-
7/27/2019 Method Validation Application Protein Biomarkers
1/14
1461ISSN 1757-6180Bioanalysis(2009) 1(8),
1461147410.4155/BIO.09.130 2009 Future Science Ltd
Review
Recent drug development is based on the mech-anism of action of
the drug on specific biologicaltargets and pathways. Biomarkers
reflective ofthese pathways have been linked to physiologi-
cal data to aid drug-development decisions [1].There are high
expectations in the pharmaceu-tical industry that biomarker
applications willdrive faster and more successful drug develop-ment
[24], as exemplified in the multitudes ofbiomarker conferences and
publications devotedto this relatively new application of
biomarkers.Biomarkers expressed in disease-specific path-ways can
provide evidence that a drug hits itstarget to exert functional
changes. Studies ofthe target and proximal biomarkers can
providepharmacodynamic (PD) information for expo-
sure/effect modeling. Data from downstreamdistal biomarkers can
provide proof-of-biologyof the drugs effect on disease progression
[5].
Protein therapeutics via target-mediatedmechanisms have been
successfully devel-oped. The bioanalysis of protein therapeuticsis
based on the evolving practices from smallto large molecules: the
US FDA issued guid-ance for bioanalytical method validation
tosupport pharmacokinetic (PK) studies with thefocus on
conventional small-molecule drugs,mainly by LCMS methods [6].
Additional
White papers on ligand-binding assays(LBAs)
that are widely used to study biotherapeuticshave been published
[79]. At the 3rd AmericanAssoc iation of Pharmaceutical Scientis
ts(AAPS)/FDA Bioanalytical Workshop, the
validation and implementation of bioanalyticalmethods for both
small- and macro-moleculeswere discussed and consensus reports were
sub-sequently published in a themed issue of theAAPS journal
[911].
The terminology of GLP compliance has beengenerally used in the
pharmaceutical industryto indicate bioanalysis in support of
PK/toxico-kinetic (TK) studies that are conducted accord-ing to
guidance from the FDA and/or otherregulatory agencies [6]. It is
not uncommon tosee analysts working in the PK/TK arena adopt-
ing the same guidance for biomarker methodvalidation. At the
same time, since biomarkerkits approved by the FDA or other
regulatoryagencies have been routinely used for diseasediagnosis,
clinical chemists also participate inbiomarker analysis for drug
development andperform the assays under regulations fromagencies
such as the Clinical Lab ImprovementAmendments in the USA. The
end-users of thedata also come from two camps: PK/PD scien-tists
who are familiar with PK-type data andphysicians/clinicians who are
comfortable with
the routine clinical chemistry output.
Method validation and application of protein
biomarkers: basic similarities anddifferences from
biotherapeutics
Protein drug development and biomarkers share common
bioanalytical technologies that are applied for different
purposes. A t-for-purpose approach should be used for biomarker
assays at various stages of novel biomarker
development and their application to drug development. Biomarker
quantications can be absolute or relative,
depending upon the characteristics of the standard curve, which
include the reference standard, substituted matrix
and parallelism. Appropriate method-validation experiments
should be carried out on sample collection, relative
accuracy and precision, range nding, parallelism, selectivity,
specicity and stability in order to meet the need for
exploratory or advanced application that is specied for a study.
The interaction of a biotherapeutic with the targetligand or
inter-related biomarkers should be taken into consideration for
method platform choice and validation.
Direct adoption of commercial diagnostic kits can produce
confounding data. Therefore, kit comparison, modication
and appropriate validation experiments are often carried out to
meet the specic purpose for drug development.
Multiplex assays and physicochemical methods can complement the
single-analyte ligand-binding assay for protein
drugs and biomarkers.
Jean W Lee
Pharmacokinetics and Drug
Metabolism, Amgen Inc.,
One Amgen Center Drive30E-3-B, Thousand Oaks,
CA 91320, USA
Tel.: +1 805 447 9463
Fax: +1 805 499 9027
E-mail: [email protected]
BiotheRapeutics
Therapeutics derived frombiological products or processes
Ligand-Bindingassay
Analytical methods thatdetermine the analyte using thesignal
resulting from the binding
reaction of the reagent andthe analyte
-
7/27/2019 Method Validation Application Protein Biomarkers
2/14
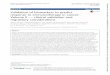
DemonstrationDiscovery Characterization Qualification
Surrogacy
Studies of cells, animal model
or human with tight patient control
Confirmatory with small
human population atmultiple sites
Multiple sites
Large sample size Extended populations Multiple drugs of similar
mechanism
Exploratory method validation Advanced method validation (GLP
similar)
Biomarker development
Drug development
NonclinicalLead optimization Pivotal clinical Post-approvalEarly
clinical
Nonregulated Regulated (GLP) PK bioanalysis
Post-approval
surveillance Safety and
efficacy biomarkers
Patient stratification
Other therapeutic indications
Market differentiation
Safety biomarkers
Efficacy biomarkers Proof of Biology
Protocol design
PK/PD modeling
Dose selection
Biomarker panel
selection
Target and
candidate selection
Candidate attrition and refinement
A
B
Review|Lee
Bioanalysis(2009) 1(8)1462 future science group
The inconsistency in adaptations of regula-tions in either
bioanalytical or clinical laborato-ries and a lack of regulatory
guidance contribute
to the confusion regarding biomarker data qual-ity required for
drug development. A positionpaper proposed that biomarker assay val
idationand implementation should be fit-for-purposeto produce
reliable data appropriate for theapplication [12]. Biomarker
applications are verydifferent from those of diagnosis, which
pro-hibit the direct adoption of clinical laboratory
practices. The intended use of biomarker datashould be
considered in order to determine therigor of method validation and
implementation
for the specified purpose. Biomarker analysis tosupport PDs
should be similar to that for PKstudies with differences based upon
the uniqueendogenous nature of the heterogeneous bio-marker [1215].
This review focuses on the simi-larities and differences of protein
biomarkerassays compared with those from PK bioanalysisfor
biotherapeutic development.
Table 1. Comparison of pharmacokinetic and biomarker
bioanalysis.
Intendedapplication
Method types Pre-analyticsample
collection
Referencestandard
Analytes Calibratormatrix
Validationsample and
QC preparation
Accuracy
PK study
PK parameters
of BA and BE
Mostly definitive
quantification
methods
Test with spiked
standard
Well
characterized
and pure
Exogenous
and well
defined
Analyte-free
biological
matrix
Spiked reference
standard into
biological matrix
Absolute
accuracy
Biomarker study
PD: safety and
efficacy
Definitive,
relative,
quasiquantitative
or qualitative
methods
Consider
pathway
conversion and
artifact from
cell activation;
diurnal effect
Many are not
well
characterized or
pure, may not
be the same
as endogenous
Endogenous,
not well
defined
Substituted
matrix
Spiked reference
standard for VS
and QC, pooled
authentic samples
for sample
controls
Mostly
relative
accuracy
BA: Bioavailability; BE: Bioequivalance; LLOQ: Lower limit of
quantification; PD: Pharmacodynamic; PK: Pharmacokinetic; QC:
Quality control; VS: Validation samples.
Figure 1. (A) Biomarker- and (B) drug-development processes.PD:
Pharmacodynamics; PK: Pharmacokinetics.
pRoteinBiomaRkeR
Protein that is objectivelymeasured and evaluated as anindicator
of normal biologicprocesses, pathogenic processesor pharmacologic
response to atherapeutic intervention
-
7/27/2019 Method Validation Application Protein Biomarkers
3/14
Method validation & application of protein biomarkers |
Review
www.future-science.com 1463future science group
Intended purposes of biomarker
bioanalysis are different
from biotherapeutics
Development of novel biomarkers follows phasesof discovery,
characterization and clinical qual-
ification/validation (FiguRe 1a) analogous tothose of drug
development (FiguRe1B) [5,16,17].Initially, biomarkers are
discovered for explor-atory studies in cell systems, animal models
orwell-controlled human studies. The data providecharacterization
of the biomarker in the path-way for internal decision making.
Application toadvanced studies will test the linkage to
clinicaloutcome using small sets of patient populationsat multiple
sites. Some biomarker results mayshow negative or uninterpretable
linkage. Otherswith promising results may advance to clinical
qualification (validation) studies where extensivedata are
collected from multiple sites (with largepatient numbers and
extended populations) onmultiple drugs and for multiple
indicationsinvolving the same pathway. Surrogacy can onlyoccur
after the accumulation of a huge amountof data before the biomarker
can replace theclinical outcome. The level of rigor of
methodvalidation and documentation increases fromexploratory to
advanced use.
The drug-development phases are depicted inFiguRe1B. The drug
exposure data determinedin animal TK and PK and human PK help
to
define the therapeutic window and decide theproper dose and
dosing frequency. The use ofbiomarkers at various phases of drug
develop-ment is depicted in the boxes below the bar.These include
go/no-go decision making oncandidates, PK/PD modeling to decide
dose andfrequencies, patient stratification and safety, andefficacy
monitoring [3,4,1319].
The objective of method validation is todemonstrate that a
particular method is reli-able for the intended application [6].
Thus, afit-for-purpose approach for method validationand sample
assays is suitable for both drug and
biomarker bioanalysis. During discovery andlead optimization of
drug development, fastdecision making on multiple drug candidates
issupported by methods with or without minimalprestudy
characterization by non-GLP methods.To support TK and PK studies,
GLP methodswith rigorous method validation are usuallyrequired
(FiguRe1B).
The purposes of biomarkers are more diversethan those of drug
development; however,method-validation approaches can be
roughlycategorized into those of exploratory or advanced
application(FiguRe1a)
.FiguRe2
depicts the basicconcept of fit-for-purpose biomarker method
val-idation and how the exploratory and advancedvalidations are
used during biomarker develop-ment. The rigor of method
development, valida-tion and documentation for advanced
applica-tion is more intense and GLP similar, except fora few
distinguishing features. The basic similari-ties and major
differences are listed in taBLe1and further discussed later in this
article.
Pre-analytic considerations that
impact biomarker analysis
Pre-analytic considerations for biomarkers arerepresented in
FiguRe3. It is necessary to selectthe potential biomarkers and the
correspond-ing biological matrix, define the intended pur-pose and
decide the type of method validationthat suits the application. A
work plan maybe used to clarify the purpose and lay out
theexperiments to be conducted [20].
Table 1. Comparison of pharmacokinetic and biomarker bioanalysis
(cont.).
Selectivity Specificity Assay acceptancecriteria
Stability Reproducibility
PK study
Spike recovery test on
~six matrix lots at LLOQ
and one other level
Test against target ligand, in addition to
similar structures; measurement of free
drug preferred over total
4-6-X rule Use QC samples Incurred sample
repeats
Biomarker study
Spike sufficient amount
over basal level, test
more lots from healthy
and disease populations
Test against drug molecule(s), precursor
and downstream molecules;
measurement of total target biomarker
may be the pragmatic option over the
free form
Depends on drug effect,
disease and biological
modulation and method
performance
Use sample
controls and trend
analysis for long
term storage
Sample controls
BA: Bioavailability; BE: Bioequivalance; LLOQ: Lower limit of
quantification; PD: Pharmacodynamic; PK: Pharmacokinetic; QC:
Quality control; VS: Validation samples.
-
7/27/2019 Method Validation Application Protein Biomarkers
4/14
Discovery
Demonstration
Characterization
Qualification
Surrogacy
Novel biomarker
development
Advanced method validationExploratory method validation
In-study method validation
Prevalidation
Pre-analytical and analytical method
feasibility method optimization
Review|Lee
Bioanalysis(2009) 1(8)1464 future science group
Stability of the analytes in stock solution andbiological matrix
during the processes of samplecollection, storage, shipping,
freezing/thawingand throughout the last assay should be
evaluatedfor drug compounds and biomarkers [21]. Thesample
collection stability for monoclonal anti-body drugs in serum has
been well established;however, the stability of peptides often
dependsupon blood collection, anticoagulants and timeof exposure to
high temperatures. Therefore,plasma or serum sample collection for
novel
peptide biotherapeutics and biomarkers shouldbe investigated for
possible stability issues.
Pre-analytic variables have hindered datautility in proteomic
biomarker discovery andvalidation [22,23]. Errors from variable
specimencollection can be higher than those arising fromsample
analysis itself. The conversion of precur-sors to the biomarker of
interest will lead tooverestimation, while degradation will
resultin underestimation of the analyte. Inhibitorsof relevant
activation or proteolysis should beincluded in the collection
syringe or added to
the sample promptly. Some biomarkers can onlybe quantified in
plasma so as to avoid prote-olysis or platelet activation of the
coagulationpathway during serum collection. Bulk serumcollected
into a bag can result in lower recoverythan in serum from
venipuncture used in a clin-ical study for some biomarkers [24].
The shear-ing effect through a small bore needle or theuse of
high-speed centrifugation on blood cellsmay cause endothelial cell
act ivation, resultingin analytical artifacts. For biological
fluids ofrelatively low protein content (e.g., urine and
cerebral spinal fluid), collection tubes, transfer
pipettes and storage containers must be evalu-ated to minimize
adsorption of a peptide/protein to the contact surfaces.
It is important to standardize techniques forall sample
collection and handling and to keepthese consistent throughout the
duration of theuse of the assay [25,26]. For example, the
G-forceand revolution per minute conversion shouldbe defined for
each laboratorys centrifuge inorder to avoid mistakes. The standard
processesof collections from multiple sites, barcodes and
transports to the analytical laboratory shouldbe followed.
Inappropriate collection time and otheradverse conditions often
lead to confoundingor uninterpretable data. If there is a
diurnaleffect, it is prudent to pool samples or to collectthem at
the same time of the day. The initialsurvey of healthy and patient
samples providea rough idea of biological variability. The
clini-cal question is the comparison of the treatmentversus
placebo. Appropriate clinical (placeboand/or predose samples) and
assay control
(sample control or QC) data can be assessedfor analyt ical and
biological variability, to pro-duce unbiased clinical answers.
However, forcancer studies, placebo or baseline samples maynot be
available to provide data to parse outthe true drug effect versus
the biological andassay variability.
Reference standard: the
basic yardstick
The basic requirements of reference standardshold true for both
PK and biomarker assays. The
standard is required to be:
Figure 2. Concept of fit-for-purpose method validation of
biomarkers at variousdevelopment phases.
-
7/27/2019 Method Validation Application Protein Biomarkers
5/14
Method development and validation:
Reference standard Relative accuracy and precision
Sensitivity, selectivity and specificity Stability
Quality and sample controls
Pre-analytical
sample integrity
Define purpose of study and
biomarker measurements: Which biomarker(s) to be
included in the study? Exploratory or advanced
application?
Choose the rightbiological matrix and
collection time forbiomarker assay
Method validation & application of protein biomarkers |
Review
www.future-science.com 1465future science group
n Purified and well characterized
n Representative of the analyte in the unknownsamples
n Available in a large quantity to support thedevelopment
program
n Stable under the defined conditions
n Accessible to the participating laboratories
Small-molecule biomarkers are well definedand pure reference
standards can be procuredin large quantities to meet these
requirements.Absolute, definitive quantification methods canbe
developed and validated, similar to those of PKassays [12].
Examples are the regulatory peptides(e.g., insulin), steroid
hormones and metabolites.However, since most biomarkers are large
pep-tides or proteins with molecular weights greaterthan 5000 Da
and generally heterogeneous in
nature, reference standard characterization andprocurement can
be challenging.
Biotherapeutics may also be heterogeneous.The reference
standards are purified and char-acterized extensively by
physicochemical andbiological methods. For example, intact
molec-ular weights are determined by SDSPAGE orMALDITOFMS. The
primary structure ofthe protein is assessed by LCMS peptide
map-ping and Edman degradation. Higher orderstructures are defined
by Fourier transfer infra-red spectroscopy, near UV circular
dichroism,
fluorescence spectroscopy and surface plasmonresonance. Surface
hydrophobicity is defined byaniline naphthalene sulfonate binding.
Thermalstability and stressed data are obtained fromdifferential
scanning calorimetry and dynamiclight scanning. Potency is defined
by the specificcellular bioactivity of the drug. Specificationsare
defined to assure lot-to-lot reproducibility.Storage and shipping
conditions and boundariesare also specified to assure stability.
Aggregationis detected by differential scanning calorimetry,size
exclusion LC, SDS, capillary and isoelec-
trofocusing electrophoresis and analytical
ultracentrifugation. Storage degradation isdetected by peptide
mapping and SDSPAGE.Documents of the characterization and
stabil-ity of a standard, such as a certificate or recordof
analysis and stability, are available to thebioanalytical
laboratory.
Protein biomarker reference standards rarelymeet the
requirements of drug compounds; oftenthe standard is impure, poorly
characterized, notfully representative of the endogenous analyte
oravailable only in limited quantities. Generally, nodocument of
certification is provided from eitherinternal or commercial
suppliers. It is doubtfulthat the same kind of extensive
characteriza-tion for biotherapeutic reference standards willever
be used for a protein biomarker unless ithas achieved qualification
or surrogacy [27,28].In addition, the reference material may
differsubstantially between lots and manufacturers,
which is a major problem contributing to datainconsistency
[29,30]. This issue must be addressedwith a collaborative effort
from pharmaceuticaland diagnostic manufacturers in the future.
Many biomarker standards are obtained fromrecombinant expression
in noneukaryotic cells;they may differ from the endogenous forms
inimmunoreactivity and bioactivity. The recom-binant reference
standard serves as a relativeyardstick of measurement, assuming
that theimmunoreactivity of the endogenous form isproportional to
that of the recombinant form
(parallelism) [13]. Thus, such methods providerelative
quantification. If there is no refer-ence standard or
proportionality between theendogenous form or the reference
standard doesnot exist, the methods are quasiquantitativein
nature.
Standard calibrator matrix &
selectivity: matrix effect matters
n Standard calibrator matrixThe preparation of standard
calibrators ina substituted matrix is a major difference
between therapeutic and biomarker analysis.
Figure 3. Process of biomarker selection, pre-analytic decisions
and method validation to supportdrug development.
-
7/27/2019 Method Validation Application Protein Biomarkers
6/14
Review|Lee
Bioanalysis(2009) 1(8)1466 future science group
Most biomarkers are endogenous compoundswith measurable levels
in the biological matrix.Standard calibrators are preferably
prepared inthe intended analyte-free sample matrix [6]; how-ever,
it is difficult to find analyte-free biological
matrix for biomarkers. The alternative optionis to use a
substituted matrix, such as a proteinbuffer, a corresponding
biological matrix fromanother species without the biomarker or
todeplete the biomarker in the biological matrixby stripping with
affinity adsorption or char-coal. The use of a substituted matrix
would avoidthe need for continual screening and testingof numerous
lots of samples to identify blankcontrols for standard
preparation.
When the calibrators are prepared in a substi-tuted matrix by
spiking a reference standard that
may not be in the same form as the endogenousbiomarker, two
types of experiments should beperformed to demonstrate method
validity:
n Comparison of spike recovery from the samplematrix and the
substituted matrix to show thatthe concentrationresponse
relationshipsare similar;
n Performance of parallelism tests on authenticsamples to show
that the endogenous biomar-ker behaves in a similar
immunochemicalmanner to the standards.
If the results fail to show similarities, the
method is considered to be quasiquantitative [20].
n Selectivity & matrix effectSelectivity is the ability of
the method to deter-mine the analyte unequivocally in the
presenceof components that may be expected to be pres-ent in the
sample. For small peptides, extrac-tion procedures similar to those
of small drugmolecules can be used to isolate, concentrate
andanalyze the peptides by LCMS/MS. A stablelabeled isotope
internal standard is added to thesamples to correct for recovery
and ionization
variability. For protein molecules, the extrac-tion step would
denature the protein and aninternal standard for LBAs would not be
avail-able. Usually, a simple buffer dilution would bethe
pretreatment step, the lack of an extractionprocess and internal
standard dictates that LBAspecificity and selectivity are solely
dependentupon the ligand-binding reagents [31]. Therefore,the
selection of reagents is of utmost importancefor both
biotherapeutic and biomarker LBAs.
With no process to remove matrix com-ponents, the LBA would be
prone to matrix
interferences (the matrix effect). Unrelated
compounds in the matrix, such as heterophilicantibodies,
rheumatoid factor and proteases,may inhibit or enhance the binding
of proteinanalytes to the reagents. Often, the immuno-reactive
signal would be suppressed, resulting in
decreased sensitivity and a negative bias.When carrying out
method development for
biotherapeutics, standard matrix curves frommultiple individual
lots are assessed for theirperformance closeness to a buffer
standardcurve. Reagents and incubation conditions canbe manipulated
so that the readouts from thematrix lots converge to those of the
buffer curve.Dilution with a high salt buffer and/or chaotropicor
chelating agent may reduce the matrix effect.The amount of dilution
required to sufficientlyremove the matrix effect is referred to as
the min-
imal required dilution (MRD)[7]
. Since bindingprotein types and levels are affected by the
healthstatus and collection conditions, selectivity testsare
conducted by spike recovery at the LLOQand at a higher level from
at least six matrix lots.Thus, accurate spike recovery at the LLOQ
con-firms assay sensitivity beyond the single matrixpool used for
standard/QC preparations duringaccuracy and precision
experiments.
For biomarkers, the matrix effect would alsobe tested by spike
recovery. However, the basallevels of the individual lots are
determined firstagainst the standard curve in the substituted
matrix. Then, the reference material is spikedinto each matrix
lot, at a level comparable tothat of the basal concentration. The
spike con-centration cannot be substantially lower thanthe baseline
and the spiked volume should notexceed 5% of the individual matrix
volume [20].Spike recovery is calculated after subtraction ofthe
basal value and compared with the nominalspike concentration or the
mean of the test lots.If most of the endogenous levels are
relativelyhigh, the LLOQ of the buffer standard wouldnot be
established for the biological samples. As
a result of biological variability, more than thesix lots from
various populations required forbiotherapeutics should be tested
for biomarkers(e.g., more than ten from each population)
[29,30].
The relative concentrations of the analyte/ inter-ferent will
vary with dose, subject and time point.Combination therapies may
change the amountof target and binding proteins or
bioavailabilityof the drug if there is a drugdrug interaction.One
option would be to pool incurred samplesfrom previous studies of
concomitant drugs fromaround the T
maxand trough levels and use these as
test samples for specificity and selectivity tests [31].
-
7/27/2019 Method Validation Application Protein Biomarkers
7/14
Method validation & application of protein biomarkers |
Review
www.future-science.com 1467future science group
n ParallelismParallelism is a dilutional linearity test of
anauthentic sample. The objective is to show thatthe endogenous
analyte in the unknown sample,which may be different from the
standard and/
or vary with subjects, behaves similarly, regard-less of
dilution by the standard matrix (or asubstituted matrix in the case
of a biomarker).The experiments are performed for both
bio-therapeutics and biomarkers. However, theexperimental design
and results interpretationare slightly different.
For biotherapeutics, incurred samples fromseveral subjects are
diluted with the standardblank matrix and analyzed. Therefore,
theexperiment can only be conducted after thein-study commences.
Several dilutions are
performed to dilute high concentration studysamples into the
standard-curve range for quan-tification. Each result (the
regressed value of thediluted sample multiplied by the dilution
fac-tor) is compared with the mean of the quantifi-able results and
should be within the acceptancecriteria. Although metabolites or
drugdruginteractions may cause nonparallelism, failedresults may be
caused by errors from multipledilutions of the high concentration
samplesrather than real interferences.
For biomarkers, parallelism is an importantcomponent and should
be performed during
prestudy validation if possible [30]. Severalindividual samples,
with concentrations atthe high end of the standard curve from
theinitial screening, are chosen. They are analyzedundiluted and
with a dilution factor of threeto four. The ratio of the calculated
results(observed concentration dilution factor)divided by the mean
of the results are plot-ted against the inverse of the dilution
factor.Parallelism is demonstrated if the ratio is notaffected by
dilution.
When para llelism cannot be per formed
because samples of sufficiently high concen-tration are not
available, dilutional linearitycan be performed in a manner similar
to para l-lelism, using high-concentration spike samplesin place of
the authentic samples. The failureto demonstrate parallelism may
mean that themethod is only quasiquantitative [12,29]. In thiscase,
longitudinal comparison within a sub-ject would become important,
using the pre-dose baseline as a reference point. The clinica
lstudy design may need to col lect more predosedsamples for within
subject comparison to the
predose baseline.
Specicity: the uncertainty of what is
being measured
n Analyte versus structurallysimilar moleculesSpecificity is the
ability of the assay to distin-
guish between the analyte and other structur-ally related
components. Crossreaction withassay-binding reagents from
structurally similarmolecules, such as metabolites, would lead
tooverestimation. In contrast to small molecules,the catabolic
species of macromolecule drugs arenot always known and are not
purified for theinvestigations into their biological activity
orassay interference. It is often assumed that theintact structure
of a protein is required for thepharmacological action; however,
this may notbe true for novel biotherapeutics.
For definitive quantitative methods for smallpeptides, the
metabolites can be identified,purified and tested for
pharmacological bio-activity. Purified metabolites can be tested
forcrossreactivity against the standard curve.
Theconcentrationsignal relationship of a LBA isnonlinear and often
the magnitude of metaboliteinterference is not monodispersed over
the entireassay range. The estimate of interference is notas
straightforward as that of a chromatographicmethod, which uses a
single percentage interfer-ence factor to cover the entire range.
Usually,percentage crossreactivity is expressed as the
ratio of midpoint concentration of the bind-ing curve of the
standard versus that of a givenmetabolite (ED
50). In addition, QC samples
should be spiked with the known metabolitesto confirm
specificity [32].
For most protein therapeutics, it is difficult toascertain the
metabolic species, due to hetero-geneity; this is even more
difficult for biomark-ers. Multitudes of isoforms and truncated
piecesthat can cause specificity problems may exist inthe matrix.
Ligand binding coupled with MShas been used as a novel approach for
identify-
ing the truncated forms and guiding methoddevelopment for LBAs
[33].
For biomarkers, structurally related moleculesinclude the dosed
drug, the precursor moleculesand homologs of the same family
[29,30]. In con-trast to drug assays, the goal of specificity tests
forbiomarkers is not to demonstrate absolute speci-ficity. Instead,
the intended purpose is to provideinformation regarding what is
being measuredfor proper data treatment and interpretation.
There is no specific guideline or consensusin the pharmaceutical
sector on how speci-
ficity and selectivity experiments should be
-
7/27/2019 Method Validation Application Protein Biomarkers
8/14
Review|Lee
Bioanalysis(2009) 1(8)1468 future science group
conducted in method validation. The NationalCommittee for
Clinical Laboratory StandardsWorking Group defines interference a s
beingfrom a known source and the matrix effectas being from an
unidentified source [34].
Discussions on selectivity, specificity and freeversus bound
tests for LBAs have been occur-ring at the AAPS meetings organized
by theLigand Binding Assay Bioanalytical FocusGroup [12,31,35] .
LCMS methods can be usedas an orthogonal technique to confirm
speci-ficity for an LBA method, as well as to detectdifferences in
isoforms of a biomarker in diseasepopulations [3640].
n Free or bound target biomarkerto biotherapeutics
Many protein drugs bind soluble ligand tar-gets. Depending on
the binding kinetics, freeand bound forms of the biotherapeutics
and thetarget ligand coexist in the biological samplesat very
different levels. It is necessary to knowwhat the method should be
measuring in orderto confirm assay specificity [35].
Proper PK/PD models are based upon avail-able data for free
and/or bound (e.g., IgE andomalizumab), bound (e.g., VEGF and
VEGF-TRAP) or total (e.g., IL-6 and canakinumab)forms [4144].
Knowledge of the free drug levelsis preferred since it reflects the
species that is
biologically active in vivo. The ligand may existin a soluble
form in the plasma at high (e.g.,IgE) or low (e.g., IL-6)
abundance. If the drugis a monoclonal antibody against an
abundantsoluble target, the ligand may cause interferencein the
drug assay if the LBA reagent binds tothe same or overlapping
epitope. Vice versa, thepresence of a high-concentration drug
wouldinterfere with the target biomarker assay.
Depending on the mechanism of action,the driver of the PD effect
can be the freebiomarker or the drugreceptor complex, for
which data would be desirable. In addition,the total
concentration of the target may pro-vide information on possible
compensatoryrise due to induction or membrane shedding.However,
issues of protein binding for bio-markers and the relevant PD data
requiredhave rarely been discussed, due to the lack ofadequate
analytical tools to provide data for thethorough understanding of
the physiology andbinding kinetics.
In the case of small-molecule biomarkers, anextraction method
using organic solvents or a
solid phase can dissociate protein binding prior
to LCMS/MS analysis. The method wouldprovide total (free plus
bound) quantificationof the small biomarker.
For a LBA of biotherapeutics and protein bio-markers, multiple
configurations present options
to measure different forms of the drug and tar-get. The data are
valuable for the understand-ing of binding kinetics. For example,
free andtotal drug can be measured by the appropriatechoice of
binding reagent, coating density of thecapture reagent, incubation
time, buffers andsample dilution. In addition, alkaline or
acidicpretreatment can be used to dissociate drugligand binding and
then be neutralized beforeLBA analysis for an assay that measures
totalligand [45,46]. Since it may be difficult to mea-sure the free
biomarker, a consistent method for
measuring the total or bound form may be apragmatic option.
These techniques and applica-tions to PK/PD at different
drug-developmentstages are being discussed by a work team inthe
AAPS Ligand Binding Bioanalytical FocusGroup in preparation of a
manuscript.
Accuracy, precision & assay range:
quantication characteristics
To ensure data quality, assay performance isevaluated during
method validation with valida-tion samples (VSs) and monitored
during sampleanalysis with QC samples prepared by spiking
known amounts of reference standard into thebiological matrix.
VSs are used in method vali-dation to define intra- and inter-run
accuracy,precision and sample stability. The prestudyvalidation
accuracy and precision data of the VSdemonstrate the suitability of
the standard curveassay range and performance characteristics
forits intended application. QC samples are used forrun acceptance
during sample analysis.
Accuracy and precision experiments andacceptance criteria for
macromolecule drugswere discussed at the AAPS FDA-sponsored
workshop [9]. Briefly, results from multiple runsof VS over the
entire span of a standard curvewould establi sh accuracy and
precision, theLLOQ and the ULOQ. Total error is the sumof the
systemic error (bias from nominal valueor percentage relative error
[%RE]) and randomerror (imprecision or percentage coefficient
ofvariation [%CV]). In-study run acceptance cri-teria are set based
on the total-error information.At least two thirds of all QC
results for a runshould be within a specific percentage (e.g.,
20%for most LBAs) of the nominal values, with at
least 50% accepted for each QC level. A 4-6-X
-
7/27/2019 Method Validation Application Protein Biomarkers
9/14
Method validation & application of protein biomarkers |
Review
www.future-science.com 1469future science group
rule was proposed for LBA: four out of six QCs(three QC levels,
each in duplicate) should bewithin x%, as determined by method
validationtotal error.
For most biomarkers, since the VSs are pre-
pared by spiking the reference standard into apool of authentic
matrix with unknown concen-tration, the nominal values are not
known. Aninitial target mean can be determined from a fewruns and
used to monitor the assay trend with-out a rigid acceptance
criteria. The true value ofthe QC may be determined after multiple
runs,using an approach similar to the Westgard Rule.For relative or
quasiquantitative methods, thereis no accuracy assessment and,
therefore, therandom error component (%CV) of the assayis more
important.
n Sensitivity: low limit of assay rangeSensitivity for drug
analysis is determined bythe LLOQ, which is the lowest
concentrationdemonstrated to be measurable with accept-with
accept-with accept-able accuracy and precision, during
methodvalidation. Extrapolation beyond the LLOQ isprohibited
[6].
Sensitivity is often defined by the limit ofdetection (LOD) for
diagnostics kits, which isthe lowest amount of analyte in a sample
thatcan be detected with 95% confidence intervalsor other stated
probability [47]. For exploratory
biomarkers, variability data at the region belowthe LLOQ but
measurable above the LODmay be needed. Samples from subjects
fromthe intended populations are surveyed by themethod for range
finding (see later). If too manysample concentrations fall below
the LLOQ, themethod is not considered sensitive enough forthe
intended application. In addition, a sensitivemethod is required if
the drug effect is expectedto suppress the level of biomarker. In
somecases, it is tolerable to have some subject samplesbelow the
LLOQ and yet above the LOD. If
these data were to be used, one should be awareof the higher
variability in the LODLLOQrange and interpret the data with
caution. Forexample, serum C-terminal telopeptides of type1
collagen (CTx) is a bone resorption biomarker.Clinical effects of
antiresorptive therapeutics,such as bisphosphonates and denosumab,
onCTx is expressed as the percentage change ofpostdose
concentrations over that of predose. Acommercial kit was used to
monitor CTx changefor denosumab drug development. The kitLOD was
0.02 ng/ml without defined accuracy
and precision; the LLOQ of 0.049 ng/ml was
defined with accuracy of -6.6%RE, precisionof 20.1 %CV and total
error of 26.7%. As themethod was for advanced application in
drugdevelopment, we chose to only report data thatwere above the
LLOQ and not those above the
LOD [48].
n Assay range & sample dilutionThe FDA guidance stated that
concentrationsof standards should be chosen on the basis ofthe
concentration range expected in a particu-lar study [6]. However,
the working range of aLBA is governed mainly by the binding
reac-tion with assay reagents. Most methods are verysensitive at
picogram or nangram per milliliterlevel, while concentrations in a
PK study formany biotherapeutics would be in the micro-micro-
gram per milliliter range. Study samples arediluted into the
working range before the assay.Sample dilution can contribute
significantly toassay variability within and between laboratories[P
K . S -
. M
P]. Three levels of QCs (low, midand high) within the standard
curve range areused to monitor accuracy and precision perfor-mance.
There has been no common practice ofhow sample dilution should be
monitored foreach assay; however, it is prudent to have dilu-
tion QCs in an assay run and a strategy has beenproposed to
include dilution QCs in the 4-6-Xapproach [P K . S -
.
M P]. For novel biomark-For novel biomark-For novel biomark-ers,
the concentration range, modulation andbiological activity of
biomarker variants are notknown. They may vary with health status,
time(age and season) and between individuals (gen-der, genetics and
ethnicity). Biological variabil-ity should be surveyed in samples
from normal
and diseased donors, especially in samples fromanticipated
patient populations (e.g., 1020each) to determine if the assay
range would beappropriate. The data should be compared withthe
literature and the commercial kit brochure.It is not uncommon to
see discordant literaturedata due to the differences in methods
(e.g.,sample collection, reference standard material,reagents and
assay conditions). Most of the time,patient levels are unavailable
or unreliable in theresearch-grade commercial kit brochure;
there-fore, the bioanalytical laboratory is responsible
for carrying out the range-finding experiments.
-
7/27/2019 Method Validation Application Protein Biomarkers
10/14
Review|Lee
Bioanalysis(2009) 1(8)1470 future science group
In addition, the expected drug effect on thebiomarker
concentration should be consideredfor the assay range. This is
often a challengeduring the exploratory phase, as the extent ofdrug
modulation is not known. The starting
assay range should aim to cover different levelsof healthy and
disease populations and also theanticipated changes from a
desirable drug effect.
The ancillary purpose of the range-findingexperiment is to find
authentic samples of lowand high concentrations to be pooled for
use assample controls (SCs). The SC concentrationswould be
determined during method val ida-tion and monitored during in-study
runs. SCsare useful for stability trending and detectingperformance
bias due to reagent lot changes [48].
n
Data regression: curve tting &data assessmentStandard LBA
curves are usually nonlinear withnonconstant error
(heteroscedastic). Most LBAdata can be appropriately fitted to
four- and five-parameter logistic models with weighting fac-tors.
Sufficient nonzero standards (six to eight)are required to define
the regression functionparameters, with additional anchor points
out-side the range to help define the asymptotes [9,49].It is
recommended to use the residuals of back-fitted standard values,
instead of the correlationcoefficient R2, to evaluate
goodness-of-fit. The
precision profile of the VS data during accuracyand precision
experiments confirms the appro-priateness of the regression model.
The accu-racy and precision data for definitive and rela-tive
quantification methods are used to assessthe systematic and random
error components fortotal error. These error components are
furthermonitored with QCs during in-study.
n Acceptance criteriaThe 4-6-X rules are commonly used in PK
appli-cations as acceptance criteria for each in-study
run [7,10,11]. The value of X is usually 15% forLCMS methods.
For LBAs, many bioanalyti-cal laboratories use a fixed value of
20%, whileothers use a statistical approach to determine Xbased on
the accuracy and precision performancedata from method validation
[79].
No guidance or consensus has been given foracceptance of
biomarker assays. One major pur-pose of biomarker application is to
distinguishdrug effect (dosed vs placebo and/or baseline)and
disease progression (healthy vs disease). Thegap between healthy
and disease, and the desir-
able drug effect, should be considered for method
suitability and in determining acceptance cri-teria. For
example, the change in IL-6 is muchgreater for sepsis than for
asthma. A more sensi-tive method and stringent acceptance criteria
willbe required in drug development for the latter
indication. During the exploratory phase, accep-tance criteria
may be set according to the initialmethod performance. After pilot
studies, biologi-cal modulation and assay variability data can
beused to refine the initial acceptance criteria.
Stability
Stability of peptides and proteins in the stocksolution and the
intended biological matrixshould be demonstrated [6]. The analyte
mayundergo biological (e.g., proteolysis) and chem-ical (e.g.,
oxidation leading to aggregation)
changes. Adsorption to the container-vessel wallsor tubing will
result in low recovery. Essentially,stability should be evaluated
during sample col-lection and handling, after long-term (frozen
atthe intended storage temperature) and short-term (bench-top, room
temperature) storage andafter going through freezethaw cycles and
theanalytical process.
Stability tests of a biotherapeutic analytein biological matrix
are conducted on the VS,spiked with reference standards at low and
highconcentrations. For biomarkers, since the SCreflects the
authentic samples, it is preferable to
use SC over VS for stability tests. In addition,the same SC set
can be monitored during in-study runs to produce long-term
stability datafor trend analysis [48].
Reproducibility demonstrated by sample
control data
The screening and selectivity tests for biomarkersare more
rigorous than those for biotherapeutics,with more lots of matrix
from normal and targetdisease populations. In addition, pooled SCs
areused to monitor assay reproducibility, reflecting
the authentic samples. For example, SC pools athigh and low
levels are aliquoted and their levelsdetermined during method
validation experi-ments and pilot studies from approximately
30runs. An acceptance criterion of mean 2 stan-dard deviation can
be used. The SCs are thenused as QCs in all in-study runs, as well
as partof the conformance samples for interlaboratoryperformance.
The SC data can be a commonthread to compare precision and relative
accuracyamong multiple studies by different analyticallaboratories.
In addition, SC data can be used to
detect reagent lot variability [48].
-
7/27/2019 Method Validation Application Protein Biomarkers
11/14
Method validation & application of protein biomarkers |
Review
www.future-science.com 1471future science group
Application of commercial kits
Commercial kits for diagnostic use have beencommonly adopted for
drug development sincethey are readily available. The varieties of
kitsrange from the well-established FDA-approved
(or FDA-cleared) kits to less-proven for researchuse only or for
investigational use only kits. Asthe purposes of drug development
are differentfrom that of diagnosis, it is not recommendedto
directly adopt a kit method for drug develop-ment without method
validation [30]. The valida-tion experiments should evaluate the
referencematerial and standard matrix, determine perfor-mance
characteristics, patient range and drugmodulation and set up
SCs.
The standard calibrators can be a major contrib-utor to
confounding data in research kit applica-
tion. If there are multiple commercial kit sources,it is prudent
to assay the same set of authenticsamples using various kits for
comparison. It is notsurprising to find that the results are
totally differ-ent from one kit to another because the
calibrators(yardsticks) are of different forms. In addition,
thecalibrators from one supplier can be different withtime, due to
changes in purification processes andrecalibration. If a bulk
standard material in suf-ficient quantity can be acquired from one
supplier,standard calibrators should be prepared in-house,in the
appropriate matrix, to assure calibrator con-sistency throughout an
advanced application, such
as in the example of serum CTx [48]. The bulkstandard material
also allows the preparation ofsufficient levels of calibrators with
anchor pointsfor appropriate curve fitting with weighting, aswell
as spiked QCs for accuracy and precisionexperiments to define the
assay range.
The assay range should be evaluated againstthe population range
and the desirable drugeffect. When the biomarker levels are
extremelylow, as is the case with the free soluble recep-tor
activator of NFkB ligand, many literatureresults using a research
kit reported concentra-
tions below the LLOQ. Therefore, most of thepopulation baseline
values would actually beassay noise [50].
For research-grade commercial kits, QCs orauthentic sample
controls may not be available. Itis the analysts responsibility to
set up these con-trols to characterize assay accuracy and
precisionand to monitor assay performance. For example,method
validation using commercial kits forexploratory and advanced
biomarker applica-tions have been reported for
tartrate-resistantacid phosphatase (TRACP 5b) and serum CTx
for bone resorption, respectively [24,48].
The same basic principle of fit-for-purposemethod validation
must be applied for theadoption of commercial kits for PK
bioana-lysis. Again, the responsibility resides with
thebioanalytical laboratory to establish the assay
characteristics and determine run-acceptancecriteria. Moreover,
kit comparison and rigorousspecificity tests should be conducted
[32].
Multiplex assays
Definitive quantitative methods using LCMSare capable of
multi-analyte assays using thespecific mass-to-charge ratios of
each analyte ofinterest. For peptide analytes, precursor
peptides(e.g., the prodrug or endogenous propeptide)and their
potential metabolites can be quanti-fied simultaneously [38,51].
The data handling
of multiple analytes would be similar to that ofconventional
small molecules and metabolites.Multiplex LBA platforms and
applications have
been developed for biomarkers. Multiple analyteprofiling can be
bead-based (e.g., Luminex) orplanar (e.g., MesoScale Discovery)
formats.Multiplexing saves time and requires less sam-ple volume. A
panel of potential biomarkers istested during early phase to find
those that indi-cate drug effect. The disproportionate
variablebiological ranges of the biomarker analytes andnonlinearity
of the assays should be considered inmethod design and assay
development [52]. After
the selection of the few relevant biomarkers, thedecision can be
made to use either several single-analyte methods or a multiplex of
fewer analytesfor robust assays in later phases.
Future perspective
The position paper on fit-for-purpose biomarkerassay validation
briefly discussed the differ-ences between biomarker applications
and drugbioanalysis [12]. Protein drug development andbiomarkers
share common bioanalytical tech-nologies but are being applied for
different pur-
poses. This review explains in detail the basicsimilarities and
differences between assays forbiomarkers and assays for
biotherapeutics tosupport PK/PD studies.
The intended applications of bioanalysis oftherapeutics are
usually well defined in eachstudy protocol. Method validation and
bioana-lysis are performed in a GLP-compliant labo-ratory, with
clear guidance from regulatoryagencies and consensus
recommendations fromposition publications. The recent discussions
onbiotherapeutics, in relation to their correspond-
ing biomarkers, are total and free analyte assays
Fit-FoR-puRpose
methodvaLidationProcess of dening study intentand establishing
withexperimental data whether theassay performancecharacteristics
are reliable forthe intended application
-
7/27/2019 Method Validation Application Protein Biomarkers
12/14
Review|Lee
Bioanalysis(2009) 1(8)1472 future science group
Executive summary
Intended purposes of biomarker & biotherapeutic
bioanalysis
n The purposes are well defined for pharmacokinetic (PK)
bioanalysis in a study.
n The intended applications of biomarkers are more diverse and
may not be well-defined.
n A fit-for-purpose approach for biomarker method validation and
analysis is needed; the rigor of method validation and assay
documentation depend upon exploratory or advanced
application.
Pre-analytic considerations
n Pre-analytic considerations include the choice of biomarkers
and corresponding biological matrix, the intended application and
method
validation plan based on the need for the specified exploratory
or advanced application.
Reference standards
n The reference standards of PK assays and definitive biomarkers
are well defined.
n The reference standards are not the same as, but represent,
the endogenous analytes in relative quantitative methods.Standard
calibrator matrix & selectivity
n Biomarker standard curves often use a substituted matrix
devoid of the analyte.
n More extensive matrix tests are required for biomarkers
compared with biotherapeutics.
n Parallelism of authentic samples diluted with standard matrix
are required for a relative method.
Specificity
n Method should be specific for analyte versus structurally
similar molecules (including precursors and pathway
metabolites).
n Specify if free, bound or total target biomarker or
biotherapeutic will be measured by the method.
Accuracy, precision & assay range
n Accuracy and precision validation data are used to
characterize method performance.
n Sensitivity: limit of detection may be used in addition to the
LLOQ with caution.
n Assay range is extended by sample dilution.n Nonlinear curve
fitting is used for data regression. It is necessary to assess data
variability from various sources.
n Acceptance criteria: 4-6-X rule for biotherapeutic PK;
flexible for biomarkers, depend upon the pathological modulation
and drug effect,
in addition to method performance.
Stability
n Sample controls are used to reflect authentic samples.
Reproducibility demonstrated by incurred sample or sample
control data
n There is no requirement for incurred sample with thorough
matrix tests and sample control tracking.
Application of commercial kits
n No direct adoption; users are responsible for appropriate
validation.
Multiplex assays
n Saves sample volume and time at the early stages.
[35,46] . The information is important for theunderstanding of
drugtarget interactions andperforming robust PK/PD modeling
[4144].
The intended applications of biomarkersare more diverse than
those of biotherapeu-
tics. There are applications at various stagesof novel biomarker
development (FiguRe1a),which are intertwined with drug develop-ment
[29]. The rough category of exploratoryor advanced application is a
wide spectrumthat demands flexibility in method validationrigor.
Appropriate experiments should be car-ried out on sample
collection, relative accuracy,precision, range finding,
parallelism, selectivity,specificity and stability [20]. Biomarker
bioana-lysis and method validation are continuous
processes for accumulating knowledge throughthe development of
inter-related biomarkers andunderstanding proteinprotein
interactionsof biomarkers and biotherapeutics of similarmechanisms.
Multiplex assays and other physi-
cochemical methods are evolving to enhancethis knowledge.
The development of companion diagnosticswil l open up
collaborative opportunities forthe pharmaceutical and diagnostic
sectors foreffective development and applications of
novelbiomarkers in drug development and prognosis.
Acknowledgements
The author thanks Michael Hall for critical review of
the manuscript.
-
7/27/2019 Method Validation Application Protein Biomarkers
13/14
Method validation & application of protein biomarkers |
Review
www.future-science.com 1473future science group
BibliographyPapers of special note have been highlighted as:nof
interestnn of considerable interest
1 Bild AH, Yao G, Chang JTet al.Oncogenic
pathway signatures in human cancers as a
guide to targeted therapies. Nature
439(7074), 353357 (2006).
2 Jadhav PR, Mehta MU, Gobburu JVS.
How biomarkers can improve clinical drug
development.Am. Pharm. Rev. 7, 6264
(2004).3 Kummar S, Kinders R, Rubinstein Let al.
Compressing drug development timelines in
oncology using Phase 0 trials. Nat. Rev.
Cancer7(2), 131139 (2007).
4 Wong R, Cunningham D. Using pred ictive
biomarkers to select patients with advanced
colorectal cancer for treatment with epidermal
growth factor receptor antibodies.J. Clin.
Oncol. 26(35), 56685670 (2008).
5 Wagner JA, Wi lliams SA, Webster CJ.
Biomarkers and surrogate end points for
fit-for-purpose development and regulatory
evaluation of new drugs. Clin. Pharmacol.
Therapeutics81(1), 104107 (2007).
6 US FDA. Guidance for Industry on
Bioanalytical Method Validation: Availability.
Center for Drug Evaluation and Research,
Rockville, MD, USA (2001).
n US FDA guidance that most bioanalyses are
based on.
7 DeSilva B, Smith W, Weiner Ret al.
Recommendations for the bioanalytical
method validation of ligand-binding assays to
support pharmacokinetic assessments of
macromolecules.Pharm. Res.20(11),
18851900 (2003).
n Consensus paper on the ligand-binding assay
of pharmacokinetic bioanalysis.
8 Smolec J, DeSilva B, Smith Wet al.
Bioanalytical method validation for
macromolecules in support of
pharmacokinetic studies. Pharm. Res. 22(9),
14251431 (2005).
9 Kelley M, DeSilva B. Key elements of
bioanalytical method validation for
macromolecules.AAPS J. 9(2), E156E163
(2007).
10 Viswanathan CT, Bansal S, Booth Bet al.
Quantitative bioanalytical methods va lidation
and implementation: best practices for
chromatographic and ligand binding assays.
Pharm. Res. 24(10), 19621973 (2007).
11 Bansal S, DeStefano A. Key elements of
bioanalytical method validation for small
molecules.AAPS J. 9(1), E109E114 (2007).
12 Lee JW, Devanarayan V, Barrett YCet al.
Fit-for-purpose method development and
validation for successful biomarker
measurement. Pharm. Res. 23(2), 312328
(2006).
n First White Paper on fit-for-purpose
method validation and applicationfor biomarkers.
13 Lee JW, Weiner RS, Sailstad JM et al.
Method validation and measurement of
biomarkers in nonclinical and clinical
samples in drug development: a conference
report. Pharm. Res. 22(4), 499511 (2005).
14 Chau CH, Rixe O, McLeod H, Figg WD.
Validation of analytic methods for
biomarkers used in drug development. Clin.
Cancer Res.14(19), 59675976 (2008).
nn Good overall rev iew of biomarkers for
drug development.
15 Cummings J, Ward TH, Greystoke A,
Ranson M, Dive C. Biomarker method
validation in anticancer drug development.
Br. J. Pharmacol. 153(4), 646656 (2008).
16 Goodsaid F, Frueh F. Biomarker
qualification pilot process at the US Food
and Drug Administration.AAPS J.9(1),
E105108 (2007).
17 Goodsaid FM, Frueh FW, Mattes W.
Strategic paths for biomarker qualification.
Toxicology245(3), 219223 (2008).
18 US FDA. Using disease, placebo, and drug
prior knowledge to improve decisions in
drug development and at FDA. Case StudiesAcros s Companies
Disease Models at FDA:
Overview and Case Studies (Diabetes and
Obesity).FDA, Rockville, MD, USA (2006).
19 Stoch SA, Wagner JA. Biomarker analysis as
a decision-making tool in drug discovery and
development: implications for peroxisome
proliferator-activator receptors. Int. J. Pharm.
Med. 21, 271277 (2007).
20 Lee J, Pan Y, OBrian P, Xu R.
Development and validation of
ligand-binding assays for biomarkers.
In:Ligand-Binding Assays: Development,
Validation and Implementation in the Drug
Development.Khan MN, Findlay WA (Eds).
John Wi ley & Sons, NY, USA, 129161
(2010).
21 Nowatzke W, Woolf E. Best practices
during bioanalytical method validation
for the characterization of assay reagents
and the evaluation of analyte stability in
assay standards, quality controls, and
study samples.AAPS J. 9(2), E117E122
(2007).
22 Banks RE. Preanalytical influences in
clinical proteomic studies: raising awareness
of fundamental issues in sample banking.Clin. Chem. 54(1), 67
(2008).
23 Ferguson RE, Hoschstrasser DF, Banks RE.
Impact of preanalytical variable on the
analysis of biological fluids in proteomic
studies. ProteomicsClin. Appl. 1, 739746
(2007).
24 Wu Y, Lee JW, Uy L et al.Tartrate-resistant
acid phosphatase (TRACP 5b): a biomarker
of bone resorption rate in support of drug
development: modification, validation and
application of the BoneTRAP kit assay.
J. Pharm. Biomed . Anal .49(5), 12031212
(2009).
25 Clinical and Laboratory Standards Institute.
Procedures for the Handling and Processing of
Blood Specimens; Approved Guideline (Third
Edition). Document number H18-A3
(2004).
26 Clinical and Laboratory Standards Institute.
Procedures for the Collection of Diagnostic
Blood Specimens by Venipuncture: Approved
Standard (Sixth Edition).Document number
H3-A6 (2007).
27 American Diabetes Associat ion, European
Associat ion for the Study of Diabetes,
International Federation of Clinical
Chemistry and Laboratory Medicine,International Diabetes
Federation.
Consensus statement on the worldwide
standardisation of the HbA1c
measurement. Diabetologia50(10),
20422043 (2007).
28 Khatami M. Standard izing cancer biomarkers
criteria: data elements as a foundation for a
database. Inflammatory mediator/M-CSF as
model marker. Cell Biochem. Biophys. 47,
187198 (2007) .
29 Lee JW, Figeys D, Vasilescu J. Biomarker
assay translation from discovery to clinical
studies in cancer drug development:
Financial & competing interests disclosure
The author has no relevant affiliations or financial
involve-
ment with any organization or entity with a financial inter-
est in or financial conflict with the subject matter or
materi-
als discussed in the manuscript. This includes employment,
consultancies, honoraria, stock ownership or options, expert
testimony, grants or patents received or pending,
or royalties.
No writing assistance was utilized in the production of
this manuscript.
-
7/27/2019 Method Validation Application Protein Biomarkers
14/14
Review|Lee
Bioanalysis (2009) 1(8)1474 f t i
quantification of emerging protein biomarkers.
In: Genomics in Cancer Drug Discovery and
Development.Hampton GM, Sikora K (Eds).
Elsevier, London, UK, 269298 (2007).
30 Lee JW, Hall M. Method validation of protein
biomarkers in support of drug development or
clinical diagnosis/prognosis.J. Chromatogr. B877(13), 12591271
(2009).
nn Overall review of method validation and
implementation approaches for the
exploratory and advanced application
of biomarkers.
31 Lee JW, Ma H. Specificity and selectivity
evaluations of ligand binding assay of protein
therapeutics against concomitant drugs and
related endogenous proteins.AAPS J. 9,
E164E170 (2007).
32 Sukovaty RL, Lee JW, Fox J et al.
Quantification of recombinant human
parathyroid hormone (rhPTH(184)) inhuman plasma by immunoassay:
commercial
kit evaluation and validation to support
pharmacokinetic studies.J. Pharm. Biomed .
Anal. 42, 261271 (2006).
33 Hall M, Lee JW, Spahr C, Lu H, Ortiz R.
Ligand bindingmass spectrometry methods
for understanding macromolecular drug
biotransformation and impact on
immunoassay quantification. Presented at:
56th Annual ASMS Conference on Mass
Spectrometry and Allied Topics. Denver, CO,
USA, 15 June 2008.
34 Clinical and Laboratory Standards Institute.
Evaluation of Matrix Effects; Approved
Guideline (Second Edition).Document number
EP14A2 (2005).
35 Lee J, Quarmby V, Yang J, Ahene A,
Salimi-Moosavi H. Why do we care whether a
LBA is measuring total or free protein
therapeutic or biomarker?AAPS Ligand
Binding Assay Bioanalytical Focus Group
Newsletter.July (2008).
36 Oe T, Ackermann BL, Inoue K et al.
Quantitative analysis of amyloid bpeptides in
cerebrospinal fluid of Alzheimer's disease
patients by immunoaffinity purification and
stable isotope dilution liquid chromatography/negative
electrospray ionization tandem mass
spectrometry. Rapid Commun. Mass Spectrom.
20(24), 37233735 (2006).
37 Barnidge DR, Goodmanson MK, Klee GG,
Muddiman DC. Absolute quantification of
the model biomarker prostate-specific antigen
in serum by LC-MS/MS using protein
cleavage and isotope dilution mass
spectrometry.J. Proteome Res.3(3), 644 652
(2004).38 Li H, Rose MJ, Tran Let al.Development of
a method for the sensitive and quantitative
determination of hepcidin in human serum
using LC-MS/MS.J. Pharmacol. Toxicol .
Meth. 59(3), 171180 (2009).
39 Kemna EH, Tjalsma H, Podust VN,
Swinkels DW. Mass spectrometry-based
hepcidin measurements in serum and urine:
analytical a spects and clinical implications.
Clin. Chem. 53(4), 620628 (2007).
40 Kiernan UA, Nedelkov D, Nelson RW.
Multiplexed mass spectrometric immunoassay
in biomarker research: a novel approach to the
determination of a myocardial infarct.
J. Proteome Res. 5(11), 29282934 (2006).
41 Hayashi N, Tsukamoto Y, Sallas WM,
Lowe PJ. A mechanism-based binding model
for the population pharmacokinetics and
pharmacodynamics of omalizumab.
Br. J. Clin. Pharmacol. 63(5), 548561
(2007).
42 Rudge JS, Holash J, Hylton Det al.Inaugural
article: VEGF trap complex formation
measures production rates of VEGF,
providing a biomarker for predicting
efficacious angiogenic blockade. Proc. Natl
Acad. S ci. USA 104(47), 1836318370(2007).
43 Lowe PJ, Gautier A. On the ability to predict
free ligand suppression when free l igand
assays are not available or impossible.
Presented at:Annual Meeting of the Populat ion
Approach Group in Europe. St Petersburg,
Russia, 2326 June 2009.
44 Tannenbaum S, Gautier A, Lowe PJ.
A mechanism based binding model for the
population pharmacokinetics and
pharmacodynamics of canakinumab, a
monoclonal antibody in development for
rheumatoid arthritis. Presented at:AmericanAssociation of
Pharmaceutical Scientis ts
Meeting. Atlanta, GA, USA, 1719 November
2008.
45 Moxness M, Tatarewicz S, Weeraratne D
et al. Immunogenicity testing by
electrochemiluminescent detection for
antibodies directed against therapeutic
human monoclonal antibodies. Clin Chem.
51(10), 19831985 (2005).
46 Salimi-Moosavi H, Burns Det al.A novelapproach for the
measurement of total and
free target proteins in serum samples in the
presence of antibody therapeutics. Presented
at:AAPS National B iotechnolog y Conference.
Toronto, ON, Canada, 2225 June 2008.
47 Wayne PA. Protocols for determination of
limits of detection and limits of quantitation;
proposed guideline. Clinical and Laboratory
Standards Institute. Document number
EP17-A (2004).
48 Wang J, L ee J, Burns Det al.Fit-for-
purpose method validation and application
of a biomarker (C-terminal telopeptides of
type 1 collagen) in denosumab clinical
studies.AAPS J. 11(2), 385394 (2009).
n Practical illustration of modification
and implementing a commercial kit
for the advanced application of a
pharmacodynamic biomarker for
drug development.
49 Findlay JWA, Dillard RF. Appropriate
calibration curve fitting in ligand binding
assays.AAPS J. 9, E260267 (2007).
50 Bowsher RR, Sailstad JM. Insights in the
application of research-grade diagnostic kits
for biomarker assessments in suppor t ofclinical drug
development: bioanalysis of
circulating concentrations of soluble receptor
activator of nuclear factor kB ligand.
J. Pharm. Biomed. Anal. 48(5), 12821289
(2008).
51 Tubbs KA, Kiernan UA, Niederkofler EE,
Nedelkov D, Bieber AL, Nelson RW.
Development of recombinant-based mass
spectrometric immunoassay with application
to resistin expression profiling.Anal . Chem.
78(10), 32713276 (2006).
52 Ray CA, Dumaual C, Willey Met al.
Optimization of analytical and pre-
analytical variables associated with an ex vivocytokine
secretion assay.J. Pharm. Biomed.
Anal. 41(1), 189195 (2006).