Embed Size (px)
Citation preview

Ckwcicd Engineerhg Schxce. Vol. 41. No. 6, pp. 1631-1645. 1986. OOO!-X09/86 13.00 + 0.00 Printed in Great Britain. Pergamon Journals Ltd.
MATHEMATICAL MODELLING OF DIFFUSION AND REACTION IN BLOCKED ZEOLITE CATALYSTS
S. SUNDARESAN? and C. K. HALL* Department of Chemical Engineering, Princeton University, Princeton, NJ 08544, U.S.A.
(Received 4 December 1985)
Abstract-A mathematical model for diffusion and reaction in blocked zeolites is developed which takes into account nonidealities arising from interaction between sorbed molecules as well as the effect of pore and surface blocking. The model combines a microscopic approach, in which expressions for chemical potential and diffusive fluxes are calculated within the lattia-gas framework, with the more traditional continuum approach which takes into account the effect of surface blocking. The effect of pore blocking on the diffusive fluxes is accounted for through an effective medium approximation. The effects of crystal size and blocking on the activity-selectivity characteristics of the crystal are illustrated through an example that is qualitatively similar to industrially important reactions such as alkylation of toluene in blocked ZSM-5 zeolite.
1. INTRODUCTION Molecular shape selectivity in catalysis by zeolite crystals, which are regular grids of intersecting pores of typically molecular dimensions, has been described in several studies (Chen et al., 1979; Dejaifue et al., 1981; Haag et al., 1982; Kaeding ef al., 1981a,b, 1984a, b; Olson and Haag, 1984; Vedrine et al., 1982; Yashima et al., 197Oa, b; Wei, 1982; Weisz, 1980). It is generally agreed that shape selectivity arises from one or both of the following two mechanisms: First, certain reaction complexes (transition states) are too large to be formed in the zeolite cavities and hence are excluded (Haag et al., 1982; Olson and Haag, 1984). For example, the high selectivity of ZSM-5 in xylene isomerization relative to larger-pore acid catalysts is attributed to the selective retardation of the undesired transalkylation of xylenes in ZSM-5 (Olson and Haag, 1984). The second mech- anism for selectivity is due to the large differences in the diffusivities of the variously sized molecular species in the intracrystalline channels (Haag et al., 1982; Olson and Haag, 1984; Wei, 1982). For example, small, unmodified crystals of HZSM-5 do not exhibit pro- nounced para-selectivity toward xylene isomerization, alkylation of toluene and toluene disproportionation, at low temperatures. Catalyst modifications such as coking (Dejaifue et al., 1981; Kaeding et al., 1981a, 1984a,b; Olson and Haag, 1984), treatment with phosphorus compounds (Kaeding et al., 1981a, 1984a,b; Vedrine et al., 1982), impregnation with boron or magnesium compounds (Chen et al., 1979, Kaeding et al., 1981a, b, 1984% Young et al., 1982) and surface coating with polymers (Kaeding et al., 1981a), as well as increasing the crystallite size lead to an enhancement of para-selectivity. Modification by boron or magnesium compounds causes the pores within the crystal to be blocked while treatment with
tTo whom corresvondence should be addressed. ~Present address:-Department of Chemical Engineering,
North Carolina State University, Raleigh, NC 276957905, U.S.A.
phosphorus compounds, surface coating and coking results in a random blocking of pore entrances along the surface of the crystal (Theodorou and Wei, 1983). All of these methods result in increased diffusional resistance, which, in large and/or modified crystals is believed to be responsible for this increase in para- selectivity (Olson and Haag, 1984; Wei, 1982).
The problem of diffusion and reaction in blocked zeolites has been studied theoretically by Theodorou and Wei (1983) within the framework of the lattice-gas model. Using the Monte Carlo computer simulation technique they investigated the effects of pore and surface blocking on the diffusion process but neglected the effects of interaction between molecules. Our work builds on the analysis of Theodorou and Wei. A mathematical model for diffusion and reaction in blocked zeolites is developed which takes into account the nonidealities arising from direct and indirect interactions between sorbed molecules as well as the effect of pore blockage due to added impurities. A model of broad generality is obtained by combining a microscopic approach, in which expressions for chemi- cal potential and diffusive fluxes are calculated within the lattice-gas framework, with the more traditional continuum approach which takes into account the effect of surface blocking. The effect of pore blocking on the diffusive fluxes is accounted for through an effective medium approximation (Kirkpatrick, 1973). The effects of crystallite size and blocking on the activity-selectivity characteristics of the crystal are illustrated through an example that is qualitatively similar to industrially important reactions such as alkylation of toluene in blocked ZSM-5 zeolite.
2. MODEL FORMULATION Diffusion and reaction in zeolite crystals will be
analysed within the framework of the latticegas model of statistical mechanics. This allows us to treat the effects of pore blocking and of interactions be- tween sorbed molecules on a molecular level. In the lattice-gas approach, the zeolite crystal is modelled as
1631

1632 S. SLINDARESAN and C. K. HALL
an array of sites, each of which is connected to z nearest neighbouring sites by pores of equal length. In our model, z is assumed to be either 4 or 6 depending on whether the grid of intersecting channels is two- or three-dimensional. Though the above representation is an over-simplification of the actual channel networks in zeolites (Kokotailo et al., 1978; Meier and Olson, 1978), it retains those features, regularity of the crystal and interconnectivity of the pores, which are most important in the present context. Such representations have been used previously to analyse diffusion in zeolites (Barrer and Jost, 1949; Barrer and Rees, 1954a,b, Ruthven, 1974; Theodorou and Wei, 1983).
We shall derive a continuum model for diffusion and reaction in the crystal from this lattice-gas model. A continuum model is justified only for crystals contain- ing many sites; accordingly we confine our treatment to large crystals.
In our continuum model, hindered diffusion within the interior of the crystal will be modelled using pore blocking alone, with surface-site blocking entering the analysis through boundary conditions. This is reason- able since, for a large crystal the extent of surface-site blocking should have little effect on the details of the diffusion and reaction processes in the interior of the crystal (far away from the surface), and similarly, the extent of pore blocking far away from the surface is not likely to have a significant effect on the process of sorbate transfer across the surface layer.
Let us first consider the interior of the crystal. The sorbed molecules are typically found only in the
sites. Each site can be occupied by at most one sorbate molecule. The diffusion of sorbed molecules may be viewed as a succession of discrete jumps from site to site, with jumps to occupied sites forbidden. Molecules move through the pores in single file, thus two molecules cannot cross each other moving in opposite directions in a pore. The migration of a molecule from one site to an adjacent unoccupied site through the pore connecting these sites is an activated process. An isolated sorbate molecule of species i executes kmi jumps per unit time in the absence of pore blocking, with an activation energy Emi.
k,j = k”,, exp ( - E,,JR T).
A sorbate will move from an occupied site to any of the unoccupied adjacent sites connected by unblocked pores with equal probability_ Following Theodorou and Wei (1983), we assume that pore blocking does not completely exclude transport through a pore, but diminishes the probability of migration through it by a factor S. In the configurational diffusional regime encountered in diffusion through zeolite pores, 6 4 1 since even a small change in the pore radius as a result of blocking can be expected to lead to a large decrease in the diffusivity through it (Theodorou and Wei, 1983).
The migration of a sorbed molecule in the crystal may be modified by the presence of other sorbates, beyond the simple steric effects discussed above. For example, it is quite conceivable that a sorbed molecule
would be more likely to jump to an adjacent unoc- cupied site if some or all of the remaining (z - 1) adjacent sites were occupied, than if all the adjacent sites were unoccupied. This is equivalent to saying that the sorbed molecules in the adjacent sites tend to push the molecule under consideration out of its site. Such an effect can be rationalized as follows: A sorbed molecule occupying site I does not know a priori whether an adjacent site II is occupied or empty. It appears more likely that the sorbed molecule in site I will have to enter the pore structure leading to site II and traverse at least a part of the distance between the two sites before it can determine if the site at the other end of the pore (site II) is occupied. If site II is already occupied, then the molecule cannot enter that site and will be repelled back to its original site. During the course of this event, it is reasonable to expect that the occupant of site II would have experienced a push from the occupant of site I, encouraging it to vacate its site. Such a tendency of the sorbed molecules to push on one another may be modelled as repulsive interactions between molecules occupying nearest-neighbour sites. Weak long-range interactions between the sorbates may also exist, but will not be considered here.
We assume that the total interaction energy of the sorbates can be treated as a sum of two-body interac- tions which are nearest-neighbour repulsions as de- scribed above. The activation energy for the migration of a species i molecule from a site to an adjacent empty site is equal to
E, - c (tnjqj + m;Ei’i) i
where .zij(~$) is the interaction energy exerted on the species i molecule by a speciesj molecule occupying an adjacent site with the pore joining these sites being unblocked (blocked), mj(mf) is the number of neigh- bouring sites occupied by speciesj molecules with the pores leading to thesej molecules from the i molecule under consideration being unblocked (blocked) and the sum is over all speciesj. Repulsive interactions are assigned positive values. The energetics of sorption are also modified in a similar fashion. In general, sr, # eij and et # eJ+i, for i #j.
Clearly, any realistic model of sorption equilibrium and sorbate diffusion which takes intoaccount sorbate interactions will depend on how the sorbed molecules distribute themselves in the lattice (the configurations). A rigorous lattice-gas treatment of this problem would require that we be able to calculate the partition function for the multicoponent, nearest-neighbour lattimgas model exactly, a formidable problem which has yet to be solved. Instead we treat the problem using a simple approximation in which all pairs of sites are treated independently, the so-called quasi-chemical approximation. The quasi-chemical approximation for the chemical potential will be given in Section 2.1. In addition to being able to treat the configuration of sorbed molecules, we must be able to treat the configuration of the blocked pores, particularly since we are interested in diffusion. We will assume here that

Diffusion and reaction in blocked zeolite catalysts 1633
a fraction, &, of the pores are blocked and that the blocked pores are distributed randomly.
The broad lattice--gas framework described above is adequate to derive expressions for the chemical poten- tials of sorbed molecules, their diffusive fluxes and the rates of chemical reactions. These expressions, when combined suitably, form the continuum model for diffusion and reaction.
For the sake of illustration, we consider the forma- tion of a product having two isomers A and B from a set of reactants, R denoting the limiting reactant. We assume that the product molecules are large and that only one product molecule can fit in a site at a time. The reactant molecules are assumed to be small compared to the radius of the pores in the zeolite crystal and so move in and out of the zeolite crystal easily without being blocked by the product molecules or by the pore- blocking agents. The formation of the product mol- ecules takes place in the sites, provided that these sites are not already occupied by product molecules. In addition to the formation reaction, the isomerization reaction A P B takes place in the crystal. We wish to study the effect of diffusional resistance on the ob- served selectivity, if the intrinsic mobilities of the two isomers A and B through the zeolite pore structure are different. Industrially important reactions such as alkylation of toluene in blocked ZSM-5 zeolite are qualitatively similar to this example.
Under reaction conditions there will be gradients in the chemical potentials of A and B in the crystal which provide the driving force for the diffusion of these species. Though it is possible that a gradient in the chemical potential of one species can give rise to a diffusional flux of other species, we have no reason to believe that such an effect is important in the problem under study. Hence we assume that the diffusive flux of a species depends only on its chemical potential gradient. We also assume that local thermodynamic equilibrium prevails at every location in the zeolite (from a continuum viewpoint). This assumption allows us to convert the chemical potential gradients into composition gradients_ However, it is important to note the limitations of this assumption. It can be shown that in the context of a single species diffusing in an unblocked lattice, the assumption of local equilib- rium is an acceptable approximation if L %- 1, where L and 1 denote the characteristic length of the crystal and lattice spacing, respectively. In binary systems, it can be shown that the local equilibrium assumption requires
kmi(L/l)’ a k,, i, j = A, B.
In situations where kmA u lo3 k,, [which is typical for the diffusion of xylene isomers in ZSM-5 (Wei, 1982)], the above requirement is satisfied for lattices with L N 100 1 or larger. This corresponds to crystals of 0.1 pm or larger (corresponding to I _ 10 A). Industrial catalysts contain crystals as large as 1 pm. Thus the assumption of local equilibrium is acceptable for large unblocked zeolite crystals of practical interest.
A further assumption inherent in our model con- cerns the fraction of pores blocked. At low levels of
pore blocking (i.e. very small f,), the blocked pores occur singly or in small isolated clusters of adjacent blocked pores. Now, as the fraction of pores that are blocked increases, larger clusters are formed and the mean size of the clusters increases monotonically. As fP approaches a critical value & (called the percolation threshold). the large clusters begin to merge creating a few extremely large clusters, so that the mean cluster size approaches the size of the crystal itself (Kirkpatrick, 1973). It is important to note that in a continuum model for diffusion and reaction in a zeolite crystal, the composition of sorbates at any location on the crystal usually refers to an average over a sample volume (containing several sites) in the vicinity of the given location. This sample volume must be much larger than the mean size of the blocked pore clusters for the averaging process to be meaningful. It then follows that a continuum model is justifiable only if fP is well below fpc, which we assume. It has been established in the literature that in this region the effect of pore blocking can be treated through an effective medium approximation (Kirkpatrick, 1973). We shall discuss this in greater detail in Section 2.2.
2.1. Expressions for the chemical potentials A brief description of the quasi-chemical approxi-
mation for the chemical potential in terms of com- positions is presented. Consider a lattice containing M sites. Let N* and N, be the number of molecules of A and 3 in the lattice. The number of unoccupied sites, No, is equal to M -N, -IV,. Let N, (Nt) denote the number of il pairs occupying adjacent lattice sites connected by an unblocked (blocked) pore. It is convenient to define the following dimensionless variables:
Bi = NJM; uij = NdJ/zM i, j = 0, A, B
u$ = Nt/zM
where 0 denotes unoccupied sites. All the aii and c$ are not independent as
uij = aji; c$ = a$, i,j = 0, A, B (I)
2aCij+ C (2) j=O.A.B
f&j = (1 -f&l,
j#i
2u;+ c afj = fpei i = 0, A, B. (3) j=O,A.B
j#i
The quasi-chemical approximation for the configur- ations of molecules sorbed in an unblocked lattice has been obtained through kinetic arguments by Sundaresan and Kaza (1985). A straightforward exten- sion of their analysis to include pore blocking yields
aioviQi = 2 aiiqii 0,, .i = A, B (4)
aFoyiei = 2agqfeo. i=A,B (5)
Cm.40 + ~azol(~*Y-2 [aBO~A/eO-%B&d%l +
(kmB/kmA)~uBO + sa'Cggl(Yg)z-2CQao~~leo -aAB4deB1 = 0 (6)

1634 S. SUNDARESAN~II~ C. K. HALL
dimensional grid of intersecting channels, NI in every grid can be expressed as
x CaXOfdeO - diX~&A/eBl = o t7) where
4ri = exp (aii/RT); 48 = exp (s$IRT) (8)
Yt = Ch4ri + aim1 + ai0Y(l -fp>ei (9)
r: = (2azqg +atqt + a:o)/f,& (10)
ri =fpY? + (1 -fp)Yi, 4j = A B (11)
j # i.
For specified eA and 0, values, eqs (l)-(1 1) determine the configuration of the sorbed molecules. Note that the sorbate configuration depends on the ratio between the mobilities of the two species (k&k,), the parameter 6 describing the effect of pore blocking on mobility and the interaction energies.
Once the adsorbate configuration has been de- termined, the chemical potential of species i, pi, can be determined from
p&7,, eB, T) = p,o(T)+RTln (el/eo)+zRTlnj+, i = A,B (12)
where @i(T) is the standard chemical potential of sorbed species i molecules. In the absence of interac- tions between sorbed molecules, yi = y: = Pr = 1 and the Langmuir isotherm for sorption is obtained.
Equations (4 j( 12) assume that the sorbed molecules are present as a single phase. Under certain conditions it may be thermodynamically more favourable for the sorbed molecules to separate into two or more phases than to remain as a single phase (Fowler and Guggenheim, 1956). It is straightforward to show that such phase separation will not occur if for given values of pA, pa and T, there can be only one set of 8, and 0, values. Such uniqueness is guaranteed if
+a &(, - - de, de, a/b a&3
#O (13) -- ae, ae,
for any combination of BA and 8, values. There has been no documented evidence for thermodynamic phase separation in the context of sorption in zeolites, to the best of our knowledge. Hence we restrict our attention to only those situations where inequality (13) is true. The Langmuir isotherm, for example, satisfies this inequality.
2.2. Expressions for diflusional jhxes For zeolite crystals consisting of a three-
dimensional grid of intersecting channels, the flux of species i under isothermal conditions, Ni, can be expressed as
Ni = L, & ( >
V(&RT) mol/m2*s (14a) a”
where LI denotes the Onsager coefficient for species i. In a similar way, for zeolite crystals consisting of a two-
V(p,/RT) mol/m-s. (14b)
The Onsager coefficients, in general, depend on the sorbate composition, lateral interaction energies be- tween sorbates and the fraction of blocked pores. In the absence of lateral interactions between sorbates, it can be easily shown that, for a random walk in an unblocked lattice (Reed and Ehrlich, 1981),
Li = k,,lz z aio- (15)
Following the phenomenological argument of Reed and Ehrlich (198 1) or the kinetic argument of Zhdanov (1985), the quasi-chemical approximation for the Onsager diffusion coefficients in an unblocked lattice in the presence of lateral interactions between sorbed molecules is given by
k .I= L, = mr z-1 Z
afOyi
where y1 in a binary system is given by eq. (9). A straightforward extension of the phenomeno-
logical approach of Reed and Ehrlich (1981) to include the presence of randomly distributed blocked pores yields
&,I2 Li = - h0 + 6 &!3)(XY-’ (17) z
which, in the absence of lateral interaction between sorbates, becomes
Li = y (1 -r, + Sfp)l,O (18)
where di,, = aio + are is the total (dimensionless) number of i0 pairs in the lattice. However, in reality, the above expression overestimates L, since, even with a random arrangement of blocked and unblocked pores certain groups of unblocked pores contained within blocked pore clusters will be less available for transport than is assumed in the phenomenological approach. The effect of pore blocking on the Onsager coefficients has been studied for noninteracting mol- ecules as a percolation problem (Kirkpatrick, 1973). For fp well below fpcr the percolation threshold, an effective medium approximation for the percolation problem yields (Kirkpatrick, 1973)
Li = k,i12 _ ~ aiOQ,(4
i!
where
an@) = 1 Z(l--fP) _l+s(+_ 1>)tz_2)-1 2
+ -K
z(l -f 1 _zl+,(+)] 2
I l/2
+2S(z -2) (z-2)-i. (20)

Diffusion and reaction in blocked zeolite catalysts 1635
Expanding g,(S) in a Taylor series (since 6 Q 1) gives
&@) = 9mo+9m,~ (21)
where
gmo = 1 -&(1-2/z)- r
gm, = (zfr - 2)(2 - 2)- l + {z(l --f,)/2 - l} - i. (22)
Equation (19) can be now rewritten as
k J2 Li = * {a,,~,/(1 -f,)+a?0hdS/fp). (23)
The diffusion of sorbates in a partially blocked lattice in the presence of lateral interactions does not appear to have been studied previously. We propose the following approximation for Li in such systems:
Li = v {aiOsdd(l -_&I + G3Gd~/f,l(Yi),-’
(24)
where & in a binary system is given by eq. (11). Equation (24) reduces to eq. (23) in the absence of lateral interactions and to eq. (16) in the absence of pore blocking.
The quasi-chemical approximation for the adsorb- ate configuration described by eqs (4~(7) treats pairs of sites as strictly independent and does not consider the issue of connectivity of unblocked pores at all. To include the effects of lattice connectivity directly in the derivation of eqs (6) and (7) would require modifi- cation of the terms [aAo + 6a&] and [aBo + 6a Qo] in these equations. It can, however, be argued that for 6 < 1 and fP well below fF, the error involved in eqs (6) and (7) is probably very small.
2.3. Expressions for the reaction rates The rate of formation of the two product isomers
from the set of reactants is assumed to be first order in the concentration of the limiting reactant, Cx. As it has been assumed that the diffusional resistance for the reactant molecules is negligible, C, will be essentially uniform throughout the crystal. Noting that the products can be formed only in unoccupied sites, the rate of formation of product i(i = A, B) from the reactants, Ri, can be written as
Ri = kpiC,130/(N,P) mol/md-s
where d is the dimensionality of the system. Here pi denotes the probability that product isomer i is produced during the reaction,
PA+PB= 1.
The rate of isomerization of A to B, RI,, can be expressed as
R 1A = k,B,/(N,,P) mol/md. s.
Similarly, the rate of isomerization of B to A, R,,, is given by
R,, = k,8B/(iV,,ld) mol/md* s
where the rate constants for isomerizaion are assumed
to be the same in both directions. We have chosen to ignore the effect of lateral interactions between the sorbed molecules on the rates of the chemical reaction, as there is no documented evidence in the literature to indicate that the rates of chemical reactions in zeolite crystals are appreciably modified by the lateral interac- tions. [We should point out, however, that such an effect can be taken into account within the framework of our theory (Sundaresan and Kaza, 1985)].
2.4. Diflusion and reaction in the interior of the zeolite crystal
The diffusion and reaction in the interior of the zeolite crystal are described by the usual conservation equations,
V.NA+RA+RRIB-RIA=O (25)
V-NB+RB+RIA-RR,B = 0. (26)
For the sake of illustration, we assume that the zeolite crystal is a slab of thickness 2L. Assuming symmetry about the centre line, x = 0, we have
Ni(x=O)=O, i=A,B. (27)
It then follows from eqs (13), (14) and (27) that
g (x = 0) = 0, i = A, B. (28)
At the surface of the crystal, x = L, a different kind of blocking, namely surface site blocking, has to be taken into account. We shall include the effect of surface blocking on the diffusion and reaction in the crystal through the boundary conditions at the surface. To derive the boundary conditions at the surface, we view the surface layer as a dividing surface separating the interior of the crystal from the ambient fluid phase. We then postulate that the expressions for pi, Ni and the reaction rates in the interior of the crystal, de- scribed earlier, are valid all the way up to the surface layer. This is a reasonable assumption for large zeolite crystals. It then follows that the flux of species i from the interior of the crystal to the surface layer located at x = L is given by Ni(x + L-) - < where < denotes the unit outward normal to the surface at x = L. Similarly, the assumption of local equilibrium everywhere in the crystal implies that the chemical potential of species i in the surface layer, &, should be equal to pi(x + L-). An expression for & will be derived in the next section.
2.5. Expressions for the chemical potentials in the surface layer
A brief description of the quasi-chemical approxi- mation for p~ in terms of the composition in the surface layer is given below. Consider a surface layer containing MS sites, each site having z, neighbouring surface sites. The surface contains Nj impurities of species I which are deposited prior to exposing the crystal to the reactive environment under study and which block surface sites from occupation by A or B. Thus we are modelling surface blocking as due to immobile impurities confined to surface sites, while

1636 S. SUNDARESAN and C. K. HALL
interior blocking is due to blockage of pores. Let Ng and Nb be the number of molecules of A and B in this surface layer. The number of unoccupied surface sites, NS,, isequal to MS-N; - N& -NT. Let Nb denote the number of ij pairs occupying adjacent surface sites and define the following dimensionless variables:
where
l?” = Nf/M”; tlb = Nb/z,MS, i,j = 0, A, B, I
where 0 denotes unoccupied sites. All the afj are not independent as
2uf+ c usj = e;‘, i = 0, A, B, I. (29) j=O,A,B,I
j+i
The impurities blocking the surface sites are assumed to be distributed randomly. Thus,
ai1 = (0$/2. (30)
In general, a molecule of species i sorbed on a surface site interacts with the occupants of an adjacent surface site as well as with the occupant of the neighbouring interior site. It is not at all obvious how to incorporate the latter interaction into a treatment in which the surface layer is a dividing surface separating the bulk of the crystal from the external environment. Hence we shall take into account only the interaction between sorbates within the surface layer and neglect the interaction between a sorbate in the surface layer with a sorbate in the adjacent interior site. It should be pointed out that, for every surface site, there is only one interior neighbouring site while there are two or more adjacent surface sites. Hence the effect of interactions with the occupants of neighbouring surface sites is likely to be more important than that with the occupant of the neighbouring interior site. The lateral interaction energy between species i and species j molecules occupying adjacent surface sites will be denoted by E?j.
As discussed earlier, the lateral interaction energy &ij (E&) is a convenient way of quantifying the tendency of a sepcies j molecule to push an adjacent molecule of species i out of its site. Since the impurity molecules are considered to be frozen in their sites, we set E:, = 0, i = A, B. As the jump of a sorbate from a surface site to an adjacent unoccupied surface site does not involve transport through a pore structure, the rate constants for the surface migration of the two isomers A and B are likely to be comparable. Thus it is reasonable to expect that &h = E s = constant, i, j = A, B. The quasi- chemical approximation for the configuration of the sorbates in the surface layer is then independent of the surface mobilities and is given by
and
@g (aXI + abI) = a;oy*(Ok + O”,) (33)
qS = exp (@/RT)
ys = (2aX*qS+“XBqS+a”AI+uXo)/eX
= (2aiBq s + cxftB qs + cYg, + a!$,)/B;, .
For specified values of @L, @g, of and T, eqs (29k(33) determine the configuration of the sorbed molecules. Finally ,$, i = A, B, is given by
p; = &(T)+RTln (0;/@) +z,RTln(y*). (34)
We assume that the J&‘(T) in eqs (12) and (34) are equal. This is equivalent to assuming that the energies of isolated molecules of sorbates in the interior and on the surface are the same. In the absence of lateral interactions, ys = 1 and the Langmuir isotherm for sorption is obtained. As in Section 2.1, we restrict our attention to situations where the following inequality holds for any combination of 0~. 0; and @q values:
It can be shown that, within the framework of the quasi-chemical approximation, the rate of desorption of species i(i = A, B) from the surface layer to the surrounding fluid phase is equal to (Sundaresan and Kaza, 1985)
k,,~9~(y”)~~/N,,l~- l mol/md- ’ . s
while the rate of adsorption is equal to
k,,Cisi/N,,ld- ’ mol/md- ’ . s
where si denotes the sticking probability of a molecule of species i in the fluid phase upon collision with the surface. In our analysis, we make the commonly invoked assumption that si = 0%. It then follows that the boundary conditions at the surface of the crystal can be written as
k,,B;(yS)‘~/NJ’+ 1 - k,,Cisi/N,ld-’
= N,(x + L-)*t (35)
p;(W) = pLI(x + L-), i = A, B. (36)
Equation (35) equates the flux of species i from the interior of the crystal to the surface layer, to the net flux of that species away from the surface layer into the surrounding fluid phase. Equation (36) expresses the continuity of the chemical potential of species i at the surface and in the interior.
2.6. Model equations in dimensionless form Letting p = x/L, eqs (25) and (26) can be written for
O<ptl
+ +~(@B-QA) = 0 (37)

Diffusion and reaction in blocked zeolite catalysts 1637
d,*f$+dBBd8” dp 1 + dJ2YIteoPB
where
+ g(t),- e,) = 0 (38)
fl= L,Jk,a; Ya = CR/c”
42 = kL2zCo/k,,12 (39) 4: = k,L2z/k,,12 W) Et = Ca,gmO(l -Q-l +~bhd~f,‘lGW-l (41) d, = E,a(pJRT)/a(e,), i,j = A, B. (42)
Here C”, a characteristic concentration, is chosen as the concentration of the reactant Rat the reactor inlet. The chemical potentials are related to the local com- position through eqs (l)-(12), and the effect of pore blocking on the permeability is described by eq. (22). The boundary conditions for eqs (37) and (38) come from eqs (28), (35) and (36), and are:
de, atp=O, -_=O dp
i = A,B (43)
at p = 1, &(P -+ I-) = Pt, i=A,B (44)
K&ef(r*)=s- K,,cOy,eb] + i d,,% = 0, i=1
i = A, B (45)
where
K, = kai/kdi; yi = C,/C” Kdi = kd, z-Vk,,L i = A, B. (46)
The chemical potentials & are related to the com- position in the surface layer by eqs (29~(34).
Since A and B are two isomers of the same species, it is reasonable to expect that k, = kme, k, = k,, and K, = K,, = K,, which we assume to keep the number of parameters to a minimum_ Equation (45) can then be written as
de, ABdp 1 psi =o (47)
de, dm$+dBB- dp 1 = 0. (48)
p=l
For the case of negligible lateral interactions, i.e. sr, = .si:_ = ss = 0, the sorbate configuration at any location within the crystal is independent of S, k, and k mB, and is given by
cC*j = (1 -_&)ei~~; a$ = f,e,e,, j z i aii = (1 -f,)e:/2; at = f,e:/a yi = y: = yt = 1, i,j =O,A,B
CES 41:6-R
and, similarly, in the surface layer
at = egeg, j # i afi = (e;)2/2, i, j = 0, A, B, I.
It can be shown from eqs (12), (34) and (44) that
eg = (1 -et) ei(p = I), i = 0, A, B.
The differential equations and the boundary con- ditions describing diffusion and reaction in the zeolite crystal can be simplified to obtain
+ ($d’ teB - eA) = o (49)
eBf$+(i-eA)~ 1
+(~)2yReopB
At p = 0,
+ (&2(eA-eB) = 0. (50)
!!!!!6=() i=AB
dp ’ 7 (51)
At p = 1,
+-e,)% dp +eAdBs +~o[eA-K,CoyA~o]
1 = 0 (52)
‘.2+ cl-eA)$$ 1 +~n[eB-KccoyBeO] = 0
_ - (53)
where
$=@lJs, (54)
&^I= W./z (55)
G = K,, (1 - es)ig,. (56)
It is useful to define the following dimensionless groups:
K, = C&‘&= = C9,/+12 (57)
K, E K,,/$ = (1 - e;)[k&,z/k,,kC0g,]“2. (58) Increasing the size of the zeolite crystal is equivalent
to increasing 4, holding K,, #?, K, and K, constant [in eqs (49)-(53)]. At a particular crystal size, if the extent of pore blocking is changed, K,, K, and /? are unaffected while $ and K, change. Similarly, when the extent of surface-site blocking is changed, K, is altered at constant 4, K,, K, and /L If the operating pressure is changed, Co changes, resulting in a change in t$ andK, at constant 8, K, and $r. Thus one can expect the effects of L, &, 0; and Co on the rate of consumption of the reactant R and selectivity to be quite different. Since the dependence of various dimensionless groups on temperature is difficult to generalize, we focus our attention on a study of the effects of L, &, Sf and Co on the rates and selectivity under isothermal conditions_

1638 S. SUNDARESAN and C. K. HALL
Assuming intracrystalline diffusion to be the only significant mass-transfer resistance, a plug flow model for an isothermal fixed-bed reactor of uniform cross- section can be written as
dYtt 1
-=
di’ -YR fJo (~1 dp
dy, -= dC s o1 bAYReO(P) + K,(%(P) - e,(P))] dp
(W
YE(~) = 1 - Y,(c) - YR(c) (61) where c is the dimensionless length along the reactor, defined as
c = Xk(l - Eb)/t’-Navld. (62)
At each location in the reactor, the diffusion and reaction equations described earlier will have to be solved in order to determine the right-hand side of eqs (49) and (50). At any location in the reactor, we define
s 1
rR = YR cob) dp 0
(63)
as a dimensionless measure of the rate of consumption of R and
U
1 s, = bAyR&,+K1(+b)idp *R (64)
cl >I
as the selectivity towards A.
3. RESULTS AND DISCUSSION
The differential equations describing diffusion and reaction in the crystal were solved by the method of orthogonal collocation (Villadsen and Michelsen, 1978). Without loss of generality, we assume that /?l > 1 and that A is the desired product. We first consider the case of negligible lateral interactions between the
sorbates. The requirements for maximizing the selec- tivity, S,, while sacrificing as little of rR as possible will, in general, be different at different locations in the reactor.
3.1. Reactor inter Let us first consider the reactor inlet, where yA = ya
= 0 and y, = 1. The values of rR and S, are plotted against 3 for various values of K,, K, and /3 in Figs 1 and 2, respectively. Increasing the crystallite size (i.e. increasing 4 at fixed K,, K, and /? values) leads to a decrease in rR and an increase in S,. At large crystal sizes, S, apparently reaches an asymptotic value which depends on K,, K, and /3. Thus increasing the size of the crystal may not, in general, be an adequate strategy for achieving high selectivity towards A.
Increasing the value of /l increases both rR and S, [see Figs 1 (a) and 2(a)]. Thus selectivity improvement by a modification of’the diffusional resistance is more easily achieved if /3 is very large. In the remainder of the analysis we assume that jl = 1000, which, for example, appears to be the case for alkylation of toluene in ZSM-5 zeolites (Wei, 1982).
It is clear from Figs 1 (b) and 1 (c) that rR increases as K, increases. However, Figs 2(b) and 2(c) show that for large 6, as K, increases S, increases at first, reaches a maximum and then decreases with any further increase in K,. Though it is difficult to generalize, the optimum value of K, seems to increase as K, increases [com- pare Figs 2(b) and 2(c) at large 61.
The effects of K, on rR and S, are shown in Figs l(d) and 2(d), respectively. K, may be viewed as the characteristic time for the production of isomer B in the interior of the crystal under diffusion-limited conditions divided by the characteristic time for its desorption from the surface. As described in the previous section, the characteristic time for desorption from the surface is assumed to be the same for both isomers. Thus, while the intracrystalline diffusion
Ke=lO I9 = 000
I I 1.1 IO
Fig. I. The dimensionless rate of CO~s~ptiOn of the mactant R. rR3 is plotted against the effective Thiele modulus, 4. Pa = pB = 0.5; b = 0.01; y, = ya = 0.

Diffusion and reaction in blocked xeolite catalysts 1639
Fig. 2. The selectivity towards A, S,, is plotted against the effective Thiele modulus, tj% pA = pB = 0.5; 6 = 0.01; y* = ys = 0.
favours selectivity towards A, the resistance for de- sorption from the surface tends to equalize the selec- tivities toward the two isomers. Thus it would be desirable to have a large value for Ku in order to maximize S,. This is confirmed by Fig. 2(d).
In summary, near the reactor inlet, surface-site blocking is undesirable (as it leads to a decrease in K,) and pore blocking is desirable (as it increases both 4 and K,). In general, a high degree of pore blocking and a moderately large crystal size appear to be the best combination.
Qualitatively, the model considered here differs
from that of Wei (1982) in the way in which the surface blocking is brought into the analysis and in the expressions for the diffusive fluxes. In Wei’s analysis (1982), the flux of a species is taken as proportional to its concentration gradient, while a more accurate model would relate the fluxes to gradients in the chemical potentials. The significance of this difference is illustrated in Fig. 3, where the @,@) and 0,(p) profiles obtained by solving eqs (49~(53) for a typical condition near the reactor inlet are shown. Note that near the surface of the crystal, the diffusion of the fast- moving species (A) takes place against its own concen-
0.3 - s”
8
0’ 0.2- %
B =I000 o-l_ +=K,=lO
K,=l
Fig. 3. The profiles of eA and 8, in the crystal. PA = Pa = 0.5; 6 = 0.01; YA = yg = 0.
1640 S. SLJNDMESAN and C. K. HALL
tration gradient. This feature, which was found to occur over a wide range of parameter values, could not be predicted by Wei’s analysis.
As there is little A in the fluid phase at the inlet of the reactor, the only concern in the choice of the catalyst has been selective conversion of R to A. However, as the fluid moves through the reactor, the concentration of A increases and is, in general, larger than that of B. Therefore, the inhibition of the undesirable isomeriz- ation of A in the fluid phase becomes a more and more important consideration as one moves downstream in the reactor.
3.2. Isomerization Let us consider the isomerization of A and examine
what requirements one would place on the crystal in order to minimize the rate of isomerization. We set y, = ye = 0, yA = 1 and define the following quantity, r,, as a measure of the rate of isomerization:
s
1 r, = ceA@) - %&)I dp. (65)
0
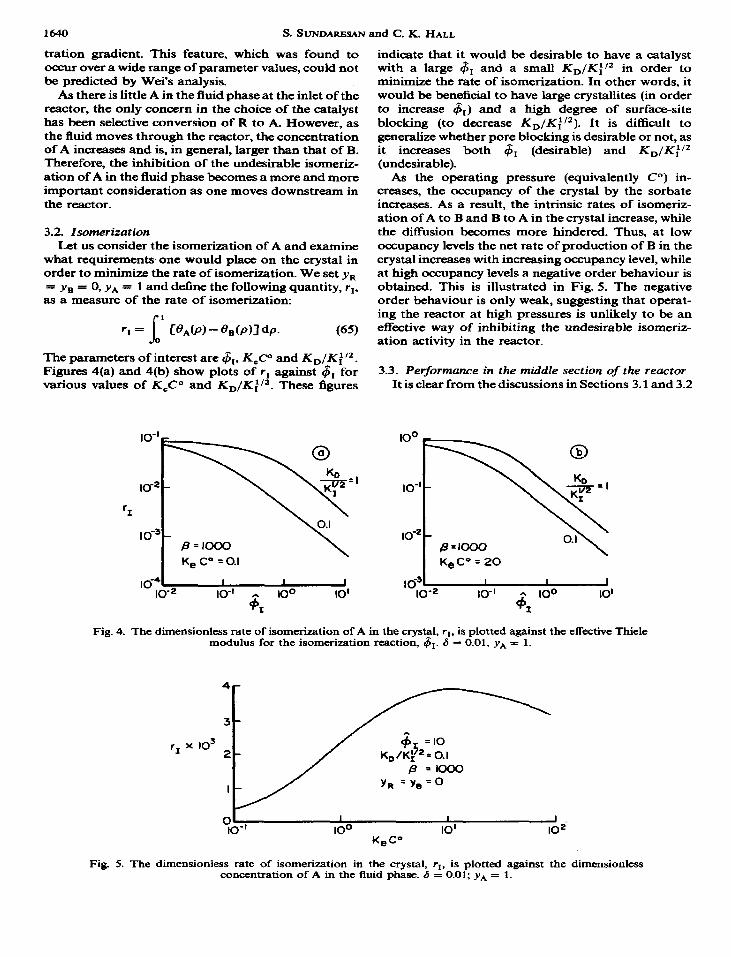
The parameters of interest are $r, Kg? and KD/Ki”. Figures 4(a) and 4(b) show plots of r, against 6, for various values of K&Y and KD/Ki12. These figures
@- P=lOoo K, C” = 0.1
indicate that it would be desirable to have a catalyst with a large 4, and a small KD/Kif2 in order to minimize the rate of isomerization. In other words, it would be beneficial to have large crystallites (in order to increase 6,) and a high degree of surface-site blocking (to decrease KD/Ki’2). It is difficult to generalize whether pore blocking is desirable or not, as it increases both $r (desirable) and KD/Kii2 (undesirable).
As the operating pressure (equivalently Co) in- creases, the occupancy of the crystal by the sorbate increases. As a result, the intrinsic rates of isomeriz- ation of A to B and B to A in the crystal increase, while the diffusion becomes more hindered. Thus, at low occupancy levels the net rate of production of B in the crystal increases with increasing occupancy level, while at high occupancy levels a negative order behaviour is obtained. This is illustrated in Fig. 5. The negative order behaviour is only weak, suggesting that operat- ing the reactor at high pressures is unlikely to be an effective way of inhibiting the undesirable isomeriz- ation activity in the reactor.
3.3. Performance in the middle section of the reactor It is clear from the discussions in Sections 3.1 and 3.2
10-2 - p=Kwo K,C*=ZO
Fig. 4. The dimensionless rate of isomerization of A in the crystal, ri, is plotted against the effective Thiele modulus for the isomerization reaction, 41. 6 = 0.01, yA = 1.
rI X IO3
Fig. 5. The dimensionless rate of isomerization in the crystal, rI, is plotted against the dimensionless concentration of A in the Auid phase. 6 = 0.01; yA = 1.
Diffusion aad reaction in blocked zeolite catalysts 1641
that a highly pore-blocked catalyst with little surface- site blocking is desirable at the inlet of the reactor, while surface-site blocking is beneficial near the reactor exit (assuming that our goal is to convert the limiting reactant nearly completely in a single pass). A combi- nation of pore and surface-site blocking may be expected to be desirable in the middle section of the reactor (from the point of view of maintaining both rR and S, as high as possible), the extent to which each type of blocking is imposed being different at different locations in the reactor. The determination of optimal levels of pore and surface-site blocking at various locations in the reactor in order to maximize the yield of A, though intriguing, is beyond the scope of this publication. We simply point out that the model framework presented here is a convenient way of addressing such optimization problems.
3.4. Eflect of interactions between sot-bates Considering the origin of the interactions (described
in Section 2), we set &ALl = ‘%aB - -q EC= 0 i,j=A,B
EM = EBA = E
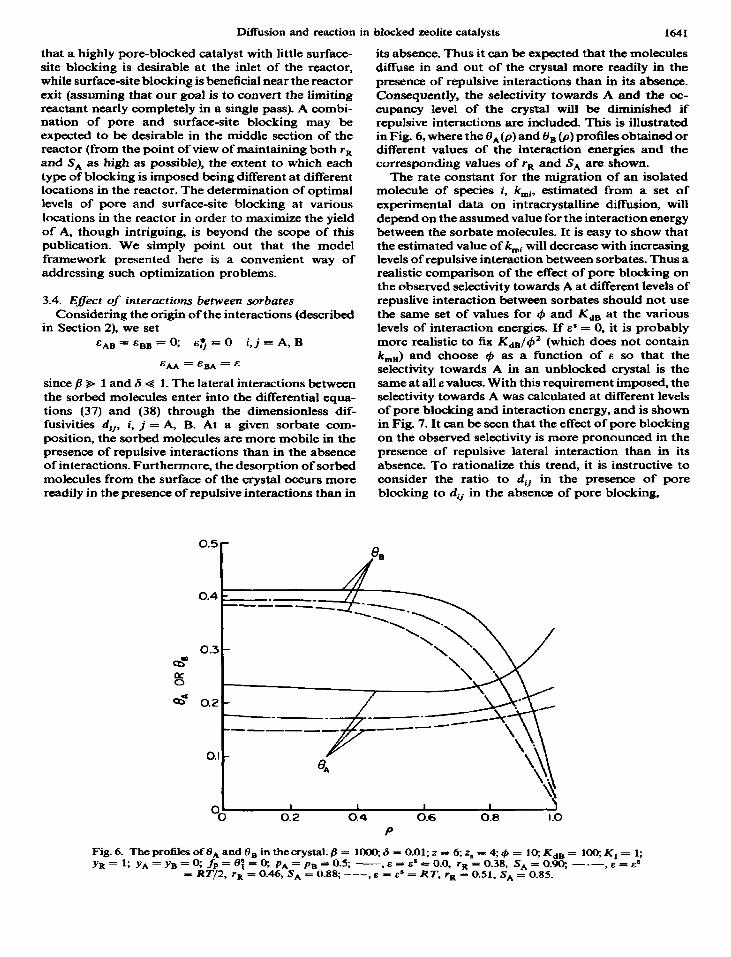
since fl* 1 and 6 G 1. The lateral interactions between the sorbed molecules enter into the differential equa- tions (37) and (38) through the dimensionless dif- fusivities dij, i, j = A, B. At a given sorbate com- position, the sorbed molecules are more mobile in the presence of repulsive interactions than in the absence of interactions. Furthermore, the desorption of sorbed molecules from the surface of the crystal occurs more readily in the presence of repulsive interactions than in
its absence. Thus it can be expected that the molecules diifuse in and out of the crystal more readily in the presence of repulsive interactions than in its absence. Consequently, the selectivity towards A and the oc- cupancy level of the crystal will be diminished if repulsive interactions are included. This is illustrated in Fig. 6, where the 0,(p) and 8, @) profiles obtained or different values of the interaction energies and the corresponding values of ra and S, are shown.
The rate constant for the migration of an isolated molecule of species i, kmir estimated from a set of experimental data on intracrystalline diffusion, will depend on the assumed value for the interaction energy between the sorbate molecules. It is easy to show that the estimated value of kmi will decrease with increasing levels of repulsive interaction between sorbates. Thus a realistic comparison of the effect of pore blocking on the observed selectivity towards A at different levels of repuslive interaction between sorbates should not use the same set of values for 4 and K,, at the various levels of interaction energies. If ss = 0, it is probably more realistic to fix K&c$’ (which does not contain k,,,a) and choose Cp as a function of E so that the selectivity towards A in an unblocked crystal is the same at all E values. With this requirement imposed, the selectivity towards A was calculated at different levels of pore blocking and interaction energy, and is shown in Fig. 7. It can be seen that the effect of pore blocking on the observed selectivity is more pronounced in the presence of repulsive lateral interaction than in its absence. To rationalize this trend, it is instructive to consider the ratio to d, in the presence of pore blocking to dii in the absence of pore blocking,
Fig. 6. The profiles of OA and OB in the crystal. fi = 1OCQ 6 = 0.01; z = 6; 2, = 4; Q = 10; K,, = 100; K, = 1; yR= 1; yA=yB=Q fp=tl;=0; pa=p*=0.5; -,E=E s=O.O,
= RT/2, r’R = 0.46, S, = ra=0.38. S,=O.90, --.-•.e=9
0.88; ---, e = es = RT, rR = 0.51, SA = 0.85.

1642 S. SUNDARESAN and C. K. HALL
0.7
S,
0.6 ,
f P Fig. 7. The effect of pore blocking on the observed selectivity towards A. y, = y, = 0; yR = 8; = ,o;~‘;=“=l~ ?I
l;KI=lq~=looo;s=o.ol; ; = 6; zs = 4; pa = p,, = 0.5; Kde/@ = 1;
; -.---,&=1.8, e=O.SRT; ---,$ = 4.1; E = RT.
Dij = dij(E, f-,, eA, &j)/d,(E> 0, e,, e,), i,i = A, B 056)
at different levels of interaction energy (since pore blocking enters our analysis through the d& Figure 8 shows plots of D,, i, j = A, B, against & for various interaction energies at eA = 0, = 0.3. The Dijs de- crease with increasing &, as one would expect. Note that the decrease in the Dijs with increasing fP becomes more pronounced as the magnitude of the repulsive lateral interaction energy increases. This can be attri- buted to the shielding of the interactions by pore blocking.
In a similar fashion, the rates of isomerization in the crystal at different levels of surface-site blocking were calculated for different values of interaction energies in the surface layer, E’. A sample set of results is presented in Table 1. In these computations, the values of &, &CO, fl and 6 were held constant while K,, was chosen as a function of .sS so that the rate of isomeriz- ation in an unblocked crystal was the same for all .sS values. This appears to be reasonable as the rate of desorption of the two isomers from the surface layer is the quantity most directly affected by (the chosen value of) E*. The rate of isomerimtion decreases with increas- ing levels of surface-site blocking, as one would expect. Furthermore, the difference between the results at different values of Es is very small. Thus, it appears that the lateral interaction between sorbates in the surface layer may be neglected with little sacrifice in the qualitative as well as in the quantitative predictions of the model.
In industrially important reactions such as toluene alkylation, small unmodified (unblocked) HZSM-5 crystals do not exhibit pronounced para-selective properties, while large HZSM-5 crystals display moderate para-selectivity (Chen et al., 1979). On the other hand, modification of the catalyst by blocking agents results in a tremendous enhancement of para- selectivity (Chen et al., 1979, Kaeding et al., 1981a, b). These experimental results and our result that the effect of blocking is more pronounced in the presence of repulsive lateral interactions between sorbates than in its absence suggest that sorbate-sorbate interactions in the interior of the crystal may be an important factor in diffusion and reaction in zeolite catalysts. It is needless to emphasize that this is hardly evidence for the importance of sorbate-sorbate interactions and that more direct experimental evidence is required to
Fig. 8. Variations of D,, DA,, DBA and D,, with the fraction of pores blocked, j& eA = 8, = 0.3; z = 6; p - looo; 6 = 0.01. p, E=Q----,&=RTp---,e-~~.

Diffusion and reaction in
Table 1. E&et of surface-site blocking on the rate of isomerization of A
Dimensionless rate of isomerization, rr
&= = 0 E= = OSRT 2=RT et K dB = 0.9 K,, = 0.69 K,, = 0.63
0.0 0.0205 0.0205 0.0205 0.15 0.0179 0.0176 0.0175 0.30 0.0152 0.0145 0.0144 0.45 0.0123 0.0114 0.0112 0.60 0.0092 0.0082 0.0080 0.75 0.0059 0.0050 0.0048 0.90 0.0025 0.0020 0.0018
fi = looo; d = 0.01; z = 6; z, = 4; e = 0; K,C” = 1; r#q = 3; yg=Ya=o;y*=l;fp=O.
establish the importance (or lack of it) of such interactions in the analysis of diffusion and reaction in zeolites.
The model described in this paper contains a number of parameters which need to be estimated before the model can be used predictively. A possible approach to estimating these parameters is described below. Considering the origin of the interactions visualized in this study, it appears reasonable to expect that
&Ii = Eli = . _. =&ii = . . . = Ej -_ &Fj = &fj = . . . * * = Ejj = . _ _ 3 Ej
(67)
i.e. the interaction energies sij and E$ are expected to depend on the identity of species j only, in which case one can obtain all the information necessary to describe multicomponent diffusion by simply studying the sorption of each of the species under consideration separately. It appears reasonable to expect that the adsorption of sorbates from the fluid phase onto the surface layer is non-activated, while the desorption from the surface layer into the fluid phase and migration in the interior of the crystal are activated. Further, in most of the cases of interest, the activation energy for desorption will be larger than that for the migration in the interior of the crystal. Now, consider a zeolite crystal in equilibrium with a fluid phase con- taining the sorbate species at some concentration. If the fluid-phase concentration of this species is in- creased, the amount of sorbate in the zeolite will increase and the rate of this increase is dictated by intracrystalline diffusion. If the fluid-phase concen- tration of this species is decreased, the amount of sorbate in the zeolite crystal will decrease and the rate of this decrease is governed by the rate of desorption from the surface layer. Thus, these two types of transient experiment provide a convenient way of isolating the effects of intracrystalline diffusion and desorption from the surface layer. From the steady-state and transient sorption measurements in unblocked zeolite crystals of uniform size (for each of the sorbate species under consideration separately), one should be able to estimate the equilibrium constant (K,,), the interaction energies (.sJ, sj) and the rate constant for intracrystal- tine migration (k,). Repeating the experiments with
blocked zeolite catalysts 1643
zeolite crystals having different levels of pore blocking, one can estimate sf and the value of 6 for the j-th species (m 6,). A few multicomponent sorption ex- periments can then be performed to verify if eq. (67) is indeed a satisfactory approximation.
4. SUMMARY
A mathematical model for diffusion and reaction in blocked zeolites has been developed which takes into account nonidealities arising from interaction between sorbed molecules as well as the effect of pore and surface blocking. The zeolite crystal was modelled as a regular array of sites, and expressions for chemical potential and diffusive fluxes were calculated by corn- bining the lattice-gas approach with an effective medium approximation for the percolation problem. The effect of surface blocking was brought into the analysis by viewing the surface layer as a dividing surface separating the interior of the crystal from the surrounding fluid phase.
An example that is qualitatively similar to indus- trially important reactions such as alkylation of toluene in blocked ZSM-5 zeolite was studied in detail in order to elucidate the effects of crystal size, blocking and sorbate-sorbate interactions on the activity- selectivity characteristics of the crystal.
Acknowledgement-This manuscript is based on a paper presented at the Annual Meeting of the A.1.Ch.E. held in Chicago, November 1985.
CO
ci
d dij
Dij %i
Bm B m0’ gml k
k,
kai
k,
km,
K,, K, Kdi
;:
Ki
NOTATION
inlet concentration of the limiting reactant concentration of species i, i = A, 3, R dimensionality of the lattice dimensionless diffusion coefficients (i, j = A, B), see eq. (42) see eq. (66) activation energy for the migration of an isolated molecule of species i(i = A, B) through the pores fraction of pores blocked fraction of pores blocked at the perco- lation threshold permeability of the lattice [see eq. (20)] see eqs (21) and (22) rate constant for the consumption of R rate constant for the ismerization reaction A#B rate constant for the adsorption of species i (i = A, B) rate constant for the desorption of species i(i=A,B) rate constant for the migration of species i(i = A, B) in the lattice equilibrium constants for sorption see eq. (46) see eq. (58) see eq. (56) see eq. (57)

S. SUNDARESAN and C. K. HALL 1644
L 1 Li
4
Ni
N&W
Pi
Qij *
zi
R si
sA T
V
X
X
Yi
half-thickness of the zeolite slab distance between adjacent sites Onsager coefficient (i = A, B) see eq. (41) diffusive flux of species i(i = A, B) Avogadro’s number probability that isomer i(i = A, B) is pro- duced from the reactants see eq. (8) exp (E’/ R T) rate of formation of species i(i = A, B) from the reactants rate of consumption of species i (i = A, B) by the isomerization reaction dimensionless rate of isomerization rsee
$1 f: Pi
Pi
eq. (WI dimensionless rate of consumption of R Subscripts
[see eq. (63)] o
gas constant i orj
sticking probability for species i (i = A, B)
see discussion preceding eq. (31) see eqs (9), (lo), (11) and (33), respectively (i = A, B) dimensionless distance in the crystal dimensionless distance along the reactor [see eq. (WI see eqs (39) and (40), respectively see eqs (54) and (SS), respectively chemical potential of species i at any location within the crystal (i = A, B) chemical potential of species i on the surface of the crystal (i = A, B) standard chemical potential of the sorbed molecules of species i(i = A, B)
unoccupied site species i or j (i, j = A, B, R, I)
selectivity towards A [see eq. (64)] temperature superficial velocity of the fluid in the reactor spatial variable describing location inside the crystal spatial variable describing location in the reactor dimensionless concentration of species i at any location in the reactor (i = A, B, R) number of nearest neighbouring sites for every site in the interior of the crystal number of neighbouring surface sites for every site on the surface of the crystal
Superscripts 0 reference state S surface layer * blocked pore
REFERENCES Barrer. R. M. and Jost, W., 1949, A note on interstitial
diffusion. Trans. Faraday Sot. 45,928-930. Jkwrer, R. M. and Rees, L. V., 1954a, Sorption of mixtures:
3. Polar sorbates as modifiers of zeolitic crystals. Trans. Faraday Sot. 50, 852-863.
Barrer, R. M. and Rees, L. V.. 1954b, Sorption of mixtures: 4. Molecular diffusion ib crystals modified by Polar sor- bates. Trans. Faraday Sot. SO, 989-999.
Chen, N. Y., Kaeding, W. W. and Dwyer, F. G., 1979, Para- Greek letters directed aromatic reactions over shape-sele&ve molecular
dimensionless number of ij pairs con- sieve zeolite catalysts. J. Am. Gem. Sot_ 101, 6783-6784.
Deiaifue, P.. Auroux. A. A.. Gravelle. P. C.. Vedrine. J. C.. netted by unblocked (blocked) pores at Gabe.l&, i. and D&o&e, E. G., 198 1, M-ethanol &nverl anv location within the crvstal li. i = 0. sion on acidic ZSM-5, offretite and mordenite zeolites: a
k-W dimensionless number of ij pairs on the surface of the crystal (i, j = 0, A, B, I) fraction of sites at any location within the crystal occupied by species i(i = 0, A, B) fraction of sites on the surface of the crystal occupied by species i(i = 0, A, B, I)
k&k,, ratio of the rate constants for the mig- ration of a molecule through blocked and unblocked pores void fraction in the catalyst bed interaction energy exerted on a species i molecule by a species j molecule occupy- ing an adjacent site with the pore joining these sites being unblocked (blocked) (i, j = A, B) interaction energy exerted on a species i molecule occupying a surface site by a species j molecule in a neighbouring sur- face site (i, j = A, B, I)
comparative study of the formation and stability of coke deposits. J. CataZysis 70, 123-136.
Fowler, R. and Guggenheim, E. A., 1956, Statistical Thermodynamics, p. 426. Cambridge University Press, Cambridge.
Haag, W. O., Lago, R. M. and Weisz, P. B., 1982. Transport and reactivity of hydrocarbon molecules in shape-sele.ctive zeolite. Faraday Discuss. 72, 317-330.
Kaeding, W. W.,-Chu. C., Y&g, L. B. and Butler, 5. A., 1981a, Selective alkylation of toluene with methanol to produce para-xylene. J. Catalysis 67, 159-174.
Kaeding, W. W., Chu, C., Young, L. B. and Butler, S. A., 198lb, Shape-selective reactions with zeolite catalysts: II. Selective disproportionation of toluene to produce be-e and p-xylene. J. Catalysis 69, 392-398.
Kaeding, W. W., Young L. B. and Chu, C., 1984a, Shape- selective reactions with zeolite catalvsts: IV. Alkvlation of toluene with ethylene to prod&e p-ethyltoiuene. J. Catalysis 89, 267-273.
Kaeding, W. W., Barile, G. C. and Wu, M. M., 1984b, Mobil zeolite catalysts for monomers. Catal. Reu.-Sci. Engng 26, 597612.
Kirkpatrick. S.. 1973, Percolation and conduction. Rev. mod. Phvs. 45. 574-588.
Kok&ilo,-G. T., Lawton, S. L.,Olson, D. H. and Meier. W. M., 1978, Structure of synthetic zeolite ZSM-5. Nature. Lord 272,437.

Diiusion and reaction in blocked xeolite catalysts 1645
Meier. W. M. and Olson, D. H.. 1978, Atlas of Zeolite
Theodorou, D. and Wei, J., 1983, Diffusion and reaction in
Structure
blocked and high occupancy xeolite catalysts, J. Catalysis
Types. Structure
83, 205224.
Commission of the International Zeolite Association, Zurich.
Olson, D. H. and Haag, W. 0.. 1984, Structure-selectivity relationship in xylene isomerixation and selective toluene disproportionation. ACS Symp. Ser. No. 248, 275-307.
Reed, D. A. and Ehrlich, G., 1981, Surface diffusion, atomic jump rates and thermodynamics. Surf. Sci. 102, 588-609.
Ruthven, D. M., 1974, Diffusion in partially ion exchanged molecular sieves. Can. J. Chem. 52, 3523-3528.
Sundaresan, S. and Kaxa, K. R., 1985, Nonrandom distri- bution of adsorbates on catalytic surfaces: the role of adsorbate mobilities on reaction rates. Chem. Engng Commun. 35, l-22.
Vedrine, J. C., Auroux, A., Dejaifue. P., Ducarme, V.. Hoser, H. and Zhou. S.. 1982, Catalytic and physical properties of phosphorus-modified ZSM-5 xeolite. J. Catalysis 73, 147-160.
Villadsen, J. and Michelsen, M. L., 1978, Solution of Diflmential Equation Models by Polynomial
Approximation, pp. 212-213. Prentice-Hall, Englewood Cliffs, NJ.
Yashima, T, Ahmad, H.. Yamam ki. K.. Katsuta, M. and Ham, N., 197Oa, Alkylation on synthetic xeolites-I. Alkylation of toluene with methanol. 1. Catalysis 16, 27%280.
Yash&a, T., Yamamki, K., Ahmad, H., Katsuta, M. and Ham, N., 197Ob, Alkylation on synthetic xeolitcs-II. Selectivity of p-xylene formation. J. Catalysis 17. 151-156.
Young, L. B., Butler, S. A. and Kaeding, W. W., 1982, Shape- selective reactions with xeolite catalysts-III. Selectivity in xylene isomerixation, toluene-methanol alkylation and toluene disproportionation over ZSM-5 xeolite catalysts. J. Catalysis 76, 418432.
Wei, J., 1982, A mathematical theory ofenhanced para-xylene selectivity in molecular sieve catalysts. J. Catalysis 76, 433439.
Weisz, P. B., 1980, in New Horizons in Catalysis (Edited by Seiyama, T. and Tanabe, K.), No. 7A, p. 3. Elsevier, Amsterdam.
Zhdanov, V. P., 1985, General equations for description of surface diffusion in the framework of the lattiegas model. Surf. Sci. 149, L13-L17.





























