson, H. B. Streeter, J. Verhagen, C. A. Sabatos-Peyton,D. J. Morgan, and D. C. Wraith. 2009. Negative feedback control of theautoimmune response through antigen-induced differentiation of IL-10-secreting Th1 cells. J. Exp. Med. 206: 1755–1767.
53. Chabaud, M., J. M. Durand, N. Buchs, F. Fossiez, G. Page, L. Frappart, andP. Miossec. 1999. Human interleukin-17: A T cell-derived proinflammatory cy-tokine produced by the rheumatoid synovium. Arthritis Rheum. 42: 963–970.
54. Stamp, L. K., M. J. James, and L. G. Cleland. 2004. Interleukin-17: the missinglink between T-cell accumulation and effector cell actions in rheumatoid arthri-tis? Immunol. Cell Biol. 82: 1–9.
55. Nakae, S., A. Nambu, K. Sudo, and Y. Iwakura. 2003. Suppression of immuneinduction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 171:6173–6177.
56. Hultqvist, M., P. Olofsson, K. A. Gelderman, J. Holmberg, and R. Holmdahl.2006. A new arthritis therapy with oxidative burst inducers. PLoS Med. 3: e348.
57. Rosloniec, E. F., K. B. Whittington, D. M. Zaller, and A. H. Kang. 2002. HLA-DR1 (DRB1*0101) and DR4 (DRB1*0401) use the same anchor residues forbinding an immunodominant peptide derived from human type II collagen. J.Immunol. 168: 253–259.
58. von Delwig, A., D. M. Altmann, J. D. Isaacs, C. V. Harding, R. Holmdahl,N. McKie, and J. H. Robinson. 2006. The impact of glycosylation on HLA-DR1-restricted T cell recognition of type II collagen in a mouse model. ArthritisRheum. 54: 482–491.
The Journal of Immunology 2709
on August 19, 2010
ww
w.jim
munol.org
Dow
nloaded from
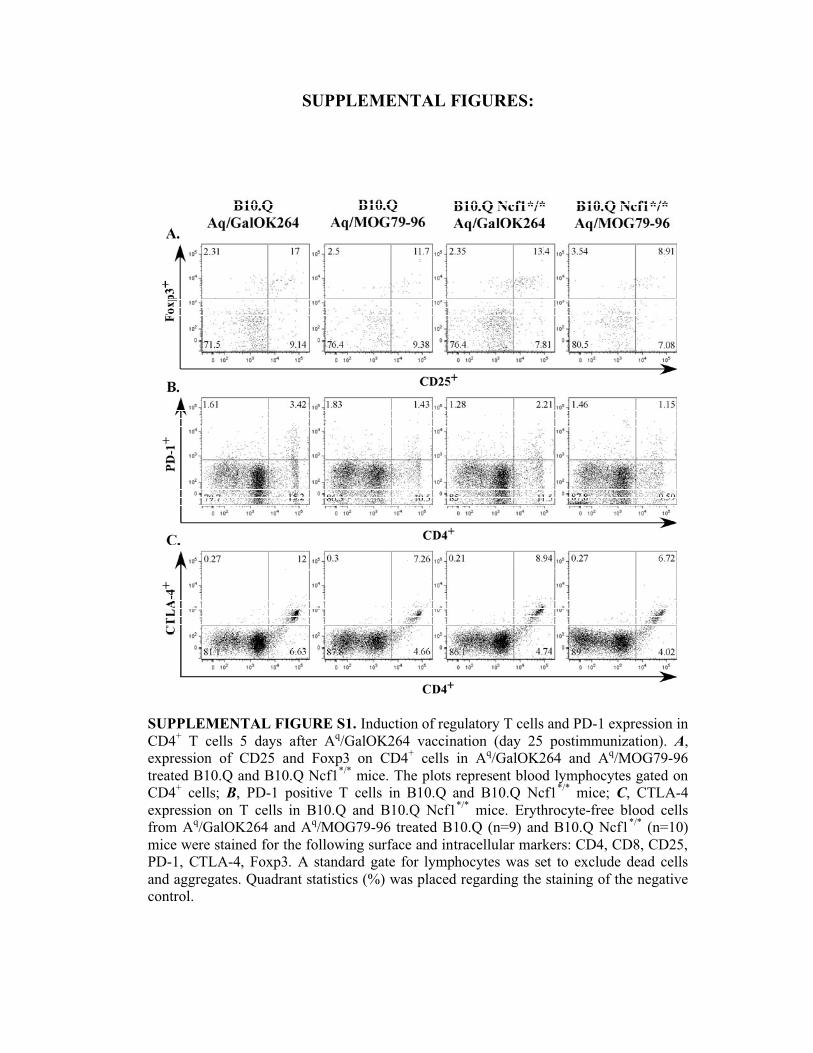
SUPPLEMENTAL FIGURES:
SUPPLEMENTAL FIGURE S1. Induction of regulatory T cells and PD-1 expression in
CD4+ T cells 5 days after A
q/GalOK264 vaccination (day 25 postimmunization). A,
expression of CD25 and Foxp3 on CD4+ cells in A
q/GalOK264 and A
q/MOG79-96
treated B10.Q and B10.Q Ncf1*/*
mice. The plots represent blood lymphocytes gated on
CD4+ cells; B, PD-1 positive T cells in B10.Q and B10.Q Ncf1
*/* mice; C, CTLA-4
expression on T cells in B10.Q and B10.Q Ncf1*/*
mice. Erythrocyte-free blood cells
from Aq/GalOK264 and A
q/MOG79-96 treated B10.Q (n=9) and B10.Q Ncf1
*/* (n=10)
mice were stained for the following surface and intracellular markers: CD4, CD8, CD25,
PD-1, CTLA-4, Foxp3. A standard gate for lymphocytes was set to exclude dead cells
and aggregates. Quadrant statistics (%) was placed regarding the staining of the negative
control.
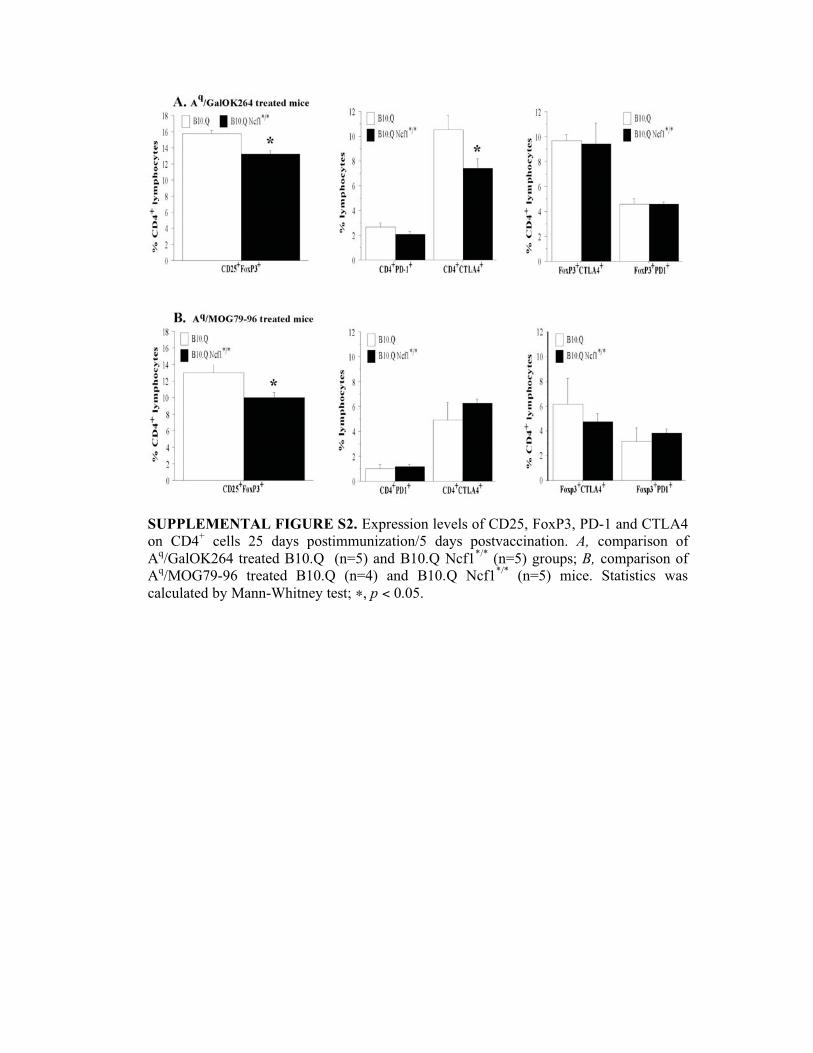
SUPPLEMENTAL FIGURE S2. Expression levels of CD25, FoxP3, PD-1 and CTLA4
on CD4+ cells 25 days postimmunization/5 days postvaccination. A, comparison of
Aq/GalOK264 treated B10.Q (n=5) and B10.Q Ncf1
*/* (n=5) groups; B, comparison of
Aq/MOG79-96 treated B10.Q (n=4) and B10.Q Ncf1
*/* (n=5) mice. Statistics was
calculated by Mann-Whitney test; �, p < 0.05.
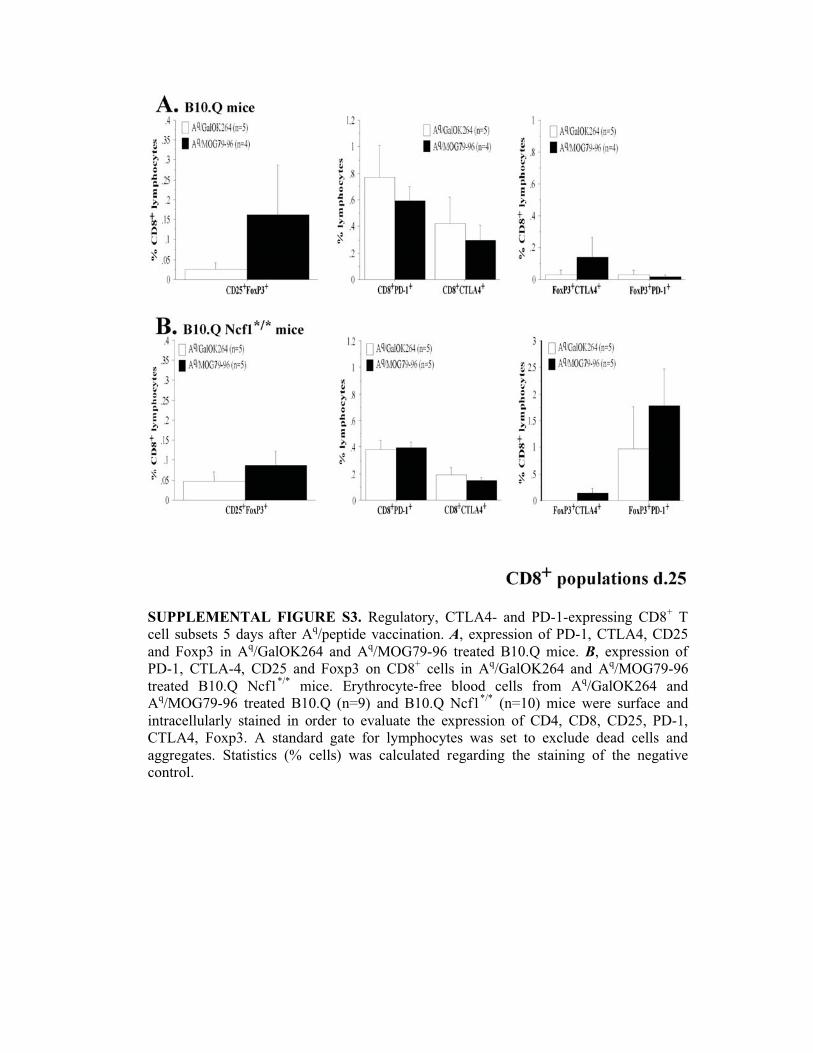
SUPPLEMENTAL FIGURE S3. Regulatory, CTLA4- and PD-1-expressing CD8+ T
cell subsets 5 days after Aq/peptide vaccination. A, expression of PD-1, CTLA4, CD25
and Foxp3 in Aq/GalOK264 and A
q/MOG79-96 treated B10.Q mice. B, expression of
PD-1, CTLA-4, CD25 and Foxp3 on CD8+ cells in A
q/GalOK264 and A
q/MOG79-96
treated B10.Q Ncf1*/*
mice. Erythrocyte-free blood cells from Aq/GalOK264 and
Aq/MOG79-96 treated B10.Q (n=9) and B10.Q Ncf1
*/* (n=10) mice were surface and
intracellularly stained in order to evaluate the expression of CD4, CD8, CD25, PD-1,
CTLA4, Foxp3. A standard gate for lymphocytes was set to exclude dead cells and
aggregates. Statistics (% cells) was calculated regarding the staining of the negative
control.
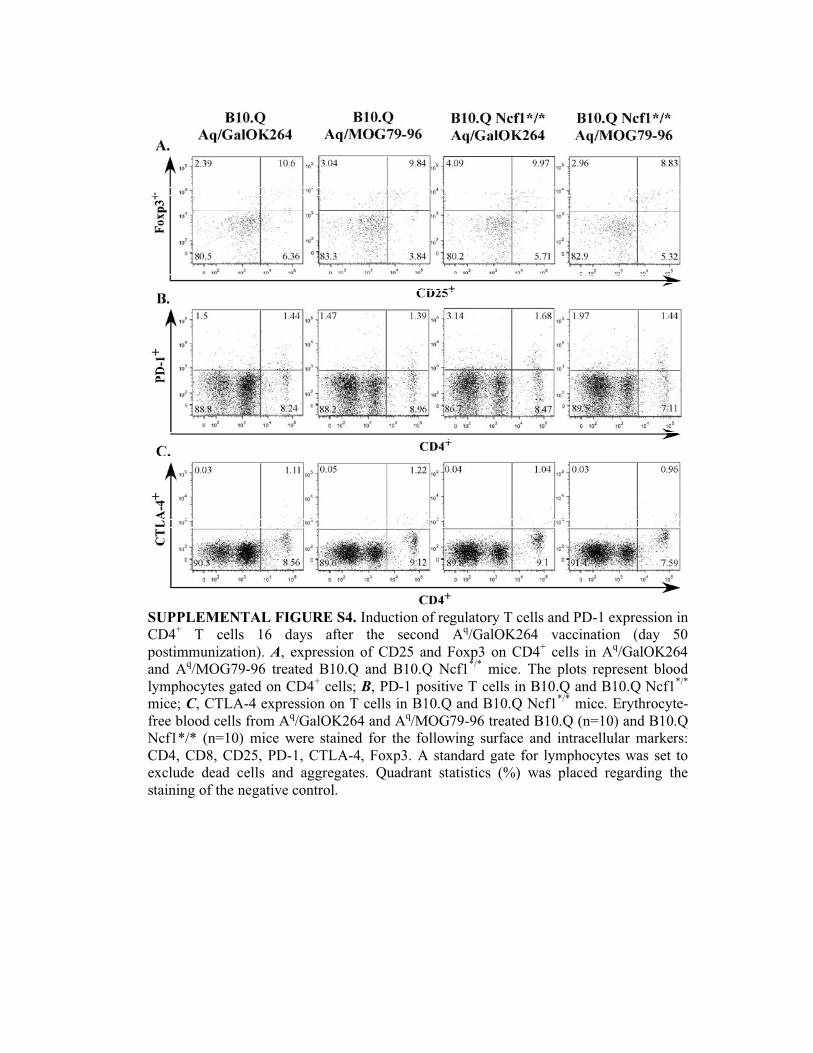
SUPPLEMENTAL FIGURE S4. Induction of regulatory T cells and PD-1 expression in
CD4+ T cells 16 days after the second A
q/GalOK264 vaccination (day 50
postimmunization). A, expression of CD25 and Foxp3 on CD4+ cells in A
q/GalOK264
and Aq/MOG79-96 treated B10.Q and B10.Q Ncf1
*/* mice. The plots represent blood
lymphocytes gated on CD4+ cells; B, PD-1 positive T cells in B10.Q and B10.Q Ncf1
*/*
mice; C, CTLA-4 expression on T cells in B10.Q and B10.Q Ncf1*/*
mice. Erythrocyte-
free blood cells from Aq/GalOK264 and A
q/MOG79-96 treated B10.Q (n=10) and B10.Q
Ncf1*/* (n=10) mice were stained for the following surface and intracellular markers:
CD4, CD8, CD25, PD-1, CTLA-4, Foxp3. A standard gate for lymphocytes was set to
exclude dead cells and aggregates. Quadrant statistics (%) was placed regarding the
staining of the negative control.
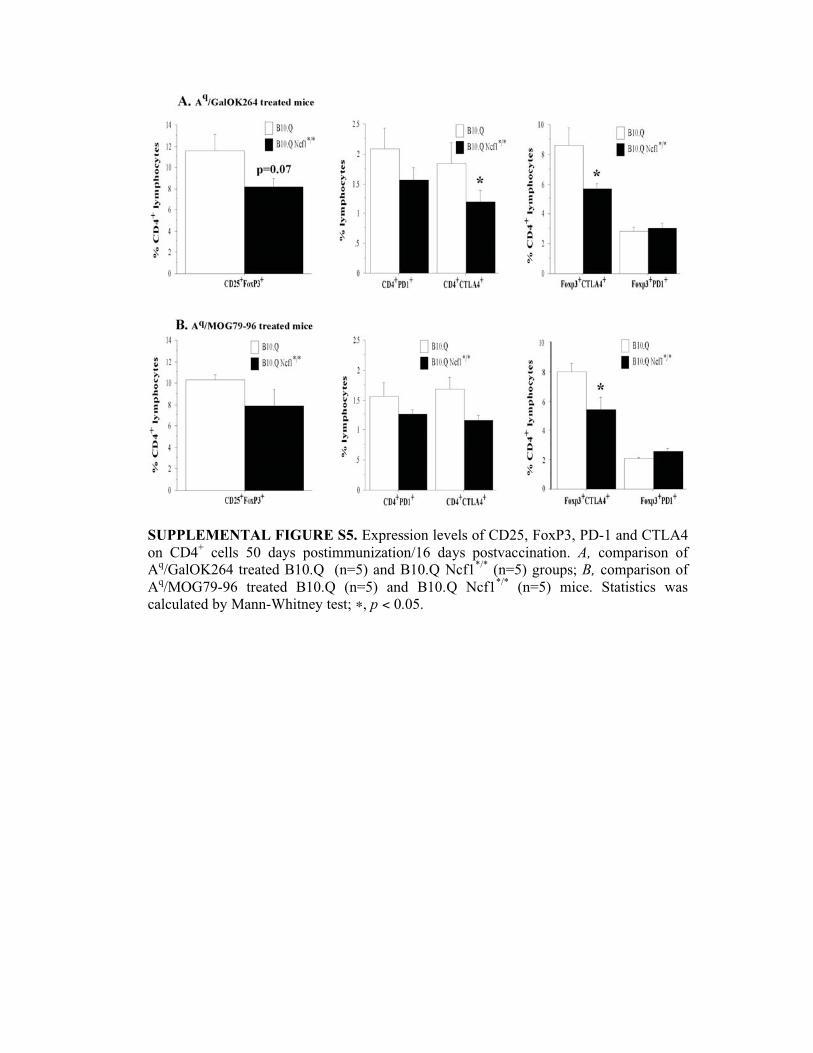
SUPPLEMENTAL FIGURE S5. Expression levels of CD25, FoxP3, PD-1 and CTLA4
on CD4+ cells 50 days postimmunization/16 days postvaccination. A, comparison of
Aq/GalOK264 treated B10.Q (n=5) and B10.Q Ncf1
*/* (n=5) groups; B, comparison of
Aq/MOG79-96 treated B10.Q (n=5) and B10.Q Ncf1
*/* (n=5) mice. Statistics was
calculated by Mann-Whitney test; �, p < 0.05.
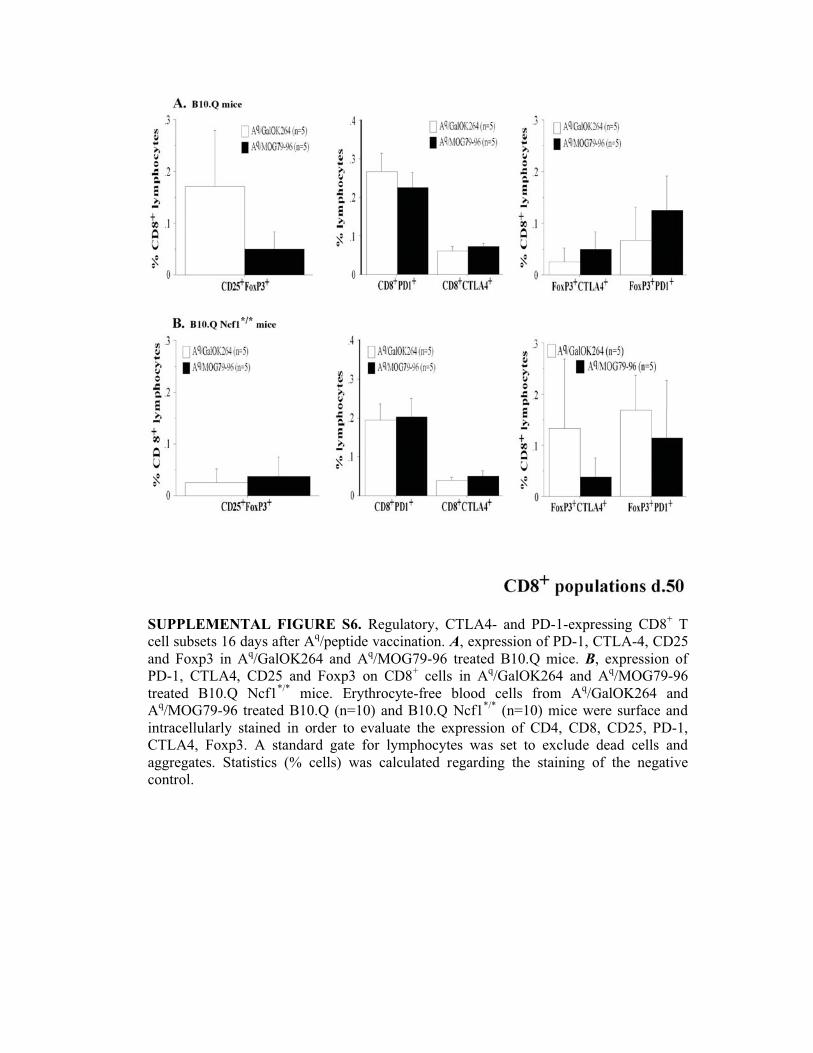
SUPPLEMENTAL FIGURE S6. Regulatory, CTLA4- and PD-1-expressing CD8+ T
cell subsets 16 days after Aq/peptide vaccination. A, expression of PD-1, CTLA-4, CD25
and Foxp3 in Aq/GalOK264 and A
q/MOG79-96 treated B10.Q mice. B, expression of
PD-1, CTLA4, CD25 and Foxp3 on CD8+ cells in A
q/GalOK264 and A
q/MOG79-96
treated B10.Q Ncf1*/*
mice. Erythrocyte-free blood cells from Aq/GalOK264 and
Aq/MOG79-96 treated B10.Q (n=10) and B10.Q Ncf1
*/* (n=10) mice were surface and
intracellularly stained in order to evaluate the expression of CD4, CD8, CD25, PD-1,
CTLA4, Foxp3. A standard gate for lymphocytes was set to exclude dead cells and
aggregates. Statistics (% cells) was calculated regarding the staining of the negative
control.