Embed Size (px)
Citation preview

Supplementary information to
De novo loss-of-function mutations in WAC cause a recognizable intellectual disability syndrome and are associated with learning deficits in Drosophila
1

CASE REPORTS
Individual 1 was reported before. 1 She is a women, born after 42 weeks of an uncomplicated pregnancy, as the second of two children, to non-consanguineous parents. Family history for developmental delay was negative. From the start she had severe developmental delay and started to walk independently at 2 years and 10 months of age but with toe walking. At 22 years of age, she had a lack of speech and severe ID. She showed behaviour problems including self-mutilation, sleep disturbances and feeding problems. Due to a diaphragmatic hernia, she had oesophageal reflux. In her teens, she developed absence seizures, which were not well controlled by various anti epileptic drugs. Physical examination showed facial dysmorphism including a square shape of the face, deep set eyes, epicanthus, long palpebral fissures, short philtrum, broad mouth and broad chin. She also had breathing dysregulation. Previous investigations consisting of a CT cerebrum and metabolic screening showed no abnormalities. Genetic testing included a 250k SNP genomic microarray analysis, Angelman syndrome methylation testing and MECP2, RAI1 and TCF4 targeted sequencing, but all were normal. Whole exome sequencing revealed two de novo mutations, in WAC (NM_016628.3:c.139C>T; p.(Arg47*)) and MIB1 (NM_020774.2:c.521G<A; p.(Arg147His)), respectively (Table 1).
Individual 2 is a boy, and single child born to non-consanguineous parents. There was no family history of developmental delay. He was born at term after ventouse extraction after an uncomplicated gestation with a birth weight of 3580 gram (0 SDS). His motor development was delayed with walking at the age of 24 months. He had a normal language development, but at the age of 5 years, there was temporary regression of speech. Psychological assessment at the age of 10 years showed mild ID with an IQ of 58. He had behavioural problems consisting of autism, concentration problems, anxiety and a sleeping disorder. His breathing pattern was disturbed, possibly due to laryngomalacy. Other respiratory problems included asthma and recurrent respiratory infections. From the age of 11 years on he suffers from gender dysphoria. Physical examination at the age of 12 years showed a height of 149.7 cm (-1.1 SDS), a high weight of 48.8 kg (2.1 SDS) and a head circumference of 56.3 cm (1.1 SDS). At the age of 18 years he developed hypertrichosis, necessitating laser treatment. Currently he is 23 years old. He has frequent outbursts of aggression. Due to problems with balance, he has developed extreme anxiety for walking stairs and cycling. Facial characteristics included a square shape of the head, low posterior hairline, simple ears, low set and full eyebrows with synophrys, hypertelorism, deep set eyes, long palpebral fissures, deep nasal bridge, flat nose, broad chin, broad mouth, broad gums and bifid tip of the tongue. He had a hoarse voice. His extremities were hypotonic and he had broad first digits. Previous investigations consisting of genetic tests, including karyotyping, analysis of FMR1, subtelomeric regions and RAI1 were all normal. Metabolic screening and an MRI of the brain were normal. In blood, a low cholesterol was measured (2.09 and 2.2 mmol/L (N: 3.9-7.3 mmol/L), but there was no defect in cholesterol biosynthesis. Using a 250k genomic SNP microarray, a de novo partial deletion of WAC was identified hg19 chr10:g.(?_288422777)_(28929097_?)del (ISCN nomenclature: [hg19] arr snp 10p11.23p11.23(288422777-28929097)x1 dn) (Table 1).
Individual 3 is a boy and the second of two children born to non-consanguineous parents. There was no family history of developmental delay. Birth after 40+5 weeks of gestation was uncomplicated with a birth weight of 3600 gram (0 SDS). His development was delayed with the first steps at the age of 2 years, and first words at the age of 2 years and 6 months. At the age of 9 years, he spoke only 3
2

to 4-word sentences, used supportive sign language and had a hypotonic dysarthria. In childhood, he developed concentration problems and hyperactivity. Asthma and recurrent upper respiratory tract infections were present. Moreover, there were frequent febrile convulsions. He was easily fatigued and had unclear reduced vision during movements. Surgery was necessary to correct phimosis. Physical examination at the age of 9 years showed a height of 131.3 cm (-1 SDS), a weight of 31.5 kg (+1.25 SDS) and a head circumference of 52 cm (-0.5 SDS). Facial dysmorphisms included a square shape of the face, brachycephaly, broad forehead, prominent antihelix, posterior ear creases, low set and full eyebrows with synophrys, long palpebral fissures, prominent teeth, broad mouth and broad chin. Brachydactyly of fingers and toes, a unilateral Simian crease, fetal finger pads, a sandal gap and a pedes planus valgi were present. He had hypotonic limbs and walked on his toes. Previous investigations consisting of karyotyping, metabolic screening in urine and blood, analysis of subtelomeric regions, 250k SNP array, OPA1, POLG and RYR1 analysis were normal. An MRI cerebrum showed megacisterna magna and in a muscle biopsy, low mitochondrial activity was measured. Whole exome sequencing revealed a de novo mutation, NM_016628.3:c.329C>A; p.(Ser110*), in WAC (Table 1).
Individual 4 is a male and the only child of non-consanguineous healthy parents. There was no family history of developmental delay. He was born after 37 weeks of an uncomplicated gestation. His birth weight was normal (>3500gr) and he had large frontal fontanel. His development was delayed with an axial hypotonia from the beginning, delayed achievement of motor milestones and absence of speech at the age of 1.5 years. At the age of 1.5 years, there was remarkable hypotonia. He could only walk with support on both hands and showed poor facial expression with poor eye contact. Eye movements were normal. Further evaluation of his vision has started. The first four months there was a disturbed sleep pattern with predominant crying probably due to gastro-oesophageal reflux. Physical examination at the age of 1.5 years showed a height of 75.4 cm (-2 SDS), a weight of 10 kg (-2 SDS) and a head circumference of 48.5 cm (0 SDS). Facial dysmorphisms included bilateral epicanthus, slight ptosis and broad nasal bridge. He had broad upper legs and small curious planovalgus feet. Previous investigation consisting of Cytoscan array, revealed a maternally inherited 9p22.1 gain and a Xp22.31 gain. Extensive metabolic screening showed no abnormalities. MRI scan of the brain was normal. Whole exome sequencing revealed a de novo mutation, NM_016628.3:c.1885_1886del; p.(Leu629fs), in WAC (table 1).
Individual 5, a girl, was born as the first child of non-consanguineous parents. Mother had ulcerative colitis and used azathioprine, mesalazin and prednisone during pregnancy. Family history was otherwise unremarkable. She was born at 37+2 weeks gestation via C-section because of breech position. Birth weight was 2920 g (-0.1 SD), length 44 cm (-1,9 SD), Apgar scores 9 and 10 after 1 and 5 minutes, respectively. As a neonate she had mild feeding difficulties and hypotonia was noted. She was referred at the age of 15 months because of axial hypotonia and motor delay. She could roll, but could not sit unsupported. Speech development was normal with 10 single words. On physical examination length was 72.5 cm (-1.8 SDS), weight 8.5 kg (0 SDS for length), and head circumference 47 cm (0 SDS). She had a relative large head, with prominent forehead, open anterior fontanel, square face, bilateral epicanthic folds, flat nasal bridge, and hypertelorism. She had small hands and small, puffy feet with short toes. On re-evaluation at the age of 3y 2m height was 87 cm (-3 SDS), weight 13 kg (0.7 SDS for height), head circumference 50.4 cm (-0.6 SDS). She had started walking unsupported at the age of 2.5 years. At the age of 3 years sleeping problems (waking up in the
3

middle of the night, rising early) became apparent. At the age of 41 months motor skills were measured (BSID-x): fine motor skills were at a level of 27 months, and gross motor skills at a level of 18 months. At the age of 4 years IQ had been tested (SON-R), she scored a disharmonious profile: performance 66, reasoning 91. She got glasses because of refraction error (+3.75 D). Currently, at the age of 6 years her concentration is poor, and she scores high on ADHD-scales. She has articulation problems due to hypotonia in the oral region. Exome sequencing identified a de novo mutation, NM_016628.3:c.356dup; p.(Asn119fs), in WAC (Table 1).
Individual 6 is a female and first of two children of non-consanguineous parents. There was no family history of developmental delay. She was born after 41+2 weeks with ventouse extraction after an uncomplicated gestation, at a birth weight of 3070 gram (-1 SD). Her Apgar scores after 1, 5 and 10 minutes were 5, 8 and 10, respectively. She was referred at the age of 3.5 years because of attacks of general paralysis. No cause for these attacks was identified, but were under control after the start of acetazolamide. Her development was delayed: she both walked her first steps and spoke the first words at the age of 2 years. There were articulation problems due to hypotonia in the oral region. Her IQ was measured several times and the last psychological assessment at the age of 16 years revealed a moderate ID with an IQ of 44. Moreover, she was diagnosed with autism spectrum disease with attention and a concentration disorder. There were sleeping problems due to anxiety and hypersensitivity for noise. Evaluation of her vision showed visual impairment of unknown origin and strabism. At the age of 3.5 years, she became obese despite normal food intake and different strategies to lose weight were ineffective. As child, she had recurrent respiratory infections. Physical examination at the age of 20 years showed a obesity (75.5 kg;+3.5 SD), a short height of 156.5 cm (-2.5 SD) and a low-normal head circumference of 54 cm (-1 SD). She had a square face, prominent antihelix, frontal bossing, deep set eyes, broad mouth, dental crowding, broad teeth, high palatum and a broad chin. Her hands were broad with short, tapering fingers. She had brachydactyly of her toes and pedes plano valgi. Previous investigations including karyotyping, Angelman and fragile X screening, CACNA1A analysis and array analysis were normal. Blood analysis, MRI of the brain and muscle biopsy revealed no abnormalities. WES identified a de novo mutation NM_016628.3:c.1648C>T, p.(Arg550*) in WAC (Table 1).
Individual 7 was reported before.2 The girl, a white female, was born vaginally at 41 weeks following augmentation via Pitocin administration. At birth, the Apgar score was normal. There are no reported language delays. She first used words at 20 months of age and phrase speech by 30 months of age and currently uses fluent phrase speech. No history of regression. Currently, she is 6-year and 10-month-oldand diagnosed with an autism spectrum disorder and clinical judgment using the DSM. Quantitative assessment of autism symptom severity by clinician report is 7 (out of 10 on the Calibrated Severity Score) and by parent report is a T score of 90 (90 is ceiling on the Social Responsiveness Scale). She has a full scale IQ of 89 with a nonverbal IQ of 89 and verbal IQ of 94, falling in the average range. Her overall adaptive ability falls at 70 (2 standard deviations below the norm). Parent report of childhood internalizing and externalizing symptomatology falls in the normal range although on indices of affective problems (T=70) and thought problems (T=71) she falls in the clinical range. She has challenges with motor coordination (diagnosed with excessive clumsiness). She has macrocephaly, and is righthanded. Vision and hearing are within normal limits. There is no history of seizures or other diagnosed medical or neurological problems. No history of sleep disturbances. Physical measurements: HC Z=+2.9; Height Z=+0.86; BMI Z=+1.7. Whole exome
4

sequencing revealed a de novo mutation, NM_016628.3:c.523_524del; p.(Lys175fs), in WAC (Table 1).
Individual 8 was reported before.2 The child, a White female, was born vaginally at 38 weeks following induction via Pitocin administration after an uncomplicated pregnancy. She had nuchal cord. She was diagnosed with cerebral palsy at birth. There are noted physical abnormalities (although these are unspecified in the data). She has reported language delays. She first used words and phrase speech at 60 months of age and currently uses phrase speech. No history of regression. EEG conducted at 18 months was normal, but brain MRI at the same age was abnormal (no details available in the data). At the last report she was 9-year, 4-month old and also diagnosed with an autism spectrum disorder and clinical judgment using the DSM and a diagnosis of intellectual disability based on cognitive and adaptive testing. Quantitative assessment of autism symptom severity by clinician report is 8 (out of 10 on the Calibrated Severity Score) and by parent report is a T score of 90 (90 is ceiling on the Social Responsiveness Scale). She has a full scale IQ of 61 with a nonverbal IQ of 57 and verbal IQ of 64, falling in the extremely low range. Her overall adaptive ability falls at 69 (-2SDS). Parent report of childhood internalizing symptomatology falls in the normal range while externalizing symptomatology falls in the clinical range (T=71). Additionally, on indices of affective problems (T=70), conduct problems (T=73), oppositional behaviour (T=70), and thought problems (T=73) she falls in the clinical range. She is righthanded. Hearing is within normal limits and her vision is corrected with glasses. There is no history of seizures but she has a history of kidney problems and respiratory problems. Physical measurements: HC Z =+0.53; Height Z=+2.88; BMI Z=-2.9. Whole exome sequencing revealed a de novo mutation NM_016628.3:1209_1212del; p.(His404fs) in WAC (table 1).
Individual 9 is a girl and the third of three children of non-consanguineous parents. There was no family history of developmental delay. She was born after 39+1 weeks with ventouse extraction after an uncomplicated pregnancy, at a birth weight of 3380 gram (0 SD). Her development was delayed with independent walking at the age of 22 months, and speaking first words at the age of 5 years. Psychological assessment at the age of 12 years showed mild ID with an IQ of 65. During childhood, she developed a hypotonic dysarthria and she used supportive sign language. She suffered from cerebral visual impairment. She had behaviour problems including anxiety and regular sleep disturbances. Feeding difficulties and recurrent upper respiratory tract infections in childhood were present. As child, she was diagnosed with a left hip dysplasia. Physical examination at the age of 12 years showed a height of 161 cm (+0.75 SDS), a weight of 61.2 kg (+2 SDS) and a head circumference of 56 cm (+1.5 SDS). Facial dysmorphisms included a square shape of the face, a prominent antihelix, hypertelorism, deep set eyes, long palpebral fissures, broad mouth and broad chin. She had brachydactyly of her fifth finger, Simian creases, fetal finger pads and pedes planus valgi. Previous genetic investigations including karyotyping, analysis of FMR1 and subtelomeric regions were normal. Metabolic screening was normal too. An MRI of the cerebrum showed a mild asymmetry in the right frontotemporal lobe. Targeted sequencing using molecular inversion probes identified a de novo mutation NM_016628.3:c.1415del; p.(Pro472fs) in WAC (Table 1).
Individual 10 is a boy, and single child born to non-consanguineous parents. Except for a maternal half-sister with PDD-NOS and mild ID, there was no familial history of developmental delay. The boy was born at term after an uncomplicated gestation with a high birth weight of 4500 gram
5

(+2.5 SDS). His development was delayed, with walking at the age of 2 years and 3 months and speaking first words at the age of 18 months. At the age of 7 years, he spoke 3 to 5-word sentences. Psychological assessment at the age of 7 years showed mild ID with an IQ of 61. During infancy, he developed concentration problems. Additionally, he received special treatment for toe walking. Physical examination at the age of 7 years showed a height of 114.8 cm (-1.75 SDS) and a high normal head circumference of 54.9 cm (+2 SDS). With a weight of 26.2 kg (+3 SDS), he was considered obese. Facial dysmorphisms included a square shape of the face, broad forehead, frontal bossing, deep set eyes, low set eyebrows, epicanthus, long palpebral fissures, broad mouth with prominent teeth and broad chin. He had a pedes planus valgi, fetal finger pads, strabismus and inverted nipples. His limbs were hypotonic. Previous investigations including metabolic screening and 250k genomic SNP microarray were both normal. An MRI cerebrum showed mildly enlarged ventricles. Blood analysis revealed a slightly increased Creatine Kinase (245 IU/L) level. Targeted re-sequencing using molecular inversion probes identified a de novo mutation, NM_016628.3:c.1648C>T; p.(Arg550*) in WAC (Table 1).
6

SUPPLEMENTARY METHODS
FM1-43 dye uptake assayFM1-43 labeling was performed as described.3 Third instar larvae were dissected in HL-3. Subsequently, synaptic boutons were stimulated for 1 min in HL-3 with 90 mM KCl and 1.5 mM CaCl 2
in the presence of FM 1-43 (4 µM) (Invitrogen). Next, non-internalized dye was removed by washing several times with HL-3. Images were captured using a confocal microscope (A1R, Nikon) with a 60x NA 1.0 water dipping lens. The boutonic FM1-43 intensity was quantified using ImageJ and was corrected for background labeling in the muscle.
TMRE labelingTetramethylrhodamine ethyl ester (TMRE) was used to label the mitochondrial membrane potential , as described before.4 In brief, third instar larvae were dissected in HL3 and incubated with 50 mM TMRE for 15 minutes. Next, fillets were washed with HL3 and images were captured using a confocal microscope (A1R, Nikon) with a 60x NA 1.0 water dipping lens. The TMRE labeling intensity was quantified using ImageJ and was corrected for background labeling in the muscle. First, the muscle background was substracted from the TMRE images, next 32-bit images were thresholded to set the background to NaN. Subsequently, the TMRE labeling intensity was measured at the neuromuscular junction.
ImmunohistochemistryImmunohistochemistry was performed as described.5 Antibodies used: rabbit anti-HRP 1:1000 (Jackson ImmunoResearch Laboratories), mouse anti-ATP synthase (Complex V) subunit beta (MitoSciences Inc.) 1:500. Alexa 488 and 555-conjugated secondary antibodies (Invitrogen) were used at 1:1000. Images of ventral nerve cords were captured using a confocal microscope (A1R, Nikon) with a 60x NA 1.4 oil lens.
7

Supplementary Figure 1: Knock-down of the Drosophila WAC orthologue CG8949 does not affect synaptic vesicle cycling, mitochondrial function and morphology.
(A) Representative image for FM1-43 labeling in control and mutant flies. (B) Quantification of the boutonic FM1-43 labeling intensity. Control larvae (control, genotype: w1118 UAS-Dicer-2/w1118;; nSyb-Gal4/+) and larvae neuronally expressing RNAi against CG8949 (CG8949vdrc48307; genotype: w1118 UAS-Dicer-2/w1118; UAS-CG8949vdrc48307/+; nSyb-Gal4/+) were stimulated with 90mM KCl in the presence of FM1-43. FM1-43 labels newly internalized synaptic vesicles at boutons of the neuromuscular junction. Knock-down of the Drosophila WAC orthologue CG8949 does not affect synaptic vesicle cycling. Bars and error bars represent the mean and SEM of 5 larvae (3-4 images per larvae, 2-11 boutons per image). (C) Quantification of TMRE labeling intensity to assess the mitochondrial membrane potential at the neuromuscular junction. Control larvae (control, genotype: w1118 UAS-Dicer-2/w1118; nSyb-Gal4/+) and larvae neuronally expressing RNAi against CG8949 (CG8949vdrc48307; genotype: w1118 UAS-Dicer-2/w1118; UAS-CG8949vdrc48307/+; nSyb-Gal4/+) after incubation with 50mM TMRE. Knock-down of the Drosophila WAC orthologue CG8949 does not affect mitochondrial functioning. Bars and error bars represent the mean and SEM of 5 larvae (3-10 neuromuscular junctions per larvae). (D) Maximum intensity projections of ventral nerve cords of control larvae (control, genotype: w1118 UAS-Dicer-2/w1118; nSyb-Gal4/+) and larvae neuronally expressing RNAi against CG8949 (CG8949vdrc48307; genotype: w1118 UAS-Dicer-2/w1118; UAS-CG8949vdrc48307/+; nSyb-Gal4/+) labelled with anti-HRP (presynaptic area) and anti-complexV (mitochondria).
8

9

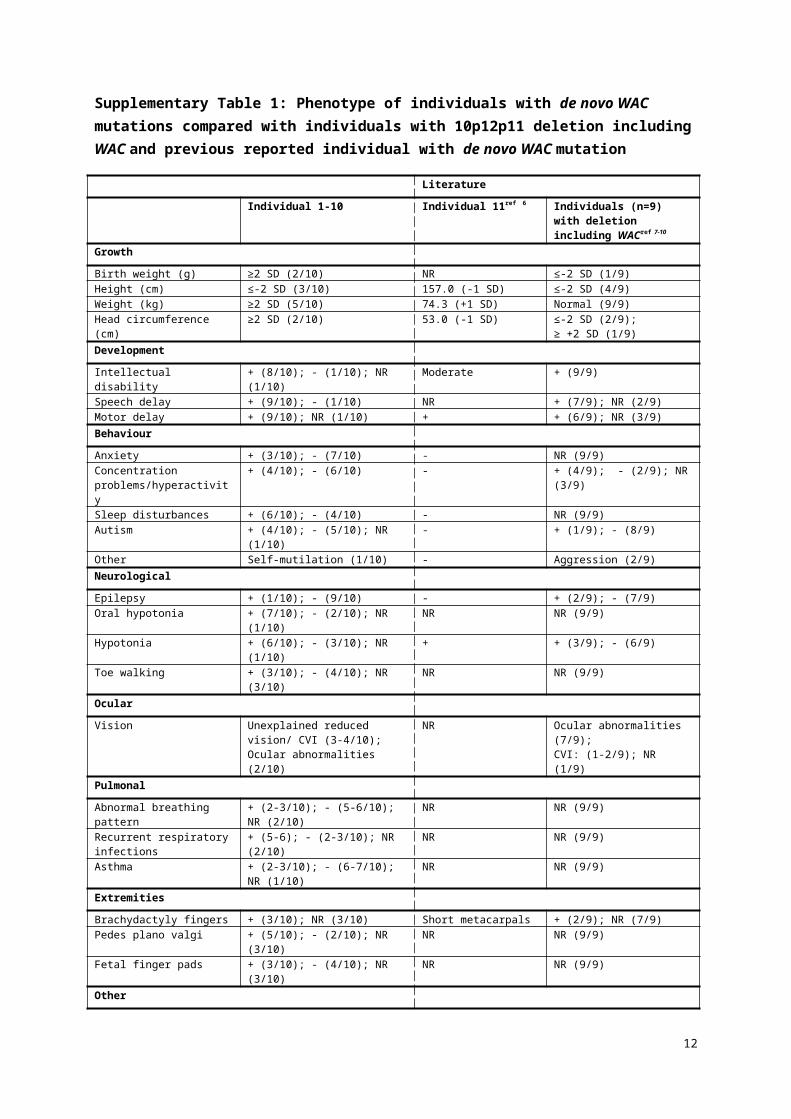
Supplementary Table 1: Phenotype of individuals with de novo WAC mutations compared with individuals with 10p12p11 deletion including WAC and previous reported individual with de novo WAC mutation
LiteratureIndividual 1-10 Individual 11ref 6 Individuals (n=9) with deletion
including WACref 7-10
Growth
Birth weight (g) ≥2 SD (2/10) NR ≤-2 SD (1/9)Height (cm) ≤-2 SD (3/10) 157.0 (-1 SD) ≤-2 SD (4/9)Weight (kg) ≥2 SD (5/10) 74.3 (+1 SD) Normal (9/9)Head circumference (cm) ≥2 SD (2/10) 53.0 (-1 SD) ≤-2 SD (2/9);
≥ +2 SD (1/9)DevelopmentIntellectual disability + (8/10); - (1/10); NR (1/10) Moderate + (9/9)Speech delay + (9/10); - (1/10) NR + (7/9); NR (2/9)Motor delay + (9/10); NR (1/10) + + (6/9); NR (3/9)BehaviourAnxiety + (3/10); - (7/10) - NR (9/9)Concentration problems/hyperactivity
+ (4/10); - (6/10) - + (4/9); - (2/9); NR (3/9)
Sleep disturbances + (6/10); - (4/10) - NR (9/9)Autism + (4/10); - (5/10); NR (1/10) - + (1/9); - (8/9)Other Self-mutilation (1/10) - Aggression (2/9)NeurologicalEpilepsy + (1/10); - (9/10) - + (2/9); - (7/9)Oral hypotonia + (7/10); - (2/10); NR (1/10) NR NR (9/9)Hypotonia + (6/10); - (3/10); NR (1/10) + + (3/9); - (6/9)Toe walking + (3/10); - (4/10); NR (3/10) NR NR (9/9)OcularVision Unexplained reduced vision/ CVI (3-
4/10); Ocular abnormalities (2/10)NR Ocular abnormalities (7/9);
CVI: (1-2/9); NR (1/9)PulmonalAbnormal breathing pattern + (2-3/10); - (5-6/10); NR (2/10) NR NR (9/9)Recurrent respiratory infections + (5-6); - (2-3/10); NR (2/10) NR NR (9/9)Asthma + (2-3/10); - (6-7/10); NR (1/10) NR NR (9/9)ExtremitiesBrachydactyly fingers + (3/10); NR (3/10) Short metacarpals + (2/9); NR (7/9)Pedes plano valgi + (5/10); - (2/10); NR (3/10) NR NR (9/9)Fetal finger pads + (3/10); - (4/10); NR (3/10) NR NR (9/9)OtherNeonatal feeding difficulties + (4/10); - (6/10) - - (9/9)CVI: cerebral visual impairment; NR: Not reported
10

Supplementary Table 2: Quantification of relative expression level of CG8949 upon RNAi-mediated knock-down
Raw data of biological and technical replicatesCG8949** BetaCop ***
Genotype Exp* Ct Ct avg Sd Ct Ctav Sd dCT dCTav Sd ddCT % rel. expr. p-val.
CG8949vdrc48307 1.1 28.1328.07 0.09
27.0127.07 0.09 1.00 0.93 0.2
7 0.80 57.54 0.01CG8949vdrc48307 1.2 28.00 27.13
CG8949vdrc48307 2.1 28.1828.09 0.13
26.9526.92 0.04 1.17
CG8949vdrc48307 2.2 28.00 26.90
CG8949vdrc48307 3.1 27.4827.41 0.10
26.7326.77 0.06 0.64
CG8949vdrc48307 3.2 27.34 26.82
control 1.1 27.5027.41 0.13
27.2927.21 0.12 0.21 0.14 0.1
1control 1.2 27.32 27.12
control 2.1 26.8926.88 0.02
26.7226.69 0.04 0.19
control 2.2 26.86 26.66
control 3.1 26.7926.79 0.00
26.7426.77 0.04 0.01
control 3.2 26.78 26.80
CG8949vdrc107328 1.1 26.8626.86 0.00
26.4826.59 0.17 0.27 0.23 0.0
9 0.19 87.75 0.16CG8949vdrc107328 1.2 26.86 26.71
CG8949vdrc107328 2.1 26.2826.23 0.06
26.1126.11 0.00 0.12
CG8949vdrc107328 2.2 26.19 26.11
CG8949vdrc107328 3.1 27.2227.23 0.01
26.9426.94 0.00 0.29
CG8949vdrc107328 3.2 27.23 26.94
control 1.1 26.8726.86 0.01
27.0526.94 0.16 -0.07 0.04 0.1
7control 1.2 26.85 26.82
control 2.1 26.9626.96 0.00
27.0227.00 0.03 -0.04
control 2.2 26.96 26.98
control 3.1 27.7827.67 0.16
27.4427.44 0.00 0.23
control 3.2 27.56 27.44
Summary CG8949 knock down
Genotype dCT avg SD ddCT Relative expression CG8949 (%) p-val. Conclusion
CG8949vdrc48307 0.93 0.270.80 57.54 0.01 Significant
knock downcontrol 0.14 0.11
CG8949vdrc107328 0.23 0.090.19 87.75 0.16 Insufficient
knock downcontrol 0.04 0.17
The ubiquitous actin-Gal4 driver w1118; P(w[+mC]=Act5c-Gal4)/CyO (Bloomington Drosophila Stock Center11) was used to generate RNAi-mediated knock-down for quantitative PCR. *Experiment refers to ‘biological experiment and technical replicate’, e.g. 3.2 refers to the replicate of the second biological experiment. **Primer sequences used for amplification of CG8949: 5’-TGGAATTACGACAACGATGG-3’ and 5’-TAACTGGCTTCCGAGGTAGG-3’. ***BetaCop was used as reference gene, using primer sequences 5’-AACTACAACACCCTGGAGAAGG-3’ and 5’-ACATCTTCTCCCAATTCCAAAG-3’. Ct = threshold cycle, dCt = CtCG8949 – CtBetaCop, ddCt = dCtRNAi – dCtcontrol, p-value calculated with student’s t-test.
11
References
1. de Ligt J, Willemsen MH, van Bon BW et al: Diagnostic exome sequencing in persons with severe intellectual disability. The New England journal of medicine 2012; 367: 1921-1929.
2. Iossifov I, O'Roak BJ, Sanders SJ et al: The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014; 515: 216-221.
3. Verstreken P, Ohyama T, Bellen HJ: FM 1-43 labeling of synaptic vesicle pools at the Drosophila neuromuscular junction. Methods in molecular biology 2008; 440: 349-369.
4. Shidara Y, Hollenbeck PJ: Defects in mitochondrial axonal transport and membrane potential without increased reactive oxygen species production in a Drosophila model of Friedreich ataxia. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010; 30: 11369-11378.
5. Kasprowicz J, Kuenen S, Miskiewicz K, Habets RL, Smitz L, Verstreken P: Inactivation of clathrin heavy chain inhibits synaptic recycling but allows bulk membrane uptake. The Journal of cell biology 2008; 182: 1007-1016.
6. Hamdan FF, Srour M, Capo-Chichi JM et al: De novo mutations in moderate or severe intellectual disability. PLoS genetics 2014; 10: e1004772.
7. Mroczkowski HJ, Arnold G, Schneck FX, Rajkovic A, Yatsenko SA: Interstitial 10p11.23-p12.1 microdeletions associated with developmental delay, craniofacial abnormalities, and cryptorchidism. American journal of medical genetics Part A 2014; 164A: 2623-2626.
8. Okamoto N, Hayashi S, Masui A et al: Deletion at chromosome 10p11.23-p12.1 defines characteristic phenotypes with marked midface retrusion. Journal of human genetics 2012; 57: 191-196.
9. Shahdadpuri R, de Vries B, Pfundt R, de Leeuw N, Reardon W: Pseudoarthrosis of the clavicle and copper beaten skull associated with chromosome 10p11.21p12.1 microdeletion. American journal of medical genetics Part A 2008; 146A: 233-237.
10. Wentzel C, Rajcan-Separovic E, Ruivenkamp CA et al: Genomic and clinical characteristics of six patients with partially overlapping interstitial deletions at 10p12p11. European journal of human genetics : EJHG 2011; 19: 959-964.
11. Bloomington Drosophila Stock Center; www.flystocks.bio.indiana.edu
12

![WAC 296 - 46B CHAPTERlawfilesext.leg.wa.gov/law/WACArchive/2014/WAC 296... · (11/5/13) [ch. 296-46b wac p. 1] chapter 296-46b chapter 296-46b wac electrical safety standards, administration,](https://img.pdfslide.us/doc/110x75/5f937088d75d77697316c60c/wac-296-46b-296-11513-ch-296-46b-wac-p-1-chapter-296-46b-chapter.jpg)





![WAC 182 - 12 CHAPTER - lawfilesext.leg.wa.govlawfilesext.leg.wa.gov/law/WACArchive/2014/WAC 182 - 12 CHAPTE… · (10/28/13) [Ch. 182-12 WAC p. 1] Chapter 182-12 Chapter 182-12 WAC](https://img.pdfslide.us/doc/110x75/5f937086d75d77697316c603/wac-182-12-chapter-182-12-chapte-102813-ch-182-12-wac-p-1-chapter.jpg)





















