Embed Size (px)
Citation preview

J . Basic Microbiol. 31 (1991) 3, 177-188
(Department of Chemistry BG-10, University of Washington, Seattle, Washington 98 195, USA; Department of Chemistry, Ohio State University, Columbus, Ohio 43210, USA; Department of
Medicinal Chemistry and Pharmacognosy, Purdue University, West Lafayette, Indiana 47907, USA; and Laboratory for Microbial Biochemistry, Institute for Chemical Research, Kyoto University, Uji, Kyoto 61 I , Japan)
Mechanistic studies of two amino acid racemases of broad substrate specificity from Pseudomonas striata and Aeromonas caviae KEVIN REYNOLDS, JENNIFER MARTIN SHU-JANE SHEN ’, NOBUYOSHI ESAKI ’, KENJI SODA and HEINZ G. FLOSS
(Received 3 December 1990/Accepted 12 December 1990)
The conversion of ~-[a-~H]alanine in H 2 0 and unlabeled L-alanine in ’H,O into D-alanine, under nearly irreversible conditions, with the amino acid racemase from Pseudomonas striata showed significant internal transfer of the cc-hydrogen. This result has been interpreted as being indicative of a single base mechanism for the racemization.
The relative rates of deuterium incorporation into unlabeled D- and L-methionine by the two amino acid racemases of broad substrate specificity from P . striata and Aeromonas cauiue, were measured in ’ H 2 0 . The results showed a markedly different pattern, dependent upon the configuration of the initial substrate; with D-methionhe as substrate deuterium is incorporated into both enantiomers at approximately the same rate, but with L-methionine as substrate deuterium is incorporated considerably faster into the D than the L enantiomer. These results argue against a single base mechanism of racemization for these enzymes and are best rationalized in terms of a double base model where only one of the bases undergoes proton (deuterium) exchange with the solvent while the amino acid is enzyme-bound. The interpretation of the earlier experiment needs to be considered in light of these results.
Most of the amino acid racemases isolated and characterized to date (ADAMS 1972) exhibit moderately high substrate and binding specificity and all have been tentatively classified into two groups, the first group being those that utilize a cofactor for activation of the C,-H bond (pyridoxal phosphate has been found to be a requisite for many primary amino acid racemases) and the second those that have no cofactor requirement.
In 1968 CARDINALE and ABELES first proposed two alternative modes of action for amino acid racemases. These have become known as the “one-base’’ and “two-base’’ models. To date, only a handful of mechanistic studies of amino acid racemases have been reported. Proline (CARDINALE and ABELES 1968, RUNICK and ABELES 1975, ALBERY and KNOWLES 1986) and diaminopimelic acid (DAP) racemases (WISEMAN and NICHOLS 1984), which are known not to require cofactors, have been shown to operate via a two-base mechanism. The case is not so clear with pyridoxal phosphate-dependent enzymes. ADAMS (1976) has proposed a two-base mechanism for the alanine racemases from Pseudomonasputida and Bacillus subtilis, based on the extensive asymmetry observed in comparisons of the relative rates of exchange and racemization for D- and L-alanine. The pyridoxal phosphate-dependent a-amino-E-caprolactam racemase reaction is believed to proceed (AHMED et al. 1986) via a one-base mechanism based on evidence for the internal transfer of the cr-hydrogen of the amino acid. Based on the same criterion, in a preliminary report of data presented below, we (SHEN et al. 1983) proposed a one-base mechanism for a broad specificity amino acid
I2 J. Basic Microbiol. 31 (1991) 3

178 K. REYNOLDS et al.
racemase from P . striata. SODA and coworkers have reported (SAWADA et al. 1984) mechanistic studies of the same racemase from P. striata in which the rates of racemization and a-hydrogen exchange in H,O, Z H 2 0 and H3H0 were measured and the results also interpreted in terms of a one-base mechanism. To date, however, there has been no rigorous proof of a one-base mechanism for this racemase.
CARDINALE and ABELES (1968) proposed and subsequently conducted an experiment to differentiate between a one-base and a two-base mechanism. In this experiment, the incorporation of deuterium into the a-hydrogen of unlabeled proline by proline racemase was measured. When the reaction was carried out in 'H20 with L-proline as substrate, then essentially all of the deuterium was initially incorporated into D-proline. In the case where D-proline was the substrate, all the deuterium was incorporated into L-proline. This result is considered consistent with a two-base mechanisfn where there are no protons of the enzyme-bound intermediate that can exchange with the solvent. A one-base mechanism is predicted to give the same pattern of deuterium incorporation regardless of which enantiomer is used as substrate. This clearly was not the case for proline racemase.
Herein we report the results of two sets of experiments aimed at distinguishing between a one-base and a two-base mechanism for the broad specificity amino acid racemases from P. striata (SODA and OSUMI 1969) and Aeromonas caviae (INAYAKI et al. 1987). The first set of experiments was designed to determine if racemization occurred with internal transfer of the a-hydrogen from substrate to product (as seen with a-amino-8-caprolactam racemase) while the second set of experiments measured the relative rates of deuterium incorporation from 'H,O as solvent into the separate enantiomers of the amino acid (as was done by CARDINALE and ABELES for proline racemase).
Materials and methods
The two broad substrate specific amino acid racemases, obtained from P. striata and A . cauiae, were purified as previously described (SODA and OSUMI 1969, INAYAKI et al. 1987) and were stored frozen (-70 "C) in potassium phosphate in 'H,O (20 mM, pZH = 8.5). Amino acids and biochemicals were obtained from either SIGMA or ALDRICH Chemical Company. L-[ '3C2H,]Methionine was prepared by methylation of L-homocysteine with [' 'CZHJmethyl iodide using a procedure provided by Professor R. W. WOODARD of the University of Michigan. Reagent grade inorganic and organic chemicals were purchased from either ALDRICH or MALLINCKRODT Chemical Company and ion exchange resins were obtained from BIO-RAD Laboratories. Deuterium oxide (99.75 atom% deuterium) and ~,~-[a-'H]alanine were obtained from MERCK SHARP and DOAME Ltd.
Amino acids were analyzed by ClMS using a FINNIGAN 4023 mass spectrometer with isobutane gas for chemical ionization. Derivatized amino acids were analyzed by GC-MS on a HEWLETT-PACKARD 5970/5790A series gas chromatograph-mass selective detector equipped with a CHIRAsIL-VaI IIZ chiral capillary column from ALLTECH Associates (ABE et al. 198 1). Radiochromatograms were obtained using a PACKARD model 7021 radiochromatogram scanner.
Experiments probingfor in te rna l t ransfer of the a -hydrogen ofL-alanine t o D-alanine: Preparation of ~-[a-'H]alanine: A solution of ~ , ~ - [ a - ~ H ] a l a n i n e (270.3 mg, 3 mmol) in 0.5 N sodium hydroxide (4 ml) was treated sequentially, at 0 "C, with freshly distilled acetic anhydride (350 pl, 3.7mmol) and 0.5 N sodium hydroxide (2ml). The solution was then allowed to warm to room temperature over 15 minutes. The reaction was terminated by neutralizing the solution with 5 N hydrochloric acid, followed by a further adjustment to pH 5. N-Acetyl-~,~-[a-~H]alanine was purified in quantitative yield on a Dowex 50W-X8 (hydrogen form) column. A portion of this material ( 1 31.1 mg, 1 mmol) was dissolved in water (8 ml) and the pH of the resulting solution was adjusted to 7.1 with a 25% (w/v) solution of ammonium hydroxide. This was treated with 200 pl of a solution containing 200 pg of acylase I from porcine kidney (2000 IU/mg) and the volume made up to 10 ml with water. This solution was incubated at 38 "C for 24 hours and subsequently adjusted to pH 4 with 1 N hydrochloric acid. The precipitated protein was removed by filtration and the filtrate was loaded onto a Dowex 50W-X8 (hydrogen form) column. N-Acetyl-~-[a-~H]alanine was eluted with water and

Mechanistic studies of amino acid racemases 179
~-[a-~H]alanine was subsequently eluted from the resin with a 1 N ammonium hydroxide solution. The eluate was repeatedly evaporated to dryness and redissolved in water. Recrystallization from 80% ethanol afforded ~-[cx-~H]alanine: CIMS, m/z (relative intensity): 90 (MH', 22.6), 91 ( M H + + 1, 100); 'H NMR (D'O, 80 MHz) S 1.46 (s, 3H), 1.47 (d, J = 7.2 Hz, 3H, trace amount), 4.04 (q, J = 7.2 Hz, 1 H, trace amount); specific optical rotation (5 N HC1)
Enantiomeric purity of ~-[cx-~H]alanine: ~-[cx-'H]Alanine (13.3 mg, 150 pmol, 93.4 1.1% 'H) and D-alanine (2.68 mg, 30 pmol) were incubated together with acetyl-CoA (14 mg, 15 pmol) and D-amino acid acetyltransferase (1.1 IU) in Tris . HCI buffer (0.15 M, pH 8.4, 2.0ml) for 10 hr at 39 "C. N-Acetyl-D-alanine was isolated and purified from the enzyme system in the manner described below and was subsequently analyzed by CIMS for deuterium content. CIMS, m/z (relative intensity): 132 (MH', loo), 133 ( M H + + l , 7.4
Purification of acetyl-CoA : D-amino acid-cx-N-acetyltransferase: Acetyl-CoA : D-amino acid-a-N- acetyltransferase was isolated and purified 2000 fold from baker's yeast (Mary Lou Donuts, Inc., Lafayette, IN), following essentially the method of ZENK and SCHMITT (1965). The enzyme was assayed in the following manner: To a solution of acetyl-CoA (0.3 pmoles) and D-amino acid acetyltransferase (0.02-0.03 IU) in Tris . HCI (0.15 M, pH 8.4) was added D-alanine (40 pmol) in a final volume of 2.0 ml. Control experiments were conducted with either L-alanine (40 pmol) or no amino acid. The solution was incubated at 39 "C prior to addition of the amino acid. Enzyme activity was monitored by following the decrease in absorbance at 232 nm which corresponds to the cleavage of the thioester bond of acetyl-CoA [cZ3' = 8700 (prior to cleavage of the thioester), dcZ3' = 4500 (absorption change due to cleavage)].
Assay of amino acid racemase: Lactate dehydrogenase (15 mg, 700 IU/mg), NADH (0.16 pmol), pyridoxal phosphate (0.5 pmol), D-amino acid oxidase (100 pg, 20 IU/mg), L-alanine (40 pmol), and amino acid racemase (0.02 -0.05 IU) were made up to 2.0 ml in Tris . HCl buffer (0.15 M, pH 8.4). The solution was brought to 38 "C prior to addition of the L-alanine substrate. Control experiments were conducted in the absence of either the L-amino acid or the amino acid racemase. Activity was monitored by following the decrease in absorption at 340 nm resulting from the consumption of NADH.
Reaction conditions for probing internal transfer of the a-hydrogen of L-alanine during its conversion to D-alanine: Table 1 outlines the conditions for conversion both of ~-[cx-~H]alanine in H,O and unlabeled L-alanine in 'H,O into D-alanine. The in situ acetylation of the o-alanine produced insured that the reaction proceeded under nearly irreversible conditions. Deuterated buffer was prepared as follows: Tris . HCI buffer (5.0 ml, pH 8.4, 0.15 M) was lyophilized to dryness, redissolved in 5 ml of 'H20 (99.75% 'H), then lyophilized. This exchange procedure was repeated three times. In the experiments conducted with deuterated buffer it was also necessary to exchange the o-amino acid acetyl transferase solution with 'H,O. Accordingly, this enzyme solution was concentrated to less than 1 ml by rotary evaporation, 10 ml of 'H,O was added and the solution reduced to less than 1 ml again. This exchange procedure was repeated four times and resulted in no significant loss of enzyme activity. The N-acetyl-D-alanine produced in all of the enzyme incubations was isolated and purified
Table 1 Reaction conditions for conversion of L-alanine into N-acetyl-D-alanine Amino acid racemase (120 IU/mg) from Pseudomonas striata was incubated with t-alanine, pyridoxal phosphate (PLP), acetyl-CoA and acetyl-CoA: D-amino acid-cc-N-acetyltransferase in Tris . HCI buffer (0.15 M, pH 8.4) at 39 "C for either 3 hrs*) or 6 hrs').
= + 14.3 & 0.1".
1.1).
Expt. Substrate No.
Racemase: acetyl pmoles of pmoles of Total transferase (IU) acetyl-CoA PLP volume
1 80 pmoles ~-[a-'H]alanine 1.16:4.06 27 1 .o 2.10 ml')
2 120 pmoles L-alanine 1.16 : 4.06 41 1.5 3.1 ml*)
3 40 pmoles L-alanine 0.6 : 2.0 16 0.5 1.0 ml*)
4 120 pmoles ~-alanine 1.1 6 : 2.0 38 1.5 3.4 ml*)
(93% 'H) in H,O
in 'H20 (98% 'H)
in 'H,0/H20 (90% 'H)
in 'H20/H20 (65% 'H)

180 K. REYNOLDS et al.
in the following manner. The reaction was terminated by acidifying the solution to pH 4.5 with 1 N hydrochloric acid. The solution was then passed through a Dowex 50W-X8 (hydrogen form) column, and the N-acetyl-D-alanine product which does not bind was collected in the water eluate. The eluate was evaporated to dryness and the product purified by paper chromatography (solvent system: n-butanol/88% formic acid/water = 2: 1 : 1; R , = 0.83). The product was eluted with water and purified further on a Dowex 1-X8 (acetate form) column using an acetate gradient (300 ml 0.5 N acetic acid/300 m12 N acetic acid). The N-acetyl-D-alanine was then evaporated to dryness and analyzed by CIMS and NMR. CIMS, m/z (relative intensity): 131 (M', 0), 132 (MH', loo), 133 ( M H t + l , 5.58 5 0.12), 134 (MH++2, 0.68 f: 0.05); 'H NMR (CD,OD, 80 MHz) 6 1.36 (d, J = 7.2 Hz, 3H), 1.96 (s, 3H), 4.35 (4. J = 7.2 Hz, 1H). The trapping efficiency of the coupled enzyme system used in these studies was measured by including a trace amount of ~-[ '~C]alanine in the incubation and subsequently measuring the partitioning of 14C between N-acetyl-D-alanine and unreacted alanine.
Calculation of isotope effects: The theoretical quantity of protonated D-alanine formed as a result of a combination of internal transfer of the a-hydrogen and a deuterium isotope effect during the racemization of unlabeled L-alanine in deuterated buffer was calculated using the following formula:
A is defined as the percentage of a-protonated N-acetyh-alanine that is obtained from the enzyme incubation, I is the percentage internal transfer of the a-hydrogen and F is the fraction of protium in the deuterated water.
Experiments measuring t h e relat ive ra tes of deuter ium incorpora t ion i n t o D - a n d L-methionine: Enzyme incubation procedure: The amino acid (15 mg, 0.1 mmol) was first dissolved in potassium phosphate in 'H,O (1.0 ml, 20 mM) and then dried by lyophilization. The resulting residue was redissolved in 'H,O (2.0 ml) and dried again. This was then dissolved in 'H,O (0.96 ml) with the amino acid racemase solution (0.04 ml) and was subsequently incubated at 30 "C. Samples (0.04 ml) were removed at various time intervals and heated at 100 "C for 5 minutes in hydrochloric acid (0.2 N, 0.60 ml). Each of these samples was then evaporated to dryness and stored at 4 "C prior to derivatization.
Amino acid derivatization and analysis: The amino acid hydrochloride residues from the enzyme incubations were derivatized, with slight modifications, according to the method recommended by ALLTECH to form the N-pentafluoropropionyl isopropyl ester of the amino acid (ABE et al. 198 I ) . Acetyl chloride (0.10 ml, 1.4 mmol) was added dropwise to isopropanol (0.25 ml, 3.3 mmol) at 0 "C. Each amino acid residue was suspended in this mixture and subsequently heated in a WHEATON vial equipped with a teflon faced septum at 87 "C for 45 minutes. The product was dried and treated with chloroform (0.25 ml) and pentafluoropropionic anhydride (0.1 ml, 0.51 mmol) at 97 "C for 15 minutes. The derivatized amino acid was evaporated to dryness and then resuspended in spectral grade methylene chloride prior to GC-MS analysis. The GC conditions were as follows: split vent ratio: 1/15; carrier gas: helium; carrier gas flow rate: 1.0 ml/min. The mass spectrometer parameters were as follows: mass sweep width: 200-400 atomic mass units; sweep rate 1.07 scans/sec; scan threshold: 20. Using the CmusIL-Val I l l capillary column the L- and D-aminO acids were readily separated. The percentage of deuterium enrichment at each time point for each enantiomer was calculated according to the method of BIEMANN (1962). The number of millimoles of each amino acid enantiomer present at a given time, t , was determined by integration from the total ion current chromatogram. Multiplication of these two numbers afforded the number of millimoles of each deuterated enantiomer, and a plot of these values against time allowed the relative rates of deuterium incorporation into the D- and L-amino acid to be determined.
Measurement of rate of a-carbon-hydrogen exchange and racemization: These measurements were carried out according to the method described by SODA and coworkers (SAWADA et al. 1984), using 100 mM solutions of D- and L-methionine with the P. striuta amino acid racemase.
Results
~-[cr-~H]Alanine, prepared by acetylation of the racemate under basic conditions followed by enzymatic resolution of the N-acetyl-~~-[a-~H]alanine by porcine kidney acylase, was shown by CIMS to contain 93.4 & 1.1% deuterium. The enantiomeric purity of this
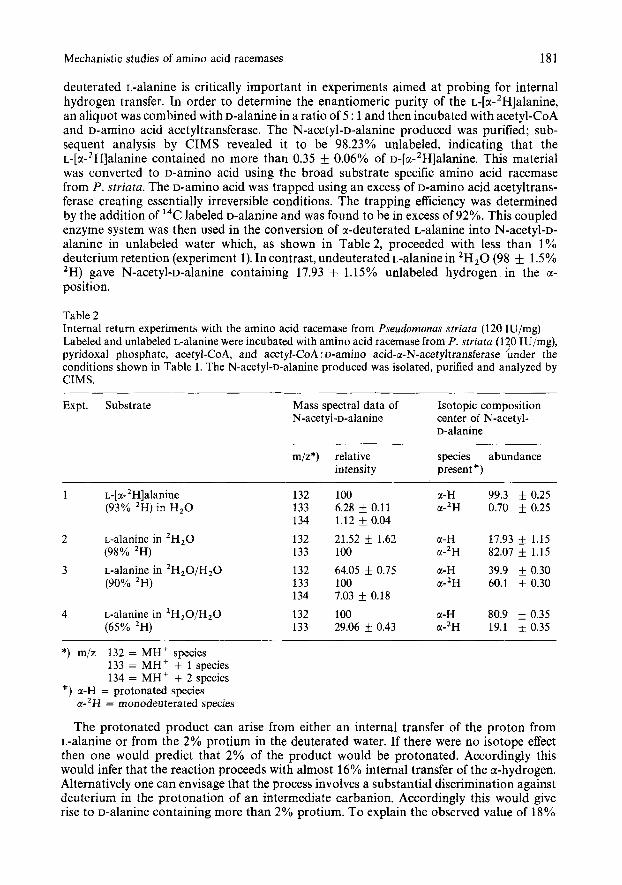
Mechanistic studies of amino acid racemases 181
deuterated L-alanine is critically important in experiments aimed at probing for internal hydrogen transfer. In order to determine the enantiomeric purity of the ~-[a-'H]alanine, an aliquot was combined with D-alanine in a ratio of 5 : 1 and then incubated with acetyl-CoA and D-amino acid acetyltransferase. The N-acetyl-D-alanine produced was purified; sub- sequent analysis by CIMS revealed it to be 98.23% unlabeled, indicating that the ~-[a-'H]alanine contained no more than 0.35 0.06% of ~-[a-'H]alanine. This material was converted to D-amino acid using the broad substrate specific amino acid racemase from P . striata. The D-amino acid was trapped using an excess of D-amino acid acetyltrans- ferase creating essentially irreversible conditions. The trapping efficiency was determined by the addition of 14C labeled D-alanine and was found to be in excess of 92%. This coupled enzyme system was then used in the conversion of a-deuterated L-alanine into N-acetyl-D- alanine in unlabeled water which, as shown in Table 2, proceeded with less than 1% deuterium retention (experiment 1). In contrast, undeuterated L-alanine in 'H,O (98 f 1.5% 'H) gave N-acetyl-D-alanine containing 17.93 + 1.15% unlabeled hydrogen. in the a- position.
Table 2 Internal return experiments with the amino acid racemase from Pseudomonus striata (120 IU/mg) Labeled and unlabeled L-alanine were incubated with amino acid racemase from P. striutu (120 IU/mg), pyridoxal phosphate, acetyl-CoA, and acetyl-CoA : D-aminO acid-a-N-acetyltransferase h d e r the conditions shown in Table 1. The N-acetyl-D-alanine produced was isolated, purified and analyzed by CIMS.
Expt. Substrate Mass spectral data of N-acet yl-D-aianine
Isotopic composition center of N-acetyl- D-alanine
m/z*) relative in tensity
species abundance present ")
1 ~-[a-~H]alanine (93% 'H) in H,O
2 L-alanine in 'H,O (98% 'H)
3 L-alanine in 2 ~ , ~ / ~ , ~
(90% 'H)
4 L-alanine in 2 ~ , ~ / ~ , ~
(65% 'H)
132 133 134 132 133 132 133 134 132 133
100 6.28 & 0.1 1 1.12 f 0.04 21.52 f 1.62 100 64.05 f 0.75 100 7.03 f 0.18
100 29.06 .t 0.43
a-H a-*H
a-H a-'H
a-'H a-H
a-H a-2H
99.3 rt 0.25 0.70 & 0.25
17.93 & 1.15 82.07 & 1.15 39.9 0.30 60.1 f 0.30
80.9 f 0.35 19.1 .t 0.35
*) m/z 132 = MH' species 133 = MH' + 1 species 134 = MH' + 2 species
") a-H = protonated species a-'H = monodeuterated species
The protonated product can arise from either an internal transfer of the proton from L-alanine or from the 2% protium in the deuterated water. If there were no isotope effect then one would predict that 2% of the product would be protonated. Accordingly this would infer that the reaction proceeds with almost 16% internal transfer of the a-hydrogen. Alternatively one can envisage that the process involves a substantial discrimination against deuterium in the protonation of an intermediate carbanion. Accordingly this would give rise to D-alanine containing more than 2% protium. To explain the observed value of 18%

182 K. REYNOLDS et al.
protium, one would have to invoke a deuterium isotope effect (kH/kD) of approximately 11. To obtain a measure of the actual isotope effect the experiment was repeated in 2H,0-Hz0 mixtures containing 90% and 65% deuterium (experiments 3 and 4, respectively). From the results with a solvent system containing 98% deuterium, a set of theoretical internal hydrogen transfer values for each different k,/k, value was calculated using the method described in the Experimental Section. Using the assumption that the level of internal hydrogen transfer is constant in all experiments, the data from experiments 2 ,3 and 4 would indicate that k H / k D for the reaction is 4.1 f 0.6. This result predicts that in experiment 2, conducted in 98% deuterated buffer, of the 17.9% of protonated product, 7.6% arises from the protium impurity in the solvent and the remaining 10.3% is due to an internal transfer of the a-hydrogen. The large difference between the degree of transfer of the a-'H of ~-[a-~H]alanine in an H,O environment us the a-H of the unlabeled amino acid in a 2H,0 environment can be explained by an isotope effect in the transfer of the hydrogen from the enzyme base back to the a-carbon of the amino acid, if the base if polyprotic. On the other hand the observed difference may simply be due to conformational differences between the enzyme in H,O and 'H20, resultingin different rates ofexchange with solvent.
While this interpretation of the data, pointing to an internal transfer of the amino acid a-hydrogen, favors a one-base mechanism for the P. striata amino acid racemase, an alternative interpretation consistent with a two-base mechanism cannot be ruled out. The above interpretation in terms of an internal hydrogen transfer is highly dependent on the result of the experiment in 98% 'H,O. The 'H enrichment of the solvent is a calculated value with a significant error. If the true value were to lie at the lower end of the error range, the percentage of protium appearing in the N-acetyl-D-alanine could be accounted for entirely by a discrimination against deuterium of about 7 f 1 in a two-base mechanism without internal hydrogen transfer.
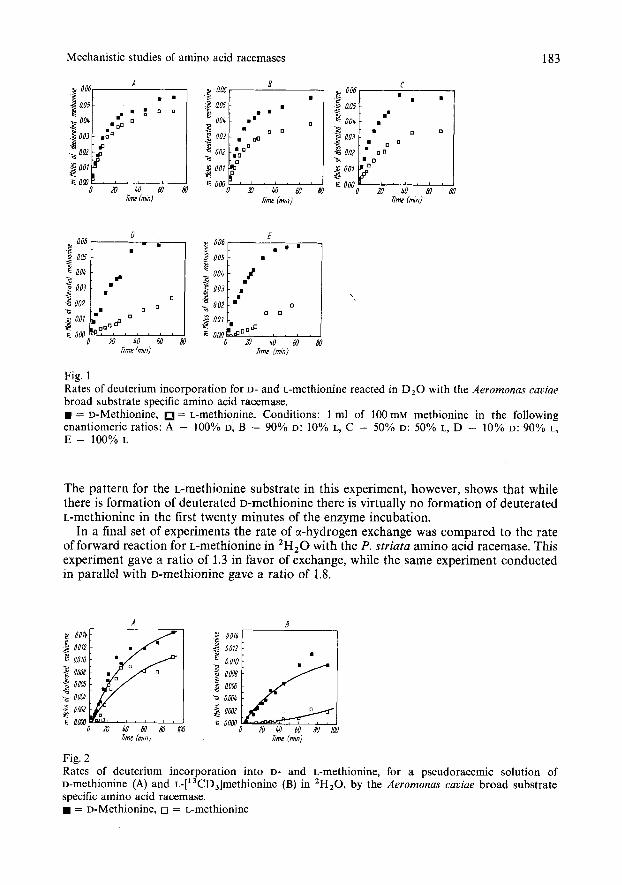
In an attempt to apply a second criterion to distinguish a one-base from a two-base mechanism, we determined the dependence of the rates of deuterium incorporation from 'H,O into L- and D-methionine on substrate configuration. Graphs of the number of millimoles of deuterated L- and D-methionine formed versus time, starting with various different enantiomeric ratios of unlabeled methionine, using the A . caviae amino acid racemase in 'H,O are shown in Fig. 1. It can be seen that with 100% D-methionine as substrate the rates of formation of deuterated D- and L-methionine are very similar, but as the fraction of L-methionine in the substrate increases, then a decrease in the rate of formation of the deuterated L-enantiomer is observed. Indeed, at the initial time points with 100% L-methionine as substrate there is virtually no formation of the deuterated L enantiomer although there is rapid formation of the deuterated D enantiomer. A very similar pattern was observed for the same experiments conducted with the P . striata amino acid racemase. Further studies were carried out using leucine and lysine as substrates for both amino acid racemases, and similar trends dependent upon the initial configuration of the substrate were observed in all of these cases (data not shown).
To determine whether the influence of substrate configuration on deuterium incorporation into D- and L-methionine is an allosteric or an active site effect, the rates of deuterium incorporation into D- and L-methionine by A . caviae amino acid racemase were observed using as substrate a pseudo-racemic mixture of D-methionine and ~-['~C'H,]methionine (this study was conducted using 75% less enzyme than in the previous experiments). Under these conditions it was possible to follow deuterium incorporation into L- and D-methionine formed from the ~-['~C~H,]methionine, independently from that into L- and D-methionine formed from the D-methionine. In these experiments the calculations of number of millimoles of deuterated D- and L-methionine included a further factor: the percentage integral area (from the GC-MS analyses) of each deuterated enantiomer arising from either L-['~C'H,]- methionine or D-methionhe. As shown in Fig. 2, the rates of deuterium incorporation into D- and L-methionine for the D-methionhe substrate in the racemic mixture, are similar.
Mechanistic studies of amino acid racemases 183
Fig. 1 Rates of deuterium incorporation for D- and L-methionine reacted in D,O with the Aeromonas cuuiue broad substrate specific amino acid racemase. H = D-Methionine, = L-methionine. Conditions: 1 ml of 100 mM methionine in the following enantiomeric ratios: A - 100% D, B - 90% D: 10% L, c - 50% D: 50% L, D - 10% D: 90% L, E - 100% L
The pattern for the L-methionine substrate in this experiment, however, shows that while there is formation of deuterated D-methionine there is virtually no formation of deuterated L-methionine in the first twenty minutes of the enzyme incubation.
In a final set of experiments the rate of a-hydrogen exchange was compared to the rate of forward reaction for L-methionine in 'H,O with the P. striata amino acid racemase. This experiment gave a ratio of 1.3 in favor of exchange, while the same experiment conducted in parallel with D-methionhe gave a ratio of 1.8.
Fig. 2 Rates of deuterium incorporation into D- and L-methionine, for a pseudoracemic solution of D-methionine (A) and ~-['~CD,]methionine (B) in 'H,O, by the Aeromonas cauiae broad substrate specific amino acid racemase.
= D-Methionine, 0 = L-methionine

184 K. REYNOLDS rt al.
Discussion
The conversion of unlabeled L-alanine to D-alanine in deuterated buffer by the P. striata amino acid racemase under nearly irreversible conditions was shown to give product containing significant amounts of 'H at the a-position. This result had previously been interpreted to suggest that racemization of L-alanine by the racemase from P. striata proceeds via a one-base mechanism (SHEN et al. 1983). It is also clear that while the a-hydrogen of the amino acid is bound to the enzyme base it exchanges extensively with the medium.
From the experiments measuring the rates of deuterium incorporation, it is seen that both amino acid racemases produce neither the result predicted (CARDINALE and ABELES 1968) for a one-base nor a two-base mechanism. A one-base mechanism should give the same pattern of deuterium incorporation into D- and L-methionine, regardless of the initial substrate configuration. These broad specificity amino acid racemases, however, show radically different patterns of formation of deuterated D- and L-methionine which are dependent upon the initial substrate configuration. It could be that these enzymes contain an allosteric site to which either L or D amino acids bind to produce a conformational change in the active site, thereby giving rise to these substrate configuration-dependent deuterium incorporations. Such a possibility was tested using a pseudo-racemic mixture of D-methionine and ~-['~C'H,]methionine. Under these conditions the deuterium incorpo- ration patterns for the L- and D-methionine formed from the ~-['~C'H,]methionine of the racemic mixture are the same as observed with 100% L-methionine as substrate. Similarly, the result obtained for the D-methionine in the mixture is similar to that obtained when only D-methionine is the substrate. If there was an allosteric effect one would predict that the deuterium incorporation patterns for the D- or L-methionine in this racemic mixture would differ from those observed with the pure D- or L-methionine as substrate and would be the same as those seen with racemic methionine as substrate. These results clearly rule out an allosteric effect as the source of the observed dependence of deuterium incorporation on substrate configuration.
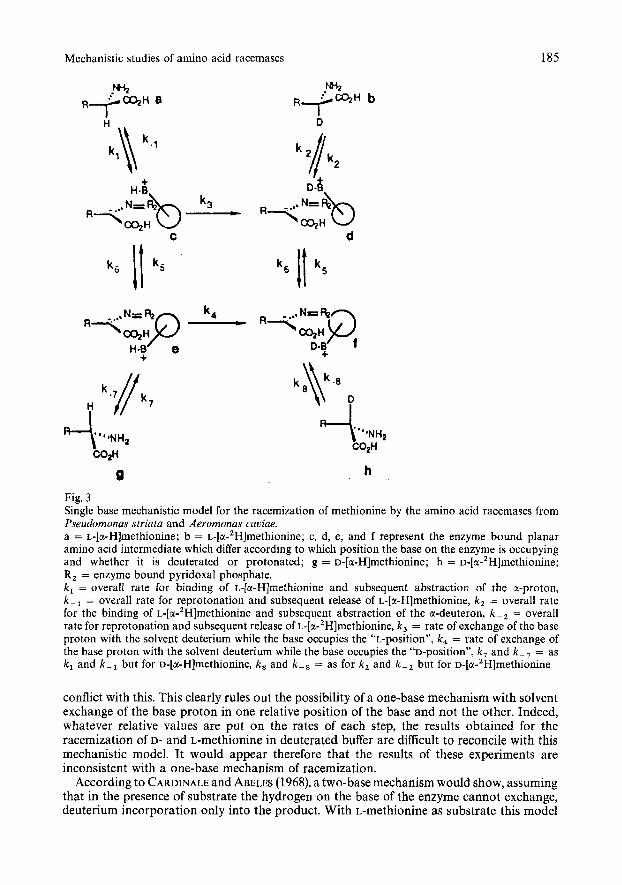
HENDERSON and JOHNSTON (1976) suggested on the basis of their results for alanine racemase from Bacillus subtilis a one-base, swinging door mechanism where one position of the door is more stable than the other. In such a model, as shown in Fig. 3, this would correspond to a difference in the values of k, and k,. This cannot be the case with the P. striata and A. caoiae amino acid racemases because it has been observed in the present work that when D-methionine is racemized, deuterated L- and D-methionine are formed at nearly the same rate. This could only arise if the values of k , and k , are similar. Along similar lines, a single base model can be considered where the base proton is able to exchange with the solvent while the door is in one position but is unable to do so when the door occupies the other position. In Fig. 3 this would correspond to k , being very small and k, being very large. Since, at best, only a small level of internal transfer of the a-proton is observed during the conversion of L-alanine to D-alanine this would indicate that k - , must be considerably smaller than k,. D-Methionine has also been shown to undergo conversion to L-methionine in deuterated buffer with almost no internal transfer of the a-hydrogen (MARTIN et al., unpublished results). This would indicate that either k, is larger than k , or alternatively k , is larger than k - In either case, it can be predicted that if D-methionhe is now racemized using this mechanistic model then it will be converted very quickly to intermediate f. The result of the studies with D-methionhe revealed that under these conditions deuterated L- and D-methionhe (compounds b and h) are formed at the same rate. This would again indicate that the relative values of k , and k , are similar. If L-methionine as substrate is analyzed by the same mechanistic model under the same constraints, it also will undergo conversion predominantly through intermediate f in order to produce deuterated amino acids. Accordingly, one would again predict that the deuterated D- and L-methionine will form at the same rate. The experimental evidence, however, is in direct
Mechanistic studies of amino acid racemases 185
.EH2 +rC4”
C d
h
Fig. 3 Single base mechanistic model for the racemization of methionine by the amino acid racemases from Pseudomonas striata and Aeromonas caviae. a = L-[a-Hlmethionine; b = ~-[a-~H]methionine; c, d, e, and f represent the enzyme bound planar amino acid intermediate which differ according to which position the base on the enzyme is occupying and whether it is deuterated or protonated; g = ~-[a-H]methionine; h = ~-[a-’H]methionine; R, = enzyme bound pyridoxal phosphate. k , = overall rate for binding of L-[a-Hlmethionine and subsequent abstraction of the a-proton, k - , = overall rate for reprotonation and subsequent release of L-[a-Hlmethionine, k , = overall rate for the binding of ~-[a-’H]methionine and subsequent abstraction of the a-deuteron, k - , = overall rate for reprotonation and subsequent release of ~-[a-~H]methionine, k , = rate of exchange of the base proton with the solvent deuterium while the base occupies the po position", k, = rate of exchange of the base proton with the solvent deuterium while the base occupies the “D-position”, k , and k - , = as k , and k - , but for D-[a-Hlmethionine, k , and k - , = as for k z and k - , but for ~-[a-’H]methionine
conflict with this. This clearly rules out the possibility of a one-base mechanism with solvent exchange of the base proton in one relative position of the base and not the other. Indeed, whatever relative values are put on the rates of each step, the results obtained for the racemization of D- and L-methionine in deuterated buffer are difficult to reconcile with this mechanistic model. It would appear therefore that the results of these experiments are inconsistent with a one-base mechanism of racemization.
According to CARDINALE and ABELES (1968), a two-base mechanism would show, assuming that in the presence of substrate the hydrogen on the base of the enzyme cannot exchange, deuterium incorporation only into the product. With L-methionine as substrate this model
186 K. REYNOLDS et al.
predicts that deuterium will be incorporated faster into D-methionine than L-methionine, exactly as observed in our work. Such a model, however, also predicts that with D-methionine as substrate there will be a faster formation of deuterated L-methionine compared to D-methionine. This was not observed, and in actual fact the rates of formation of the deuterated D- and L-methionine in this case are very similar. The latter pattern would be observed, however, if there were rapid exchange of the hydrogen on the base of the enzyme while the substrate is bound (this exchange being faster than the reprotonation of the enzyme-bound intermediate). This then would explain the results obtained for D-methionine but not those for L-methionine as substrate.
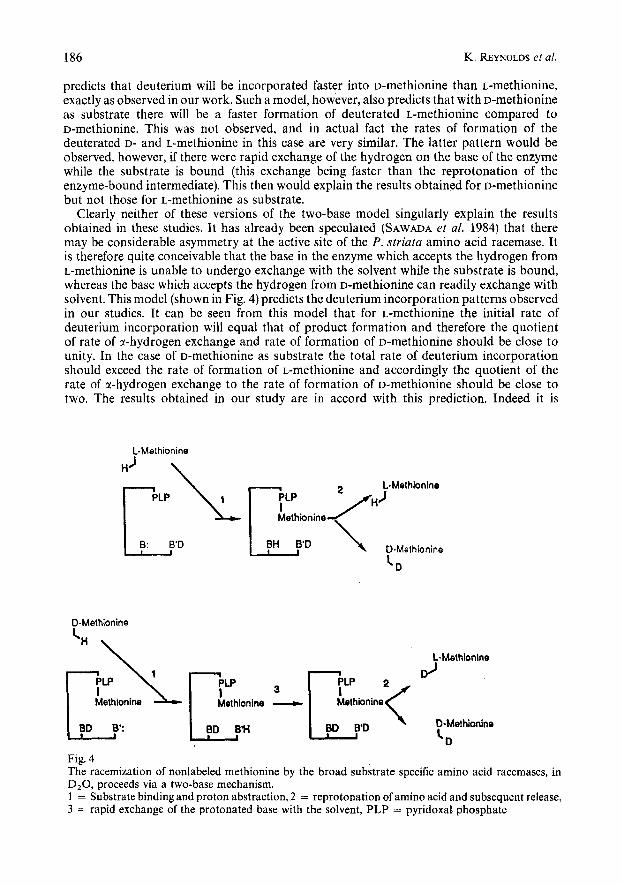
Clearly neither of these versions of the two-base model singularly explain the results obtained in these studies. It has already been speculated (SAWADA et al. 1984) that there may be considerable asymmetry at the active site of the P. striata amino acid racemase. It is therefore quite conceivable that the base in the enzyme which accepts the hydrogen from L-methionine is unable to undergo exchange with the solvent while the substrate is bound, whereas the base which accepts the hydrogen from D-methionine can readily exchange with solvent. This model (shown in Fig. 4) predicts the deuterium incorporation patterns observed in our studies. It can be seen from this model that for L-methionine the initial rate of deuterium incorporation will equal that of product formation and therefore the quotient of rate of a-hydrogen exchange and rate of formation of D-methionhe should be close to unity. In the case of D-methionine as substrate the total rate of deuterium incorporation should exceed the rate of formation of L-methionine and accordingly the quotient of the rate of a-hydrogen exchange to the rate of formation of D-methionhe should be close to two. The results obtained in our study are in accord with this prediction. Indeed it is
H;.Methionine
\ >jMethbnine
D-Methionine
t o BH B D \
0-Methionine
Methionine - BD B'D
Fig. 4 The racemization of nonlabeled methionine by the broad substrate specific amino acid racemases, in D,O, proceeds via a two-base mechanism. 1 = Substrate binding and proton abstraction, 2 = reprotonation of amino acid and subsequent release, 3 = rapid exchange of the protonated base with the solvent, PLP = pyridoxal phosphate

Mechanistic studies of amino acid racemases 187
pertinent to note that ADAMS (1976) obtained similar values for these measurements with alanine racemase and speculated that the results might reflect a two-base mechanism where a difference in the environment of the two postulated basic groups might permit exchange with the solvent (while the alanine is enzyme bound) in one case and not the other. It is particularly encouraging that this model predicts (CARDINALE and ABELES 1968) that in the racemization of nonlabeled L-methionine in 'H,O by the P . striuta amino acid racemase, an initial overshoot to D-methionhe, followed by a return to a racemic mixture should be observed. D-Methionine under the same conditions should exhibit no such behavior. Such a phenomenon has indeed been reported (SAWADA et al. 1984).
In summary, some of the results reported here are inconsistent with a one-base mechanism for the two pyridoxal phosphate-dependent amino acid racemases from P . striata and A. cavine, but these data can be accommodated by a two-base mechanism. The experiments measuring the relative rates of deuterium incorporation into D- and L-methionine argue strongly for a two-base mechanism with the enzyme containing an asymmetric active site in which one base can exchange more rapidly with the solvent than the other. However, this model does not explain any internal return of the a-hydrogen from substrate to product, if that is the explanation for the observations in the first set of experiments. A two-base mechanism would predict internal (but intermolecular) @-hydrogen transfer only in the unlikely event that at She end of a catalytic cycle at proton migrates from the protonated to the unprotonated base in the active site without the intervention of water molecules. Further experiments will be necessary to resolve the apparent discrepancy between the two sets of experiments.
Acknowledgements
We thank R. TSUCHIYA for preparation of the ~-['~C~H,]methionine and Professor R. W. WOODARD, University of Michigan, for providing an improved procedure for this synthesis, and Professor R. WEINKAM, then Purdue University, and R. HERROLD, Ohio State University for a large number or careful mass spectral analyses. We also are indebted to Profs. J. R. KNOWLES and C. WALSH, Harvard University, and Dr. W. S. FARACI, PFIZER, for useful comments on an earlier draft of this manuscript. Financial support by the US Public Health Service (GM 32333) is gratefully acknowledged.
References
ABE, I., IZUMI, K., KURAMOTO, S. and MUSHA, S., 1981. GC resolution of various D,L-aminO acid derivatives on a chiral-val capillary column. J. High Res. Chromatogr. and Chromatogr. Commun.,
ADAMS, E., 1972. Amino acid racemases and epimerases. In: The Enzymes VI (P. BOYER, Editor),
ADAMS, E., 1976. Catalytic aspects of enzymatic racemization. Adv. Enzymol. Relat. Areas Mol. Biol.,
AHMED, S. A,, ESAKI, N., TANAKA, H. and SODA, S., 1986. Mechanism of a-amino-e-caprolactam
ALBERY, J. and KNOWLES, J., 1986. Energetics and mechanism of proline racemase. Biochemistry, 25,
BIEMANN, K., 1962. In : Mass Spectroscopy: Organic Chemical Applications, p. 204- 227. McGraw
CARDINALE, G. J. and ABELES, R. H., 1968. Purification and mechanism of action of proline racemase.
HENDERSON, L. L. and JOHNSTON, R. B., 1976. Inhibition studies of the enantiomers of 8-chloroalanine
4, 549 - 552.
p. 281 -320. Academic Press New York.
44, 69-138.
racemase reaction. Biochemistry, 25, 385 - 388.
2572 - 2577.
Hill, New York.
Biochemistry, 7, 3979 - 3987.
on purified alanine racemase from B. subtilis. Biochem. Biophys. Res. Commun., 68, 793 - 798.

188 K. REYNOLDS et al.
INAYAKI, K., TANIZAWA, K., TANAKA, H. and SODA, S., 1987. Purification and characterization of amino acid racemase with very broad substrate specificity from Aeromonus cuuiue. Agric. Biol. Chem.,
RUNICK, G. and ABELES, R. H., 1975. Reaction mechanism and structure of the active site of proline racemase. Biochemistry, 14, 4515 - 5422.
SAWADA, S., ESAKI, N., YAGI, T. and SODA, K., 1984. Reaction mechanism of low substrate specific amino acid racemase of Pseudomonas striuta. Bull. Kyoto Univ. Education Series, B64, 21 -23.
SEN, S., FLOSS, H. G., KUMAGI, H., YAMADA, H., ESAKI, N., SODA, K., WASSERMANN, S. and WALSH, C., 1983. Mechanism of pyridoxal phosphate-dependent enzymatic amino-acid racemization. J. Chem. SOC., Chem. Commun., 82- 83.
SODA, K . and OSUMI, T., 1969. Crystalline amino acid racemase with low substrate specificity. Biochem. Biophys. Acta, 243, 363 - 368.
WISEMAN, J . S. and NICHOLS, J . S., 1984. Purification and properties of diaminopimelic acid epimerase from Escherichiu coli. J. Biol. Chem., 259, 8907 - 8914.
ZENK, M. H. und SCHMITT, J. H., 1965. Reinigung und Eigenschaften von Acetyl-CoA-D-Aminosaure- a-N-Acetyltransferase aus Hefe. Biochem. Z., 342, 54 - 65.
51, 173- 180.
Mailing address: Prof. Dr. H. G. FLOSS, Department of Chemistry BG 10, University of Washington, Seattle, Washington 98 195, USA







![Clinical and Therapeutic Implications of Aeromonas Bacteremia: … · 2016-12-28 · Aeromonas bacteremia are malignancy and hepatobiliary dis-eases [5]. Aeromonas spp. tend to produce](https://img.pdfslide.us/doc/110x75/5ec79db8c2bd727c0b32cc58/clinical-and-therapeutic-implications-of-aeromonas-bacteremia-2016-12-28-aeromonas.jpg)