Embed Size (px)
Citation preview

540 VOLUME 25 NUMBER 5 MAY 2007 NATURE BIOTECHNOLOGY
CO2, its ability to generate energy distinguishes it from the endogenous photorespiratory pathway that consumes reductant and ATP5. Sidestepping the mitochondrial and peroxisomal photorespiratory reactions generates NADH while eliminating the expenditure of ATP and reductant normally required to refix ammonia by the glutamine synthetase/glutamate synthase cycle6 (Fig. 1). However, as high fluxes of the engineered shunt might lead to redox imbal-ance within the chloroplast, a trade-off for the superior nitrogen-use efficiency and reduced energy consumption might be that it diminishes the role that photorespiration could play in the dissipation of excess energy as a stress response to adverse environmental conditions5.
The work of Kebeish et al. is the latest of sev-eral efforts to diminish the impact of photores-piration. These include attempting to improve the efficiency of rubisco by rational engineer-ing or by prospecting for more efficient natu-ral variants7 and concentrating CO2 around rubisco by emulating C4 plants or expressing a cyanobacterial bicarbonate transporter8. However, the engineering of C4 photosynthe-sis into C3 plants, such as rice9,10, is inherently more ambitious than introducing the glyco-late shunt as it requires the engineering of a photosynthetic pathway in two cell types, as well as structural modifications to reduce CO2 leakage from the vicintity of rubisco, a problem that must always be considered in engineering CO2-concentrating mechanisms.
The potential rewards of taming photorespi-ration are enormous. Enabling photosynthesis to operate at lower leaf CO2 concentrations would increase the efficiency of using water (through reduced stomatal transpiration) and nitrogen (through more efficient use of rubisco, the most abundant protein in leaves). Improved CO2 fixation could, therefore, not only increase the productivity of crops to meet the world’s ever-increasing demands for food and fuel, but also simultaneously decrease consumption of water, fertilizer and land9,10.
COMPETING INTERESTS STATEMENTThe author declares no competing financial interests.
1. Sharkey, T.D. Physiol. Plant. 73, 147–152 (1988).2. Kebeish, R. et al. Nat. Biotechnol. 25, 593–599
(2007).3. von Caemmerer, S. Plant Cell Environ. 26, 1191–1197
(2003).4. Eisenhut, M. et al. Plant Physiol. 142, 333–342
(2006).5. Wingler, A. et al. Phil. Trans. R. Soc. Lond. B 355,
1517–1529 (2000).6. Keys, A.J. et al. Nature 275, 741–743 (1978).7. Andrews, T.J. & Whitney, S.M. Arch. Biochem. Biophys.
414, 159–169 (2003).8. Lieman-Hurwitz, J. et al. Plant Biotechnol. J. 1, 43–50
(2003).9. Sheehy, J.E., Mitchell, P.L. & Hardy, B. Redesigning
Rice Photosynthesis to Increase Yield (International Rice Research Institute/Elsevier, Los Baños, Philippines, 2000).
10. Normile, D. Science 313, 423 (2006).
Metagenomic sorcery and the expanding protein universeEugene V Koonin
A metagenomic study of the Atlantic and Pacific oceans reveals dominant bacterial species, remarkable microdiversity and a wealth of new proteins.
The magisterial series of three articles pub-lished recently by J. Craig Venter and col-leagues1–3 in PLOS Biology establishes a new milestone for metagenomics. The previous groundbreaking work from this4 and other groups5,6 notwithstanding, the scale of the new study is such that for the first time we can begin to contemplate the planet-wide diversity of prokaryotic life in an important habitat.
The Sorcerer II expedition traveled from Nova Scotia in the Northwestern Atlantic to Hawaii in the tropical Pacific, producing an unprecedented 6.25 gigabases of prokaryotic DNA sequence from surface water samples collected at 41 locations. Fittingly, Venter and colleagues note that they drew their inspira-tion from the famous HMS Challenger survey of 1872–1876, which explored the worldwide diversity of macroscopic marine life. As 10 of their 41 samples were collected off the Galapagos Islands, one cannot help but think also of the voyage of the HMS Beagle. It took Darwin 20 years to transform the observations made during this voyage, particularly in the Galapagos Islands, into his sweeping vision of evolution by natural selection. Similarly, work-ing out the full implications of the Sorcerer II study will take time. Nevertheless, the wealth of information and analytic results in the three papers already convey the general significance of these studies.
The overarching theme is diversity—within bacteria, viruses and protein families. The first paper1 addresses genomic diversity. At the level of bacterial ribotypes (that is, distinct classes of prokaryotic sequences defined by 16S rRNA comparison and corresponding roughly to species) and, especially, higher taxa, the extent of diversity found was, in fact, relatively low. Altogether, 811 ribotypes were observed, about half of which are novel. However, only 60 ribo-types were abundant (together accounting for >70% of the 16S RNA sequences), and all but one of these had been described previously.
Furthermore, ~70% of the sequence reads aligned to one or more of the 584 prokaryotic genomes available from public databases at the time of the analysis.
Considering the relatively low sensitivity of the assignment method (BLAST with nucleo-tide sequences7), it therefore seems that, at the level of the ribotype, most of the ocean’s microbial diversity is already represented in genomics databases. That unknown prokary-otes, at least near the ocean’s surface, largely fall within known groups might come as a sur-prise to many microbiologists given that most bacteria cannot currently be cultivated.
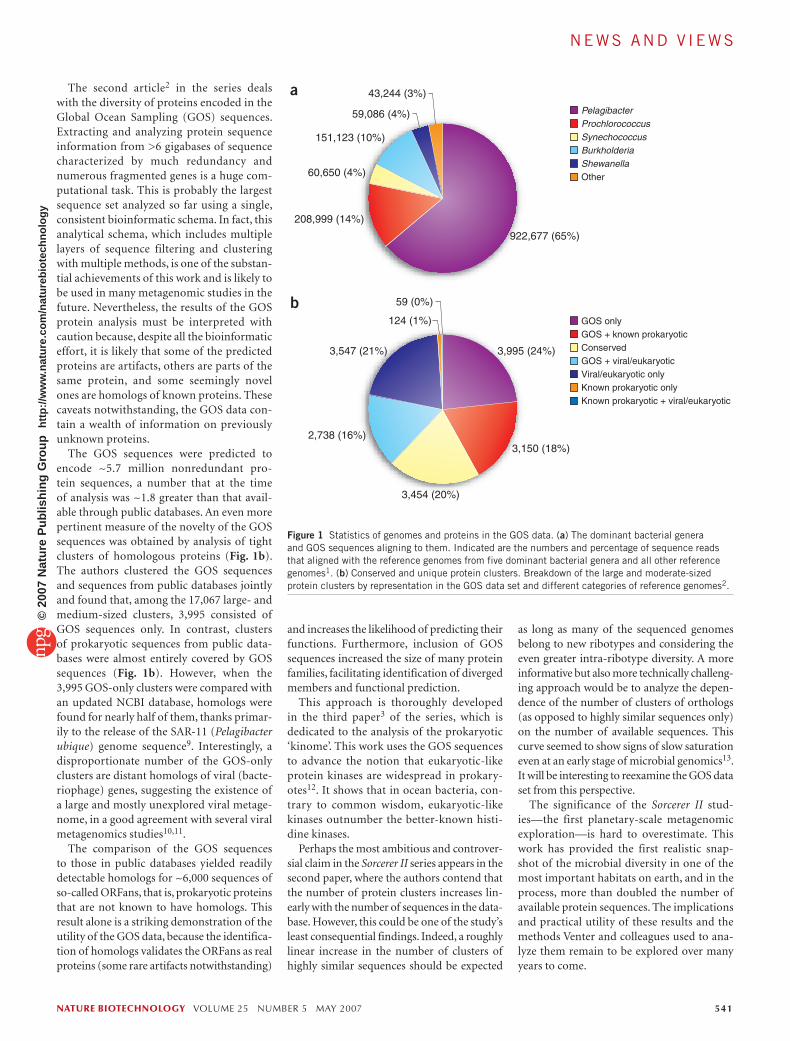
The Sorcerer II results suggest that the dis-tribution of identifiable bacteria is strikingly nonuniform. The entire genomic landscape is dominated by only five bacterial genera, which together account for ~97% of the sequences that can be aligned to available genomes. More than 60% of these sequences are from Pelagibacter alone—a striking case of domina-tion of the microbial communities by a select few (Fig. 1a).
The relative lack of diversity at the higher taxonomic levels contrasts with the extensive diversity within each ribotype. Quite strik-ingly, even though sequences aligning to many parts of the reference genomes of the abundant bacteria were sequenced hundreds of times over, very few identical sequences were found, suggesting that no two cells that contribute to the metagenome have identical genome sequences. Nevertheless, the diversity within ribotypes was shown by phylogenetic analysis to have a clear structure of distinct subtypes. Furthermore, there seems to be con-siderable variability in the gene composition of the abundant genomes, and each includes gaps (regions to which no or few environmen-tal sequences are assigned) that are likely to be hotspots of horizontal gene transfer. Taking their cue from the Beagle naturalist and in accord with the theoretical predictions of intense selection acting on large populations8, Venter and colleagues suggest that the diver-sity is largely adaptive: the different subtypes have distinct roles in the ecosystem and their combined presence provides the system with stability and robustness.
Eugene V. Koonin is at the National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, 8600 Rockville Pike, Bethesda, Maryland 20894, USA. e-mail: [email protected]
NEWS AND V IEWS©
2007
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
NATURE BIOTECHNOLOGY VOLUME 25 NUMBER 5 MAY 2007 541
The second article2 in the series deals with the diversity of proteins encoded in the Global Ocean Sampling (GOS) sequences. Extracting and analyzing protein sequence information from >6 gigabases of sequence characterized by much redundancy and numerous fragmented genes is a huge com-putational task. This is probably the largest sequence set analyzed so far using a single, consistent bioinformatic schema. In fact, this analytical schema, which includes multiple layers of sequence filtering and clustering with multiple methods, is one of the substan-tial achievements of this work and is likely to be used in many metagenomic studies in the future. Nevertheless, the results of the GOS protein analysis must be interpreted with caution because, despite all the bioinformatic effort, it is likely that some of the predicted proteins are artifacts, others are parts of the same protein, and some seemingly novel ones are homologs of known proteins. These caveats notwithstanding, the GOS data con-tain a wealth of information on previously unknown proteins.
The GOS sequences were predicted to encode ~5.7 million nonredundant pro-tein sequences, a number that at the time of analysis was ~1.8 greater than that avail-able through public databases. An even more pertinent measure of the novelty of the GOS sequences was obtained by analysis of tight clusters of homologous proteins (Fig. 1b). The authors clustered the GOS sequences and sequences from public databases jointly and found that, among the 17,067 large- and medium-sized clusters, 3,995 consisted of GOS sequences only. In contrast, clusters of prokaryotic sequences from public data-bases were almost entirely covered by GOS sequences (Fig. 1b). However, when the 3,995 GOS-only clusters were compared with an updated NCBI database, homologs were found for nearly half of them, thanks primar-ily to the release of the SAR-11 (Pelagibacter ubique) genome sequence9. Interestingly, a disproportionate number of the GOS-only clusters are distant homologs of viral (bacte-riophage) genes, suggesting the existence of a large and mostly unexplored viral metage-nome, in a good agreement with several viral metagenomics studies10,11.
The comparison of the GOS sequences to those in public databases yielded readily detectable homologs for ~6,000 sequences of so-called ORFans, that is, prokaryotic proteins that are not known to have homologs. This result alone is a striking demonstration of the utility of the GOS data, because the identifica-tion of homologs validates the ORFans as real proteins (some rare artifacts notwithstanding)
and increases the likelihood of predicting their functions. Furthermore, inclusion of GOS sequences increased the size of many protein families, facilitating identification of diverged members and functional prediction.
This approach is thoroughly developed in the third paper3 of the series, which is dedicated to the analysis of the prokaryotic ‘kinome’. This work uses the GOS sequences to advance the notion that eukaryotic-like protein kinases are widespread in prokary-otes12. It shows that in ocean bacteria, con-trary to common wisdom, eukaryotic-like kinases outnumber the better-known histi-dine kinases.
Perhaps the most ambitious and controver-sial claim in the Sorcerer II series appears in the second paper, where the authors contend that the number of protein clusters increases lin-early with the number of sequences in the data-base. However, this could be one of the study’s least consequential findings. Indeed, a roughly linear increase in the number of clusters of highly similar sequences should be expected
as long as many of the sequenced genomes belong to new ribotypes and considering the even greater intra-ribotype diversity. A more informative but also more technically challeng-ing approach would be to analyze the depen-dence of the number of clusters of orthologs (as opposed to highly similar sequences only) on the number of available sequences. This curve seemed to show signs of slow saturation even at an early stage of microbial genomics13. It will be interesting to reexamine the GOS data set from this perspective.
The significance of the Sorcerer II stud-ies—the first planetary-scale metagenomic exploration—is hard to overestimate. This work has provided the first realistic snap-shot of the microbial diversity in one of the most important habitats on earth, and in the process, more than doubled the number of available protein sequences. The implications and practical utility of these results and the methods Venter and colleagues used to ana-lyze them remain to be explored over many years to come.
43,244 (3%)
59,086 (4%)
59 (0%)
124 (1%)
3,547 (21%)
2,738 (16%)
3,454 (20%)
3,150 (18%)
3,995 (24%)
151,123 (10%)
60,650 (4%)
208,999 (14%)
922,677 (65%)
PelagibacterProchlorococcusSynechococcusBurkholderiaShewanellaOther
GOS onlyGOS + known prokaryoticConservedGOS + viral/eukaryoticViral/eukaryotic onlyKnown prokaryotic onlyKnown prokaryotic + viral/eukaryotic
a
b
Figure 1 Statistics of genomes and proteins in the GOS data. (a) The dominant bacterial genera and GOS sequences aligning to them. Indicated are the numbers and percentage of sequence reads that aligned with the reference genomes from five dominant bacterial genera and all other reference genomes1. (b) Conserved and unique protein clusters. Breakdown of the large and moderate-sized protein clusters by representation in the GOS data set and different categories of reference genomes2.
NEWS AND V IEWS©
2007
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy

542 VOLUME 25 NUMBER 5 MAY 2007 NATURE BIOTECHNOLOGY
COMPETING INTERESTS STATEMENTThe author declares no competing financial interests.
1. Rusch, D.B. et al. PLoS Biol. 5, e77 (2007).2. Yooseph, S. et al. PLoS Biol. 5, e16 (2007).3. Kannan, N., Taylor, S.S., Zhai, Y., Venter, J.C. &
Manning, G. PLoS Biol. 5, e17 (2007).4. Venter, J.C. et al. Science 304, 66–74 (2004).5. Tringe, S.G. et al. Science 308, 554–557 (2005).6. Langer, M. et al. Biotechnol. J. 1, 815–821 (2006).7. Altschul, S.F., Gish, W., Miller, W., Myers, E.W. &
Lipman, D.J. J. Mol. Biol. 215, 403–410 (1990).8. Giovannoni, S.J. & Stingl, U. Nature 437, 343–348
(2005).9. Giovannoni, S.J. et al. Science 309, 1242–1245
(2005).10. Suttle, C.A. Nature 437, 356–361 (2005).11. Angly, F.E. et al. PLoS Biol. 4, e368 (2006).12. Leonard, C.J., Aravind, L. & Koonin, E.V. Genome Res.
8, 1038–1047 (1998).13. Tatusov, R.L. et al. Nucleic Acids Res. 29, 22–28
(2001).
Fungal genomics goes industrialNicholas J Talbot
A functional genomics study of the rice blast fungus unravels the genetic basis of pathogenicity.
Rice blast is one of the plant world’s most dev-astating diseases, each year destroying enough rice to feed 60 million people1,2. In a recent issue of Nature Genetics, Jeon et al.1 present a high-throughput functional genomics study of the rice blast fungus, Magnaporthe oryzae. Their results, which involved the generation of >20,000 mutants of M. oryzae, constitute the largest individual study of fungal pathoge-nicity published to date and reveal many new gene functions required for rice blast disease.
Fungi are responsible for the rusts, mildews, blasts and wilts that afflict many of our most important crop species, including the cere-als that sustain much of the planet’s human population. In addition, fungal diseases of humans are on the rise as a consequence of the widespread use of immunosuppressive therapies for treating chronic illnesses and the prevalence of autoimmune disorders. Taken together, the human, social and economic costs of fungal infections provide a powerful incentive to investigate the mechanisms that diverse fungi deploy to cause disease.
There are, however, significant obstacles to studying pathogenic fungi using the tools of molecular genetics and genomics3. Many fungal species lack sexual reproductive stages, which prevents the use of conventional for-ward genetics, and most fungi exhibit very low frequencies of DNA-mediated trans-formation, prohibiting complementation studies3. Other important pathogenic spe-cies—the rust and powdery mildew fungi, for example—cannot be cultured at all outside a living host.
Despite these difficulties, much has been learned about fungal pathogens in recent years, mainly by the careful deployment of reverse genetic analysis. Researchers have begun to define the major signaling pathways associated with initial plant infection4,5, the processes by which fungi are perceived by resistant plant varieties6 and the manner in which fungi invade plant and animal tissues to cause disease4,7. These studies have relied on selective study of model fungal species for which genetic resources have been developed and have primarily used targeted gene replace-ment to define gene function3. Above all, the study of fungal biology has been revolution-ized by the rapid sequencing of >30 species8. These sequencing efforts have presented a new challenge: how to determine the functions of each of the ~10,000 genes of a fungal species in a high-throughput manner.
The work of Jeon et al. represents a sig-nificant step forward in achieving this aim for M. oryzae. A large library of mutants was generated using insertional mutagenesis with Agrobacterium tumefaciens (Fig. 1). A. tume-faciens is, of course, the workhorse of plant molecular biology, but it has become clear in recent years that it can be a highly efficient method for introducing DNA into filamen-tous fungi9, with reported frequencies of up to 800 transformants per experiment (compared with <40 transformants per µg of DNA in a conventional fungal-transformation experi-ment). Although insertional mutagenesis in fungi has been used previously to identify novel determinants of pathogenicity2,5, the scale of this study is unprecedented.
Jeon et al. carefully cataloged the mutants and stored them in multiwell plates. A set of high-throughput screens was then deployed to characterize growth- and virulence-associated
phenotypes. These included the ability of each mutant to sporulate, to form specialized infection structures called appressoria4 and to cause rice blast disease when inoculated on rice leaves. Although analysis of plant infection used a detached leaf assay in single tubes rather than whole plants, this was still a large-scale effort, because extensive technical replication was necessary to confirm the ability of each mutant to cause rice blast disease symptoms.
The real power of the method, however, lies in the analysis of insertion sites, which show good coverage of T-DNA insertions across the M. oryzae genome10 and saturation of ~61% of the genome1. A relational database was gen-erated to provide a key to all of the insertion lines of M. oryzae created. Two hundred two new pathogenicity loci have so far been identi-fied. This is about twice as many as were pre-viously known, with the existing knowledge having been gained during the previous 18 years of molecular genetic studies.
What are the lessons learned from this work, and how might others who study fungal dis-eases benefit? First and foremost, the existing mutant collection has been replicated and safely archived so that it can be made avail-able to the worldwide community of research-ers studying rice blast. Second, the informatic resources generated are now being interlinked in an effective way with the ongoing annota-tion program for the M. oryzae genome10, making it a seamless process to go from ana-lyzing a novel gene sequence to finding the corresponding insertion mutant.
The study of Jeon et al. highlights the need for similar industrial-scale studies of other important cereal pathogens for which genome sequence information is becoming available and in which there are well-organized com-munities of researchers already working. This is a key priority given the diversity of mecha-nisms deployed by fungal pathogens to invade their plant and animal hosts and the impor-tance of developing durable disease control strategies. The ability to validate a virulence factor quickly in a large number of pathogenic species would allow academic research to be far more easily translated into target identifi-cation and validation for new antifungal drug development.
There are other obstacles that hinder prog-ress in studying pathogenic fungi. One prior-ity is increasing the ease of gene replacements to validate gene functions. Only in this way can rapid analysis of signaling and metabolic pathways be carried out from the leads identi-fied through insertional mutagenesis. Useful tools in this regard are fungal strains that can carry out only homologous recombination because the nonhomologous DNA repair
Nicholas J. Talbot is at the School of Biosciences, University of Exeter, Geoffrey Pope Building, Exeter EX4 4QD, UK.e-mail: [email protected]
NEWS AND V IEWS©
2007
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy





























