Embed Size (px)
Citation preview

49
MATERIAL AND METHODS
TISSUE CULTURE STUDIES:
This section presents the details of material and methods which were employed
for undertaking the present study.
PLANT MATERIAL: The mother plant was collected from the Tri Shakti
farms, Sanathnagar, Hyderabad. The explants used were the Shoot Apex, Nodal
Region, and Leaf Segments.
Glassware :
All glassware used during the course of experiments like culture tubes, beakers,
pipettes and funnels were produced from m/s. Borosil India Ltd., Mumbai Luxbro
India Ltd., supplied polypropylene caps for culture tubes, while caps of bottles
were obtained from Varsha Storage Racks.
Chemicals and other media ingredients:
Chemicals required for media preparation were of analytical grade obtained from
Sigma, BDH, Hi-media and LOBA chemical companies, sugar used as a source of
sucrose was obtained from local market.
Preparation of Explants:
Shoots of 10-15 cm lengths of Stevia auxillary buds were detached and brought
from the field to the Tissue Culture Laboratory. The shoots, leaves and nodal
regions were cut into 1-2 cm. Length as shoot tips and nodal cuttings and were

50
used as explants to establish in vitro cultures. The procedure of surface
sterilization followed in given here under.
Explants were washed under running tap water with 5.1 Teepol solution
immediately they were soaked in solution of 2% Bavistin and K-Cycline which
served as fungicide and bactericide respectively. Two explants were soaked for
1hrs. Then the explants are transferred the laminar air flow cabinet for surface and
aseptic sterilization.
Explants surface sterilization:
Explants washed with sterile water.
Explants washed with 70% alcohol for 30 seconds.
Washed with sterile distilled water for 2 or 3 minutes.
The explants washed with 0.01% mercuric chloride + Tween 20 (1 or 2
days ) for 10 minutes .
Then washed with sterile distilled water four times.
First time - 4 minutes
Second Time - 4 minutes
Third Time - 4 minutes
Fourth Time - 12 minutes
Explants surface sterilization is over. Then the explants were inoculated in the
appropriate media.
51
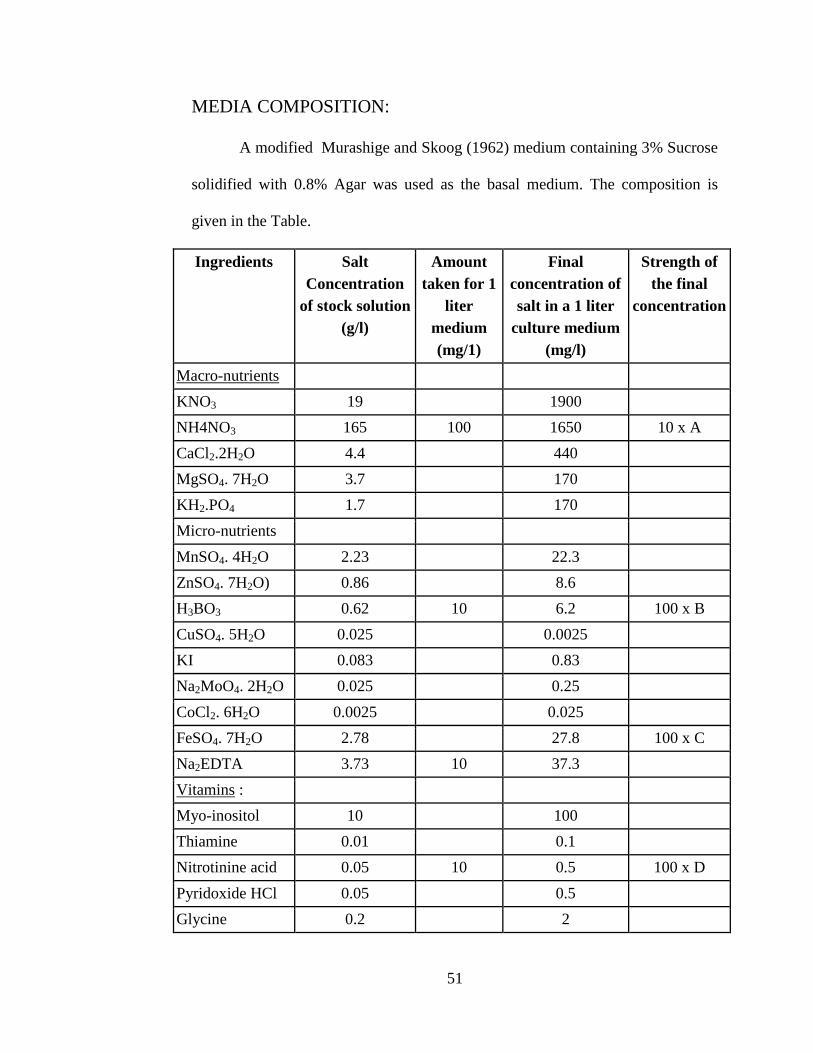
MEDIA COMPOSITION:
A modified Murashige and Skoog (1962) medium containing 3% Sucrose
solidified with 0.8% Agar was used as the basal medium. The composition is
given in the Table.
Ingredients Salt
Concentration
of stock solution
(g/l)
Amount
taken for 1
liter
medium
(mg/1)
Final
concentration of
salt in a 1 liter
culture medium
(mg/l)
Strength of
the final
concentration
Macro-nutrients
KNO3 19 1900
NH4NO3 165 100 1650 10 x A
CaCl2.2H2O 4.4 440
MgSO4. 7H2O 3.7 170
KH2.PO4 1.7 170
Micro-nutrients
MnSO4. 4H2O 2.23 22.3
ZnSO4. 7H2O) 0.86 8.6
H3BO3 0.62 10 6.2 100 x B
CuSO4. 5H2O 0.025 0.0025
KI 0.083 0.83
Na2MoO4. 2H2O 0.025 0.25
CoCl2. 6H2O 0.0025 0.025
FeSO4. 7H2O 2.78 27.8 100 x C
Na2EDTA 3.73 10 37.3
Vitamins :
Myo-inositol 10 100
Thiamine 0.01 0.1
Nitrotinine acid 0.05 10 0.5 100 x D
Pyridoxide HCl 0.05 0.5
Glycine 0.2 2

52
Growth Regulators :
Benzylamino purine 2 mg /l
Kinetin 1 mg /l
Napthalene acetic acid 1 mg /l
2,4-Dichloro acetic acid 2 mg /l & 3 mg /l

53
Preparation of culture media:
The macro nutrients, micro nutrients and vitamins drawn from the stock
solution were mixed in the required quantity. The growth substances were added
as necessary. Sucrose as a source of carbon was dissolved at the rate of 30 g/l
(3%). The final volume of known quantity was obtained by adding double
distilled water. The pH was adjusted to 5.0 by a addition of in HCl or NaOH as
required.
Agar-Agar was added to the boiling media at a rate of 8 g/l (0.8%) slowly
and gradually with constant stirring to avoid formation of any clumps. Then the
medium was dispossed into culture vessels i.e. glass bottles (baby jars of 250 ml.
Capacity) or test tubes (25 x 150mm. Size) @ 40 ml. respectively. These vessels
were plugged with polypropylene caps and were then autoclaved along with other
surgical instruments required for transfer operation at 121 º C and at a pressure of
15 lbs for 20 minutes.
Stock Solution of Potassium Iodide KI (1000X):
0.0830 (83mg) of KI was dissolved in 100 ml distilled water: usage =1ml/l
Stock Solution of Na2EDTA (100X):
Dissolve 37.2mg of Na2EDTA (Ethylene di amine tetra acetic acid, di sodium
salt) in 50 ml DH20. Boil Na2EDTA solution and dissolve 27.8 mg of FeSo4.7H2o
gently by stirring and final volume is made up to 100ml. To prepare 1000 ml of
culture medium. Usage- 1ml/l.

54
Stock Solution of Calcium chloride (100X):
4.4 g of Calcium Chloride was dissolved in 80 ml of sterile distilled water and the
final volume is made up to 100 ml. Usage-10 ml/l.
Stock Solution of Myoinositol (10X):
1000 mg of myoinositol was dissolved in 100 ml distilled water. Usage-1ml /l.
ROLE OF PLANT GROWTH REGULATORS:
Some chemicals occurring naturally within plant tissue (i.e. endogenously) have a
regulatory rather than a nutritional role in growth and development. Growth as
well as differentiation of tissues in vitro is controlled by various growth regulators
Auxins, Cytokinins, Gibberellins, Ethylene, Abscisic acid etc.
Auxins :
Auxins (IAA, NAA, 2,4-D, IBA) are phytohormones that influence cell
enlargement, root initiation, adventitious bud formation. They suppress the
initiation of lateral buds.
2,4-D: (2,4 dichlorophenoxy acetic acid: C8 H6 C12 O3) is a synthetic
auxin known primarily as a weedicide. It is used for callus induction.
IAA (Indole – 3 – acetic acid: C10 H9 NO2) It is a promoter rooting of
cuttings.
IBA (indole – 3 – butyric acid:C12 H13 NO2) It is a rooting hormone and
more stable than IAA.

55
NAA ( Napthalene acetic acid : C12 H16 O2 ) :
It is also a root-inducing hormone and also promotes callus kinins.
Cytokinins: The cytokinin (KN, BAP, 2-ip and Zeatin) is adenine a derivatives
and is formerly called Kinins.
BAP (Benzyl amino Purine: C12 H11N5 ) : It is used to promote auxillary
bud growth.
2-ip (i PA N6 – (2-iso pentyl adenine: C10 N5): H13 it causes rapid cell
division and consequent irregular growth.
Kinetin (6- Furfuryl aminopurine: (C10 H9 N5 O): promote cell division.
Zeatin (6-[4-hydroxy – 3 – methyl but – 2 – enly aminopurine: C10 H13
N5 O): it is a common alternative cytokinin to 2-ip.
Cytokinins are even effective in promoting direct or indirect shoot
initiation Wickson and Thimann (1958) discovered that cytokinin could release
the lateral buds from apical dominance in an intact stem tip.
The role of cytokinin in shoot organogensis is well-established (Flick et al.
1983). Cytokinin in combination with an auxins appear essential for the onset of
growth and the induction of embryogenisis (Fujimure and Komamine 1980).
Gibberellins: Gibberellins (gibberellic acid, GA3 C19 H22 O6): Influence
cell enlargement and stem elongation.
Abscisic Acid: It is useful in embryo culture and somatic embryogenesis.
Ethylene (C2H4): It is a gas produced by plants. It is involved with fruit
ripening, flowering and leaf abscission.

56
GROWTH REGULATORS:
Plant Growth Regulators incurred in the preparation of culture medium include
6´-benzyl adenine (BAP), Kinetin (Kn) Adenine Sulphate (AS), 2,4-dichloro
phenoxy acetic acid (2,4-D),1-Napthalene acetic acid (NAA).These were prepared
in desired concentrations (to be maintained as Stock Solution) to induce plant
regeneration from explants.
6´-benzyl adenine(BAP):
6´-benzyl adenine(BAP) is a cytokinin [ A synthetic N- (Phenyl)-1H-Purine-6-
amine compound ].
10 mg of 6´-benzyl adenine(BAP) was dissolved in 1 ml of 1N NaOH was
adjusted to 8 ml with distilled water, then final volume was made up to 10 ml
with distilled water, filter sterilized and stored at 4°C.
Kinetin (Kn):
Kinetin (Kn) is a cytokinin [ a synthetic N-(2- furanyl methyl) 1H –Purine -6-
amine compound].
10 mg of Kinetin was dissolved in 1 ml of 1N NaOH, volume was adjusted top
8ml with distilled water, then final volume was made up to 10 ml with distilled
water, filter sterilized and stored at 4°C.
Adenine Sulphate (AS):
Adenine Sulphate (AS) is a synthetic cytokinin, and its formula is
(C5H5N5)2H2SO4).

57
10 mg of Adenin Sulphate was dissolved in 1 ml of 1 N NaOH, volume was
adjusted to 8 ml with distilled water. Then final volume was made up to 10 ml
with distilled water, filter sterilized and stored at 4°C.
2,4-Dichloro phenoxy acetic acid (2,4-D):
10 mg of 2,4-D was dissolved in 1 ml of 1N NaOH, the volume was adjusted to 8
ml with distilled water. Then final volume was made up to 10 ml with distilled
water, filter sterilized and stored at 4°C.
Naphthalene acetic acid (NAA):
10 mg of NAA was dissolved in 1 ml of 1 N NaOH, volume was adjusted to 8 ml
with distilled water. Then final volume was made up to 10 ml with distilled water,
filter sterilized and stored at 4°C.
Indole butyric acid (IBA): [a synthetic 4 –(indole-3-yl) butyric acid
compound].
10 mg of IBA was dissolved in 1 ml of 1N NaOH, the volume was
adjusted to 8 ml with distilled water, then final volume was made up to 10 ml
with distilled water, filter sterilized and stored at 40° C.
Preparation of transfer area for aseptic culture:
Aseptic culture works like final surface sterilization of plant material,
preparation and inoculation of explants and further sub culturing of in vitro
cultures were carried out in a laminar Air flow cabinet. Initially before the use of
the cabinet, the working surface was sterilized by swabbing the surface of the

58
cabinet with 70% ethyl alcohol. Later the cabinet was ensured sterile by switching
on UV light 2500 Aº for about 15 minutes before use. Then the sterile airflow was
switched on and left for at least 10 minutes before use. During the course of
transfer, between each transfer of explants to culture bottles, or tubes, the surgical
instruments were dipped in absolute ethyl alcohol followed by dipping them in
glass head sterilization for 15-20 seconds, where the temperature was maintained
at 250 ºC and cooled before use. After the completion of sterile transfer operation,
the laminar airflow was cleaned and sprayed with 70% ethyl alcohol and kept
closed.
Incubation room:
The culture was incubated in an air-conditioned room at the
temperature of 25 ± 2ºC under a micro propagation region of 16 hours light and 8
hours dark cycle.
Growth room:
Each growth room is fitted with mobile culture incubation racks fitted
with 40 watts cool day white fluorescent tube lights for providing light for
photosynthesis of tissues. Growth room is maintained clean was of CL 1,00,000
and temperature of 25± 27ºC for temperature crops 16 hours photoperiod and
8hours darkness are provided in each growth room. The photoperiod and
temperature is maintained.

59
MAINTENANCE OF CULTURES IN LABORATORY:
The in vitro cultures of Stevia rebaudiana Bertonii or all the
experiments were maintained at 25±2°C and 3000 Lux Illumination comprising a
16 hour photoperiod provided by cool flourescent light and with a relative
humidity of 50±20%.
INITIATION OF CULTURES :
After Surface Sterilization the explants were cut into a specific size with a
scalpel on pre sterilized petri plates and embedded on to the media with various
concentrations of hormones. The cultures were maintained for 4 weeks and after
proper callusing they were Sub cultured on different media.
All the experiments were repeated 3 times and 12 replications (Test
Tubes/culture vessels) for each treatment were used each time. The culture were
observed regularly to watch the growth and regeneration and recorded the data
for various parameters in each experiment.
MICRO PROPAGATION:
Micro propagation became one of the most ways to produce crops
that are difficult to propagate by conventional methods like cuttings or seeds. The
application of tissue culture technique to produce virus free plants initially led to
subsequent micro propagation of a great diversity of plants in vitro.

60
Days taken For the Establishment Of Explants in vitro:
Murashige and Skoog medium (1962) along with different concentrations of
cytokinin BAP (0.25mg/l, 0.5mg/l, 1.0mg/l, 1.5mg/l, 2.0mg/l, 2.5mg/l) were used.
The following data were collected during this experiment.
1. Average number of days taken for the initiation of shoot bud, first
leaf, and second leaf:
The mean time taken for establishment i.e., the initiation of shoot bud, first
leaf, second leaf varied with different concentrations of growth regulators. The
response of Initiation of Shoot bud, first leaf, second leaf was early i.e., 15 days.
In the medium supplemented with different concentrations of BAP.
The mean number of days taken for the emergence of first leaf varied
with different concentrations of growth regulators in the basal media. The early
initiation of first leaf was observed within 5 days in medium supplemented with
three different concentrations of BAP.
MULTIPLE SHOOTS:
Multiple shoots formation was observed in MS medium with different
concentration of BAP. Maximum numbers of multiple shoots were obtained in
media supplemented with 1.0 mg/l and significant difference was not found in
low concentration of 0.5 mg/l. The mean number of days taken for the multiple
shoots induction was recorded. Frequency of multiple shoots can be measured by
using the formula.

61
No. of explants that responded
(by producing multiple shoots)
Frequency of Multiple Shoots = --------------------------------------- × 100
Total No. of explants inoculated
1. Shoot length in cms
The shoot length of the plantlet was recorded after the 90 days of initiation and
the tip of the top most leaf formed to the initiation point of roots were taken into
consideration.
2. Root length in cms
The root length of the plantlet was recorded by measuring the longest root of the
plantlet this was taken into records before the plantlet bottles were shifted to
green house for acclimatization after 90 days of initiation.
3. Number of leaves
The total number of freshly surviving healthy green leaves both long and short
were counted before the cultures bottles were shifted to green house 90 days for
further process of acclimatization the leaf at the tip of the plantlet to the lower
point near the rooting area were taken into consideration.
4. Number of roots
The total number of roots per each plantlet after 90 days was taken into account
before shifting them to green house all the roots were thoroughly washed with
water and roots were cleaned from medium in which it was inoculated.
No. of rooted shoots
Frequency of Rooting = -------------------------------------------- ×100
Total No. of inoculated explants

62
5. Fresh weight
The in vitro rooted plantlets after 90 days were removed from the culture vessels,
and the agar was removed with tissue paper. And fresh weight was recorded in
(mg).
6. Percentage of survival during Hardening of the plantlets
In vitro rooted plantlets from different treatments after 90 days were taken to
green house from the laboratory for acclimatization. The plantlets removed from
the culture bottles, washed off the agar and planted in portray having sterilized
potting mixture of decomposed coco peat + vermiculite at 1:1 ratio. After placing
the portrays with plants transferred to high Relative humidity sheds in the green
house for 30 days at 80 -90 % (RH) Relative humidity and temperature of
30ºC±2ºC. After 30 days plants transferred to shade house and taken the data on
percentage of survival.
No. of surviving plants
in the shade house
Frequency percentage of survival in shade house =-------------------------------×100
Total No. of transferred plants
ORGANOGENESIS:
In the present study the full strength MS media was supplemented with two plant
growth regulators for the further growth of the plant material before its
acclimatized. It was found that BAP 0.5 mg/l + KN 0.4 mg/l showed more results
of organogenesis.

63
CALLUS INDUCTION:
In the present study full strength MS medium has been used. The response of
Initiation and callus formation was early i.e., 15 days. In the medium
supplemented with different concentrations of BAP, 2,4-D mg/l.
No. of explants showing response
Callus Induction Frequency = --------------------------------------------- ×100
Total No. of explants inoculated
1. Mean days taken for callus initiation
The mean number of days taken for the initiation of callus i.e. formation of
undifferentiated tissue in the cultured vessels/bottles of the experiment
2. Fresh weight of the callus
Fresh weight of the callus was recorded after 60 days of callus initiation.
3. Nature of the callus
Nature of the callus, colour of the callus was recorded in different treatments i.e.
globular, smooth, green, light green, brown.
GROWTH REGULATORS:
The growth regulator used in callus initiation was BAP, and 2,4-D
with different concentrations (0.25 mg/l, 0.5 mg/l, 1.0 mg/l, 1.5 mg/l, 2.0 mg/l,
2.5 mg/l, 3.0 mg/l) for both the explants (i.e.) nodal region and leaf segment. The
shoots developed from the embryos of the explants were transferred to rooting
medium of MS medium with 1mg NAA along with activated charcoal for rooting
of plantlets.

64
SURFACE STERILIZATION:
The Callus was removed from the culture bottles and placed on sterile
paper. Dried callus, hard callus and dead tissue and if any multiple clamps were
removed by using sterile forceps and blades. Then the callus were placed on the
fresh media if it is in the shoot development stage for further growth of the
plantlets from callus media comprising of 1 mg/l BAP.
The following observations were made during the experiment.
After the first sub culture of the callus obtained from the nodal
region weight about 2.0 mg in 2, 4-D 1.5 mg/l. The colour of the callus was Light
green smooth in nature whereas the callus obtained from the leaf segment weight
about 1.8 mg in 1.5 mg/l 2,4-D here also the colour of the callus was Light green
and smooth in nature.
SUB CULTURE OF PLANTLETS FROM GROWN CALLUS
The plantlets were removed from the culture bottles and placed on sterile
paper. Dried leaves, hard callus and dead tissue and if any multiple clamps were
removed by using sterile forceps and blades. Then the callus were placed on the
fresh media if it is in the shoot development stage and later on transferred to the
rooting medium comprising of 1mg/l NAA. The survivals of plantlets during
acclimatization after transferring all the fully developed plantlets to field were
monitored.
To study the effect of different growth regulators on callus initiation and
regeneration through somatic embryogenesis from the callus of nodal and Leaf of
Stevia rebaudiana Bertonii.

65
In the present study the full strength MS medium was used along with two
different plant growth regulators (i.e.) 2,4-D, and 2-ip with different
concentrations.
The growth regulators along with basal medium used for the Initiation of
Somatic embryos takes about 3 months of time. The concentrations used for both
the callus obtained from nodal and leaf callus were 2,4-D (0.01mg/l,0.25mg/l,
0.5mg/l, 1.0mg/l and 2ip (0.01mg/l, 0.05mg/l, 1.0mg/l,1.5mg/l). The number of
days taken for the initiation of somatic embryos is 90 days and it was found that
2,4-D with 1.0mg/l and 2-ip with 1.5mg/l showed more number of somatic
embryos. (as evidenced by the development of globular callus on the nodal callus)
was recorded.
Nature of the callus:
Nature of the callus, colour of the callus was recorded in different
treatments i.e. single spherical cell, early globular shape with conspicuous
suspensor, typical globular shape, elongated shape, early heart shape, typical heart
shape, torpedo shape ( as evidenced by SEM photos).
MATURATION OF SOMATIC EMBRYOS:
After four weeks of induction, calli bearing somatic embryos were shifted
to the full strength MS media was supplemented with two plant growth regulators
for the further growth. It was found that BAP 0.5 mg/l + KN 0.4 mg/l showed
more results.

66
The cultures were maintained at 25±2°C and 3000 Lux Illumination
comprising a 16 hour Photoperiod provided by cool fluorescent light and with a
relative humidity of 50±20%. After four weeks the frequency of maturation of
somatic embryos (as evidenced by the transition of globular stage to torpedo stage
of embryo) was recorded.
Mean days taken for the organogenesis from the somatic embryos:
During the above mentioned maturation stage of somatic embryos. The
mean days taken for the organogenesis from somatic embryos were recorded.
Number of embryos formed, Percentage of somatic embryos and shoot formation
was taken into consideration.
Germination of somatic embryos:
The somatic embryos after attaining to the torpedo stage of development
on maturation medium were transferred to the rooting medium comprising of MS
medium supplemented with NAA for both the 2,4-D and 2-ip grown somatic
embryo genetically grown plants for germination.
The cultures were incubated for 2 weeks at 25±2°C and 3000 Lux
Illumination comprising a 16 hour Photoperiod provided by cool fluorescent light
and with a relative humidity of 50±20%.
1. Shoot length in cms
The shoot length of the plantlet was recorded after the 90 days of initiation and
the tip of the top most leaf formed to the initiation point of roots were taken into
consideration.

67
2. Root length in cms
The root length of the plantlet was recorded by measuring the longest root of the
plantlet this was taken into records before the plantlet bottles were shifted to
green house for acclimatization after 90 days of initiation.
3. Number of leaves
The total number of freshly surviving healthy green leaves both long and short
were counted before the cultures bottles were shifted to green house 90 days for
further process of acclimatization the leaf at the tip of the plantlet to the lower
point near the rooting area were taken into consideration.
4. Number of roots
The total number of roots per each plantlet after 90 days was taken into account
before shifting them to green house all the roots were thoroughly washed with
water and roots were cleaned from medium in which it was inoculated.
No. of rooted shoots
Frequency of Rooting = -------------------------------------------- ×100
Total No. of inoculated explants
5. Fresh weight
The in vitro rooted plantlets after 90 days were removed from the culture vessels,
and the agar was removed with tissue paper. And fresh weight was recorded in
(mg).
6. Percentage of survival during hardening of the plantlets
In vitro rooted plantlets from different treatments after 90 days were taken to
green house from the laboratory for acclimatization. The plantlets removed from
the culture bottles, washed off the agar and planted in portray having sterilized

68
potting mixture of decomposed coco peat + vermiculite at 1:1 ratio. After placing
the portrays with plants transferred to high Relative humidity sheds in the green
house for 30 days at 80 -90 % (RH) Relative humidity and temperature of
30ºC±2ºC. After 30 days plants transferred to shade house and taken the data on
% ge of survival.
No. of surviving plants
in the shade house
Frequency percentage of survival in shade house = ------------------------------×100
Total No. of transferred plants
SEM (Scanning electron microscope) :
The embryogenic callus at different stages was used for the SEM studies.
The callus samples were processed by fixing 4% (v/v) glutaraldehyde in 0.1 M
phosphate buffer (pH6.8) for about 3 hours of duration. The samples were washed
thoroughly in distilled water 3 times. Then the samples were dehydrated by
passing through acetone series from low to high concentrations
[20,40,60,80,100% (w/v)] each lasting about 30 minutes of duration .The samples
were dried to critical point for gold coating, they were mounted onto the stubs
using “JEOL – JFC – 1100E/JEOL – 100 CX –BALZAR‟S - 4 CD – Ion
Sputtering device”. After gold coating, the material was observed under” JEOL –
JSM – 5200/JEOL – 100CX – Scanning electron microscope” and different stages
of somatic embryogenesis were photographed.
The fine structure of somatic embryos was clearly identified during the
following sequential developmental stages: single spherical cell, early globular

69
shape with conspicuous suspensor, typical globular shape, elongated shape, early
heart shape, typical heart shape, and torpedo shape.
High performance liquid chromatography (HPLC) analysis:
Material and Method:
Dried leaves of Stevia plant [ex vitro grown plant]- 1 g
Dried leaves of Stevia plant [in vitro grown plant]-1g
Stevioside standard solution [mg/ml]
Acetonitrile [Mercks company]
Water.
Equipments used:
Soxhlet extractor apparatus.
High Performance Liquid Chromatography (Preparative HPLC Waters Auto
PurificationTM
System).
SAMPLE PREPARATION:
1g leaves of each of ex vitro and in vitro grown plants were dried and
ground to fine powder using a dry blender. The sample of dried Stevia leaves
were put into extraction thimble and place sample into Soxhlet extractor.
Extraction was made using a solvent [Dichloromethane and methanol (1:1)].
[HPLC grade Merck].

70
STANDARD SOLUTION PREPARATION:
The 2.5 mg of each two standards solution were prepared by diluting
with 80% methanol in water into volumetric flask respectively in prior to sonicate
them for 15 minutes.
The separation of stevioside from the leaves was carried out by
preparatory HPLC [Waters HPLC purification System]. The HPLC operating
conditions and parameters are as follows: The column used was C18, with
dimensions of 250 X 4.6mm, 5μ. The mobile phase used was Acetonitrile : water
[80:20 v/v] the Wavelength of detection was at 254nm.
HPLC analysis of S. rebaudiana crude extract was carried out to
determine the stevioside content found in the leaves of S. rebaudiana in
comparison with a reference standard. Acetonitrile - water (80:20 v/v) was used
as the mobile phase. Stevioside was separated by means of a reversed phase
Agilent technologies HPLC system comprised of quaternary pump, a column
oven, sample freeze and UV detector. A ZORBAX Eclipse XDB-C18 column
(4.6x150 mm, 5 µm) was employed, at 30ºC. Separation was made in isocratic
mode, using Acetonitrile : water (80:20 v/v) at a flow rate of 1ml/min with 20 µl
injection volume; detector and column temperature were set at 30ºC. The
detection wave length was 210 nm (Ahmed and Smith, 2002).

71
Sample preparation for HPLC analysis:
Silica (25 g) was added to a column. The column was conditioned with
hexane (80 ml) and not allowed to dry. The crude extract (250 mg) dissolved in
methanol (20 ml), was mixed with silica (5 g) and dried under vacuum. Silica
adsorbed concentrated extract was then applied to the column. The analytes were
eluted with chloroform: ethyl acetate: water (65:25:4, 100 ml) (Wagner and Bladt
1996). The eluate was concentrated almost to dryness under vacuum. The residue
was re-dissolved in methanol (20 ml). A light yellow solution was obtained and
then decolorized using activated charcoal. Determination of the content of
stevioside in plant material was performed by external standard method. A stock
solution of standard stevioside (Menge Germany) was used and solutions with
concentrations of 100, 200, 300, 500 and 800 mg/l were used to draw calibration
curve. Triplicate determinations were carried out and the average taken in
drawing the calibration curve.
ISOLATION OF GENOMIC DNA:
Extraction Buffer:
2% CTAB, 100mM Tris, 20mM EDTA and 1.4 M NaCl
♦ Preheat 15 ml of CTAB buffer at 600C (Add 50mg PVP and 200 ul β-
mercaptoethanol)
♦ Grind the leaf samples (2 to 5 gm) in liquid Nitrogen and add into
preheated buffer

72
♦ The contents were mixed gently by swirling and inverting the tube and
incubated at 600C water bath for 45 min. with occasional mixing.
♦ Keep the samples at room temperature, and add equal volume of
chloroform-Isoamyl alcohol (24:1)
♦ Mix the contents by inversion for 10 minutes and centrifuged at 8000 rpm
for 10 min.
♦ Transfer the clear aqueous upper layer into a new centrifuge tube, add two
volumes of cold isopropanal and mixed by inversion and placed in -200C
for 30 min.
♦ Pellet the genomic DNA by centrifuging at 10000 rpm for 20 min at 100C
♦ Wash the pellet with 70% ethanol twice and air dry the DNA pellet
♦ Dissolve the DNA in T10E1 and use 25 – 50 ng/ul DNA for PCR analysis.
1M Tris (100ml):12.11g of Tris was dissolved in 70ml of sterile distilled water
and adjust pH to 8.0 with 1N HCl and final vol7ume made to 100ml.
0.5M EDTA (100ml): 18.612g of EDTA was dissolved in 70ml of sterile
distilled water and adjust pH to 8.0, adding NaOH pellets and final volume made
to 100ml.

73
5M NaCl (100ml): 29.22g of NaCl was dissolved in 70ml of sterile distilled
water and final volume made to 100ml and autoclaved.
CTAB Buffer (2%;500ml): 50ml of 1M TRIS, 20ml of 0.5 M EDTA,140ml of
5M NaCl,10g of CTAB,10g of PVP,10ml of beta mercapto ethanol were added in
about 250ml sterile distilled water and final volume make up to 500ml.
Testing of Clonal fidelity in tissue cultured plants using RAPDs
Isolation and quantification of genomic DNA :
Genomic DNA was isolated from the field grown plant and regenerated
plants on MS medium supplemented growth regulators. The quantification of
extracted DNA was done by Spectrophotometer and through comparison with
defined concentrations of DNA. The quantity of DNA in different samples
when compared with DNA and was found to be ranging from 200-700ng/l. Four
plant samples were taken for variability assessment.
RAPD analysis :
The results were scored as patterns of bands obtained from in vitro
regenerated plants grown on MS medium supplemented growth regulators. 20
OPA RAPD primers tested produced amplification products that were
monomorphic across all regenerants. The size of the monomorphic DNA
fragments, produced by these primers was shown. For each primer major bands
were scored and the size of the amplification products ranged between 500 bp-2.5
kb. A total of 92 bands were scored from PCR amplification of genomic DNA

74
from four Samples. No polymorphism was detected after amplification by PCR
among regenerated plants grown on different hormones. Thus, all regenerated
plants showed 100% similarity with the field grown plants.