Embed Size (px)
Citation preview

1
1100 1200 1300 1400 1500 1600 1700 1800
[M+K]+
[M+Na]+
Mass (m/z)
[M+H]+
900 1000 1100 1200 1300 1400 1500 1600
[M+H]+
Mass (m/z)
1160 1180 1200 1220 1240 1260 1280
[M+H]+
Mass (m/z)
420 440 460 480 500
[M+H]+
Mass (m/z)
500 520 540 560 580 600
[M+K]+[M+Na]+
[M+H]+
Mass (m/z)
600 640 680 720 760 800
[M+K]+[M+Na]+
[M+H]+
Mass (m/z)
a
c
e
d
b
f
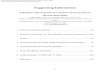
Supplementary Figure S1 ǀ Characterization of molecular weight identity. MALDI-
TOF mass spectra of molecule 2 (a), molecule 3 (b), molecule 5 (c), molecule 8 (d),
molecule 9 (e), and molecule 10 (f).

2
2600 2800 3000 3200 3400 3600
0.0
0.5
1.0
1.5
2.0
3480 cm-1
3480 cm-1
Wavenumber ( cm -1 )
Ab
sorp
tion
Supplementary Figure S2 ǀ FT-IR spectra (2500-3700 cm-1). Black-solid line from
dried solution of self-assembled 1 in methanol and water (2 : 3) and Red-dash line from
dried solution of self-assembled 2 in water.

3
0
400
800
300 350 400 450 500
0.0
0.4
0.8
U
V in
ten
sity F
L in
tensity
Wavelength ( nm )100 1000 10000
1 2
Diameter ( nm )
Inte
nsi
ty
0.004 0.008 0.012 0.016
0
2
4
q ( nm -1 )
Iq-2
( KH
z n
m2
)
a b
1 2 3
0.00
0.06
0.12
Dap
p×
10 -7
( cm
-2 / s
)
q2× 10 10 ( cm -2 )
c d
Supplementary Figure S3 ǀ Characterization of supramolecular nanotubes of 1. (a)
Absorption and emission spectra of 0.01 wt% of 1 in CHCl3 (black and solid line) and
mixed solution (H2O : MeOH = 7 : 3 red and dashed line). (b) Size distribution graphs
of 0.01 wt% of 1 in MeOH-Water (30 % MeOH) solution and 0.01 wt% of 2 in aqueous
solution. (c) Kratky plot and linear fit of 0.01 wt% of 1 in MeOH-Water (30 % MeOH)
solution was confirmed the cylindrical micelle in solution. (d) Angular dependence of
the apparent diffusion coefficient of 0.01 wt% of 1, Dapp ~ 0.024 for the cylindrical
micelle in solution.

4
0.0000 0.0001 0.0002 0.0003
-5.5
-5.0
-4.5
1 2 3
0.45
0.60
0.75
q2× 10 10 ( cm -2 )
Dap
p×
10 -7
( cm
-2 / s
)
a b
0.005 0.010 0.015
1
2
3
4
5
6
q ( nm -1 )
Iq-2
( KH
z n
m2
)
q2 ( nm -2 )
ln( I
q/
nm-1
)
1 2
11
20
10
q ( nm -1 )
Inte
nsi
ty
c d
Supplementary Figure S4 ǀ Characterization of supramolecular nanofibers of 2. (a)
AFM image of 2 in aqueous solution (0.01 wt%) transformed on mica. (b) Small-angle
X-ray diffraction pattern of 30 wt% of 2 in aqueous solution showed tetragonal
columnar structure with lattice constants a = 9.4 nm implying that the cross section of a
cylinder consists of 14 molecules. (c) The linear fit of Kratky plot and the Holzer plot in
the inset of 0.01 wt% of 2 in aqueous solution was confirmed the cylindrical micelle in
solution. (d) Angular dependence of the apparent diffusion coefficient of 0.01 wt% of 2,
Dapp ~ 0.035 for the cylindrical micelle in aqueous solution.

5
V
iabi
lity
%
a
b c
Supplementary Figure S5 ǀ Cell culture in gels and on tissue-culture plastic. (a)
Optical images of C2C12 cells in nanofibers (Left: immediately after seeding, Middle:
after 1 day, Right: after 2 days). (b) The viability of C2C12 cells using trypan blue assay
in three different conditions 1 wt% (blue), 1.5 wt% (red) and 2 wt% (green) for 5 days.
(c) The control image of C2C12 cells grown as a monolayer on tissue-culture plastic
(2D). The scale bars in all images are 100 μm. The error bars in b mean the standard
deviations of the measured cell viability from four replicate experiments.

6
Supplementary Figure S6 ǀ Optical images of grown C2C12 cells. The control image
of C2C12 cells grown in 4 wt% of collagen gel (a), 0.5 wt% of 2 in the presence of 10
mol% of 3 (b), and 1 wt% of 2 in the presence of 10 mol% of 3 (c). The scale bars in all
images are 100 μm.

7
Supplementary Figure S7 ǀ Cell grown in gel and released from gel. Optical images
of C2C12 cells in nanofiber gel with the concentration of 2 1 wt% (a), 1.5 wt% (b), and
2 wt% (c). The grown image of C2C12 cells after release from 1 wt% (d), 1.5 wt% (e),
and 2 wt% (f) of 2 in gel on culture dish for 8 hours. The scale bars in all image are 100
μm.

8
Supplementary Figure S8 ǀ Movement of cells after release from gel. Time-lapse
optical images of C2C12 cells as cooling the gel with a cold cell media (scale bar in all
images are 100 μm).

9
Supplementary Methods
Materials and instruments
Materials. 1, 2-diamino benzene, triethylene glycol monomethyl ether, diethylene
glycol monomethyl ether, and 4-hydroxy benzaldehyde from Aldrich were used as
received. NaH (60 %) and p-toluenesulfonyl chloride (98 %) from TCI and Tokyo Kasei
were used as received. Unless otherwise indicated, all starting materials were obtained
from commercial suppliers (Aldrich, TCI, Acros, etc.) and were used without
purification. Hexane, dichloromethane, and ethyl acetate were distilled before use.
Visualization was accomplished with UV light, iodine vapor. Flash chromatography was
carried out with Silica Gel 60 (230-400 mesh) from EM Science. Dry THF was obtained
by vacuum transfer from sodium and benzophenone. The synthesis of compound 1
based on diethylene oxide 2nd generation dendrimer has been reported elsewhere.31 2,5-
dibromophenol,32 4-biphenylboronic acid,40 mono-tosylated tetraethylene glycol,41 and
tosylated tri ethylene 2nd generation dendrimer42 were prepared according to the similar
procedures described previously.
Instruments. 1H NMR and 13C NMR spectra were recorded from CDCl3 or DMSO
solutions on a Bruker AM 300 spectrometer. The purity of the products was checked by
thin-layer chromatography (TLC; Merck, silica gel 60). Recycling preparative high-
pressure chromatography (HPLC) was performed for further purification by using
HITACHI model pump L-7110, JAI model UV detector 310 and JAI model RI detector
RI-7S. MALDI TOF-MS spectroscopy (MALDI-TOF-MS) was performed on a Bruker
Microflex LRF20 using α-cyano-4-hydroxy cinnamic acid (CHCA) as matrix. The

10
dynamic light scattering experiment was performed by DLS-8000 from Otsuka
Electronics with a 632.8 nm He-Ne laser. The Uv/vis spectra was obtained from a
Hitachi U-2900 Spectrophotometer. The fluorescence spectra was obtained from a
Hitachi F-7000 Fluorescence Spectrophotometer. The rheology experiment was
performed by ARES-LS from TA Instrument. The transmission electron microscopy
(TEM) was performed at 120 kV using JEOL-JEM 2100.
Synthesis of compound 2
Reagents and conditions : (a) K2CO3, CH3CN, reflux; (b) 4-biphenylboronic acid, 2M K2CO3, tetrakis-
triphenylphosphine palladium(0), THF, reflux.
Scheme 1. Synthesis of compound 2.

11
Compound 5. Compound 4 (0.2 g, 0.54 mmol), Tosylated tri-ethylene 2nd generation
dendrimer (0.5 g, 0.5 mmol) and K2CO3 (1.1g, 7.0 mmol) were dissolved in 20 mL of
CH3CN. The mixture was heated at reflux for 48 hours and then cooled to room
temperature. The solvent was removed in a rotary evaporator, and the resulting mixture
was poured into water and extracted with ethyl acetate. The ethyl acetate solution was
dried over anhydrous magnesium sulfate and filtered. After the solvent was removed in
a rotary evaporator, the crude products were purified by column chromatography (silica
gel) using methanol : ethyl acetate (1:8 v/v) as eluent to yield 70% (0.43 g) of colorless
liquid.
Compound 5. 1H-NMR (300MHz, CDCl3, δ, ppm): 10.49 (s, 1H), 8.06 (d, J = 7.5 Hz,
2H), 7.2 (m, 2H), 6.95 (d, J = 7.5 Hz, 2H), 4.05 (d, J = 4.8 Hz, 2H), 3.62-3.35 (m, 78H),
2.39-2.04 (m, 3H). 13C-NMR (100 MHz, CDCl3, δ, ppm): 160.9, 135.4, 129.1, 128.5,
127.5, 123.6, 122.1, 114.9, 71.7, 70.6, 70.3, 69.8, 69.7,67.6, 54.7, 40.2.
Compound 2. Compound 5 (0.4 g, 0.33 mmol) and 4-biphenylboronic acid (0.26 g,
1.31 mmol, 4 eq.) were dissolved in degassed THF (30 mL). Degassed 2M aqueous
K2CO3 (30 mL) was added to the solution and then tetrakis(triphenyl-phosphine)
palladium (0) (0.06 g, 0.05 mmol, 0.15 eq.) was added. The mixture was heated at
reflux for 48 hours with vigorous stirring under argon. Cooled to room temperature, the
layers were separated, and the aqueous layer was then washed twice with methylene
chloride. The combined organic layer were dried over anhydrous magnesium sulfate and
filtered. The solvent was removed in a rotary evaporator, and the crude product was

12
purified by column chromatography (silica gel) using ethyl acetate : methanol (8:1 v/v)
as eluent and then further purified by prep-HPLC to yield 0.34 g (75 %) of colorless
liquid.
Compound 2. 1H-NMR (300 MHz, CDCl3, δ, ppm): 10.15 (s, 1H), 8.30 (d, J = 8.6 Hz,
2H), 7.79-7.38 (m, 20H), 7.06 (d, J = 8.6 Hz, 2H), 4.05 (d, J = 7.5 Hz, 2H), 3.62-3.35
(m, 78H), 2.40-2.04 (m, 3H).; 13C-NMR (100 MHz, CDCl3, δ, ppm): 157.3, 141.8,
136.7, 135.4, 129.1, 128.5, 127.9, 127.7, 123.4, 122.1, 114.7, 71.9, 70.6, 70.5, 69.8,
69.7,67.6, 54.7, 40.2; MALDI-TOF-MS [M+H]+, [M+Na]+ and [M+K]+ calcd. for
C69H90N2O15: m/z 1364.67, 1387.67 and 1403.67; Found: 1364.78, 1388.06 and 1403.11.

13
Synthesis of compound 3
Reagents and conditions : (a) K2CO3, ACN, reflux; (b) I TsCl, Et3N, DCM; II NaN3, DMF 110 oC; (c) I
Pa/C, H2, EtOH; II Succinic anhydride, Dioxane 80 oC; (d) 4-biphenylboronic acid, 2M K2CO3, tetrakis-
triphenylphosphine palladium(0), THF, reflux. (e) I 4-Nitrophenol, CHCl3; II L-RGD, DIPEA, DCM,
DMF.
Scheme 2. Synthesis of compounds 3.
Compound 7. Compound 6 (0.4 g, 1.6 mmol), mono-tosylated tetraethylene glycol
(0.44 g, 1.4 mmol) and K2CO3 (1.1g, 7.0 mmol) were dissolved in 20 mL of CH3CN.
The mixture was heated at reflux for 12 hours and then cooled to room temperature. The

14
solvent was removed in a rotary evaporator, and the resulting mixture was poured into
water and extracted with ethyl acetate. The ethyl acetate solution was dried over
anhydrous magnesium sulfate and filtered. After the solvent was removed in a rotary
evaporator, the crude products were purified by column chromatography (silica gel)
using ethyl acetate as eluent to yield 70 % (0.37 g) of colorless liquid.
Compound 7. 1H-NMR (300MHz, CDCl3, δ, ppm): 7.37 (d, J = 8 Hz, 1H), 7.05 (s, 1H),
6.96 (d, J = 8 Hz, 1H), 4.15 (t, J = 4.6 Hz, 2H), 3.90 (t, J = 4.6 Hz, 2H), 3.79-3.65 (m,
12H) ; 13C-NMR (100 MHz, CDCl3, δ, ppm): 141.9, 133.9, 132.0, 131.6, 121.8, 120.7,
71.0, 70.7, 61.5.
Compound 8. P-Toluenesulfonylchloride (0.38 g, 2 mmol) was added to Compound 7
(0.57 g, 1.34 mmol) and triethylamine 4 mL in CH2Cl2 20 mL at room temperature. The
reaction mixture was stirred at room temperature for 6 h. Water (20 mL) was then added
to the reaction mixture, the organic layer was separated and the aqueous layer was
extracted with CH2Cl2 (3×20 mL). The combined organic layers were dried over
MgSO4 and concentrated under vacuum. The residue was then purified by column
chromatography on silica gel using ethyl acetate as eluent to yield 90 % (0.7 g) of
colorless liquid. A solution of Sodium azide (86 mg, 1.32 mmol) in ethanol 20 mL was
added at room temperature. The reaction mixture was stirred overnight at 70 °C. The
reaction was then quenched by addition of water (50 mL), and concentrated under
vacuum. The aqueous layer was extracted with ethyl acetate (3×50 mL). The combined
organic layers were then dried over MgSO4 and concentrated under vacuum. The
residue was then purified by column chromatography on silica gel using ethyl acetate as
eluent to yield 95 % (0.52 g) of colorless liquid.

15
Compound 8. 1H-NMR (300MHz, CDCl3, δ, ppm): 7.38 (d, J = 8 Hz, 1H), 7.1 (s, 1H),
6.96 (d, J = 8 Hz, 1H), 4.15 (t, J = 4.6 Hz, 2H), 3.90 (t, J = 4.6 Hz, 2H), 3.79-3.65 (m,
10H), 3.37 (t, J = 4.6 Hz, 2H); 13C-NMR (100 MHz, CDCl3, δ, ppm): 141.6, 133.8,
132.0, 131.4, 121.6, 120.5, 71.1, 70.6, 44.7.
Compound 9. To a solution of 8 (0.46 g, 1 mmol) in 12 mL of methanol, 10% Pd/C (55
mg) was added. The reaction suspension was stirred at room temperature for 12 h under
hydrogen. After removing the Pd/C by filtration, the filtrate was concentrated to dryness.
The residue was purified by silica-gel column chromatography using
chloroform/methanol = 7/1, v/v) as eluent to give colerless liquid. The liquid was
dissolved in 5 mL of dioxane and then slowly added to a solution of Succinic anhydride (1
mmol) in dioxane 10 mL. The mixture was heated to 80 oC and stirred 6 h. The reaction
was then quenched by addition of water (50 mL), and concentrated under vacuum. The
aqueous layer was extracted with ethyl acetate (3×50 mL). The combined organic layers
were then dried over MgSO4 and concentrated under vacuum. The residue was then
purified by flash column chromatography on silica gel using chloroform/methanol = 7/1,
v/v) as eluent to give 91 % (0.48 g) colerless liquid.
Compound 9. 1H-NMR (300MHz, CDCl3, δ, ppm): 7.35 (d, J = 8 Hz, 1H), 7.04 (s, 1H),
6.90 (d, J = 8 Hz, 1H), 4.15 (t, J = 4.6 Hz, 2H), 3.88 (t, J = 4.6 Hz, 2H), 3.72-3.60 (m,
10H), 3.41 (t, J = 4.5 Hz, 2H), 2.61 (m, 2H), 2.46 (m, 2H); 13C-NMR (100 MHz, CDCl3,
δ, ppm): 173.8, 173.1, 141.9, 133.7, 132.0, 131.6, 121.8, 120.7, 71.0, 70.7, 43.3, 30.5,

16
29.3.
Compound 10. Compound 9 (0.16 g, 0.3 mmol) and 4-biphenylboronic acid (0.13 g,
1.2 mmol, 4 eq.) were dissolved in degassed THF (30 mL). Degassed 2M aqueous
K2CO3 (30 mL) was added to the solution and then tetrakis(triphenyl-phosphine)
palladium (0) (0.06 g, 0.05 mmol, 0.15 eq.) was added. The mixture was heated and
reflux for 48 hours with vigorous stirring under argon. Cooled to room temperature, the
layers were separated, and the aqueous layer was then washed twice with methylene
chloride. The combined organic layer were dried over anhydrous magnesium sulfate and
filtered. The solvent was removed in a rotary evaporator, and the crude product was
purified by column chromatography (silica gel) using chloroform/methanol = 5/1, v/v)
as eluent to give 80 % (0.16 g) colerless liquid.
Compound 10. 1H-NMR (300 MHz, CDCl3, δ, ppm): 7.73-7.45 (m, 20H), 6.85 (d, J =
8 Hz, 1H), 4.23 (t, J = 4.6 Hz, 2H), 3.82 (t, J = 4.6 Hz, 2H), 3.68-3.35 (m, 12H), 2.82-
2.50 (m, 4H); 13C-NMR (100 MHz, CDCl3, δ, ppm): 173.8, 173.1, 156.3, 141.1, 139.7,
137.5, 131.6, 130.1, 129.7, 128.9, 128.0, 127.6, 127.4, 126.7, 120.1, 71.9, 70.6, 69.5,
43.5, 30.3, 29.1.
Compounds 3. The compound 10 (50 mg, 0.063 mmol), DCC (14.3 mg) and 4-
nitrophenol (8.8 mg, 0.063 mmol) was dissolved in CHCl3 (5 mL). After stirring for 12
hours, the solvent was removed in a rotary evaporator, and the resulting mixture was

17
poured into water and extracted with ethyl acetate. The ethyl acetate solution was dried
over anhydrous magnesium sulfate and filtered. After the solvent was removed in a
rotary evaporator, the crude products were purified by column chromatography (silica
gel) using ethyl acetate as eluent to yield colorless liquid. Then, the liquid was dissolved
in anhydrous DMF (5 mL). To the solution L-RGD (38 mg, 0.063 mmol), DIPEA (25
mg) were added. After stirring for 24 hours, the solvent was removed. The residue was
recrystallized by methanol gave 3 as a white solid. MALDI-TOF-MS [M+H]+ calcd. for
C69H90N2O15: m/z 1263; Found: 1262.62.

18
Supplementary References
40. Kim, B.-S., Hong, D.-J., Bae, J., Lee, M. Controlled Self-Assembly of
Carbohydrate Conjugate Rod Amphiphiles for Supramolecular Multivalent
Ligands. J. Am. Chem. Soc. 127, 16333-16337 (2005).
41. Sanders, B. C. Friscourt, F., Ledin, P. A., Mbua, N. E., Arumugam, S., Guo, J.,
Boltje, T. J., Popik, V. V., Boon, G.-J. J. Am. Chem. Soc. 133, 949-957 (2011).
43. Kim, J.-K., Lee, E. Huang, Z., Lee, M. Nanorings from Self-Assembly of
Amphiphilic Molecular Dumbbells. J. Am. Chem. Soc. 128, 14022-14023 (2006).