Embed Size (px)
Citation preview
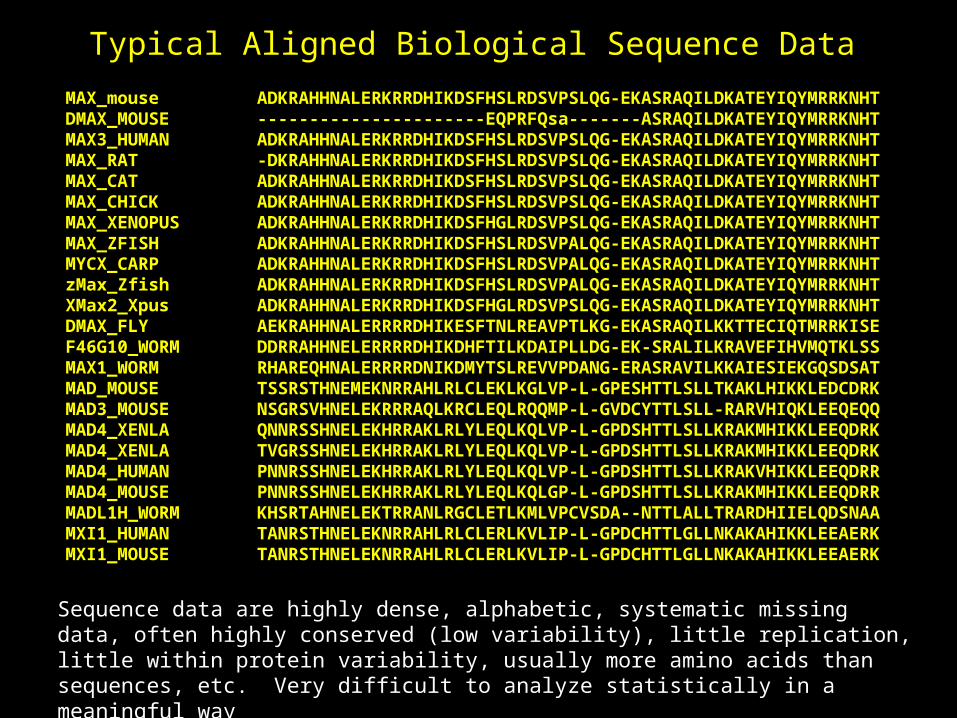
MAX_mouse ADKRAHHNALERKRRDHIKDSFHSLRDSVPSLQG-EKASRAQILDKATEYIQYMRRKNHTDMAX_MOUSE ----------------------EQPRFQsa-------ASRAQILDKATEYIQYMRRKNHTMAX3_HUMAN ADKRAHHNALERKRRDHIKDSFHSLRDSVPSLQG-EKASRAQILDKATEYIQYMRRKNHTMAX_RAT -DKRAHHNALERKRRDHIKDSFHSLRDSVPSLQG-EKASRAQILDKATEYIQYMRRKNHTMAX_CAT ADKRAHHNALERKRRDHIKDSFHSLRDSVPSLQG-EKASRAQILDKATEYIQYMRRKNHTMAX_CHICK ADKRAHHNALERKRRDHIKDSFHSLRDSVPSLQG-EKASRAQILDKATEYIQYMRRKNHTMAX_XENOPUS ADKRAHHNALERKRRDHIKDSFHGLRDSVPSLQG-EKASRAQILDKATEYIQYMRRKNHTMAX_ZFISH ADKRAHHNALERKRRDHIKDSFHSLRDSVPALQG-EKASRAQILDKATEYIQYMRRKNHTMYCX_CARP ADKRAHHNALERKRRDHIKDSFHSLRDSVPALQG-EKASRAQILDKATEYIQYMRRKNHTzMax_Zfish ADKRAHHNALERKRRDHIKDSFHSLRDSVPALQG-EKASRAQILDKATEYIQYMRRKNHTXMax2_Xpus ADKRAHHNALERKRRDHIKDSFHGLRDSVPSLQG-EKASRAQILDKATEYIQYMRRKNHTDMAX_FLY AEKRAHHNALERRRRDHIKESFTNLREAVPTLKG-EKASRAQILKKTTECIQTMRRKISEF46G10_WORM DDRRAHHNELERRRRDHIKDHFTILKDAIPLLDG-EK-SRALILKRAVEFIHVMQTKLSSMAX1_WORM RHAREQHNALERRRRDNIKDMYTSLREVVPDANG-ERASRAVILKKAIESIEKGQSDSATMAD_MOUSE TSSRSTHNEMEKNRRAHLRLCLEKLKGLVP-L-GPESHTTLSLLTKAKLHIKKLEDCDRKMAD3_MOUSE NSGRSVHNELEKRRRAQLKRCLEQLRQQMP-L-GVDCYTTLSLL-RARVHIQKLEEQEQQMAD4_XENLA QNNRSSHNELEKHRRAKLRLYLEQLKQLVP-L-GPDSHTTLSLLKRAKMHIKKLEEQDRKMAD4_XENLA TVGRSSHNELEKHRRAKLRLYLEQLKQLVP-L-GPDSHTTLSLLKRAKMHIKKLEEQDRKMAD4_HUMAN PNNRSSHNELEKHRRAKLRLYLEQLKQLVP-L-GPDSHTTLSLLKRAKVHIKKLEEQDRRMAD4_MOUSE PNNRSSHNELEKHRRAKLRLYLEQLKQLGP-L-GPDSHTTLSLLKRAKMHIKKLEEQDRRMADL1H_WORM KHSRTAHNELEKTRRANLRGCLETLKMLVPCVSDA--NTTLALLTRARDHIIELQDSNAAMXI1_HUMAN TANRSTHNELEKNRRAHLRLCLERLKVLIP-L-GPDCHTTLGLLNKAKAHIKKLEEAERKMXI1_MOUSE TANRSTHNELEKNRRAHLRLCLERLKVLIP-L-GPDCHTTLGLLNKAKAHIKKLEEAERK
Typical Aligned Biological Sequence Data
Sequence data are highly dense, alphabetic, systematic missing data, often highly conserved (low variability), little replication, little within protein variability, usually more amino acids than sequences, etc. Very difficult to analyze statistically in a meaningful way

A major goal of biological research is to provide general models or systematic
principals to explain complex phenomena. In proteins, this involves
the molecular architecture of their component parts their origins,
regulation, interrelationships and evolution.

MDALQLANSAFAVDLFKQLCEKEPLGNVLFSPICLSTSLSLAQVGAKGDTANEIGQVLHFENVKDIPFGFQTVTSDVNKLSSFYSLKLIKRLYVDKSLNLSTEFISSTKRPYAKELETVDFKDKLEETKGQINNSIKDLTDGHFENILADNSVNDQTKILVVNAAYFVGKWMKKFPESETKECPFRLNKTDTKPVQMMNMEATFCMGNIDSINCKIIELPFQNKHLSMFILLPKDVEDESTGLEKIEKQLNSESLSQWTNPSTMANAKVKLSIPKFKVEKMIDPKACLENLGLKHIFSEDTSDFSGMSETKGVALSNVIHKVCLEITEDGGDSIEVPGARILQHKDELNADHPFIYIIRHNKTRNIIFFGKFCSP
Serine proteinase inhibition
(Ovalbumin)
Computational Biology Holy GrailSequence → Structure → Function → Evolution
O
BC
D
E
O
B
C
D
E
Interrelationships with other proteins –
the “Network” Problem
Evolution of the network

Are phylogenetic trees the best
way to describe
protein sequence
variability and evolution?

Phylogenetic Trees: Pros
Trees are good for• describing hierarchical patterns• clustering taxa• describing extent of lineage divergence• estimating ancestral relationships• summarizing overall changes in data.

Phylogenetic Trees: cons
Trees are NOT very good for:• understanding dimensionality• analyzing covariation• describing the biological basis of evolutionary divergence• elucidating the components upon which selection might operate

Significance of Covariation
• Understanding covariation is fundamental to modeling protein structure and evolution• Evolutionary and structural change constrained by covariation among amino acids • Accurate prediction of protein structure requires knowledge of covariance structure• Covariation reduces the dimensionality of phylogenetic information• Analytical procedures (like ML) make strong assumptions about covariances

Structural constraints
Decomposition of Covariance Among Amino Acid sites i
and jCij = CP + CS + CF + CI + e
Phylogenetic
constraints
Functionalconstraints
Interactions between model
components
Unexplainedeffects

abcdef
abcDeF
aBcdeF
abCDeF abCDeF
AbCdeF
abcDeF
abcDef
aBCdeF
ABcdeF
aBCdEF
ABcdeF
ABcdeF
Geneduplication(paralogy)
orthology
evolutionary time
Phylogeneticconstraints
(Taxa related by a common evolutionary history)

Compensatoryinteractions
7
1
2
34
5
6
Proximityeffects
Constraints dueto folding
Structural constraints – associationsamong amino acids arise from the
3-dimensional geometry or “folding” pattern of proteins.

Some Structural Associations in Proteins
Hydrophobic interactions: associations among amino acids with non-polar side chains
Salt bridges: correlations between charged residues
Hydrogen bonds: correlations between electron donors and receptors
Size constraints: correlations between large and small side chains

bHLH transcriptional regulators control a diverse array of developmental processes.
Basic (B) region binds to hexanucleotide E-box and controls gene expression.
Helix-loop-helix (HLH) region involved in protein – protein interactions (dimerization)
At least 5 different DNA binding groups in bHLH proteins based on how basic region interacts with E-Box
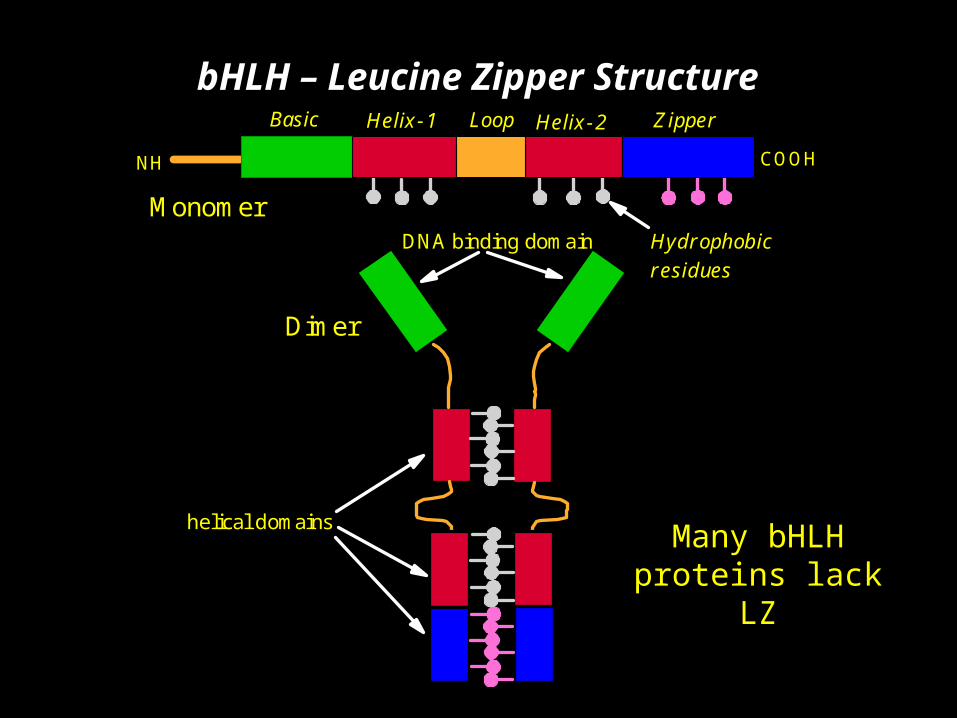
bHLH – Leucine Zipper StructureBasic Helix-1 Loop Helix-2 Zipper
COOH
Hydrophobic residues
Monomer
Dimer
DNA binding domain
helical domains
NH
Many bHLH proteins lack
LZ

i j k . n
i Ei
j Mij Ej
k Mik Mjk Ek
. . . . .
n Min Mjn Mkn . En
Generalized Covariance MatrixAmino acid sites
E reflects amino acid diversity at each siteM describes mutual information between sites

Entropy Dynamics in bHLH Domain
B-2
B-6
B-7
B-8
B-11
B-12
H1-14H1-21
H1-25
H1-26
H2-64
H2-63
H2-62
H2-61
H2-60
H2-59H2-56
H2-55
H2-52
H2-57
H2-54H2-50
H2-53
H2-51
L-49L-48
L-47
L-31
L-46
L-45
L-29 L-30
H1-28
H1-24
H1-27
H1-20
B-4
B-5
H1-22
H1-23
H1-17
H1-18
H1-15
B-9
B-10B-13
H1-16B-1
B-3
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
0.90
1.00
B-1
B-2
B-3
B-4
B-5
B-6
B-7
B-8
B-9
B-1
0B
-11
B-1
2B
-13
H1-
14H
1-15
H1-
16H
1-17
H1-
18H
1-19
H1-
20H
1-21
H1-
22H
1-23
H1-
24H
1-25
H1-
26H
1-27
H1-
28L
-29
L-3
0L
-31
L-4
5L
-46
L-4
7L
-48
L-4
9H
2-50
H2-
53H
2-54
H2-
55H
2-56
H2-
57H
2-58
H2-
59H
2-60
H2-
61H
2-62
H2-
63H
2-64
H2-
65H
2-66
Amino Acids
No
rma
lize
d E
ntr
op
y
Dynamic pattern indicative of amphipathic α-helix with highly variable hydrophilic face alternating with conserved hydrophobic core. Spectral analysis confirms periodicity of ~3.6.
Zhi Wang
Packed sites in Max defining hydrophobic core are in red Packed sites in Max defining hydrophobic core are in red
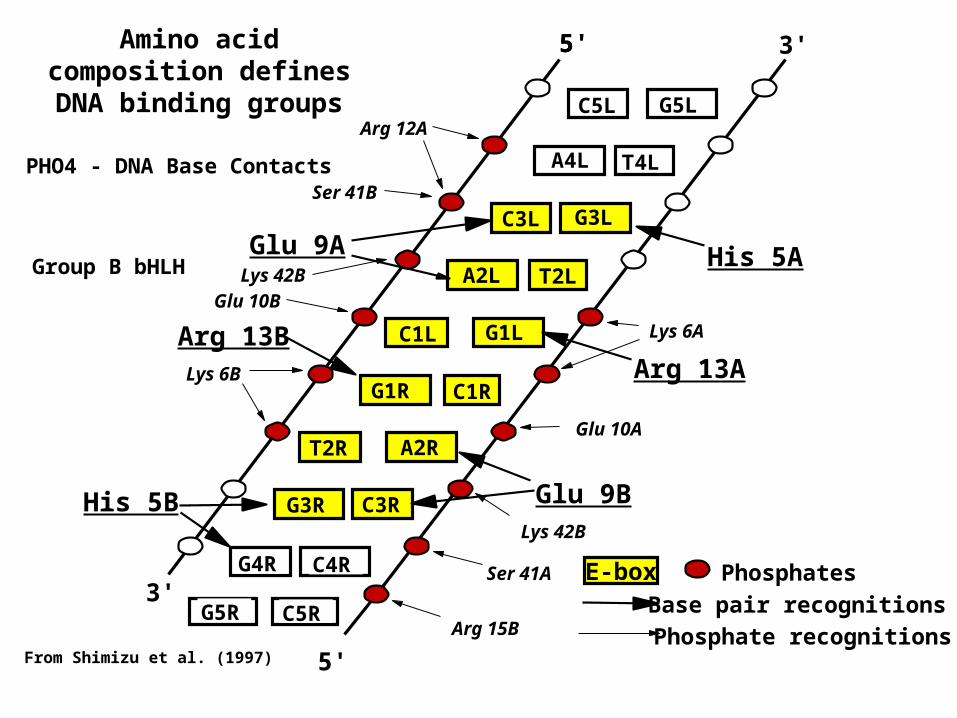
Amino acid composition defines DNA binding groups
C3L
A2L
C1L
G4R
G1R
A4L
G3R
T2R
C5L G5L
T4L
G3L
T2L
G1L
C1R
A2R
C3R
C4R3'
5' 3'5'
5'
Glu 9A
Arg 13B
His 5B
His 5A
Arg 13A
Glu 9B
Arg 12A
Ser 41B
Lys 42B
Lys 6B
Lys 6A
Glu 10B
Glu 10A
Ser 41A
Lys 42B
Arg 15BC5RG5R
From Shimizu et al. (1997)
E-box Phosphates
Base pair recognitions
Phosphate recognitions
PHO4 - DNA Base Contacts
Group B bHLH

Packed sites in Max are underlined
Entropy Dynamics in bHLH Domain
B-2
B-6
B-7
B-8
B-11
B-12
H1-14H1-21
H1-25
H1-26
H2-64
H2-63
H2-62
H2-61
H2-60
H2-59H2-56
H2-55
H2-52
H2-57
H2-54H2-50
H2-53
H2-51
L-49L-48
L-47
L-31
L-46
L-45
L-29 L-30
H1-28
H1-24
H1-27
H1-20
B-4
B-5
H1-22
H1-23
H1-17
H1-18H1-15
B-9
B-10B-13
H1-16B-1
B-3
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
0.90
1.00B
-1B
-2B
-3B
-4B
-5B
-6B
-7B
-8B
-9B
-10
B-1
1B
-12
B-1
3H
1-14
H1-
15H
1-16
H1-
17H
1-18
H1-
19H
1-20
H1-
21H
1-22
H1-
23H
1-24
H1-
25H
1-26
H1-
27H
1-28
L-2
9L
-30
L-3
1L
-45
L-4
6L
-47
L-4
8L
-49
H2-
50H
2-53
H2-
54H
2-55
H2-
56H
2-57
H2-
58H
2-59
H2-
60H
2-61
H2-
62H
2-63
H2-
64H
2-65
H2-
66
Amino Acids
No
rma
lize
d E
ntr
op
y
Contacts backbone in some groups
Contacts base
Contacts both
Contacts phosphate backbone

Dotted lines describe packing interactions
Solid arrows contacts with phosphate backbone
Dotted arrows contact with backbone and DNA
Intersite interactions in bHLH Domain
B-2
B-6
B-7
B-8
B-11
B-12
H1-14H1-21
H1-25
H1-26
H2-64
H2-63
H2-62
H2-61
H2-60
H2-59H2-56
H2-55
H2-52
H2-57
H2-54H2-50
H2-53
H2-51
L-49L-48
L-47
L-31
L-46
L-45
L-29 L-30
H1-28
H1-24
H1-27
H1-20
B-4
B-5
H1-22
H1-23
H1-17
H1-18
H1-15
B-9
B-10B-13
H1-16B-1
B-3
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
0.90
1.00
B-1
B-2
B-3
B-4
B-5
B-6
B-7
B-8
B-9
B-1
0B
-11
B-1
2B
-13
H1-
14H
1-15
H1-
16H
1-17
H1-
18H
1-19
H1-
20H
1-21
H1-
22H
1-23
H1-
24H
1-25
H1-
26H
1-27
H1-
28L
-29
L-3
0L
-31
L-4
5L
-46
L-4
7L
-48
L-4
9H
2-50
H2-
53H
2-54
H2-
55H
2-56
H2-
57H
2-58
H2-
59H
2-60
H2-
61H
2-62
H2-
63H
2-64
H2-
65H
2-66
No
rma
lize
d E
ntr
op
y
H1-16 packs against H1-20
H1-20 has many van der Waal contacts with H2 side chains
H1-23 packs against H2-53, H2-57
H-27 packs against H2-60, H2-61
H-28 terminates Helix 1
L-47 stabilizes loop path
H2-50 starts Helix 2, packs with H1-20, anchors Helix 2 to DNA
H2-53 packs against H2-50 and H2-54’
H2-54 packs against H2-50 and H2-53
H2-61 packs against symmetry mate 61 and 60
H2-64 interacts with symmetry mate and terminates helix 2
Max protein
HLH x HLH interaction
region

Factor Analysis of Mutual Information Matrix of Amino Acids
• 64 amino acids of bHLH domain, 288 sequences• 64 x 64 matrix of standardized MI matrix elements• Maximum Likelihood factor analysis used• 7 factors extracted that accounted for all of the common
information• Multivariate patterns of amino acid covariation described
and then related to known 3-D structure of bHLH domain from crystal structure studies

Factor Analysis of bHLH Domain Covariances
• Analyses involving covariances among 49 amino acid sites in bHLH domain• 288 separate bHLH domain sequences• Normalized mutual information values used• Seven significant eigenvectors • Each reflected significant multivariate components of covariation• Each eigenvector represented important phylogenetic, structural and functional information
Michael Buck

Flow of Statistical Analyses
Multiple alignment of sequences
Compute E, MI matrix for sequence elements
Factor analysis of MI matrix. patterns of covariation
Project factor coefficients onto RasMol models.
Interpret
Factor Analysis of 500 amino acid
attributes. Compute factor
scores
Transform alphabetic amino acid
codes to numericalfactor scores.
(5 datasets)
Compute R matrices.of each data set. Factor analysis on
each dataset
Determine patterns of
physiochemicalvariation within
proteins
Model underlyingcauses of
multidimensional protein variation
Model underlyingcauses of
multidimensional protein variation
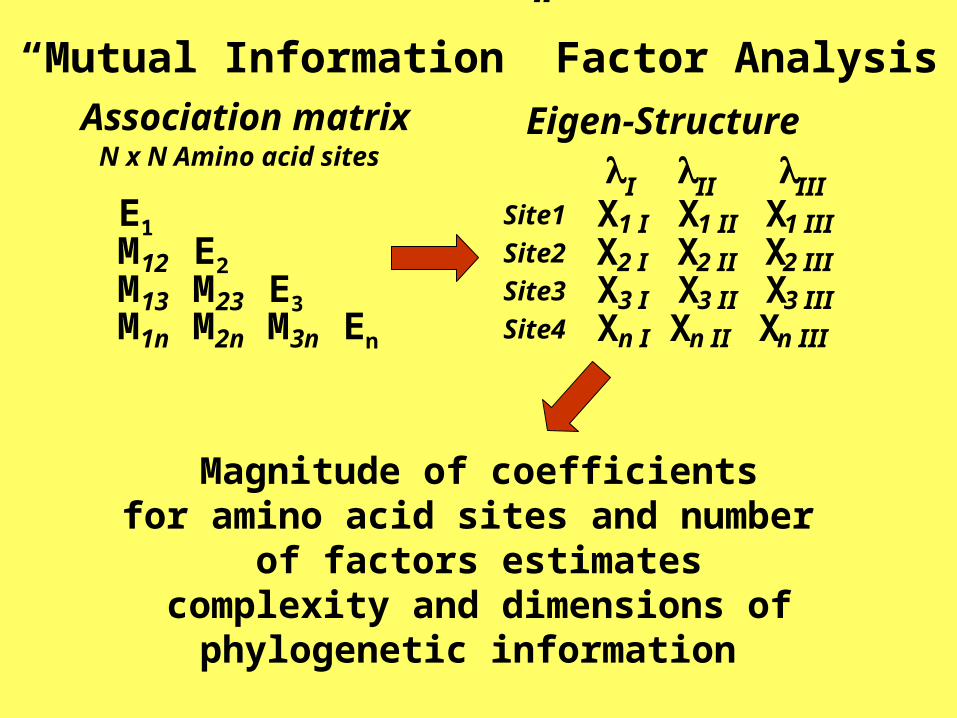
I II IIISite1 X1 I X1 II X1 IIISite2 X2 I X2 II X2 IIISite3 X3 I X3 II X3 IIISite4 Xn I Xn II Xn III
Eigen-Structure
E1M12 E2M13 M23 E3M1n M2n M3n En
Association matrixN x N Amino acid sites
“Mutual Information” Factor Analysis
Magnitude of coefficientsfor amino acid sites and number
of factors estimatescomplexity and dimensions of
phylogenetic information

Site Domain Factor1 Factor2 Factor3 Factor4 Factor5 Factor6 Factor7 Comm
site30 L-2 0.646 0.161 0.112 0.254 0.066 0.138 0.079 0.549site31 L-3 0.646 0.136 0.108 0.042 0.344 -0.131 -0.137 0.604site29 L-1 0.599 0.113 0.054 0.069 0.162 0.077 0.063 0.415site63 H2-14 0.548 0.190 0.108 0.152 0.039 0.085 0.099 0.389site4 B-4 0.541 0.173 0.078 0.081 0.120 0.135 0.189 0.404
site56 H2-7 0.524 0.237 0.259 0.287 0.004 -0.001 0.171 0.509site21 H1-8 0.502 0.129 0.233 0.210 0.081 0.205 0.158 0.440site58 H2-9 0.501 0.120 0.323 0.062 0.065 -0.009 0.041 0.380site3 B-3 0.491 0.174 0.160 0.190 0.091 0.162 0.144 0.389
site45 L-4 0.488 0.100 0.081 0.153 0.233 0.052 0.102 0.345site15 H1-2 0.486 0.136 0.234 0.122 0.216 0.063 0.151 0.398site11 B-11 0.479 0.196 0.171 0.198 0.026 0.178 0.143 0.389site14 H1-1 0.472 0.252 0.344 0.135 0.133 0.107 0.121 0.467site62 H2-13 0.471 0.123 0.207 0.103 0.138 0.024 0.129 0.327site18 H1-5 0.470 0.123 0.009 0.111 0.037 0.072 0.143 0.275site59 H2-10 0.453 0.186 0.163 0.106 0.019 0.025 0.135 0.297site25 H1-12 0.450 0.104 0.091 0.115 0.159 0.143 0.182 0.314site7 B-7 0.446 0.167 0.158 0.207 0.149 0.177 0.083 0.355
site52 H2-3 0.443 0.056 0.338 0.190 0.254 0.141 0.130 0.451site46 L-5 0.422 0.100 0.161 0.134 0.323 0.100 0.180 0.378site26 H1-13 0.416 0.063 0.404 0.153 0.076 0.064 0.126 0.389site51 H2-2 0.402 0.271 0.362 0.197 0.245 -0.009 -0.029 0.466site55 H2-6 0.392 0.228 0.310 0.260 0.155 -0.016 0.164 0.421site5 B-5 0.391 0.276 0.333 0.190 0.265 0.202 0.122 0.502
site22 H1-9 0.386 0.213 0.129 0.175 0.069 0.030 0.048 0.250site19 H1-6 0.379 0.273 0.334 0.078 0.238 0.237 0.140 0.469site8 B-8 0.375 0.405 0.296 0.031 0.255 0.223 0.198 0.547
site13 B-13 0.370 0.267 0.452 0.051 0.163 0.042 0.030 0.445site48 L-7 0.337 0.174 0.266 0.256 0.062 -0.022 0.374 0.424site27 H1-14 0.305 0.115 0.091 0.396 0.016 0.084 0.051 0.281site6 B-6 0.299 0.495 0.193 0.314 0.223 0.244 0.011 0.579
site16 H1-3 0.294 0.344 0.345 0.190 0.062 0.228 -0.025 0.417site1 B-1 0.285 0.262 0.053 0.058 0.046 0.040 0.292 0.245
site49 L-8 0.263 0.148 0.300 0.314 0.466 0.113 0.062 0.514site20 H1-7 0.262 0.440 0.287 0.060 0.128 -0.077 0.015 0.371site47 L-6 0.238 0.050 0.100 0.407 0.031 0.272 0.076 0.315site28 H1-15 0.234 0.078 0.037 0.552 0.219 -0.054 0.068 0.423site24 H1-11 0.226 0.088 0.275 0.340 0.155 0.234 -0.019 0.329site64 H2-15 0.204 0.096 0.055 0.074 0.297 0.350 0.209 0.313
Eigenvalue 14.582 1.982 0.993 0.841 0.647 0.591 0.523
Cumulat % 0.723 0.822 0.871 0.913 0.945 0.974 1.000
Portion of Factor matrix of MI values for 64 amino acid sites of bHLH domain. Varimax rotation of ML factor solution. High coefficients on each vector shown in yellow.

Factor analyses describe:
• Dimensionality of shared and unique covariation
• Major patterns of amino acid covariation among all major bHLH lineages• A model for structural and sequence evolution in bHLH• An understanding of the biological bases of simultaneous changes among amino acid sites

DNA
Factor 1
> 0.5> 0.4> 0.3< 0.3
Factor Coefficients
bHLH monom
er
Accounts for 72% of sequence common covariance in 288 proteins
22 of 49 sites with factor coefficients > 0.4
Most sites with high coefficients occur on exposed or hydrophilic face of helices
High correlation between factor coefficients and site by site entropy values, clade membership and loop length
Sequence variation reflected by Factor 1 has strong phylogenetic signal

Factor 1Showing the orientation of the side-chains of the amino acids on the
hydrophilic surface and awayfrom DNA

Use dummy variables for classification codes
Estimate phylogenetic tree from well-aligned sequences
Define clades (monophyletic lineages)
Delimiting clades uses both biological and
statistical information -- clade definition can be somewhat subjective
Assign dummy variable to all sequences in each clade
Covariance between given site and dummy variable measures phylogenetic signal
Prediction of clade membership by multivariate statistics used to define “sequence signatures”
“Group membership” approach useful for structural and functional variables also
?
A
C
D
E
F
B
G
H
Estimating phylogenetic signal in any amino acid?

Fact1 Fact2 Fact3 Fact4 Fact5 Fact6 Fact7
clade 0.705 0.078 0.725 0.244 0.219 -0.039 -0.131
group 0.168 0.414 0.584 0.092 0.424 0.161 -0.070
loop-len 0.741 0.146 0.531 0.277 0.233 -0.147 -0.319
comm 0.276 0.598 0.234 0.086 0.164 0.150 -0.180
entropy 0.938 -0.149 0.298 0.153 0.030 -0.088 -0.038
Correlations of Factor Coefficients of Pair-wise Mutual Information Values with Extrinsic Variables
Clade = monophyletic lineages of proteins with equivalent functions
Group = DNA Binding Groups based upon E-Box binding patterns
Loop-length = number of residues in the loop region separating helices 1 and 2
Comm = Communality from factor analysis; amount of variability at site explained by 7 factors
Entropy = extent of uncertainty (variability) at each site in the bHLH sequence domain
Critical correlation coefficient to reject Ho at P < 0.01 = 0.43

B-2
B2-57
B-9
H1-12
B-10
B-9
B-6
H1-20
H1-16
B-8
B-6
H1-16
Factor 2
10% of sequence variability Large factor coefficients for 8 sites:
6 from DNA binding region, 1 from each helix
B2, B6, B8, B10 and B12 involved in protein side-chain – phosphate backbone contacts. B9 also contacts DNA base
Site H1-20 buried site with many van der Waal contacts with Helix 2. H2-57 important structurally.
Sites important to maintain structural “geometry”
All sites with high coefficients occur at nadirs of entropy dynamics plots. Highly conserved but intrinsic variability covarys among these 8 sites
7 of 8 sites components of “sequence signature” that identifies all bHLH proteins

Packed sites in Max are underlined
Entropy Dynamics in bHLH Domain
B-2
B-6
B-7
B-8
B-11
B-12
H1-14H1-21
H1-25
H1-26
H2-64
H2-63
H2-62
H2-61
H2-60
H2-59H2-56
H2-55
H2-52
H2-57
H2-54H2-50
H2-53
H2-51
L-49L-48
L-47
L-31
L-46
L-45
L-29L-30
H1-28
H1-24
H1-27
H1-20
B-4
B-5
H1-22
H1-23
H1-17
H1-18
H1-15
B-9
B-10B-13
H1-16B-1
B-3
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
0.80
0.90
1.00B
-1B
-2B
-3B
-4B
-5B
-6B
-7B
-8B
-9B
-10
B-1
1B
-12
B-1
3H
1-14
H1-
15H
1-16
H1-
17H
1-18
H1-
19H
1-20
H1-
21H
1-22
H1-
23H
1-24
H1-
25H
1-26
H1-
27H
1-28
L-2
9L
-30
L-3
1L
-45
L-4
6L
-47
L-4
8L
-49
H2-
50H
2-53
H2-
54H
2-55
H2-
56H
2-57
H2-
58H
2-59
H2-
60H
2-61
H2-
62H
2-63
H2-
64H
2-65
H2-
66
Amino Acids
No
rma
lize
d E
ntr
op
y
High coefficients – Factor 1
High coefficients – Factor 2

H1-13
H1-14
B-5 Factor 3
H1-26
H1-14
H1-26
H2-49H1-16
H1-19 H2-51
H2-55
H2-58
B-5
H2-58
8 sites with large factor coefficients
Sites involved with interrelationships between variable and conserved sites. Each site adjacent to highly conserved “packed” site.
Suggests role in compensatory variation
Potentially important to maintain geometry of hydrophobic core
Strong phylogenetic content.

Factor 4H1-28
B-6 B-6
L-47
L-49L-49
L-47
H1-24
H1-27
H2-60
H1-28 – P in 75% of sequences, initiates loop
H1-27 – packs against H2-60, H2-61
L-47 – stabilizes loop path back into groove
H2-60 – interaction with helix 1 (H1-27)
Definition of the Loop Region

MyoD 3-D Structure
DNA Binding Group - A

SREBP
Max
USF
MyoD
Models of bHLH-DNA Structure
PHO4
• Structures available for 6 proteins
• Good fit of all to canonical structure
• Function of bHLH domain well- understood
• Simple domain structure
• Phylogeny well-documented





























