Embed Size (px)
Citation preview

Mapeo, Ensamble y Comparación del Genoma
José Antonio Molinet Berenguer
CINVESTAV
30 de julio del 2013
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 1 / 45

1 Mapeo, Ensamble y Comparación del GenomaIntroducciónMapeo del genomaSecuenciación del GenomaEnsamble de la secuencia del genomaProgramas de base-calling y ensambleGenómica comparativaConclusiones
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 2 / 45

Mapeo, Ensamble y Comparación del Genoma Introducción
1 Mapeo, Ensamble y Comparación del GenomaIntroducciónMapeo del genomaSecuenciación del GenomaEnsamble de la secuencia del genomaProgramas de base-calling y ensambleGenómica comparativaConclusiones
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 3 / 45

Mapeo, Ensamble y Comparación del Genoma Introducción
Introducción
La Genómica es el estudio de los genomas. Estos estudios secaracterizan por el análisis simultáneo de un gran número degenes obtenidos con herramientas automatizadas de recolecciónde datos.
Los temas de genómica van desde el mapeo del genoma, lasecuenciación y el análisis genómico funcional, hasta el análisisgenómico comparativo.
El advenimiento de la genómica y la explosión de informaciónsobre secuencias son la principal fuerza impulsora del rápidodesarrollo de la bioinformática en la actualidad.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 4 / 45

Mapeo, Ensamble y Comparación del Genoma Introducción
Introducción
El estudio de la Genómica se puede dividir en genómicaestructural y genómica funcional.
La Genómica estructural se refiere a la fase inicial de análisis delgenoma, que incluye la construcción de mapas genéticos y físicosde un genoma, la identificación de genes, la anotación de lascaracterísticas de los genes, y la comparación de las estructurasdel genoma.
La Genómica estructural ha sido utilizada para determinarestructuras tridimensionales de las proteínas en una célula(proteómica estructural).
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 5 / 45
Mapeo, Ensamble y Comparación del Genoma Introducción
Introducción
En esta presentación, de la estructura genómica nos ocupaprincipalmente las estructuras de secuencias del genoma.
La Genómica funcional se refiere al análisis de la expresióngenética global y funciones de los genes en un genoma.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 6 / 45
Mapeo, Ensamble y Comparación del Genoma Mapeo del genoma
1 Mapeo, Ensamble y Comparación del GenomaIntroducciónMapeo del genomaSecuenciación del GenomaEnsamble de la secuencia del genomaProgramas de base-calling y ensambleGenómica comparativaConclusiones
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 7 / 45

Mapeo, Ensamble y Comparación del Genoma Mapeo del genoma
Mapeo del genoma
El primer paso para la comprensión de la estructura de ungenoma es a través de mapeo del genoma, siendo este elproceso de identificar las localizaciones de los genes, mutacioneso rasgos específicos en un cromosoma.
Un enfoque de baja resolución al mapeo de genomas es describirel orden y la distancia relativa de marcadores genéticos en uncromosoma.
Los marcadores genéticos son partes identificables de uncromosoma, cuyos patrones hereditarios pueden ser seguidos.Para muchas células eucariotas, los marcadores genéticosrepresentan fenotipos morfológicos.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 8 / 45
Mapeo, Ensamble y Comparación del Genoma Mapeo del genoma
Mapeo del genoma
Además de los mapas de relación genética, existen otros tipos demapas del genoma, tales como los mapas físicos y los mapascitológicos, que describen los genomas en diferentes niveles deresolución.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 9 / 45
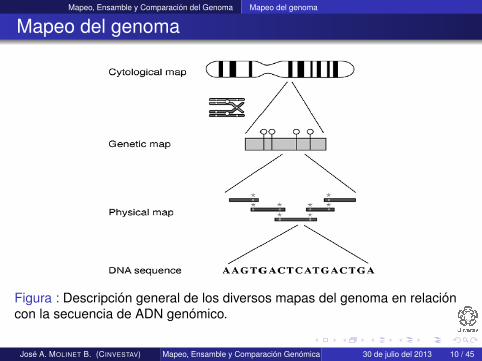
Mapeo, Ensamble y Comparación del Genoma Mapeo del genoma
Mapeo del genoma
Figura : Descripción general de los diversos mapas del genoma en relacióncon la secuencia de ADN genómico.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 10 / 45
Mapeo, Ensamble y Comparación del Genoma Mapeo del genoma
Mapeo del genoma
Los mapas genéticos identifican las posiciones de los marcadoresgenéticos en un cromosoma y se basan en la frecuencia con quelos marcadores se heredan juntos.
La razón detrás del mapeo genético es que cuanto más cercaestán dos marcadores, lo más probable es que se hereden juntosy no se separan en un evento de cruce genético.
La distancia entre dos marcadores genéticos se mide encentiMorgans (cM), que es la frecuencia con que sonrecombinados, o sea, cuando se observa separación de los dosmarcadores genéticos en un experimento de cruce genético.
Un centiMorgan es de aproximadamente 1 Mb en humanos y 0.5Mb en Drosophila
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 11 / 45
Mapeo, Ensamble y Comparación del Genoma Mapeo del genoma
Mapeo del genoma
Los mapas físicos representan localizaciones de puntos dereferencia en el ADN genómico, independientemente de lospatrones de herencia. La distancia entre los marcadoresgenéticos se mide directamente en Kb o Mb.
Los mapas citológicos se refieren a los patrones de bandas vistosen los cromosomas teñidos, que pueden ser observadosdirectamente bajo un microscopio. La luz visible y las bandasoscuras son los marcadores visualmente distintos en uncromosoma.
Un marcador genético puede estar asociado con una banda oregión cromosómica específica. Los patrones de bandas, sinembargo, no son siempre constantes y están sujetos a cambios.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 12 / 45
Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
1 Mapeo, Ensamble y Comparación del GenomaIntroducciónMapeo del genomaSecuenciación del GenomaEnsamble de la secuencia del genomaProgramas de base-calling y ensambleGenómica comparativaConclusiones
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 13 / 45

Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
Secuenciación del Genoma
La más alta resolución del mapa del genoma es la secuencia deADN genómico, que puede ser considerado como un tipo demapa físico describiendo un genoma al nivel de pares de bases.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 14 / 45

Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
Secuenciación del Genoma
Método de Sanger para la secuenciación de ADN:El uso de ADN polimerasas para sintetizar cadenas de ADN dediferentes longitudes.
La síntesis de ADN se detiene mediante la adición dedidesoxinucleótidos. Los didesoxinucleótidos son etiquetados concolorantes fluorescentes, que terminan la síntesis de ADN en lasposiciones que contienen todas las cuatro bases, lo que resultaen fragmentos anidados que varían en longitud por una sola base.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 15 / 45

Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
Secuenciación del Genoma
Los rastros fluorescentes de las secuencias de ADN son leídospor un programa de computadora que asigna bases para cadapico en un cromatograma. Este proceso automatizado sedenomina base-calling. Durante el base-calling se puedengenerar errores y la intervención humana es a menudo necesariapara corregirlos.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 16 / 45

Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
Secuenciación del Genoma
Hay dos estrategias principales para la secuenciación de todo elgenoma: el enfoque shotgun y el enfoque jerárquico.
El enfoque shotgun secuencia al azar los clones, desde ambosextremos del ADN clonado. Este enfoque genera un gran númerode fragmentos de ADN secuenciados.
El número de fragmentos al azar tiene que ser muy grande, tangrande que los fragmentos de ADN se solapen lo suficiente comopara cubrir todo el genoma.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 17 / 45

Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
Secuenciación del Genoma
No requiere conocimientos del mapeo físico de los fragmentosclonados, sino más bien un robusto programa de ensamblaje paraunir las piezas de fragmentos aleatorios en una única secuenciade todo el genoma.
Está diseñado para reducir al mínimo los errores desecuenciación y asegurar el montaje correcto de una secuenciacontigua.
Es por esto que normalmente se obtienen secuencias solapadascon una longitud total de seis a diez veces el tamaño del genoma.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 18 / 45

Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
Secuenciación del Genoma
El enfoque de la secuenciación jerárquica es similar al enfoqueshotgun, pero en una escala más pequeña. Los cromosomas semapean inicialmente utilizando la estrategia del mapeo físico.
Fragmentos más largos de ADN genómico (100 a 300 kB) seobtienen y se clonan en un vector bacteriano de alta capacidadllamado cromosoma artificial bacteriano (CAV).
Basándose en los resultados de mapeo físico, las ubicaciones yórdenes de los clones CAV en un cromosoma pueden serdeterminados.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 19 / 45

Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
Secuenciación del Genoma
Mediante la secuenciación sucesiva de fragmentos de clonesCAV adyacentes todo el genoma puede ser cubierto.
La secuencia completa de cada clon CAV individual puede serobtenida usando el enfoque de shotgun. La superposición declones CAV posteriormente se ensambla en una secuenciacompleta del genoma.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 20 / 45

Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
Secuenciación del Genoma
Hay ventajas y desventajas en ambos enfoques:El enfoque jerárquico es más lento y más costoso que el enfoquede shotgun porque implica una etapa inicial de mapeo físico delclon. Sin embargo, una vez que se genera el mapa, el montaje detodo el genoma se hace relativamente fácil y menos propenso aerrores.
Por el contrario, el enfoque de shotgun puede producir unborrador de secuencia muy rápidamente, ya que se basa en lasecuenciación directa. Sin embargo, es computacionalmente muycostoso ensamblar los cortos fragmentos aleatorios que obtiene.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 21 / 45

Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
Secuenciación del Genoma
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 22 / 45

Mapeo, Ensamble y Comparación del Genoma Secuenciación del Genoma
Secuenciación del Genoma
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 23 / 45

Mapeo, Ensamble y Comparación del Genoma Ensamble de la secuencia del genoma
1 Mapeo, Ensamble y Comparación del GenomaIntroducciónMapeo del genomaSecuenciación del GenomaEnsamble de la secuencia del genomaProgramas de base-calling y ensambleGenómica comparativaConclusiones
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 24 / 45

Mapeo, Ensamble y Comparación del Genoma Ensamble de la secuencia del genoma
Ensamble de la secuencia del genoma
Las reacciones de secuenciación de ADN generan secuenciascortas leídas a partir de clones de ADN. La longitud media de laslecturas es de aproximadamente 500 bases.
Para ensamblar una secuencia de todo el genoma, estosfragmentos cortos se unen para formar fragmentos más grandesdespués de la eliminación de las superposiciones.
Estas secuencias más largas, fusionadas se denominan contigs,que son por lo general 5.000 a 10.000 bases de longitud.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 25 / 45

Mapeo, Ensamble y Comparación del Genoma Ensamble de la secuencia del genoma
Ensamble de la secuencia del genoma
Un número de contigs superpuestos pueden ser aún másfusionados formando los llamados supercontigs (30.000-50.000bases), que son orientadas unidireccionalmente a lo largo de unmapa físico de un cromosoma.
La superposición de los supercontigs se conectan entonces paracrear el mapa final de más alta resolución del genoma.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 26 / 45

Mapeo, Ensamble y Comparación del Genoma Ensamble de la secuencia del genoma
Ensamble de la secuencia del genoma
La correcta identificación de superposiciones y montaje decontigs es como unir un rompecabezas, que puede sercomputacionalmente muy costoso cuando se trata de datos anivel de todo el genoma
Los principales retos en el montaje del genoma son los errores desecuenciación, la contaminación por vectores bacterianos y lasregiones de secuencias repetitivas.
Los errores de secuenciación a menudo se pueden corregirmediante la elaboración de un consenso de un alineamiento demúltiples secuencias solapadas.
Las secuencias de vectores bacterianos pueden ser eliminadasmediante programas de filtrado antes del montaje.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 27 / 45

Mapeo, Ensamble y Comparación del Genoma Ensamble de la secuencia del genoma
Ensamble de la secuencia del genoma
Para eliminar el problema de las repeticiones de secuencias,programas tales como RepeatMasker se pueden utilizar paradetectar y eliminar repeticiones.
Una técnica comúnmente usada para evitar errores causados porlas repeticiones de secuencia es la denominada restricciónforward–reverse.
Esta técnica se basa en que al generar una secuencia de ambosextremos de un único clon, la distancia entre los dos fragmentosopuestos de un clon se fija a un cierto rango, lo que significa quesiempre están separados por una distancia definida por lalongitud del clon (normalmente 1.000 a 9.000 bases).
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 28 / 45
Mapeo, Ensamble y Comparación del Genoma Programas de base-calling y ensamble
1 Mapeo, Ensamble y Comparación del GenomaIntroducciónMapeo del genomaSecuenciación del GenomaEnsamble de la secuencia del genomaProgramas de base-calling y ensambleGenómica comparativaConclusiones
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 29 / 45

Mapeo, Ensamble y Comparación del Genoma Programas de base-calling y ensamble
Programas de base-calling y ensamble
El primer paso hacia el ensamble del genoma es realizar elproceso de base-calling y asignar puntajes de calidad.
El siguiente paso es ensamblar las secuencias leídas ensecuencias contiguas. Este paso incluye la identificación de lossolapamientos entre fragmentos de secuencias, asignando elorden de los fragmentos y obteniendo una secuencia deconsenso general.
El ensamble de todos los fragmentos obtenidos por shotgun en ungenoma completo es un paso computacionalmente muy difícil.Hay una variedad de programas disponibles para este proceso. Lasiguiente es una selección de programas de montaje comúnmenteutilizados en los proyectos de secuenciación del genoma.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 30 / 45

Mapeo, Ensamble y Comparación del Genoma Programas de base-calling y ensamble
Programas de base-calling y ensamble
Phred (www.phrap.org/) es un programa de UNIX para realizardetección de bases. Se utiliza un análisis de Fourier para resolverlos rastros de fluorescencia y predecir posiciones de pico realesde bases. También da una puntuación de probabilidad para cadabase, que puede ser atribuible a un error. Si el valor de lapuntuación está por debajo del umbral, es necesaria laintervención humana.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 31 / 45

Mapeo, Ensamble y Comparación del Genoma Programas de base-calling y ensamble
Programas de base-calling y ensamble
VecScreen (www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html) esuna aplicación web que ayuda a detectar la contaminación porsecuencias de vectores bacterianos. Se analiza una secuencia denucleótidos de entrada y la compara con una base de datos desecuencias de vectores conocidos usando el programa BLAST.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 32 / 45

Mapeo, Ensamble y Comparación del Genoma Programas de base-calling y ensamble
Programas de base-calling y ensamble
EULER (http://nbcr.sdsc.edu/euler/) es un algoritmo de montajeque utiliza el enfoque del camino de Euler, que es un algoritmopolinomial para resolver los puzzles, como el famoso "problemadel viajante de comercio". En este enfoque, un fragmento desecuencia se descompone en tuplas de veinte nucleótidos.
Las tuplas se distribuyen en un diagrama con numerosos nodosque están todos interconectados. Las tuplas son convertidas avectores binarios en los nodos. Mediante el uso de un algoritmode Viterbi, el camino más corto entre los vectores se puedeencontrar, que es la mejor manera de conectar las tuplas en unasecuencia completa.
Debido a que este enfoque no se basa directamente en ladetección de solapamientos, puede ser ventajoso en el montajede secuencias con motivos repetitivos.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 33 / 45
Mapeo, Ensamble y Comparación del Genoma Programas de base-calling y ensamble
Programas de base-calling y ensamble
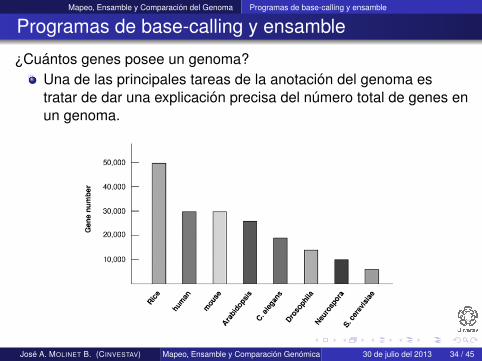
¿Cuántos genes posee un genoma?Una de las principales tareas de la anotación del genoma estratar de dar una explicación precisa del número total de genes enun genoma.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 34 / 45

Mapeo, Ensamble y Comparación del Genoma Genómica comparativa
1 Mapeo, Ensamble y Comparación del GenomaIntroducciónMapeo del genomaSecuenciación del GenomaEnsamble de la secuencia del genomaProgramas de base-calling y ensambleGenómica comparativaConclusiones
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 35 / 45

Mapeo, Ensamble y Comparación del Genoma Genómica comparativa
Genómica comparativa
La genómica comparativa es la comparación de genomascompletos de diferentes organismos, que incluye la correlacióndel número de genes, la localización de los genes, y el contenidode los genes.
La comparación ayuda a revelar el grado de conservación entrelos genomas, lo cual aporta conocimientos sobre el mecanismode la evolución del genoma y la transferencia de genes entre losgenomas.
Además, permite entender el patrón de adquisición de genesextraños a través de la transferencia lateral de genes y a revelar elconjunto básico de los genes comunes entre diferentes genomes,que deben corresponder a los genes que son cruciales para lasupervivencia.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 36 / 45
Mapeo, Ensamble y Comparación del Genoma Genómica comparativa
Genómica comparativa
La genómica comparativa incluye la alineación de todo elgenoma, la comparación del orden de genes entre los genomas,la construcción de genomas mínimos, y la transferencia lateral degenes entre los genomas.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 37 / 45

Mapeo, Ensamble y Comparación del Genoma Genómica comparativa
Genómica comparativa
La alineación del genoma completoPara comprender la conservación de la secuencia entre losgenomas se puede realizar una comparación directa entregenomas o el alineamiento del genoma.
La alineación del genoma no difiere en gran medida de laalineación de secuencias básicas. Sin embargo, la alineación delas secuencias extremadamente grandes presenta nuevascomplejidades.
Los programas de alineamiento regulares tienden a ser propensoa errores e ineficientes al tratar con largos tramos de ADN quecontengan cientos o miles de genes.
No se puede realizar una visualización efectiva de los resultadosde la alineación.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 38 / 45

Mapeo, Ensamble y Comparación del Genoma Genómica comparativa
Genómica comparativa
MUMmer (Maximal Unique Match,www.tigr.org/tigr-scripts/CMR2/webmum/
El programa es esencialmente un BLAST modificado que, en laetapa de siembra, encuentra los partidos aproximados más largosque incluyen desajustes en lugar de encontrar exactamente k-mercoincidencias como en BLAST regular.
El resultado de la alineación de los genomas enteros se muestracomo un gráfico de puntos con líneas de puntos conectados paraindicar colinealidad de los genes. Está optimizado para lacomparación por pares de genomas microbianos estrechamenterelacionados mumplot es un programa libre de TIGR para laalineación de dos secuencias completas del genoma y lacomparación de las ubicaciones de ortólogos.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 39 / 45

Mapeo, Ensamble y Comparación del Genoma Genómica comparativa
Genómica comparativa
BLASTZ (http://bio.cse.psu.edu/) es un programa modificado deBLAST para la alineación de grandes secuencias de ADNgenómico.
El programa BLAST modificado primero enmascara secuenciasrepetitivas y busca "palabras"muy similares que se definen comodoce coincidencias idénticas con un tramo de diecinuevenucleótidos.
Las palabras sirven como semillas para la extensión de laalineación en ambas direcciones hasta que las puntuacionescaen por debajo de un cierto umbral.
Realiza el alineamiento de regiones cercanas y estas se unenmediante el uso de un esquema de peso que emplea un sistemade penalización por hueco único que tolera variaciones menores.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 40 / 45

Mapeo, Ensamble y Comparación del Genoma Genómica comparativa
Genómica comparativa
Búsqueda de un genoma mínimoUno de los objetivos de la comparación del genoma es entenderlo que constituye un genoma mínimo, que es un conjunto mínimode genes necesarios para el mantenimiento de un organismocelular de vida independiente.
Este análisis consiste en la identificación de genes ortólogoscompartidos entre un número de genomas divergentes.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 41 / 45
Mapeo, Ensamble y Comparación del Genoma Genómica comparativa
Genómica comparativa
Búsqueda de un genoma mínimoCoregenes (http://pasteur.atcc.org:8050/CoreGenes1.0//) es unprograma basado en la web que determina un núcleo de genessobre la base de comparación de cuatro genomas pequeños. Elusuario proporciona los números de acceso NCBI de losgenomas de interés.
El programa realiza una comparación BLAST iterativo paraencontrar genes ortólogos mediante el uso de un genoma comouna referencia y la otra como una consulta.
Se lleva a cabo esta comparación por pares de los cuatrogenomas. Como resultado de ello, los genes comunes secompilan como un conjunto básico de los genes de los genomas.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 42 / 45
Mapeo, Ensamble y Comparación del Genoma Conclusiones
1 Mapeo, Ensamble y Comparación del GenomaIntroducciónMapeo del genomaSecuenciación del GenomaEnsamble de la secuencia del genomaProgramas de base-calling y ensambleGenómica comparativaConclusiones
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 43 / 45

Mapeo, Ensamble y Comparación del Genoma Conclusiones
Conclusiones
El mapeo del genoma utilizando las posiciones relativas de losmarcadores genéticos sin el conocimiento de la secuencia dedatos, es un enfoque de baja resolución para describir lasestructuras del genoma.
Un genoma se puede describir a la máxima resolución mediantela secuencia completa del genoma.
La secuenciación de todo el genoma puede llevarse a caboutilizando el enfoque shotgun o el enfoque jerárquico.
La anotación del genoma incluye la búsqueda de genes y laasignación de la función de estos genes.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 44 / 45
Mapeo, Ensamble y Comparación del Genoma Conclusiones
Conclusiones
El número de genes, sin embargo, no dicta complejidades de ungenoma.
Los genomas se pueden comparar sobre la base del contenido desus genes y de su orden. Se han desarrollado varios programasespecializados en comparación de genomas mediante laalineación transversal.
La comparación de genomas a través del orden de los genes amenudo ayuda a descubrir potenciales operones y asignarfunciones.
José A. MOLINET B. (CINVESTAV) Mapeo, Ensamble y Comparación Genómica 30 de julio del 2013 45 / 45