Embed Size (px)
Citation preview

Mechanism of Phenolic Polymer Dissolution: Importance of Acid-BaseEquilibria
Lewis W. Flanagin,† Christopher L. McAdams,† William D. Hinsberg,‡Isaac C. Sanchez,† and C. Grant Willson*,†
Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712, andIBM Almaden Research Center, 650 Harry Road, San Jose, California 95126
Received February 23, 1999; Revised Manuscript Received June 5, 1999
ABSTRACT: The dissolution phenomena that are the basis of microlithography are largely dependenton the acid-base equilibrium of phenolic polymers in aqueous base. Fundamental equations are derivedto relate the probabilistic quantities of the critical-ionization model to experimentally measurable acid-base properties in such polymer systems: solution pH, polymer pKa, degree of polymerization, and averagedegree of ionization. Model predictions for the dependence of the dissolution rate on these propertiessupport previous experimental observations. A method for estimating the pKa of phenolic polymers as afunction of the average degree of ionization is developed, and the results of this approach for novolac andpoly(hydroxystyrene) agree with the observed differences in the dissolution rates of these two species.These results also corroborate the hydrogen-bonding dissolution inhibition model previously reported.The change in dissolution rate accompanying the substitution of deuterium for hydrogen in the phenolgroup is interpreted in terms of the deuterium isotope effect on pKa.
Introduction
Improved photoresists have enabled extraordinaryadvancements in photolithography and rapid growth inthe semiconductor industry. The enhanced performanceof photoresists arises from modifications to all compo-nent polymer and organic materials that make up resistformulations. Nearly every commercially available pho-toresist operates on the principle of a photoinducedchange in the rate at which the photoresist dissolves inaqueous base. The most widely used photoresists employa base-soluble phenolic resin, novolac, and a photoactiveadditive, diazonaphthoquinone.1 The diazonaphtho-quinone reduces the rate at which novolac dissolves inaqueous base. Exposure to 365 nm radiation photo-chemically converts diazonaphthoquinone to a base-soluble indenecarboxylic acid that accelerates dissolu-tion.
The drive toward ever-smaller features inspired thedevelopment of more sensitive, “chemically amplified”resists that operate at wavelengths of 248 and 193 nm.2Like novolac-based photoresists, most chemically ampli-fied resists also contain acidic polymers that providesolubility in aqueous-base developers. The majority ofchemically amplified resist systems utilize poly(hy-droxystyrene), another phenolic polymer. Although no-volac and poly(hydroxystyrene) are both phenolic poly-mers, they dissolve in aqueous base at substantiallydifferent rates. Difficulties encountered in the transitionto new photoresists manifest the value of understandingthe dissolution kinetics of acidic polymers in aqueousbase.
According to the critical-ionization model that wedescribed in 1997,3 a critical fraction of acidic sites onthe polymer chain must ionize for the chain to dissolvein aqueous base. In the first stage of developing thecritical-ionization model, the dissolution rate was ex-
plained in terms of the probability that an individualpolymer chain meets the solubility criterion.3 Thisapproach offers qualitative insight into the dissolutionbehavior of these phenolic polymer systems, but a fullyquantitative understanding of the dissolution raterequires a clear, mathematical expression relating theparameters of the probabilistic model to precise, fun-damental, and experimentally measurable quantities.The current paper addresses this need by consideringthe acid-base equilibria between the phenolic polymerand the developer solution. Because knowledge of thepKa of the polymer is crucial to such an approach, amethod for estimating the pKa of phenolic polymers isdeveloped, and the pKa is analyzed as a function of thedegree of ionization. Finally, the experiment of Kim andReiser4 that quantifies the effect of substituting deute-rium for hydrogen on the phenol group is discussed interms of the deuterium isotope effect on polymer pKaand the critical-ionization model.
Acid-Base Equilibria of Phenolic PolymersFor a solid film consisting of an acidic polymer with
X protons that can be ionized per molecule in contactwith an aqueous solution of hydroxide ion, the followingset of deprotonation steps can occur at the liquid-solidinterface:
Here, PxHx-ii- represents the polymer chain, which
* Author to whom correspondence should be addressed.† The University of Texas at Austin.‡ IBM Almaden Research Center.
OH- + PxHx y\zK1
PxHx-1- + H2O
OH- + PxHx-1- y\z
K2PxHx-2
2- + H2O
l
OH- + PxHx-i+1(i-1)- y\z
KiPxHx-i
i- + H2O
l
OH- + PxH1(X-1)- y\z
KXPxH0
X- + H2O (1)
5337Macromolecules 1999, 32, 5337-5343
10.1021/ma990271y CCC: $18.00 © 1999 American Chemical SocietyPublished on Web 07/23/1999

began with X ionizable protons per chain but now hasX - i ionizable protons remaining on the chain. Anequilibrium constant
can be written for each reaction in eq 1. Under theconditions of interest, the OH- concentration can beconsidered to be constant. Having a single equilibriumconstant, Ki, for each degree of ionization implies thatthe equilibrium constant of ionization is the sameregardless of the particular arrangement of ionized andun-ionized sites on a given polyion.
The set of constants, K1, ..., KX, can be related to oneanother by a statistical factor. The statistical factorarises from a difference in the number of pathways forprotonation and deprotonation for each equilibriumexpression.5 If Kh is defined as the geometric mean ofthe constants, K1, ..., KX, then these constants arerelated by the following expression:
By material balance, the sum of the surface concen-trations of all polymer species is a constant equal to theinitial polymer surface concentration, [PXHX]0, prior tocontact with the liquid:
If the initial surface concentration of polymer, the valuesof K1, ..., KX (calculable from the pKa of the polymericspecies), and the OH- concentration are all known, thesurface concentrations of all polymer species can becalculated:
Using eq 3, eq 5 may be simplified to the form
where
Combining eq 4 with eq 6 results in
Dissolution Rate of Phenolic PolymersThe overall process of dissolution is composed of three
steps: base transport (diffusion) into the polymer film,equilibration of the acid-base reactions, and polymertransport into aqueous solution. All models for thedissolution of phenolic polymers in aqueous base assumethat one of these steps determines the observed dis-
solution rate. Some of the earliest of these models (e.g.,models by Hinsberg and Gutierrez6 and by Garza et al.7)proposed that the ionization of the phenolic groups atthe interface between the solvent and polymer limitsthe rate of dissolution. In the membrane model of Arcus,“the formation and stabilization of the phenolate anionis the major chemical reaction for the dissolution ofphotoresist films.”8 Arcus suggested that ionizationoccurs within a thin boundary layer, or membrane,between the polymer and solvent, and that “the onlyway for this [ionization] to occur is for the base in thedeveloper to be transported into the membrane via somemechanism such as diffusion or infiltration into voids.”8
Reiser and co-workers9,10 developed this concept of aboundary layer further by using percolation theory todescribe the diffusion of developer base into the filmthrough a thin penetration zone. The percolation modeloriginally defined the rate-determining step in thedissolution process as the “phenolate-phenol transfer [ofbase] which occurs at the front of the penetrationzone.”10 More recently, the critical-ionization model,3which assumes that the ionization step determines thesolubility of the polymer, has demonstrated very inter-esting and useful predictive capabilities. After thecritical-ionization model had been introduced, Kim andReiser4 conducted an important study of the effect ofthe base cation on the dissolution rate. The results ofthis study led the authors to shift the emphasis of thepercolation model from base transport to the ionizationstep. Kim and Reiser concluded, “the rate-determiningstep of Novolak dissolution is not the diffusion of basewithin the penetration zone but the deprotonation stepat the interface of the zone with the virgin matrix.”4 Analternative approach is taken in the chain disentangle-ment model of Peppas and co-workers,11 which is anexcellent description of the dissolution of polystyreneand other high molecular weight polymers. The chaindisentanglement model, however, is inappropriate fordescribing novolac, which has a Mw on the order of 1000,far below the entanglement molecular weight. The realrate-determining step is, in all likelihood, dependent onthe molecular weight of the polymer. For low-molecular-weight polymers, ionization probably controls the dis-solution rate, while for high-molecular-weight polymers,polymer disentanglement is the limiting step.
The dissolution rate of the polymer film can be relatedto the rate at which each polymer species dissolves. Thetransfer of each ionized species, PXHX-i
i-, from the solidstate to aqueous solution is now assumed to be anirreversible first-order process characterized by a rateconstant kd,i:
The polymer film dissolves on a molecular basis at arate R given below:
While the values for Kh and the rate constants, kd,1,..., kd,X, are not yet determined for the phenolic polymersystems of interest here, this description can be usedto test the hypothesis that film dissolution kinetics are
[PXHX-ii-] 98
kd,iPXHX-i(aq)
i-;
ri )d[PXHX-i(aq)
i-]dt ) kd,i[PXHX-i
i-] (9)
R ) ∑i ) 0
X
kd,i[PXHX-ii-] (10)
Ki )[PXHX-i
i-]
[PXHX-i+1(i-1)-][OH-]
) Ka/Kw (2)
Ki ) (X - i + 1)Kh /i (3)
∑i ) 0
X
[PXHX-ii-] ) [PXHX]0 (4)
[PXHX-ii-] ) [PXHX][OH-]i ∏
j ) 1
i
Kj (5)
[PXHX-ii-] ) [PXHX][OH-]i(Xi )Kh i (6)
(Xi ) ) X!i!(X - i)!
(7)
[PXHX-ii-] )
[PXHX]0[OH-]i(Xi )Kh i
∏j ) 0
X
[OH-]j(Xj )Kh j
(8)
5338 Flanagin et al. Macromolecules, Vol. 32, No. 16, 1999
controlled by the degree of surface ionization. Thishypothesis is reasonable in the molecular weight regimeof engineering interest for both novolac and poly-(hydroxystyrene). It may not be accurate for highermolecular weight polymers. The dissolution rate con-stant, kd,i, is assumed to depend on the extent ofionization but not on the chain length. The rate constantkd,i is assumed to be zero unless the fraction of phenolategroups on the chain exceeds a critical value F. For allchains in which the phenolate fraction is greater thanF, the rate constant kd,i is assumed to have a singular,nonzero value, kd. Under these assumptions, the rateequation reduces to the following:
Earlier, the dissolution rate of phenolic polymers wasdescribed by the probability of satisfying a criterion forsolubility based on the degree of ionization.3 In thiswork, a similar criterion is invoked in an analysis ofthe thermodynamics and kinetics of polymer dissolution.The probabilistic and thermodynamic/kinetic approaches,although different, are complementary. Both modelsrest on the premise of a critical degree of ionizationrequired for solubility.
A direct, quantitative correspondence exists betweenthe microscopic quantities that appear in the probabi-listic model and the macroscopic observable propertiesthat appear in the acid-base equilibria model. Thefraction Y of polymers meeting the solubility criterionmay be expressed in terms of polymer concentrationsas
Combining eq 12 with eq 11 yields the following relationbetween the observable dissolution rate, R, and theprobabilistic quantity Y:
According to this result, Y may be viewed appropriatelyas a dimensionless dissolution rate. The function R isdefined as the deprotonated fraction of phenol groups(i.e., the ratio of phenolate concentration to the initialphenol concentration); R may be expressed in terms ofpolymer concentrations as
Substitution of eq 8 into eq 14 leads to
where z ) [OH-]Kh . When one recognizes that ∑j)0X (j
X)zj
) (1 + z)X and from this that ∑i)1X i(i
X)zi ) Xz(1 + z)X-1,this seemingly complex expression for R reduces to therelatively simple result
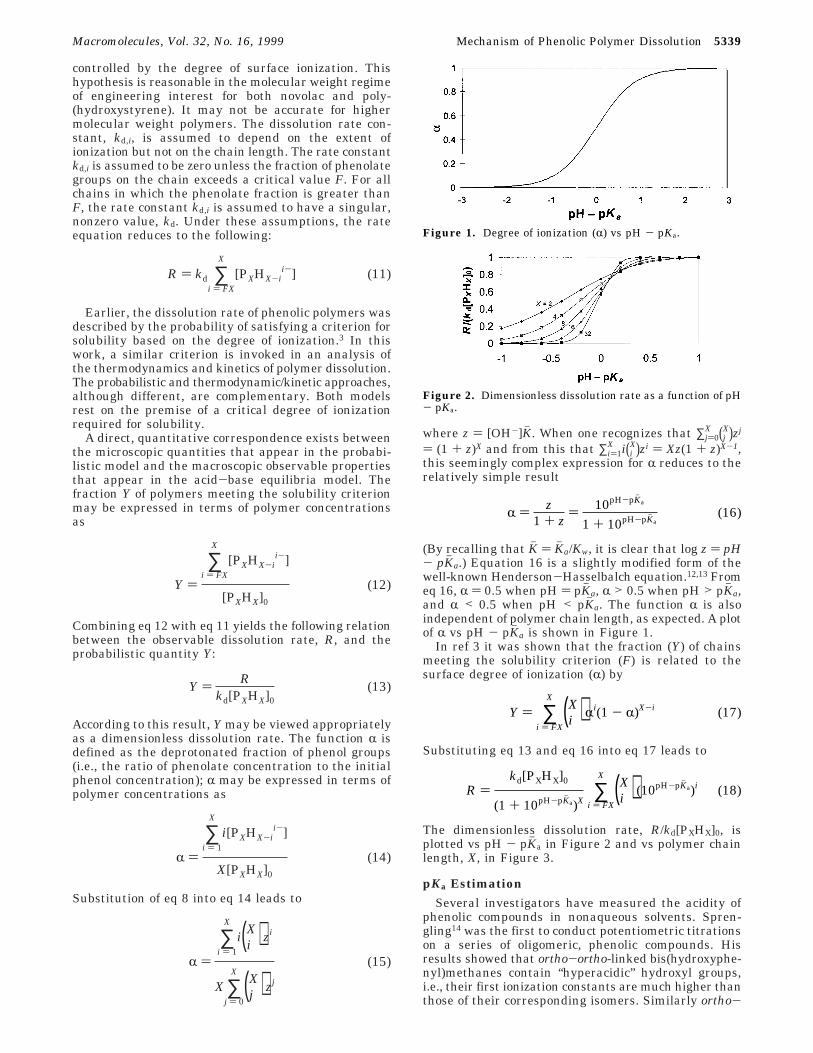
(By recalling that Kh ) Kh a/Kw, it is clear that log z ) pH- pKh a.) Equation 16 is a slightly modified form of thewell-known Henderson-Hasselbalch equation.12,13 Fromeq 16, R ) 0.5 when pH ) pKh a, R > 0.5 when pH > pKh a,and R < 0.5 when pH < pKh a. The function R is alsoindependent of polymer chain length, as expected. A plotof R vs pH - pKh a is shown in Figure 1.
In ref 3 it was shown that the fraction (Y) of chainsmeeting the solubility criterion (F) is related to thesurface degree of ionization (R) by
Substituting eq 13 and eq 16 into eq 17 leads to
The dimensionless dissolution rate, R/kd[PXHX]0, isplotted vs pH - pKh a in Figure 2 and vs polymer chainlength, X, in Figure 3.
pKa EstimationSeveral investigators have measured the acidity of
phenolic compounds in nonaqueous solvents. Spren-gling14 was the first to conduct potentiometric titrationson a series of oligomeric, phenolic compounds. Hisresults showed that ortho-ortho-linked bis(hydroxyphe-nyl)methanes contain “hyperacidic” hydroxyl groups,i.e., their first ionization constants are much higher thanthose of their corresponding isomers. Similarly ortho-
R ) kd ∑i ) FX
X
[PXHX-ii-] (11)
Y )
∑i ) FX
X
[PXHX-ii-]
[PXHX]0
(12)
Y ) Rkd[PXHX]0
(13)
R )
∑i ) 1
X
i[PXHX-ii-]
X[PXHX]0
(14)
R )
∑i ) 1
X
i(Xi )zi
X∑j ) 0
X (Xj )zj
(15)
Figure 1. Degree of ionization (R) vs pH - pKa.
Figure 2. Dimensionless dissolution rate as a function of pH- pKa.
R ) z1 + z
) 10pH-pKh a
1 + 10pH-pKh a(16)
Y ) ∑i ) FX
X (Xi )Ri(1 - R)X-i (17)
R )kd[PXHX]0
(1 + 10pH-pKh a)X∑
i ) FX
X (Xi )(10pH-pKh a)i (18)
Macromolecules, Vol. 32, No. 16, 1999 Mechanism of Phenolic Polymer Dissolution 5339

ortho-linked trimers and tetramers demonstrate evengreater hyperacidity, but ortho-para-linked compoundsare less acidic.14 The apparent reason for the hypera-cidity of the ortho-ortho-linked compounds is theirability to form hydrogen bonds and to distribute theanionic charge efficiently over several atoms.
Bohmer et al.15 measured the first dissociation con-stants of ortho-ortho-linked oligomeric phenolic com-pounds. In each of these compounds, the researchersincorporated a p-nitrophenol unit, which is much moreacidic than the remaining alkylphenol units. They werethus able to assign the first dissociation unequivocallyto the nitrophenol group. Substituent effects on theacidity of the nitrophenol group were studied rigorouslyand systematically. The first dissociation constantincreases (i.e., the pK1 decreases) as the number ofortho-linked alkylphenol units increases from 0 to 3.This result provides evidence that a chain of up to threeintramolecular hydrogen bonds stabilizes the mono-anion and that this effect grows as the number ofphenolic units participating in the distribution of chargeincreases. Bohmer et al. found that those compoundshaving their dissociating unit in the middle of the chainare more acidic because the monoanion is stabilized bya second intramolecular hydrogen bond to the negativelycharged oxygen.15 The UV spectra of the monoanionsshow that the absorption coefficient and wavelength ofthe absorption maximum related to the nitrophenol unitdecrease as the strength of the intramolecular hydrogenbond increases.15
Over the course of about 20 years, Chatterjee and co-workers performed a systematic and thorough study ofortho-ortho-linked phenolic compounds through poten-tiometric and conductometric nonaqueous titrations.The initial study demonstrated that intramolecularhydrogen bonding and ion association between the basecation and the partially neutralized polyphenol stronglyinfluence the titration curves.16 The potentiometrictitration curves of pentameric phenolic compounds inpyridine with sodium methoxide as titrant showed fourinflections, the first of which was very sharp, and theconductometric titration curves for the same compoundsdisplayed five breaks.17 Each inflection or break coin-cides with the neutralization of a hydroxyl group. Thefirst, sharp inflection, therefore, corresponds to theneutralization of a hyperacidic hydroxyl group. Hydro-gen bonding, electrostatic interactions, and ion associa-tion all evidently play a role in the stepwise neutral-ization of the hydroxyl groups. The ability to distinguishbetween hydroxyl groups was demonstrated further inmixtures of dimeric and trimeric phenolic compounds.18
Titrations of oligomers containing six to eight phenolic
groups showed as many distinct breaks in the conduc-tometric curves as there were hydroxyl groups on thecompound.19 The potentiometric curves, however, showedonly two to three inflections, indicating that some of thehydroxyl groups on each chain are hyperacidic, whileothers have roughly the same acidity. A similar studyof a nonameric phenolic compound showed as well thatthe hydroxyl groups are neutralized in mostly a stepwisemanner in the conductometric titration, whereas theyare lumped together in the potentiometric titration.20
Chatterjee attributes this difference to homoconjuga-tion, which potentiometry cannot detect. Later studiesconsidered the influence of additional functional groupson the acidity of novolac-like oligomers.21,22
The nonaqueous titrations reviewed thus far provideclear evidence for the stepwise neutralization of phenolicpolymers, but they are unable to produce precise valuesfor the pKa of phenolic polymers in aqueous base. ThepKa of novolac and poly(hydroxystyrene) in aqueoussolution can be estimated using the acidity function forphenol given by Biggs et al.23 and Perrin et al.5 Thisapproach allows one to estimate the pKa of phenolicoligomers and polymers by the following equation:
Here ∑σ is the sum of all Hammett sigma constants,which account for substituent effects, and log(A/B) isthe statistical correction needed for polybasic acids. Theconstant A is the number of energetically equivalentways to deprotonate a given polybasic acid, and theconstant B is the number of reverse protonation path-ways. Thus, a dibasic acid, such as bis(2-hydroxyphen-yl)methane (Figure 4), has two equivalent protons thatmay be deprotonated, but the reverse protonation mayoccur at only one location. In this case, the statisticalfactor is equal to log(2/1), or ∼ 0.3. For the secondionization (pK2) the statistical factor is log(1/2). Ifsubstituent effects are ignored, then pK1 and pK2 arecalculated to be 9.62 and 10.22, respectively. Themeasured values24 (Figure 4) of pK1 (7.5) and pK2 (11.6)show that substituent effects in this case are very largeand cannot be ignored. Perrin5 has calculated theHammett ortho σ constant (σortho ) 0.95) for the effectof a (2-hydroxyphenyl)methyl group on a phenol. Sincethe pK2 is known to be 11.60, σortho for the phenolate iscalculated to be -0.62.
If one assumes that only nearest neighbor effects needto be considered for novolac and that substituent effects
Figure 3. Dimensionless dissolution rate plotted as a functionof polymer chain length.
Figure 4. Calculation of the pKa of a dibasic acid.
pKa ) 9.92-2.23∑σ - log(A/B) (19)
5340 Flanagin et al. Macromolecules, Vol. 32, No. 16, 1999

are additive, then one may use this acidity function (eq19) to estimate the pKa of any ortho-ortho-linkednovolac oligomer or polymer (Figure 5). Nonnearest-neighbor effects may be ignored because the energy ofthe electrostatic, Coulombic interaction varies inverselywith the square of the distance between the ions.25
Additivity of substituent effects is essentially the basisof linear free-energy relationships.
Assuming additive substituent effects, the acidityfunction (eq 19) predicts that the pKa of a phenoliccompound decreases as the number of neighboringphenolic groups increases. However, the pKa of a phenolgroup vicinal to an already existing phenolate group islarger than the pKa of an unsubstituted phenol becauselike charges repel.
Consider, for example, the phenolic pentamer shownin Figure 6. Each of the three energetically “equivalent”,ionizable phenols in the middle of the pentamer has twonearest-neighbor hydroxyl groups (∑σ ) 1.90). Eachterminal group, however, has only one adjacent phenolgroup (∑σ ) 0.95) and subsequently requires moreenergy to deprotonate. Thus, the first deprotonation ismost likely to occur at any of the three middle phenolgroups (A ) 3). After the first deprotonation, only onereverse pathway is possible (B ) 1). For the seconddeprotonation, with which pK2 is associated, only onehydroxyl group with two adjacent phenols remains (∑σ) 1.90, A ) 1, B ) 2). The most likely candidate for thethird deprotonation, associated with pK3, is a terminalgroup adjacent to a phenolate (∑σ ) -0.62, A ) 2, B )1). The fourth ionization, associated with pK4, occursat the other end group, and either end group mayreaccept a proton (∑σ ) -0.62, A ) 1, B ) 2). The finalionization, associated with pK5, can occur at only one
site, which is adjacent to two ionized sites (∑σ ) -1.24,A ) 1). Three equivalent choices for the reverse proto-nation exist (B ) 3). In each of the above steps, theactual phenolic site that ionizes may be different fromthe one indicated, but in this case, the molecule wouldquickly rearrange to the lowest-energy configuration bythe classic Grotthuss proton-transfer mechanism.26
For completely ortho-ortho-linked novolac systemswith degree of polymerization X, it is possible tocalculate the pKi (ith pKa) using the same logical steps(Table 1).
Conformations available to poly(hydroxystyrene) al-low the hydroxyl groups to be separated much furtherin space (Figure 5);27 thus, nearest-neighbor effects canbe ignored, and all of the acidic sites can be treated asenergetically equal. However, the alkyl-substituent ef-fect (σ ) -0.15) is included (eq 20).
The degree of ionization for a single polymer chain isdefined as the number of ionized sites divided by thenumber of total sites (R ) i/X). In Figure 7, pKi is plottedvs R (Figure 7 is the example for X ) 20). Clearly, asthe degree of ionization increases, the pKi also increases.However, our model for ortho-ortho-linked novolacpredicts a sharp change in the slope at R ) ∼0.5, wherefor the first time, deprotonation creates adjacent anions.This type of behavior has been observed in otherpolyelectrolytes. For example, Kitano et al.28 observedthat the pKa has this sharp increase as a function ofthe degree of ionization for poly(maleic acid) but not forpoly(acrylic acid) (Figure 8). The increased distance
Figure 5. Optimized gas-phase structures of (a) ortho-ortholinked p-cresol novolac trimer, (b) poly(4-hydroxystyrene)trimer, and (c) the trianion of the novolac trimer depicted inpart a. Energies calculated using the HyperChem PM3 self-consistent field are (a) -5459, (b) -6005, and (c) -5224 kcal/mol, respectively. The distances between nearest oxygen atomsare (a) 2.75, (b) 10.59, and (c) 5.58 Å, respectively.
Figure 6. Calculations of pK1 through pK5 for an ortho-ortho-linked phenolic pentamer.
Table 1. Expressions for the pKa of Novolac
i pKi
<X/2 9.92 - 2.23(1.90) - log((X - 2i)/i)X/2 (for even X) 9.92 - 2.23(0.95) - log(1/1)(X + 1)/2 (for odd X) 9.92 - 2.23(-0.62) - log(2/1)X/2 + 1 (for even X) 9.92 - 2.23(-0.62) - log(1/1)(X + 3)/2 (for odd X) 9.92 - 2.23(-0.62) - log(1/2)>(X + 3)/2 9.92 - 2.23(-1.24) -
log((X - i + 1)/(2i - X - 2))
pKi ) 9.92-2.23(-0.15) - log((X + 1 - i)/i) (20)
Macromolecules, Vol. 32, No. 16, 1999 Mechanism of Phenolic Polymer Dissolution 5341

between the nearest neighbors in poly(acrylic acid)results in a lower charge density.
Honda et al.29 demonstrated that adding ortho-ortho-linked diad and triad phenolic moieties to poly(hydroxy-styrene) lowers the pKa significantly during the earlystages of neutralization. At later stages of neutraliza-tion, the pKa finally reaches the same value as that ofunmodified poly(hydroxystyrene). In agreement withour model, these results indicate that ortho-ortho-linkages initially facilitate proton dissociation throughintramolecular hydrogen bonding, but eventually elec-trostatic repulsion dominates.
Equation 16, which expresses the degree of ionizationin terms of the difference between pH and pKh a, wasderived under the assumption that each Ki differs fromKh a by only a statistical factor (eq 3). The followingequation, which is a general form of eq 16 that allowsfor differences such as substituent effects, may be usedto calculate a predicted degree of ionization at any pH(R vs pH in Figure 9) when individual values for pKiare known:
Note that the pH of the 0.26 N OH- solution is drawnin as a reference to the strength of standard developersolution. In this model, R is only about 0.75 in thedeveloper solution. The model predicts that, in this pHrange, novolac is not completely ionized. Since thedissolution rate is a function of R, the prediction is inagreement with the observation that higher molecularweight poly(hydroxystyrene) dissolves more quicklythan lower molecular weight novolac in 0.26 N tetram-ethylammonium hydroxide.
It should be noted in this discussion that the resultsin Figures 7 and 9 pertain only to ortho-ortho-linkednovolac systems. Because novolacs used in photolithog-raphy are synthesized from a combination of m-cresoland p-cresol monomers, both ortho-para and ortho-ortho linkages exist in these systems. Because ortho-para linkages disrupt the hydrogen-bonded network anddistribute the charge density differently, the pKa forcomplex novolacs probably changes more gradually thanthe pKa for completely ortho-ortho-linked novolac.
The concept that the dissolution rate depends on thepKa of the polymer helps in explaining the function ofdissolution inhibitors. Our previous work30 has shownthat “a primary mode of dissolution inhibition is a strongpolarization of the intramolecular hydrogen bondingclusters found in novolac. The polarization increases theeffective pKa of novolac [by stabilizing the acid form ofthe phenol] and decreases the ratio of ionized to un-ionized phenolic sites.” The higher pKa induced by thedissolution inhibitor decreases the rate of dissolution(Figure 2). Because poly(hydroxystyrene) does not havea high degree of intramolecular hydrogen bonding, theeffect of dissolution inhibitors is much weaker on poly-(hydroxystyrene) than on novolac. The hydrogen-bond-ing interaction between the inhibitor and the polymercan be strengthened by introducing resonance-stabiliz-ing electron donating groups or by substituting sulfonylesters in place of carbonyl esters in the inhibitor.30
Deuterium Isotope Effect
Recent experimental data4 for the effect of isotopicsubstitution on the dissolution rate of novolac supportthe assumption of the critical-ionization model that thepKa of a polymer controls the dissolution rate. Kim andReiser4 found that the dissolution rate of deuteratednovolac in deuterated base developer is about a factorof 2 less than the dissolution rate of the correspondingprotonated system. This observation is consistent withthe solvent isotope effect on pKa as stated by Lowry:31
“Weak acids are stronger in H2O than in D2O by about0.5 pK unit.” If the pKa of novolac in deuterateddeveloper is approximately 0.5 pK units higher than inprotonated developer, Figure 2 implies that the deuter-ated system dissolves at a slower rate than the proto-nated system because each rate curve is shifted rightby 0.5 pK units.
Conclusions
Through equations describing the acid-base equilib-ria of phenolic polymers in aqueous base, the predictionsof the critical-ionization model can now be understoodin terms of experimentally measurable properties: solu-tion pH, polymer pKa, degree of polymerization, andaverage degree of ionization. The model has been usedto predict the dependence of the dissolution rate on eachof these properties. The faster rate at which poly-(hydroxystyrene) dissolves compared to novolac is at-
Figure 7. Predicted pKa vs degree of ionization (R) for poly-(hydroxystyrene) and ortho-ortho-linked novolac.
Figure 8. Poly(maleic acid) (left) and poly(acrylic acid) (right).
Figure 9. Predicted degree of ionization (R) vs pH for poly-(hydroxystyrene) and ortho-ortho-linked novolac.
R )
∑i ) 1
X
10∑j)1i pH-pKji
X[1 + ∑i)1
X
10∑j)1i pH-pKj]
(21)
5342 Flanagin et al. Macromolecules, Vol. 32, No. 16, 1999

tributed to its greater acidity, whereas the function ofdissolution inhibitors is explained as an effective de-crease in the polymer acidity. Finally, the change indissolution rate accompanying the substitution of deu-terium for hydrogen in the phenol group has beeninterpreted in terms of the deuterium isotope effect onpKa.
Acknowledgment. The authors gratefully acknowl-edge valuable input from Pavlos Tsiartas and Prof. CliffHenderson. We have also benefited greatly from dis-cussions with Ralph Dammel and Prof. Arnost Reiser.We are grateful to Hoang Vi Tran for helping us togenerate the optimized structures of novolac and poly-(4-hydroxystyrene). We thank the Semiconductor Re-search Corp. (96-LP-409) for its financial support.
References and Notes
(1) Dammel, R. R. Diazonaphthoquinone-based Resists; SPIEPress: Bellingham, WA, 1993.
(2) Willson, C. G. In Introduction to Microlithography, 2nd ed.;Thompson, L. F., Willson, C. G., Bowden, M. J., Eds.;American Chemical Society: Washington, DC, 1994; Chapter3.
(3) Tsiartas, P. C.; Flanagin, L. W.; Henderson, C. L.; Hinsberg,W. D.; Sanchez, I. C.; Bonnecaze, R. T.; Willson, C. G.Macromolecules 1997, 30, 4656-4664.
(4) Kim, M. S.; Reiser, A. Macromolecules 1997, 30, 4652-4655.(5) Perrin, D. D.; Dempsey, B.; Serjeant, E. P. pKa Prediction
for Organic Acids and Bases; Chapman and Hall: London,1981.
(6) Hinsberg, W. D.; Gutierrez, M. L. Proc. SPIE-Int. Soc. Opt.Eng. 1984, 469, 57-64.
(7) Garza, C. M.; Szmanda, C. R.; Fischer, R. L. J. Proc. SPIE-Int. Soc. Opt. Eng. 1988, 920, 321-328.
(8) Arcus, R. A. Proc. SPIE-Int. Soc. Opt. Eng. 1986, 631, 124-131.
(9) Huang, J.-P.; Kwei, T. K.; Reiser, A. Macromolecules 1989,22, 4106-4112.
(10) Yeh, T.-F.; Shih, H.-Y.; Reiser, A. Macromolecules 1992, 25,5345-5352.
(11) Peppas, N. A.; Wu, J. C.; von Meerwall, E. D. Macromolecules1994, 27, 5626-5638.
(12) Henderson, L. J. J. Am. Chem. Soc. 1908, 30, 954-960.(13) Hasselbalch, K. A. Biochem. Z. 1916, 74, 18-47.(14) Sprengling, G. R. J. Am. Chem. Soc. 1954, 76, 1190-1193.(15) Bohmer, V.; Schade, E.; Antes, C.; Pachta, J.; Vogt, W.;
Kammerer, H. Makromol. Chem. 1983, 184, 2361-2376.(16) Mitra, R. P.; Chatterjee, S. K. J. Sci. Ind. Res., Sect. B 1961,
20, 310-315.(17) Mitra, R. P.; Chatterjee, S. K. Indian J. Chem. 1963, 1, 62-
67.(18) Mitra, R. P.; Chatterjee, S. K. Indian J. Chem. 1964, 2, 85-
91.(19) Chatterjee, S. K. Indian J. Chem. 1969, 7, 605-610.(20) Chatterjee, S. K. Can. J. Chem. 1969, 47, 2323-2326.(21) Chatterjee, S. K.; Chatterjee, N. Makromol. Chem. 1978, 179,
1031-1039.(22) Chatterjee, S. K.; Pandith, R. L.; Pachauri, L. S. J. Macromol.
Sci.sChem. 1981, A16, 717-727.(23) Biggs, A. I.; Robinson, R. A. J. Chem. Soc. (London) 1961,
388-393.(24) Francis, D. J.; Yedanapalli, L. M. Indian J. Chem. 1964, 2,
166-167.(25) Reed, C. E.; Reed, W. F. J. Chem. Phys. 1992, 96, 1609-1620.(26) Huggins, M. L. J. Am. Chem. Soc. 1931, 53, 3190-3191.(27) Templeton, M. K.; Szmanda, C. R.; Zampini, A. Proc. SPIE-
Int. Soc. Opt. Eng. 1987, 771, 136-147.(28) Kitano, T.; Kawaguchi, S.; Anazawa, N.; Minakata, A.
Macromolecules 1987, 20, 2498-2506.(29) Honda, K.; Slater, S.; Beauchemin, Jr., B.; Toukhy, M.;
Tadros, S.; Aoai, T.; Kawabe, Y. Proc. SPIE-Int. Soc. Opt. Eng.1994, 2195, 524-541.
(30) McAdams, C. L.; Yueh, W.; Tsiartas, P.; Hsieh, D.; Willson,C. G. In Micro- and Nanopatterning Polymers; Ito, H.,Reichmanis, E., Eds.; ACS Symposium Series 706; AmericanChemical Society: Washington, DC, 1998; pp 292-305.
(31) Lowry, T. H.; Richardson, K. S. In Mechanism and Theoryin Organic Chemistry, 3rd ed.; Berger, L. S., Ed.; Harper &Row: New York, 1987; pp 143-159.
MA990271Y
Macromolecules, Vol. 32, No. 16, 1999 Mechanism of Phenolic Polymer Dissolution 5343





























