Embed Size (px)
Citation preview

MECHANISMS OF EPHB2 MEDIATED OPIATE-DEPENDENT TOLERANCE AND LEARNING
By
Sofia Huroy
A thesis submitted in conformity with the requirements for the degree of Master of Science
Graduate Department of Pharmaceutical Sciences Faculty of Pharmacy, University of Toronto
© Copyright by Sofia Huroy 2012

ii
Mechanisms of EphB2 Mediated Opiate-Dependent Tolerance and Learning
Master of Science, 2012
Sofia Huroy
Department of Pharmaceutical Sciences, University of Toronto
ABSTRACT
The underlying mechanism of morphine tolerance remains unclear. EphB2 regulates synaptic
efficiency with respect to learning and memory. Previously, we demonstrated that loss of EphB2
significantly accelerates the rate of morphine tolerance and alters behavioural responses to
morphine following tolerance. However, EphB2 null mice exhibit no significant alteration in
their metabolism of morphine compared to littermate controls, or altered mu opioid receptor
expression levels within the spinal cord or brain compared to littermate controls. Therefore, we
investigated whether loss of EphB2 alters learned responsiveness to morphine through
modification of hippocampal function. Interestingly, results indicate that electrolytic lesions of
the dorsal hippocampus of wild-type mice display similar behavioural responses seen in EphB2
null mice compared to sham operated controls. These findings suggest that loss of EphB2
function within the hippocampus is a critical feature in mediating morphine-dependent
tolerance, and suggests a novel role for EphB2 receptor signaling in opiate-dependent learning.

iii
ACKNOWLEDGEMENTS
What a memorable two years it has been. First and foremost, I would like to thank my
supervisor, Dr. Jeffrey Henderson, who has mentored, challenged and motivated me throughout
my graduate studies. I am grateful and appreciate all the training I have learned and acquired
throughout these past two years.
I would also like to thank my thesis advisory committee members, Dr. ZhengPing Jia and Dr.
Peter Wells for their valuable suggestions and constructive comments throughout my study. I
would also like to acknowledge the following individuals who took time to assist me with my
research:
• Dr. Derek van der Kooy for his insightful suggestions and feedback, as well as providing
us with a generous supply of morphine.
• Dr. Ryan Ting-A-Kee for his statistical assistance.
• Dr. Carolyn Cummins and Lilia Magomedova for their assistance with optimization of the
LC-MS/MS protocol (adapted from Shu Chen).
• Dr. Sandy Pang for use of her lab’s C18 column to conduct our LC-MS/MS analyses, and
for providing us with a supply of morphine-3-glucuronide.
• Dr. James Eubanks for use of the activity monitors and Dr. Richard Logan for his
assistance in troubleshooting problems related to the activity monitors.
I would also like to thank my past and present lab members: Yanshan Cao, Ashlin Kanawaty,
Maya Latif, Mary Shan, William Tang and Zoe Winterton-Perks. Your support and friendship
was very much appreciated.

iv
Finally, I would like to thank my parents, my siblings, and my dearest friends for their
unconditional support, encouragement and love throughout these years. Thank you for all the
laughter and joy. I could not have done it without you by my side. Thank-you!

v
TABLE OF CONTENTS
ABSTRACT __________________________________________________________ ii
ACKNOWLEDGEMENTS ______________________________________________ iii
TABLE OF CONTENTS _________________________________________________ v
LIST OF FIGURES ____________________________________________________ ix
LIST OF TABLES _____________________________________________________ xi
SUMMARY OF ABBREVIATIONS _____________________________________ xii
CHAPTER 1: INTRODUCTION __________________________________________ 1
1.1 History of Opioids __________________________________________________ 2
1.2 Opioid Receptors ____________________________________________________ 3
1.2.1 Mu Opioid Receptor Structure ______________________________________ 5
1.3 Ligands for Opioid Receptors _________________________________________ 9
1.3.1 Endogenous Ligands _____________________________________________ 9
1.3.2 Exogenous Ligands _____________________________________________ 10
1.3.2.1 Morphine _________________________________________________ 14
1.4 Mu Opioid Receptor Signaling _______________________________________ 17
1. 5 Morphine Tolerance _______________________________________________ 19
1.6 Opiates and NMDA Receptors ________________________________________ 22
1.7 Animal Model: Mu Opioid Receptor Knockout Mice ______________________ 24
1.8 Eph Receptors and Ephrins ___________________________________________ 25
1.8.1 Eph/Ephrin Structure ____________________________________________ 25
1.9 Eph-ephrin Interactions and Signaling __________________________________ 30

vi
1.9.1 Forward Signaling ______________________________________________ 33
1.9.2 Reverse Signaling ______________________________________________ 34
1.9.3 Termination of Eph-ephrin Signaling _______________________________ 35
1.10 EphB2 Expression in the CNS _______________________________________ 35
1.11 EphB2 and Synaptic Plasticity _______________________________________ 37
1.12 EphB and Pain Modulation _________________________________________ 39
1.13 Learning and Memory: Overview ____________________________________ 41
1.13.1 Hippocampus _________________________________________________ 44
1.14 Opiate-Dependent Tolerance and Learning _____________________________ 46
1.14.1 EphB2 and Opiate Tolerance _____________________________________ 47
1.15 Thesis Rationale __________________________________________________ 54
1.15.1 Thesis Hypotheses _____________________________________________ 54
CHAPTER 2: MATERIALS AND METHODS _____________________________ 55
2.1 Animals _________________________________________________________ 56
2.2 Chemicals ________________________________________________________ 56
2.3 Morphine Tolerance Tests: EphB2 Wild-type and Null Mice _______________ 56
2.3.1 Sensory Analyses: Tail Pinch and Tail Flick Assay _____________________ 57
2.4 Pharmacokinetic Analyses of Morphine Metabolism ______________________ 57
2.4.1 Preparation of LC-MS/MS Standard Solutions ________________________ 57
2.4.2 Collection of Blood and Brain Samples ______________________________ 60
2.4.3 Purification of Blood and Brain Samples _____________________________ 60
2.5 LC-MS/MS Analyses _______________________________________________ 61
2.6 Immunohistochemistry ______________________________________________ 61

vii
2.7 Stereotactic Surgeris ________________________________________________ 62
2.7.1 Kainic Acid Induced Lesion _______________________________________ 62
2.7.2 Electrolytic Lesion ______________________________________________ 63
2.8 Behavioral Analyses ________________________________________________ 63
2.8.1 Passive Avoidance ______________________________________________ 63
2.8.2 Activity Monitor ________________________________________________ 65
2.9 Morphine Related Behavior __________________________________________ 65
2.9.1 Morphine Induced Hyperactivity ___________________________________ 65
2.9.2 Morphine Tolerance Tests: Lesioned and Sham Operated Control Animals __ 65
2.10 Statistical Analyses _______________________________________________ 66
CHAPTER 3: RESULTS ________________________________________________ 67
3.1 Morphine related responses of EphB2 null mice __________________________ 68
3.2 Distribution of mu opioid receptors in the CNS __________________________ 70
3.3 Pharmacokinetic Analysis of Morphine in EphB2 Null Mice ________________ 75
3.4 EphB2 null mice display deficits in hippocampal learning ___________________ 81
3.4.1 Behavioral assessments of animals with bilateral electrolytic lesions in the
dorsal hippocampus __________________________________________________ 83
3.5 Morphine related responses in animals with bilateral electrolytic lesions in the
dorsal hippocampus ____________________________________________________ 91
CHAPTER 4: DISCUSSION ____________________________________________ 105
4.1 Changes in morphine responsiveness seen in EphB2-null mice are mediated via
modification of cortical influences on sensory function. ______________________ 106

viii
4.2 EphB2 null mice exhibit deficiencies in contextual learning similar to that seen
in wild-type animals containing bilateral electrolytic lesions of the dorsal
hippocampus. ________________________________________________________ 109
4.3 Impaired opiate-dependent responses seen in EphB2 null mice arise from
hippocampal-dependent deficiencies in contextual learning. ___________________ 111
4.4 Concluding remarks and future studies _________________________________ 114
REFERENCES _______________________________________________________ 116

ix
LIST OF FIGURES
CHAPTER 1: INTRODUCTION
Figure 1.1 Alternative splicing of mouse Oprm1 gene
Figure 1.2 Crystal structure of the mu-opioid receptor bound to morphinan antagonist β-FNA
Figure 1.3 Structures of common MOR ligands
Figure 1.4 Metabolism of morphine
Figure 1.5 Pre- and postsynaptic MOR signaling
Figure 1.6 Proposed pathways for MOR regulation
Figure 1.7 Eph ligand and receptor structure
Figure 1.8 Eph-ephrin Signaling
Figure 1.9 Morphine tolerance in EphB2 null mice on Day 1
Figure 1.10 Morphine tolerance in EphB2 null mice on Day 3
Figure 1.11 Morphine tolerance in EphB2 null mice on Day 6
Figure 1.12 Antinociceptive responses of EphB2 wild-types upon removal to novel
environment.
Figure 1.13 Antinociceptive responses of EphB2 null mice upon removal to novel environment.
CHAPTER 2: MATERIALS AND METHODS
Figure 2.1 Schedule of morphine dosing
Figure 2.2 Schedule of analysis following stereotactic surgery.
CHAPTER 3: RESULTS
Figure 3.1 Expression of EphB2 in the CNS

x
Figure 3.2 Distribution of mu opioid receptor in dorsal spinal cord of EphB2 null mice and
controls
Figure 3.3 Distribution of mu opioid receptors in striatum and hippocampus of EphB2 null mice
and wild-type littermates
Figure 3.4 LC/MS/MS analyses of morphine and metabolites
Figure 3.5 LC/MS/MS analyses of brain morphine metabolism in EphB2 null mice and controls
Figure 3.6.LC/MS/MS analyses of blood morphine metabolism in EphB2 null mice and
controls
Figure 3.7 LC/MS/MS analyses of brain and blood morphine metabolism in EphB2 null mice
and controls
Figure 3.8 Passive avoidance responses of EphB2 mice.
Figure 3.9 Example of an electrolytic lesion of the dorsal hippocampus.
Figure 3.10 Serial views of electrolytic lesions of the dorsal hippocampus
Figure 3.11 Passive avoidance responses of lesioned wild-type animals
Figure 3.12 Active time of EphB2 null mice and lesioned wild-types
Figure 3.13 Motor activity of EphB2 null mice and lesioned wild-types
Figure 3.14 Morphine induced hyperactivity in wild-type mice.
Figure 3.15 Morphine induced hyperactivity in EphB2 null mice.
Figure 3.16 Spontaneous motor activity of saline injected animals.
Figure 3.17 Morphine induced hyperactivity in sham operated control animals.
Figure 3.18 Morphine induced hyperactivity in lesioned animals.
Figure 3.19 Antinociceptive responses of sham and lesioned animals in home versus novel
environments

xi
LIST OF TABLES
CHAPTER 1: INTRODUCTION
Table 1.1 Overview of opioid receptors.
Table 1.2 Binding affinities of endogenous opioid peptides to Mu, Delta and Kappa opiate
receptors in nanomolar
Table 1.3 Binding affinities of ephrin-Fc’s for EphB2 ligand binding domain.
Table 1.4 Calculated binding affinities of ephrin ligands to EphA4.
CHAPTER 3: RESULTS
Table 3.1 Analyses of day 1, 3 and 6 antinociceptive responses by 3-way ANOVA
Table 3.2 Analyses of day 7 antinociceptive responses by 3-way ANOVA

xii
SUMMARY OF ABBREVIATIONS
AC Anterior commissure
ACpa Pars anterior branch of the AC
ACpp Pars posterior branch of the AC
ADAM A-disintegrin and metalloprotease
AMPA 2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl)propanoic acid
ANOVA Analysis of variance
CGRP Calcitonin gene related peptide
CNS Central nervous system
DAB 3,3’-diaminobenzidine
DAMGO [D-Ala2, N-MePhe4, Gly-ol]-enkephalin
DAPI 4',6-diamidino-2-phenylindole
DOR Delta opioid receptor
DRG Dorsal root ganglion
ECL Extracellular loop
Eph receptor Erythropoietin producing hepatocellular receptor
Ephexin Eph-interacting exchange proteins
Ephrin Eph family receptor interacting protein
ERK Extracellular signal-regulated kinase
GABA γ-Aminobutyric acid
GDP Guanosine diphosphate
GEF Guanine nucleotide exchange factor
GFAP Glial fibrillary acidic protein

xiii
GPI Glycosyl-phosphatidyl-inositol
Grb Growth factor receptor-bound protein
GIRK channel G protein-coupled inwardly-rectifying potassium channel
GPCR G protein coupled receptor
Grb4 Growth factor receptor-bound protein 4
GRIP Glutamate receptor-interacting protein1
GRK G protein-coupled receptor kinase
GTP Guanosine triphosphate
HRP Horseradish peroxidase
IB4 Isolectin B4
ICL Intracellular loop
JMR Juxtamembrane region
KD Kinase domain
KOR Kappa opioid receptor
LBD Ligand binding domain
LC-MS/MS Liquid chromatography–tandem mass spectrometry
LTD Long term depression
LTP Long term potentiation
M3G Morphine-3-glucuronide
M6G Morphine-6-glucuronide
MAPK Mitogen-activated protein kinase
MOR Mu opioid receptor
MRM Multiple Reaction monitoring
M/Z Mass-to-charge ratio

xiv
N-WASP Neural Wiskott-Aldrich syndrome protein
NMDA N-methyl-D-aspartate
PBS Phosphate buffered saline
PDZ postsynaptic density protein (PSD95)/Drosophila disc large
tumour suppressor (Dlga)/ zonula occludens-1 protein (zo-1)
PFA Paraformaldehyde
PKA Protein kinase A
PKC Protein kinase C
RT Retention time
RTK Receptor tyrosine kinase
SAM Sterile α motif
SEM Standard error of the mean
SFK Src family kinase
SH2 Src-homology 2 domain
TLT Transfer latency time
TM Transmembrane

CHAPTER 1: INTRODUCTION
1

1.1 History of Opioids
For thousands of years, crude opium extracts from poppy seeds (Papaver somniferum)
have been used for medicinal and recreational purposes, and are among the oldest known agents
to treat pain [1]. Despite their significant worldwide usage clinically, use of opioids is
complicated by a variety of side effects including the risk of respiratory depression, sedation,
and gastrointestinal dysfunction [2, 3]. As well, repeated use of opioids increases the risks for
the development of tolerance, physical dependence and addiction [2, 3]. Opium is a mixture of
plant alkaloids comprised primarily of benzylisoquinoline and phenanthrene class-alkaloids [3].
In 1805, morphine, a phenanthrene alkaloid, became the first pure alkaloid isolated from opium
extracts. The name morphine is derived from Morpheus, the Greek god of sleep [1]. Over the
next few decades, additional alkaloids were isolated from opium extracts and as a class the drug
group became known as opiates [3].
In 1973, three independent research laboratories determined that opiates bound to
specific receptors within the brain [4-6]. Given the nature of these receptor responses,
researchers hypothesized that these receptors must normally bind endogenous forms of these
ligands. In 1975 Hughes et al., determined the first of these endogenous ligands now termed
enkephalins [5]. When introduced, this short polypeptide exhibited morphine-like properties
with binding to opiate receptors within the brain [7]. Shortly thereafter, two additional peptide
classes with morphine-like properties were discovered and classified as endorphins and
dynorphins [8, 9]. Collectively, these ligands are now termed opioids, distinguishing them from
opiates, compounds found naturally in opium [3, 10]. Historically, opioid receptors were named
and classified based their most selective agonists. As such, receptors were named mu (µ) for
morphine, kappa (κ) for ketocyclazocine, and sigma (σ) for SKF 10,047 (or N-
allylnormetazocine) [11]. However, the sigma opioid receptor is no longer considered a true
opioid receptor and has been replaced by the delta (δ) opioid receptor [12]. My thesis focuses
2

on understanding the interaction between a specific EphB-family receptor and the mu opioid
receptor.
1.2 Opioid Receptors
As indicated above, three primary types of opioid receptors exist: mu (µ), delta (δ), and
kappa (κ). In recent years, a fourth subtype of opioid receptors has also been reported, named
the Opioid Receptor Like-1 (ORL1) [13-15]. This receptor subclass, although its amino acid
sequence is similar to that of other opioid receptors, it does not bind to classical opioid ligands
[15, 16]. All opioid receptors are encoded by separate structural genes. The receptor genes for
mu, kappa, delta, and ORL1 are Oprm1, Oprk1, Oprd1 and Oprl1 respectively. Binding sites
within opiate receptors share several structural similarities. Thus, opioid receptors can exhibit
significant overlap with respect to binding of particular ligands, despite noted profiles of
selectivity. The majority of opioids and opiates can bind to multiple receptor subtypes, but may
exhibit noted selectivity toward a particular receptor subtype [1]. Also, a few variants exist for
each opioid receptor subtype: µ1/ µ2, δ1/δ2, and κ1/ κ2/ κ3 [3, 12]. Table 1.1 summarizes the
prototypic ligands and important physiological effects of each receptor subtype [3, 17-19].
Structurally, the genes of opioid receptors are highly homologous to one another, with their
coding regions divided over three exons. Exon 1 codes for the extracellular domain and for
transmembrane domain I. Exon 2 codes for transmembrane domains II–IV. Exon 3 codes for
transmembrane domains V–VII followed by the cytoplasmic C-terminal region. The only
variation currently known is for the Oprm1 gene where the last 12 codons of exon 3 are actually
found on a fourth coding exon [20], and in recent years a number of additional exons having
been reported (see below).
Research over the past several decades has confirmed all opioid receptors display classic
G-protein coupled receptor (GPCR) signaling characteristics [12, 21, 22]. Like other GPCRs,
3
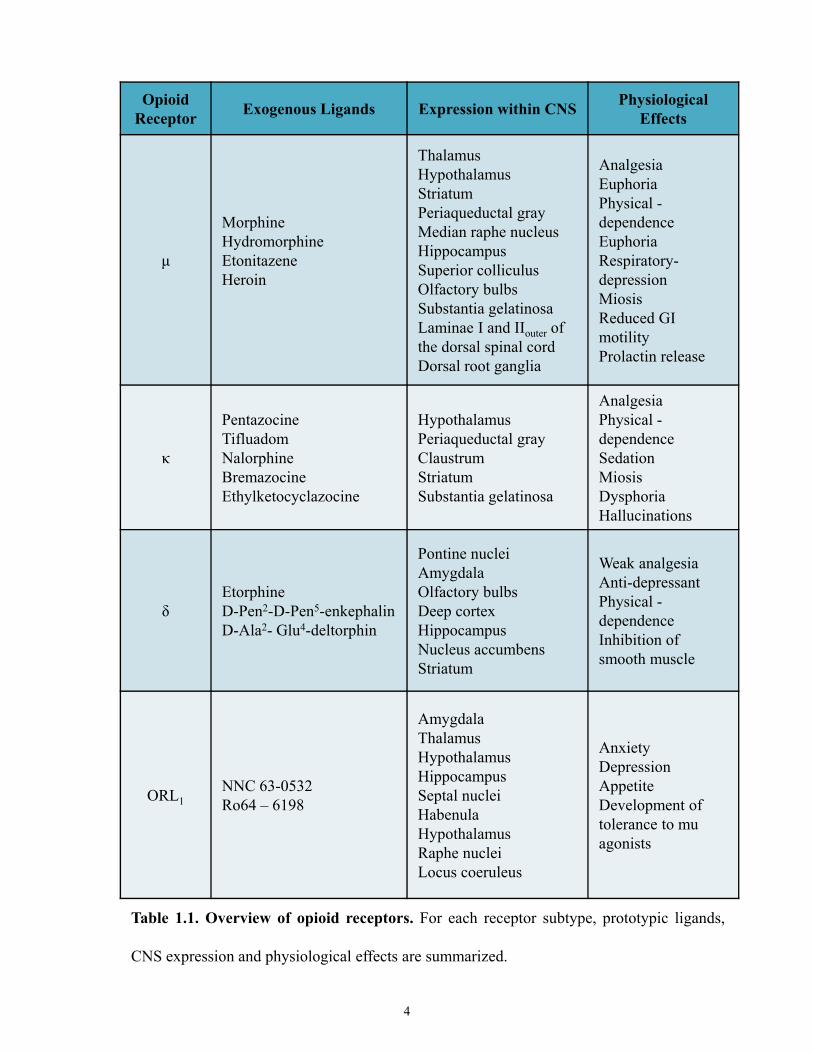
OpioidReceptor Exogenous Ligands Expression within CNS Physiological
Effects
μ
MorphineHydromorphineEtonitazeneHeroin
ThalamusHypothalamusStriatum Periaqueductal gray Median raphe nucleusHippocampusSuperior colliculusOlfactory bulbsSubstantia gelatinosaLaminae I and IIouter of the dorsal spinal cordDorsal root ganglia
AnalgesiaEuphoriaPhysical -dependenceEuphoriaRespiratory-depressionMiosisReduced GI motilityProlactin release
κ
PentazocineTifluadomNalorphineBremazocineEthylketocyclazocine
HypothalamusPeriaqueductal gray ClaustrumStriatumSubstantia gelatinosa
AnalgesiaPhysical -dependenceSedationMiosisDysphoriaHallucinations
δEtorphineD-Pen2-D-Pen5-enkephalinD-Ala2- Glu4-deltorphin
Pontine nucleiAmygdalaOlfactory bulbsDeep cortexHippocampusNucleus accumbensStriatum
Weak analgesiaAnti-depressantPhysical -dependenceInhibition of smooth muscle
ORL1NNC 63-0532Ro64 – 6198
AmygdalaThalamusHypothalamusHippocampusSeptal nucleiHabenulaHypothalamusRaphe nuclei Locus coeruleus
AnxietyDepressionAppetiteDevelopment of tolerance to mu agonists
Table 1.1. Overview of opioid receptors. For each receptor subtype, prototypic ligands,
CNS expression and physiological effects are summarized.
4

opioid receptors are believed to be capable of hetero or homodimerization [21, 23]. GPCRs can
be classified into six primary groups based on their relative sequence homology and functional
similarities. Opioid receptors belong to Class A, the Rhodopsin-like receptor family. Within this
class, opioid receptors are categorized under the γ subfamily [12, 22, 24]. Like many GPCRs,
opioid receptors are composed of 7 transmembrane (TM) spanning regions [12, 25]. These
opioid receptors display high sequence homology with highly conserved intracellular loops and
transmembrane (TM) domains. In particular TM helices II, III and VII display the highest
sequence homology (75%) among the different classes of opioid receptors. Variations among
opioid receptors classes include the N-terminus, extracellular loops (especially ECL 2 and 3), C-
terminus and the number and location of surface glycosylation sites (5 on MORs, 2 on DORs
and 2 on KORs) [12, 26]. These differences are thought to explain the different affinities and
actions of opiate ligands on opioid receptors.
1.2.1 Mu Opioid Receptor Structure
Cloning and characterization of MORs [27-29] provided much insight into their
structure-function relationships. Initially, the MOR gene Oprm1 was believed to contain one
promoter and four exons, all of which encoded one protein [30]. However, pharmacological
binding studies revealed multiple isoforms of MORs existed: mu1, mu2, and morphine-6-B-
glucuronide [31]. However, given that a mouse has a single Oprm gene, researchers began to
investigate how multiple subtypes of the MOR could exist. Researchers hypothesized that
alternative pre-mRNA splicing may be playing a role [30-32]. Multiple studies have shown that
that the mouse Oprm1 gene is more complex than initially thought, and in fact comprises two
promoters generating up to 27 splice variants [30] (Figure 1.1). These splice variants have been
established to exist either through differential 3′-splicing or 5’-splicing. Variants of 3′-splicing
are typically generated through the exon1 promoter and are traditional G protein coupled
5

Figure 1.1. Alternative splicing of mouse Oprm1 gene. Two major classes of splice
variants exist, those generated by the promoter associated with exon 1 (white), and those
generated by the promoter in exon 11 (yellow).
Adapted from Majumdar et al., Proc Natl Acad Sci USA , 2011
6

receptors with 7-TM regions [32]. These variants differ in their carboxyl termini, which create
functional differences in their agonist-induced G-protein activation, adenylyl cyclase activity,
receptor internalization and agonist-induced receptor phosphorylation [30, 31]. As well, some
variants display different desensitization properties, where some MOR isoforms are more
resistant to agonist-induced desensitization [33, 34]. In contrast, 5′ splice variants which are
believed to be generated through exon 11 promoter, comprise traditional full-length receptors
but also truncated 6-TM and 1-TM variants [32]. These exon 11-associated variants are believed
to mediate drug specific actions such as analgesia without the common side effects typically
observed [32]. Specifically, it has been shown that exon 11 knockout mouse exhibited normal
morphine and methadone induced analgesia, but failed to display analgesic response following
heroin, M6G, and fentanyl administration [35]. These variants also displayed differences in
regional distributions within the CNS [36].
Until recently, the crystal structure of opioid receptors was extrapolated from closely
related GPCRS such as the β-adrenergic receptor [37]. Recently Manglik et al., (2012)
crystallized the 2.8A˚ mouse MOR complexed with the irreversible morphinan antagonist β-
FNA (β-Funaltrexamine) [38] (Figure 1.2). As expected, the structure consisted of seven TM α-
helices connected by three extracellular loops (ECL1–3) and three intracellular loops (ICL1–3).
TM3 is connected to ECL2 by a conserved disulphide bridge [38]. The intracellular surface of
the MOR closely resembled that of the related GPCR, rhodopsin, with respect to the relative
positions of TM3, TM5 and TM6. The MOR crystallized structure was found arranged in
dimmers, with the association found between the TM5 and TM6 interface. Another, dimer
structure was also found to exist between the interface of TM1 and TM2, and helix 8.
Normally, ligand binding pocket within GPCRs is buried within a helical bundle [38]. In
contrast, the binding pocket for β-FNA in the MOR was largely exposed to the extracellular
surface, and made contact with TM3, TM5, TM6 and TM7 [38]. The authors suggest that this
7
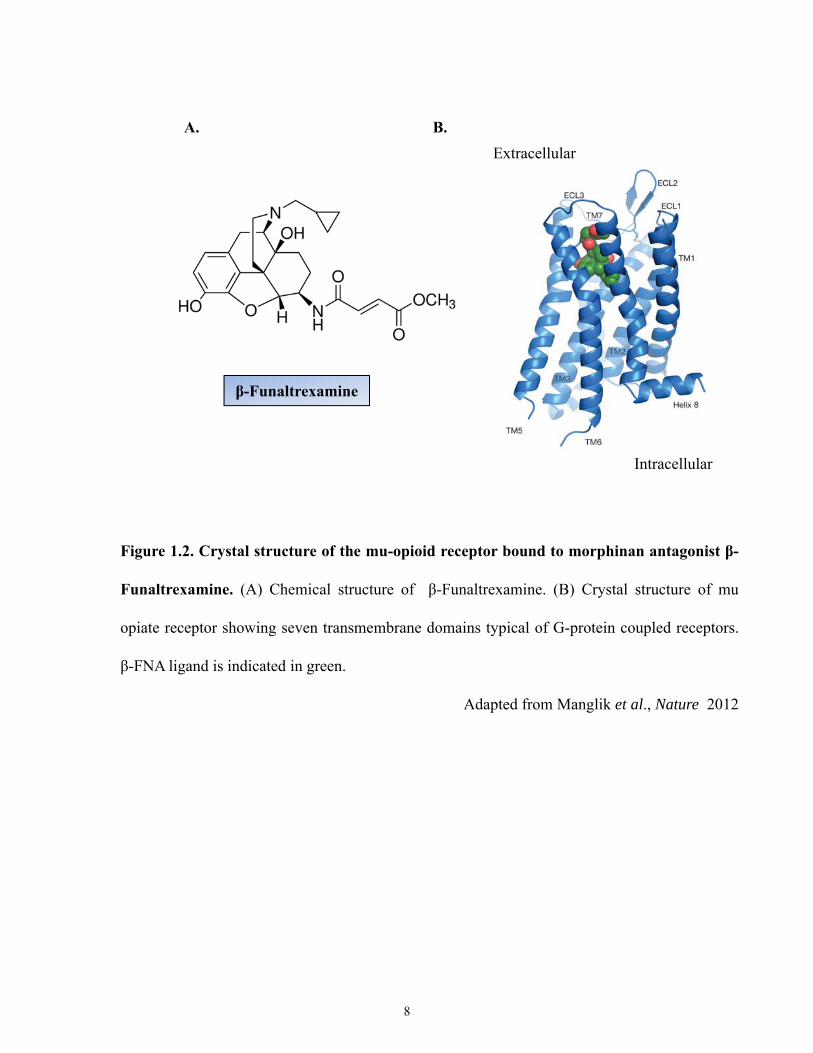
Figure 1.2. Crystal structure of the mu-opioid receptor bound to morphinan antagonist β-
Funaltrexamine. (A) Chemical structure of β-Funaltrexamine. (B) Crystal structure of mu
opiate receptor showing seven transmembrane domains typical of G-protein coupled receptors.
β-FNA ligand is indicated in green.
Adapted from Manglik et al., Nature 2012
β-Funaltrexamine
A. B.Extracellular
Intracellular
8

type of exposed ligand binding pocket may provide a basis for fast dissociation kinetics of
opiate ligands, and thus could be used to explain some of the unique pharmacologic and
physiologic properties of distinct opioid ligands. For instance, potent opioids such as
buprenorphine and etorphine have an inhibition constant (Ki) of 740pM and 270pM,
respectively; and rapid dissociation half-lives of 44 min and 1 min, respectively [38]. Thus, the
authors of the study suggested this may explain why heroin overdoses are rapidly reversible by
naloxone (highly selective competitive MOR antagonist); given that the MOR binding pocket is
largely exposed to the extracellular surface [38].
1.3 Ligands for Opioid Receptors
1.3.1 Endogenous Ligands
Opioid receptors differ in their physiological responses, tissue distribution and relative
affinity for various opioid ligands. All endogenous opioid peptides share a common NH2-
terminal Tyr-Gly-Gly-Phe sequence which interacts with the opioid receptor [1, 39]. Opioids act
both centrally and peripherally, and depending on the class of opioid receptors activated, they
mediate different physiological responses. Opioid peptides are initially synthesized as part of a
larger precursor molecule. Each opioid peptide arises from a unique precursor, which has
prepro- and pro- forms, from which the active opioid peptide and other neuroendocrine peptides
are derived from [10].
Enkephalins are short pentapeptides cleaved from the precursor pro-enkephalin A, and
was first identified in the adrenal medulla [10]. Subtypes of this peptide include: Met-
enkephalin, Leu-enkephalin, Met-enkephalin-Arg6-Phe7, Met-enkephalin-Arg6-Gly7-Leu8 and
peptide E. Met-enkephalin and Leu-enkephalin possess high selectivity for DORs [10, 39]. Met-
enkephalin-Arg6-Phe7 and Met-enkephalin-Arg6-Gly7-Leu8 display comparable affinities for
9

MORs and DORs, with lower affinities for KORs. Peptide E shows high affinity to MORs but
also for KORs [10]. β-endorphins are 31-amino acids long peptides, which are cleaved from
pro-opiomelanocortin (POMC), found in the pituitary [10]. β-endorphins are the main parent
peptide of the endorphin family, although shorter cleavage products of β-endorphins have been
reported. They display binding selectivity for MORs over DORs, with negligible affinity for
KORs [10, 40]. Dynorphins are 17-amino acids long peptides. They are cleaved from pro-
dynorphin (also known as pro-enkephalin B). This family of peptides is comprised of:
dynorphinA (1-17), dynorphinA (1-8), dynorphin B, α-neo-endorphin, and β-neo-endorphin
[10]. The dynorphin family of peptides display greater preference for KORs. However,
dynorphinA (1-8) display some binding affinities for DORs and dynorphinA (1-13) is potent at
both KORs and MORs [10, 39]. For the ORL1 receptor, the only known ligand is Orphanin FQ
(OFQ or Nociceptin). Structurally it resembles other opioid peptides particularly dynorphin A.
Similar to other peptides described, it is also derived from a preproOFQ precursor to yield a
peptide that is 17-amino acids long [14-16]. Table 1.2 summarizes the relative binding affinities
of these peptides to their respective opioid receptors [41].
1.3.2 Exogenous Ligands
Exogenous ligands for opioid receptors comprise a spectrum of drugs that includes
opiates (derived from opium) but also semi-synthetic and synthetic compounds (Figure 1.3).
Examples of opiates include morphine and codeine (3-methylmorphine) [3]. Semi-synthetic
morphine derivatives display morphine-like pharmacology and consist of a morphine-like
chemical structure. For example, heroin is a diacetylated morphine, which is twice as potent as
morphine; or oxycodone which has a methoxy substituent replacing the C3 hydroxyl group and
has better oral bioavailability than morphine [3]. Fully synthetic opioids also display morphine-
like pharmacology but do not possess a morphine-like chemical structure. For example,
10
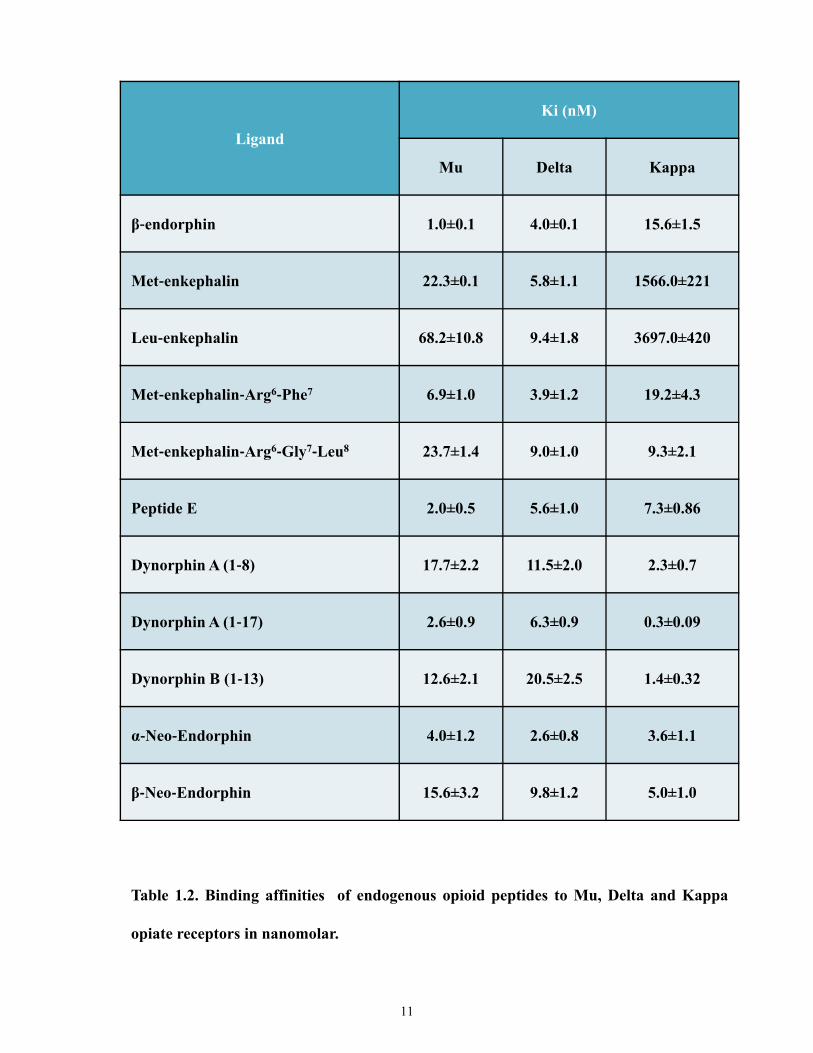
Ligand
Ki (nM)
Mu Delta Kappa
β‐endorphin 1.0±0.1 4.0±0.1 15.6±1.5
Met‐enkephalin 22.3±0.1 5.8±1.1 1566.0±221
Leu‐enkephalin 68.2±10.8 9.4±1.8 3697.0±420
Met‐enkephalin‐Arg6‐Phe7 6.9±1.0 3.9±1.2 19.2±4.3
Met‐enkephalin‐Arg6‐Gly7‐Leu8 23.7±1.4 9.0±1.0 9.3±2.1
Peptide E 2.0±0.5 5.6±1.0 7.3±0.86
Dynorphin A (1‐8) 17.7±2.2 11.5±2.0 2.3±0.7
Dynorphin A (1‐17) 2.6±0.9 6.3±0.9 0.3±0.09
Dynorphin B (1‐13) 12.6±2.1 20.5±2.5 1.4±0.32
α‐Neo‐Endorphin 4.0±1.2 2.6±0.8 3.6±1.1
β‐Neo‐Endorphin 15.6±3.2 9.8±1.2 5.0±1.0
Table 1.2. Binding affinities of endogenous opioid peptides to Mu, Delta and Kappa
opiate receptors in nanomolar.
11

Figure 1.3. Structures of common MOR ligands. (A) Example of an endogenous MOR
agonist β-endorphin. (B) Common MOR opiates and semi-synthetic ligands. (C) Synthetic
ligands. (D) Common antagonists and partial agonist (buprenorphine).
12
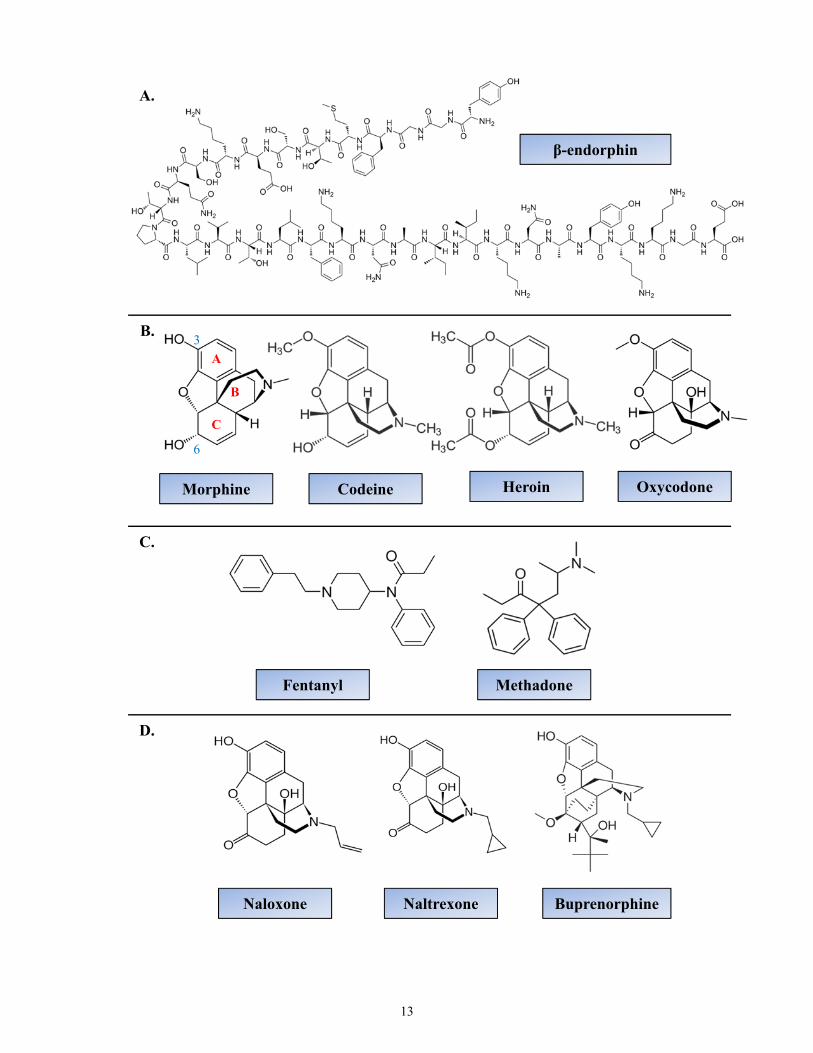
β-endorphin
Morphine Codeine Heroin
Naloxone Naltrexone
Oxycodone
Buprenorphine
Fentanyl Methadone
A.
C.
D.
B.
A
C
B
3
6
13

methadone which is as potent as morphine but has a longer duration of action, and fentanyl
which has a rapid onset but a short duration of action [3]. In addition, over the years researchers
have designed several synthetic opioids demonstrating greater class specificity. Examples of
these highly selective agonists for the mu, delta and kappa receptor sites are DAMGO (D-
Ala2,N-MePhe4,Gly-ol5-enkephalin), DPDPE (D-Pen2,D-Pen5-enkephalin) and U69,593,
respectively [42-44]
Structurally, opiates can exist in either the levorotatory (-) or dextrorotatory (+) form.
The biologically active isomer of morphine is the levo isomer. The dextro isomers do not
possess analgesic properties [3]. Within morphine (Figure 1.3), the presence of the phenolic OH
groups in position 3 is considered important for opiate action [39]. Substitution of the methyl
group on the nitrogen atom on morphine determines the agonistic or antagonistic nature of the
ligand. For instance a methyl group results in agonistic activity, whereas substitutions of allyl/
cyclopropylmethyl/ or propyl groups results in antagonistic pharmacological activity [39].
Examples of opioid antagonists include naloxone, naltrexone, and buprenorphine (Figure 3).
Naloxone which has an allyl substitution has the greatest affinity for MORs [39]. Naltrexone
which has a cyclopropylmethyl is similar to naloxone, but has a longer half-life [3, 39].
Buprenorphine has a cyclopropylmethyl substituent on the N-atom like naltrexone. However,
because it also has a methoxy group on C6 similar to codeine, it displays mixed agonistic and
antagonistic properties [3].
1.3.2.1 Morphine
Morphine is a prototypical MOR opiate [3]. Its C-ring adopts a boat conformation,
which places the 6α-hydroxyl group in an equatorial position (Figure 1.3). This hydroxyl group
is believed to confer the principle component of selectivity [25]. Morphine can also bind with
lower affinities to KORs and DORs and produce diminished analgesic responses, as examined
14

through MOR knockout mice [45]. Since MORs are expressed in the CNS and peripherally,
treatment with morphine frequently provokes undesirable side effects such as respiratory
depression, constipation, nausea and vomiting [2, 3, 46].
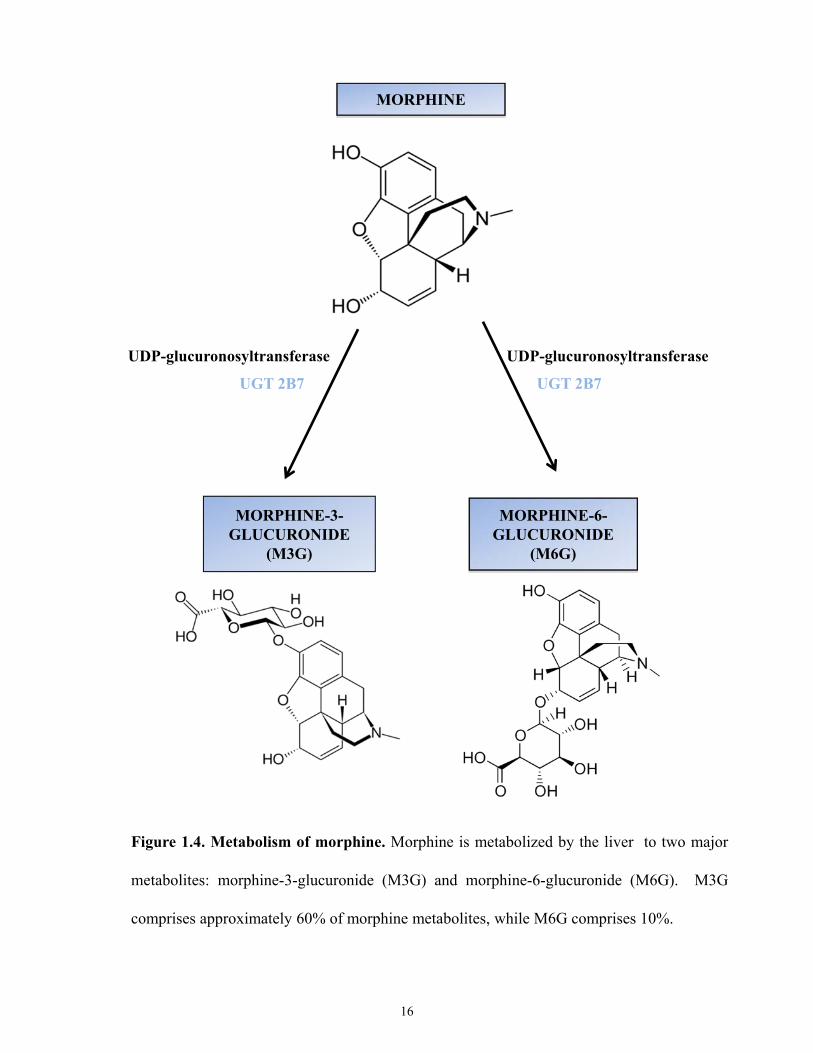
Metabolism of morphine is primarily catalyzed by uridine 5’-diphospho-
glucuronosyltransferase (UGT) enzymes [47]. Morphine exhibits a systemic half life of
approximately two hours in humans and 50 minutes in mice [48]. In humans, morphine is
primarily metabolized by UGT2B7 to morphine-3-glucuronide (M3G) and morphine-6-
glucuronide (M6G) (Figure 1.4) [47, 49, 50]. In rodents, UGT2B1 has also been shown to
catalyze the formation of M3G [49]. Glucuronidation occurs on free hydroxyl groups. Aromatic
hydroxyl groups (position 3 of morphine) are glucuronidated more readily than alicyclic
hydroxyl groups (position 6 of morphine) [47, 50]. In humans, approximately 60% of morphine
is converted to M3G and 10% to M6G [51]. The remaining 30% of morphine is converted to
compounds such as normorphine, 3-acetylmorphine, morphine-6-sulfate, morphine-6-sulfate,
and 6-acetylmorphine [51].
A significant fraction of morphine binding occurs within the CNS. The exact
mechanism of transport of morphine and its metabolites into and out of the brain is still unclear.
However, the primary blood brain barrier transporter of morphine is believed to be the P
glycoprotein (Pgp) (also known as ABCB1 transporter), which is an ATP-dependent active
efflux pump [47, 52]. This transporter has been shown to limit the rate of morphine
accumulation within the brain [52]. However, this transporter has not been shown to transport
morphine-glucuronides across the blood brain barrier [47, 53].
In humans, both M3G and M6G are produced. Mice also metabolism morphine to M3G,
however whether they metabolize morphine to M6G is still a matter of some debate. Several
studies suggest that little to no M6G is produced in rodents [54, 55]. However, the issue is
clouded by the relative low levels of M6G compared to other morphine metabolites. M3G is
15
MORPHINE
MORPHINE-6-GLUCURONIDE
(M6G)
UGT 2B7 UGT 2B7
Figure 1.4. Metabolism of morphine. Morphine is metabolized by the liver to two major
metabolites: morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G). M3G
comprises approximately 60% of morphine metabolites, while M6G comprises 10%.
MORPHINE-3-GLUCURONIDE
(M3G)
UDP-glucuronosyltransferaseUDP-glucuronosyltransferase
16

biologically inactive and does not bind to the MORs [47]. Treatment with M3G alone does not
illicit analgesic activity [47]. However, some studies have reported it can antagonize some
pharmacological actions of morphine [56], although this is still a matter of some debate [57-59].
In contrast, M6G possesses distinct analgesic properties such as exhibiting a slower onset of
action and longer duration of action compared to morphine. M6G has also been reported to
exhibit equal or greater potency than morphine for MORs [56, 60-62]. Notably, M6G bypasses
many of the common side effects seen with morphine such as respiratory depression [47]. This
has created much interest in M6G as a possible therapeutic alternative to morphine. A downside
of this is that M6G is more hydrophilic than morphine and hence exhibits lower blood brain
barrier permeability [47, 63].
1.4 Mu Opioid Receptor Signaling
Activation of MOR signalling mediates many physiological functions such as analgesia,
respiratory depression, state of euphoria, sedation, reduced gastrointestinal mobility, nausea, and
miosis [2, 3, 46]. As with other GPCRS, opioid receptors convey their signals through activation
of heterotrimeric G-proteins via exchange of bound GDP for GTP, and interaction with the
inhibitory (Gi/Go) G-proteins [37, 64-66]. Such conformational modulation is achieved through
interaction of the MOR cytoplasmic domain with the G-protein heterotrimer. Specifically,
ligand binding causes a re-arrangement of transmembrane domains 3, 6, and 7 from the inactive
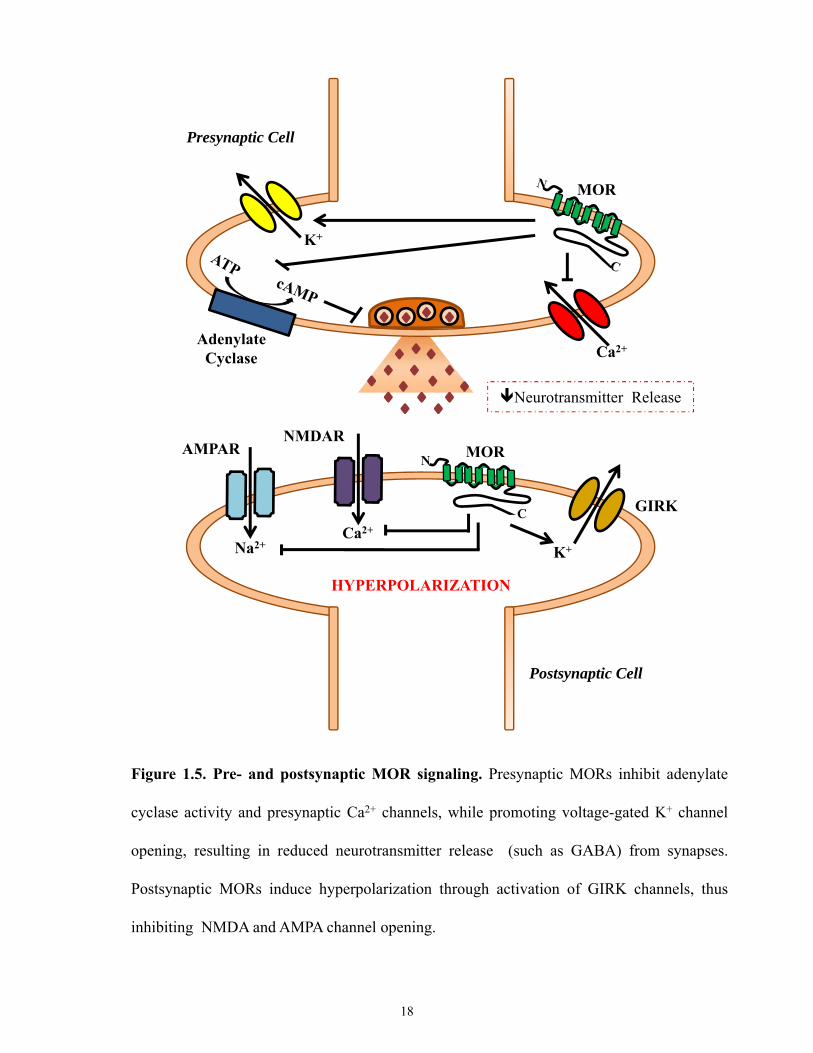
to the active conformation. In the CNS, MORs are found presynaptically and postsynaptically.
Presynaptically, MOR mediated G-protein activation results in inhibition of adenylyl cyclase,
which decreases cAMP production and inhibition of Ca2+ influx [67]. This decrease in calcium
influx ultimately results in decreased neurotransmitter release (Figure1.5). Postsynaptically,
MORs work through G-proteins to enhance cellular K+ efflux through G protein-coupled
17
Ca2+
MOR
Adenylate Cyclase
AMPARNMDAR
GIRK
Presynaptic Cell
K+
Neurotransmitter Release
MOR
Ca2+
Na2+ K+
Postsynaptic Cell
HYPERPOLARIZATION
Figure 1.5. Pre- and postsynaptic MOR signaling. Presynaptic MORs inhibit adenylate
cyclase activity and presynaptic Ca2+ channels, while promoting voltage-gated K+ channel
opening, resulting in reduced neurotransmitter release (such as GABA) from synapses.
Postsynaptic MORs induce hyperpolarization through activation of GIRK channels, thus
inhibiting NMDA and AMPA channel opening.
18

inwardly-rectifying potassium channels (GIRKs). This action hyperpolarizes the neuron,
inhibiting activity of both NMDA and AMPA receptors [68, 69] (Figure 1.5).
Activation of the dopaminergic mesolimbic system plays a role in mediating opioid
reward, and development of opioid dependence and addiction. The current accepted neuronal
circuit model suggests morphine excites and activates dopamine neurons in the ventral
tegmental area (VTA) indirectly through disinhibition of GABAergic neurons in the VTA [70-
72]. Specifically, morphine activation of MORs hyperpolarizes local GABAergic interneurons
in the VTA, thereby decreasing GABA release onto dopamine neurons [70-72]. This decreased
GABA release removes the inhibition on the VTA neurons. As a result, VTA neurons can then
project back to nucleus accumbens and increase dopamine release from dopaminergic neurons
[70-72]. This has been shown experimentally through numerous studies where opioids such as
DAMGO microinjected directly into the VTA caused an increase of dopamine release in the
nucleus accumbens following activation of MORs in the VTA [70-72]. Opioid injections into
the VTA have also been demonstrated to mediate rewarding effects through numerous self-
administration and place conditioning studies [70-72].
1. 5 Morphine Tolerance
Like many opiates, morphine exhibits significant susceptibility toward the development
of tolerance and physical dependence which limits its use in several contexts of chronic pain.
Following the development of tolerance, increasing doses of the opiate must be administered to
maintain an equivalent level of analgesia. Tolerance poses significant limitations to opiate use as
increasing morphine concentration may cause toxic effects, physical dependence and addiction
[73]. Traditionally, tolerance had been viewed as a consequence of decreased number of
functional receptors at the cell membrane. However, recent studies have shown that simple
down-regulation of opioid receptors does not occur unanimously with all opioid agonists.
19

Following activation, MORs may undergo receptor desensitization and endocytosis
(Figure 1.6). Following endocytosis, receptors can be recycled from endosomes back to the
plasma membrane, thus allowing resensitization. Alternatively, receptors can be retained
intracellularly or targeted to lysomoes/proteosomes for degradation thus down regulating
receptor numbers [74]. MOR regulation following activation is suspected to be ligand
dependent. Various ligands such as β-endorphin, fentanyl, methadone and DAMGO are
believed to promote rapid MOR internalization and recycling [75, 76]. In contrast, morphine,
pentazocine and buprenorphine promote less MOR internalization [76-80]. Thus, it is believed
that morphine has a greater tendency to induce development of tolerance and dependence
compared to other MOR agonists such as DAMGO and fentanyl, since morphine activation of
MORs does not promote removal of MORs from the plasma membrane [74, 78, 81].
The MOR specific agonist DAMGO promotes MOR internalization and induces rapid
desensitization, endocytosis and recycling (Figure 1.6) [75, 76]. DAMGO activates MORs in a
similar pattern through the heterotrimeric G-protein. Repeated exposure results in MOR
phosphorylation through the G-protein coupled receptor kinase (GRK). It is this phosphorylation
that attracts arrestin to the MOR, thereby uncoupling the MOR-G-protein complex, and
interrupting activation of downstream signaling cascades [82]. The arrestin-MOR complex
becomes endocytosed through clathrin coated pits and fuse with early endosomes [79]. It is in
the early endosomes that DAMGO unbinds MORs and the MORs become dephosphorylated
and return to the cell surface for another round of activation [79].
Unlike DAMGO, morphine does not promote MOR internalization [77, 79]. Therefore,
researchers have hypothesized that it is this feature that makes morphine administration prone to
the development of tolerance. Morphine induces weak desensitization with minimal
endocytosis. That is because morphine activated MORs fail to undergo sufficient receptor
phosphorylation by GRKs [77, 79]. Without phosphorylation, arrestin fails to be recruited to
20

Figure 1.6. Proposed pathways for MOR regulation. Regulation of MORs have been
proposed to be ligand specific. Following receptor activation, MORs may become
phosphorylated by GRKs which then recruits arrestins to the receptor. Arrestins subsequently
uncouple MORs from associated G-proteins. Following uncoupling, MORs frequently
undergo endocytosis and are internalized. Once internalized, MORs enter the degredative
pathway or are recycled. In the recycling pathway, phosphatases dephosphorylate the MOR,
thereby re-sensitizing it and allowing its return the cell surface.
Adapted from Connor et al., British Journal of Pharmacology, 2004
21

MORs thereby limiting clathrin-dependent endocytosis. Therefore, differential agonist
regulation of MORs has been suggested to occur due to differences in regulation of receptor
phosphorylation. MORs possess approximately 20 serine, threonine, and tyrosine residues on
their intracellular loops and carboxyl terminal tail [73, 83]. Following activation, these amino
acids on MORs may become phosphorylated by kinases depending on the agonist [84]. For
example, G-protein coupled receptor kinase (GRK2 or 3) may regulate the activity of MORs
through phosphorylation of its intracellular domains, and subsequently recruiting arrestin 2 or
arrestin 3 binding (depending on the agonist induced activation) to the phosphorylated residues
of MORs [82]. Thus, agonist-induced receptor phosphorylation is believed to be important for
regulation of opioid tolerance.
Few studies have shown that morphine can induce increased receptor internalization
following over expression of GRK2 [81, 85]. Overexpression of GRK-2 increased
phosphorylation and hence desensitization of MORs [85]. Similarly, overexpression of β-
arrestin (arrestin 2) has been shown to increase MOR internalization in vitro [81, 85].
Alternatively, loss of the β-arrestin-2 (arrestin 3) gene in mice strongly impaired agonist
induced desensitization of MORs, and enhanced analgesic response following morphine
treatment [86]. As well, mice lacking β-arrestin-2 displayed reduced development to tolerance
to MOR opioids such as morphine, suggesting that MOR regulation requires β-arrestin-2 [86].
Although, mice lacking β-arrestin-2 failed to develop tolerance, they were able to develop
physical dependence [87]. This is consistent with other studies that have suggested that
tolerance and dependence maybe mediated by separate mechanisms [86-88].
1.6 Opiates and NMDA Receptors
NMDA receptors are vital for synaptic plasticity. The connections between MOR
signaling and hippocampal LTP have also been examined upon opiate administration. Studies
22

have revealed that repeated exposure to morphine could impair LTP in the hippocampal CA1
region [89, 90]. However, this LTP could be restored following re-exposure to morphine [89,
90]. Thus, numerous studies have implicated a role for NMDA receptor signalling in promoting
the development of morphine tolerance [91]. Consistent with this, administration of non-
competitive NMDA antagonists such as MK-801, has been shown to attenuate the development
of morphine tolerance [92, 93]. Although studies have attempted to examine the role of NMDA
in morphine tolerance, the specific mechanisms governing these effects remain unclear. The
current accepted model of opioid dependence is that repeated opiate administration causes
adaptive increases through the influx of Ca2+ into the synapse [94]. The increased Ca2+ has been
shown to be mediated through the activation of NMDA receptors which leads to the opening of
the receptor-gated ion channels, allowing Ca2+ to enter the neuron [94]. The increased Ca2+
influx results in elevated expression of Ca2+/calmodulin-dependent protein kinase (CaMKII).
CaMKII in turn phosphorylates CREB, which increases c-Fos mRNA expression [94]. Gene
expression is thought to play an important role in many forms of neuronal plasticity. Blocking
NMDA receptors, which is known to be required for development of morphine tolerance,
inhibits the activation of CaMKII and thus negatively regulates gene expression.
The exact mechanism underlying the role of NMDA receptors in morphine tolerance
remains unclear. Provided that morphine activated MORs fail to undergo GRK phosphorylated-
arrestin mediated internalization, researchers have investigated whether there are other
mechanisms regulating MOR desensitization. A number of studies have demonstrated that
protein kinase C (PKC) is involved in opioid tolerance or desensitization. Specifically, PKC has
been shown to mediated inhibition of MOR internalization and thus play a role in the
development of acute tolerance through desensitization of MORs [95, 96]. It has been suggested
that perhaps PKC phosphorylates MORs directly or indirectly through phosphorylation of other
proteins involved in receptor desensitization [95, 96]. Treatment with PKC inhibitors has been
23

shown to induce morphine-mediated MOR internalization, and thus attenuate morphine
tolerance [95, 96]. As previously mentioned, chronic morphine treatment has been shown to
enhance NMDA activity and thus elevate intracellular levels of Ca2+. MOR activation induced
Ca2+ influx has been shown to activate protein kinases such as PKC [97, 98]. Increased
intracellular PKC has been shown to potentiate NMDA activated currents, by increasing the
probability of channel openings and by reducing the voltage-dependent Mg2+ block of NMDA-
receptor channels [97, 98]. PKC inhibitors blocked this potentiation of NMDA receptors.
1.7 Animal Model: Mu Opioid Receptor Knockout Mice
Over the years several strains of mice lacking the mu opioid receptor have been
generated. These knockouts were created through either the deletion of exon 1 [99, 100],
insertion of a Neo cassette in exon 2 [101], or deletion of exons 2 and 3 [102]. All MOR null
mutants exhibit normal growth and are fertile, indicating that MORs are not essential for
survival. No morphologic or behavioural abnormalities were detected in MOR null mice [99,
100]. Loss of MORs did not alter expression or distribution of other opioid receptors, or alter
transcription regulation of genes encoding for endogenous ligands. Moreover, administration of
morphine in MOR null mice did not induce analgesia, hyperlocomotion, reward, physical
dependence, withdrawal, induction of drug-dependent place preference activity, or other
peripheral effects (e.g. respiratory depression, inhibition of gastric motility) [99, 100]. Loss
MORs was also functionally confirmed using [3H] DAMGO binding [20]. Similar to morphine,
administration of DAMGO induced no analgesic response in these animals [20]. Although
morphine is known to bind with lower affinity to receptors other than the MORs, no major
changes in physiological responses were observed suggesting no major compensatory changes
occurred within the opiate receptors. These findings confirm that the major physiologic effects
of morphine are mediated through MORs [99, 100].
24

1.8 Eph Receptors and Ephrins
Erythropoietin-producing hepatocellular carcinoma (Eph) receptors represent the largest
known family of mammalian receptor tyrosine kinases [103]. They were discovered while
attempting to identify oncogenic kinases within the carcinoma cell line, ETL-1 [103]. Eph
receptors are classified into two major sub-groups, EphA and EphB, depending on their ligand
binding preferences [104, 105]. A total of 14 Eph receptors have been characterized: EphA1-8,
10 and EphB1-4,6 [106]. Eph receptors bind to Eph receptor interacting ligands named ephrins.
Ephrins are similarly divided into 2 major sub-classes: ephrinAs which are bound to the cell’s
outer membrane via a glycophosphatidylinositol (GPI) linkage, and ephrinBs which are
transmembrane proteins with their own intracellular signaling capabilities [104, 105]. In
mammals, 8 ephrins have been identified, ephrin-A1–5 and ephrin-B1–3 [106, 107].
This ligand-receptor system is unusual in that both the receptor and ligand are membrane
bound. This allows cell signaling to be propagated bidirectionally through Eph receptor
mediated (forward) signaling or through ephrin mediated (reverse) signalling [108, 109]. In
most instances, EphA receptors preferentially bind to ephrinA ligands, while EphB receptors
bind to ephrinB ligands. However, known cases of receptor promiscuity exist, such as the
binding of EphA4 to ephrinB2. Similarly EphB2 demonstrates significant affinity for ephrinA5
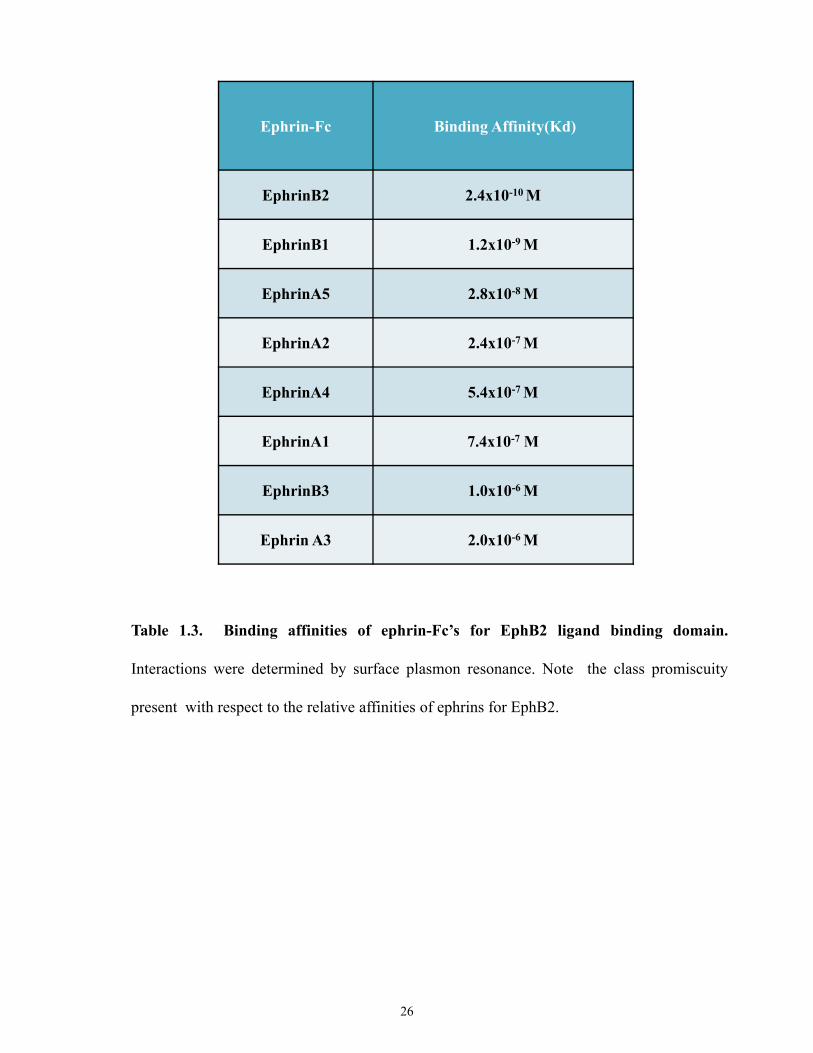
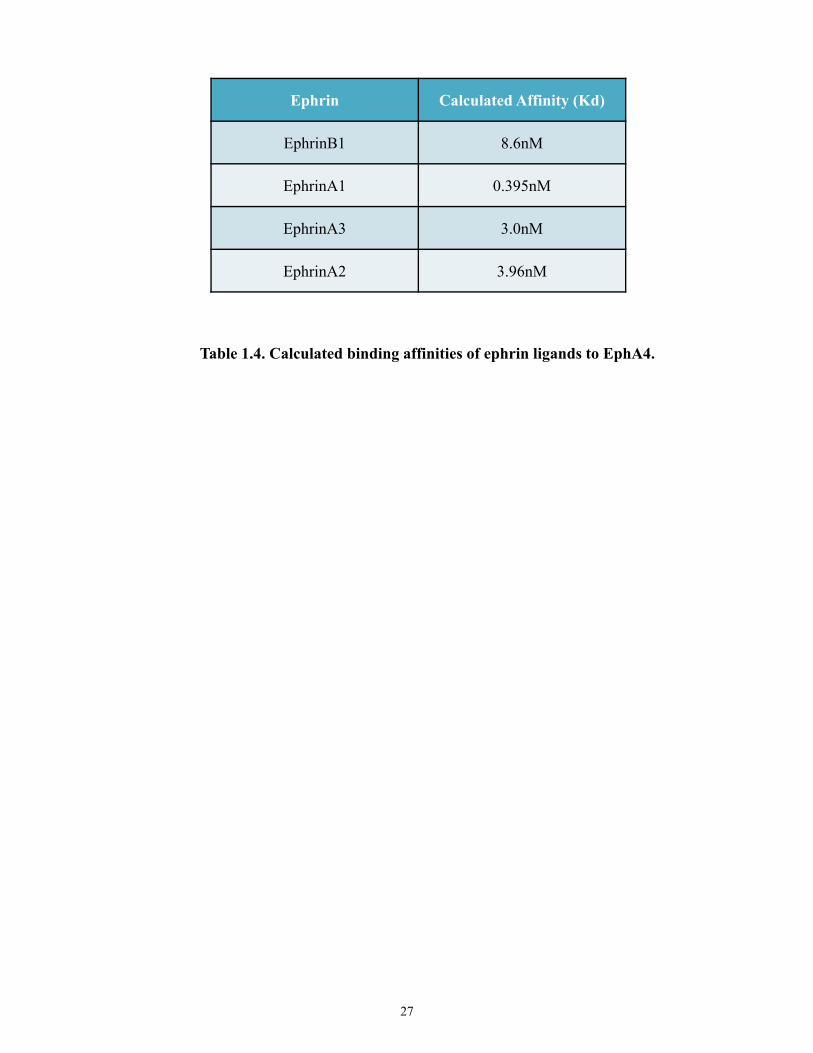
[106, 110]. Tables 1.3 and 1.4 summarize the relative binding affinities of EphB2 and EphA4
[104, 111]. Ligand binding results in receptor auto-phosphorylation. Following receptor
activation, signaling cascades are initiated (see below).
1.8.1 Eph/ Ephrin Structure
EphA and EphB share similar basic structural motifs, differing principally in the amino
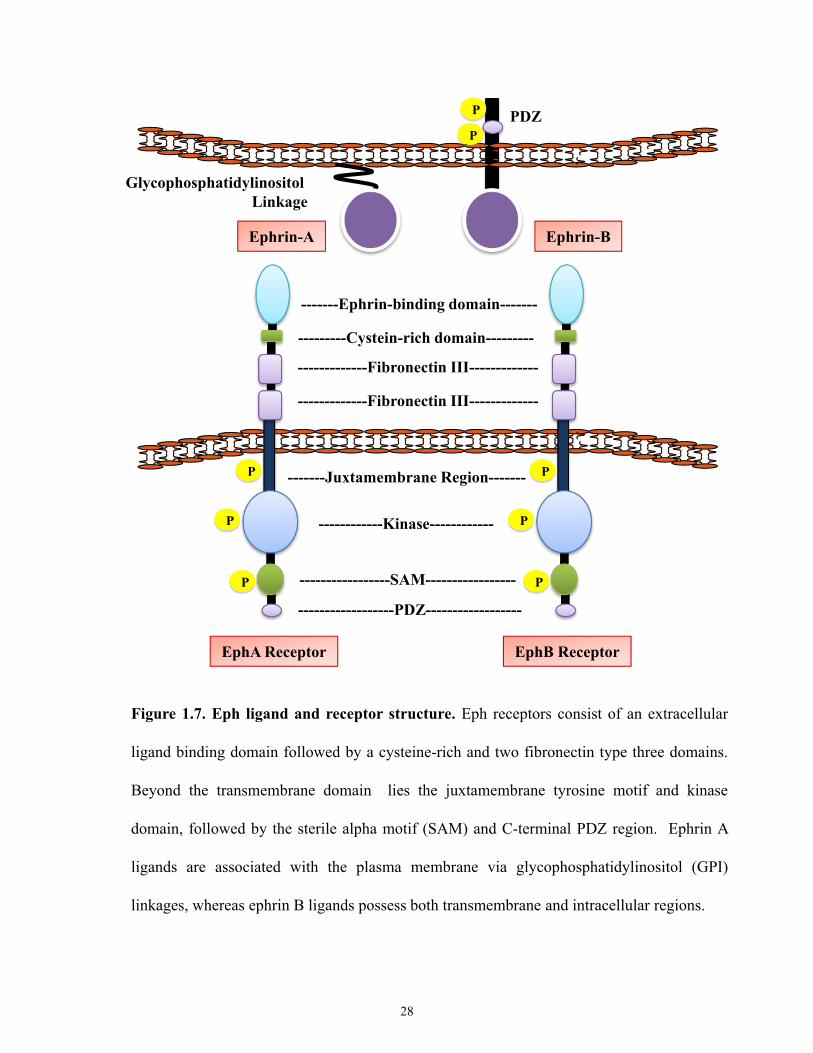
acid sequences governing the ligand binding site (Figure 1.7). The ectodomain region consists
25
Ephrin-Fc Binding Affinity(Kd)
EphrinB2 2.4x10-10 M
EphrinB1 1.2x10-9 M
EphrinA5 2.8x10-8 M
EphrinA2 2.4x10-7 M
EphrinA4 5.4x10-7 M
EphrinA1 7.4x10-7 M
EphrinB3 1.0x10-6 M
Ephrin A3 2.0x10-6 M
Table 1.3. Binding affinities of ephrin-Fc’s for EphB2 ligand binding domain.
Interactions were determined by surface plasmon resonance. Note the class promiscuity
present with respect to the relative affinities of ephrins for EphB2.
26
Ephrin Calculated Affinity (Kd)
EphrinB1 8.6nM
EphrinA1 0.395nM
EphrinA3 3.0nM
EphrinA2 3.96nM
Table 1.4. Calculated binding affinities of ephrin ligands to EphA4.
27
P
P
P
P
P
P
Ephrin-B
PDZ
Ephrin-A
P
P
EphA Receptor EphB Receptor
-----------------SAM-----------------
------------Kinase------------
-------Juxtamembrane Region-------
-------------Fibronectin III-------------
-------------Fibronectin III-------------
---------Cystein-rich domain---------
-------Ephrin-binding domain-------
------------------PDZ------------------
GlycophosphatidylinositolLinkage
Figure 1.7. Eph ligand and receptor structure. Eph receptors consist of an extracellular
ligand binding domain followed by a cysteine-rich and two fibronectin type three domains.
Beyond the transmembrane domain lies the juxtamembrane tyrosine motif and kinase
domain, followed by the sterile alpha motif (SAM) and C-terminal PDZ region. Ephrin A
ligands are associated with the plasma membrane via glycophosphatidylinositol (GPI)
linkages, whereas ephrin B ligands possess both transmembrane and intracellular regions.
28

of an amino-terminal ligand-binding globular domain, a cysteine-rich region, and two
fibronectin type III repeats, followed by a single transmembrane domain [107, 111-113]. The
intracellular domain is comprised of a short juxtamembrane region with several conserved
tyrosine residues, a tyrosine kinase domain, a sterile-alpha-motif (SAM) protein domain, and a
C-terminal PDZ (postsynaptic density protein/disc large/zona occludens) binding motif [106,
107]. Between EphA and B receptors, sequence identities are approximately 30–70% in the
extracellular domains, and approximately 65–90% in the kinase domains [111].
Conserved tyrosine residues within the juxtamembrane region have been shown to
regulate the early stages of receptor activation. In the unphosphorylated state these
juxtamembrane tyrosine residues fold to inhibit kinase activity [114, 115]. Phosphorylation of
these residues creates charge repulsion which opens the kinase domain, thus allowing
subsequent phosphorylation of tyrosine residues within the kinase motif to occur, and thereby
prompting full activation [114, 115]. Subsequently, the phosphorylated juxtamembrane region
serves as docking sites for Src homology 2 (SH2)-domain containing proteins [106, 116-118].
Furthermore, the SAM domain which may homo- and hetero-oligomerize with other SAM
domains, has been suggested to aid in receptor oligomerization by serving to stabilize receptor
clustering [119-121] . As well, the SAM domain may modulate active Eph receptors at the cell
surface by regulating endocytosis and receptor degradation [122]. Similarly, the PDZ domain
aids in stabilizing Eph/ ephrin clustering [106], but may also serve as target site for many
cytoplasmic scaffolding proteins, such as the recruitment of the Ras family proteins through
interaction with PDZ domains of other intracellular proteins [123].
EphrinA and ephrinBs share 30–70% identity within their core sequence which
comprises approximately 125 amino acids, including 4 invariant cysteine residues [111].
Ephrins are membrane bound ligands (Figure 1.7). The ephrinA subclass is anchored to the cell
membrane through a GPI linkage, which is uncommon among ligand families of other types of
29

receptor tyrosine kinases [111]. Ephrin-B ligands possess a single-pass transmembrane domain
and a short cytoplasmic tail of approximately 80 amino acids long containing five conserved
tyrosine residues. This followed by a C-terminal PDZ binding motif, which is important for
ephrin signaling (see below) [124, 125].
1.9 Eph-Ephrin Interactions and Signaling
The unique feature of Eph receptors and ephrins both being membrane bound, allows for
two different types of Eph-ephrin interactions: trans or cis interactions. When Eph receptors and
ephrins are expressed on opposing cells this is known as trans interactions. This type of
interaction results in bidirectional signaling (Figure 1.8). In contrast, when Eph receptors and
ephrins are expressed in the same cell this is known as cis interactions. This type of interaction
is believed not to mediate any active signaling [108].
Eph receptors require oligomerization for biological activity. Crystal structural analysis
of EphB2-ephrinB2 complex, revealed two distinct ephrin binding sites [126]. One site is
believed to be the high affinity binding site to ephrinB ligand, while the other is a lower affinity
binding site to another ephrinB ligand following dimerization with another EphB-ephrinB
complex [126]. EphBs and ephrinBs first bind with high affinity and specificity to form
heterodimers. Then at high concentrations, two EphB-ephrinB complexes (two heterodimer
complexes) form a circular tetramer [126]. In the tetramer complex, each ligand interacts with
two receptors and similarly each receptor interacts with two ligands [126]. This has been
suggested to promote higher-order clustering and initiation of bidirectional signalling [126].
However, this tetramer complex is believed to exist only with EphB-ephrinB complexes. That is
because, studies examining EphB2 bound to ephrinA5 showed that this complex exists solely as
a dimer [110, 127].
30

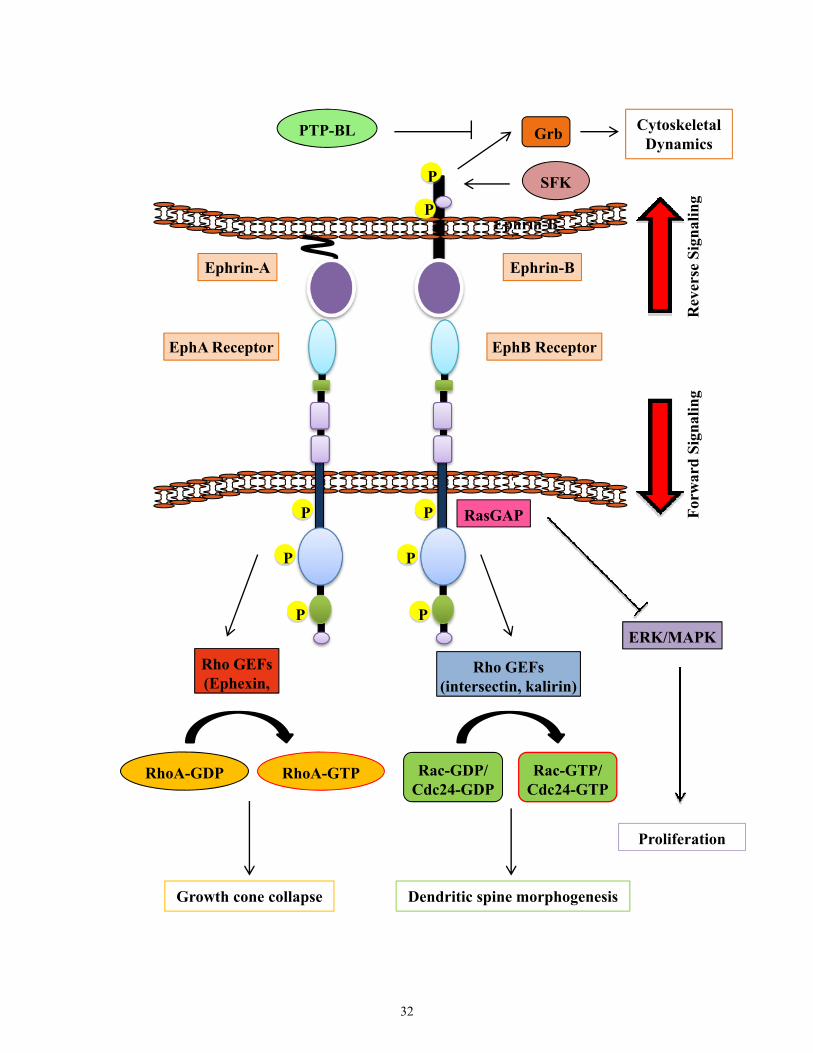
Figure 1.8. Eph-ephrin Signaling. Both Eph receptors and ephrins are membrane bound,
thus allowing a variety of cell-cell interactions and the potential for bidirectional signaling.
Signaling mediated by the Eph receptors is referred to as forward signaling, while that
mediated via ephrins is termed reverse signaling. Activation of Eph receptors alters Rho-
family (RhoA, Rac, and Cdc42) GTPases activity influencing actin polymerization and
ultimately growth cone and dendritic spine morphology. In addition, Eph receptors can
signal through Ras GTPase members influencing ERK/MAPK signaling to regulate cell
proliferation. EphrinBs illicit signaling cascades following phosphorylation by Src-family
kinases (SFKs). Following activation, signaling adaptors such as Grb are recruited to these
sites of phosphorylation, regulating features such as spine and synapse formation.
Phosphatase such as PTP-BL terminate ephrinB signaling cascades.
31
Ephrin-B
P
P
P
P
P
P
P
P
Rho GEFs(intersectin, kalirin)
Rac-GTP/Cdc24-GTP
Dendritic spine morphogenesis
Rho GEFs(Ephexin,
RhoA-GDP
Growth cone collapse
RasGAP
ERK/MAPK
Proliferation
SFK
Grb CytoskeletalDynamics
PTP-BL
Rev
erse
Sig
nalin
gFo
rwar
d Si
gnal
ing
Ephrin-A Ephrin-B
EphA Receptor EphB Receptor
RhoA-GTP Rac-GDP/Cdc24-GDP
32

When an ephrin binds to an Eph receptor, receptor auto-phosphorylation occurs. Soluble
ephrins do not trigger receptor auto-phosphorylation unless artificially pre-clustered [109]. Non-
clustered forms can act as functional antagonists [108]. This limits signaling of this nature to
cell-to-cell communication. Thus, as mentioned above, Eph-ephrin signaling is bidirectional.
Signaling may mediate and proceed through the receptor-bound membrane (forward signaling)
or through the ligand bound membrane (reverse signaling). Both signaling events can happen
simultaneously, and the relative contributions of Eph forward and ephrin reverse signaling can
vary depending on cellular context [108].
1.9.1 Forward Signaling
The majority of Eph-mediated effects on repulsive axon guidance and synaptic plasticity
are mediated through forward signaling via the Rho family of GTPases. The Rho family is
comprised of RhoA, Cdc42 and Rac. Cycling between the active GTP-bound form and the
inactive GDP-bound conformation is mediated through guanine exchange factors (GEFs). GEFs
exchange bound GDP for GTP thus activating Rho GTPases. EphA and EphB receptors activate
a unique subset of GEFs [128] (Figure 1.8). For example, the Ephexin family of Rho GEFs are
EphA specific. Ephexin promotes the activation of RhoA, while it inhibits activation of Rac and
cdc42. Activation of RhoA downstream of EphA receptors mediates growth cone collapse in
neurons through shifts in actin cytoskeleton dynamic to increase contraction and reduce
extension [129, 130]. In contrast, EphB specific Rho GEFs such as Kalirin and Intersectin
activate and signal through Rac and cdc24. Intersectin activates Cdc42 (filopodia extension) and
Kalirin activates Rac (dendritic spine extension) to regulate spine morphology [131-133].
However, both Kalirin and Intersectin regulate signalling downstream of EphB receptors
differentially. Kalirin binds to activated EphB2 receptors. In contrast, Intersectin binds to the
33

kinase domain of EphB2 (which is associated with the neural Wiskott-Aldrich syndrome protein
(N-WASP)), independent of receptor activation [132].
In addition, Eph receptors also regulate the Ras family of GTPases, such as H-Ras to
regulate cell proliferation. Upon activation, H-Ras subsequently activates a cascade of
serine/threonine kinases including Raf1, Mek1, and the MAP kinases Erk1 and Erk2 [128]. Eph
receptors can act as positive and negative regulators of the MAPK pathway. For instance,
EphB1 receptors have been shown to activate the MAPK cascade through recruitment of the
Grb2/Sos complex, which acts as a Ras-specific GEF [134]. This leads to activation of Ras
followed by activation of downstream serine/threonine kinases such as Raf1, Mek1, and the
MAP kinases Erk1/2. This induces cell proliferation. In contrast, in neuronal cells, EphB2 has
been shown to negatively regulate MAPK, through recruitment of p120RasGAP, which
suppresses H-Ras [135, 136]. This inhibits Ras activation, and thus inhibits activation of Raf1,
Mek1, and Erk1/2, and suppresses proliferative response (Figure 1.8).
1.9.2 Reverse Signaling
Following EphB binding and clustering, activated ephrinB subunits recruit Src-family
kinases (SFKs) which phosphorylate the tyrosine residues on the cytoplasmic tail of ephrinB
ligands [137]. These phosphorylated tyrosine residues induce conformational changes that
disrupt the β-hairpin structure. This allows for the binding of Src-homology 2/3 (SH2/SH3)
domain-containing signaling proteins, such as Grb4 [138, 139]. EphrinB reverse signaling
through Grb4 in neurons has been shown to play a role in a diverse set of activities including
spine maturation, synaptic plasticity and synaptogenesis [140, 141]. Signaling is terminated
upon dephosphorylation of ephrins by phosphatases such as PTP-BL [137] (Figure 1.8).
Moreover, reverse signaling mediated through ephrinA is at still unclear. Unlike ephrinB,
ephrinA ligands lack a cytoplasmic tail to transduce signalling cascades. However, it has been
34

postulated that perhaps ephrinA, through interactions with lipid-raft-associated protein
complexes recruit the Src family kinases, and mediate downstream signaling [142].
1.9.3 Termination of Eph-Ephrin Signaling
Proteases such as the ADAM family of metalloproteases and γ-secretase proteases have
been reported to cleave Eph receptors and ephrins thereby disrupting oligomer complexes and
terminating signaling [143, 144]. Eph-ephin complexes may also undergo trans-endocytosis,
whereby the entire complex is internalized into either the Eph or ephrin expressing cell [145,
146]. The mechanism underlying trans-endocytosis are still unclear, however there is evidence
to suggest the endocytosis is mediated by clathrins [147]. As well, ongoing research suggests
that direction of the endocytosis is bidirectional, mediated and dependent on the type of
signalling occurring: reverse or forward signaling. That is because, studies examining EphB2
receptors lacking the cytoplasmic region, which fail to mediate forward signaling when in
complex with ephrinB1s, were found to be internalized into the ephrin expressing cell.
Similarly, an EphB2 interaction with a truncated ephrin-B1 resulted in internalization into the
receptor expressing cell, while truncation of both EphB2 and ephrin-B1 prevented
internalization [145]. Endocytosis of the EphB-ephrinB complex is necessary to mediate cell-
cell repulsion to guide migrating cells and axons.
1.10 EphB2 Expression in the CNS
Eph-ephrin interactions regulate many functions such as axon guidance, cell
proliferation, cell migration, synaptic plasticity, and oncogenesis [106, 124]. During the
development of the CNS, the expression of Eph and ephrins fluctuate throughout different
regions of the CNS [148]. EphB2 which is also known as Cek5, Nuk and Sek3 prior to
standardization of Eph nomenclature in 1997 [105], has been shown to be expressed during
35

development and adulthood in the CNS. To investigate the expression pattern and role of EphB2
in the CNS, two mutant alleles of EphB2 null mice were generated [149]. One represented a null
mutation of the EphB2/Nuk gene. This was generated through homologous recombination in
embryonic stem cells by deletion of the 5′ segment of the Nuk locus and insertion of a neomycin
resistance cassette. In the second strain, an EphB2-β-galactosidase fusion was generated just
past the juxtamembrane tyrosines, hence eliminating the kinase, SAM and C-terminal PDZ
domains [149]. The generation of this NuklacZ allele allowed localization of EphB2 to by
dynamically tracked (in heterozygotes) and allowed differentiation of forward versus reverse
signaling processes.
During development, expression of EphB2 is largely within the developing nervous
system with higher expression among axonal tracts versus neuronal dendrites [150]. The earliest
expression of EphB2 in mice has been reported to be at E8.5, in the neuroectodermal cells of the
neural plate, as well as the ventral midbrain and hindbrain rhombomeres r3 and r5 [149]. At
E9.25 EphB2 expression is observed within the hypothalamic region of the diencephalon and
the tegmental region of the midbrain [149]. During embryonic development, EphB2 expression
is highest in sensory and motor neuronal axons. By E13.5, EphB2 becomes defined in a dorsal-
ventral gradient within the retinal ganglion cells [151]. EphB2 expression increases in other
regions such as the hypothalamus and preoptic area at E14.5. In addition, regions ventral to the
anterior commissure (AC) begin to express EphB2 around this period. By E15.5, the pars
posterior AC (ACpp) tracts are observed to cross the midline. In EphB2 null mice the ACpp
axons fail to migrate and project toward the floor of the forebrain instead [149]. As the
embryonic development nears the end, EphB2 expression within the brain largely shuts down
except in areas such as the superior colliculus and ventral forebrain [150]. By postnatal day 7,
EphB2 expression in the CNS begins to remerge, but expression pattern also reverses. In
contrast to embryonic development, postnatally, EphB2 expression is high in neuronal dendrites
36

[150]. Expression of EphB2 remerges in areas such as the CA3 and dentate gyrus of the
hippocampus. By postnatal day 10, EphB2 expression pattern widens and increases into the
hippocampus, neocotex, amygdala, and thalamic centers, as well as Purkinje cells. This
expression continues into adult hood [149, 150]. Therefore, the continued expression of EphB2
in regions undergoing continual synaptic modification signifies the important role of EphB2 in
regulating and modulating synaptic function.
1.11 EphB2 and Synaptic Plasticity
The sustained expression of Eph receptors and ephrins in the adult brain, especially in
regions associated with synaptic remodeling such as the hippocampus, olfactory bulb and
cerebellum suggests that EphB-ephrin signaling may play a role in regulating synaptic plasticity.
The importance of the Eph family in learning and memory has been examined using multiple
animal models. Comparisons between different Eph/ephrin knockouts and wild-type mice have
shown that pre- and post-synaptic Eph/ephrins mediate dendritic spine formation, synapse
formation and synaptic plasticity, all of which is required for learning and memory [107, 152,
153]. For instance, EphA4 null mice display morphologically disorganized, long and
overlapping dendritic spines [107]. While EphA6 knockout mice exhibit behavioural deficits in
learning and memory. Specifically, loss of EphA6 produced impairments in the fear
conditioning paradigm where EphA6 null mice displayed less freezing; and in the Morris Water
Maze where they failed to learn quickly in the hidden platform task compared to control
littermates [154].
EphB receptors have been showed to be localized at excitatory synapses, suggestive of
their role in synapse formation and regulation [155]. The specific synaptic roles of EphB
receptors have also been extensively studied using animal models such as the
EphB1/EphB2/EphB3 triple knockout mice [156]. The EphB family of receptor tyrosine
37

kinases, which is enriched at excitatory synapses, is important during synapse and spine
formation and maintenance. Triple knockout mice lacking EphB1/B2/B3 displayed fewer
excitatory synapses and decreased number of immature dendritic spines. However, this was
absent in the single or double knockout mice [156]. As well, hippocampal neurons cultured from
EphB1/EphB2/EphB3 triple knockout mice were abnormally long and thin in morphology, and
failed to produce mature spines [156]. This suggested that EphB-ephrinB signalling is required
for spine formation and maturation [150, 156, 157].
Excitatory synapses contain both NMDA and AMPA receptors. At these synapses, EphB
receptors have been shown to associate and cluster with NMDA receptors to indirectly regulate
Ca2+ influx [158]. EphB receptors phosphorylate NR2B in a Src-dependent manner to potentiate
Ca2+ influx through NMDA receptors. Elevated levels of intracellular Ca2+ phosphorylates
Ca2+/cAMP-responsive element binding protein (CREB), which induces the immediate early
gene c-Fos. Studies have shown that EphB2 receptors which are strongly expressed in the
hippocampus specifically associate with NMDA receptors [158, 159]. EphB2 plays a role in
stabilizing NMDA-dependent synaptic plasticity and synapse formation in regions such as the
hippocampus [158, 159]. EphB2, through its extracellular domains also directly interacts with
the NR1 subunit of NMDA receptors [155]. As such, EphB2 receptors have been implicated in
LTP generation through the NMDA receptors [150, 157]. Loss of EphB2 in mice has been
shown to reduce LTP and reduce localization of NMDA receptors in the cell membrane of the
synapse [150, 157]. Such findings indicate that EphB2 signalling plays a role in modulating
NMDA-mediated signaling to regulate synaptic plasticity which is essential processing in
learning and memory. Furthermore, EphB2 receptors have also been shown to associate with
and regulate AMPA receptors localization through PDZ domain containing proteins such as
GRIP [141].
38

The role of EphB, especially EphB2 in learning and memory has been a hot area of
research in the past few years. Loss of EphB2 and EphA4 receptors has been shown to precede
memory decline in a murine model of Alzheimer’s disease [160]. As well, recent research has
suggested that amyloid-β oligmoemers bind to the fibronectin repeats domain of EphB2, which
signals EphB2 for degradation. Loss of EphB2 in an Alzheimer model was found to display
reduced NMDA currents and impaired LTP in the dentate gyrus [161]. This is consistent with
previous findings regarding the role of EphB2 in modulating NMDA receptor activity.
1.12 EphB and Pain Modulation
A new area of research has emerged over the past few years examining the role of EphB-
EphrinB signaling in pain processing. As previously mentioned, EphB receptors have been
shown to regulate synaptic plasticity through interaction with NMDA receptors. Battaglia et al.,
was first to report and investigate pain processing through EphB-ephrin interactions in the rat
spinal cord. In the rat lumbar spinal cord, exogenous activation of EphB receptors using the
dimeric chimeric molecule, ephrinB2-Fc, decreased analgesic response by about 50% and
increased thermal hyperalgesia upon exposure to noxious thermal stimuli [162]. However, this
induced hyperalgesia was blocked if pretreated with NMDA receptor antagonist, MK-801[162].
Thus, this suggested that EphB activation through interaction of NMDA receptors was
mediating neuropathic pain [162].
Given that EphB and ephrinB expression is present within the DRG and spinal cord
[163, 164], a few studies examined whether expression of EphB/ephrinB increased following
injury. One study, through immunohistochemical methods reported that ephrinB2 expression
was enhanced in the DRG and spinal cord following a spinal nerve crushing injury model
[163]. To examine the exact involvement of the Eph system following neuropathic pain, the
researchers administrated ephrinB2 siRNA to reduce ephrinB2 expression. This resulted in
39

reduced mechanical allodynia [163]. Similarly, Song et al. also reported upregulated expression
of EphB1 and ephrin in the DRG and spinal cord following a chronic constriction injury model
[164]. As well, blocking EphB-receptors, through administration of EphB1-Fc and EphB2-Fc
chimeras, inhibited induction and maintenance of nerve injury-induced thermal hyperalgesia and
mechanical allodynia [165]. These blockers also prevented and suppressed the nerve injury-
induced hyperexcitability of nociceptive small DRG neurons, and reduced LTP induction at
synapses between C fibers and postsynaptic dorsal horn neurons in the spinal cord [165]. These
findings indicated that EphB- ephrinB receptor signaling contributes to the regulation of
neuropathic pain.
Given the role of EphB receptors in synaptic plasticity, researchers began to examine the
role of EphB in the processing of neuropathic pain and alterations of morphine responses. A
study by Han and colleagues reported that peripheral nerve injury unlike in wild-type animals
did not induce thermal hyperalgesia in EphB1 null mice [166]. As well, while intrathecal
injections of EphB receptor blocking reagent EphB2-Fc diminished behavioral responses to
morphine withdrawal, EphB1 null mice failed to exhibit development of physical dependence to
morphine compared to control littermates [166]. These findings indicated that the EphB1
receptor was necessary for the development of neuropathic pain and physical dependence on
morphine [166]. Interestingly, Liu et al. examined the role of NMDA receptor subunit NR2B
following morphine exposure. They reported that chronic morphine exposure significantly
increased phosphorylation of NR2B [167]. However, intrathecal administration of EphB2-Fc
inhibited NR2B phosphorylation. Therefore, these findings suggested that EphB receptor
signaling through interaction with NMDA receptors played a role in the development of opioid
physical dependence [167]. Another recent study by Liu and colleagues examined the role of
EphB1 signaling in regulating morphine tolerance with respect to bone cancer pain. They
40

reported that blocking EphB1 receptor activation using EphB2-Fc rescued the analgesic effect of
morphine and prevented the development of morphine tolerance [168].
The aforementioned studies indicate that EphB‐family signaling plays a role in
modulating nociceptive sensory responses and morphine responses. Research over the years has
also implicated a role for NMDA receptors in regulating morphine tolerance through its
influences on opiate signaling. However, the exact mechanism through which EphB receptors
such as EphB2 interact with NMDA receptors to mediate morphine related responses and
regulate morphine dependent tolerance remains unknown.
1.13 Learning and Memory: An overview
Cases such as H.M. (Henry Gustav Molaison 1926-2008) and others have provided
valuable insights into the organization and execution of processes which mediate human
learning and memory [169]. In an attempt to cure H.M.’s epilepsy, an experimental bilateral
medial temporal-lobe resection was performed, where components of his hippocampus,
parahippocampal gyrus, and amygdala were removed [170]. As a result of this procedure the
patient experienced severe anterograde amnesia, and demonstrated deficiencies in acquisitions
of new episodic and semantic knowledge [171]. He also suffered moderate retrograde amnesia
and could not remember the majority of events 3 years prior to his surgery [170]. However, his
working and procedural memory were intact, as he could learn and retain new motor skills for
short periods of time [170, 171]. In this respect, H.M.’s short-term and procedural memories
were largely unimpaired; however his long-term memory exhibited severe deficits. This and
other similar cases lead scientists to postulate that more than one region of the brain was
involved in the learning and execution of memory, thereby supporting the concept that multiple
memory systems existed [172].
41

Over the years, research has defined several different forms of memory. Memory can be
classified into short-term and long-term memory. Short-term memory is the temporary storage
of information for a short period of time; working memory is a form of short-term [173]. Long-
term memory has been broadly categorized into declarative (explicit) and non-declarative
(implicit) components [169, 172]. Through the imposition of specific brain lesions, researchers
have been able to interrogate which regions are critical for specific forms of memory. Studies
from H.M. and of other patients highlighted the importance of structures within the medial
temporal lobe and medial thalamic regions in establishing and maintaining declarative
memories, which represent explicit descriptions of facts, events, places and objects, [174-176].
However, integration of non-declarative or implicit (sub-conscious) memory is believed to lie
largely outside of these temporal structures, in regions such as the basal ganglia, cerebellum and
components of the limbic system [169, 172]. Non-declarative memory has been defined as the
facilitation of performance on a task due to previous experience without conscious recollection
or awareness of that experience [177]. Non-declarative memory is further subdivided into
priming, procedural, non-associative learning, and associative/classical conditioning learning
[172]. Priming represents the recognition of objects/words due to prior exposure and the
neocortex has been suggested to processes these types of memories. Procedural memories are
implicit memories that become skills and habits, such that once acquired these memories are
evoked unconsciously and automatically [175]. Through localized brain lesion and
pharmacological blocking approaches, research has shown that the dorsal striatum mediates a
component of this type of procedural memory [178, 179]. The cerebellum has also been shown
to be involved in mediating procedural memory [175] and is an important region for motor
learning together with constituents of the basal ganglia [180]. Furthermore, non-associative
learning is learning to a single stimulus that evokes a reflex [172]. In contrast, associative
learning is based on responses to multiple stimuli. Classic conditioning (common examples
42

include fear conditioning or conditioned place preference) represent forms of associative
learning [172]. Lesion studies in the context of fear conditioning have revealed a role for the
amygdala together with the hippocampus [172, 179]. The conditioned place preference
paradigm is used to measure the rewarding properties of drugs such as opiates, where animals
develop a preference for the drug paired environment. Studies have shown that components of
the hippocampus and the limbic system such as the amygdala are necessary for the development
of a conditioned place preference to a drug [181, 182].
Though distinct forms of learning may be mediated throughout different regions of the
brain, the basic cellular mechanisms mediating these forms of learning are believed to be
fundamentally similar. Because of the nature of its structural organization and central role in
learning and memory, studies of synaptic plasticity have focused on the hippocampus to identify
the cellular features of learning and memory. Research has demonstrated several forms of
cellular plasticity within this structure consistent with the Hebbian properties seen in learning.
These include activity dependent models of synaptic plasticity termed long-term potentiation
(LTP) and long-term depression (LTD) [183-185]. Induction of LTP involves coincident
activation of NMDA receptors with subsequent activation of intracellular calcium-dependent
signaling. Specifically, there is a transient increase in postsynaptic calcium through the NMDA
receptor, and through voltage-gated calcium channels, or through calcium permeable AMPA
receptor channels. This increase in calcium influx leads to activation of protein kinases such as
calcium-calmodulin dependent protein kinase II (CaMKII), protein kinase A (PKA) and protein
kinase C (PKC) [183]. However, LTP has been shown not to be confined solely to synapses of
the hippocampus, but can be induced within numerous CNS regions associated with learning
[183]. Though, LTP has been a primary cellular model of learning and memory, recent evidence
suggests that LTD may also play a similar role, particularly for motor learning at sites such as
the cerebellum [175, 180, 186].
43

1.13.1 Hippocampus
The hippocampus is a component of the medial temporal lobe and consists of a largely
unidirectional neural circuit. The main input into the hippocampus is from the entorhinal cortex,
which projects to the dentate gyrus via the perforant path [187, 188]. Inputs from dentate gyrus
project to the CA3, at which point Schaffer collaterals from the CA3 project to the CA1 region
of the hippocampus. From here the outputs flow to the subiculum which serves as the main
output for the hippocampus [187, 188]. Originally the hippocampus was believed to be uniform
in function, however over the years studies have suggested functional dissociations exists
within hippocampal sub-regions, specifically along the septotemporal axis [189-191]. Numerous
studies over the past three decades have highlighted the critical role of the hippocampus in
spatial and contextual learning. Studies demonstrate that loss of hippocampal function
substantially impairs acquisition and retention of spatial learning in rodent paradigms such as
the water and radial mazes [179, 192]. Specifically, lesions confined to the dorsal hippocampus
were shown to impair spatial learning in the water and the eight-arm radial maze tasks [189,
191]. As well, recent studies also suggest the CA1 region of the dorsal hippocampus is
important for acquisition of spatial memories, as mutant mice lacking the NMDA receptors in
the CA1 synapses exhibit impaired synaptic currents and LTP and impaired spatial learning
[193]. In contrast, lesions confined to the ventral hippocampus were comparable to controls with
respect to spatial learning. Ventral hippocampal lesions instead appeared to be involved in the
processing of emotional responses to fear [189-191]. Such findings are consistent with studies
demonstrating that the dorsal hippocampus receives greater sensory input from visual, auditory
and somateosensory association areas via connections from the entorhinal cortex, while the
ventral hippocampus is more strongly connected to sub-cortical loci [194, 195]. As well, the
dorsal hippocampus contains a higher proportion of place cells [194]. Place cells are believed to
44

be hippocampal pyramidal cells that are important for processing of spatial information. Studies
done in rats reveal that a collection of place cells represent specific regions within the
environment, and when the rat is in a particular location in the environment (place field) these
cells fire in their place fields. The firing of these cells thus provides the spatial information of a
specific location within the environment [196, 197].
Moreover, lesion studies reveal a requirement for intact hippocampal function for the
processing of contextual cues [192]. Researchers have utilized tests such as the Pavlovian fear
conditioning task to monitor hippocampal involvement in contextual learning and memory
[192]. In classical fear conditioning, a conditioned stimulus is paired with an aversive
unconditioned stimulus such as an electrical foot shock. This typically elicits conditioned fear
responses, such as freezing [198]. Studies have revealed that the hippocampus is required for the
processing of the contextual cues associated with the unconditioned stimulus. Lesions to the
hippocampus, specifically, animals with lesions on the dorsal hippocampus fail to develop fear
and exhibit impairment in freezing when tested in a contextual fear conditioning task [198, 199].
Morris water and radial maze studies have been previously used to examine hippocampal
dependent spatial and contextual learning. However, in my own studies passive avoidance and
open field tests were utilized to examine hippocampal function. Passive avoidance represents an
associative/contextual task, where animals are conditioned to avoid the dark compartment paired
with the aversive stimulus (foot-shock). Thus, the passive avoidance task tests an animals’
ability to process and link a specific contextual cue (dark compartment) with an aversive
stimulus (foot-shock). It has been shown that animals with lesions to the dorsal hippocampal
CA1 and CA3 regions are impaired in this conditioned learning, and are likely to enter the dark
compartment [200]. In fact, CA1 lesioned animals performed as poorly as CA3 lesioned animals
despite less cellular damage evident in the CA1 lesioned animals versus the CA3 lesioned
animals [200]. Furthermore, open field tests allow measurement of exploratory activity, which is
45

dependent upon interaction of the animal with a novel environment [201]. Studies have shown
that hippocampal lesions, correlate with increased locomotor hyperactivity [202]. Thus, this
open field activity has been utilized as a measure of exploration, but also habituation and
learning due to the environmental cue [201].
1.14 Opiate-Dependent Tolerance and Learning
In an attempt to understand the mechanisms underlying drug tolerance, researchers have
begun to explore how environmental variables might influence drug response following
administration. Early theories of opiate tolerance suggest that tolerance was primarily due to
decreases in the sensitivity or number of active opiate receptors within a given interval [203-
205]. However, evidence subsequently emerged that the phenomena of morphine tolerance
could be viewed as a form of Pavlovian conditioning, where the environment represents the
conditioned stimulus, and the pharmacological actions of a drug represents the unconditioned
stimulus [203]. As such, associations between the systemic effects of the drug and local
environmental cues might influence experience-dependent drug response; promoting a
conditioned response. Key studies by Shepard Siegel revealed that if rats were made tolerant to
morphine in a given environment, those animals continued to display analgesic tolerance in the
environment in which they had previously been administered morphine [203, 204]. However,
tolerant rats failed to display analgesic tolerance in an alternative environment for which they
were exposed to for the first time [203, 204]. Thus, environmental cues associated with
morphine administration continued to establish tolerance, where as environmental cues not
linked to morphine attenuated tolerance [203-205]. Therefore, the phenomena of morphine
tolerance may include a component of experience-dependent learned response. However, the
exact molecular mechanisms by which such actions occur remain uncertain.
46

1.14.1 EphB2 and Opiate Tolerance
Previous work done in our laboratory, examined the basic motor and sensory intrinsic
responses between EphB2 null mice and wild-type litter mates. Motor tasks, such as the
hindlimb extension, edge performance, hindlimb grip response, grip strength, wire platform
locomotion and 90º incline climb were compared between EphB2 null and wild type animals
(Stephanie Ho, Thesis 2009). For all motor tasks with the exception of wire platform, EphB2
null mice performed identical to wild-type controls. In addition, sensory responses were
examined using Von Frey filament, tail pinch and tail flick tests. Similar to motor responses
examined, EphB2 null mice exhibited wild-type like sensory responses for the above tests.
Our laboratory examined and compared sensory responsiveness to the opiate morphine
between EphB2 null and wild-type animals. Examinations of the analgesic response of EphB2
null and wild-type littermates following morphine administration using tail pinch and tail flick
(data not shown) assays was performed by a previous graduate student (Ashlin Kanawaty,
Thesis 2011). As shown on Figure 1.9, upon initial exposure to morphine, both EphB2 null and
control littermates exhibit similar patterns of morphine-induced analgesia as measured by tail
pinch latency during the first 30 minutes. However, at time periods greater than 45 minutes
following morphine injection on day 1 EphB2null mice demonstrated significantly lower tail
pinch latencies compared to wild-type controls. Repeated morphine administration over the next
several days induced a reproducible pattern of tolerance to morphine as demonstrated by the
reduced analgesic response. On Day 3 (Figure 1.10), consistent with the effects seen on day one,
EphB2 null mice continued to display accelerated tolerance compared to wild type littermates,
as shown by significantly reduced tail pinch latencies compared to controls. By day 6 (Figure
1.11), both EphB2 null mice and control littermates are substantially tolerized to the effects of
morphine as demonstrated with similar tail pinch latencies. These results suggest that while loss
47
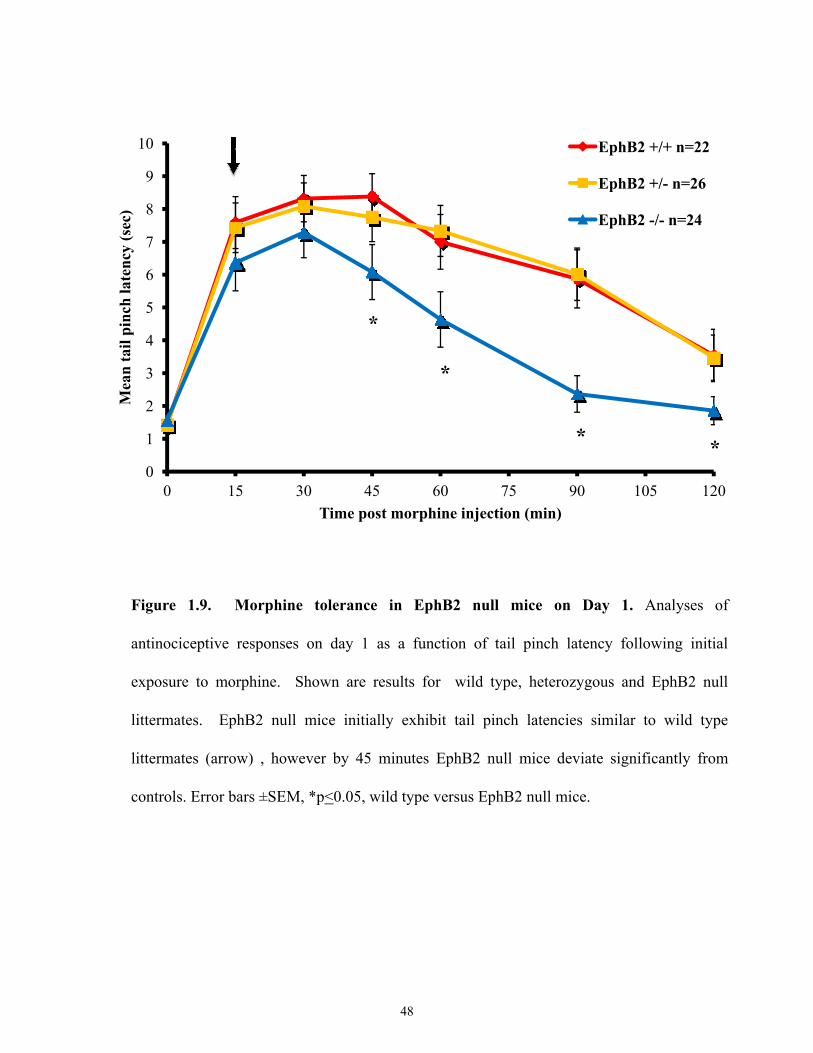
0
1
2
3
4
5
6
7
8
9
10
0 15 30 45 60 75 90 105 120
Mea
n ta
il pi
nch
late
ncy
(sec
)
Time post morphine injection (min)
EphB2 +/+ n=22
EphB2 +/- n=26
EphB2 -/- n=24
*
*
**
Figure 1.9. Morphine tolerance in EphB2 null mice on Day 1. Analyses of
antinociceptive responses on day 1 as a function of tail pinch latency following initial
exposure to morphine. Shown are results for wild type, heterozygous and EphB2 null
littermates. EphB2 null mice initially exhibit tail pinch latencies similar to wild type
littermates (arrow) , however by 45 minutes EphB2 null mice deviate significantly from
controls. Error bars ±SEM, *p<0.05, wild type versus EphB2 null mice.
48
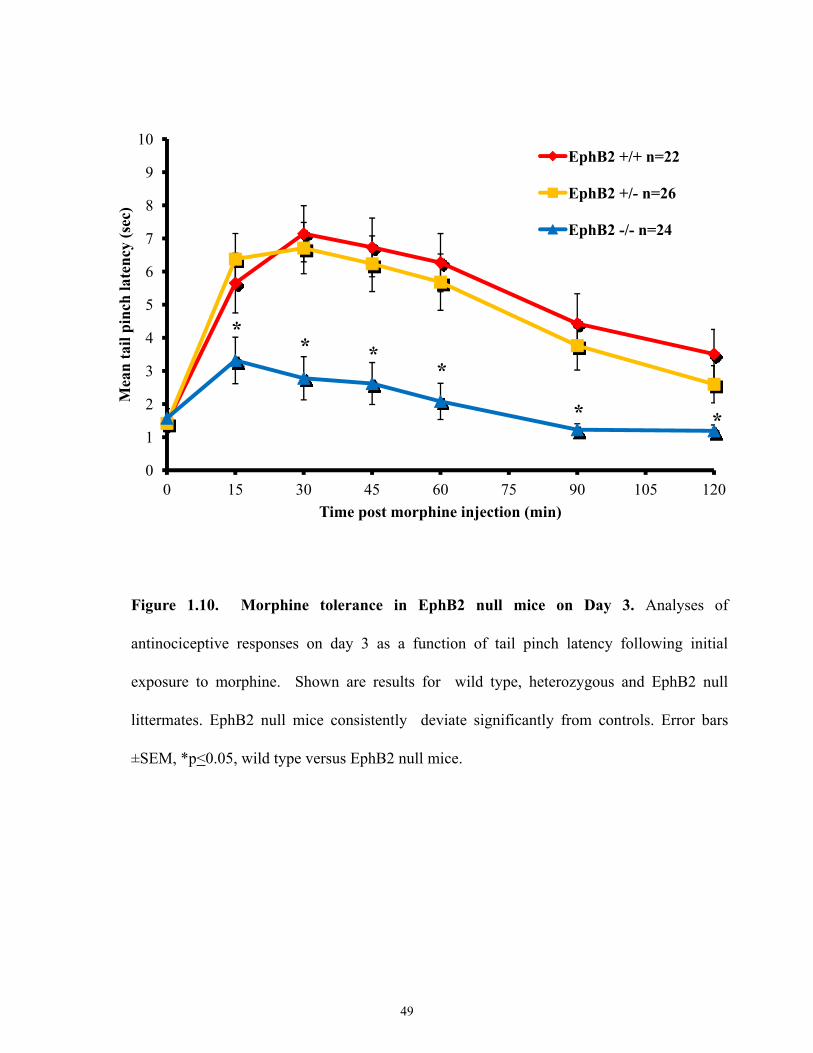
0
1
2
3
4
5
6
7
8
9
10
0 15 30 45 60 75 90 105 120
Mea
n ta
il pi
nch
late
ncy
(sec
)
Time post morphine injection (min)
EphB2 +/+ n=22
EphB2 +/- n=26
EphB2 -/- n=24
** *
*
* *
Figure 1.10. Morphine tolerance in EphB2 null mice on Day 3. Analyses of
antinociceptive responses on day 3 as a function of tail pinch latency following initial
exposure to morphine. Shown are results for wild type, heterozygous and EphB2 null
littermates. EphB2 null mice consistently deviate significantly from controls. Error bars
±SEM, *p<0.05, wild type versus EphB2 null mice.
49
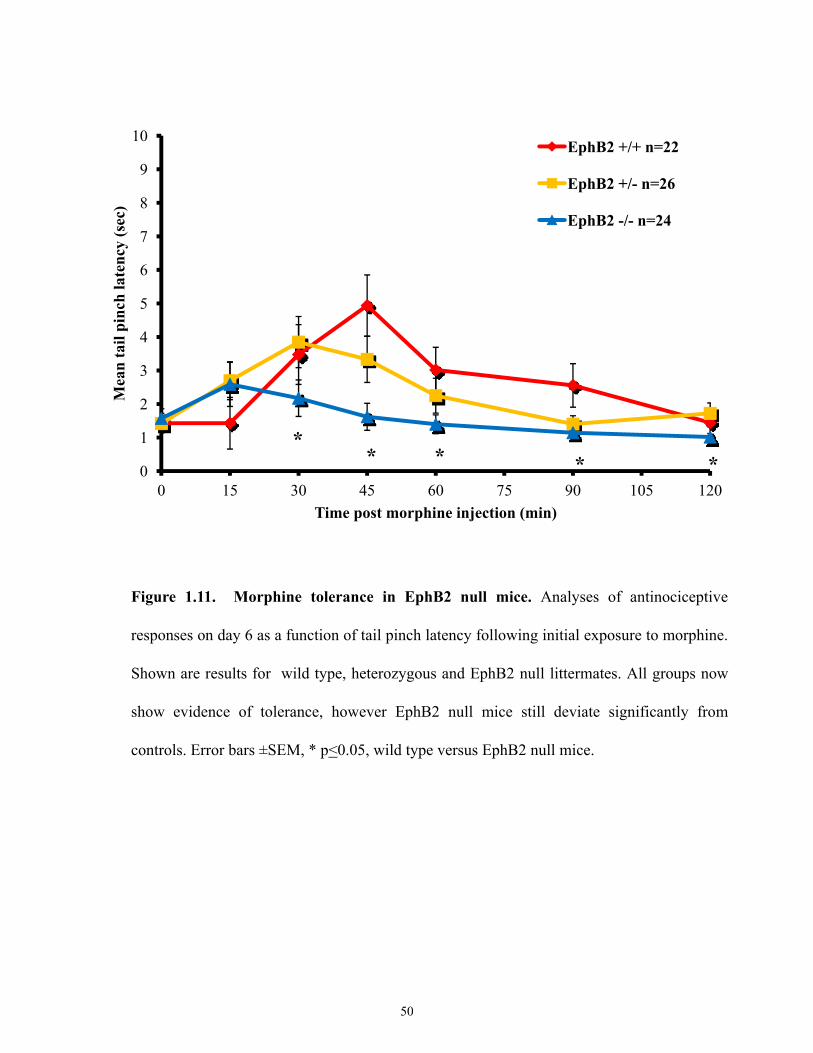
0
1
2
3
4
5
6
7
8
9
10
0 15 30 45 60 75 90 105 120
Mea
n ta
il pi
nch
late
ncy
(sec
)
Time post morphine injection (min)
EphB2 +/+ n=22
EphB2 +/- n=26
EphB2 -/- n=24
** * * *
Figure 1.11. Morphine tolerance in EphB2 null mice. Analyses of antinociceptive
responses on day 6 as a function of tail pinch latency following initial exposure to morphine.
Shown are results for wild type, heterozygous and EphB2 null littermates. All groups now
show evidence of tolerance, however EphB2 null mice still deviate significantly from
controls. Error bars ±SEM, * p<0.05, wild type versus EphB2 null mice.
50

of EphB2 does not alter baseline sensory responses, it significantly alters the rate of morphine
tolerance.
To determine the degree to which the differences in morphine tolerance seen in EphB2
null mice was a function of modified higher cortical input as opposed to direct changes in opiate
receptor signal transduction, the opiate-dependent hippocampal behavior of EphB2 null mice
and control littermates was examined. Previous work by ourselves and others has demonstrated
that loss of EphB2 receptors disrupts the stability of postsynaptic NMDA receptors, reducing the
efficiency of hippocampal dependent LTP [150, 157]. We therefore examined the analgesic
response of EphB2 null mice and controls following a change in environmental setting. As
described in the materials and methods, following six days of twice daily morphine exposure,
treatment groups were divided on day 7. One group remained in the home environment, while
the other was transferred to a novel environmental setting. Consistent with previously reported
results [203], wild-type mice tolerized to morphine exhibited an increase in tail pinch latency
when placed in a novel environment compared to those retained in their home environment
(Figure 1.12) (Ashlin Kanawaty, Thesis 2011). In contrast, EphB2 null mice exhibited no such
enhancement in sensory analgesia upon placement into a novel environment on day 7 (Figure
1.13). Thus, loss of EphB2 alters these morphine-dependent learned responses, suggesting that
EphB2 null mice lack the ability to integrate these sensory-dependent associative cues.
51
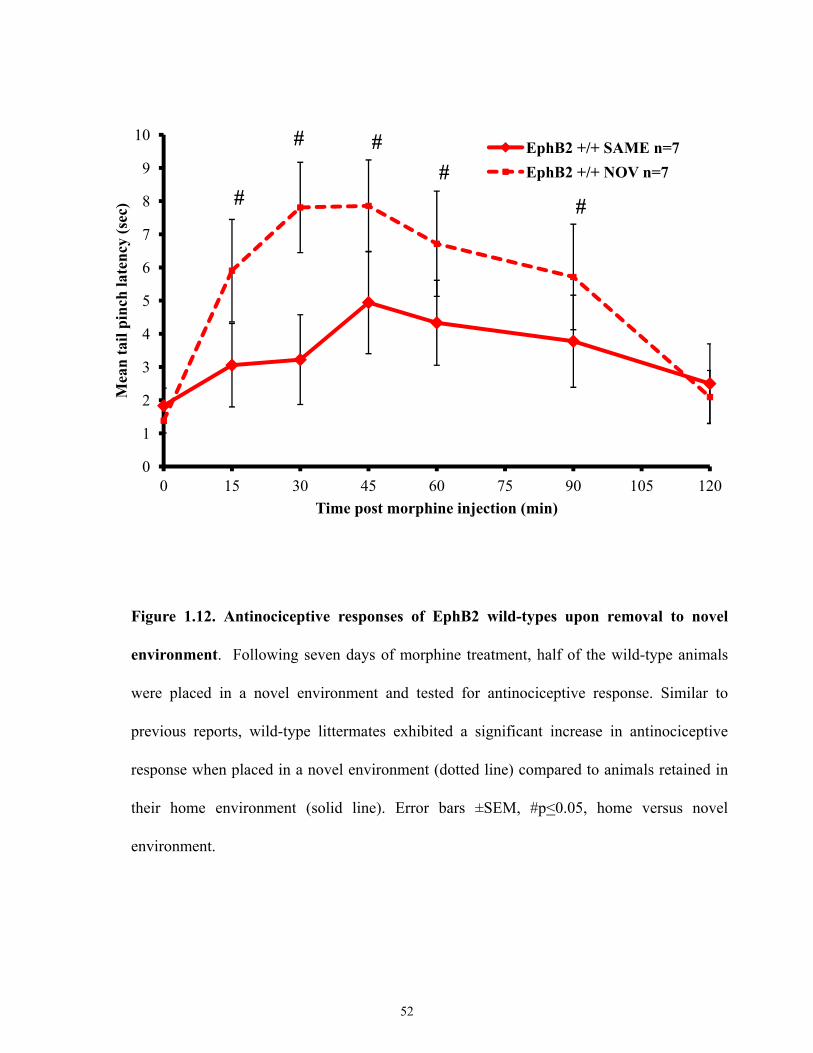
0
1
2
3
4
5
6
7
8
9
10
0 15 30 45 60 75 90 105 120
Mea
n ta
il pi
nch
late
ncy
(sec
)
Time post morphine injection (min)
EphB2 +/+ SAME n=7EphB2 +/+ NOV n=7
#
# ##
#
Figure 1.12. Antinociceptive responses of EphB2 wild-types upon removal to novel
environment. Following seven days of morphine treatment, half of the wild-type animals
were placed in a novel environment and tested for antinociceptive response. Similar to
previous reports, wild-type littermates exhibited a significant increase in antinociceptive
response when placed in a novel environment (dotted line) compared to animals retained in
their home environment (solid line). Error bars ±SEM, #p<0.05, home versus novel
environment.
52
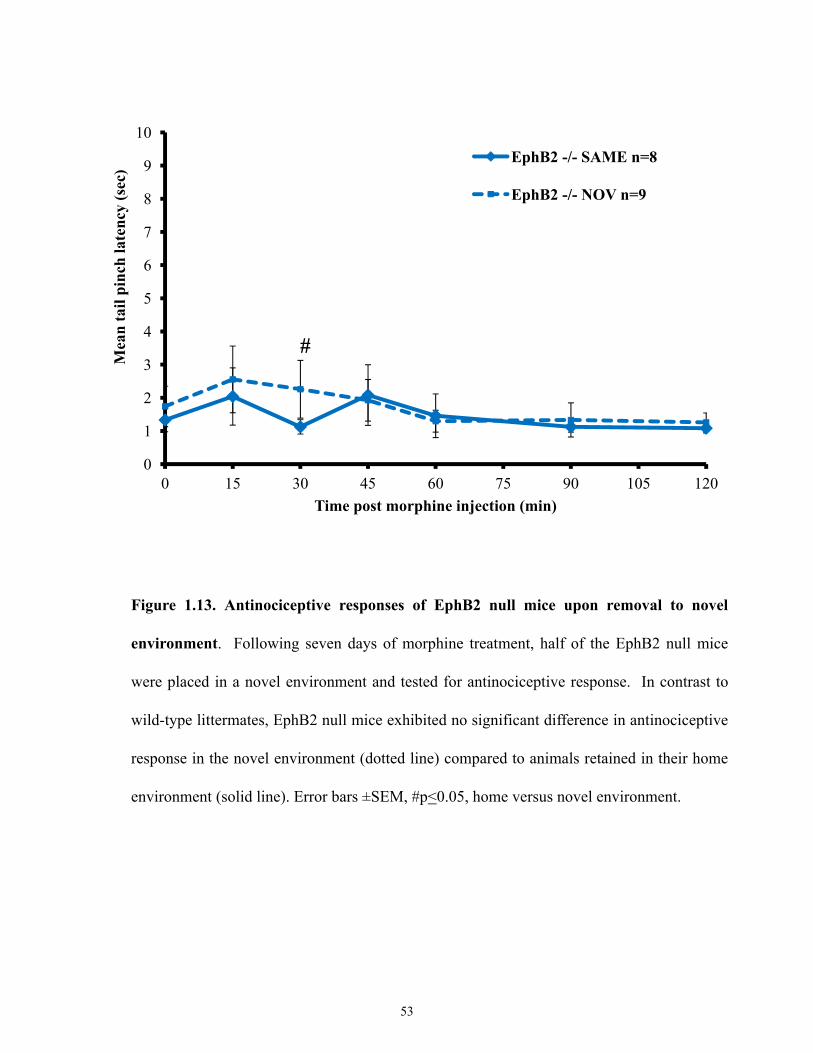
0
1
2
3
4
5
6
7
8
9
10
0 15 30 45 60 75 90 105 120
Mea
n ta
il pi
nch
late
ncy
(sec
)
Time post morphine injection (min)
EphB2 -/- SAME n=8
EphB2 -/- NOV n=9
#
Figure 1.13. Antinociceptive responses of EphB2 null mice upon removal to novel
environment. Following seven days of morphine treatment, half of the EphB2 null mice
were placed in a novel environment and tested for antinociceptive response. In contrast to
wild-type littermates, EphB2 null mice exhibited no significant difference in antinociceptive
response in the novel environment (dotted line) compared to animals retained in their home
environment (solid line). Error bars ±SEM, #p<0.05, home versus novel environment.
53

1.15 Thesis Rationale
Following the development of opiate tolerance, the degree of physiologic analgesia can
be significantly modified by alterations in previously unlinked environmental stimuli. Such
effects demonstrate that there is a significant learned component in the perception and execution
of opiate dependent analgesia. With respect to such learned behaviors, numerous studies have
demonstrated the central role by which NMDA-mediated signaling plays in regulating the
development of behaviors such as LTP. Inhibition of such signaling is known to inhibit
development of morphine tolerance. Despite such progress, the molecular mechanisms linking
these features remain elusive.
Previously we and others have demonstrated that the EphB receptor, EphB2, interacts
with NMDA receptors to prolong their activity, thereby enhancing features such as LTP. As
well, EphB2 is persistently expressed within the adult CNS at sites involved in sensation and
processing of sensory stimuli, and EphB signalling has been shown to modulate sensory
responses. In these present studies, I am examining the relationship between EphB2, morphine
tolerance, and the role of hippocampal learning in modification of this process.
1.15.1 Thesis Hypotheses
The specific hypotheses to be addressed in my thesis are:
1) EphB2-mediated signaling regulates the development of morphine-dependent opiate
tolerance.
2) Such EphB2-mediated actions are mediated through a direct influence on neuronal
signaling, as opposed to secondary influences such as modifications of morphine
metabolism or changes in receptor distribution.
3) The effects of EphB2 on morphine tolerance are mediated via an influence on
hippocampal learning.
54

CHAPTER 2: MATERIAL AND METHODS
55

2.1 Animals
EphB2 wild type, heterozygous, and null littermate mice were generated as previously described
[150] and bred and maintained at University of Toronto Ramsay Wright Zoological
Laboratories. Mice used for studies were two to four months of age, weighing between 25 and
35 grams. Outbred male CD1 wild-type mice (aged two to three months, weighing between 35-
42 grams) from Charles River Laboratories (Wilmington, MA, USA) were used for
hippocampal lesion experiments. A mu opioid receptor knockout mouse (Oprm-/-) (The Jackson
Laboratory, Bar Harbor, Maine, USA) served as a negative control for immunohistological
studies.
2.2 Chemicals
Drugs: Morphine sulphate was obtained from the laboratory of Dr. Van der Kooy (Wiler PCCA,
London, ON) and was dissolved at a concentration of 1.5 mg/mL in 0.9% saline and
administered at a dose of 10 mg/kg intraperitoneally (i.p.). Morphine-3-glucuronide (M3G) was
obtained from the laboratory of Dr. Sandy Pang (National Institutes on Drug Abuse, Rockville,
MD, USA). All surgeries were done under anesthesia with 2.5% Avertin (Sigma-Aldrich)
injected i.p. at a dose of 0.2 mL/10g. Reagents used for LC-MS/MS analysis: Acetonitrile HPLC
Grade (Caledon), Methanol HPLC Grade (Caledon), Caffeine Anhydrous (Bioshop), Formic
Acid reagent grade (Sigma Aldrich), Perchloric Acid (Sigma Aldrich).
2.3 Morphine Tolerance Tests: EphB2 Wild-type and Null Mice
Animals were injected with 10 mg/kg morphine sulphate i.p. twice per day in the morning and
afternoon at intervals of 8 hours for a period of six days. Sensory tests (see below) were done
after morning injections on Day 1, 3 and 6. On day 7, animals were split into two groups. They
either remained in their home environment, or alternatively were placed in a novel environment
56

prior to receiving their daily injection of morphine and subsequent behavioral assessment
(Figure 2.1). This was previously performed on EphB2 wild-type and null mice by a previous
graduate student, Ashlin Kanawaty. These studies were repeated with lesioned and sham
operated control animals (see below).
2.3.1 Sensory Analyses: Tail Pinch and Tail Flick Assay
To test the analgesic response of morphine on animals, mechanoceptive (tail pinch) and
thermoceptive (tail flick) tests following morphine injection were conducted. Tail pinch assay
was performed using a flat forcep, and briefly the closing force was applied to the proximal
third of the animal’s tail. Nociceptive responses were indicated by the latency required for the
animal to respond to the pressure. To avoid tissue damage, a cut-off time of 10 sec was set. The
forcep was applied every 15, 30, 45, 60, 90 and 120 minutes following morphine administration.
Tail flick assay was performed at 55ºC. Animals were placed in a Plexiglas mouse retainer
allowing free tail movement. The distal third of a mouse tail was immersed in the warm water
bath and nociceptive responses were measured by the latency required for the animal to respond
to the temperature. To avoid tissue damage, a cut-off time of 15 sec was set. The tail was
immersed 30 min following morphine administration.
2.4 Pharmacokinetic Analyses of Morphine Metabolism
2.4.1 Preparation of LC-MS/MS Standard Solutions. Stock solutions of morphine and M3G
were prepared in 0.9% saline. All subsequent working standard solutions were prepared using
serial dilutions in acetonitrile, and stored at -20ºC. Whole blood and brain homogenates were
obtained from non-injected morphine naive mice. Standard solutions of morphine and M3G
were used to spike blank blood and blank brain homogenates with known concentrations to
57

Figure 2.1. Schedule of morphine dosing. Animals were injected i.p. with 10mg/kg
morphine twice per day at 8 hour intervals. Analgesic tests were conducted following the
morning injection on day 1, 3 and 6. Experiments on days 1-6 were conducted in the same
environmental setting (purple). For tests performed on day 7, animals were divided into 2
groups, with half remaining in the home environment (purple) and the other half placed in a
novel environment (pink).
58
Day 0
Day 1
Day 2
Day 3
Day 4
Day 5
Day 6
Day 7
TEST
10 am
TEST
6 pm
10 am
6 pm
10 am
TEST
6 pm
10 am
6 pm
10 am
6 pm
10 am
TEST
6 pm
10 am
TEST
6 pm
10 am
TEST
6 pm
Day 7
59

create a desired concentration range. Caffeine was used as an internal standard and prepared in
water at a stock concentration of 3 μg/mL.
2.4.2 Collection of Blood and Brain Samples. EphB2 wild-type and null animals were given a
bolus injection of 10 mg/kg morphine sulphate. Blood was then collected terminally by heart
puncture at 30, 60, and 90 min after injection. Blood samples were frozen immediately and
stored at -80ºC until analyzed. Brain samples (excluding the cerebellum) were homogenized in
0.1N perchloric acid, to a final concentration of 0.33g tissue/mL homogenate, vortexed and then
stored at -80ºC. Following one freeze-thaw cycle, brain homogenates were sonicated using the
Misonix 3000 water bath sonicator for 10-12 min at high speed in ice cold water. Samples were
then centrifuged at 15,000 rpm for 10 min. The supernatants were collected in new
microcentrifuge tubes and neutralized with 2M NaOH and stored at -80ºC until analyzed.
2.4.3 Purification of Blood and Brain Samples. A 10 μl and 20 μl aliquot of the internal
standard caffeine (3 μg/ml) was added to each 100 μL blood sample or to each 200 μL brain
sample, respectively. Blood and brain samples were vortexed for 60 seconds with 400 μL and
800 μL of an equal mixture of methanol and acetonitrile, respectively. Samples were then
centrifuged at 13,000×g for 10 min to precipitate the proteins. The supernatants were transferred
into Sep-Pak Vac C18 3 cc cartridges (200 mg; Waters, Milford, MA, USA). Each cartridge was
pre-conditioned with 2 mL of acetonitrile followed by 2 mL of Millipore water. After loading of
the sample, the samples were eluted with 2 mL of acetonitrile. The eluent was pooled and dried
under a stream of nitrogen at room temperature.
60

2.5 LC-MS/MS Analyses
The residue from blood and brain samples was reconstituted with 200 μL and 100 μL,
respectively, of the mobile phase (70% water with 0.1%v/v formic acid and 30% acetonitrile
with 0.1%v/v formic acid). For blood samples 1 μL of the reconstituted sample, and for brain
samples 5 μL of the reconstituted sample, was injected into the LC–MS/MS system for analysis.
Samples were analyzed by LC-MS/MS using 6410 Triple Quad LC/MS instrument (Agilent
Technologies) with ESI source in positive ion modes. Samples were separated on a C18 column
at 1 mL/min. The mobile phase consisted of HPLC grade water (A) and acetonitrile (B) both
containing 0.1% formic acid. The following gradient was run: 0-1 min, 4% (B); 4-5 min, 4%
(B); 5-9 min 4% to 100% (B); 9-10 min, 100% (B); 10-11min 100% to 4% B; 11-16 min, 4%
(B). MS parameters were as follows: gas temperature 350°C, nebulizer pressure 50 psi, drying
gas (nitrogen) 11 L/min and VCap 3500V. Using MRM monitoring (in positive-ion mode) the
following transitions were observed: morphine-3-glucoronide (m/z 462 286, RT 6.7 min),
morphine (m/z 286.1 165, RT 7.8 min) and caffeine (m/z 195 138, RT 11.4 min). Fragmentor
voltage settings used for morphine, M3G and caffeine were 155V, 160V and 85V, respectively.
Collision energy settings used for morphine, M3G and caffeine were 42V, 32V and 20V,
respectively.
2.6 Immunohistochemistry
Brain and spinal sections from wild-type, heterozygous, EphB2 null, and MOR knockout mice
(Oprm-/-) were prepared following intracardial perfusion of saline followed by 4%
paraformaldehyde in 0.1 M phosphate buffered saline pH 7.4 (PBS). Following overnight
fixation, samples were embedded in paraffin wax and 7 μm thick sections were sliced.
To probe sections with MOR antibody, antigen retrieval was performed. Following dewaxing,
sections were immersed in 10 mM sodium citrate (pH 6.0) and heated in a pressure cooker for
61

10 min to bring the water to a boil, followed by an additional two minutes at a boil. Following
cooling, sections were then pre-incubated in blocking solution (5% goat serum, 0.25% Tween-
20 dissolved in PBS) for 30min. Sections were then incubated with primary antibody, anti-MOR
(1:1000,Immunostar, rabbit, polyclonal) diluted in blocking solution overnight at 4ºC. Next day,
slides were washed (3x5 minutes in blocking buffer), then incubated in biotinylated goat anti-
rabbit secondary antibody (1:200, Vector Labs) for two hours at room temperature, followed by
tertiary reagent (avidin-horseradish peroxidase, Vector Labs) for 45 minutes, and visualized
using 3,3-diaminobenzidine (DAB).
2.7 Stereotactic Surgeries
Bilateral lesions of the dorsal hippocampus were performed as follows:
2.7.1 Kainic Acid Induced Lesion. Adult male CD1s mice (2-3 months of age) were
anaesthetized with 2.5% Avertin (0.2 mL/kg), and the scalp was incised along the sagittal
suture. At a position located 60% from the bregma, and 1.7 mm lateral of the sagittal suture,
burr holes were made in the skull using a carbon dioxide laser. The needle was inserted 1.15
mm below the dura and the kainic acid (Tocris Biosciences) was injected bilaterally into the
hippocampus at a concentration of 20 mM and a total volume of 300 nL. Thus, each mouse
received 6 nmoles of kainic acid per hippocampus. Following surgery, scalp was closed and the
animals were left to recover for 72 hours prior to start of behavioural assessments. Since
previous attempts at this experimental procedure resulted in seizures, a dose of 10mg/kg of
Diazepam immediately prior to surgery was administered. However, due to the continued high
mortality rate observed with kainic acid injections, this procedure was discontinued and
electrolytic lesions were performed as an alternative method.
62

2.7.2 Electrolytic Lesion. Adult male CD1s mice (2-3 months of age) were anaesthetized with
2.5% Avertin (0.2 mL/kg), and the scalp was incised along the sagittal suture. At the midpoint
between the lambda and bregma sutures, two 0.2 mm burr holes using a carbon dioxide laser
were drilled on either side of the sagittal midline at a displacement of 1.8 mm. A 0.1 mm
diameter platinum-iridium electrode with an exposed tip of 0.5 mm was then inserted to a depth
of 1.5 mm at each site. Electrolytic lesions were produced with a single constant direct current
discharge of 3 mA for 3 sec at each site of the hippocampus (Figure 2.2). Following surgery,
scalp was closed and the animals were left to recover for 72 hours prior to start of behavioural
assessments (Figure 2.2). Sham operated controls underwent the same exact procedure minus
the electrode insertion. After the completion of all behavioural testing, all mice were perfused
transcardially with 4% PFA. Brain sections were embedded in paraffin wax and 7 μm thick
sections were collected. Sections were stained with thionin dye to examine the full extent of the
lesion. Paraffin sections we also stained with GFAP (1:400, Dako) and DAPI.
2.8 Behavioral Analyses
2.8.1 Passive Avoidance. The passive avoidance apparatus consists of light and dark chambers.
Mice were initially placed in the light chamber for 30 seconds. Following this, the door to the
dark chamber is opened and the time required to enter the dark chamber was recorded as the
transfer latency time (TLT). This was recorded as the TLT-acquisition. Following 10 seconds in
the dark chamber, EphB2 wild-type and null mice were subjected to a one time shock of 0.5 mA
for 5 seconds (or 0.7 mA for 5 seconds for CD1 animals). After a total time of 30 sec in the dark
chamber, the animal was free to return to the light chamber, and subsequently returned to its
home cage. Twenty-four hours following acquisition, mice were re-introduced into the light
chamber. The time required for the animal to enter the dark chamber was recorded as TLT-
63
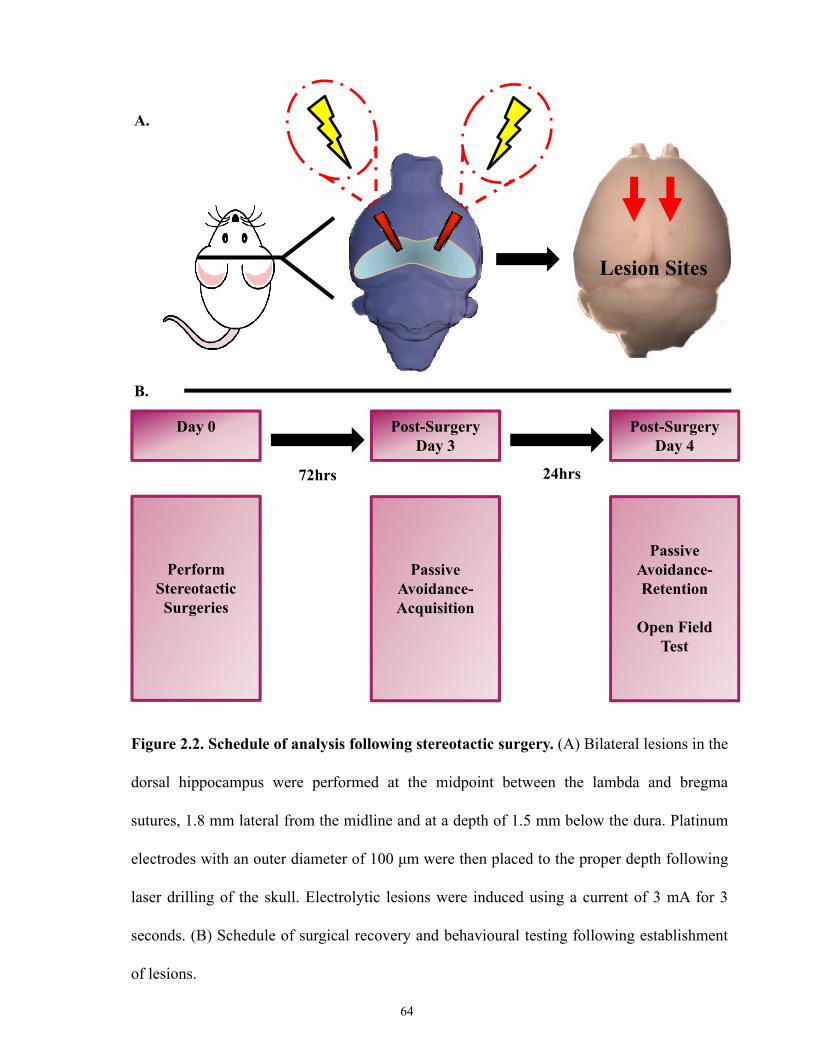
Lesion Sites
Day 0
Perform Stereotactic Surgeries
Post-SurgeryDay 3
72hrs
Passive Avoidance-Acquisition
Post-SurgeryDay 4
Passive Avoidance-Retention
Open Field Test
24hrs
A.
B.
Figure 2.2. Schedule of analysis following stereotactic surgery. (A) Bilateral lesions in the
dorsal hippocampus were performed at the midpoint between the lambda and bregma
sutures, 1.8 mm lateral from the midline and at a depth of 1.5 mm below the dura. Platinum
electrodes with an outer diameter of 100 μm were then placed to the proper depth following
laser drilling of the skull. Electrolytic lesions were induced using a current of 3 mA for 3
seconds. (B) Schedule of surgical recovery and behavioural testing following establishment
of lesions.
64

retention. A cut-off time of 300 sec was used if the animal failed to enter the dark chamber. This
test was previously examined on wild-type and EphB2 null animals by a previous graduate
student, Ashlin Kanawaty. Studies were repeated with lesioned and sham operated control
animals.
2.8.2 Activity Monitor. The activity monitor was used to conduct open field tests. EphB2 wild-
type and null mice were examined in a 25 cm by 42 cm open field for a period of one hour.
Spontaneous motor activity was recorded using an automated movement detection system
(AM1053 activity monitors; Linton Instrumentation, UK). The apparatus consisted of 24 (16 by
8) infrared beams forming a grid. Animal movement resulted in beam breakage and the resulting
activity and type of movement was registered. Studies were repeated with lesioned and sham
operated control animals.
2.9 Morphine Related Behaviour
2.9.1 Morphine Induced Hyperactivity
To determine morphine naïve performance, EphB2 wild-type and null animals were examined
on the activity monitor for a period of one hour. Twenty-four hours later they were injected with
a single dose of 10 mg/kg of morphine and analyzed for activity for a period of 60 minutes
as described above. Animals injected with 0.9% saline served as control. This was repeated with
lesioned and sham operated control animals.
2.9.2 Morphine Tolerance Tests: for Lesioned and Sham Operated Control Animals
Lesioned and sham operated control animals following the 72 hour recovery period and
assessment of behavioural functions were investigated for their response to morphine tolerance.
65

As described above, animals were injected with 10 mg/kg morphine sulphate i.p. twice per day
in the morning and afternoon at intervals of 8 hours for a period of six days. On day 7, mice
were split into two groups. They either remained in their home environment, or alternatively
were placed in a novel environment prior to receiving their injection of morphine and
subsequent behavioral assessment via tail flick. The tail flick assay (as described above) was
performed on day 7 at 30, 60, 90 and 120 min following the morning morphine injection.
2.10 Statistical Analyses
Two-way ANOVA with repeated measurement (time) using GraphPad Prism software V5.0
(GraphPad Software Inc, La Jolla, CA, USA) was used to assess latency and concentration
differences among animal groups overtime. Three-way ANOVA was conducted to compare
latency differences among animal groups overtime and over days using SPSS 16 (SPSS Inc.,
Chicago, IL, USA). Simple t-test was used to compare differences between each test group and
its corresponding control group using Microsoft Excel. All data are presented as mean±SEM.
Statistical results are considered significant if P ≤0.05.
66

CHAPTER 3: RESULTS
67

3.1 Morphine related responses of EphB2 null mice
Previously, our laboratory examined the baseline motor and sensory responses between
EphB2 null mice and wild-type littermates (Stephanie Ho, Thesis 2009). Similar to motor
responses examined, EphB2 null mice exhibited wild-type like sensory responses. Interestingly,
temporal and spatial distribution of EphB2 exists within the sensory nervous system (Figure
3.1). However, given that EphB2 null mice demonstrate no differences in baseline motor and
sensory responses from that seen in littermate controls, we began to examine whether other
elements of sensory response might be altered in EphB2 null animals. Thus, we examined
sensory responsiveness to the opiate morphine. Examinations of the analgesic response of
EphB2 null and wild-type littermates following morphine administration using tail pinch assay
was performed by a previous graduate student (Ashlin Kanawaty, Thesis 2011). As shown on
Figures 1.9-1.11, EphB2 null displayed accelerated morphine tolerance compared to control
littermates. These results suggested that while loss of EphB2 does not alter baseline sensory
responses, it significantly alters the rate of morphine tolerance. Consistent with this, analysis of
day 1 data using two-way ANOVA (genotype versus time with repeated measurement)
demonstrated a significant effect of both genotype [F(1,40)=6.85, p<0.05], and time
[F(6,240)=50.81, p<0.05], with a significant interaction between these factors [F(6,240)=2.91,
p<0.05]. Analysis of Day 3 data using two-way ANOVA also revealed a significant effect of
both genotype [F(1,40)=21.91, p<0.05], and time [F(6,240)=18.98, p<0.05], with significant
interaction between these factors [F(6,240)=6.79, p<0.05]. Similarly, analysis of Day 6 data by
two-way ANOVA again demonstrated a significant effect of both genotype [F(1,40)=11.39,
p<0.05], and time [F(6,240)=15.97, p<0.05], with significant interaction between these factors
[F(6,240)=7.34, p<0.05]. Analysis of day 1, 3 and 6 data using three-way ANOVA examining
genotype (wild-type versus EphB2 null) as between-subject factors, with time (0, 15, 30, 45, 60,
68
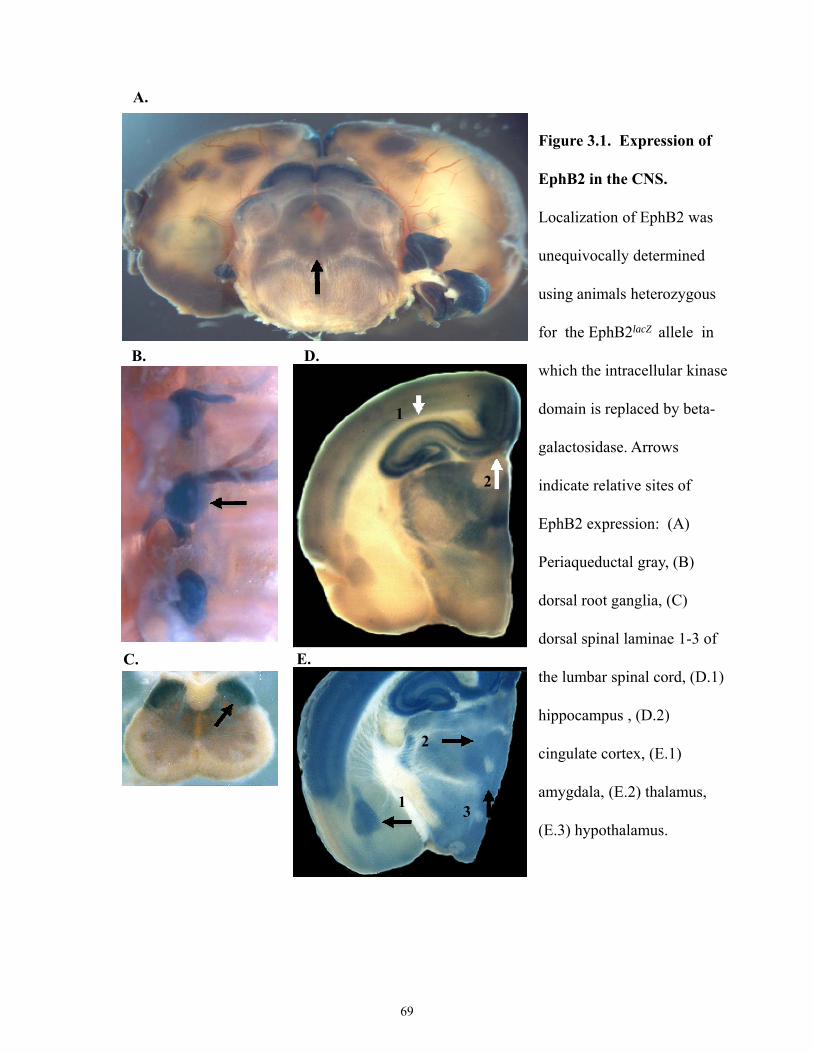
Figure 3.1. Expression of
EphB2 in the CNS.
Localization of EphB2 was
unequivocally determined
using animals heterozygous
for the EphB2lacZ allele in
which the intracellular kinase
domain is replaced by beta-
galactosidase. Arrows
indicate relative sites of
EphB2 expression: (A)
Periaqueductal gray, (B)
dorsal root ganglia, (C)
dorsal spinal laminae 1-3 of
the lumbar spinal cord, (D.1)
hippocampus , (D.2)
cingulate cortex, (E.1)
amygdala, (E.2) thalamus,
(E.3) hypothalamus.
B.
C.
A.
D.
E.
1
2
1 3
2
69

90 and 120 minutes) and day of analysis (1, 3 and 6) as the within-subjects factors, found all
effects and interactions to be significant (Table 3.1).
Our lab also previously performed Shepard Siegel’s experiment [203], to investigate
whther EphB2 null mice exhibited any alterations in learning/ behavioural responses to
morphine following the development of tolerance (Ashlin Kanawaty, Thesis 2011). Consistent
with previously reported results [203], wild type mice tolerized to morphine exhibited an
increased analgesic response when placed in a novel environment compared to those retained in
their home environment (Figure 1.12). In contrast, EphB2 null mice failed to display enhanced
analgesic response upon placement into a novel environment on day 7 (Figure 1.13). This
suggested that Eph B2 null mice lack the ability to integrate these sensory-dependent associative
cues. Analysis of this context-dependent learning using three-ANOVA using genotype (EphB2
null or control) and environment (novel or same) as between-subjects factors, and time (0, 15,
30, 45, 60, 90 and 120 minutes) as with within-subjects factors, demonstrated all effects and
interactions to be significant with the exception of the interaction between genotype and
environment (Table 3.2).
3.2 Distribution of mu opioid receptors in the CNS
We examined whether the accelerated development of morphine tolerance was attributed
to the direct loss of EphB2 receptors and its direct affect on MOR expression levels or
distribution pattern in the CNS. We examined MOR expression and distribution at several key
CNS sites using immunohistochemistry (Figures 3.2-3.3). EphB2 null mutants and control
littermates exhibited similar distributions of MORs within the dorsal spinal cord, which is
known to be enriched with MORs (Figure 3.2). A MOR null mouse (Oprm-/- mouse) served as a
negative control. As well, EphB2 wild-types, heterozygous and null mutants exhibited similar
patterns of MOR distribution within regions of the striatum and the hippocampus (Figure 3.3).
70
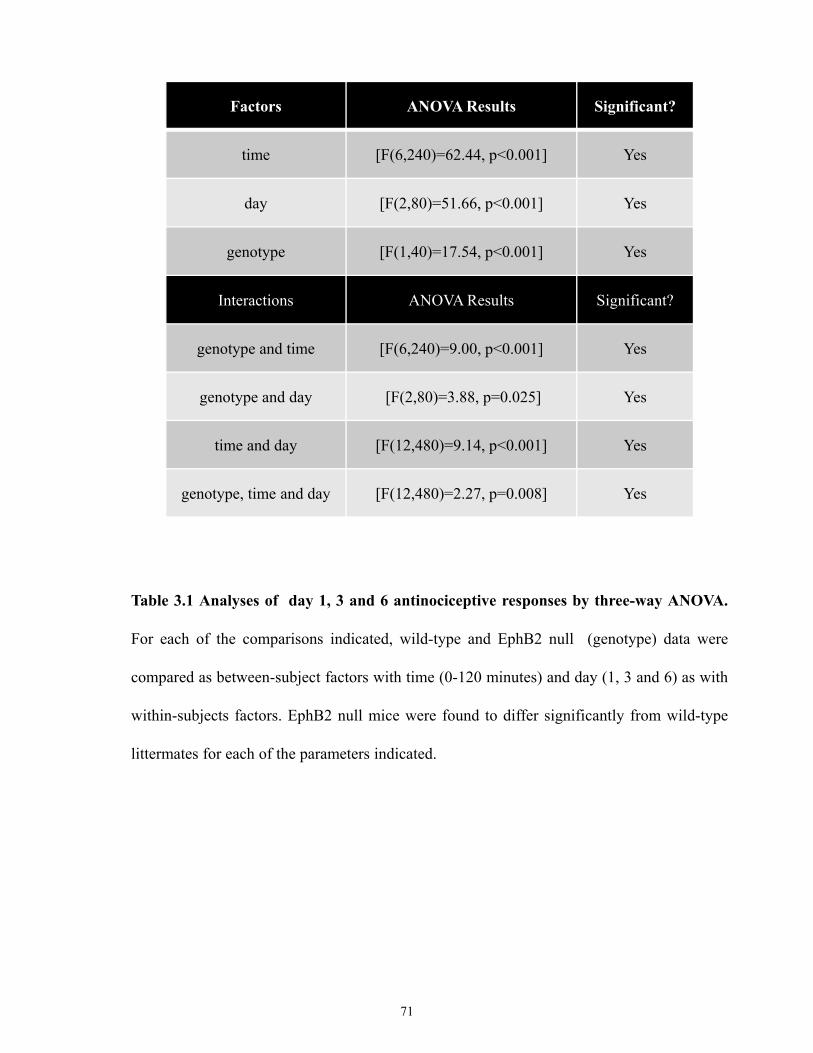
Factors ANOVA Results Significant?
time [F(6,240)=62.44, p<0.001] Yes
day [F(2,80)=51.66, p<0.001] Yes
genotype [F(1,40)=17.54, p<0.001] Yes
Interactions ANOVA Results Significant?
genotype and time [F(6,240)=9.00, p<0.001] Yes
genotype and day [F(2,80)=3.88, p=0.025] Yes
time and day [F(12,480)=9.14, p<0.001] Yes
genotype, time and day [F(12,480)=2.27, p=0.008] Yes
Table 3.1 Analyses of day 1, 3 and 6 antinociceptive responses by three-way ANOVA.
For each of the comparisons indicated, wild-type and EphB2 null (genotype) data were
compared as between-subject factors with time (0-120 minutes) and day (1, 3 and 6) as with
within-subjects factors. EphB2 null mice were found to differ significantly from wild-type
littermates for each of the parameters indicated.
71
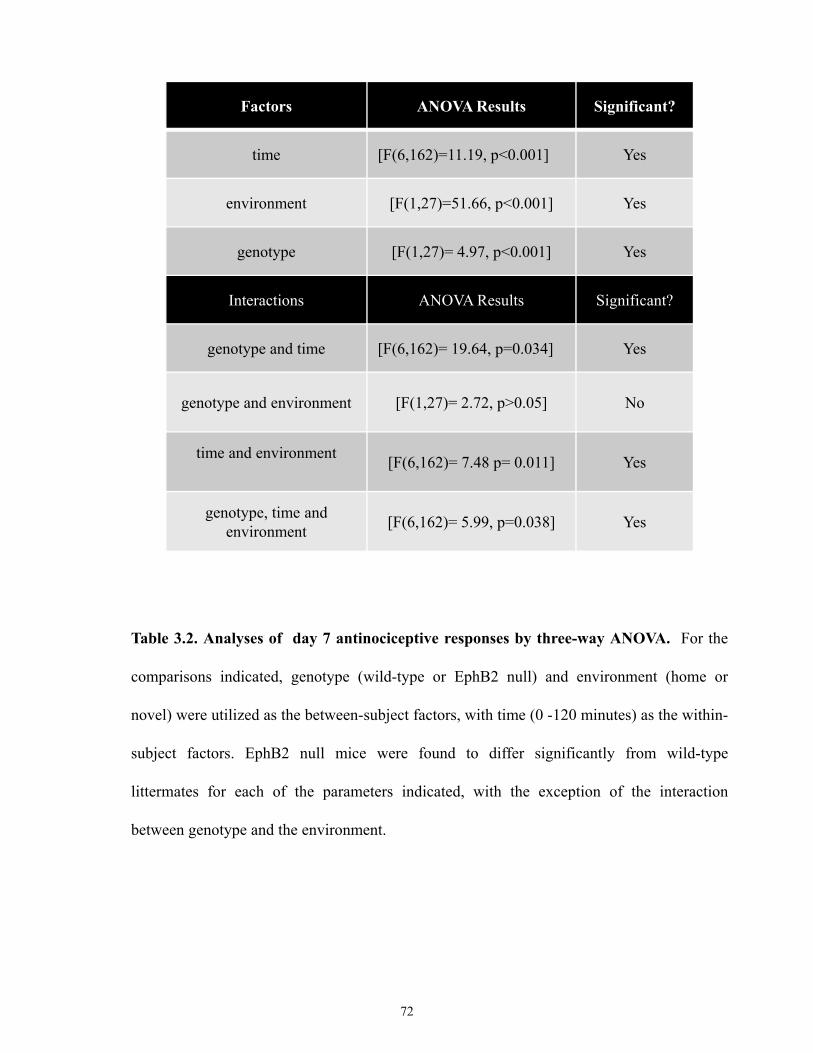
Factors ANOVA Results Significant?
time [F(6,162)=11.19, p<0.001] Yes
environment [F(1,27)=51.66, p<0.001] Yes
genotype [F(1,27)= 4.97, p<0.001] Yes
Interactions ANOVA Results Significant?
genotype and time [F(6,162)= 19.64, p=0.034] Yes
genotype and environment [F(1,27)= 2.72, p>0.05] No
time and environment [F(6,162)= 7.48 p= 0.011] Yes
genotype, time andenvironment [F(6,162)= 5.99, p=0.038] Yes
Table 3.2. Analyses of day 7 antinociceptive responses by three-way ANOVA. For the
comparisons indicated, genotype (wild-type or EphB2 null) and environment (home or
novel) were utilized as the between-subject factors, with time (0 -120 minutes) as the within-
subject factors. EphB2 null mice were found to differ significantly from wild-type
littermates for each of the parameters indicated, with the exception of the interaction
between genotype and the environment.
72
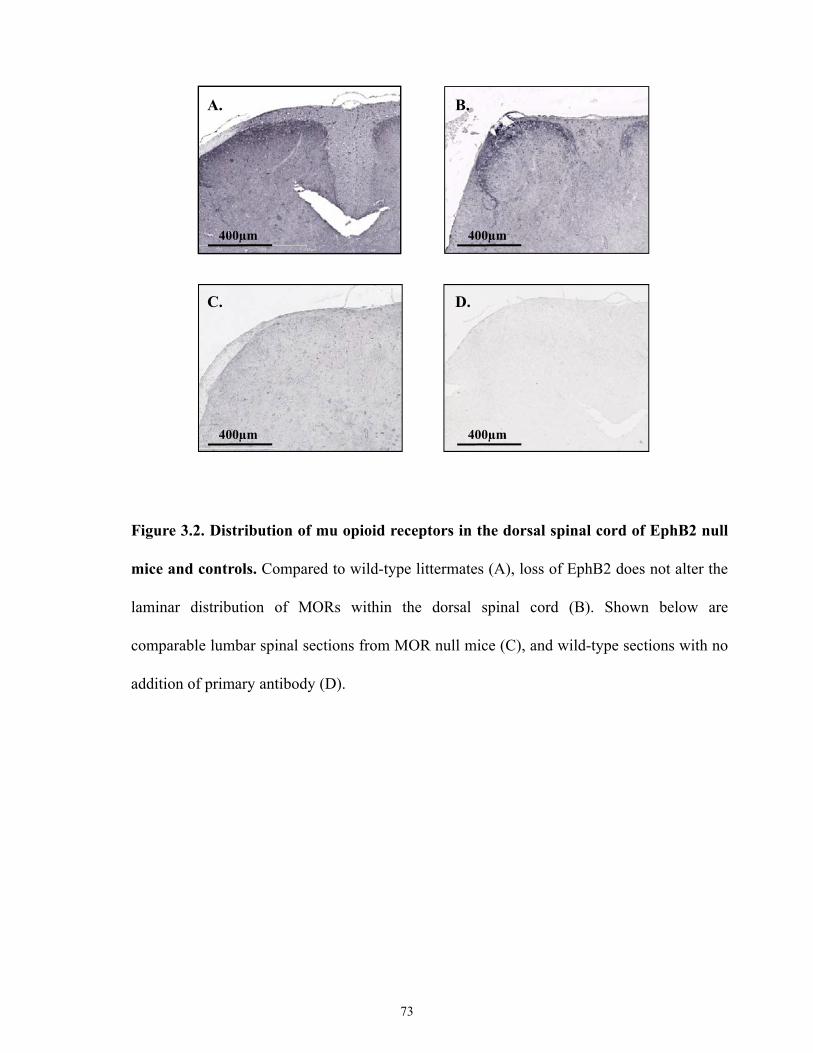
A. B.
C. D.
400µm 400µm
400µm 400µm
Figure 3.2. Distribution of mu opioid receptors in the dorsal spinal cord of EphB2 null
mice and controls. Compared to wild-type littermates (A), loss of EphB2 does not alter the
laminar distribution of MORs within the dorsal spinal cord (B). Shown below are
comparable lumbar spinal sections from MOR null mice (C), and wild-type sections with no
addition of primary antibody (D).
73
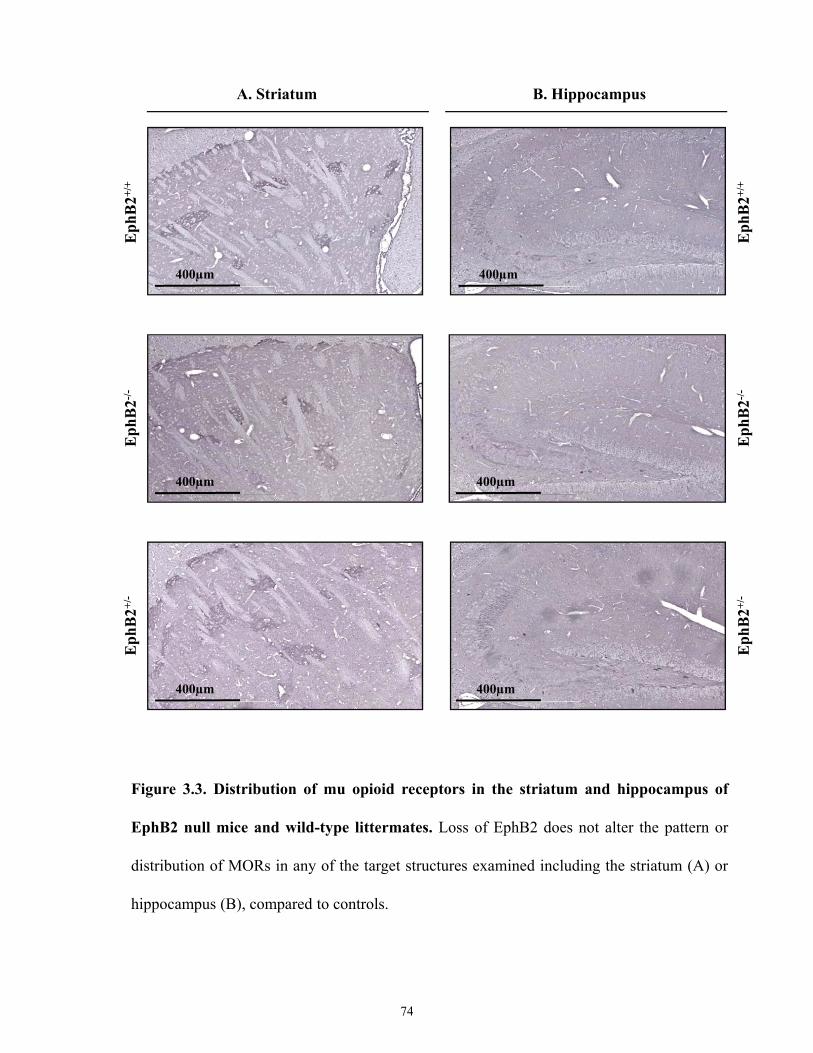
Eph
B2-/-
Eph
B2+/
+E
phB
2+/-
400µm 400µm
400µm 400µm
400µm 400µm
A. Striatum B. Hippocampus
Eph
B2-/-
Eph
B2+/
+E
phB
2+/-
Figure 3.3. Distribution of mu opioid receptors in the striatum and hippocampus of
EphB2 null mice and wild-type littermates. Loss of EphB2 does not alter the pattern or
distribution of MORs in any of the target structures examined including the striatum (A) or
hippocampus (B), compared to controls.
74

Therefore, loss of EphB2 does not significantly alter the distribution of MORs in the spinal cord
and brain regions examined, compared to wild-type littermates.
3.3 Pharmacokinetic analysis of morphine in EphB2 null mice
One possible explanation for the accelerated morphine tolerance observed in EphB2 null
mice compared to wild-type littermates is that these animals may possess differences in their
relative level of morphine metabolism. For instance, if EphB2 null mice metabolized morphine
at a higher rate compared to wild-type controls, they might exhibit lower relative levels of
morphine analgesia at specific periods post-injection. Therefore, we investigated whether loss of
EphB2 significantly altered the kinetics of morphine metabolism. Initially, levels of morphine
and its main metabolite M3G were examined in a time-dependent manner in brain and whole
blood following first time exposure to morphine using LC-MS/MS. Concentrations of morphine
and M3G were determined in brain and whole blood at 30, 60 and 90 min following a single
injection of morphine. LC-MS/MS analyses revealed sufficient separation of primary
compounds within 15 min (Figure 3.4). Examination of morphine and M3G levels in the brain
demonstrated no significant differences between EphB2 null and wild-type animals at all time
points measured (Figure 3.5). Examination of morphine and M3G concentration in whole blood
was also found to be not significantly different between EphB2 null and wild-type animals at 60
and 90 min, but did differ at 30 min (Figure 3.6). In addition, I examined brain and whole blood
levels of morphine and M3G following repeated morphine exposure for 7 days. Consistent with
the above results, EphB2 null and wild-type animals display no significant differences in brain
or blood levels of morphine or M3G following chronic morphine exposure (Figure 3.7). Thus,
loss of EphB2 therefore does not induce major alterations in morphine metabolism.
Analysis of morphine brain levels using two-way ANOVA (genotype versus time with
repeated measurement) revealed no significant effect of both genotype [F(1,10)=0.64, p>0.05],
75

Figure 3.4. LC/MS/MS analyses of morphine and M3G. Elution profiles of morphine
and M3G compared to caffeine (internal calibration standard) in whole blood and brain
extracts. Using multiple reaction monitoring (MRM) the following transitions were
observed: M3G (m/z 462 286, RT 6.7 min), morphine (m/z 286.1 165, RT 7.8 min) and
caffeine (m/z 195 138, RT 11.4 min).
76
Morphine‐3‐Glucuronide
Morphine‐3‐Glucuronide
77
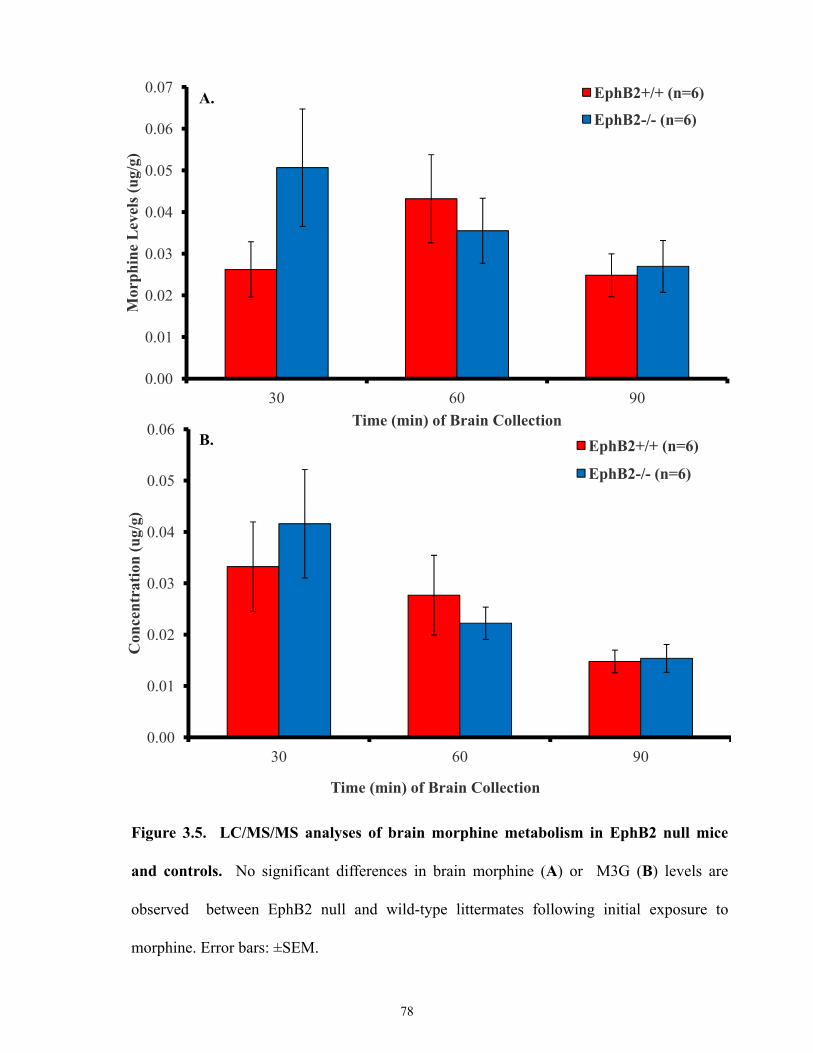
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
30 60 90
Mor
phin
e L
evel
s (ug
/g)
Time (min) of Brain Collection
EphB2+/+ (n=6)
EphB2-/- (n=6)
Figure 3.5. LC/MS/MS analyses of brain morphine metabolism in EphB2 null mice
and controls. No significant differences in brain morphine (A) or M3G (B) levels are
observed between EphB2 null and wild-type littermates following initial exposure to
morphine. Error bars: ±SEM.
A.
0.00
0.01
0.02
0.03
0.04
0.05
0.06
30 60 90
Con
cent
ratio
n (u
g/g)
Time (min) of Brain Collection
EphB2+/+ (n=6)
EphB2-/- (n=6)
B.
78
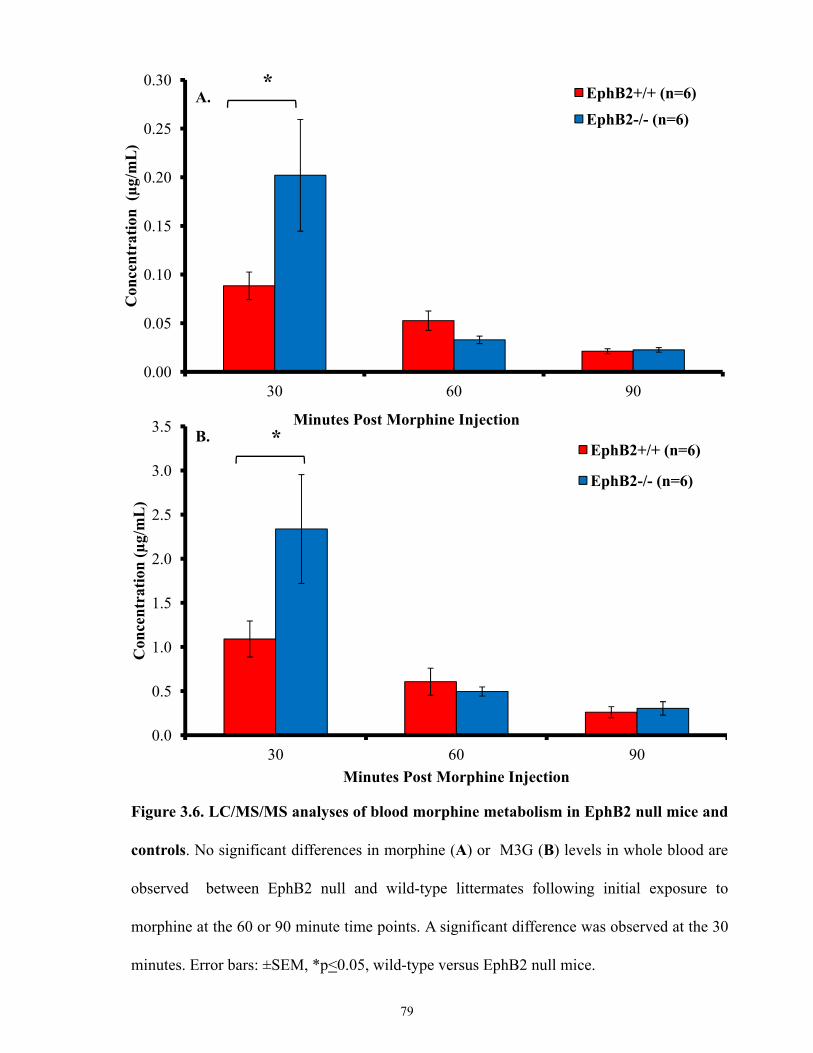
0.00
0.05
0.10
0.15
0.20
0.25
0.30
30 60 90
Con
cent
ratio
n (µ
g/m
L)
Minutes Post Morphine Injection
EphB2+/+ (n=6)
EphB2-/- (n=6)
*
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
30 60 90
Con
cent
ratio
n (µ
g/m
L)
Minutes Post Morphine Injection
EphB2+/+ (n=6)
EphB2-/- (n=6)
*
A.
B.
Figure 3.6. LC/MS/MS analyses of blood morphine metabolism in EphB2 null mice and
controls. No significant differences in morphine (A) or M3G (B) levels in whole blood are
observed between EphB2 null and wild-type littermates following initial exposure to
morphine at the 60 or 90 minute time points. A significant difference was observed at the 30
minutes. Error bars: ±SEM, *p<0.05, wild-type versus EphB2 null mice.
79
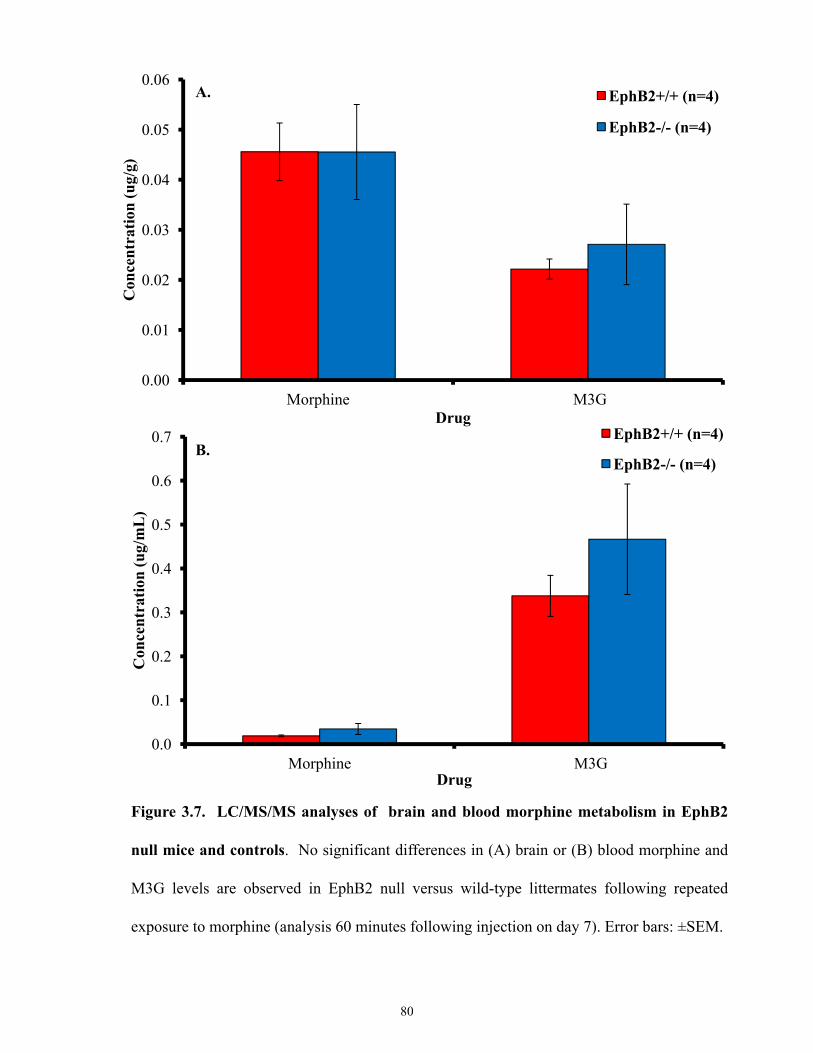
0.00
0.01
0.02
0.03
0.04
0.05
0.06
Morphine M3G
Con
cent
ratio
n (u
g/g)
Drug
EphB2+/+ (n=4)
EphB2-/- (n=4)
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
Morphine M3G
Con
cent
ratio
n (u
g/m
L)
Drug
EphB2+/+ (n=4)
EphB2-/- (n=4)
A.
B.
Figure 3.7. LC/MS/MS analyses of brain and blood morphine metabolism in EphB2
null mice and controls. No significant differences in (A) brain or (B) blood morphine and
M3G levels are observed in EphB2 null versus wild-type littermates following repeated
exposure to morphine (analysis 60 minutes following injection on day 7). Error bars: ±SEM.
80

and time [F(2,20)=2.48, p>0.05], with no significant interaction between these factors
[F(2,20)=1.79, p>0.05]. Analysis of M3G brain levels using two-way ANOVA revealed no
significant effect of both genotype [F(1,10)=0.40, p>0.05], and time [F(2,20)=6.10, p>0.05],
with no significant interaction between these factors [F(2,20)=0.18, p>0.05]. Analysis of
morphine blood levels using two-way ANOVA revealed no significant effect of genotype
[F(1,10)=2.83, p>0.05], but a significant effect of time [F(2,20)=14.18, p<0.05], however no
significant interaction between these factors [F(2,20)=4.06, p>0.05]. Analysis of M3G blood
levels using two-way ANOVA also revealed no significant effect of genotype [F(1,10)=4.14,
p>0.05], but a significant effect of time [F(2,20)=14.42, p<0.05], however no significant
interaction between these factors [F(2,20)=3.21, p>0.05].
3.4 EphB2 null mice display deficits in hippocampal learning.
Previous work in the lab (Ashlin Kanawaty, Thesis 2011) demonstrated that EphB2 null
mice exhibit significantly reduced transfer latency times compared to wild type controls in the
task of learned passive avoidance. In this task, animals experience single-pass training to an
aversive stimulus in a darkened chamber. Wild-type animals exhibited similar average transfer
latency times during acquisition as EphB2 null mice (Figure 3.8). However, following single
pass training, wild-type animals exhibited an expected increase in transfer latency times
indicating a level of learning had occurred. In contrast, EphB2 null mice exhibited significantly
lower transfer latency times into the dark chamber following single‐pass training relative to
littermate controls. These data support the contention that EphB2 null mice exhibit a deficit in
hippocampal-dependent tasks.
As previously mentioned, EphB2 null animals display deficits in NMDA-dependent
synaptic plasticity in the hippocampus as a result of reduced LTP in CA1 neurons of the
hippocampus. Therefore, to determine if the deficiencies in hippocampal learning observed in
81
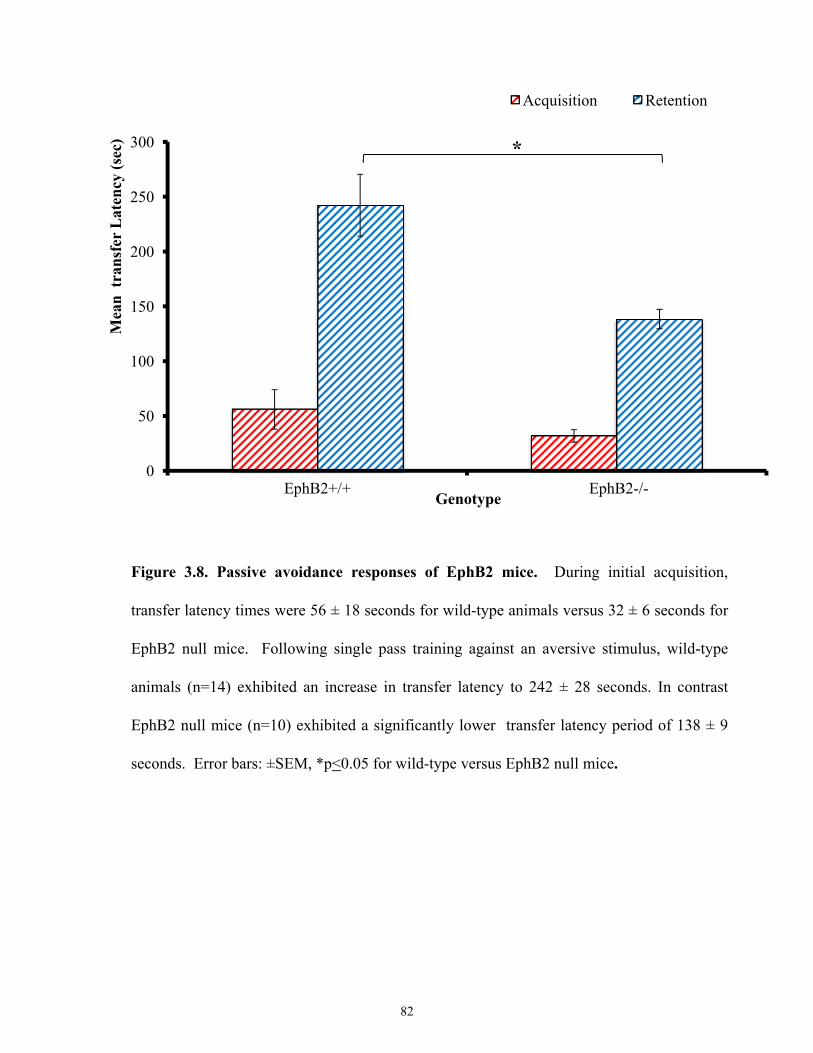
0
50
100
150
200
250
300
EphB2+/+ EphB2-/-
Mea
n tr
ansf
er L
aten
cy (s
ec)
Genotype
Acquisition Retention
*
Figure 3.8. Passive avoidance responses of EphB2 mice. During initial acquisition,
transfer latency times were 56 ± 18 seconds for wild-type animals versus 32 ± 6 seconds for
EphB2 null mice. Following single pass training against an aversive stimulus, wild-type
animals (n=14) exhibited an increase in transfer latency to 242 ± 28 seconds. In contrast
EphB2 null mice (n=10) exhibited a significantly lower transfer latency period of 138 ± 9
seconds. Error bars: ±SEM, *p<0.05 for wild-type versus EphB2 null mice.
82

EphB2 null mice are directly related to the changes in morphine-dependent behavior seen in
these animals, I performed focal bilateral lesions of the dorsal hippocampus in wild-type mice in
an attempt to duplicate the effects seen following the ablation of EphB2. An example of such
lesion is shown in Figure 3.9. Lesions were localized to the dorsal hippocampus and serial
thionin stained sections showing the extent of these lesions is shown in Figure 3.10. Following
surgical recovery, animals were examined in a series of behavioral tests and compared with the
behavioural responses seen in EphB2 null mice.
3.4.1 Behavioural assessments of animals with bilateral electrolytic lesions in the dorsal
hippocampus
We investigated whether lesioned animals displayed altered responsiveness to learned
behaviours using the learned passive avoidance test. The passive avoidance task tests an
animal’s ability to learn the contextual cue (dark compartment) is paired with an aversive
stimulus (foot-shock). As shown in Figure 3.11, sham animals exhibited average transfer latency
times of 9.5±1.3 seconds in the acquisition phase, while lesioned animals had a latency time of
15±1 seconds. Following single pass training sham animals exhibited an expected increase in
transfer latency times to 84±23 seconds, indicating a level of learning had occurred. In contrast,
lesioned animals exhibited significantly lower transfer latency times into the dark chamber
following single‐pass training relative to sham animals (39.5±9 seconds). This is the same
pattern that was observed between EphB2 null and wild-type mice.
EphB2 null mice due to impaired hippocampal learning may be unable to habituate
quickly upon exposure to a novel environment. Thus, we also measured spontaneous
exploratory activity of animals upon placement into a novel environment using the Activity
Monitor. As shown in Figures 3.12-3.13, both EphB2 null and wild-type animals display
elevated levels of exploratory activity that decreased overtime throughout continuous exposure
83
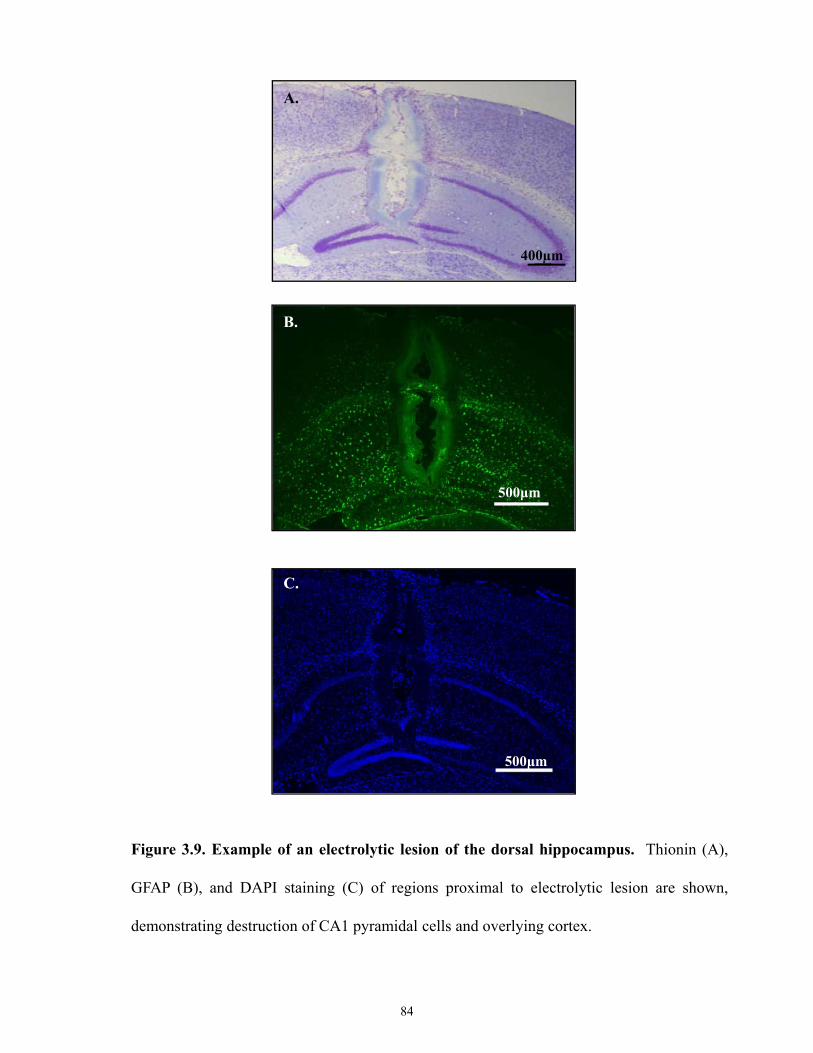
Figure 3.9. Example of an electrolytic lesion of the dorsal hippocampus. Thionin (A),
GFAP (B), and DAPI staining (C) of regions proximal to electrolytic lesion are shown,
demonstrating destruction of CA1 pyramidal cells and overlying cortex.
400µm
500μm
500μm
A.
B.
C.
84

Figure 3.10. Serial views of electrolytic lesions of the dorsal hippocampus. Shown are
serial thionin stained sections taken at 200 µm intervals from (A) a non-stimulated control
hippocampus, and (B) a hippocampus containing a typical electrolytic lesion. Lesions were
generated bilaterally.
1mm
1mm
A.
B.
85
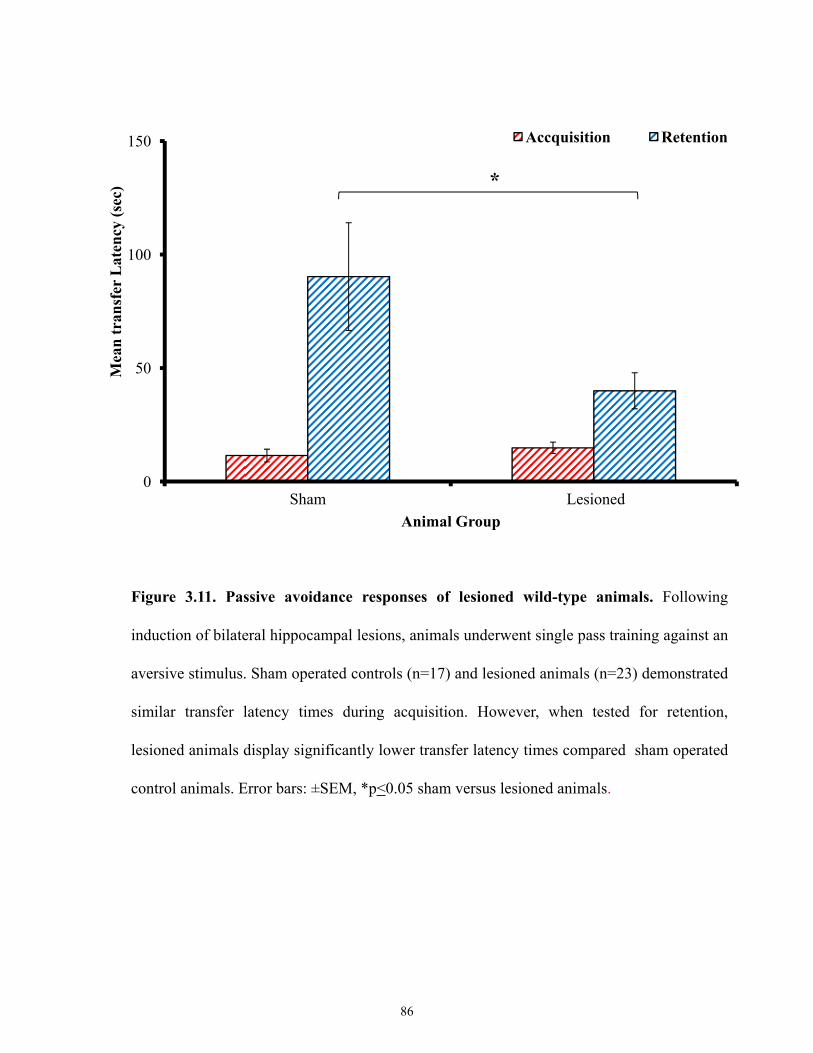
0
50
100
150
Sham Lesioned
Mea
n tr
ansf
er L
aten
cy (s
ec)
Animal Group
Accquisition Retention
*
Figure 3.11. Passive avoidance responses of lesioned wild-type animals. Following
induction of bilateral hippocampal lesions, animals underwent single pass training against an
aversive stimulus. Sham operated controls (n=17) and lesioned animals (n=23) demonstrated
similar transfer latency times during acquisition. However, when tested for retention,
lesioned animals display significantly lower transfer latency times compared sham operated
control animals. Error bars: ±SEM, *p<0.05 sham versus lesioned animals.
86

Figure 3.12. Active time of EphB2 null mice and lesioned wild-types. Spontaneous
activity as measured by mean active time per five minute period, was monitored for 60
minutes following introduction to a novel environment. (A) EphB2 null mice versus wild-
type littermates, (B) lesioned animals versus sham operated controls. Both EphB2 null mice
and lesioned wild-types exhibit higher levels of activity compared to wild-type controls.
Error bars: ±SEM, *p<0.05, comparison of control versus experimental animals.
87
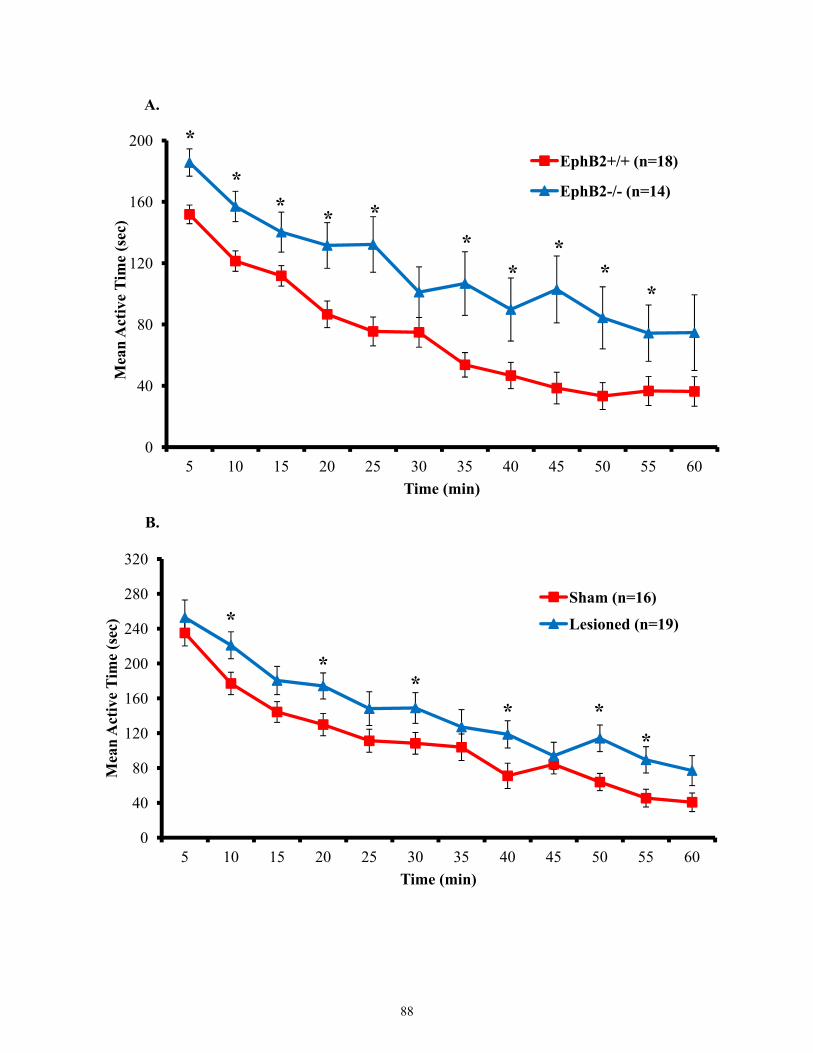
0
40
80
120
160
200
5 10 15 20 25 30 35 40 45 50 55 60
Mea
n A
ctiv
e Ti
me
(sec
)
Time (min)
EphB2+/+ (n=18)
EphB2-/- (n=14)
*
** * *
*
**
**
0
40
80
120
160
200
240
280
320
5 10 15 20 25 30 35 40 45 50 55 60
Mea
n A
ctiv
e Ti
me
(sec
)
Time (min)
Sham (n=16)
Lesioned (n=19)*
**
**
*
A.
B.
88

Figure 3.13. Motor activity of EphB2 null mice and lesioned wild-types. Motor activity
as defined by mean distance traveled per five minute period, was monitored for 60 minutes
following introduction to a novel environment. (A) EphB2 null mice versus wild-type
littermates, (B) lesioned animals versus sham operated controls. Both EphB2 null mice and
lesioned wild-types exhibit greater levels of distance traveled compared to wild-type
controls. Error bars: ±SEM, *p<0.05, comparison of control versus experimental animals.
89
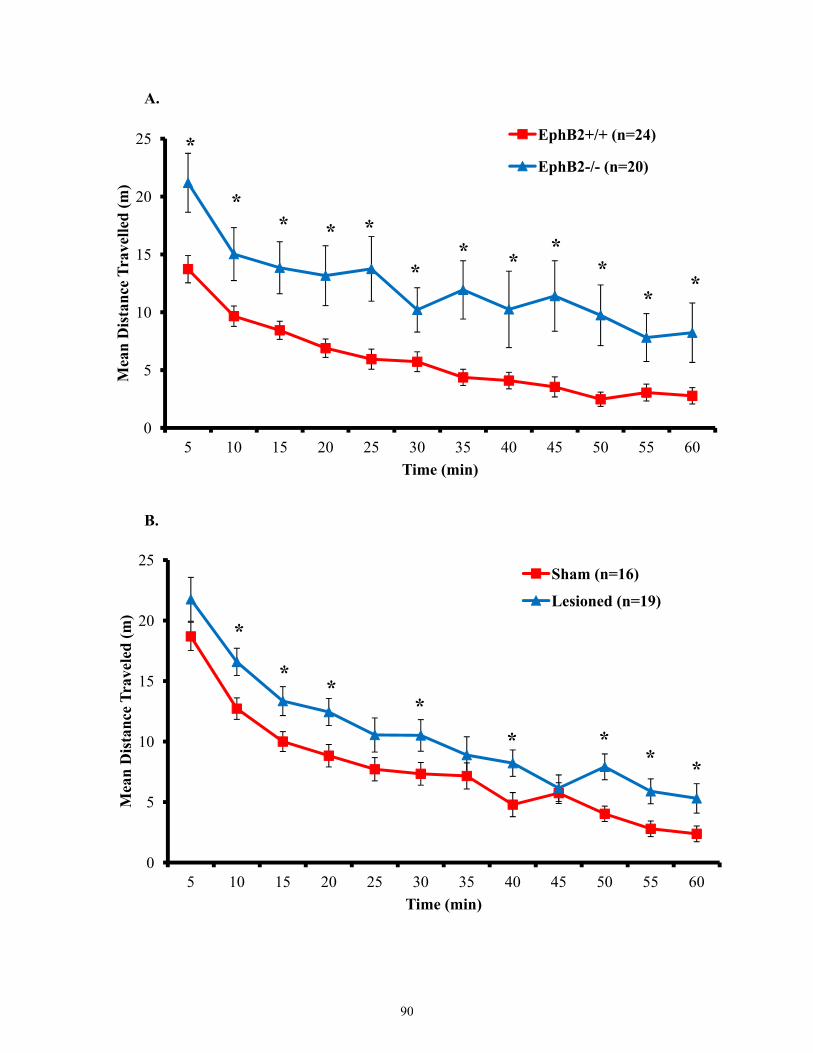
0
5
10
15
20
25
5 10 15 20 25 30 35 40 45 50 55 60
Mea
n D
ista
nce
Trav
elle
d (m
)
Time (min)
EphB2+/+ (n=24)
EphB2-/- (n=20)*
** * *
** *
**
**
0
5
10
15
20
25
5 10 15 20 25 30 35 40 45 50 55 60
Mea
n D
ista
nce
Trav
eled
(m)
Time (min)
Sham (n=16)
Lesioned (n=19)
*
**
* ** *
*
A.
B.
90

of the novel environment. However, EphB2 null mice display significantly elevated exploratory
activity following introduction into the novel environment compared to wild type controls.
Therefore, we investigated wither lesioned animals also displayed the same pattern of
exploratory activity as EphB2 null mice. As shown in Figures 3.12-3.13 both lesioned and sham
operated control animals display elevated exploratory activity that decreased overtime.
However, lesioned animals like the EphB2 null animals displayed elevated spontaneous
exploratory activity compared to sham operated controls. These results indicate that bilateral
electrolytic lesions of the dorsal hippocampus of wild-type animals display strikingly similar
behavioural responses seen in EphB2 null mice compared to sham operated controls.
3.5 Morphine related responses in animals with bilateral electrolytic lesions in the dorsal
hippocampus
We have shown that both EphB2 null mice and animals with bilateral electrolytic lesions
in the dorsal hippocampus exhibit similar behavioural responses. Thus, this suggests EphB2 null
possess deficits in hippocampal learning. EphB2 null mice also displayed accelerated morphine
tolerance and failed to display enhanced analgesic response in a novel environment following
development of morphine tolerance. Thus, we were interested in examining whether
impairments in hippocampal learning had an effect on morphine-specific learning. Multiple
studies have reported that morphine induces hyperactivity known as a running fit in multiple
strains of mice [206, 207]. Therefore, we examined the spontaneous motor activity of EphB2
null and wild-type mice upon first time exposure to morphine. We observed that both EphB2
null and wild-type mice both display morphine induced hyperactivity about 25min post
morphine injection. The key difference in this observable running fit is the duration. EphB2
wild-type mice display sustained morphine induced hyperactivity throughout the duration of the
Activity Monitor recording (Figure 3.14), while EphB2 null mice display morphine induced
91

Figure 3.14. Morphine induced hyperactivity in wild-type mice. Shown are measures of
spontaneous activity 24 hours prior to (left panel) and immediately following (right panel)
morphine injection. Arrow represents time of morphine injection. Dashed red line represents
averaged morphine-induced activity during first 15 minutes of observation. Dashed black
line represents average of motor activity during final 15 minutes of observation period 24
hours prior to morphine injection. Note morphine-induced motor hyperactivity is maintained
throughout the 60 minute observation period. N≥15, Error bars +SEM, *p≤0.05 compared to
baseline values (dashed black line).
92
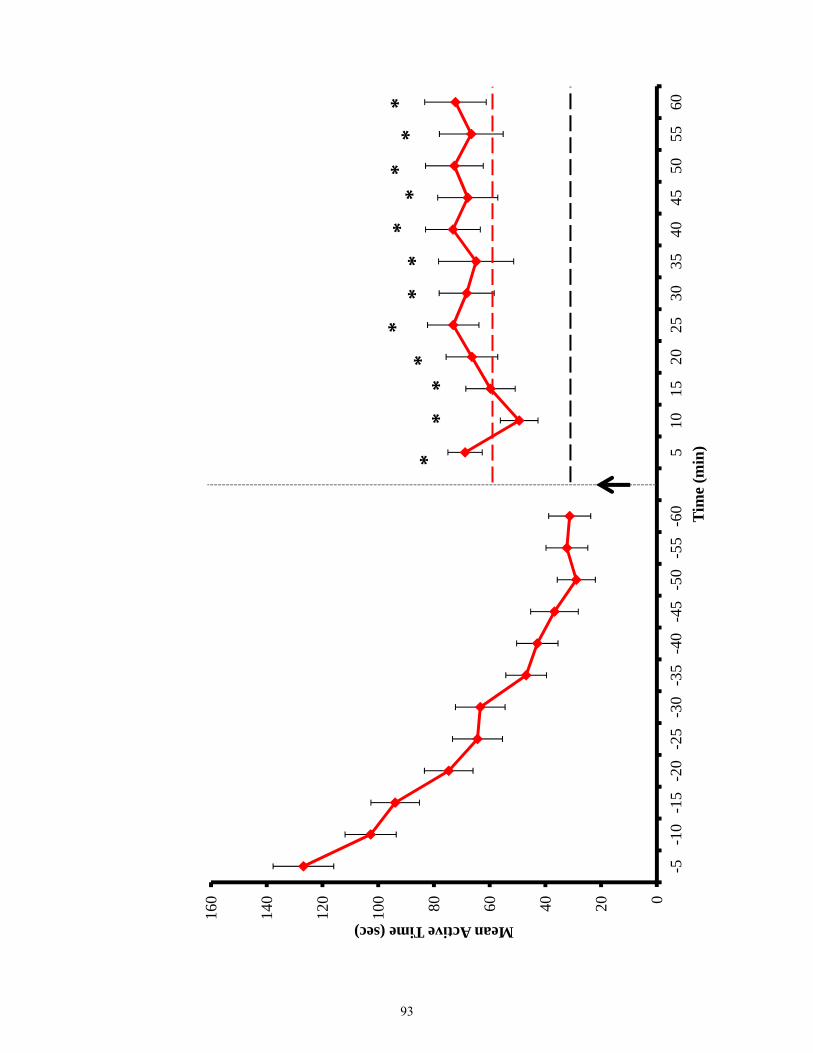
020406080100
120
140
160
-5-1
0-1
5-2
0-2
5-3
0-3
5-4
0-4
5-5
0-5
5-6
05
1015
2025
3035
4045
5055
60
Mean Active Time (sec)
Tim
e (m
in)
**
**
**
**
**
**
93

hyperactivity for about 10min but then the elevated spontaneous motor activity decays overtime
as seen in the non-morphine state (Figure 3.15). As a control, we injected animals with saline
and this did not illicit the morphine induced hyperactivity behaviour, thus confirming the effect
we observed was due to morphine (Figure 3.16).
We also examined the morphine induced hyperactivity for our lesioned and sham
operated control animals. As shown in Figures 3.17-3.18, we observed that both our sham and
lesioned animals display morphine induced hyperactivity about 20 and 25min post morphine
injection, respectively. The difference once again is the duration. Sham animals like wild-type
animals display relatively sustained morphine induced hyperactivity throughout the duration of
the Activity Monitor recording (Figure 3.17). In contrast, lesioned animals like the EphB2 null
mice display morphine induced hyperactivity for about 15min and then the elevated spontaneous
motor activity starts to decline overtime as seen in the non-morphine state (Figure 3.18).
As indicated above, unlike wild-type animals EphB2 null mice do not display enhanced
morphine dependent analgesic response following transfer to a novel environment following
development of morphine tolerization on day 7. To determine whether these findings were
hippocampal dependent, and a result of impairment in learning and memory, we performed the
same study in our lesioned and sham operated control animals. Following twice daily injection
of morphine for 6 days, on day 7 animals were divided and injected in either the same
environment or a novel environment and tested for their antinociceptive response using tail
flick. Although, antinociceptive responses of EphB2 animals were tested using tail pinch and
tail flick (data not shown) assays, lesioned and sham operated control animals were only tested
using tail flick assay. Perhaps due to differences in the genetic background between EphB2 null
mice and lesioned animals, we were unable to obtain robust response measurements using the
tail pinch assay, thus we only measured analgesic responses using the tail flick assay. Consistent
with previous results, lesioned animals like EphB2 null mice, failed to display enhanced
94

Figure 3.15. Morphine induced hyperactivity in EphB2 null mice. Shown are measures
of spontaneous activity 24 hours prior to (left panel) and immediately following (right panel)
morphine injection. Arrow represents time of morphine injection. Dashed blue line
represents averaged morphine-induced activity during first 15 minutes of observation.
Dashed black line represents average of motor activity during final 15 minutes of
observation period 24 hours prior to morphine injection. Morphine-induced motor
hyperactivity is only maintained transiently during the observation period. N≥13, Error bars
+SEM,*p≤0.05 compared to baseline values (dashed black line).
95
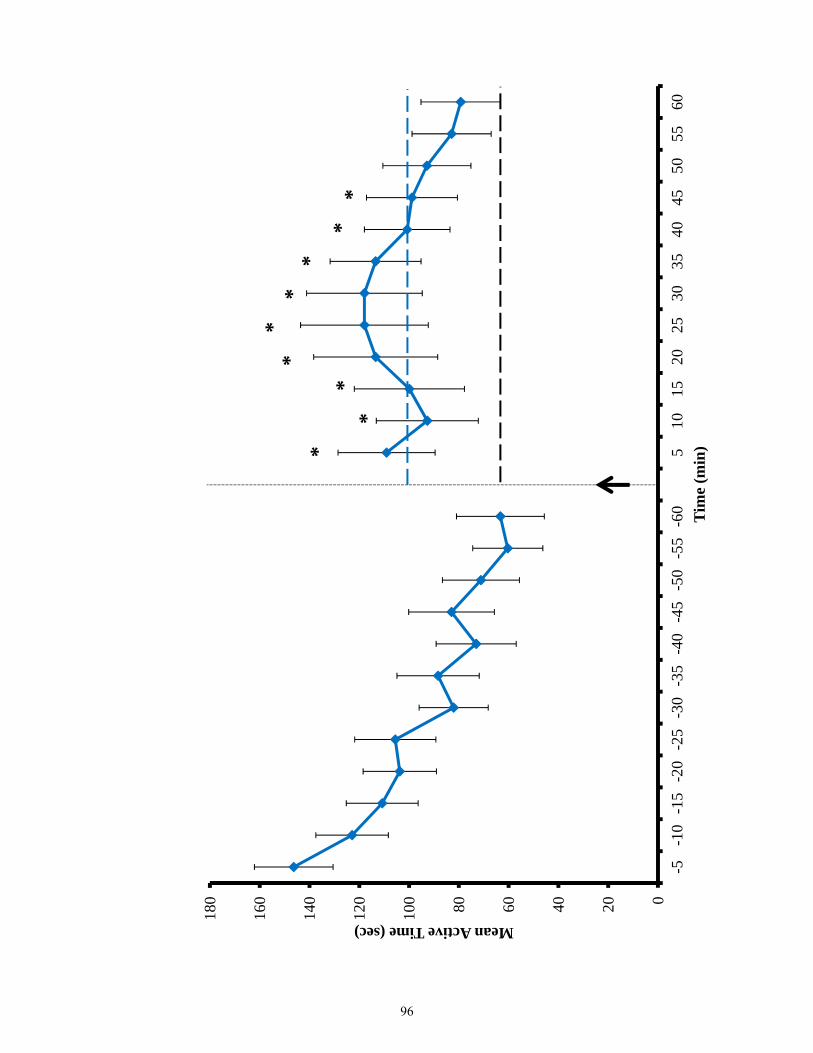
020406080100
120
140
160
180
-5-1
0-1
5-2
0-2
5-3
0-3
5-4
0-4
5-5
0-5
5-6
05
1015
2025
3035
4045
5055
60
Mean Active Time (sec)
Tim
e (m
in)
**
**
**
**
*
96

Figure 3.16. Spontaneous motor activity of saline injected animals. Shown are measures
of spontaneous activity 24 hours prior to (left panel) and immediately following (right panel)
saline injection. Arrow represents time of saline injection. Dashed yellow line represents
averaged activity during first 15 minutes of observation. Dashed black line represents
average of motor activity during final 15 minutes of observation period 24 hours prior to
saline injection. Standard depression in exploratory activity is observed. N≥11, Error bars
+SEM, *p≤0.05 compared to baseline values (dashed black line).
97
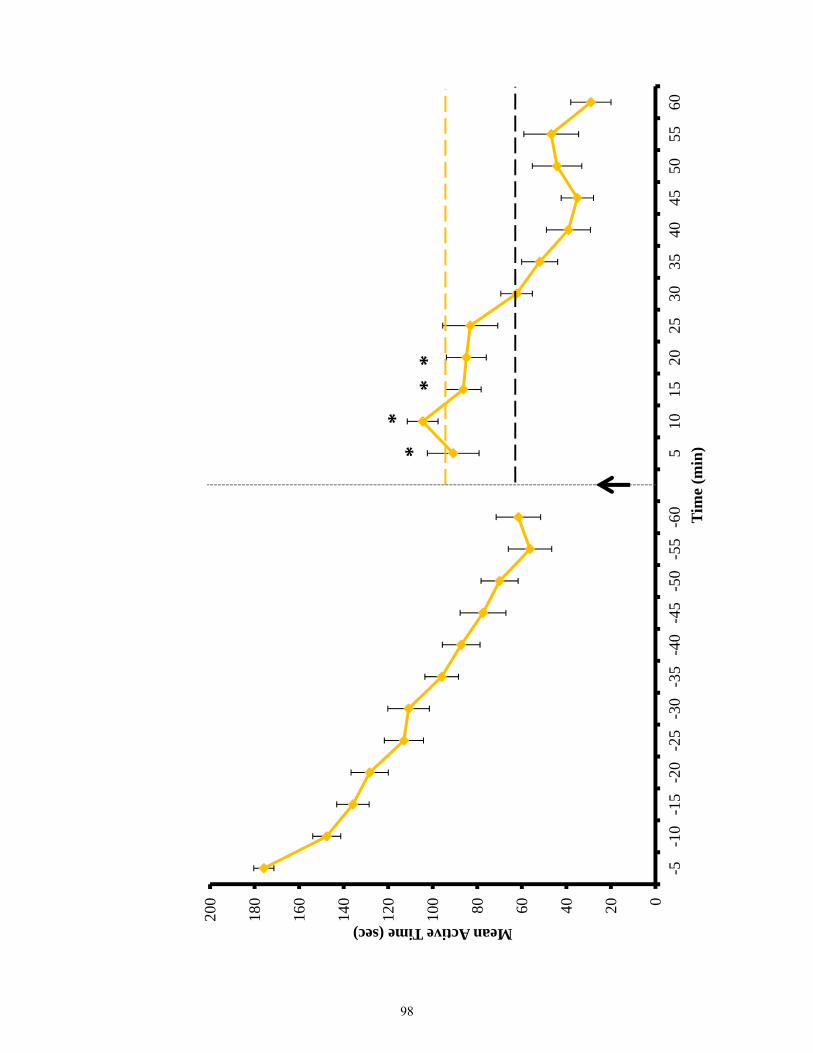
020406080100
120
140
160
180
200
-5-1
0-1
5-2
0-2
5-3
0-3
5-4
0-4
5-5
0-5
5-6
05
1015
2025
3035
4045
5055
60
Mean Active Time (sec)
Tim
e (m
in)*
**
*
98

Figure 3.17. Morphine induced hyperactivity in sham operated control animals. Shown
are measures of spontaneous activity 24 hours prior to (left panel) and immediately
following (right panel) morphine injection. Arrow represents time of morphine injection.
Dashed red line represents averaged morphine-induced activity during first 15 minutes of
observation. Dashed black line represents average of motor activity during final 15 minutes
of observation period 24 hours prior to morphine injection. Note morphine-induced motor
hyperactivity is maintained throughout the 60 minute observation period. N≥7, Error bars
+SEM, *p≤0.05 compared to baseline values (dashed black line).
99
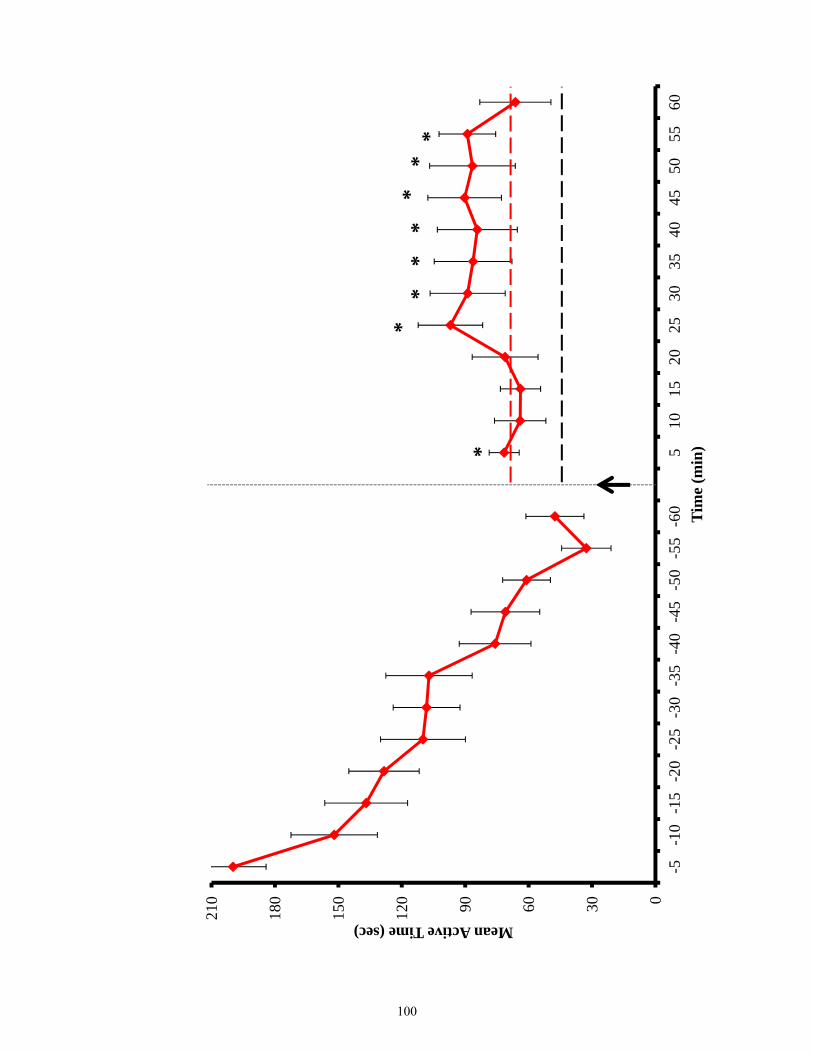
0306090120
150
180
210
-5-1
0-1
5-2
0-2
5-3
0-3
5-4
0-4
5-5
0-5
5-6
05
1015
2025
3035
4045
5055
60
Mean Active Time (sec)
Tim
e (m
in)
**
**
**
*
*
100

Figure 3.18 . Morphine induced hyperactivity in lesioned animals. Shown are measures
of spontaneous activity 24 hours prior to (left panel) and immediately following (right panel)
morphine injection. Arrow represents time of morphine injection. Dashed blue line
represents averaged morphine-induced activity during first 15 minutes of observation.
Dashed black line represents average of motor activity during final 15 minutes of
observation period 24 hours prior to morphine injection. Morphine-induced motor
hyperactivity is only maintained transiently during the observation period. N≥8, Error bars
+SEM,*p≤0.05 compared to baseline values (dashed black line).
101
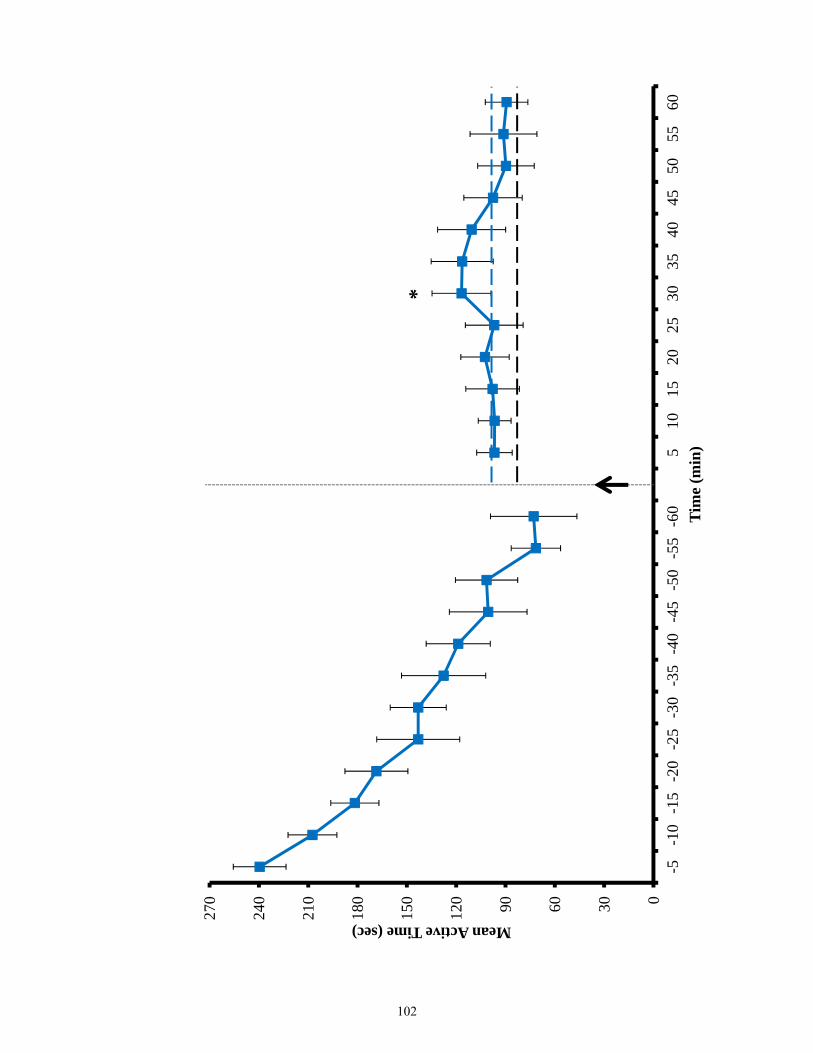
0306090120
150
180
210
240
270
-5-1
0-1
5-2
0-2
5-3
0-3
5-4
0-4
5-5
0-5
5-6
05
1015
2025
3035
4045
5055
60
Mean Active Time (sec)
Tim
e (m
in)
*
102

analgesic response upon transfer to novel environment following development of morphine
tolerization on day 7 (Figure 3.19). In contrast, sham operated controls like EphB2 wild-type
animals displayed some enhanced analgesic response upon transfer to a novel environment
(Figure 3.19). Thus, animals with hippocampal lesions localized to the dorsal hippocampus
mimicked EphB2 null morphine related responses. This suggests that EphB2 null mice as a
result of hippocampal learning deficits, lack the ability to integrate these sensory-dependent
environmental cues associated with morphine exposure.
103
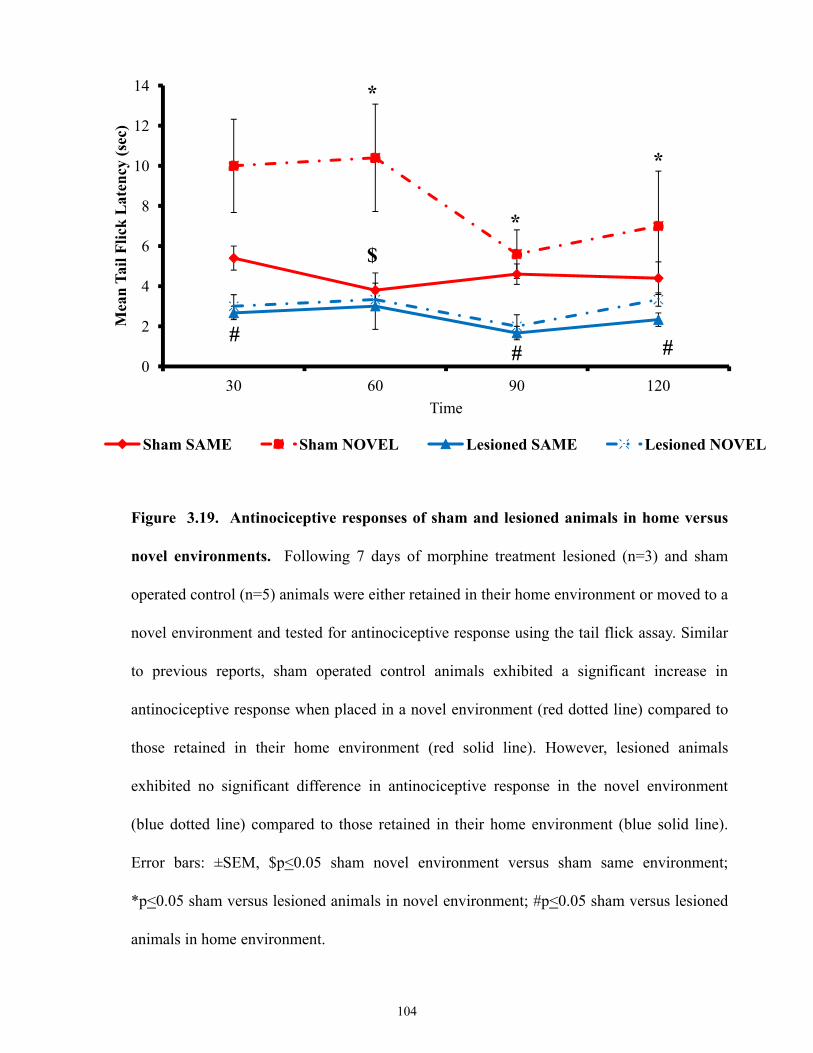
Figure 3.19. Antinociceptive responses of sham and lesioned animals in home versus
novel environments. Following 7 days of morphine treatment lesioned (n=3) and sham
operated control (n=5) animals were either retained in their home environment or moved to a
novel environment and tested for antinociceptive response using the tail flick assay. Similar
to previous reports, sham operated control animals exhibited a significant increase in
antinociceptive response when placed in a novel environment (red dotted line) compared to
those retained in their home environment (red solid line). However, lesioned animals
exhibited no significant difference in antinociceptive response in the novel environment
(blue dotted line) compared to those retained in their home environment (blue solid line).
Error bars: ±SEM, $p<0.05 sham novel environment versus sham same environment;
*p<0.05 sham versus lesioned animals in novel environment; #p<0.05 sham versus lesioned
animals in home environment.
0
2
4
6
8
10
12
14
30 60 90 120
Mea
n Ta
il Fl
ick
Lat
ency
(sec
)
Time
Sham SAME Sham NOVEL Lesioned SAME Lesioned NOVEL
$
*
*
*
## #
104

CHAPTER 4: DISCUSSION
105

4.1 Changes in morphine responsiveness seen in EphB2-null mice are mediated via
modification of cortical influences on sensory function.
EphB2 receptors are persistently expressed within the adult CNS at sites involved in
sensation and processing of sensory stimuli. Although the expression of EphB2 typically
diminishes at a given site following the establishment of primary synaptic connections, EphB2
expression does remerge at several locations following late embryogenesis [150]. Specifically,
EphB2 expression remerges at sites which undergo significant activity-dependent modification
including the hippocampus, amygdala, thalamus, and cingulate cortex [150]. Within the murine
spinal cord, EphB2 expression also undergoes a significant shift during the late embryonic/early
postnatal period, becoming prominently expressed within sensory ganglia, dorsal rexed laminae
and ascending sensory pathways [150]. Despite this, EphB2 null mice display no significant
differences in baseline motor or sensory function compared to control littermates, and EphB2
null mutants displayed similar analgesic responses to littermate controls upon initial exposure to
morphine. However over time, EphB2 null mutants exhibit a substantially accelerated pattern of
morphine tolerance compared to wild-type controls. This suggests that loss of EphB2 may alter
the rate of morphine metabolism, or alter the availability of morphine binding to MORs, or alter
the extent of MOR signaling, or modify some central response regulating this form of sensory
input.
The primary site of morphine metabolism is the liver. However, EphB2 is never
expressed in the liver during developmental or adulthood. Nevertheless some degree of
morphine metabolism is known to occur locally within the CNS. Thus to investigate and
confirm loss of EphB2 does not alter morphine metabolism, we examined levels of morphine
and its metabolite M3G in the blood and brain of EphB2 null and control littermates using LC-
MS/MS. In depth pharmacokinetic studies of morphine in mice are few, and few studies have
used LC-MS/MS for quantification. However, our values are within range of what has
106

previously been reported [208, 209]. The results of both naïve and morphine-tolerized mice
demonstrated that the rates of morphine metabolism are not significantly altered in the absence
of EphB2. The metabolite M3G is inactive and not believed to bind to MORs or illicit analgesic
response [56]. M3G does however represent the main metabolite of morphine, with about 60%
of morphine metabolized to this compound. In the brain, morphine and M3G levels were found
not to differ significantly between EphB2 null and control littermates at all time points
measured. Similarly in whole blood, morphine and M3G concentrations were found not to differ
significantly between EphB2 null and control animals at all time points; excluding at 30
minutes. At this time point, EphB2 null mutants appear to exhibit significantly higher
concentrations of morphine and M3G (differences within the brain at this time point were not
statistically significant). These differences at 30min may be attributed to higher variability
observed following the initial distribution of morphine injected i.p. As well, it appears unrelated
to the previous functional findings observed in EphB2 null mice. EphB2 null mice would if
anything be expected to exhibit lower levels of morphine in the brain and whole blood since
they display significantly lower tail pinch latencies compared to wild-type mice. As well, two-
way ANOVA analysis of the data indicated no statistical effect of genotype is present. Morphine
and M3G concentrations were also quantified following repeated exposure of morphine for 7
days. As expected, EphB2 null mice displayed no significant difference in morphine or M3G
concentration in the blood and brain compared to wild-type controls. This indicates morphine
tolerance data observed for day 7 in EphB2 null mice is independent of differences in morphine
metabolism, but perhaps due to modification of central response. All together, loss of EphB2
receptors does not appear to significantly alter morphine metabolism in mice.
With respect to loss of EphB2 altering the extent of mu opiate receptor binding or
capacity, previous studies performed in the laboratory have determined that affinity and total
binding capacity of mu opioid receptors is unaltered in EphB2 null mice within the tissues
107

examined (spinal cord, and superior colliculus) (Ashlin Kanawaty, Thesis 2011).
Immunohistological studies have previously examined MOR expression and distribution in
rodents, within various regions of the brain such as the cortex, caudate-putamen, hippocampus,
superior/inferior colliculus, and dorsal horn of the spinal cord [210, 211]. We also examined and
demonstrated that the distribution of MORs within the nervous system (lumbar dorsal root
ganglia, spinal cord, superior colliculus, and striatum) is unaltered in EphB2 null mice compared
to littermate controls. Within the spinal cord, EphB2 null mutants and control littermates exhibit
similar laminar distributions of MORs within the dorsal laminae I-III. These positions were
verified by previous work in the lab using known markers such as IB4 or CGRP (Ashlin
Kanawaty, Thesis 2011). We also examined the striatum; since EphB2 receptors are not
expressed in the striatum this region of the brain served as a good control. Within the striatum,
the distribution of mu opioid receptors within the striatal and matrix compartments examined,
demonstrated no significant morphologic differences between EphB2 null and control
littermates. We hypothesized that our morphine tolerance observations may be hippocampal
dependent (see below). Thus we examined MOR distribution within the hippocampus, where no
difference in distribution was observed between EphB2 null and control littermates. Overall, no
morphological difference was observed in the distribution of MORs between EphB2 null and
wild-types at all CNS regions examined. Thus, loss of EphB2 receptors did not significantly
alter MOR distribution and expression in the CNS.
Together, these aforementioned findings indicate loss of EphB2 does not alter rate of
morphine metabolism or affect MOR distribution in the CNS. Thus the action of EphB2 to alter
morphine-dependent signaling may be mediated through higher-order influences that modify
morphine dependent contextual learning (see below).
108

4.2 EphB2 null mice exhibit deficiencies in contextual learning similar to that seen in wild-
type animals containing bilateral electrolytic lesions of the dorsal hippocampus.
The hippocampus plays a central role in learning and memory. Numerous lesion studies
in a variety of mammalian systems have demonstrated the important role of the hippocampus in
formation of new episodic memories, short-term memory consolidation, and cognitive features
involved in learning to novel environmental stimuli [170, 192, 200, 212-215]. However, the
hippocampus is not a uniform structure [187, 189, 194]. Localized lesions have shown that
different sub-regions of the hippocampus mediate different forms of learning and memory.
Studies comparing dorsal hippocampal lesions to ventral hippocampal lesions have shown that
the dorsal hippocampus is important for processing of spatial memory and learning of
conceptual cues; whereas the ventral hippocampus is involved with stress, fear and emotional
aspects of memory [187, 190, 191, 194, 216, 217]. Physiologically, the phenomena of long-term
potentiation (LTP) and long-term depression (LTD), along with NMDA signaling are thought to
currently represent our best cellular and molecular models of the processes involved in
acquisition and consolidation of learned behavior [183, 218, 219]. Incoming sensory
information arising from primary centers in the CNS are subsequently transferred to higher
cortical stations [220]. Such incoming information is subject to consolidation and processing
within the hippocampus, which allows contextual modification of sensory experiences [221,
222]. Therefore, alterations in the consolidation or processing of spatial and/or sensory-related
memories within the hippocampus can result in significant modification of learned behaviors.
Previous work by Siegel and colleagues [203-205, 223] have demonstrated that context
dependent cues and Pavlovian conditioning exert significant functional influences over the
strength of morphine-dependent analgesia and tolerance. Re-examination of Siegel's context
dependent paradigm of morphine tolerance in EphB2 null mutants and littermate controls
demonstrated findings similar to those previously published. Morphine tolerized wild-type
109

animals demonstrated an apparent enhancement in morphine analgesia following the transfer to
a novel environment. In contrast, morphine tolerized EphB2 null mice demonstrated a complete
inhibition of response in this context dependent paradigm of learning. This suggests that EphB2
null mice exhibit impairment in morphine-dependent contextual learning. This is consistent with
previously published results by our lab and others, that found EphB2 null mice exhibit both an
attenuation in the magnitude and stability of hippocampal LTP, compared to littermate controls
[150, 157]. These effects on LTP have been demonstrated to be due to EphB2 influences on the
relative synaptic localization and lifetime of NMDA receptors on the postsynaptic membrane of
hippocampal CA1 neurons [150, 157]. Thus, we investigated whether the defects in contextual
learning observed in EphB2 null mice were in fact dependent upon an inhibition of dorsal
hippocampal function, by performing bilateral electrolytic lesions to the rostral component of
the dorsal hippocampus in wild-type mice. We examined the behavioural response of EphB2
null mice and controls in several paradigms of context dependent learning. We compared these
behavioural responses to those of lesioned animals, to determine whether such hippocampal
inhibition could explain the modified morphine responses seen in EphB2 null mice.
One behavioural assessment employed was the passive avoidance, which is a well-
characterized paradigm of context-dependent learning that tests hippocampal learning [200]. It
examines an animal's ability to distinguish between two distinct contextual frameworks [224-
226]. Hippocampal dependence of this task (and others such as the Morris water maze) has been
confirmed using animals with bilateral lesions to the dorsal hippocampus, which demonstrated a
significant attenuated capacity to discriminate between the two contextual cues [224-226].
When examined on this task, EphB2 null mice performed poorly relative to controls, similar to
the performance seen in animals with bilateral lesions to the dorsal hippocampus. In both cases,
EphB2 null and lesioned animals demonstrated significantly lower transfer latency times into
the aversive environment compared to that observed in wild type and sham operated control
110

littermates. These findings suggest that loss of EphB2 impairs contextual/spatial memory in a
hippocampal-dependent manner.
The other behavioural assessment employed was the open field test to measure
exploratory activity upon placement into a novel environment. Studies have suggested the open
field test represents measures of contextual learning, novelty seeking and anxiety [201, 227].
Thus, we have utilized such activity measurements to assess the relative exploratory activity of
EphB2 null mice compared littermate controls. Although, no baseline differences in motor and
sensory performance were observed between EphB2 null and control littermates; EphB2 null
mice exhibit consistently higher levels of exploratory activity compared to wild type littermates.
The inability of EphB2 null mice to habituate to the novel environment in comparison to
controls suggests they may exhibit problems in contextual learning, and/or increased levels of
anxiety. Both such behavioral responses could be linked to problems in hippocampal dependent
learning, consistent with known deficiencies of EphB2 null mice in CA1 dependent LTP. Thus,
upon examination of the bilateral lesioned animals on the open field test, lesioned animals
similar to EphB2 null mice also exhibit higher levels of exploratory activity compared to sham
operated control animals. Taken together, the above findings suggest that EphB2 null mice
exhibit deficiencies in contextual learning which are similar to that seen in animals following
disruption of dorsal hippocampal function.
4.3 Impaired opiate-dependent responses seen in EphB2 null mice arise from
hippocampal-dependent deficiencies in contextual learning.
Several studies have recently examined the role of the EphB-ephrinB signalling in
modulating the perception of neuropathic pain [162, 164, 165, 167, 168], and the role of EphB
family receptors in influencing opiate dependence and withdrawal [167, 168]. In general, these
studies have postulated a model where interruption of the Eph-ephrin signaling by receptors
111

such as EphB1 induces responses similar to that seen following NMDA inhibition (i.e.
impediment in the development of morphine tolerance) [93, 168]. However, loss of EphB2
induces the opposite behavior; actually accelerating the development opiate tolerance.
Therefore, EphB2 plays a role in the regulation of morphine dependent response, distinct from
direct enhancement of NMDA function and signalling. In contrast to published results, EphB2
null mice fail to exhibit enhancement in morphine analgesia following the transfer to a novel
environment compared to littermate controls. As well, consistent with our previous description
of impairments in hippocampal LTP in EphB2 null mice [150, 157], our analysis of contextual
learning in EphB2 null mice also demonstrated impairments in hippocampal dependent learning.
Therefore, we investigated whether EphB2 null mice exhibit impairment in morphine-dependent
contextual learning.
In a morphine naive state, we demonstrated that animals with bilateral dorsal
hippocampal lesions exhibit similar learning behavioural impairments to the EphB2 null
animals. Thus, we were interested in investigating whether opiate-dependent learning was also
hippocampal dependent. Morphine has been shown to induce hyperactivity/ hyperlocomotion
following injection. This morphine induced motor hyperexcitability, termed “running fit,” is
species specific [206, 207]. Not all strains of mice exhibit this behavioral characteristic [206,
207]. EphB2 null mice display normal motor and sensory responses, and their initial response
upon morphine exposure is identical to that seen in wild-type mice. Upon examination of
morphine induced hyperactivity, both EphB2 null and wild-type mice exhibit morphine induced
hyperactivity following initial exposure to morphine. However, despite the similar baseline
responses between EphB2 null and wild-type mice, EphB2 null mice display distinct and
abnormal response to opiates which may be attributed to their hippocampal learning
impairments. For example, although EphB2 null animals displayed morphine induced
hyperactivity, this behaviour was observed for a short period of time before it begins to decay.
112

This in contrast to EphB2 wild-type controls which exhibited morphine induced hyperactivity
for a longer sustained duration. Similarly, in morphine induced state bilateral lesioned animals
display strikingly similar characteristics to EphB2 null mutants, while the sham operated
controls behaved as the wild-types. Interestingly, the decay of morphine induced hyperactivity
observed in EphB2 null mice and lesioned animals is reminiscent of the induction of LTP in
EphB2 null mice. EphB2 null mice induce LTP that is not stable and which decays rapidly
compared to control littermates [150]. As well, the pattern displayed by the EphB2 null animals
and wild-types is consistent with the analgesic pattern of morphine tolerance displayed by the
animals on day 1. On day 1 analysis of morphine tolerance initially both groups of animals
displayed similar analgesic responses, however, EphB2 null mutants quickly display decreased
analgesic response as observed with the morphine induced hyperactivity. The similar behavioral
findings between EphB2 null and lesioned animals suggest EphB2 null mice exhibit impairment
in morphine-dependent contextual learning due to inhibition of dorsal hippocampal processing.
To further investigate EphB2 null mice impairment in morphine-dependent contextual
learning, we performed Siegel's context-dependent paradigm of morphine tolerance in the
animals with bilateral lesions in the dorsal hippocampus and the sham operated controls.
Environmental cues have been shown to be important in predicting the analgesic actions of
morphine following the development of tolerance. That is because opiate tolerance has been
shown to involve a learned response of the association between the systemic actions of an opiate
and the environmental cues. Thus, contextual learning can modulate an animal’s response to a
drug depending on the environment. In contrast, to previously described results [203-205]
lesioned animals that developed morphine tolerance like the EphB2 null mice, failed to display
enhancement in morphine analgesia following the transfer to a novel environment compared to
sham operated controls. All together, the findings suggest that lesioned animals exhibit similar
behavioral responses in morphine naive state, and altered pattern of responses in morphine
113

induced state, similar to those observed of EphB2 null mice. Therefore, this suggests that EphB2
null mice exhibit impairment in hippocampal-dependent learning that fail to undergo contextual
learning of the environmental cues associated with morphine exposure. Hence, both EphB2 null
mice and lesioned animals display impairment in morphine-dependent contextual learning. This
suggests that loss of CA1 neurons in the dorsal hippocampus is important for learning of
contextual cues in opiate-dependent learning paradigms. This is consistent, with previous work
in the lab that demonstrated that loss of EphB2 results in reduced levels of active postsynaptic
NMDA receptor expression in CA1 neurons of the hippocampus, resulting in reduced LTP at
this site within the hippocampus [150, 157]. Thus, EphB2-dependent changes in the
hippocampus may be mediating the observed opiate dependent learning.
4.4. Concluding Remarks & Future Studies
The mechanisms underlying physiologic responses to opiates are complex and varied.
They not only depend on signaling interactions triggered by binding of opiate ligands to their
respective opiate receptors, but also the integration of information from higher-order cortical
centers. Consistent with previous studies, in this study we have shown that contextual cues
associated with exposure to opiates can modulate and exert an influence over physiologic
responses such as sensory analgesia and motor excitability. In the present study, I have tried to
understand the role by which EphB2, a receptor tyrosine kinase, plays in regulating opiate-
dependent function. Despite overlapping expression of EphB2 and MORs at several CNS loci
directly involved in mediating the sensory effects of opiates, EphB2 appears to exert a majority
of its effects by modifying the perception of contextual cues associated with morphine exposure.
The complexity of these contextual associations is highlighted by the fact that loss of EphB2
does not simply inhibit NMDA receptor function. That is because, based on previously
published studies, one might expect the effects of EphB2 ablation to mimic those seen following
114

the infusion of the non-competitive NMDA receptor antagonist, MK-801, which in contrast to
EphB2 actually inhibit the development of morphine tolerance rather than accelerate it [93].
Instead inhibition of EphB2 appears to alter morphine-dependent response at sites such as the
hippocampus, through modification of contextual learning.
The data presented in the current thesis demonstrates an important role for EphB2
signaling in the hippocampus in regulating opiate-dependent learning. Analyses of the EphB2
receptor mutants in which the kinase domain is inactivated further demonstrate that this effect is
dependent upon reverse signaling through EphB2 ligands (unpublished data). Our findings
suggest that that loss of EphB2 receptors does not alter MOR avidity or distribution in the CNS,
nor does it significantly alter morphine metabolism. However, this study is one of many more
studies to come in order to establish a thorough understanding of the physiologic interactions
regulating time-dependent morphine response. Further studies will be required to examine
whether loss of EphB2 alters other aspects of morphine-dependent signaling such as β-arrestin
translocation within primary neurons, or phosphorylation of MORs. With respect to the
hippocampal role of EphB2 in regulating morphine-dependent response, a strong test of this
model would be local inhibition of EphB2 within the hippocampus through means such as
protein-based EphB2 blocking receptor bodies, or via local lentiviral inhibition of EphB2
mRNA using siRNA approaches.
115

References
1. Le Merrer, J., et al., Reward processing by the opioid system in the brain. Physiological reviews, 2009. 89(4): p. 1379-412.
2. McQuay, H., Opioids in pain management. Lancet, 1999. 353(9171): p. 2229-32. 3. Kalant, H., D.M. Grant, and J. Mitchell, Principles of medical pharmacology. 7th
ed2007, Toronto: Saunders Elsevier. xvii, 1022 p. 4. Pert, C.B. and S.H. Snyder, Opiate receptor: demonstration in nervous tissue. Science,
1973. 179(4077): p. 1011-4. 5. Simon, E.J., J.M. Hiller, and I. Edelman, Stereospecific binding of the potent narcotic
analgesic (3H) Etorphine to rat-brain homogenate. Proceedings of the National Academy of Sciences of the United States of America, 1973. 70(7): p. 1947-9.
6. Terenius, L., Characteristics of the "receptor" for narcotic analgesics in synaptic plasma membrane fraction from rat brain. Acta pharmacologica et toxicologica, 1973. 33(5): p. 377-84.
7. Hughes, J., et al., Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature, 1975. 258(5536): p. 577-80.
8. Goldstein, A., Opioid peptides endorphins in pituitary and brain. Science, 1976. 193(4258): p. 1081-6.
9. Goldstein, A., et al., Dynorphin-(1-13), an extraordinarily potent opioid peptide. Proceedings of the National Academy of Sciences of the United States of America, 1979. 76(12): p. 6666-70.
10. Akil, H., et al., Endogenous opioids: biology and function. Annual Review of Neuroscience, 1984. 7: p. 223-55.
11. Martin, W.R., et al., The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. The Journal of pharmacology and experimental therapeutics, 1976. 197(3): p. 517-32.
12. Knapp, R.J., et al., Molecular biology and pharmacology of cloned opioid receptors. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 1995. 9(7): p. 516-25.
13. Quigley, D.I., et al., Orphanin FQ is the major OFQ1-17-containing peptide produced in the rodent and monkey hypothalamus. Peptides, 1998. 19(1): p. 133-9.
14. Bunzow, J.R., et al., Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a mu, delta or kappa opioid receptor type. FEBS letters, 1994. 347(2-3): p. 284-8.
15. Meunier, J.C., et al., Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature, 1995. 377(6549): p. 532-5.
16. Reinscheid, R.K., et al., Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science, 1995. 270(5237): p. 792-4.
17. Thomsen, C. and R. Hohlweg, (8-Naphthalen-1-ylmethyl-4-oxo-1-phenyl-1,3,8-triaza-spiro[4. 5]dec-3-yl)-acetic acid methyl ester (NNC 63-0532) is a novel potent nociceptin receptor agonist. British journal of pharmacology, 2000. 131(5): p. 903-8.
18. Mansour, A., et al., Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends in neurosciences, 1995. 18(1): p. 22-9.
19. Higgins, G.A., et al., Influence of the selective ORL1 receptor agonist, Ro64-6198, on rodent neurological function. Neuropharmacology, 2001. 41(1): p. 97-107.
20. Kieffer, B.L. and C. Gaveriaux-Ruff, Exploring the opioid system by gene knockout. Progress in neurobiology, 2002. 66(5): p. 285-306.
116

21. Alfaras-Melainis, K., et al., Modulation of opioid receptor function by protein-protein interactions. Frontiers in bioscience : a journal and virtual library, 2009. 14: p. 3594-607.
22. Lagerstrom, M.C. and H.B. Schioth, Structural diversity of G protein-coupled receptors and significance for drug discovery. Nature reviews. Drug discovery, 2008. 7(4): p. 339-57.
23. van Rijn, R.M., J.L. Whistler, and M. Waldhoer, Opioid-receptor-heteromer-specific trafficking and pharmacology. Current opinion in pharmacology, 2010. 10(1): p. 73-9.
24. Lord, J.A., et al., Endogenous opioid peptides: multiple agonists and receptors. Nature, 1977. 267(5611): p. 495-9.
25. Kane, B.E., B. Svensson, and D.M. Ferguson, Molecular recognition of opioid receptor ligands. The AAPS journal, 2006. 8(1): p. E126-37.
26. Mansour, A., et al., The cloned mu, delta and kappa receptors and their endogenous ligands: evidence for two opioid peptide recognition cores. Brain research, 1995. 700(1-2): p. 89-98.
27. Chen, Y., et al., Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Molecular Pharmacology, 1993. 44(1): p. 8-12.
28. Wang, J.B., et al., mu opiate receptor: cDNA cloning and expression. Proceedings of the National Academy of Sciences of the United States of America, 1993. 90(21): p. 10230-4.
29. Thompson, R.C., et al., Cloning and pharmacological characterization of a rat mu opioid receptor. Neuron, 1993. 11(5): p. 903-13.
30. Doyle, G.A., et al., Identification of five mouse mu-opioid receptor (MOR) gene (Oprm1) splice variants containing a newly identified alternatively spliced exon. Gene, 2007. 395(1-2): p. 98-107.
31. Pan, Y.X., Diversity and complexity of the mu opioid receptor gene: alternative pre-mRNA splicing and promoters. DNA and cell biology, 2005. 24(11): p. 736-50.
32. Majumdar, S., et al., Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proceedings of the National Academy of Sciences of the United States of America, 2011. 108(49): p. 19778-83.
33. Zimprich, A., T. Simon, and V. Hollt, Cloning and expression of an isoform of the rat mu opioid receptor (rMOR1B) which differs in agonist induced desensitization from rMOR1. FEBS letters, 1995. 359(2-3): p. 142-6.
34. Schulz, S., et al., Immunolocalization of two mu-opioid receptor isoforms (MOR1 and MOR1B) in the rat central nervous system. Neuroscience, 1998. 82(2): p. 613-22.
35. Pan, Y.X., et al., Involvement of exon 11-associated variants of the mu opioid receptor MOR-1 in heroin, but not morphine, actions. Proceedings of the National Academy of Sciences of the United States of America, 2009. 106(12): p. 4917-22.
36. Pan, Y.X., et al., Generation of the mu opioid receptor (MOR-1) protein by three new splice variants of the Oprm gene. Proceedings of the National Academy of Sciences of the United States of America, 2001. 98(24): p. 14084-9.
37. von Zastrow, M., Mechanisms regulating membrane trafficking of G protein-coupled receptors in the endocytic pathway. Life sciences, 2003. 74(2-3): p. 217-24.
38. Manglik, A., et al., Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature, 2012. 485(7398): p. 321-6.
39. Borsodi, A. and G. Toth, Characterization of opioid receptor types and subtypes with new ligands. Annals of the New York Academy of Sciences, 1995. 757: p. 339-52.
117

40. Hollt, V., Opioid peptide processing and receptor selectivity. Annual review of pharmacology and toxicology, 1986. 26: p. 59-77.
41. Garzon, J., et al., Endogenous opioid peptides: comparative evaluation of their receptor affinities in the mouse brain. Life sciences, 1983. 33 Suppl 1: p. 291-4.
42. Lahti, R.A., et al., [3H]U-69593 a highly selective ligand for the opioid kappa receptor. European journal of pharmacology, 1985. 109(2): p. 281-4.
43. Handa, B.K., et al., Analogues of beta-LPH61-64 possessing selective agonist activity at mu-opiate receptors. European journal of pharmacology, 1981. 70(4): p. 531-40.
44. Mosberg, H.I., J.R. Omnaas, and A. Goldstein, Structural requirements for delta opioid receptor binding. Molecular Pharmacology, 1987. 31(6): p. 599-602.
45. Matthes, H.W., et al., Activity of the delta-opioid receptor is partially reduced, whereas activity of the kappa-receptor is maintained in mice lacking the mu-receptor. The Journal of neuroscience : the official journal of the Society for Neuroscience, 1998. 18(18): p. 7285-95.
46. Al-Hasani, R. and M.R. Bruchas, Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology, 2011. 115(6): p. 1363-81.
47. De Gregori, S., et al., Morphine metabolism, transport and brain disposition. Metabolic brain disease, 2012. 27(1): p. 1-5.
48. Ishikawa, K., et al., A sensitive procedure for determination of morphine in mouse whole blood by high performance liquid chromatography with electrochemical detection. Japanese journal of pharmacology, 1982. 32(5): p. 969-71.
49. King, C.D., et al., The glucuronidation of exogenous and endogenous compounds by stably expressed rat and human UDP-glucuronosyltransferase 1.1. Archives of biochemistry and biophysics, 1996. 332(1): p. 92-100.
50. Coffman, B.L., et al., The glucuronidation of opioids, other xenobiotics, and androgens by human UGT2B7Y(268) and UGT2B7H(268). Drug metabolism and disposition: the biological fate of chemicals, 1998. 26(1): p. 73-7.
51. Frolich, N., et al., Distinct pharmacological properties of morphine metabolites at G(i)-protein and beta-arrestin signaling pathways activated by the human mu-opioid receptor. Biochemical pharmacology, 2011. 81(10): p. 1248-54.
52. Xie, R., et al., The role of P-glycoprotein in blood-brain barrier transport of morphine: transcortical microdialysis studies in mdr1a (-/-) and mdr1a (+/+) mice. British journal of pharmacology, 1999. 128(3): p. 563-8.
53. Zelcer, N., et al., Mice lacking multidrug resistance protein 3 show altered morphine pharmacokinetics and morphine-6-glucuronide antinociception. Proceedings of the National Academy of Sciences of the United States of America, 2005. 102(20): p. 7274-9.
54. Handal, M., et al., Morphine-3-glucuronide inhibits morphine induced, but enhances morphine-6-glucuronide induced locomotor activity in mice. Pharmacology, biochemistry, and behavior, 2007. 86(3): p. 576-86.
55. Handal, M., et al., Pharmacokinetic differences of morphine and morphine-glucuronides are reflected in locomotor activity. Pharmacology, biochemistry, and behavior, 2002. 73(4): p. 883-92.
56. Frances, B., et al., Further evidence that morphine-6 beta-glucuronide is a more potent opioid agonist than morphine. The Journal of pharmacology and experimental therapeutics, 1992. 262(1): p. 25-31.
57. Lipkowski, A.W., et al., Morphine-3-glucuronide: silent regulator of morphine actions. Life sciences, 1994. 55(2): p. 149-54.
118

58. Hewett, K., A.H. Dickenson, and H.J. McQuay, Lack of effect of morphine-3-glucuronide on the spinal antinociceptive actions of morphine in the rat: an electrophysiological study. Pain, 1993. 53(1): p. 59-63.
59. Suzuki, Y., et al., Susceptibility to Chronic Infection with Toxoplasma-Gondii Does Not Correlate with Susceptibility to Acute Infection in Mice. Infection and Immunity, 1993. 61(6): p. 2284-2288.
60. Osborne, R., et al., The Analgesic Activity of Morphine-6-Glucuronide. British journal of clinical pharmacology, 1992. 34(2): p. 130-138.
61. Gong, Q.L., et al., Antinociceptive and Ventilatory Effects of the Morphine Metabolites - Morphine-6-Glucuronide and Morphine-3-Glucuronide. European Journal of Pharmacology, 1991. 193(1): p. 47-56.
62. Paul, D., et al., Pharmacological Characterization of Morphine-6-Beta-Glucuronide, a Very Potent Morphine Metabolite. Journal of Pharmacology and Experimental Therapeutics, 1989. 251(2): p. 477-483.
63. Bourasset, F., et al., Evidence for an active transport of morphine-6-beta-d-glucuronide but not P-glycoprotein-mediated at the blood-brain barrier. Journal of neurochemistry, 2003. 86(6): p. 1564-7.
64. Barchfeld, C.C. and F. Medzihradsky, Receptor-mediated stimulation of brain GTPase by opiates in normal and dependent rats. Biochemical and Biophysical Research Communications, 1984. 121(2): p. 641-8.
65. Childers, S.R. and S.H. Snyder, Guanine nucleotides differentiate agonist and antagonist interactions with opiate receptors. Life sciences, 1978. 23(7): p. 759-61.
66. Connor, M. and M.D. Christie, Opioid receptor signalling mechanisms. Clinical and experimental pharmacology & physiology, 1999. 26(7): p. 493-9.
67. Sharma, S.K., W.A. Klee, and M. Nirenberg, Opiate-dependent modulation of adenylate cyclase. Proceedings of the National Academy of Sciences of the United States of America, 1977. 74(8): p. 3365-9.
68. Fields, H., State-dependent opioid control of pain. Nature reviews. Neuroscience, 2004. 5(7): p. 565-75.
69. Law, P.Y., Y.H. Wong, and H.H. Loh, Molecular mechanisms and regulation of opioid receptor signaling. Annual review of pharmacology and toxicology, 2000. 40: p. 389-430.
70. Herz, A., Endogenous opioid systems and alcohol addiction. Psychopharmacology, 1997. 129(2): p. 99-111.
71. Jalabert, M., et al., Neuronal circuits underlying acute morphine action on dopamine neurons. Proceedings of the National Academy of Sciences of the United States of America, 2011. 108(39): p. 16446-50.
72. Johnson, S.W. and R.A. North, Opioids excite dopamine neurons by hyperpolarization of local interneurons. The Journal of neuroscience : the official journal of the Society for Neuroscience, 1992. 12(2): p. 483-8.
73. Connor, M., P.B. Osborne, and M.J. Christie, Mu-opioid receptor desensitization: is morphine different? British journal of pharmacology, 2004. 143(6): p. 685-96.
74. Yu, Y., et al., Mu opioid receptor phosphorylation, desensitization, and ligand efficacy. The Journal of biological chemistry, 1997. 272(46): p. 28869-74.
75. Keith, D.E., et al., mu-Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Molecular Pharmacology, 1998. 53(3): p. 377-84.
119

76. Borgland, S.L., et al., Opioid agonists have different efficacy profiles for G protein activation, rapid desensitization, and endocytosis of mu-opioid receptors. The Journal of biological chemistry, 2003. 278(21): p. 18776-84.
77. Arden, J.R., et al., Phosphorylation and agonist-specific intracellular trafficking of an epitope-tagged mu-opioid receptor expressed in HEK 293 cells. Journal of neurochemistry, 1995. 65(4): p. 1636-45.
78. Finn, A.K. and J.L. Whistler, Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron, 2001. 32(5): p. 829-39.
79. Keith, D.E., et al., Morphine activates opioid receptors without causing their rapid internalization. The Journal of biological chemistry, 1996. 271(32): p. 19021-4.
80. Celver, J., et al., Distinct domains of the mu-opioid receptor control uncoupling and internalization. Molecular Pharmacology, 2004. 65(3): p. 528-37.
81. Zhang, J., et al., Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proceedings of the National Academy of Sciences of the United States of America, 1998. 95(12): p. 7157-62.
82. Lefkowitz, R.J., et al., Mechanisms of beta-adrenergic receptor desensitization and resensitization. Advances in pharmacology, 1998. 42: p. 416-20.
83. Chavkin, C., J.P. McLaughlin, and J.P. Celver, Regulation of opioid receptor function by chronic agonist exposure: constitutive activity and desensitization. Molecular Pharmacology, 2001. 60(1): p. 20-5.
84. Schulz, S., et al., Morphine induces terminal micro-opioid receptor desensitization by sustained phosphorylation of serine-375. The EMBO journal, 2004. 23(16): p. 3282-9.
85. Whistler, J.L. and M. von Zastrow, Morphine-activated opioid receptors elude desensitization by beta-arrestin. Proceedings of the National Academy of Sciences of the United States of America, 1998. 95(17): p. 9914-9.
86. Bohn, L.M., et al., Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science, 1999. 286(5449): p. 2495-8.
87. Bohn, L.M., et al., Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature, 2000. 408(6813): p. 720-3.
88. Raehal, K.M. and L.M. Bohn, The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology, 2011. 60(1): p. 58-65.
89. Salmanzadeh, F., et al., Dependence on morphine impairs the induction of long-term potentiation in the CA1 region of rat hippocampal slices. Brain research, 2003. 965(1-2): p. 108-13.
90. Bao, G., et al., Morphine and heroin differentially modulate in vivo hippocampal LTP in opiate-dependent rat. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology, 2007. 32(8): p. 1738-49.
91. Zhu, H., et al., Region-specific changes in NMDA receptor mRNA induced by chronic morphine treatment are prevented by the co-administration of the competitive NMDA receptor antagonist LY274614. Brain research. Molecular brain research, 2003. 114(2): p. 154-62.
92. Mendez, I.A. and K.A. Trujillo, NMDA receptor antagonists inhibit opiate antinociceptive tolerance and locomotor sensitization in rats. Psychopharmacology, 2008. 196(3): p. 497-509.
93. Trujillo, K.A. and H. Akil, Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science, 1991. 251(4989): p. 85-7.
94. Noda, Y. and T. Nabeshima, Opiate physical dependence and N-methyl-D-aspartate receptors. European journal of pharmacology, 2004. 500(1-3): p. 121-8.
120

95. Ueda, H., M. Inoue, and T. Matsumoto, Protein kinase C-mediated inhibition of mu-opioid receptor internalization and its involvement in the development of acute tolerance to peripheral mu-agonist analgesia. The Journal of neuroscience : the official journal of the Society for Neuroscience, 2001. 21(9): p. 2967-73.
96. Bailey, C.P., E. Kelly, and G. Henderson, Protein kinase C activation enhances morphine-induced rapid desensitization of mu-opioid receptors in mature rat locus ceruleus neurons. Molecular Pharmacology, 2004. 66(6): p. 1592-8.
97. Chen, L. and L.Y. Huang, Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature, 1992. 356(6369): p. 521-3.
98. Chen, L. and L.Y. Huang, Sustained potentiation of NMDA receptor-mediated glutamate responses through activation of protein kinase C by a mu opioid. Neuron, 1991. 7(2): p. 319-26.
99. Schuller, A.G.P., et al., Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nature Neuroscience, 1999. 2(2): p. 151-156.
100. Sora, I., et al., Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proceedings of the National Academy of Sciences of the United States of America, 1997. 94(4): p. 1544-1549.
101. Matthes, H.W.D., et al., Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature, 1996. 383(6603): p. 819-823.
102. Loh, H.H., et al., mu opioid receptor knockout in mice: effects on ligand-induced analgesia and morphine lethality. Molecular Brain Research, 1998. 54(2): p. 321-326.
103. Hirai, H., et al., A novel putative tyrosine kinase receptor encoded by the eph gene. Science, 1987. 238(4834): p. 1717-20.
104. Gale, N.W., et al., Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron, 1996. 17(1): p. 9-19.
105. Unified nomenclature for Eph family receptors and their ligands, the ephrins. Eph Nomenclature Committee. Cell, 1997. 90(3): p. 403-4.
106. Pasquale, E.B., Eph receptor signalling casts a wide net on cell behaviour. Nature reviews. Molecular cell biology, 2005. 6(6): p. 462-75.
107. Murai, K.K. and E.B. Pasquale, 'Eph'ective signaling: forward, reverse and crosstalk. Journal of cell science, 2003. 116(Pt 14): p. 2823-32.
108. Egea, J. and R. Klein, Bidirectional Eph-ephrin signaling during axon guidance. Trends in cell biology, 2007. 17(5): p. 230-8.
109. Davis, S., et al., Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science, 1994. 266(5186): p. 816-9.
110. Himanen, J.P., et al., Repelling class discrimination: ephrin-A5 binds to and activates EphB2 receptor signaling. Nature neuroscience, 2004. 7(5): p. 501-9.
111. Flanagan, J.G. and P. Vanderhaeghen, The ephrins and Eph receptors in neural development. Annual Review of Neuroscience, 1998. 21: p. 309-45.
112. Himanen, J.P., M. Henkemeyer, and D.B. Nikolov, Crystal structure of the ligand-binding domain of the receptor tyrosine kinase EphB2. Nature, 1998. 396(6710): p. 486-91.
113. Labrador, J.P., R. Brambilla, and R. Klein, The N-terminal globular domain of Eph receptors is sufficient for ligand binding and receptor signaling. The EMBO journal, 1997. 16(13): p. 3889-97.
121

114. Wybenga-Groot, L.E., et al., Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell, 2001. 106(6): p. 745-57.
115. Himanen, J.P., N. Saha, and D.B. Nikolov, Cell-cell signaling via Eph receptors and ephrins. Current opinion in cell biology, 2007. 19(5): p. 534-42.
116. Zisch, A.H., et al., Replacing two conserved tyrosines of the EphB2 receptor with glutamic acid prevents binding of SH2 domains without abrogating kinase activity and biological responses. Oncogene, 2000. 19(2): p. 177-87.
117. Holland, S.J., et al., Bidirectional signalling through the EPH-family receptor Nuk and its transmembrane ligands. Nature, 1996. 383(6602): p. 722-5.
118. Bruckner, K., E.B. Pasquale, and R. Klein, Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science, 1997. 275(5306): p. 1640-3.
119. Himanen, J.P. and D.B. Nikolov, Eph signaling: a structural view. Trends in neurosciences, 2003. 26(1): p. 46-51.
120. Smalla, M., et al., Solution structure of the receptor tyrosine kinase EphB2 SAM domain and identification of two distinct homotypic interaction sites. Protein science : a publication of the Protein Society, 1999. 8(10): p. 1954-61.
121. Stapleton, D., et al., The crystal structure of an Eph receptor SAM domain reveals a mechanism for modular dimerization. Nature structural biology, 1999. 6(1): p. 44-9.
122. Zhuang, G., et al., Regulation of EphA2 receptor endocytosis by SHIP2 lipid phosphatase via phosphatidylinositol 3-Kinase-dependent Rac1 activation. The Journal of biological chemistry, 2007. 282(4): p. 2683-94.
123. Hock, B., et al., PDZ-domain-mediated interaction of the Eph-related receptor tyrosine kinase EphB3 and the ras-binding protein AF6 depends on the kinase activity of the receptor. Proceedings of the National Academy of Sciences of the United States of America, 1998. 95(17): p. 9779-84.
124. Pasquale, E.B., Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nature reviews. Cancer, 2010. 10(3): p. 165-80.
125. Nakamoto, M., Eph receptors and ephrins. The international journal of biochemistry & cell biology, 2000. 32(1): p. 7-12.
126. Himanen, J.P., et al., Crystal structure of an Eph receptor-ephrin complex. Nature, 2001. 414(6866): p. 933-8.
127. Lackmann, M., et al., Distinct subdomains of the EphA3 receptor mediate ligand binding and receptor dimerization. Journal of Biological Chemistry, 1998. 273(32): p. 20228-20237.
128. Noren, N.K. and E.B. Pasquale, Eph receptor-ephrin bidirectional signals that target Ras and Rho proteins. Cellular Signalling, 2004. 16(6): p. 655-66.
129. Sahin, M., et al., Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron, 2005. 46(2): p. 191-204.
130. Shamah, S.M., et al., EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell, 2001. 105(2): p. 233-244.
131. Murai, K.K. and E.B. Pasquale, New exchanges in Eph-dependent growth cone dynamics. Neuron, 2005. 46(2): p. 161-163.
132. Irie, F. and Y. Yamaguchi, EphB receptors regulate dendritic spine development via intersectin, Cdc42 and N-WASP. Nature Neuroscience, 2002. 5(11): p. 1117-1118.
133. Penzes, P., et al., Rapid induction of dendritic spine morphogenesis by trans-synaptic EphrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron, 2003. 37(2): p. 263-274.
122

134. Pratt, R.L. and M.S. Kinch, Activation of the EphA2 tyrosine kinase stimulates the MAP/ERK kinase signaling cascade. Oncogene, 2002. 21(50): p. 7690-7699.
135. Tong, J.F., et al., Manipulation of EphB2 regulatory motifs and SH2 binding sites switches MAPK signaling and biological activity. Journal of Biological Chemistry, 2003. 278(8): p. 6111-6119.
136. Elowe, S., et al., Downregulation of the Ras-mitogen-activated protein kinase pathway by the EphB2 receptor tyrosine kinase is required for ephrin-induced neurite retraction. Molecular and Cellular Biology, 2001. 21(21): p. 7429-7441.
137. Palmer, A., et al., EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Molecular Cell, 2002. 9(4): p. 725-737.
138. Tanaka, M., R. Kamata, and R. Sakai, Phosphorylation of ephrin-B1 via the interaction with claudin following cell-cell contact formation. The EMBO journal, 2005. 24(21): p. 3700-11.
139. Ran, X. and J. Song, Structural insight into the binding diversity between the Tyr-phosphorylated human ephrinBs and Nck2 SH2 domain. The Journal of biological chemistry, 2005. 280(19): p. 19205-12.
140. Armstrong, J.N., et al., B-ephrin reverse signaling is required for NMDA-independent long-term potentiation of mossy fibers in the hippocampus. Journal of Neuroscience, 2006. 26(13): p. 3474-3481.
141. Kayser, M.S., et al., Intracellular and trans-synaptic regulation of glutamatergic synaptogenesis by EphB receptors. Journal of Neuroscience, 2006. 26(47): p. 12152-12164.
142. Davy, A., et al., Compartmentalized signaling by GPI-anchored ephrin-A5 requires the Fyn tyrosine kinase to regulate cellular adhesion. Genes & development, 1999. 13(23): p. 3125-35.
143. Hattori, M., M. Osterfield, and J.G. Flanagan, Regulated cleavage of a contact-mediated axon repellent. Science, 2000. 289(5483): p. 1360-1365.
144. Janes, P.W., et al., Adam meets Eph: An ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell, 2005. 123(2): p. 291-304.
145. Zimmer, M., et al., EphB-ephrinB bi-directional endocytosis terminates adhesion allowing contact mediated repulsion. Nature Cell Biology, 2003. 5(10): p. 869-878.
146. Marston, D.J., S. Dickinson, and C.D. Nobes, Rac-dependent trans-endocytosis of ephrinBs regulates Eph-ephrin contact repulsion. Nature Cell Biology, 2003. 5(10): p. 879-888.
147. Parker, M., et al., Reverse endocytosis of transmembrane ephrin-B ligands via a clathrin-mediated pathway. Biochemical and Biophysical Research Communications, 2004. 323(1): p. 17-23.
148. Goldshmit, Y., S. McLenachan, and A. Turnley, Roles of Eph receptors and ephrins in the normal and damaged adult CNS. Brain research reviews, 2006. 52(2): p. 327-45.
149. Henkemeyer, M., et al., Nuk controls pathfinding of commissural axons in the mammalian central nervous system. Cell, 1996. 86(1): p. 35-46.
150. Henderson, J.T., et al., The receptor tyrosine kinase EphB2 regulates NMDA-dependent synaptic function. Neuron, 2001. 32(6): p. 1041-56.
151. Birgbauer, E., et al., Kinase independent function of EphB receptors in retinal axon pathfinding to the optic disc from dorsal but not ventral retina. Development, 2000. 127(6): p. 1231-41.
152. Grunwald, I.C., et al., Hippocampal plasticity requires postsynaptic ephrinBs. Nature Neuroscience, 2004. 7(1): p. 33-40.
123

153. Klein, R., Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nature Neuroscience, 2009. 12(1): p. 15-20.
154. Savelieva, K.V., et al., Learning and memory impairment in Eph receptor A6 knockout mice. Neuroscience letters, 2008. 438(2): p. 205-9.
155. Dalva, M.B., et al., EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell, 2000. 103(6): p. 945-56.
156. Henkemeyer, M., et al., Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. The Journal of cell biology, 2003. 163(6): p. 1313-26.
157. Grunwald, I.C., et al., Kinase-independent requirement of EphB2 receptors in hippocampal synaptic plasticity. Neuron, 2001. 32(6): p. 1027-40.
158. Takasu, M.A., et al., Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science, 2002. 295(5554): p. 491-5.
159. Dalva, M.B., A.C. McClelland, and M.S. Kayser, Cell adhesion molecules: signalling functions at the synapse. Nature reviews. Neuroscience, 2007. 8(3): p. 206-20.
160. Simon, A.M., et al., Early changes in hippocampal Eph receptors precede the onset of memory decline in mouse models of Alzheimer's disease. Journal of Alzheimer's disease : JAD, 2009. 17(4): p. 773-86.
161. Cisse, M., et al., Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature, 2011. 469(7328): p. 47-52.
162. Battaglia, A.A., et al., EphB receptors and ephrin-B ligands regulate spinal sensory connectivity and modulate pain processing. Nature neuroscience, 2003. 6(4): p. 339-40.
163. Kobayashi, H., et al., Involvement of EphB1 receptor/EphrinB2 ligand in neuropathic pain. Spine, 2007. 32(15): p. 1592-8.
164. Song, X.J., et al., Upregulation and redistribution of ephrinB and EphB receptor in dorsal root ganglion and spinal dorsal horn neurons after peripheral nerve injury and dorsal rhizotomy. European journal of pain, 2008. 12(8): p. 1031-9.
165. Song, X.J., et al., EphrinB-EphB receptor signaling contributes to neuropathic pain by regulating neural excitability and spinal synaptic plasticity in rats. Pain, 2008. 139(1): p. 168-80.
166. Han, Y., et al., Targeted mutation of EphB1 receptor prevents development of neuropathic hyperalgesia and physical dependence on morphine in mice. Molecular Pain, 2008. 4: p. 60.
167. Liu, W.T., et al., EphB receptor signaling in mouse spinal cord contributes to physical dependence on morphine. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2009. 23(1): p. 90-8.
168. Liu, S., et al., Blocking EphB1 receptor forward signaling in spinal cord relieves bone cancer pain and rescues analgesic effect of morphine treatment in rodents. Cancer research, 2011. 71(13): p. 4392-402.
169. Squire, L.R., The legacy of patient H.M. for neuroscience. Neuron, 2009. 61(1): p. 6-9. 170. Scoville, W.B. and B. Milner, Loss of recent memory after bilateral hippocampal
lesions. Journal of neurology, neurosurgery, and psychiatry, 1957. 20(1): p. 11-21. 171. Corkin, S., What's new with the amnesic patient H.M.? Nature reviews. Neuroscience,
2002. 3(2): p. 153-60. 172. Squire, L.R., Memory systems of the brain: a brief history and current perspective.
Neurobiology of learning and memory, 2004. 82(3): p. 171-7. 173. Baddeley, A., Working memory. Current biology : CB, 2010. 20(4): p. R136-40. 174. Squire, L.R. and J.T. Wixted, The cognitive neuroscience of human memory since H.M.
Annual Review of Neuroscience, 2011. 34: p. 259-88.
124

175. Okano, H., T. Hirano, and E. Balaban, Learning and memory. Proceedings of the National Academy of Sciences of the United States of America, 2000. 97(23): p. 12403-4.
176. Mayes, A.R., et al., The relationship between retrograde and anterograde amnesia in patients with typical global amnesia. Cortex; a journal devoted to the study of the nervous system and behavior, 1997. 33(2): p. 197-217.
177. Squire, L.R., Memory and brain systems: 1969-2009. The Journal of neuroscience : the official journal of the Society for Neuroscience, 2009. 29(41): p. 12711-6.
178. Packard, M.G. and B.J. Knowlton, Learning and memory functions of the Basal Ganglia. Annual Review of Neuroscience, 2002. 25: p. 563-93.
179. McDonald, R.J. and N.M. White, A triple dissociation of memory systems: hippocampus, amygdala, and dorsal striatum. Behavioral Neuroscience, 1993. 107(1): p. 3-22.
180. Ito, M., Mechanisms of motor learning in the cerebellum. Brain research, 2000. 886(1-2): p. 237-245.
181. Meyers, R.A., A.R. Zavala, and J.L. Neisewander, Dorsal, but not ventral, hippocampal lesions disrupt cocaine place conditioning. Neuroreport, 2003. 14(16): p. 2127-31.
182. Olmstead, M.C. and K.B. Franklin, The development of a conditioned place preference to morphine: effects of lesions of various CNS sites. Behavioral Neuroscience, 1997. 111(6): p. 1313-23.
183. Bliss, T.V. and G.L. Collingridge, A synaptic model of memory: long-term potentiation in the hippocampus. Nature, 1993. 361(6407): p. 31-9.
184. Lisman, J., A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proceedings of the National Academy of Sciences of the United States of America, 1989. 86(23): p. 9574-8.
185. Collingridge, G.L., et al., Long-term depression in the CNS. Nature reviews. Neuroscience, 2010. 11(7): p. 459-73.
186. Ito, M., Long-term depression. Annual Review of Neuroscience, 1989. 12: p. 85-102. 187. Moser, M.B. and E.I. Moser, Functional differentiation in the hippocampus.
Hippocampus, 1998. 8(6): p. 608-19. 188. Andersen, P., et al., Lamellar organization of hippocampal excitatory pathways. Acta
physiologica Scandinavica, 1969. 76(1): p. 4A-5A. 189. Pothuizen, H.H., et al., Dissociation of function between the dorsal and the ventral
hippocampus in spatial learning abilities of the rat: a within-subject, within-task comparison of reference and working spatial memory. The European journal of neuroscience, 2004. 19(3): p. 705-12.
190. Bannerman, D.M., et al., Ventral hippocampal lesions affect anxiety but not spatial learning. Behavioural brain research, 2003. 139(1-2): p. 197-213.
191. Bannerman, D.M., et al., Regional dissociations within the hippocampus--memory and anxiety. Neuroscience and biobehavioral reviews, 2004. 28(3): p. 273-83.
192. Jarrard, L.E., On the role of the hippocampus in learning and memory in the rat. Behavioral and neural biology, 1993. 60(1): p. 9-26.
193. Tsien, J.Z., P.T. Huerta, and S. Tonegawa, The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell, 1996. 87(7): p. 1327-38.
194. Moser, M.B., et al., Spatial learning with a minislab in the dorsal hippocampus. Proceedings of the National Academy of Sciences of the United States of America, 1995. 92(21): p. 9697-701.
195. Moser, E., M.B. Moser, and P. Andersen, Spatial learning impairment parallels the magnitude of dorsal hippocampal lesions, but is hardly present following ventral lesions.
125

The Journal of neuroscience : the official journal of the Society for Neuroscience, 1993. 13(9): p. 3916-25.
196. Best, P.J., A.M. White, and A. Minai, Spatial processing in the brain: the activity of hippocampal place cells. Annual Review of Neuroscience, 2001. 24: p. 459-86.
197. Moser, E.I., E. Kropff, and M.B. Moser, Place cells, grid cells, and the brain's spatial representation system. Annual Review of Neuroscience, 2008. 31: p. 69-89.
198. Maren, S., Neurobiology of Pavlovian fear conditioning. Annual Review of Neuroscience, 2001. 24: p. 897-931.
199. Anagnostaras, S.G., G.D. Gale, and M.S. Fanselow, Hippocampus and contextual fear conditioning: recent controversies and advances. Hippocampus, 2001. 11(1): p. 8-17.
200. Stubley-Weatherly, L., J.W. Harding, and J.W. Wright, Effects of discrete kainic acid-induced hippocampal lesions on spatial and contextual learning and memory in rats. Brain research, 1996. 716(1-2): p. 29-38.
201. Walsh, R.N. and R.A. Cummins, The Open-Field Test: a critical review. Psychological bulletin, 1976. 83(3): p. 482-504.
202. Maren, S. and M.S. Fanselow, Electrolytic lesions of the fimbria/fornix, dorsal hippocampus, or entorhinal cortex produce anterograde deficits in contextual fear conditioning in rats. Neurobiology of learning and memory, 1997. 67(2): p. 142-9.
203. Siegel, S., Morphine analgesic tolerance: its situation specificity supports a Pavlovian conditioning model. Science, 1976. 193(4250): p. 323-5.
204. Siegel, S., Morphine tolerance acquisition as an associative process. Journal of experimental psychology. Animal behavior processes, 1977. 3(1): p. 1-13.
205. Siegel, S., Evidence from rats that morphine tolerance is a learned response. Journal of comparative and physiological psychology, 1975. 89(5): p. 498-506.
206. Brase, D.A., H.H. Loh, and E.L. Way, Comparison of the effects of morphine on locomotor activity, analgesia and primary and protracted physical dependence in six mouse strains. The Journal of pharmacology and experimental therapeutics, 1977. 201(2): p. 368-74.
207. Stevens, K.E., G.A. Mickley, and L.J. McDermott, Brain areas involved in production of morphine-induced locomotor hyperactivity of the C57B1/6J mouse. Pharmacology, biochemistry, and behavior, 1986. 24(6): p. 1739-47.
208. Andersen, J.M., et al., Increased locomotor activity induced by heroin in mice: pharmacokinetic demonstration of heroin acting as a prodrug for the mediator 6-monoacetylmorphine in vivo. The Journal of pharmacology and experimental therapeutics, 2009. 331(1): p. 153-61.
209. Karinen, R., et al., Determination of heroin and its main metabolites in small sample volumes of whole blood and brain tissue by reversed-phase liquid chromatography-tandem mass spectrometry. Journal of analytical toxicology, 2009. 33(7): p. 345-50.
210. Mansour, A., et al., Autoradiographic differentiation of mu, delta, and kappa opioid receptors in the rat forebrain and midbrain. The Journal of neuroscience : the official journal of the Society for Neuroscience, 1987. 7(8): p. 2445-64.
211. Arvidsson, U., et al., Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience, 1995. 15(5 Pt 1): p. 3328-41.
212. Lee, I., M.R. Hunsaker, and R.P. Kesner, The role of hippocampal subregions in detecting spatial novelty. Behavioral Neuroscience, 2005. 119(1): p. 145-53.
213. Morris, R.G., et al., Place navigation impaired in rats with hippocampal lesions. Nature, 1982. 297(5868): p. 681-3.
126

214. Squire, L.R., Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychological review, 1992. 99(2): p. 195-231.
215. Nyberg, L., et al., Activation of medial temporal structures during episodic memory retrieval. Nature, 1996. 380(6576): p. 715-7.
216. Fanselow, M.S. and H.W. Dong, Are the dorsal and ventral hippocampus functionally distinct structures? Neuron, 2010. 65(1): p. 7-19.
217. White, N.M. and S. Gaskin, Dorsal hippocampus function in learning and expressing a spatial discrimination. Learning & memory, 2006. 13(2): p. 119-22.
218. Moser, E.I., et al., Impaired spatial learning after saturation of long-term potentiation. Science, 1998. 281(5385): p. 2038-42.
219. Bear, M.F. and W.C. Abraham, Long-term depression in hippocampus. Annual Review of Neuroscience, 1996. 19: p. 437-62.
220. Gilbert, C.D. and M. Sigman, Brain states: top-down influences in sensory processing. Neuron, 2007. 54(5): p. 677-96.
221. Kentros, C.G., et al., Increased attention to spatial context increases both place field stability and spatial memory. Neuron, 2004. 42(2): p. 283-95.
222. Burwell, R.D., The parahippocampal region: corticocortical connectivity. Annals of the New York Academy of Sciences, 2000. 911: p. 25-42.
223. Krank, M.D., R.E. Hinson, and S. Siegel, Conditional hyperalgesia is elicited by environmental signals of morphine. Behavioral and neural biology, 1981. 32(2): p. 148-57.
224. Winocur, G. and D. Bindra, Effects of additional cues on passive avoidance learning and extinction in rats with hippocampal lesions. Physiology & behavior, 1976. 17(6): p. 915-20.
225. Lorenzini, C.A., et al., Role of dorsal hippocampus in acquisition, consolidation and retrieval of rat's passive avoidance response: a tetrodotoxin functional inactivation study. Brain research, 1996. 730(1-2): p. 32-9.
226. Munoz, C. and S.P. Grossman, Spatial discrimination, reversal and active or passive avoidance learning in rats with KA-induced neuronal depletions in dorsal hippocampus. Brain research bulletin, 1981. 6(5): p. 399-406.
227. Light, K.R., et al., General learning ability regulates exploration through its influence on rate of habituation. Behavioural brain research, 2011. 223(2): p. 297-309.
127





























