Embed Size (px)
Citation preview

IntroductionThe mapping of the human genome is of funda-mental importance, but only the beginning of a fullunderstanding of the relation between genes and thevarious external and internal factors that modifytheir expression. The epilepsies and psychiatricdisorders are two groups of diseases for whichboth nature and nurture play pathogenetic roles. Adeeper understanding of the interplay between thesetwo aspects is a prerequisite for new and bettertreatments.
To understand this complex interplay we need toknow more about the plasticity of the centralnervous system, the processes that generate andmodify normal neuronal development and activity,the genetic basis for inborn disorders and factorswhich may trigger an abnormal gene expression. Afull understanding also implies a detailed know-ledge of all the steps from gene activation to the®nal pathological condition. The many steps andmechanisms involved are potential targets of newdrugs.
The plasticity of the nervous system is much moreremarkable than thought of just a few years ago. Abetter knowledge of the factors involved in facil-itating and inhibiting regeneration may give hopesfor new treatments after injuries to the centralnervous system. Even more fascinating is thepossible use of modi®ed stem cells to compensatefor the loss of neurons in different kinds ofneurodegeneration. A better understanding of thegenetic processes involved will most likely make itpossible to interfere in a speci®c way.
The understanding of the genetic changes leadingto pathological function in excitability or behaviorcan be explored in animal models. Only a few ofthese changes, however, may be directly related tohuman disease. The animal data have to becompared with data from humans. This is dif®cult,as only a few of the actual diseases are monogeneticand dominant. Thus, the majority of epilepsies andpsychiatric disorders seems to involve both multiplegenes and many external and internal factors. The
variation in the relation between genotype andphenotype illustrates this complexity.
External factors include environmental stressorsin relation to psychiatric disorders and head traumaand febrile seizures in relation to epilepsy.Hormones are probably the most important inter-nal factors in¯uencing both the development andthe function of the central nervous system. Thus,alterations in hormones are important both inrelation to pathogenesis and symptomatology ofepilepsy and psychiatric disorders.
The full mapping of the genome will makepossible a more direct approach for drug designaimed at interfering with gene expression or evenmodifying speci®c genes. It is most likely that manyother drugs targets will be identi®ed both in epilepsyand psychiatry after mapping the genome. Evenbefore that goal is achieved, however, the analysisof single nucleotide polymorphisms will be useful inoptimizing drug treatment with respect to ef®cacyand side effects. In addition, the usefulness of ``old''drugs may well be increased by a better insight intheir mechanisms of action and by a better under-standing of pathogenesis and pathophysiology ingeneral.
The balance between nature and nurture maynever be fully explored. However, the mapping ofthe genome is ``the end of the beginning'' of a muchmore detailed understanding of the epilepsies andpsychiatric disorders. The promising future is newtreatment strategies and better care for patients.
This Supplement presents a set of papers from themeeting ``Nature and Nurture in Epilepsy andPsychiatry ± Implications for New TreatmentStrategies'' in Oslo, Norway June/July, 2000. Wethank the authors for their effort in writing thesepapers and Glaxo Wellcome for supporting thepublication.
Erik Taubùll Ole Petter OttersenRay Dingledine Leif Gjerstad.
Acta Neurol Scand 2000: Suppl. 175: 5±52Printed in UK. All rights reserved
Copyright # Munksgaard 2000
ACTA NEUROLOGICASCANDINAVICA
ISSN 0001-6314
5

Neuronal connectivity is reshaped by use ± in health and disease
P. AndersenInstitute for Basic Medical Sciences, University of Oslo
In his 1913±1914 double volume RamoÂn y Cajal (1)emphasized the absence of neurogenesis afterdamage to the mammalian central nervous systemand the limited regeneration of central, in contrastto peripheral, nerve ®bres. His in¯uence was, andstill is, so enormous that many later clinicalneurologists and experimental neuroscientists mayhave underestimated the possibilities for repairprocesses in the central nervous system.
Recently, progress on many fronts has changedthis situation such that the repair scepticism isabout to be replaced by a cautious optimism. Thechange of attitude is due to ®ndings in four majorareas: 1) the ability of central axons to grow inmany environments and conditions, 2) the ®ndingof neurogenesis in several adult nervous systems,including man, and ®nally, 3) the discovery thatembryonic stem cells may be experimentallyinduced to differentiate into various nerve andglial types and 4) the presence of neural stem cells,even in adult organisms.
We have long known that central axons from avariety of neuronal types may grow for longdistances into a guide formed by degeneratedperipheral nerves (2). In fact, when dissociatedadult dorsal ganglion cells are injected into thebrain, they grow axons for long distances on theouter lea¯et of myelinated ®bres, even along ®bresundergoing Wallerian degeneration (3). However,when central axons meet a glial scar, progress ishalted. Schwab and collaborators (4) have shownthat this is due to an inhibitory factor and thatapplication of an inactivating antibody againstcentral myelin, IN-1, improve regeneration. Thespeci®c target for the IN-1 action has recently beenidenti®ed as a novel protein Nogo-A (5±7). The cellsmust be damaged in order for suf®cient quantitiesof Nogo-A to be released. Myelin-associatedglycoprotein (MAG) is also a growth-inibitingmolecule (8).
Other candidates for inhibiting molecules asso-ciated with glial scars are two growth cone guidingmolecules, semaphorin-3A (9) and EphB3 (10). Aset of other extracellular matrix molecules andtissue adhesion molecules have also been shown toinhibit axonal growth (8). The degree of regenera-tion also depend upon possible priming of theinjured ®bres at, or shortly after, the injury.Interference with priming stimuli and moleculesmay well open up new therapeutic approaches.Finally, a variety of neurotrophins has been shown
to be supporting central regeneration, either aloneor in conjunction with guiding cells (see below).
Sprouting, or the emission of axonal side-branches, is another important mechanism under-lying central repair processes. Here, the experimen-tal data are more sparse, but there is evidence thatintense activation, as during long-term potentiation(LTP) or spatial learning in intact animals and inculture may induce new synapses with both pre-synaptic boutons and postsynaptic spines withnormal complement of glutamate receptors andexcitatory synaptic function (11±13). The growthappears to be mediated by the cAMP/CREB system(14).
Sprouting is also a likely explanation for much ofthe remarkable training-induced reduction of thefunctional adaptation after sensory alterations (15)or the reduction of the de®cit after an experimentalcortical infarct in monkeys (16). Another strikingexample of cortical plasticity is the fast learning seenin congenitally deaf kittens after a cochlear implant(17).
Spinal cord damage still represents an immensechallenge to neuroscience. However, several pro-mising avenues have given exciting results recently.Experimental removal of 3-5 spinal segments in ratshave been bridged by a template of degeneratedintercostal nerves embedded in a ®brin block dopedwith several neurotrophins, particularly aFGF.After removal of one vertebra and surgical narrow-ing of the original gap to ensure good contactbetween the proximal and distal parts of the spinalcord, the rats were given intense motor training.After several months, the best rats showed encour-aging regeneration as seen in histological controls ofthe implant and retrograde ®lling of pyramidal cellsin the contralateral motor cortex. Functionally, theparaplegia was reduced and clear signs of rhythmicstepping and walking with proper hind-forelimbreciprocal activation, suggest functional re-estab-lishment with proper neuronal targets in the distalpart of the spinal cord (18).
Even more convincing restitution was seen afterunilateral pyramidal tract lesions in rats in which asuspension of olfactory ensheathing cells (OEC),previously removed from the nasal cavity, grown inculture for several weeks was injected into thelesion, with and without neurotrophins. A sub-stantial number of ®bres grew from the upper partof the pyramidal tract through the lesion and madefunctional contact with inter- and motoneurones ofthe cervical and thoracic spinal cord, as judged by
6

the results of a pyramidal tract-speci®c tests offorelimb activity (19).
The recent description of cytokine-driven differ-entiation of embryonic stem cells to variousneuronal and glial phenotypes has created enor-mous interest, for which the therapeutic potential isthe most obvious reason. Of particular interest isthat the brain and spinal cord retain a small numberof neural stem cells, which may support neurogen-esis. We have known for some decades that a smallnumber of dentate granule cells may be formed inadult rats, and that the amount depends upon theactivity level and the presence of cAMP and certainhormones. However, a recent report showed thepresence of neurogenesis in the human dentategyrus as well (20). Apart from a normal appearanceand the presence of boutons and postsynapticspines, we have little knowledge of their possiblefunctional status ± not to say anything about theirphysiological role.
In addition to the dentate stem cells, there is athin band of other neural stem cells coursing overthe dorsomedial aspect of the caudate nucleus andproceeding towards the olfactory bulb. Althoughlittle is known about the properties and potential-ities of this system apart from their ability toundergo mitosis after proper mitogenic challenge,the recent demonstration that injection of neuralstem cells from mice into chick or mouse embryosgive rise to cells in all germ layers appears promising(21).
We no doubt can expect a ¯urry of investigationson the possible functional competence of neuralstem cells and their possible use in therapy (22).
References
1. RAMON Y CAJAL S. Estudios sobre la degeracioÂn y regeracioÂndel sistema nervioso. 1913±1914. Madrid, 2 vol.
2. BRAY GM, VILLEGAS-PEREZ MP, VIDAL-SANZ M, AGUAYO AJ.The use of peripheral nerve grafts to enhance neuronalsurvival, promote growth and permit terminal reconnectionsin the central nervous system of adult rats. J Exp Biol1987;132:5±19.
3. DAVIES SJA, FITCH MT, MEMBERG SP, HALL AK, RAISMAN G,SILVER J. Regeneration of adult axons in white matter tracts ofthe central nervous system. Nature 1997;390: 680±683.
4. SCHNELL L, SCHWAB ME. Axonal regeneration in the rat spinalcord produced by an antibody against myelin-associ-atedneurite growth inhibitors. Nature 1990;343:269±272.
5. CHEN MS, HUBER AB, vAN DER HAAR ME et al. Nogo-A is amyelin-associated neurite outgrowth inhibitor and an anti-gen for monoclonal antibody IN-1. Nature 2000;403:434±439.
6. GRANDPREÂ T, NAKAMURA F, VARTANIAN T, STRITTMATTER SM.Identi®cation of the Nogo inhibitor of axon regeneration as aReticulon protein. Nature 2000;403:439±444.
7. PRINJHA R, MOORE SE, VINSON M et al. Inhibitor of neuriteoutgrowth in humans. Nature 2000;403:383±384.
8. FAWCETT JW, ASHER RA. The glial scar and central nervoussystem repair. Brain Res Bull 1999;49:377±391.
9. PASTERKAMP RJ, GIGER RJ, RUITENBERG MJ et al.Expression of the gene encoding the chemorepellentsemaphorin III is induced in the ®broblast componentof neural scar tissue formed following injuries of adultbut not neonatal CNS. Mol Cell Neurosci1999;13:143±166.
10. MIRANDA JD, WHITE LA, MARCILLO AE, WILLSON CA, JAGID
J, WHITTEMORE SR. Induction of Eph B3 after spinal cordinjury. Exp Neurol 1999;156:218±222.
11. TROMMALD M, HULLEBERG G, ANDERSEN P. Long-term poten-tiation is associated with new excitatory spine syn-apses onrat dentate granule cells. Learning & Memory 1996;3:218±228.
12. ENGERT F, BONHOEFFER T. Dendritic spine changes asso-ciated with hippocampal long-term synaptic plasticity.Nature 1999;399:66±70.
13. MALETIC-SAVATIC M, MALINOW R, SVOBODA K. Rapiddendritic morphogenesis in CA 1 hippocampal dendritesinduced by synaptic activity. Science 1999;283:1923±1927.
14. KORKOTIAN E, SEGAL M. Fast confocal imaging of calciumreleased from stores in dendritic spines. Eur J Neurosci1998;10:2076±2084.
15. KAAS JH. Plasticity of sensory and motor maps in adultmammals. Ann Rev Neurosci 1991;14:137±167.
16. NUDO RJ, WISE BM, SIFUENTES F, MILLIKEN G. Neural sub-strates for the effects of rehabilitative training on motorrecovery after ischemic infarct. Science 1996;272:1791±1794.
17. KLINKE R, KRAL A, HEID S, TILLEIN J, HARTMANN R. Recruit-ment of the auditory cortex in congenitally deaf cats bylong-term cochlear electrostimulation. Science 1999;285:1729±1733.
18. CHENG H, CAO Y, OLSON L. Spinal cord repair in adultparaplegic rats: partial restoration of hind limb function.Science 1996;273:510±513.
19. LI Y, FIELD PM, RAISMAN G. Regeneration of adult ratcorticospinal axons induced by transplanted olfactoryensheating cells. J Neurosci 1998;18:10514±10524.
20. ERIKSSON PS, PERFILIEVA E, BJOÈ RK-ERIKSSON T et al.Neurogenesis in the adult human hippocampus. NatureMed 1998;4:1313±1317.
21. CLARKE DL, JOHANSSON CB, WILBERTZ J et al. Generalisedpotential of adult neural stem cells. Science 2000;288:1660±1663.
22. BJOÈ RKLUND A. The use of neural stem cells for genetherapy in the central nervous system. J Gene Med1999;1:223±226.
7

Estradiol induces formation of dendritic spines in hippocampal neurons:functional correlates
M. Segal1, D. Murphy2
1Neurobiology, The Weizmann Institute, Rehovot, Israel and 2NINDS, NIH, MD, USA
Estradiol causes a transient increase in dendriticspine density in pyramidal neurons of the cyclingfemale rat hippocampus, an area of the brain notintuitively associated with sexual behavior (1). Wehave replicated these results in dissociated rathippocampal cultures, to ®nd that estradiol canincrease dendritic spine density by up to two foldover control values (2). This effect was slow todevelop and peaked within 3 days of exposure toestradiol. It was mediated by activation of theestrogen receptor, as it was blocked by tamoxifen,an estrogen receptor antagonist. In a search for themolecular cascade leading to the formation ofnovel dendritic spines by estradiol, we found thatestradiol activates CREB phosphorylation inpyramidal neurons in culture, an effect that islikely to result from the enhanced network activityproduced by estradiol. Indeed, blockade of actionpotential discharges in the culture dish withtetrodotoxin (TTX) or blockade of CREB phos-phorylation by various means blocked the effectsof estradiol on dendritic spine formation, as well ason CREB (3). We then found that estradioldownregulates the expression of the GABAsynthesizing enzyme GAD. Interestingly, alpha-estrogen receptors were localized exclusively onGABAergic interneurons in the culture. It appearsthat with the reduction in GAD activity, theinhibitory tone in culture was reduced, andnetwork excitatory activity was enhanced (4).Furthermore, enhancement of the GABAergicreceptor function in the culture with diazepamalso abolished the effects of estradiol on dendriticspine formation and CREB response. A factorwhich may link the estrogen receptor to GAD isthe brain-derived neurotrophic factor (BDNF),which was found to be regulated by exposure toestradiol and may underlie the formation of noveldendritic spines (5).
Another hormone, progesterone, was found toblock the effects of estradiol on formation ofdendritic spines. In trying to establish the site ofaction of progesterone that interacts with theestrogen action, we realized that progesterone is
converted in our culture dish to tetra-hydroprogesterone (THP), as it does in-vivo. THP has apotentiating action on GABAergic synaptic activityin our cultured neurons (6), as shown elsewhere,con®rming the involvement of GABAergic inhibi-tion in the action of estradiol.
Finally, if indeed estradiol affects networkactivity in the culture dish by reducing inhibition,we should be able to observe these effects whilemonitoring electrical activity in the presence ofestradiol. Indeed, spontaneous activity, recordedfrom clusters of cells in the dish using the calcium¯uorescent indicator, Fluo-4 imaged with a fastCCCD camera, revealed that estradiol facilitatesformation of coordinated bursts of activity, muchlike the action of the GABA antagonist bicuculline.It is suggested that estradiol reduces inhibition inhippocampal cultures, shifting the balance to anenhanced excitation, which causes an in¯ux ofcalcium into the postsynaptic cells, activation ofCREB and subsequent formation of novel dendriticspines.
References
1. WOOLLEY CS, GOULD E, FRANKFURT M, MCEWEN BS.Naturally occurring ¯uctuation in dendritic spine densityon adult hippocampal pyramidal neurons. J Neurosci1990;10:4035±4039.
2. MURPHY DD, SEGAL M. Regulation of dendritic spine densityin cultured rat hippocampal neurons by steroid hormones. JNeuroscience 1996;16:4059±4068.
3. MURPHY DD, SEGAL M. Morphological plasticity of dendriticspines in central neurons is mediated by activation of cAMPresponse element binding protein. Proc Natl Acad Sci USA1997;94:1482±1487.
4. MURPHY DD, COLE NB, GREENBERGER V, SEGAL M. Estradiolincreases dendritic spine density by reducing GABAneurotransmission in hippocampal neurons. J Neuroscience1998;18:2550±2559.
5. MURPHY DD, COLE NB, SEGAL M. BDNF mediatesestradiol- induced dendritic spine formation in hippocampalneurons. Proc Nat Acad Sci USA 1998;95:1412±1417.
6. MURPHY D, SEGAL M. Progesterone prevents estradiol-induced dendritic spine formation in cultured hippocampalneurons. Neuroendocrinology 2000, In press.
8

Genomics and epilepsy research in the new millenium
R. Dingledine, J. DohertyDepartment of Pharmacology, Emory University, Atlanta, GA 30322, USA
By the end of June, 2000 the NIH and Celera willhave jointly announced the achievement of a ®rstdraft of the complete human genome sequence. Thecomplete genome sequences of ,30 other organismshave already been published, including those ofsuch important animals as D. melanogaster and C.elegans (1), and ,30% of the mouse genome isalready sequenced. Although the ®nished humansequence will not be available immediately, it isappropriate to consider how this development willin¯uence epilepsy research in the coming years. Atleast two bene®ts are expected ± identi®cation ofnew anticonvulsant drug targets, and geneticstrati®cation of patients to improve therapy.
Commonly prescribed anticonvulsant drugs aretargeted to just ®ve known proteins ± sodiumchannels, calcium channels, GABAA receptors,GABA transporters and GABA transaminase.Given that many more receptors, channels andenzymes regulate neuronal excitability, it is highlylikely that additional drug targets exist for epilepsy.For comparison, prescription drugs for all diseasesand disorders are directed to only 500 targetproteins, whereas Drews (2) has estimated that3000±10,000 potential drug targets exist. Moreover,with the possible (but yet undemonstrated) excep-tion of levetiracitam, none of the currently availableanticonvulsant drugs prevents the appearance ofepilepsy in those at risk, for example after headinjury. That is, we have no drugs or othertherapeutic strategies that interrupt epileptogenesis.Experimental strategies that involve an unbiasedgenome scan in epilepsy models should identifypotential new drug targets as well as the geneticprograms responsible for epileptogenesis.
We have begun experiments with DNA micro-arrays to examine the expression levels of 8700mouse mRNAs in the dentate gyrus at differentstages of epileptogenesis in the mouse pilocarpinemodel (3). Mice that experience several hours ofpilocarpine-induced status epilepticus developspontaneous intermittant seizures (i.e., epilepsy)three to four weeks later. At different times (1±30days) after 3 hours of pilocarpine-induced statusepilepticus, mice were sacri®ced and mRNA wasisolated from microdissected dentate granule celllayer. Approximately 200 genes with expressionchanges more than 3 standard deviations from themean (mean=100%) were identi®ed. A neuralnetwork algorithm known as a self-organizing map(4) was used to recognize three temporal patterns
of gene expression in dentate granule cells: early-responders (118 genes), intermediate-responders(37 genes) and late-responders (12 genes). Theexpression of the intermediate-responding geneswas not appreciably changed shortly after seizuresor during the phase of spontaneous seizures,suggesting that these genes do not respond tothe seizures themselves. However, their expressionwas changed up or down during the latent phase.Some genes with this temporal pattern of expres-sion might represent potential targets for inter-rupting the process of epileptogenesis, because theydo not respond directly to the seizures themselves,but may contribute to events in the latent periodthat eventually result in epilepsy. Less than 10% ofthe mouse genome was sampled by this experi-ment, suggesting that eventually >400 genes mayfall into the ``intermediate-responding'' category.This pool of genes contains potential drug targetsfor interrupting epileptogenesis; the initial chal-lenge is to narrow the ®eld to a manageablenumber of candidates. Following that, thesecandidate genes must be validated geneticallybefore becoming bone ®de drug targets. Variousstrategies are being employed to reduce thenumber of candidate genes to a handful, such as+/- comparisons with additional epilepsy models,and the use of Rosettsa stone sequences, phylo-genetic pro®les, keyword clustering, etc. Targetvalidation will involve ``normalizing'' the expres-sion of individual genes, either by overexpressionof genes with viral vectors for genes downregulatedduring the latent period, or antisense strategies forupregulated genes.
Although the genomes of all people are extremelysimilar, most genes are polymorphic; i.e., they differamong individuals at nucleotide positions randomlyscattered throughout the genome. Allelic variationinvolving such Single Nucleotide Polymorphisms(SNPs) occurs about every 300-1000 base pairs,generating more than 3 million SNPs in the genome.Much of our individual variability, includingperson-to-person variation in responses to drugs,is thought to be accounted for by the subtlefunctional consequences of these SNPs on bothcoding regions and promoters of genes. Evenwithout knowledge of their functions, SNPs canbe useful for sorting patients genetically for drugef®cacy and toxicity (5). In one possible implemen-tation for clinical trials, genome-wide scans of100,000 or more randomly-distributed SNPs would
9

be performed during phase II clinical trials. SNPpatterns are then correlated with clinically-deter-mined drug ef®cacy and common toxicities. Asubset of several hundred informative SNPs is thenused in phase III trials to prescreen patients forthose likely to respond, but without toxicity. Thisapproach should reduce the number of patientsneeded in phase III trials. Moreover, this approachwould also permit the parallel development of astable of drugs for a particular illness, resulting inmore individualized drug treatments. Post-market-ing studies of larger populations can further re®netreatment by identifying SNP pro®les predictive ofless common toxicities. This strategy does not relyon knowledge of the functions of any of the genesinvolved, but rather depends on simple statisticalcorrelations between genotype and phenotype.Additionally, similar strategies could be used tocorrelate genetic pro®le with optimal drug treat-ment for currently-marketed anticonvulsant drugs.Such a research program would require consider-able resources and be international in scope.
Current technologies are adequate (although cur-rently prohibitively expensive) for reliable analysisof several hundred SNPs per patient on a single``SNP chip''. The challenge is to develop methodsfor higher throughput polymorphism scoring thatcan handle the workload required for phase II trials.
References
1. BRODER S, VENTER JC. Sequencing the entire genomes of free-living organisms: the foundation of pharmacology in the newmillenium. Ann Rev Pharm Toxicol 2000;40:97±132.
2. DREWS J. Genomic sciences and the medicine of tomorrow:commentary on drug development. Nature Biotechnology1996;14:1516±1518.
3. DINGLEDINE R, DOHERTY JJ. Use of gene chips to studyepileptogenesis. Soc Neurosci Abst 1999;25:1110.
4. TAMAYO P, SLONIM D, MESIROV J et al. Interpreting patterns ofgene expression with self-organizing maps: methods andapplication to hematopoietic differentiation. Proc Nat AcadSci USA 1999;96:2907±2912.
5. ROSES AD. Pharmacogenetics and the practice of medicine.Nature 2000;405:857±865.
Receptor turnover in the central nervous system
J. M. HenleyMRC Centre for Synaptic Plasticity, Department of Anatomy, School of Medical Sciences, University of Bristol, Bristol, BS8 1TD, UK
a-Amino-3-hydroxy-5-methylisoxazolepropionate(AMPA) receptors mediate most synaptic transmis-sion in the mammalian CNS, play a central role insynapse stabilisation and plasticity and theirprolonged activation is potently neurotoxic.Developmental and activity-dependent changes inthe functional synaptic expression of these receptorsare under tight cellular regulation. The molecularand cellular mechansims which control the post-synaptic insertion and arrangement of AMPAreceptors are therefore the subject of intenseinvestigation and in the last two years there hasbeen signi®cant progress towards elucidating someof the processes involved. Recent work from ourlaboratory has suggested that AMPA receptorscontaining the subunit GluR2 undergo rapidrecycling between the postsynaptic membrane andan intradendritic pool. This recycling processinvolves the protein N-ethylmaleimide sensitivefusion protein (NSF) which we and others haveshown to bind to the GluR2 subunit (1±5).
NSF is a well characterised multihomomericATPase which plays an essential role in the
membrane fusion processes underlying proteintraf®c through the Golgi apparatus and vesicularrelease of neurotransmitters at the presynapticmembrane (6). NSF is strongly expressed in theCNS and is most abundant in the hippocampus (7,8). Furthermore, despite its known presynaptic role,transient cerebral ischemia has been reported tocause an accumulation of NSF in the postsynapticdensity (9). It has also been reported recently thatinduction of long-term potentiation (LTP) isblocked by N-ethylmaleimide (NEM), a potentinhibitor of NSF (10). Thus, even before a directinteraction between GluR2 and NSF was demon-strated there were data available to suggest thatpostsynaptic NSF could play a role in activity-dependent and pathological changes in synapticfunction.
We found that NSF binds to a seemingly uniquerecognition site located at the C-terminal of theGluR2 subunit of AMPA receptors (1). This NSFinteraction with GluR2 regulates the surfaceexpression of AMPA receptors in hippocampalneurons (5). Blockade of the interaction between
10

NSF and GluR2 by infusing inhibiting peptidescorresponding to the binding domain of GluR2 oran anti-NSF antibody from a patch pipette in to thepostsynaptic neurons in hippocampal slices resultsin a rapid and substantial decrease in evokedAMPA receptor-mediated synaptic transmission.A control peptide and a control anti-NSF antibodywhich does not recognise rat NSF had no effect (2).These data suggest that NSF may regulate themembrane insertion/stabilisation, and thus func-tional expression, of GluR2-containing AMPAreceptors. We have also shown that adenoviralexpression of the inhibiting peptide but not acontrol peptide in cultured hippocampal neuronsresulted in a dramatic loss in the number of AMPAreceptor aggregates on the cell surface (5).Importantly, AMPA receptor aggregates are stillpresent close to synapses inside the dendrites andthe total amount of GluR2 immunoreactivity doesnot decrease (5). These ®ndings suggest thatblocking the interaction between NSF and GluR2does not prevent receptor synthesis or passagethrough the ER/Golgi systems but rather that it hasan effect at the synapse.
We went on to investigate whether the interactionbetween NSF and GluR2 is involved in synapticplasticity in the CA1 region of the hippocampus.Blockade of the NSF-GluR2 interaction preventedhomosynaptic, de novo long-term depression(LTD). In addition, saturation of LTD preventedthe blocking peptide-induced reduction in AMPAreceptor-mediated excitatory postsynaptic currents(EPSCs). These data suggested that there is a poolof AMPA receptors dependent on the NSF-GluR2
interaction and that LTD expression involves theremoval of these receptors from synapses (11).
References
1. HENLEY JM, NISHIMUNE A, NASH SR, NAKANISHI S. Use ofthe two-hybrid system to ®nd novel proteins that interactwith AMPA receptor subunits. Biochem Soc Trans 1997;25:838±841.
2. NISHIMUNE A, ISAAC JTR, MOLNAR E et al. NSF binding toGluR2 regulates synaptic transmission. Neuron 1998;21:87±97.
3. OSTEN P, SRIVASTAVA S, INMAN GJ et al. The AMPA receptorGluR2 C terminus can mediate a reversible, ATP-depen-dent interaction with NSF and alpha- and beta-SNAPs.Neuron 1998;21:99±110.
4. SONG I, KAMBOJ S, XIA J, DONG H, LIAO D, HUGANIR RL.Interaction of the N-ethylmaleimide-sensitive factor withAMPA receptors. Neuron 1998;21:393±400.
5. NOEL J, RALPH GS, PICKARD L et al. Surface expression ofAMPA receptors in hippocampal neurons is regulated by anNSF-dependent mechanism. Neuron 1999;23:365±76.
6. ROTHMAN JE. Intracellular membrane fusion. Adv SecondMessenger Phosphoprotein Res 1994;29:81±96.
7. HONG YG, CECHETTO DF, WEAVER LC. Spinal cordregulation of sympathetic activity in intact and spinalrats. Am J Physiol 1994;266:H1485±93.
8. PUÈ SCHEL AW, O'CONNOR V, BETZ H. The N-ethylmaleimide-sensitive fusion protein (NSF) is preferentially expressed inthe nervous system. FEBS Letts 1994;347:55±58.
9. HU B-R, PARK M, MARTONE ME, FISCHER WH, ELLISMAN
MH, ZIVIN JA. Assembly of proteins to postsynapticdensities after transient cerebral ischemia. J Neurosci1998;18:625±633.
10. LLEDO P-M, ZHANG X, SUÈ DHOF TC, MALENKA RC, NICOLL
RA. Postsynaptic membrane fusion and long-term poten-tiation. Science 1998;279:399±403.
11. LUTHI A, CHITTAJALLU R, DUPRAT F et al. Hippocampal LTDexpression involves a pool of AMPARs regulated by theNSF-GluR2 interaction. Neuron 1999;24:389±399.
Nature in the development of epilepsy
J. L. Noebels, D. L. Burgess, J. QianDevelopmental Neurogenetics Laboratory, Department of Neurology, Baylor College of Medicine, Houston, TX, 77030 USA
Heredity is the single most important determinantof epilepsy, and many genes that control thesusceptibility of the brain to seizures are nowbeing identi®ed in human families and variousexperimental model organisms (1, 2, 3). Epilepsiesthat arise from defects in a single gene have been thesource of all gene discoveries so far, however manyif not most seizure disorders appear to be theproduct of multiple genetic loci that associate bychance, and this mode of inheritance has also beenexperimentally modeled (4). Despite the largenumber of potential epilepsy disease genes and
inherent neurobiologic complexity, detailed analysisof these genetic disorders is beginning to reveal theremarkable precision and speci®city underlying thedevelopmental expression of epilepsy.
The biological functions of epilepsy genes fallinto multiple categories. The ®rst of these includesmolecules that directly control neuronal membraneexcitability and synaptic transmission by regulatingspeci®c ion channels or transmitter signaling path-ways. Although this category is the best understood,it remains biologically complex. These diverse genefamilies are numerically large, and different muta-
11

tions within any individual gene can producedistinct excitability patterns, leading to both locusand allelic heterogeneity of any particular heredi-tary seizure phenotype. Membrane ion channelsand receptors are assembled as regionally distinctheteromeric protein complexes, thus increasing thelikelihood of distinct subunit mutations affecting aspeci®c channel type differently in particularcompartments of the neuron, or entire brainpathways. Ion channel subunits also interact withother proteins inside and outside the cell, affectingmembrane excitability functions other than porekinetics.
A second broad category of epilepsy genefunction consists of the signaling molecules thatindirectly modulate the membrane excitabilityproteins over highly variable time periods rangingfrom milliseconds to days. These molecules mayactivate other genes or protein cascades that altermultiple membrane targets under different physio-logical conditions. Some genes associated withepilepsy exert major effects at critical times ofearly brain development, while others apparentlyspare these processes and act at later stages. A thirdcategory, which for now is the largest, includes``orphan'' genes with unknown functions. In each ofthese categories, even that including ion channels,the actual pathogenesis of epilepsy remains poorlyunderstood despite some knowledge of the basiccellular mechanisms.
While each of these genes for epilepsy represents aunique regulatory site in nervous system function, afundamental issue is to understand how an inheriteddefect in cellular signaling causes episodic seizuresand not some other disorder. The anatomy of thehyperexcitable circuit is a major determinant of thephenotype, and should be de®ned by the cells thatexpress the mutant gene, however this may only beclear in some cases. For example, a sodium channelthat leads to episodic arrythmias in the heart is an
excellent candidate for seizure disorders owing to itsselective distribution in brain limbic structures,including amygdala and piriform cortex (5). In thisexample, an inherited mutation that slows theinactivation kinetics of the channel will produceprolonged depolarization in cardiac muscle cells(LQT3), and in limbic circuits, in particular twobrain regions with very low thresholds for epilepto-genesis.
In contrast, many of the genes found to date arediffusely expressed throughout the brain yet pro-duce distinct patterns of epilepsy, suggesting thatonly certain neural pathways are involved, and thatthe seizure phenotype may be determined byselective vulnerability of speci®c neural circuits.Recent work in our laboratory on calcium ionchannel mutations suggests that one source of thisselective vulnerability is based not on the anatom-ical expression of the mutant gene, but on theexpression patterns of compensatory channel sub-unit genes (6, 7).
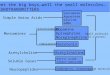
In the tottering mouse, mutation of the widelydistributed P/Q type voltage-gated calcium chan-nel reduces P/Q type currents, presumably in allcells where the current is expressed. Opticalimaging of calcium entry into presynaptic term-inals combined with electrophysiological record-ings indicating transmitter release reveal thatrelease can be rescued by the co-localization ofN-type currents at the terminal. This rescue wouldnot occur at terminals that do not co-expressfunctional N-type channels, de®ning a subpopula-tion of neurons at risk (6). In the case of thelethargic mouse, loss of the cytoplasmic betasubunit b4 calcium channel subunit leads to anearly identical spike-wave seizure phenotype inthe mouse, however the P/Q type current in somecells can be rescued by alternative pairing of theP/Q type pore-forming subunit with other avail-able (b1-3) subunits, in a process we term
Fig. 1. Differential expression of beta subunits of the voltage-gated calcium channel contributes to emergence of the spike-wave seizure phenotype in lethargic mice. (See ref. 7).
12

``reshuf¯ing'' (7). In this case, neurons without thenative b1-3 subunits, which are differentiallyexpressed in brain (Figure 1), are at risk forimpaired P/Q type channel function. Takentogether, the results provide a general mechanismfor selective excitability changes in neural circuitswithin a nervous system bearing calcium channelsubunit mutations.
While much gene discovery in epilepsy lies ahead,the biological impact of each of these molecules onthe excitability and synchronization of neuralcircuits remains to be carefully examined inmutant mouse models. The mouse mutants allowthe role of mutant gene product and developmentalplasticity to be evaluated, and can be engineered toencode gene mutations identical to those present inthe human disorder. Finally, these genetic modelscan be serially analyzed at various stages, and areideal test systems to identify and evaluate novelmolecular targets for therapy.
References
1. NOEBELS JL. The Inherited Epilepsies. In: SCRIVER, BEAUDET,SLY, VALLE, eds. The Metabolic and Molecular Basis ofInherited Disease. Chapter 230. 8th ed. New York: McGrawHill, 2000, in press.
2. STEINLEIN O, NOEBELS JL. Ion Channels and Epilepsy in Manand Mouse. Curr Opin Genet Dev 2000;10:286±291.
3. NOEBELS JL. Targeting Epilepsy Genes. Neuron1996;16:241±244.
4. FRANKEL WN, TAYLOR BA, NOEBELS JL, LUTZ CM. Geneticepilepsy model derived from common inbred mouse strains.Genetics 1994;138:481±489.
5. HARTMANN HA, COLOM LV, SUTHERLAND ML, NOEBELS JL.Selective Localization of Cardiac SCN5a Na+ Channels inLimbic Regions of Rat Brain. Nat Neurosci 1999;2:593±595.
6. QIAN J, NOEBELS JL. Presynaptic Ca2+ in¯ux at a mousecentral synapse with Ca2+ channel subunit mutations. JNeurosci 2000;20:163±170.
7. BURGESS DL, BIDDLECOME GH, MCDONOUGH SI et al. Subunitreshuf¯ing modi®es N- and P/Q-type Ca2+ channel subunitcompositions in lethargic mouse brain. Mol Cell Neurosci1999;13:293±311.
Novel insights on brain development and the deleterious and delayed effectsof early insults
Y. Ben-AriInserm U 29, INMED, 163 Route de Luminy, BP 13, 13273 Marseille Cedex 09
During development, 1012 neurons wire in thenetworks via 1015 speci®c synaptic connections toproduce our functioning, thinking brain. While thegeneral scheme of the neuronal networks is probablyencoded in genes,formation of the functional net-works requires activity-dependent re®nement of thesynaptic connections in keeping with the Hebbianconcept that ``neurons that ®re together wiretogether. Early in development, internally generatedspontaneous activities'' sculpt circuits on the basis ofthe brain's ``best guess'' at the initial con®gurationof connections necessary for function and survival.Activity-dependent processes may disrupt or alterthis normal neuronal development. In fact, there areseveral indications that although the immature brainis relatively resistant in term of neuronal lesions andcell loss to deleterious condition, it is very prone forinstance to recurrent seizures that elicit long termconsequences characterised by impairment of cog-nitive functions, anatomical reorganisations andpermanent alterations of neuronal properties. Wehave used several in vitro and in vivo hippocampalpreparations including morpho-functional studiesof primate neurons in utero and rat neonatalneurons, intact hippocampi and other componentsof the limbic system in vitro, multiple recordings in
vivo of neonatal neurons and 3D reconstruction aswell as studies in adult rats submitted during in uteroor neonatal period to various insults. I shall ®rstreview the electrical properties of the developingprimate and rat hippocampus. This is characterisedby:
i) a sequential expression of GABA and gluta-mate receptors. At birth pyramidal neurons of theCA1 region of the hippocampus are either silent (nosynaptic currents), express only GABAergic cur-rents, or express GABA and glutamatergic currents.These three different types of CA1 neurons haddifferent morphological appearances: silent neuronshad a soma and an axon, but no dendrites; GABAonly neurons had a soma, axon, and small dendrite;and GABA and glutamate neurons had a soma,axon, and extended apical and basal dendrites. Thissequence is of particular importance as GABAprovides the main excitatory drive to hippocampalneurons at early stages of postnatal developmentbecause of a high Cl- content in immature neurons.
ii) the presence of network driven synchronisedGiant Depolarising Potentials (GDPs) that providemost if not all of the synaptic activity during earlydevelopment. Initially described in the retina &hippocampus, this pattern has has now been
13

observed in a wide range of brain structures inseveral animal species including primates. GDPs areassociated with large calcium oscilations thatprovide a Hebbian pattern of stimulation thatmay participate in activity dependent formation offunctional units.
I shall then review a series of observations thatindicates that a series of seizures that do not causebrain lesions during the ®rst days and weeks of liferesult in a orphological, electrophysiological, cog-
nitive impairment and behavioral changes in therats when examined as adults in addition to thereduced seizure threshold. Taken together, thesestudies clearly demonstrate that seizures in theimmature animal result in long-term adversechanges, despite the lack of demonstrable hippo-campal cell loss. I shall also describe the ion vitroformation of a mirror focus following recurrentseizures as well as the effects of various adverseconditions on development.
Manifestation of dopamine and glutamate systems dysfunction in animalmodels of schizoid behavior
M. G. Caron, A. R. Mohn, R. R. GainetdinovHoward Hughes Medical Institute Labs, Department of Cell Biology, Duke Uni. Med. Ctr. Durham, North Carolina 27710
In the brain the major neurotransmitter systemsinvolved in the function of the cortico-basalganglia circuits include the dopaminergic, gluta-matergic and GABAergic systems. These systemsare presumably implicated in the control oflocomotion, cognition and affect. Dysregulationin the dopaminergic and glutamatergic systems, inparticular, have been postulated to contribute toseveral central nervous system disorders. In orderto gain understanding for the role of theseneuronal systems to the elaboration of physiolo-gical or pathophysiological conditions we haveused a genetic approach in the mouse by creatinganimals in which the gene for certain keycomponents of these neurotransmitter systemshave been deleted or suppressed by homologousrecombination. The dopamine transporter (DAT)calibrates the intensity and duration of dopami-nergic transmission in the brain by rapidlyrecycling dopamine (DA) back into presynapticterminals. Deletion of the DAT gene (DATKO)leads to a pronounced hyperdopaminergic statedue to the fact that DA spends 300 times longerin the extracellular space of DATKO mice thantheir wild type littermates (1). Absence of DATalso results in major changes in the homeostaticcontrol of DA transmission both pre- and post-synaptically (2, 3). Interestingly, DATKO miceare hyperactive, especially when exposed to anovel environment. Additionally, these mice areimpaired in spacial cognitive tasks and behavioralassessments of information gating, habituation,attention and memory processes. DATKO miceshow a marked decrease in locomotion in
response to psychostimulants such as, cocaine,amphetamine and methylphenidate and this effectdepends on enhanced serotoninergic transmission(4) The parallels that can be drawn between thephenotypic properties of the DATKO mice andcertain symptoms and drug responses of indivi-duals with attention de®cit hyperactivity disorderraises the possibility that common mechanismsmight underlie the pharmacological actions ofpsychostimulants if not their phenotypes.DATKO mice also recapitulate the characteristicsof the amphetamine model of psychosis in therodent in that they display marked hyperactivity,stereotypy and impaired sensory motor gating.Evidence also suggests that in the DATKO micethe interplay between the dopaminergic andserotonergic systems must involve glutamatergicneurotransmission. Treatment of DATKO micewith the NMDA receptor antagonist MK801further increases their enhanced locomotor activ-ity and antagonizes the calming effects ofpsychostimulants and serotonergic drugs. Theseresults raise the possibility that the serotonergicmodulation of the hyperactivity behavior inDATKO mice may be through regulation of theglutamatergic input to the striatal complex con-trolling locomotion.
Several lines of investigations have implicatedNMDA receptors in the pathology of psychosis butthe hypothesis has not been genetically tested. Werecently reported the generation of a mouse line thathas been genetically altered to express 5 to 10percent of the normal levels of NR1, an essentialsubunit of the NMDA receptor (5). These mice
14

display behavioral abnormalities that are consistentwith other pharmacological models of schizo-phrenia (PCP and MK801 intoxication). NR1de®cient mice display increased locomotion andstereotypy as well as de®cits in social behaviorssimilar to those elicited by PCP and MK801.Phenotypes associated with the NR1 de®cientmice are more effectively ameliorated by theatypical antipsychotic clozapine then by its typicalcounterpart haloperidol. The phenotypes of theNR1 de®cient mice as well as their responses toantipsychotics are observed without any demon-strable changes in the homeostasis of DA in theseanimals. These results demonstrate in geneticallyde®ned animals that pharmacological manipulationof a presumably normal neurotransmitter (DA orserotonin) pathway can correct the phenotypicconsequences of dysfunction in another pathway(glutamate). Studies in this animal model supportthe hypothesis that dysfunction of glutamatergictransmission may underlie some forms of schizo-
phrenia and reveal the contribution of monoami-nergic systems in this paradigm.
References
1. GIROS B, JABER M, JONES SR, WIGHTMAN RM, CARON MG.Hyperlocomotion and indifference to cocaine and ampheta-mine in mice lacking the dopamine transporter. Nature1996;379:606±612.
2. JONES SR, GAINETDINOV RR, JABER M, GIROS B, WIGHTMAN
RM, CARON MG. Profound neuronal plasticity in response toinactivation of the dopamine transporter. Proc Natl Acad SciUSA 1998;95:4029±4034.
3. JONES SR, GAINETDINOV RR, HU X-T et al. Loss ofautoreceptor functions in mice lacking the dopaminetransporter. Nat Neurosci 1999;2:649±655.
4. GAINETDINOV RR, WETSEL WC, JONES SR, LEVIN ED, JABER M,CARON MG. Role of serotonin in the paradoxical calmingeffect of psychostimulants on hyperactivity. Science1999;283:397±401.
5. MOHN AR, GAINETDINOV RR, CARON MG, KOLLER BH. Micewith reduced NMDA receptor expression display behaviorsrelated to schizophrenia. Cell 1999;98:427±436.
Cellular mechanisms in the cyclic affective disorders
R. M. Post, G. S. Leverich, S. R. B. Weiss, A. M. Speer, G. Obrocea, K. D. DenicoffBiological Psychiatry Branch, National Institute of Mental Health, NIH, Bldg. 10, Room 35239, 10 Center Drive, Bethesda, MD 20892, USA
Although there is much recent focus on geneticvulnerability factors for bipolar affective disorders,some 50% of bipolar disorders occur in the absenceof a family history of this illness in ®rst-degreerelatives, and other mechanisms such as environ-mental and experimental effects on gene expressiondeserve exploration.
Kraepelin's description of stress sensitizationphenomena have generally been supported in themodern literature (1). The data on episode sensiti-zation are now also quite compelling. The largestseries is from Kessing and associates (2) using theDanish case registry of more than 20,000 patientswho had been hospitalized for unipolar or bipolardisorder. These investigators found that the numberof prior episodes was the best predictor of thevulnerability to relapse in terms of both latency toand incidence of rehospitalization for a newepisode. In the unipolar depressive illnesses,Kendler et al. (3) have elegantly demonstrated thecombined contributions of genetics and early andmore current environmental stressors, as well asprior episodes themselves in propelling depressionrecurrence.
As part of our Stanley Foundation BipolarNetwork (SFBN), we have studied the associationof a history of childhood or adolescent physical andsexual abuse on demographic course of illnessvariables in 298 outpatients with bipolar disorder.Twenty-seven percent reported a history of child-hood or adolescent physical abuse and 28%, sexualabuse (4). Individuals with these early traumaticstressors, compared with those without, had anincreased number of Axis 1 and Axis 2 comorbid-ities including drug and alcohol abuse, a greaternumber of associated medical conditions, an earlieronset of bipolar illness, faster cycle frequencies, anda higher incidence of psychosocial stressorsreported as occurring prior to both the ®rst andthe most recent affective episodes. Physical abusewas highly associated with increased severity ofmania, whereas a history of prior sexual abuse wasassociated with an increased incidence of serioussuicide attempts.
The molecular mechanisms conveying these long-term effects of earlier life experiences on theunfolding of bipolar illness remain unknown, buta number of candidates can be derived from
15

preclinical studies. In the model pioneered byLevine et al. (5) of a single 24-hour period ofmaternal deprivation in the ten-day-old rat pup, orin the model of Plotsky & Meaney (6) of repeateddaily maternal separation for three hours (ratherthan 15 minutes) in the ®rst two weeks of life, earlystressors generate persisting behavioral and endo-crine abnormalities. These animals, subjected tosingle or repeated stressors, show greater degrees ofanxiety-related behaviors and life-long hypercorti-solemia, as well as greater vulnerability to adoptingalcohol and cocaine self-administration than theirlitter-mate controls (7). The behavioral and endo-crine abnormalities are ameliorated with mainte-nance treatment with serotonin-selective reuptakeinhibitors (SSRIs), but recur when medications arediscontinued. These ®ndings, based on environ-mental stressors, are paralleled by those generatedin a transgenic mouse model with de®cient gluco-corticoid receptor numbers and associated hyper-cortisolemia and increased anxiety-like behaviorswhich are reversible with antidepressant treatment(8). Thus, either genetic or environmental effectscan mediate these types of changes.
In the 1-day maternal deprivation paradigmthere is evidence for a doubling of the rate ofapoptosis in brain and signi®cant increases in c-fosand nerve growth factor (NGF) mRNA withdecrements in mRNA for brain-derived neuro-trophic factor (BDNF), nitric oxide synthase(NOS), and calcium calmodulin kinase-2(CaMKII) (9). In behavioral sensitization topsychomotor stimulants a different set of effectson gene expression are observed. Both paradigmsprovide models for understanding how stress andepisodes of behavioral pathology could eachincrease vulnerability to future episodes.
Based on ®ndings in the kindling model, we havegrouped effects on gene expression into two generalcategories, depending on whether they are generallyproconvulsant (and part of the primary pathologyof the kindled memory trace), or putatively, anti-convulsant (and thus potentially part of thesecondary or compensatory adaptive endogenousanticonvulsant process) (10). For example, seizure-induced increases in thyrotropin-releasing hormone(TRH) are anticonvulsant (11). There is alsoevidence that TRH is hypersecreted in somedepressed patients. Since TRH may have antide-pressant properties (12), one can also place TRH inthe presumptive group of endogenous compensa-tory (antidepressant) substances. In this way onecould conceptualize that it is the ratio of patholo-gical versus adaptive factors that determineswhether patients are in an episode or experiencinga ``well-interval'' between episodes.
Several types of the therapeutic agents currentlyused in affective illness exert signi®cant effects onneurotrophic factor gene expression and thus,potentially, on neural structure as well as biochem-istry. For example, the antidepressants have beenshown to exert opposite effects of stress on BDNFexpression, and pretreatment with antidepressantshas been shown to blunt the ability of stressors toalter gene expression (13).
At the same time, there is exciting new evidencethat lithium may have a variety of neurotrophic andneuroprotective properties in both in vitro and invivo paradigms. Chuang and associates demon-strated that lithium blocked calcium in¯ux throughthe NMDA receptor and prevented apoptotic celldeath via this mechanism in cerebellar granulecultures (14). Manji and colleagues, as well asChuang and colleagues, found that lithiumincreased expression of putative cell survival factorsBDNF and Bcl-2, while decreasing levels of putativecell death factors BAX and P53 (15, 16). Based onthese observations, Chuang and associates foundlithium neuroprotective in rat models of stroke andHuntington's chorea (17, 18).
Recent data have continued to indicate thatlithium not only markedly reduces the suicide ratein patients with unipolar and bipolar depressionmaintained on their lithium compared with thosewho discontinue it (with a 20-fold difference insuicide attempts after the ®rst year) (19), but that italso normalizes the increase in medical mortalityassociated with these affective disorders (20).Whether lithium's positive effects on medicalmortality and suicide are related to its neuropro-tective effects remains to be further delineated.
References
1. POST RM. Transduction of psychosocial stress into theneurobiology of recurrent affective disorder. Am JPsychiatry 1992;149:999±1010.
2. KESSING LV, ANDERSEN PK, MORTENSEN PB, BOLWIG TG.(1998) Recurrence in affective disorder. I. Case registerstudy. Br J Psychiatry 1998;172:23±8.
3. KENDLER KS, KESSLER RC, NEALE MC, HEATH AC, EAVES
LJ. The prediction of major depression in women: towardan integrated etiologic model. Am J Psychiatry1993;150:1139±1148.
4. LEVERICH GS, MCELROY SL, SUPPES T et al. Early physical orsexual abuse and the course of bipolar illness. Am JPsychiatry 2000, in review.
5. LEVINE S, HUCHTON DM, WIENER SG, ROSENFELD P. Timecourse of the effect of maternal deprivation on thehypothalamic± pituitary±adrenal axis in the infant rat.Dev Psychobiol 1991;24:547±558.
6. PLOTSKY PM, MEANEY MJ. Early, postnatal experience altershypothalamic corticotropin±releasing factor (CRF)mRNA, median eminence CRF content and stress±inducedrelease in adult rats. Mol Brain Res 1993;18:195±200.
7. PLOTSKY PM, EISLER JA, ANAND KJS. Long±term conse-
16

quences of neonatal stress. Eur Neuropsychopharmacol1996;6 (suppl 3),217.
8. BEAULIEU S, ROUSSE I, GRATTON A, BARDEN N, ROCHFORD J.Behavioral and endocrine impact of impaired type IIglucocorticoid receptor function in a transgenic mousemodel. Ann NY Acad Sci 1994;746:388±391.
9. XING GQ, SMITH MA, LEVINE S, YANG ST, POST RM, ZHANG
LX. Suppression of CaMKII and nitric oxide synthase bymaternal deprivation in the brain of rat pups. Society forNeuroscience Abstracts 24 (Abstract 176.9) 1998;452.
10. POST RM, WEISS SRB. A speculative model of affectiveillness cyclicity based on patterns of drug toleranceobserved in amygdala±kindled seizures. Mol Neurobiol1996;13:33±60.
11. WAN RQ, NOGUERA EC, WEISS SR. Anticonvulsant effects ofintra±hippocampal injection of TRH in amygdala kindledrats. Neuroreport 1998;9:677±682.
12. MARANGELL LB, GEORGE MS, CALLAHAN AM et al. Effects ofintrathecal thyrotropin±releasing hormone (protirelin) inrefractory depressed patients. Arch Gen Psychiatry1997;101:214±222.
13. SMITH MA, MAKINO S, KVETNANSKY R, POST RM. Stress andglucocorticoids affect the expression of brain±derivedneurotrophic factor and neurotrophin±3 mRNAs in thehippocampus. J Neurosci 1995;15:1768±1777.
14. NONAKA S, HOUGH CJ, CHUANG DM. Chronic lithium
treatment robustly protects neurons in the central nervoussystem against excitotoxicity by inhibiting N±methyl±D±aspartate receptor±mediated calcium in¯ux. Proc Natl AcadSci USA 1998;95:2642±2647.
15. CHEN RW, CHUANG DM. Long term lithium treatmentsuppresses p53 and Bax expression but increases Bcl±2expression. A prominent role in neuroprotection againstexcitotoxicity. J Biol Chem 1999;274:6039±6042.
16. CHEN G, ZENG WZ, YUAN PX, et al. The mood-stabilizingagents lithium and valproate robustly increase the levels ofthe neuroprotective protein bcl-2 in the CNS. J Neurochem1999;72:879±882.
17. CHUANG DM, WEI H, QUIN Z, WEI W, WANG Y, QIAN Y.Lithium inhibits striatal damage in an animal model ofHuntington's disease. Society for Neuroscience Abstracts1999;25:09611.
18. NONAKA S, CHUANG DM. Neuroprotective effects of chroniclithium on focal cerebral ischemia in rats. Neuroreport1998;9:2081±2084.
19. BALDESSARINI RJ, TONDO L, HENNEN J. Effects of lithiumtreatment and its discontinuation on suicidal behavior inbipolar manic±depressive disorders. J Clin Psychiatry1999;60 (Suppl 2):77±84.
20. AHRENS B, GROF P, MOLLER HJ, MULLER±OERLINGHAUSEN B,WOLF T. Extended survival of patients on long±term lithiumtreatment. Can J Psychiatry 1995;40:241±246.
On the relation between nature and nurture or the relevance of humangrowth and maturation in psychiatry
L. SaugstadBehrens gate 5, 0257 Oslo, Norway
In the jargon of the ®fties Nurture was thoughtmore important than Nature, but Nature matteredin psychiatry. Now both terms have fallen out offashion. They seem curious archaisms ± culturalconstructs rather than verities.
One may look at multifactorial inheritance asnatural selection turned around. From a focus onadaptation as the decisive factor in our survival inchallenging environments. Now our sole focus is ongenes (single or multiple) as the only factors thatmatter in development, neglecting that environmentplays an important role in multifactorial inheri-tance. This neglect is crucial when we consider thedisappointing linkage studies in mental disorder.Let us consider how the maturational theory ofbrain development explains our present geneticfailure (no single locus which raises risk by >3) andidenti®es the decisive environmental factor we arelooking for in mental disorder.
The theory holds: the central inherited factor inmental disorder is rate of physical maturation ± ageat puberty. The environmental factor is standard ofliving which affects the panorama of mental illness.This is because an association has been established
between the ®nal stage in brain development ± the3rd regressive event with pruning of some 40% ofexcitatory synapses leaving the inhibitory onesfairly unchanged, and age at puberty ± rate ofphysical maturation. Rising living standardsincrease maturational rate ± lower age at puberty,while lower standards lower rate of maturation andincrease pubertal age.
There is also a relation between rate of physicalmaturation and body-build. As far back as 1921,Kretschmer (1) posited a relation between body-build and mental illness: >90% of manic-depres-sives were broad-built pyknic and >80% ofschizophrenics were linear-leptosomic built. Therelation between the predominant body-build inmanic-depressive and schizophrenic is similar tothat between body-build in early and latematurers. The maturational heory holds thatmanic-depressive illness relates to early pubertyand schizophrenia to late puberty. The twodisorders are part of human growth and matura-tion localized at the extremes of the maturationalrate continuum with normality in between.Manic-depressive illness rises in prevalence with
17

rising living standard such as we have experiencedin this century when the about 4 years decline inpuberty has been accompanied by about 13cmincrease in mean height ± a phenotypic response.Manic-depressive illness has increased, particularlydepression, and we have seen a marked rise inother disorders in early maturers, eating disorder(anorexia nervosa and bulimia nervosa) andanxiety among others.
Concomitantly, a primary prevention of themore severe forms of schizophrenia ± the non-paranoid subgroup (catatonia, hebephrenia anddementia praecox), which is considered the mostextreme slow maturer with an excess of congenitalmalformation (1) has occurred. Now other sub-groups predominate: paranoid, acute, latent,schizo-affective and borderline conditions.
As part of human growth and maturation, weexpect the disorders manic-depressive psychosis andschizophrenia to share common susceptibility loci(those affecting growth and maturation) and todiffer in inherited vulnerability such as has beenobserved (2, 3).
The ubiquity of the two disorders despite theirreduced rate of reproduction and even childlessnessin a proportion of very late maturing schizophrenic,is explained by the fact that we have a suf®cientnumber of early and late maturers to choose from inthe population (4).
The present continuous trend to earlier age atpuberty is of special concern because it could leadto unexpected problems. This is because apartfrom the usual pruning of neural elements, in thiscase synapses, the 3rd regressive event is distin-guished by the fact that excitatory ones only areaffected. This additional characteristic means: theearlier pruning is abridged the greater is cerebralexcitability, the longer the pruning proceeds pastthe optimal the larger is the de®cit in cerebralexcitability. This particularity explains most likelythe usual cessation around puberty of ``idiopathicchildhood epilepsy''. We would actually expect anincreased proportion of pathological EEGs ofparoxysmal nature the earlier onset of puberty aswell as CNS episodic dysfunction. This has beenobserved in manic-depressive psychosis (5) whereanti-epileptics with mood stabilizing effect is thetreatment of choice. This con®rms our hypothesisthat manic-depressive illness is a disorder in earlymaturers. More particularly, investigations ofindividuals with pubertas praecox (6) haverevealed a striking excess of pathological EEGsand of seizures. It seems therefore that the mainintention of the 3rd event to secure normal brainexcitability is in danger in very early maturerwhere the risk of epilepsy is increased in addition
to a risk of disorders distinguished by episodicdysfunction of the suprachiasmatic nucleus of thehypothalamus (SCN) such as is the case in manic-depressive illness and eating disorders.
Conversely, in very slow maturers where somenever reach puberty, the de®cit in excitability is sopronounced that convulsants are the treatment ofchoice. It is well known that all neurolepticswhether atypical or typical which are used to treatschizophrenics are convulsants. The longer theduration of treatment (4) the greater the risk ofattenuation of an already attenuated CNS (5, 7).This explains tardive dyskinesia (TD) and a risingpersistency of psychopathology in chronics.
Our failure to cure is because we are unable toreverse a process which has gone too far. But wemay ``copy nature'', continue pruning with anti-epileptics which affect glutamate in manic-depres-sive illness. That we are successful in treatingmental disorders by copying CNS processes oradding a necessary factor in a disorder ofsuboptimality like schizophrenia supports thatthe disorders are deviation from the norm ingrowth and maturation.
Let us consider human growth and maturationto try to explain the decisive role of changingliving standards in mental illness. In view of ourmarine heritage it seems resonable to focus onmarine fat and fat-soluble vitamines A & D. Atheory has been presented that a diet low inpolyunsaturated fatty acids (PUFA) in the thirdtrimester of pregnancy may delay myelination andmaturation (8, 9). This underpins learning andbehaviour disorders and sudden infant death afterthe ®rst month, conditions associated with lowerthan average birthweight similar to what is oftenobserved in schizophrenia. A de®cit in PUFAespecially associated with de®cits in Vit A due toits pervasive role in brain development in utero(transcription, differentitation, migration) couldde®nitely reduce or disturb brain growth andmaturation. The fact that addition of PUFA todiet (10) is effective in schizophrenia and otherdisorders in late maturers supports this hypothesisof suboptimality. So does the more benign courseof schizophrenia in countries with a comparativelyhigh consumption of PUFA relative to saturatedfat (11). On the other hand, Wirz-Justice &Hoofdakker's (12) success with inducing euthymiawithin hours in severely depressed individualsusing a two-process model of mood regulationbased on interaction of circadian and homeostaticrhythms supports that episodic SCN dysfunctionis a primary mechanism in affective disorderwhere personality usually returns to the normalpremorbid following a psychotic episode.
18

References
1. KRETSCHMER E. Kùrperbau und Character. Heidelberg:Springer Verlag, 1921.
2. WILDENAUER DM, SCHWAB SG, MAIER W, DETERA-WADLEIGH
SD. Do schizophrenia and affective disorder share sucept-ibility genes. Schizophr Res 1999;39:107±111.
3. BERRETTINI WD. Susceptibility loci for bipolar disorderoverlap with vulnerability to schizophrenia. Biol Psychiatr2000;47:245±251.
4. SAUGSTAD LF. The central inherited factor in schizophreniaand affective disorder. In press 2000.
5. SAUGSTAD LF. Deviation in cerebral excitability: possibleclinical implications. Int J Psychophysiol 1994;18:205±212.
6. LIU N, GRUMBACH NM. Prevalence of electroencephalo-gra®c abnormalities in idiopathic precocious puberty. J ClinEndocrinol Metab 1965;25:1296±1308.
7. SAUGSTAD LF. The maturational theory of brain develop-ment and cerebral excitability in the multifactorially
inherited manic-depressive psychosis and schizophrenia.Int J Psychophysiol 1994;18:189±204.
8. SAUGSTAD LF. Optimal foetal growth in the reduction oflearning and behaviour disorder and prevention of suddeninfant death (SIDS) after the ®rst month. Int JPsychophysiol 1997;27:107±21.
9. SAUGSTAD LF. Optimality of the birth population reduceslearning and behaviour disorders and sudden infantdeath after the ®rst month. Acta Paediatr Suppl1999;88(429):9±28.
10. HORROBIN DF. The membrane phospholipid hypothesis as abiochemical basis for the neurodevelopmental concept ofschizophrenia. Schizophr Res 1998;30:193±208.
11. CHRISTENSEN O, CHRISTENSEN E. Fat consumption andschizophrenia. Acta Psychiatr Scand 1988;78:587±591.
12. WIRZ-JUSTICE A, VAN DEN HOOFDAKKER RH. Sleep depriva-tion in depression. What do we know, where do we go? BiolPsychiatr 1999;46:445±453.
Strategies to identify genes contributing to epilepsy in man
O. K. SteinleinInstitute of Human Genetics, University of Bonn, D-53111 Bonn, Germany
In the past few years the genetic analysis of epilepticdiseases has become a fast developing and interest-ing ®eld. After a long period of neglect interest isfocused on the interplay between genetic variationand neuronal excitability. Family and twin studiesshowed that the aetiology of idiopathic epilepsies ismainly genetic, but that in most syndromes themode of inheritance is complex rather than mono-genetic. Furthermore, recurrence rates are droppingmarkedly when the degree of consanguinitydecreases. Thus it is likely that the contribution ofthe underlying genes is multiplicative rather thanadditive. Most syndromes, especially the commonforms, like juvenile myoclonic epilepsy or child-hood/juvenile absence epilepsy probably have anoligogenic or polygenic background, even if somerare families suggest a major gene effect.Furthermore, genetic studies are complicated byheterogeneity within a syndrome, as well as byoverlapping genetic aetiology between differentsyndromes. The molecular lesions underlying theidiopathic epilepsies are likely to lead to subtleeffects on neuronal excitability, which might proveto be dif®cult to analyze in in vivo or in vitroexperiments. Only some idiopathic epilepsies aredue to monogenetic inheritance, but these raresyndromes can be regarded as model diseases forstudying the molecular basis of epileptogenesis.
The rare monogenic idiopathic epilepsies offerthe best chance to identify genes/gene families thatcan cause epilepsy. For three of these syndromes the
underlying genetic defects have already beendiscovered. The a4 subunit of the neuronal nACh(CHRNA4) has been identi®ed as the ®rst geneinvolved in the aetiology of idiopathic epilepsies (1,2). Mutations in this gene are associated withautosomal dominant nocturnal frontal lobe epi-lepsy. So far, three different mutations have beenreported, some of them having occurred indepen-dently in different families. This partial epilepsy ischaracterised by brief nocturnal motor seizures,which are occurring mostly during light sleep.
Recently, the genes KCNQ2 and KCNQ3, bothcoding for a formerly unknown brain-speci®cpotassium channel were found to be mutated inbenign familial neonatal epilepsy (BFNC) (3±5).BFNC patients have partial or generalized clonicconvulsions starting around day 3 after birth,which, in most cases disappear spontaneouslyafter approximately 6 weeks. More than 90% ofthe families are linked to KCNQ2, which can beregarded as the major gene for BFNC. So far onlytwo mutations have been described for KCNQ3 (6).
A point mutation in the b1 subunit (SCN1B) ofthe voltage gated sodium channel was found tochange a conserved cysteine residue in a family withgeneralized epilepsy with febrile seizures plus(GEFS+). GEFS+ has only recently beendescribed as a new epilepsy syndrome, and so farclinical descriptions are rare. The syndrome seem tobe characterized by a wide range of different seizuretypes, including typical febrile convulsions, febrile
19

seizures persisting beyond the age of 6 years as wellas different types of afebrile seizures. The mutationprobably disrupts a disul®de bridge between twoconserved cysteine residues in the extracellular partof the SCN1B protein. (7). Recently, the sodiumchannel a-subunit SCN1A was described as thesecond gene for GEFS+ (8).
Ion channel mutations obviously play an impor-tant role in the pathogenesis of idiopathic epilepsies,con®rming the concept of ion channel diseases asparoxysmal disorders (9). Searching for the geneticfactors in common idiopathic epilepsies with acomplex mode of inheritance, ion channel genestherefore are primary targets for candidate geneanalysis, including association analysis as well asdirect mutation screening. However, innumerablesubtypes of ion channels are present in themammalian brain, trying to maintain a sensitivebalance in the electrical activity of the neuronalnetwork. Theoretically, each of these ion channelscan be a candidate target for epilepsy-causingmutations. Thus, the identi®cation of genes inepilepsies with a complex genetic background,including oligo- or polygenic inheritance andgenetic heterogeneity, provides a great challengewhich can only be mastered by close collaborationbetween clinical and genetic research.
References
1. STEINLEIN O, MULLEY JC, PROPPING P et al. A missensemutation in the neuronal nicotinic acetylcholine receptoralpha4 subunit is associated with autosomal dominantnocturnal frontal lobe epilepsy. Nature Genet1995;11:201±203.
2. STEINLEIN O, MAGNUSSON A, STOODT J et al. An insertionmutation of the CHRNA4 gene in a family with autosomaldominant nocturnal frontal lobe epilepsy. Hum Mol Genet1997;6:943±947.
3. BIERVERT C, SCHROEDER BC, KUBISCH C et al. A potassiumchannel mutation in neonatal human epilepsy. Science1998;279:403±406.
4. CHARLIER C, SINGH NA, RYAN SG et al. A pore mutation in anovel KQT-like potassium channel gene in an idiopathicepilepsy family. Nature Genet 1998;18:53±55.
5. SINGH NA, CHARLIER C, STAUFFER D et al. A novel potassiumchannel gene, KCNQ2, is mutated in an inherited epilepsy ofnewborns. Nature Genet 1998;18:25±29.
6. HIROSE S, ZENRI F, AKIYOSHI H et al. A novel mutation ofKCNQ3 (c. 925T±>C) in a Japanese family with benignfamilial neonatal convulsions. Ann Neurol 2000;47:822±826.
7. WALLACE RH, WANG DW, SINGH R et al. Febrile seizures andgeneralized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nature Genet1998;19:366±370.
8. ESCAYG A, MACDONALD BT, MEISLER MH et al. Mutations ofSCN1A, encoding a neuronal sodium channel, in twofamilies with GEFS+2. Nature Genet 2000;24:343±345.
9. PTACEK LJ. Channelopathies: ion channel disorders of muscleas a paradigm for paroxysmal disorders of the nervoussystem. Neuromuscul Disord 1997;7:250±255.
Approaches to gene identi®cation in neuro-psychiatric and othercomplex disorders
P. Asherson, S. CurranSocial Genetic Developmental Psychiatry Research Centre, Institute of Psychiatry, King's College London, London, SE5 8AF
In the last two decades the classical geneticapproaches of family, twin and adoption studies,have provided considerable evidence that geneticin¯uences are important in the aetiology andpathogenesis of psychiatric disorders. At the sametime there have been remarkable advances in theapplication of molecular methods in medicinetogether with advancing technology and progressin mapping and sequencing the human genome. Asa result, many are now persuaded that the time isright to focus on the identi®cation of susceptibilitygenes, which give rise to psychiatric disorders (seeMcGuf®n et al, 1999), as well as genes that in¯uencevariation in human behaviours such as hyperactiv-ity, cognitive ability and reading ability (reviewed inAsherson and Curran, 2000).
Considerable progress has already been made incloning the genes responsible for some compara-tively rare disorders such as Huntington's Diseaseand familial forms of Alzheimer's disease andepilepsy. These all show simple inheritance in thesense of being single gene disorders with classicMendelian transmission. On the other handconditions such as schizophrenia, manic-depres-sion, autism, attention de®cit hyperactivity dis-order (ADHD), reading disability, mild mentalimpairment and common forms of idiopathicepilepsies are said to show complex inheritance.They do not conform to Mendelian patterns ofsegregation and are thought to result from thecombined effects of several genes (oligogenic), orperhaps many genes (polygenic) each of which, on
20

its own, has only a small effect. In these casesvariation of single genes are neither suf®cient nornecessary to cause the disorder, but act assusceptibility genes increasing risk for the disorder.Mapping and identifying the genes responsible forcomplex disorders represents a greater challengethan that posed by rarer Mendelian diseases but isbecoming rapidly more tractable (reviewed inRisch 2000). As a result molecular genetic studiesare underway for many psychiatric disorders andbehavioural phenotypes and have already resultedin the localisation or identi®cation of susceptibilitygenes for schizophrenia, bipolar disorder, autism,ADHD and reading disability.
Linkage analysis using multiply affected families
Early genetic studies of common neuropsychiatricdisorders were based on the assumption of singlegene inheritance. Large multiply affected familieswere identi®ed which have the appearance ofMendelian transmission and are the most likely tobe segregating genes of major effect. In some casesthis approach resulted in the identi®cation of rarefamilial forms segregating genes of major effect. Forexample, the amyloid precursor protein, presenilin-1 and presenilin-2 genes were mapped to familialforms of Alzheimer's disease and ion channel geneshave been implicated in rare familial epilepsies.Among the behavioural phenotypes there areseveral independent reports of linkage betweenmarkers on chromosome 4p near to the dopamineD5 receptor gene and bipolar and schizoaffectivefamilies. However analysis of such families usingtraditional linkage approaches have in general beenunsuccessful and are unlikely to identify genetic riskfactors for common forms of these disorders. As aresult, recent linkage studies of behavioural pheno-types have focused on alternative non-parametricstrategies, using sibling pairs rather than multiplexpedigrees.
Linkage analysis using affected sibling pairs (ASP)
A measure that is frequently used in evaluating thepower of ASP linkage is the ratio of the risk insiblings of affected probands, to population pre-valence, a parameter known as ls. Low ls valuesmay be due to a variety of factors such as polygenictransmission, genetic heterogeneity, phenocopiesand low penetrance. These may require such largesample sizes to overcome that ASP methods are nolonger feasible. Disorders such as autism may bemore amenable to this approach since the estimatedls is very large, somewhere between 100±200 and iswell within the theoretical resolution of linkage
strategies. On the other hand, disorders such asADHD have estimated ls somewhere between 2±5.Indeed, if more than one gene causes ADHD, thenthe l-value for any single gene (the gene speci®c l orlg), must be very low. As a rough guide to the size ofsamples required it has been estimated that 200ASPs would be required to detect a susceptibilitygene giving rise to a 5-fold relative risk and 700ASPs for one causing a 2-fold relative risk (Rischand Merikangas 1996, Risch 2000).
To date novel disease or susceptibility genes haveyet to be identi®ed following ASP linkage studies. Inpart this is due to the power considerationsdiscussed above so that linkage ®ndings have beendif®cult to replicate. The other problem is the poorresolution of ASP linkage methods, which can onlyidentify broad regions, often containing manyhundreds of genes. This means that even where achromosomal location has been con®rmed a con-siderable amount of additional work is required toidentify the gene itself. Linkage studies thereforeneed to be complemented by association strategies,which identify narrow chromosomal regions andhave far more power to detect genes of small effectin complex disorders.
Association Strategies
Association studies compare the frequencies ofmarker alleles in a group of affected individuals anda sample of controls without the disorder or drawnfrom the general population. A statistically sig-ni®cant difference suggests either very tight linkageresulting in linkage disequilibrium (LD) between amarker allele and the susceptibility locus, or that themarker allele itself confers susceptibility to disorder.Risch and Merikangas (1996), applied a genome-wide level of signi®cance which treats each genomescan as the test unit. Under these stringent criteria(power of 80%, P<5r10x8), they estimated that asample of 340 unrelated cases would detectassociation with a susceptibility gene with frequencyof 0.5 and lg of 2.0. This compared with 2498 ASPsrequired to detect the same gene by linkage analysis.Despite this, the usefulness of the approach hasbeen limited by the fact that the upper limit ofdistance for detection of linkage disequilibrium(LD) in out-bred populations is no more than onecentimorgan (cM) and may be considerably less,hence requiring thousands of markers to perform awhole genome search. For this reason associationapproaches are still in their infancy and in mostcases have only been applied to the analysis of a fewcandidate genes. Fortunately, the development of anew generation of very high density marker mapsand the technology to screen these in large numbers
21

is underway, so that LD mapping strategies are fastbecoming the focus of current interest in the studyof complex disorders.
Association studies in behavioural disorders suchas schizophrenia have unfortunately thrown up anumber of contradictory results (reviewed inO'Donovan and Owen, 1999). This has been inpart because of the problems of diagnosis and thequestion of comparability of patient populationsfrom different centres, and inadequate sample sizesused in many studies. A confounding factor in case-control studies is the selection of controls, whichcan result in strati®cation effects. The solution tothis problem is to sample both parents of affectedprobands and use the non-transmitted parentalalleles as control genotypes (haplotype basedhaplotype relative risk analysis; Falk andRubinstein, 1987), or look for increased transmis-sion of a speci®c allele from heterozygote parents(transmission disequilibrium test; Spielman andEwans, 1996)
Quantitative trait loci (QTL) mapping
An alternative approach to mapping genes fordisorders using categorical criteria is the analysis oftraits, which are continually distributed in thepopulation. Such quantitative traits are in¯uencedby the action and co-action of multiple genes knownas QTLs. Developmental traits such as generalcognitive ability (g), reading ability, and beha-vioural traits such as hyperactivity and anxiety/depression may be better perceived in this way.Considerable additional power can be gained by theuse of phenotypically discordant as well concordantsibling pairs and the selection of siblings within thetop and bottom deciles. Furthermore, discordantsiblings provide a powerful resource for associationmapping as well as linkage.
Current advances
The ultimate human gene map is progressing atpace as the Human Genome Project (HGP) movestowards completion, the ®rst draft of the humangenome sequence having been announced in June2000. Current developments are already turning tothe identi®cation of sequence variation across thegenome and across human populations.
My own group is currently screening the genomefor associations using several thousand markerswith a simple but ef®cient adaptation of currentgenotyping systems; using pooled DNA instead ofperforming individual genotypes. Since only 2genotypes are required per marker, initial screeningof say 3500 markers by pooling requires only 7000genotypes compared with 1,400,000 individualgenotypes, for a sample of 200 probands and 200controls. Alternative approaches, which areexpected to have more far reaching consequencesfor mapping genes in complex disorders, dependupon the detection of single-nucleotide polymorph-isms (SNPs). Further developments are aimed atscreening gene sequences directly, so that instead ofsearching for associations with anonymous mar-kers, genome scans will be able to focus on variationwithin genes that affects protein sequence andexpression, or lies very close to such functionalvariants. In parallel with identi®cation of SNPsseveral PCR-based techniques for ef®cient SNPgenotyping have been developed. The impact ofthese technologies for gene mapping is likely to beprofound, since it is becoming possible to screenvery large samples for linkage and association withvery dense marker maps, greatly increasing thepower to detect genes of small effect in complexdisorders.
References
1. ASHERSON PJ, CURRAN S. Molecular genetic approaches tochild psychiatric, behavioural and developmental disorders.British Journal of Psychiatry, 2000, in press.
2. FALK CT, RUBINSTEIN P. Haplotype relative risks: an easyreliable way to construct a proper control sample for riskcalculations. Annals of Human Genetics 1987:51(3):227±233.
3. MCGUFFIN P, OWEN M, O'DONOVAN M, THAPAR A. Seminarsin psychiatric genetics. Royal College of Psychiatrists,Gaskell, 1994.
4. O'DONOVAN MC, OWEN MJ. Candidate-gene associationstudies of schizophrenia. American Journal of HumanGenetics 1999;65:587±592.
5. RISCH N. Searching for genetic determinants in the newmillennium. Nature 2000;405:847±856.
6. RISCH N, MERIKANGAS K. The future of genetic studies ofcomplex human diseases. Science 1996;273:1516±1517.
7. SPIELMAN RS, EWENS WJ. The TDT and other family-basedtests for linkage disequilibrium and association. AmericanJournal of Human Genetics 1996;59:983±989.
22

Genetic basis for myoclonic epilepsies
A.-E. LehesjokiFolkhaÈlsan Institute of Genetics and Dept. of Medical Genetics, University of Helsinki, FIN-00014 Helsinki, Finland
Introduction
The human epilepsies are a heterogeneous groupof disorders. Genetic factors are estimated tocontribute to the etiology of epilepsy in about40% of patients. The most common familialepilepsies display a complex pattern of inheritancewith both genetic and environmental factorscontributing to the susceptibility. In contrast,single gene inherited epilepsies are rare. However,they are important model disorders in the studyof the molecular mechanisms underlying epilepsy.During the past decade signi®cant advances in theunderstanding of the molecular genetic basis ofthe single gene inherited epilepsies have beenachieved, whereas progress in the analysis ofepilepsies with complex inheritance has been lessrapid.
Myoclonic seizures occur as part of the pheno-type in more than 30 epilepsies, many of which havea genetic etiology. The majority of these myoclonicepilepsies are single gene disorders, but they alsoinclude idiopathic epilepsies displaying complexinheritance such as juvenile myoclonic epilepsy.Most progress in our understanding of the mole-cular genetic basis of these disorders has beenamongst the progressive myoclonic epilepsies(PME) some of which are now described in detail.
Progressive myoclonic epilepsies
PMEs are a clinically and etiologically heteroge-neous group of symptomatic myoclonic epilepsies,characterized by the association of epilepsy, myo-clonus and progressive neurological deterioration,in particular ataxia and dementia. Most PMEs areinherited autosomal recessively. The most commonforms of PME are: Unverricht-Lundborg disease,Lafora disease, the neuronal ceroid lipofuscinoses,myoclonus epilepsy with ragged-red ®bers(MERRF), and sialidosis. In addition, a numberof less common disorders manifest as PME. Themolecular genetics of the common forms of PMEhave been understood in the last decade (Serratosaet al., 1999a; Peltonen et al. 2000), and this hasresulted in advances in diagnostics and classi®ca-tion. Future work will be focused on functionalgenomics to elucidate the pathogenetic mechansismunderlying PMEs, which will be the basis forimproved therapy.
Unverricht-Lundborg disease
Unverricht-Lundborg disease (EPM1) is the mostcommon single cause of PME world-wide. It ischaracterized by onset at the age of 6±15 years,stimulus-sensitive myoclonus, tonic-clonic seizures,marked photosensitivity and a progressive course.In the central nervous system, widespread non-speci®c degenerative changes, but no intracellularinclusions are seen on histopathological examin-ation. EPM1 is enriched in the Finnish populationand in the western Mediterranean region. Theprevalence of EPM1 appears to be increasing due toa more benign course of the disease.
A positional cloning strategy was applied toidentify the mutated gene on chromosome 21q22.3responsible for EPM1 (Pennacchio et al., 1996). Itencodes a previously characterized protein, cystatinB (CSTB), which is an inhibitor of the papain-family of cysteine proteases. In the majority ofEPM1 patients the underlying mutation is anunstable expansion of a dodecamer repeat in thepromoter region of CSTB (Virtaneva et al., 1997;Lalioti et al., 1997). This repeat is normallypolymorphic, with 2-3 copies. However, EPM1associated expanded alleles contain at least 30repeat copies. There appears to be no correlationbetween the repeat size and the age of onset or theseverity of the clinical phenotype. Expanded allelesof the dodecamer repeat exhibit a mutation rate of47%, the highest yet observed for pathogeneticalleles of a human minisatellite. In addition to therepeat expansion, ®ve ``minor'' mutations affecting1-2 nucleotides in the CSTB gene have beenreported in EPM1 patients. These account forapproximately 10% of disease alleles in EPM1.Most patients are homozygous for the repeatexpansion mutation and all except one so farreported carry it on at least one chromosome.
Available evidence indicates that the repeatexpansion causes downregulation of CSTBmRNA expression. However, the subsequent cel-lular events and pathogenetic mechanisms remainunknown. CSTB is a 98 amino acid non-glycosy-lated protein that is ubiquitously expressed. Itbelongs to a large class of proteins that inhibitcysteine proteases. In particular, CSTB is known toinhibit lysosomal cathepsins B, H, L and S by tightreversible binding. In lymphoblastoid cells of EPM1patients the papain inhibitory activity, correspond-ing to CSTB activity in these cells, has been found tobe signi®cantly reduced or absent. This reduction is
23

correlated with a signi®cant increase in the cath-epsin activity, especially of cathepsins B, L and Ssuggesting that increased protease activity may playa role in the pathogenesis of EPM1 (Lehesjoki et al.,unpublished data).
A mouse model for Unverricht-Lundborg disease
Mice de®cient for CSTB, produced by targeteddisruption of the mouse Cstb gene, developprogressive ataxia and myoclonic seizures resem-bling the human phenotype (Pennacchio et al.,1998). The clinical features are associated withapoptotic death of cerebellar granule cells. Thus ithas been proposed that CSTB may have a role inpreventing apoptosis in speci®c mammalian cells.However, the mechanism by which CSTB blocksapoptosis is unknown. It has been proposed thatthis may occur either directly, by inhibition ofcaspases, a family of cysteine proteases that areinvolved in the initiation of apoptosis, or indirectly,by inhibition of cathepsins which activate caspases.The ®nding of increased cathepsin activity in EPM1patients would favour the latter hypothesis.However, the possibility of a direct interactionbetween CSTB and caspases remains to be inves-tigated. Further evidence supporting the hypothesisof a neuroprotective mechanism of action of CSTBarises from studies in a rat kindling model ofepilepsy. In this model, seizure activity has beenshown to induce marked and widespread upregula-tion of CSTB mRNA and protein, compatible withthe hypothesis of an endogenous neuroprotectiverole of CSTB, which can act by counteractingapoptosis (D'Amato et al., 2000).
Lafora disease
PME of the Lafora type (EPM2) is an adolescent-onset disorder characterized by epilepsy, severemyoclonus, and a rapidly progressive dementia withdeath usually ensuing within ten years after theonset. The diagnosis of EPM2 is established by thepresence, on skin biopsy, of characteristic periodicPAS-positive intracellular inclusion bodies (Laforabodies) present in various tissues. Lafora bodies
contain branched polysaccharides (polyglucosans)that have long been assumed to re¯ect disorderedcarbohydrate metabolism. A locus (EPM2A) forLafora disease is located on chromosome 6q24, buta signi®cant proportion of families is not linked tothis locus indicating genetic heterogeneity. TheEPM2A gene encodes a novel putative proteintyrosine phosphatase (PTP), named laforin(Minassian et al., 1998; Serratosa et al., 1999b). Itis widely expressed and presents alternativelyspliced forms in every tissue examined, includingbrain. Several predicted loss-of-function mutationshave been identi®ed. PTPs are a heterogeneousgroup of enzymes that regulate diverse intracellularpathways. It has been proposed that the laforin PTPis involved in glycogen metabolism, which is knownto be regulated by phosphatases.
References
1. SERRATOSA JM, GARDINER RM, LEHESJOKI AE, PENNACCHIO
LA, MYERS RM. The molecular genetic bases of theprogressive myoclonus epilepsies. Adv Neurol1999;79:383±398.
2. PELTONEN L, SAVUKOSKI M, VESA J. Genetics of the neuronalceroid lipofuscinoses. Curr Opin Genet Dev2000;10:299±305.
3. PENNACCHIO LA, LEHESJOKI AE, STONE NE et al. Mutations inthe gene encoding cystatin B in progressive myoclonusepilepsy. Science 1996;271:1731±1734.
4. VIRTANEVA K, D'AMATO E, MIAO J et al. Unstable minisa-tellite expansion causing recessively inherited myoclonusepilepsy, EPM1. Nat Genet 1997;15:393±396.
5. LALIOTI MD, SCOTT HS, BURESI C et al. Dodecamer repeatexpansion in cystatin B gene in progressive myoclonusepilepsy. Nature 1997;386:847±851.
6. PENNACCHIO LA, BOULEY DM, HIGGINS KM, SCOTT MP,NOEBELS JL, MYERS RM. Progressive ataxia, myoclonicepilepsy and cerebellar apoptosis in cystatin B-de®cient mice.Nat Genet 1998;20:251±258.
7. D'AMATO E, KOKAIA Z, NANOBASHVILI A et al. Seizures inducewidespread upregulation of cystatin B, the gene mutated inprogressive myoclonus epilepsy, in rat forebrain neurons.Eur J Neurosci 2000;12:1687±1695.
8. MINASSIAN BA, LEE JR, HERBRICK JA et al. Mutations in agene encoding a novel protein tyrosine phosphatase causeprogressive myoclonus epilepsy. Nat Genet 1998;20:171±174.
9. SERRATOSA JM, GOMEZ-GARRE P, GALLARDO ME et al. A novelprotein tyrosine phosphatase gene is mutated in progressivemyoclonus epilepsy of the Lafora type (EPM2). Hum MolGenet 1999;8:345±352.
24

Two Norwegian families illustrating the problem of genotype and phenotypein frontal lobe epilepsy
K. O. Nakken1, E. Brodtkorb2
1The National Center for Epilepsy, P.O.Box 900, 1303 Sandvika, Norway, 2Department of Neurology, Trondheim University Hospital, 7006 Trondheim, Norway
Objectives
Autosomal dominant nocturnal frontal lobe epi-lepsy (ADNFLE) was ®rst described in 1994 byScheffer et al. in one large Australian family (1).This was the ®rst report on a human partial epilepsysyndrome to follow single gene inheritance. A genelocus for ADNFLE has been assigned to thegenomic region 20q13.3 by linkage analysis of theAustralian family (2). In this family, a mutationcausing an amino acid exchange (Ser248Phe) in thesecond transmembrane domain of the neuronalnicotinic acetylcholine receptor a4 subunit wasfound.
In Norway, we have identi®ed two families withADNFLE, the ®rst in 1996 and the second in 1999(3, 4). In the ®rst Norwegian family, there was adifferent mutation (776ins3) in the same gene thanin the Australian family (CHRNA4), inserting anadditional leucine residue at the extracellular endof the second transmembrane domain. The secondNorwegian family had the same Ser248Phe-muta-tion as the original Australian family. These®ndings gave us the unique opportunity tocompare the phenotypic expressions of ADNFLEin two families with different mutations in the samegene.
Material and Methods
The two families, with 10 and 11 affected members,respectively, (Fig. 1, 2) have been thoroughlyinvestigated clinically, with interictal/ictal EEGand with CT/MRI. The molecular genetic analyseswere performed as previously described (5).
Results
Although, there were some inter- as well asintrafamilial variations in the severity of symptoms,clinical similarities between the two families werestriking. In both families the mean age of onset ofseizures was about eight years. They all had partialseizures with frontal lobe semiology; the seizureswere sleep-related, occurring most often in stage 2non-REM sleep, and had an abrupt start and end.The attacks were of short duration, 15-45 s. and inmost of the affected members in both families theseizures occurred in clusters. Typically, the seizuresbegan with arousal followed by stereotyped motorsymptoms in the form of dystonic or jerking
movements of trunk, extremities, or face. Somewere waving the arms, other displayed trashing orcycling movements with their legs, or pelvicthrusting. During the attack some individualsappeared anxious, they were shouting, crying,gasping, grunting, or moaning. The most prominentseizure type was complex partial seizures withimpaired consciousness and amnesia. In only oneaffected member in each family consciousness waspreserved during the attacks. A few patients in eachfamily (two and three patients, respectively) alsohad diurnal seizures while awake. Most often therewas little or no postictal confusion.
Many of the affected members went intoremission when reaching adulthood. Antiepilepticdrug treatment could eventually be withdrawn insome, in others the seizures were easily controlledby carbamazepine. One affected individual in the776ins3 family and several in the Ser248Phe familyhad dif®cult-to-treat seizures. In addition tointractable seizures, one patient in each familyhad subnormal intellect and behavioural problems.Most of the affected individuals had normalintellect with no abnormalities on neurologicaland neuroradiological examination. Both interictaland ictal EEGs were mostly normal or incon-clusive due to movement artifacts. Only in a fewmembers, low voltage epileptiform discharges inthe frontal regions have been demonstrated duringseizures.
The differences in the phenotypic expression inthe two ADNFLE-families were rather small.Compared to the affected members of the ®rstfamily with 776ins3 mutation, there was a strongertendency for a secondary generalization of thepartial seizures to tonic-clonic seizures and todevelop status epilepticus among individuals ofthe second family with Ser248Phe mutation. Thecourse of the epilepsy was less benign in the secondfamily in which the affected members had a strongertendency to persistent seizures in adulthood.
Discussion
We have identi®ed two Norwegian families withADNFLE with different genotypes. Althoughthere were some minor clinical differences betweenthe affected members in the two families, it seemedto be impossible to differentiate the phenotypes onclinical grounds only. The clinical similarities
25

suggest that these two mutations may have acomparable effect on the nicotinic acetylcholinereceptor function. This observation is indeedsupported by electrophysiological studies of thetwo receptors (5, 6). The Ser248Phe-mutationleads to a faster desensitization after acetylcholineactivation, and a slower recovery after desensitiza-tion. The 776ins3-mutation results in a decreasedcalcium permeability. Thus both mutations can beconsidered to render the neuronal nicotinicacetylcholine receptor less ef®cient. However,they do that by different mechanisms. Thismight be the explanation for the small variationsseen in the clinical pattern of the two families.These variations might perhaps also be due todifferences in the distribution and proportion ofmutated nicotinic acetylcholine receptors withinthe frontal lobe.
For the time being, it is unclear why a mutation inan acetylcholine receptor subunit, which is expressedin almost every part of the brain, may give rise tosleep-related frontal lobe seizures. Cholinergic neu-rons have complex modulatory effects on sleep andarousal at both thalamic and cortical levels (7). Theelectrophysiological consequences of thalamocorti-
cal input on defective receptors may be more criticalin the frontal lobe, particularly during sleep, than inother cortical areas.
Frontal lobe epilepsy is probably an under-diagnosed condition. As these seizures may have abizarre appearance with both motor and verbalautomatisms, misdiagnosis is quite common andincludes benign nocturnal parasomnias (somnam-bulism, pavor nocturnus, nightmares) (8), noctur-nal paroxysmal dystonia (9), and psychiatricdisturbance (hysterical personalities, pseudosei-zures) (10). Today there seems to be a generalagreement that some of the previously describednocturnal movement disorders, especially noctur-nal paroxysmal dystonia, actually are epilepticseizures of frontal lobe origin (11). Several of theaffected members of the two Norwegian familieswith ADNFLE had had their nocturnal symptomsfor many years before they were recognized asepileptic seizures.
Findings from families with idiopathic, mono-genic epilepsies, like the two described NorwegianADNFLE families, may constitute a step towards abetter understanding of the mechanisms by which amutated gene give rise to epileptic seizures.
Fig. 2. Pedigree of theNorwegian family with aSer248Phe mutation in theCHRNA4 gene.
Fig. 1. Pedigree of theNorwegian family with a776ins3 mutation in theCHRNA4 gene.
26

Hopefully, studies of such families also mayelucidate pathophysiological mechanisms in epi-lepsy in general.
References
1. SCHEFFER IE, BHATIA KP, LOPES-CENDES I et al. Autosomaldominant frontal epilepsy misdiagnosed as sleep disorder.Lancet 1994;343:515±517.
2. PHILLIPS HA, SCHEFFER IE, BERCOVIC SF, HOLLWAY GE,SUTHERLAND GR, MULLEY JC. Localization of a gene forautosomal dominant nocturnal frontal lobe epilepsy tochromosome 20q13.2. Nat Genet 1995;10:117±8.
3. NAKKEN KO, MAGNUSSON A, STEINLEIN OK. Autosomaldominant nocturnal frontal lobe epilepsy: An electroclinicalstudy of a Norwegian family with ten affected members.Epilepsia 1999;40:88±92.
4. STEINLEIN OK, STOODT J, MULLEY J, BERKOVIC S, SCHEFFER
IE, BRODTKORB E. Independent occurrence of the CHRNA4Ser248Phe mutation in a Norwegian family with nocturnalfrontal lobe epilepsy. Epilepsia 2000;41:529±35.
5. STEINLEIN OK, MAGNUSSON A, STOODT J et al. An insertionmutation of the CHRNA4 gene in a family with autosomal
dominant nocturnal frontal lobe epilepsy. Hum Mol Genet1997;6:943±7.
6. WEILAND S, WITZEMANN V, VILLARROEL A, PROPPING P,STEINLEIN OK. An amino acid exchange in the secondtransmembrane segment of a neuronal nicotinic receptorcauses partial epilepsy by altering its desensitizationkinetics. FEBS Letters 1996;398:91±6.
7. STERIADE M, MCCORMICK DA, SEJNOWSKI TJ.Thalamocortical oscillations in the sleeping and arousedbrain. Science 1993;262:679±85.
8. SCHEFFER IE, BHATIA KP, LOPES-CENDES I et al. Autosomaldominant nocturnal frontal lobe epilepsy. A distinctiveclinical disorder. Brain 1995;118:61±73.
9. LUGARESI E, CIRIGNOTTA F, MONTAGNA P. Nocturnalparoxysmal dystonia. J Neurol Neurosurg Psychiatry1986;49:375±80.
10. WILLIAMSON PD. Frontal lobe epilepsy: some clinicalcharacteristics. In: JASPER HH, RIGGIO S, GOLDMAN-RAKIC
PS, eds. Epilepsy and the functional anatomy of the frontallobe. (Advances in Neurology; vol 66). New York: RavenPress, 1995; 127±52.
11. HIRSCH E, SELLAL F, MATON B, RUMBACH L, MARESCAUX C.Nocturnal paroxysmal dystonia: a clinical form of focalepilepsy. Neurophysiol Clin 1994;24:207±17.
Estrogens and neuropsychiatric disorders
M. K. OÈ sterlund1,2, D. H. Overstreet3, J.-A. Gustafsson4, Y. L. Hurd1
1Dept of Clin. Neurosci., Karolinska Institute, S-171 76, Stockholm, Sweden, 2Karo Bio AB, Novum, S-141 57 Huddinge, Sweden, 3Dept of Psychiatry, University of North Carolina
at Chapel Hill, NC 27599-7175, USA, 4Dept. of Med. Nutrition, Karolinska Institute, S-141 86 Huddinge, Sweden
Objective
To give an overview of estrogen receptor geneactivity within the brain and the relation toneuropsychiatric disorders. Our recent studieswithin this topic are summarized.
Introduction
Natural ¯uctuations in gonadal hormones in women,both over the menstrual cycle and during theirlifetime, appears to in¯uence mood, cognition, andthe expression of neuropsychiatric disorders such asaffective disorders, schizophrenia, and Alzheimerksdisease (1±3). Premenstrual syndrome, postnataldepression, and postmenopausal depression areaffective disorders associated with a considerabledrop in serum estrogen levels and the two laterconditions have been shown to be improved orprevented with hormone replacement therapy.
The estrogenic effects are likely to be mediatedthrough the estrogen receptors (ERs). There are twoknown subtypes of ERs (4), ERa and ERb, whichare transcribed from two distinct genes. The ERsbelongs to the nuclear receptor superfamily and areligand activated transcription factors that regulate
gene transcription by binding to speci®c responseelements in the promoter region of target genes.
The aims of our studies were to characterize ERaand ERb gene expression and possible function inthe human and rat forebrain. In addition, estrogeniceffects on the serotonin (5-HT) system and thepossible link between estrogen and 5-HT indepression (using a genetic animal model ofdepression) were also investigated.
Methods
ER mRNA expression was detected in post mortemhuman and rat brain tissue using in situ hybridizationhistochemistry. Female Sprague Dawley ovariecto-mized rats were treated (acute or chronic) with 17b-estradiol or vehicle and the 5-HT1A receptor mRNAand binding levels in the brain were analyzed usingin situ hybridization histochemistry and radioligand(3H-WAY-100635) autoradiography respectively.To investigate the possible role of estrogen indepression studies were performed in a proposedgenetic animal model of depression, the FlindersSensitive Line (FSL) rats, and their correspondingcontrols, the Flinders Resistant Line (FRL) rats (5).Female FSL and FRL rats were ovariecomized and
27

treated with a single injection of 17b-estradiol orvehicle. The brains were analyzed for 5-HT1A and 5-HT2A receptor mRNA levels using in situ hybridiza-tion histochemistry.
Results and Discussion
Both in rat and human, the two ER subtypes weremainly expressed in limbic related brain areas andshowed distinct expression patterns (6, 7). Overall,the human ER mRNA expression in the brain wasrather low but the most abundant ERa mRNAexpressing areas were the amygdala and hypotha-lamus, suggesting a main role of ERa in estrogenmodulation of neuroendocrine and autonomicfunctions as well as emotions. In contrast, theERb subtype was predominantly expressed in areasrelated to memory and learning, such as thehippocampal formation and entorhinal cortex. Inaddition, the ERb subtype was also evident in thethalamus (ventrolateral nucleus and subthalamicnucleus). Both ERa and ERb mRNA wereexpressed in the cerebral cortex, ERa was presentin layer V and ERb in layer V and VI. In general,the ER mRNA expression patterns in the brainwere well conserved between rat and human, butsome species differences in both ERa and ERbmRNA expression were revealed. For example,high ERb mRNA expression was found in theparaventricular, supra optic and medial amygdalanucleus in the rat whereas these areas showed verylow ERb mRNA expression levels in the humanbrain. The ERs appears to be expressed in severallimbic related areas (e.g., amygdala, hippocampalformation, and temporal cortex) suggested to beinvolved in the pathophysiology of several neurop-sychiatric disorders, thus estrogen has the abilitytwo in¯uence the phenotype and function ofneuronal populations in these regions.
An imbalance in the 5-HT system is implied in theetiology of depression. Interestingly, estrogentreatment was found to down-regulate the expres-sion of both the 5-HT1A receptor mRNA andreceptor protein in several limbic areas (e.g.amygdala, hippocampus and perirhinal cortex) (8,9) of the rat brain. However, the changes in mRNAlevels were only found after acute 17b-estradioltreatment whereas the decrease in the receptor levelswere only present after the chronic treatment. Thistime shift is probably due to the long turnover of the5-HT1A receptor (several days) that has beenreported previously. The 17b-estradiol inducedeffects in the areas mentioned above suggests thatestradiol can modulate limbic functions via speci®c5-HT1A circuits.
Alterations in the 5-HT system (primarily the 5-HT2A receptor mRNA levels) were found in the
depressed FSL rats as compared to their controls(10). In the FSL animals, the 5-HT2A receptormRNA levels were lower in the amygdala andlimbic cortices, but higher in the hippocampus.Consistant with previous reports (1, 11), estradioltreatment increased the 5-HT2A receptor mRNAlevels in several of the examined areas, thusreversing the lower levels that were found inmany regions of the FSL rats up to control levels.There was no signi®cant difference between the ratlines in 5-HT1A receptor mRNA levels although atrend for higher levels were present in the perirhinalcortex and the amygdala. In line with previousresults, the 5-HT1A receptor mRNA levels weredecreased in several limbic areas after the estradioltreatment. This ®ndings suggest that the regulationby estrogens of the 5-HT2A, and possibly also the 5-HT1A receptor, might be an underlying mechanismin the hypothesized in¯uence of gonadal hormonesin the expression of affective disorders.
References
1. FINK G, SUMNER BEH, MCQUEEN JK, WILSON H, ROSIE R.Sex steroid control of mood, mental state and memory. ClinExp Pharmacol Physiol 1998;25:764±775.
2. RIECHER-ROÈ SSLER A, HAÈ FNER H. Schizophrenia and oestro-gen ± is there an association? Eur Arch Psychiatry ClinNeurosci 1993;242:323±328.
3. TARGUM SD, CAPUTO KP, BALL SK. Menstrual cycle phaseand psychiatric admissions. J Affective Disorders1991;22:49±53.
4. KUIPER GGJM, CARLSSON B, GRANDIEN K et al. Comparisonof the ligandbinding speci®city and transcription tissuedistribution of estrogen receptors alpha and beta.Endocrinology 1997;138:863±870.
5. OVERSTREET DH. The Flinders Sensitive Line rats: A geneticanimal model of depression. Neurosci Biobehav Rev1993;17:51±68.
6. OÈ STERLUND KM, KELLER E, HURD YL. The human forebrainhas discrete estrogen receptor a mRNA expression: highlevels in the amygdaloid complex. Neuroscience2000;95:333±342.
7. OÈ STERLUND M, KUIPER GGJM, GUSTAFSSON J-AÊ , HURD YL.Differential distribution and regulation of estrogen receptor-a and -b mRNA within the female rat brain. Mol Brain Res1998;101:175±180.
8. OÈ STERLUND MK, HALLDIN C, HURD YL. Effects of chronic17b-estradiol treatment on the serotonin 5-HT1A receptormRNA and binding levels in the rat brain. Synapse2000;35:39±44.
9. OÈ STERLUND MK, HURD YL. Acute 17b-estradiol treatmentdown-regulates serotonin 5-HT1A receptor mRNA expres-sion in the limbic system of female rats. Mol Brain Res1998;55:169±172.
10. OÈ STERLUND MK, OVERSTREET DH, HURD YL. The FlindersSensitive Line rats, a genetic animal model of depression,show abnormal serotonin receptor mRNA expression in thebrain that is reversed by 17b-estradiol. Mol Brain Res1999;74:158±166.
11. CYR M, BOSSE R, DI PAOLO T. Gonodal hormones modulate5-hydroxytryptamine2A receptors: emphasis on the ratfrontal cortex. Neuroscience 1998;83:829±836.
28

Hormones and epilepsy
A. G. HerzogHarvard Neuroendocrine Unit, Beth Israel Deaconess Medical Center, Boston, MA, USA
Introduction
There is considerable animal experimental andclinical evidence to suggest that gonadal steroidsin¯uence the occurrence of seizures. The repro-ductive endocrine environment of a woman withepilepsy can undergo physiological, pathologicaland pharmacological changes. Menarche, men-struation, pregnancy, and the process of meno-pause can be associated with altered seizurefrequency. Reproductive endocrine disorders areoverrepresented among women with epilepsy.Their anovulatory and inadequate luteal phasecycles often exacerbate seizures. Oral contracep-tives and menopausal hormonal replacement canexacerbate or bene®t a seizure disorder, dependingon the particular nature and circumstances of thetreatment. A knowledge of some interactionsamong hormones, epilepsy and antiepilepticdrugs, therefore, may provide the clinician witha more comprehensive basis for the effectivetreatment of women with epilepsy.
Hormonal Effects on Seizures
In many adult animal experimental models,estrogen lowers the thresholds of seizures (1).The topical brain application or intravenoussystemic administration of estradiol produces asigni®cant increase in spontaneous electricallyrecorded paroxysmal spike discharges. Theincrease is more dramatic in animals with pre-existent cortical lesions. Progesterone, on theother hand, lessens spontaneous and inducedepileptiform discharges (2). Hormones also in¯u-ence human electrical brainwave activity andepilepsy. Estrogen can activate epileptiform dis-charges and trigger clinical seizures (3), whereasprogesterone, suf®cient to produce luteal phaseserum levels, can signi®cantly decrease interictalspike frequency in some women with partialepilepsy (4).
Catamenial Epilepsy
Seizures do not occur entirely randomly inrelation to physiological reproductive endocrineevents (5). Catamenial epilepsy refers to seizureexacerbation in relation to the menstrual cycle.This occurs in approximately 70% of women withlocalization related epilepsy (6). A twofold orgreater increase in average daily seizure frequencyis noted in about one third (6). The onset of
menstruation is the reference point for day 1 withday -14 being the usual day of ovulation. Threepatterns exist: C1) seizure exacerbation during thethree days prior to menstruation and the ®rstthree days of menstruation (Days -3 to 3), C2)seizure exacerbation during the middle of thecycle between days 10 and -13, and C3) seizureexacerbation between day 10 of one cycle and day3 of the next (6).
Physiological endocrine secretion during themenstrual cycle in¯uences the occurrence of seiz-ures. In ovulatory cycles, seizure frequency shows astatistically signi®cant positive correlation with theserum estradiol/progesterone ratio (7). This ratio ishighest during the days prior to ovulation andmenstruation and is lowest during the early and midluteal phase. The premenstrual exacerbation ofseizures has been attributed to the withdrawal of theantiseizure effects of progesterone. Rapid proges-terone withdrawal is associated with a change inGABA receptor subtype to a form that is insensitiveto benzodiazepines and hence presumably toGABA (8).
Mid cycle exacerbations may be due to thepreovulatory surge of estrogen unaccompanied byany rise in progesterone until ovulation occurs.Seizures are least common during the mid lutealphase when progesterone levels are highest.
Inadequate luteal phase refers to less than normalprogesterone secretion during the second half of thecycle, regardless of whether ovulation does or doesnot occur. Serum estradiol/progesterone ratios andseizure frequencies tend to be higher than in normalovulatory cycles during the second half of thesecycles and seizure exacerbation may extend fromday 10 of one cycle to day 3 of the next cycle.
The premenstrual exacerbation of seizures maybe related to a decline in serum antiseizuremedication levels (9, 10), as well as to a withdrawalof the anticonvulsant effects of progesterone. Serumantiseizure medication levels generally decrease inthe days prior to menstruation. This decline issigni®cantly more marked in women who experi-ence premenstrual worsening of seizures. Hepaticmechanisms are implicated. Speci®cally, antiseizuremedications and gonadal steroids are metabolizedby the same microsomal enzyme systems in hepaticcells. The premenstrual decline in gonadal steroidsecretion, therefore, may permit increased metab-olism of antiseizure medications, resulting in lowerserum levels.
29

Pregnancy
Pregnancy may have variable effects on epilepsy(11, 12). A very small number of women experienceseizures for the ®rst time during pregnancy and haveseizures only during pregnancy. Most large inves-tigations throughout this century agree that preg-nancy does not affect seizure frequency in about onehalf of women. In about one third of women,seizure frequency will increase during pregnancy.Recent studies suggest that this can be attributed inthe majority of cases to a decrease in serum levels ofantiseizure medication. In some, this is due todiminished patient compliance. In others, it may berelated to a greater volume of distribution and ahigher rate of clearance. The latter may result fromincreased circulatory rate during pregnancy, altereddrug pharmacokinetics, and increased hepaticmetabolism. In about one sixth of women, seizurefrequency will decrease. This may be related, insome, to increased patient compliance with the useof antiseizure medication. In the remainder, themechanism is not known.
Menopause
The term ``menopause'' refers to a complex processand a variable end point which may differsigni®cantly among individuals (13). Early duringthe menopausal process, inadequate luteal phase/anovulatory cycles may develop and lead toincreased estrogen to progesterone ratios whichcan exacerbate seizures. At the end of the meno-pausal process, the cessation of estrogen productionby the ovary can bene®t seizures. These perimeno-pausal and menopausal effects are particularlynotable in women with catamenial epilepsy (14).
Hormonal Therapy
Progesterone
Natural progesterone therapy has been evaluatedand found to be ef®cacious in seizure management intwo open label trials. In the larger trial (15),25 womenwith catamenial exacerbation of intractable complexpartial seizures of temporal lobe origin, with orwithout secondary generalization, received naturalprogesterone in doses suf®cient to produce physio-logical luteal range progesterone serum levelsbetween 5±25 ng/ml. Seventy two percent of thewomen improved with an overall 55% decline inseizure frequency at three months. At three years, 15of the women had remained on cyclic progesteronetherapy and their original antiepileptic drugs. Threewomen were entirely seizure free. Four had totalseizure reductions of 75±99%. Eight had reductionsof 50±74%. Complex partial seizures in these 15 were
lower by a statistically signi®cant 62% compared tobaseline. Two of the 25 original women discontinuedprogesterone because of sedative and depressant sideeffects.
Synthetic Progestins
Oral synthetic progestins administered cyclically orcontinuously have not proven to be an effectivetherapy for seizures in clinical investigations (16),although individual successes with continuous dailyoral use of norethistrone and combination pills havebeen reported. The greater ef®cacy of progesterone ascompared to oral synthetic progestins may relate tothe greater degree that natural progesterone can bemetabolized to the highly neuroactive GABAergicanticonvulsant, allopregnanolone (17).
Depoprogestins
Parenteral depomedroxyprogesterone may lowerseizure frequency when it is given in suf®cientdosage to induce amenorrhea. In one open labelstudy of 13 women with refractory partial seizuresand normal ovulatory cycles, parenteral depome-droxyprogesterone administration in doses largeenough to induce amenorrhea (i.e. 120±150 mgevery 6±12 weeks) resulted in a 39% reduction inseizure frequency (18). It was unclear whether theeffect was due to direct anticonvulsant activity ofmedroxyprogesterone or to the hormonal conse-quences of the induced amenorrhea.
Clomiphene
Clomiphene acts as an estrogen antagonist toincrease gonadotropin secretion and induce ovula-tory cycles in estrogen secreting anovulatory womenwho do not have primary pituitary or ovarian failure.Normalization of reproductive endocrine functionsand menstrual cycles among women who have bothpartial seizures and menstrual disorders with docu-mented inadequate luteal phase has been demon-strated to signi®cantly and sometimes dramaticallylessen seizure frequency. In one investigation of 12women, 10 improved and seizure frequency declinedby 87% (19). Side effects can be signi®cant andinclude unwanted pregnancy, ovarian overstimula-tion syndrome, transient breast tenderness and pelviccramps. Furthermore, seizure frequency mayincrease during the enhanced preovulatory rise inserum estradiol levels in some women. Clomiphenetreatment, therefore, should be restricted to situa-tions where seizures are intractable to conventionalantiepileptic drug use, and irregular anovulatorycycles cannot be readily normalized with cyclicprogesterone use.
30

Gonadotrophin-Releasing Hormone (GnRH) Analogues
Synthetic gonadotrophin-releasing hormone (Gn-RH) analogues suppress pituitary gonadotropin andthereby gonadal steroid secretion, constituting amedical oophorectomy. In an open label trial in 10women with catamenial exacerbation of intractableseizures, three patients became seizure free and fourshowed a decrease in seizure frequency of up to 50%(20). In one, the duration of seizures was shortened;two had no therapeutic effect. These results wereattained within the ®rst two months of initiatingtreatment. All of the women became amenorrheic.Eight of the ten patients experienced hot ¯ushes,headache or weight gain. Additional experiencesuggests that during the ®rst month of treatmentwhen there is an initial stimulation rather thansuppression of gonadotropin and estrogen secretion,some women experience a marked exacerbation oftheir seizures and auras.
References
1. LOGOTHETIS J, HARNER R. Electrocortical activation byestrogens. Arch Neurol 1960;3:290±297.
2. LANDGREN S, BACKSTROM T, KALISTRATOV G. The effect ofprogesterone on the spontaneous interictal spike evoked bythe application of penicillin to the cat's cerebral cortex. JNeurol Sci 1978;36:119±133.
3. LOGOTHETIS J, HARNER R, MORRELL F, TORRES F. The role ofestrogens in catamenial exacerbation of epilepsy. Neurology1958;9:352±360.
4. BACKSTROM T, ZETTERLUND B, BLUM S, ROMANO M. Effects ofIV progesterone infusions on the epileptic discharge fre-quency in women with partial epilepsy. Acta Neurol Scand1984;69:240±248.
5. TAUBOLL E, LUNDERVOLD A, GJERSTAD L. Temporal distribu-tion of seizures in epilepsy. Epilepsy Res 1991;8:153±165.
6. HERZOG AG, KLEIN P, RANSIL BJ. Three patterns ofcatamenial epilepsy. Epilepsia 1997;38:1082±1088.
7. BACKSTROM T. Epileptic seizures in women related to plasmaestrogen and progesterone during the menstrual cycle. ActaNeurol Scand 1976;101:321±347.
8. COSTA A-MN, SPENCE KT, SMITH SS, FRENCH-MULLEN JMH.Withdrawal from the endogenous steroid progesteroneresults in GABA-A currents insensitive to benzodiazepinemodulation in rat CA1 hippocampus. J Neurophysiol 1995;74:464±469.
9. SHAVIT G, LERMAN P, KORCZYN AD, KIVITY S, BECHAR M,GITTER S. Phenytoin pharmacokinetics in catamenial epi-lepsy. Neurol 1984;34:959±961.
10. ROSCIZEWSKA D, BUNTNER B, GUZ I, ZAWISZA L. Ovarianhormones anticonvulsant drugs and seizures during themenstrual cycle in women with epilepsy. J Neurol NeurosurgPsychiat 1986;49:47±51.
11. KNIGHT AH, RHIND EG. Epilepsy and pregnancy: a study of153 pregnancies in 59 patients. Epilepsia 1975;16:99±110.
12. SCHMIDT D, CANGER R, AVANZINI G. Change of seizurefrequency in pregnant epileptic women. J Neurol NeurosurgPsychiat 1985;46:751±755.
13. HERZOG AG. Reproductive endocrine considerations andhormonal therapy for women with epilepsy. Epilepsia1991;32 (suppl 6):S27±S33.
14. HARDEN CL, PULVER MC, RAVDIN L, JACOBS AR. The effect ofmenopause and perimenopause on the course of epilepsy.Epilepsia 1999;40:1402±1407.
15. HERZOG AG. Progesterone therapy in complex partial andsecondary generalized seizures. Neurology 1995;45: 1660±1662.
16. DANAHAERI J, RICHENS A. Effect of noresthisterone on seizuresassociated with menstruation. Epilepsia 1983;24:377±381.
17. MAJEWSKA MD, HARRISON NL, SCHWARTZ RD, BARKER JL,PAUL SM. Steroid hormone metabolites are barbiturate-likemodulators of the GABA receptor. Science 1986;232:1004±1007.
18. MATTSON RH, CRAMER JA, CALDWELL BV, SICONOLFI BC.Treatment of seizures with medroxyprogesterone acetate:preliminary report. Neurology 1984;34:1255±1258.
19. HERZOG AG. Clomiphene therapy in epileptic women withmenstrual disorders. Neurology 1988;38:432±434.
20. BAUER J, WILDT L, FLUGEL D, STEFAN H. The effect of asynthetic GnRH analogue on catamenial epilepsy: a studyin ten patients. J Neurol 1992;239:284±286.
Hormones and antiepileptic drugs
E. TaubùllDepartment of Neurology, Rikshospitalet, University of Oslo, Norway
There is a complex relationship between hormonesand epilepsy in many different ways; hormonesaffects seizure thresholds, seizures affects hormonelevels, and antiepileptic drugs (AED) affects hor-mones. Considering the effect of AED on hormones,the most important issues from a clinical point ofview are related to hepatic enzyme induction, theeffect on thyroid hormone balance, and drug speci®ceffects on sex steroid hormones including thequestion of polycystic ovaries and endocrine changesafter valproate treatment.
Induction of hepatic enzymes
Most commonly used AEDs taken on a long-termbasis induces hepatic enzymes and raise the levels ofsex hormone binding globulin (SHBG) which bindmost of estrogen and testosterone. Because of that,the free and biologically active hormone consentra-tions are regularly decreased, even though the totalconcentration can be kept unchanged or evenincreased.
The most important clinical consequence ofhepatic enzyme induction is an increased metab-
31

olism of the estrogen component in the contra-ceptive pill. Especially when using contraceptivepills with a low level of estrogen, the estrogenconcentration can fall below what is necessary toavoid ovulation and pregnancy may occur. Aninteraction between the contraceptive pill andAEDs was ®rst reviewed by Coulam andAnnegers in 1979 (1) demonstrating such an effectof phenytoin, phenobarbital, primidone, car-bamazepine and ethosuximide. Later, felbamateand topiramate have also been shown to affect theef®ciency of the contraceptive pill, although themechanisms may be different. Drugs like valproate,lamotrigine and tiagabine do not have any effect onthe contraceptive pill.
The importance of reduced free androgen levelsfor potency, libido and sexual arousal has beendiscussed for years. Reduced potency and hypo-sexuality has been seen more commonly in menwith epilepsy and sexual problems are morefrequent in patients of both sexes (2). However,a clear correlation to speci®c drugs and hormonelevels has not been found. The reason for this isprobably that the etiology of these problems aremultifactorial including psychosocial, epileptic,medicational and hormonal causes.
AEDs and thyroid hormones
Changes in thyroid hormone levels after pheny-toin treatment were ®rst described byOppenheimer in 1961 (3). Since then, the interac-tion between thyroid hormones and antiepilepticdrugs has been extensively studied. In thebeginning of the eighties, i.e. Strandjord andcoworkers found in a series of studies, thatcarbamazepine reduced the concentration ofthyroxine and triiodothyronine (4, 5). The ratioof thyroxine to triiodothyronine and also the TSHlevel were unchanged, while the results for TBGvaried. Similar ®ndings have been done withphenytoin which also reduces thyroid hormonelevels (6). From a clinical point of view, it is,however, important to note that these patientsremained clinically euthyroid.
The mechanism for the effect on thyroidhormones is probably related to increased metab-olism due to induction of hepatic enzymes.However, the unchanged TSH level togetherwith low thyroid hormone levels may indicatethat central feedback mechanisms are not nor-mally activated which means that a hypothalamicinterference from the drug may be a contributingfactor.
AEDs and sex steroid hormones ± female studies
Drug speci®c effects on sex steroid hormones notdue to enzyme induction, is at the moment ®rst ofall a question of endocrine changes and polycysticovaries after valproate treatment. This issue hasbeen heavily discussed among epileptologists thelast few years and the results from our ownstudies on this topic so far will be reviewed.
Valproate affecting endocrine function was ®rstdescribed by Margraf and Dreifuss in 1981 (7)demonstrating amenorrhea developing after initia-tion of valproate treatment in eight previouslyregularly menstruating women. In 1993, IsojaÈrviand colleagues found in a group of 29 femalepatients with valproate monotherapy that 45%had menstrual disorders, 43 % had polycysticovaries, and 17% had increased testosterone levelswithout polycystic ovaries (8). These ®gures wereabout twice as high as those for patients receivingcarbamazepine monotherapy. These initial ®nd-ings were con®rmed in his later study published in1996 in which as much as 64 percent hadpolycystic ovaries and or hyperandrogenism (9).
We have just ®nished a multi-center studyincluding a total of 124 women from threeeuropean countries, Norway, Finland and theNetherlands (10). The frequency of polycysticovaries and/or hyperandrogenism was about 70%among valproate treated women, compared to20% and 19% for carbamazepine or controltreated women, respectively. The ®gures werenearly identical in each of the three countries.Obesity itself was not a necessary prerequisit fordeveloping polycystic ovaries since this was foundin as much as 65% of lean women on valproate.Polycystic ovaries could also be observed inwomen despite normal LH or insulin levels.
These ®ndings do not, however, have to berelated to valproate itself. Differences betweenpatient groups could for instance be related to theepileptic activity itself or to the selection ofpatients. One way to solve this problem is toperform studies in healthy, non-epileptic animals.The effect of the drug can then be studiedindependent of epileptic activity and withunbiased selection of subjects.
In our ®rst animal study 45 young adult femalerats were divided in three groups receiving eithercontrol solution, or valproate in doses of 50 or200 mg/kg perorally once daily for 90 days (11).The main ®nding was a signi®cant, dose-depen-dent increase in the number of cystic formationsin the ovaries at serum valproate concentrationswell within the therapeutic range. Further, therewas a dose-dependent, signi®cant decrease in
32

testosterone and a slight trend towards reductionin estrogen with unchanged progesterone.
Encouraged by these ®ndings, a second animalstudy has now been completed (12). In this study,female Wistar rats were fed perorally with either 200or 300 mg/kg valproate, 5 mg/kg lamotrigine orcontrol solution twice daily for 90 days. Serumconcentrations were within the human therapeuticrange measured 4 to 6 hours after last dose. Again, wecould demonstrate a signi®cant, dose-dependentincrease in the number of follicular cysts in theovaries after valproate, but not lamotrigine, treat-ment. This ®nding was strengthened by a concomit-tant reduction in the number of corpora lutea. Withregard to the morphological changes in the ovaries,the question of possible cancerogenic effects can beraised. To study this possibility we investigated theexpression of p53 which is the most frequentlymutated tumor suppressor gene so far described inhuman cancer. There was no pathological expressionof p53 in theovaries of either valproateor lamotriginetreated animals. As for the ®rst animal study,testosterone levels, this time the free fraction, wasreduced after valproate treatment while lamotriginedid not have any effect. Again, there was a reductionin estrogen, which this time was signi®cant. LH andinsulin, which have been concidered important forthe polycystic ovary syndrome in humans, wereunchanged or even reduced.
AEDs and sex steroid hormones ± male studies
Only few studies have been performed regardingendocrine effects of valproate in men and usuallyno or minor effects have been reported. Yerbyand McCoy (13) described, however, recently a 32year old male on valproate monotherapy withfertility problems. He had a low total number ofsperms with no motile sperms. After changing tofelbamate the total number of sperms and thenumber of motile sperms increased and he becamea father. No change was observed in LH or FSH.
In animals, a few studies have shown testicularatrophy and reduced spermatogenesis aftervalproate treatment in dogs and rats (14, 15),but the doses were high and no serum concentra-tions were given. In the study of Snyder andBadura in 1995 (16), chronic administration ofvalproate to mice seemed to slow, but not preventpubertal maturation as evaluated by slowedtesticular and skeletal growth and reduced sper-matogenesis.
On this background, we have undertaken astudy on the effect of long-term valproatetreatment on testicular morphology and endocri-nology in male rats. Male Wistar rats were fedperorally twice daily for 90 days with either
valproate 0, 200 or 400 mg/kg, or lamotrigine5 mg/kg. Serum valproate concentrations 4 and 6hours after last dose were 819 and 241 mmol/lusing 200 mg/kg, and 967 and 333 mmol/l using400 mg/kg of valproate. The respective lamotri-gine concentrations were 45.2 and 38.6 mmol/l.The main ®nding was marked testicular atrophyand reduced spermatogenesis using the highestvalproate dose, with no effect of lamotrigine. Nochanges were observed in the interstitial tissue,including the Leydig cells. Turning to the endo-crine data, there was, as for the female rat study,a marked and signi®cant reduction in freetestosterone while total testosterone showed aninsigni®cant, 17% reduction. Contrary to thefemale study, LH showed a dose-dependentincrease while FSH increased at the highestvalproate dose, only. Again, lamotrigine did nothave any effect on neither peripheral sex steroidhormones nor on FSH and LH. In conclusion,valproate but not lamotrigine induced changes ingonadal morphology and sex hormone levels inmale rats.
To study the effect of valproate in anotherspecies we have performed, together with theSchool of veterinary science, University of Oslo, astudy using male goats. Nine bucks, aged 2months at the start of the study, receivedvalproate mixture up to 125 mg/kg per daygiven in two divided doses adjusted accordingto serum concentrations for 8 months. Serumconcentration of valproate 2, 4 and 6 hours afterlast dose was 694, 485 and 301 mmol/l, respec-tively. Testicular diameter was slightly, butsigni®cantly reduced during the study comparedto controls. Semen analyses showed a signi®cantreduction in semen volume of 15% and areduction in total sperm count of 20%. Theincrease in testosterone that normally takes placeduring adolescence, was delayed, but not pre-vented. This might indicate, as in the study ofSnyder and Badura (16), that valproate possiblydelayed puberty.
It should, however, be born in mind thatseveral other antiepileptic drugs also affect semenquality and testosterone production. For instance,in an in vitro Leydig cell model, carbamazepinewas the only drug to inhibit testosterone produc-tion at therapeutic serum concentrations.Phenytoin required higher doses while valproatewas the drug with least effect (17).
Conclusion
Most antiepileptic drugs seem to affect hormonalbalance in one or another way regarding either
33

thyroid or sex steroid hormones. What is presentlymost intensively discussed is the effect of valproate.To summarize our own recent ®ndings on valpro-ate, we have found changes in hormone levels andgonadal morphology in both female and maleanimals. These effects have been found in non-epileptic animals and with what we consider astherapeutic serum concentrations. The mechanismsof action are still not settled. It could be both adirect effect and /or a centrally mediated effect. Themechanism of action may be unique and drugspeci®c and, in females, the mechanisms may well bedifferent from that commonly seen in the polycysticovarian syndrome in humans.
References
1. COULAM CB, ANNEGERS JF. Do anticonvulsants reduce theef®cacy of oral contraceptives? Epilepsia 1979;20:519±525.
2. HERZOG AG. Disorders of reproduction and fertility. In:ENGEL J JR, PEDLEY TA, eds. Epilepsy: A comprehensivetextbook. Philadelphia: Lippincott-Raven Publishers,1997:2013±2019.
3. OPPENHEIMER JH, FISCHER LV, NIELSON KM, JAILER JW.Depression of the serum protein-bound iodine levelby diphenylhydantoin. J Clin Endocrinol Metab1961;21:252±262.
4. STRANDJORD RE, JOHANNESSEN SI. Single-drug therapy withcarbamazepine in patients with epilepsy: serum levels andclinical effect. Epilepsia 1980;21:655±662.
5. STRANDJORD RE, AANDERUD S, MYKING OL, JOHANNESSEN SI.In¯uence of carbamazepine on serum thyroxine andtriiodothyronine in patients with epilepsy. Acta NeurolScand 1981;63:111±121.
6. ISOJAÈ RVI JIT, PAKARINEN AJ, MYLLYLAÈ VV. Thyroid functionwith antiepileptic drugs. Epilepsia 1992;33:142±148.
7. MARGRAF JW, DREIFUSS FE. Amenorrhea following initia-tion of therapy with valproic acid. Neurology 1981;31:159.
8. ISOJAÈ RVI JIT, LAATIKAINEN TJ, PAKARINEN AJ, JUNTUNEN
KTS, MYLLYLAÈ VV. Polycystic ovaries and hyperandrogen-ism in women taking valproate for epilepsy. New Engl JMed 1993;329:1383±1388.
9. ISOJAÈ RVI JIT, LAATIKAINEN TJ, KNIP M, PAKARINEN AJ,JUNTUNEN KTS, MYLLYLAÈ VV. Obesity and endocrinedisorders in women taking valproate for epilepsy. AnnNeurol 1996;39:579±584.
10. ISOJAÈ RVI JIT, TAUBéLL E, PAKARINEN AJ et al. Valproaterelated endocrine risks in women with epilepsy ± amulticenter study. Epilepsia 1998;39 (suppl 6):220±221.
11. TAUBéLL E, ISOJAÈ RVI JIT, FLINSTAD HARBO H, PAKARINEN AJ,GJERSTAD L. Long-term valproate treatment induceschanges in ovarian morphology and serum sex steroidhormone levels in female Wistar rats. Seizure 1999;8:490±493.
12. RéSTE LS, TAUBéLL E, BERNER Aa, ISOJAÈ RVI JIT, GJERSTAD L.Valproate, but not lamotrigine, induces ovarian morpho-logical changes in Wistar rats. Exp Toxicol Pathol 2000, inpress.
13. YERBY MS, MCCOY GB. Male infertility: possible associ-ation with valproate exposure. Epilepsia 1999;40;520±521.
14. WALKER RM, SMITH GS, BARSOUM NJ, MACALLUM GE.Preclinical toxicology of the anticonvulsant calcium valpro-ate. Toxicology 1990;63:137±155.
15. Abbott laboratories, patient information lea¯et, 1997, Ref:03±4757-R4.
16. SNYDER PJ, BADURA LL. Chronic administration of sodiumvalproic acid slows pubertal maturation in inbred DBA/2Jmice: Skeletal, histological, and endocrinological evidence.Epilepsy Res 1995;20:203±211.
17. KUÈ HN-VELTEN WN, HERZOG AG, MUÈ LLER MR. Acute effectsof anticonvulsant drugs on gonadotropin-stimulated andprecursor-supported androgen production in the rat testis.Eur J Pharmacol 1990;181:151±155.
Characterization of neural stem cells in the adult CNS
C. B. Johansson, S. Momma, D. L. Clarke, U. Lendahl, J. FrisenDepartment of Cell and Molecular Biology, Medical Nobel Institute, Karolinska Institute, SE-171 77 Stockholm, Sweden
Neurons are continuously generated in certainregions of the adult mammalian brain. Theseneurons derive from multipotent, self-renewingneural stem cells. Such stem cells can be culturedfrom the walls of the ventricular system of theadult rodent and human brain. We have found,by in vivo labeling experiments, cell sorting and invitro cultures, that ependymal cells have neuralstem cell properties in the rodent. Ependymal cellsdivide rarely to give rise to subventricular zone
progenitor cells which generate neuroblasts thatmigrate to the olfactory bulb. In response to aspinal cord injury, ependymal cells lining thecentral canal are induced to proliferate andgenerate migratory progeny which differentiateto astrocytes and contribute to scar formation.Further studies on the regulation of stem celldifferentiation may allow the development ofstrategies to stimulate neurogenesis in the adultbrain.
34

Consequences of head trauma in an animal model
I. SolteszDepartment of Anatomy and Neurobiology, University of California, Irvine
Traumatic brain injury affects over a millionpeople each year in the US alone, with anestimated annual cost to society approaching$48 billion. A large percentage of patients sufferfrom various neurological disorders includingpost-traumatic seizures, memory problems andother cognitive de®ciences.
We use the ¯uid percussion head injury (FPI)model in rats to investigate the physiological basisof post-traumatic hyperexcitability. The FPImodel, by inducing a single, controlled (duration:20 ms; amplitude: 1.8±2.0 atm), reproduciblepressure wave transient with a high-speed jet of¯uid ejected onto the exposed dura, is able toreplicate the major histological, physiological andbehavioral features of human traumatic concus-sive brain injuries. For example, there is selectivedamage to the hilus of the dentate gyrus, themajor regulator of the entorhino-hippocampalcommunication. Our data determined that thereis a long-lasting perturbation of interneuronalnetworks following impact (1, 2).
Because inhibitory interneurons synapsing ongranule cells play a central role in the regulationof the input-output functions of the hippocampus,the perturbation of dentate interneuronal net-works is likely to be an important factor in thedevelopment of post-traumatic pathological states.Speci®cally, within the hilus, the number ofparvalbumin-positive basket- and axo-axoniccells is decreased after FPI for weeks andmonths (as determined by either immunocyto-chemical or in situ hybridization methods), andthere is a concomittant decrease in the frequencyof the action potential independent, miniatureinhibitory post-synaptic currents (IPSCs) in gran-ule cells, indicating persistent disturbance of theperisomatic inhibitory control of the output fromthe principal cells of the dentate gyrus.Interestingly, neighboring interneurons situatedin the granule cell layer survive the impact,suggesting a location-speci®city of the post-trau-matic damage. Although the interneurons in thegranule cell layer survive, they show physiologicalalterations in the form of a highly reproducibledepolarization of their resting membrane poten-tial, related to the post-traumatic disturbance ofthe interneuronal electrogenic Na/K-ATPasepump (3). In contrast, granule cells in the samecell layer do not exhibit such post-traumaticdepolarization of the resting membrane potential
nor a decreased function of the sodium pump.The selective post-traumatic depolarization lastsfor days, and it increases the spontaneous ®ringrate of interneurons and augments the ef®cacy ofexcitatory inputs to discharge interneurons. Theenhanced rate of action potential ®ring ininterneurons leads to an increase in the frequencyand amplitude of the spontaneous, action poten-tial-dependent IPSCs in granule cells. Therefore,in spite of a decrease in the frequency ofminiature IPSCs (likely resulting from the lossof hilar GABAergic cells), there is an overallincrease in spontaneous inhibitory tone in granulecells, related to the post-traumatic depolarizationof the surviving interneurons. Interestingly, thetraumatic damage to the interneuronal networksof the dentate gyrus occurs instantaneously (1),indicating that purely mechanical factors mayin¯uence the pattern of initial damage afterimpact. Our data revealed that interneuronsshow a highly speci®c cytoskeletal architecture(4), which is likely to play a role in determiningthe sensitivity of interneurons to mechanicalforces. For example, parvalbumin-positivebasket- and axo-axonic cells in the dentategyrus granule cell layer show intense labelingwith antibodies raised againts the heavy neuro®la-ment (a major determinant of the mechanicalintegrity of cells), whereas neighboring granulecell dendrites appear immunonegative, indicatinga highly speci®c cell-type dependent expressionpattern of the interneuronal cytoskeleton. In fact,our data determined striking differences in theexpression levels of all three neuro®lament tripletproteins, as well as alpha-internexin and beta-tubulin III between parvalbumin-positive dentateinterneurons and dentate granule cells.Furthermore, it appears there are several othercytoskeletal proteins which are also speci®callylocalized in distinct interneuronal classes. Forexample, current research in our laboratorydetermined that NPY-positive interneurons inthe dentate and CA1 dendritic layers show highlevels of expression for the actin/NMDA receptorbinding protein alpha-actinin-2, whereas parval-bumin-positive interneurons have only low levelsof somatic and proximal dendritic immuno-reactivity to alpha-actinin-2 (Ratzliff andSoltesz, unpublished observations). Alpha-acti-nin-2 is especially interesting, since it can regulatethe rate of NMDA receptor desensitization,
35

therefore, given the potential neuroprotective rolefor NMDA receptor antagonists, the speci®c celltype dependent expression-patterns of thisNMDA-receptor/ actin-binding protein may in¯u-ence the pattern of excitotoxic damage followinghead trauma.
Finally, we carry out computer modelingstudies to test hypotheses related to the degreeof electrical stability of neuronal networks follow-ing pertubations. Our recent computer simulations(Aradi and Soltesz, unpublished data) revealedthat variability in the anatomical and/or physio-logical properties of interneurons can powerfullyregulate the electrical stability of neuronal net-works. Therefore, a crucial connection existsbetween the degree of interneuronal diversity(e.g. variability in the action potential adaptationproperties of interneurons) and network stability,a ®nding which may have important implicationsfor epilepsy research. For example, alterations inthe variability in interneuronal properties maytake place as a result of various insults, such ashead injury, which would be expected to contrib-ute to the lower degree of stability in neuronalnetworks following impact. It is an intriquingpossibility that certain naturally occuring neuro-modulators may be able to regulate the degree ofphysiological diversity (e.g. in resting membranepotential) of interneuronal networks in thehippocampus. Further detailed research aimed atthe understanding of the major regulators ofinterneuronal diversity may open novel possibi-lities in the future for controlling hyperexcitabilityfollowing traumatic brain injury.
In conclusion, the FPI model of head trauma inrats reveals a highly selective, instantaneous, long-lasting perturbation to the interneuronal networksof the dentate gyrus. The initial pattern ofdamage is likely to depend on mechanical factors(e.g. cell size, support by similarly orientedneighboring cells, cytoskeletal architecture),which is then further modulated by biologicalfactors (e.g. excitotoxic insults). Computer simu-lation data indicate that diversity in interneuronalnetworks may be a novel, major determinant ofhow inhibitory cells can regulate principal celldischarges in a dynamic, constantly changingsynaptic environment.
This research is supported by the NIH (NS35915to I.S.).
References
1. TOTH Z, HOLLRIGEL GS, GORCS T, SOLTESZ I. Instantaneousperturbation of interneuronal networks by a pressure wave-transient delivered to the neocortex. J Neurosci1997;17:8106±8117.
2. SANTHAKUMAR V, BENDER R, FROTSCHER M et al. Granule cellhyperexcitability in the early post-traumatic rat dentategyrus: the ``irritable mossy cell'' hypothesis. J Physiol2000;524:117±134.
3. ROSS ST, SOLTESZ I. Selective depolarization of interneuronsin the early posttraumatic dentate gyrus: involvement of theNa(+)/K(+)-ATPase. J Neurophysiol 2000;83:2916±2930.
4. RATZLIFF A, SOLTESZ I. Differential expression of cytoskeletalproteins in the dendrites of parvalbumin-positive interneur-ons versus granule cells in the adult rat dentate gyrus.Hippocampus 2000;10:162±168.
Perinatal insults and epilepsy
G. L. HolmesDept. of Neurology, Harvard Medical School, Children's Hospital, Boston, Massachusetts, U.S.A.
Objectives
To review the relationship between seizures occur-ring during the perinatal period with subsequentdevelopment of epilepsy.
Background
It is well recognized that children are at higher riskfor seizures than adults. In addition to the higherincidence of epilepsy in children than adults,precipitating factors such as fever are far morelikely to induce a seizure in a young child thanadult. Children also have a signi®cantly higher
likelihood of entering remission than adults furthersuggesting that the brain becomes less excitable withage. Results from animal studies parallel clinicalstudies and demonstrate that the immature brain ismore susceptible to seizures than the adult brain.Kindling, a process in which recurrent electricalstimulations which initially result only in briefelectrical discharges and mild behavioral changesbut result progressively in more prolonged andintense electrical and behavioral seizures, occurs atall ages. Young animals kindle more rapidly thanmature animals. In addition, a shorter period ofpostictal refractoriness in young animals leads to a
36

quick progression through early stages of kindlingand results in rapid generalization of seizures.Immature rats are more likely to develop seizureswith hypoxia than mature rats. Similarly, thepowerful convulsant kainic acid at very low dosesgenerates seizures shortly after birth in the rat.
Although the threshold for seizure generation islower in immature than adult brains, developingneurons are less vulnerable in terms of neuronaldamage and cell loss than adult neurons to a widevariety of pathological insults. For example,immature hippocampal neurons will continueresponding to synaptic stimuli in a fully anoxicenvironment for longer durations than adult ones;likewise, longer anoxic episodes are required toirreversibly destroy the circuit in young animals.Young animals are less vulnerable to cell lossfollowing a prolonged seizure than mature animal.Sprouting of mossy ®bers is less prominent follow-ing prolonged seizures in young animals thanseizures of similar duration in older animals.
Brain damage through excessive glutamaterelease and ``cerebral excitotoxicity'' appears to bea common mechanism for many neurologicalinsults, including hypoxia-ischemia and seizures.The immature brain appears to be more `resistant`to the toxic effects of glutamate than the maturebrain. The degree of Ca2+ entry into the hippo-campal sub®eld CA1 and subsequent damage isdirectly related to age. In postnatal days (P) 1-3neurons glutamate increases intracellular Ca2+
minimally while in P21±25 neurons glutamateresults in marked increases in intracellular Ca2+
and causes marked swelling of the cell andretraction of dendrites into the soma of theneuron (1). This relative resistance is thought tobe due to the smaller density of active synapses,lower energy consumption, and in general therelative immaturity of biochemical cascades thatlead to cell death following insults.
Behavioral consequences following status epilep-ticus are also related to age of the animal at the timeof the status; adult animals surviving statusepilepticus have signi®cant de®cits in learning,memory, and behavior whereas young rats follow-ing status epilepticus have fewer de®cits in learning,memory, and behavior. Likewise, spontaneousseizures following status epilepticus are morelikely to occur in adult animals experiencingstatus epilepticus than in young animals.
While immature rats during the ®rst two weeksof life may have less cell loss following statusepilepticus than their adult counterparts, recentstudies from our laboratory have demonstratedthat repetitive seizures during the ®rst 5 days oflife in a rat do alter subsequent brain develop-
ment (2). We have found that neonatal seizuresresult in subsequent sprouting of mossy ®bers toboth the CA3 and supragranular regions withoutany accompanying cell loss. Furthermore, ratswith neonatal seizures have reduced seizurethresholds and impairment in learning, memory,and activity level compared to controls whentested as adults. Likewise, animals with otherperinatal insults including hypoxia-ischemia andcortical dysplasias are at risk for subsequentepilepsy. Animals with seizures induced byhypoxia at P10, but not P5 or P60, have long-term changes in seizure susceptibility (3). Ratswith cortical dsyplasias induced at P1 by a freezelesion have abnormal neuronal activation, asmeasured by c-fos, and increased mossy ®bersprouting, when studied in the pubescent period(4).
We raised the question as to whether seizure-induced changes in connectivity during early braindevelopment alters subsequent risk for damageduring seizures. The goal of this study was todetermine whether neonatal seizures affects braindamage induced by status epilepticus at a later age.
Methods
Twenty-®ve seizures were induced by the inhalant¯urothyl in neonatal rats during the ®rst ®ve days oflife. Flurothyl reliably produced generalized sei-zures with concomitant electroencephalographicchanges and a low mortality rate. During adoles-cence or early adulthood animals were subjected tostatus epilepticus using either a chemical convul-sant, kainic acid, or electrical stimulation, perforantpath stimulation. We chose to use two differentmodels of status epilepticus since we wished tocreate a range of seizure-induced damage. Kainicacid typically results in major cell loss whileperforant path stimulation causes less severe cellloss.
Results
We found that ¯urothyl reliably produces beha-vioral and electroencephalographic seizures in theneonatal period. While these neonatal seizures didnot lead to cell loss, the rats had signi®cantly morecell loss following subsequent status epilepticusthan did rats without a history of neonatal seizures.
Discussion
We found that seizures during the neonatal periodsubstantially increase the degree of seizure-induceddamage later in life. In both the kainic acid and
37

perforant path stimulation models, animals withprevious ¯urothyl seizures had signi®cant increasesin cell loss compared to control rats without priorneonatal seizures. This cell loss was a result of thestatus epilepticus induced by kainic acid orperforant path stimulations since cell loss was notobserved in rats with neonatal ¯urothyl seizureswhen examined at P10, P20 or P60 days. This studyalso demonstrates that neonatal seizures predisposethe brain to further seizure-induced injury regard-less of whether the second seizure is severe, as withKA, or mild, as with perforant path stimulation.
Why neonatal seizures that result in no aparentcell loss could ``prime'' the brain for later seizure-induced cell loss is not clear. While neonatalseizures do not result in signi®cant cell loss theydo result in alterations of neuronal circuitry. Withboth recurrent ¯urothyl and pentylenetetrazolseizures during the neonatal period, there issprouting of mossy ®bers into the inner-molecularlayer of the dentate and CA3 pyramidal cell layer.Therefore, one explanation for our ®ndings mightbe that the excess number of granule cells hasresulted in an over-abundance of projections totarget cells resulting in a hyperexcitable circuit.
Since glutamate is the neurotransmitter of themossy ®bers, it is tempting to suggest that increasednumbers of glutamatergic synapses could increaseexcitability, lower seizure threshold, and increasethe amount of seizure-induced damage. However,sprouting of mossy ®bers may not necessarilyincrease glutamate release in ¯urothyl-treated rats.
Acsady et al. (5) demonstrated that terminals fromdentate granule cells are more likely to innervategamma-aminobutyric acid (GABA) inhibitoryinterneurons than excitatory pyramidal cells.
This study presents evidence that despite lack ofcell loss, neonatal seizures initiate a cascade ofchanges in the developing brain that are maladap-tive and increase the risk of subsequent damagewith a second insult. Whether children withneonatal seizures and other perinatal insults are athigher risk for neurological damage following asecond seizure later in life is unclear. It is alsounclear whether medical intervention at the time ofthe perinatal insult would alter the subsequentcourse.
References
1. MARKS JD, FRIEDMAN JE, HADDAD GG. Vulnerability of CA1neurons to glutamate is developmentally regulated. DevelopBrain Res 1996;97:194±206.
2. HOLMES GL, GAIARSA J-L, CHEVASSUS-AU-LOUIS N, BEN-ARI
Y. Consequences of neonatal seizures in the rat: morpholo-gical and behavioral effects. Ann Neurol 1998;44:845±857.
3. JENSEN FE, HOLMES GL, LOMBROSO CT, BLUME HK, FIRKUSNY
IR. Age-dependent changes in long-term seizure suscepti-bility and behavior following hypoxia in the rat. Epilepsia1992;33:971±980.
4. HOLMES GL, SARKISIAN M, BEN-ARI Y, LIU Z, CHEVASSUS-AU-LOUIS N. Consequences of cortical dysplasia during devel-opment in rats. Epilepsia 1999;40:537±544.
5. ACSADY L, KAMONDI A, SIK A, FREUND T, BUZSAÂ KI G.GABAergic cells are the major postsynaptic targets of mossy®bers in the rat hippocampus. J Neurosci 1998;18:3386±3403.
Prolonged febrile seizures: neuroanatomical and functional consequences
C. Dube, R. A. Bender, K. Chen, Z. Toth, M. Eghbal-Ahmadi, I Soltesz, T. Z. BaramAnatomy, Neurobiology and Pediatrics, University of California at Irvine. Irvine, CA., USA 92697-4475
Rationale and Objectives
Febrile seizures are common, affecting 2±5% ofinfants and young children worldwide (1±3). Therelationship of childhood febrile seizures to adulttemporal lobe epilepsy (TLE) has remained a focusof intense controversy (see 4±7 for brief recentreviews): Whereas prospective epidemiologicalstudies have not shown a progression of febrileseizures to TLE, retrospective analyses of adultswith TLE have demonstrated a high prevalence (30->60%) of a history of prolonged (longer than 10±15minutes) febrile seizures during early childhood,suggesting an etiological role for these seizures inthe development of TLE. Speci®cally, neuronaldamage induced by febrile seizures has been
suggested as a mechanism for the development ofmesial temporal sclerosis, the pathological hallmarkof TLE. However, this high correlation should notbe taken to indicate a causal relationship, andalternative mechanisms may exist for the corre-lation of prolonged febrile seizures and TLE. Theseinvolve pre-existing, genetic or acquired, functionalor structural neuronal changes, that may underlieboth the prolonged febrile seizures and the sub-sequent TLE (see diagram):
Alternative I:
Normal brainp Febrile seizuresp neuronal damagep TLE
Alternative II:
Pre-existing injury/lesionp fever-triggered seizure=®rst sign of TLE
38

These critical questions regarding the causalrelationship of prolonged febrile seizures and TLEare dif®cult to resolve in human studies. However,animal models permit induction of febrile seizuresof controlled duration, and prospective studies fordissecting out the nature of these seizures and theirconsequences. Therefore, a model of febrile seizuresin the immature rat, using animals during a brain-development age generally equivalent to that of thehuman infant and young child, has been developedand characterized (8). This model has been used todetermine the acute and long-term consequences ofprolonged hyperthermic seizures on neuronal func-tion and survival both in vivo and in vitro, and onthe development of spontaneous limbic seizures- i.e.,TLE. Hyperthermic seizures, provoked by generat-ing brain- temperatures seen physiologically in illinfants and children, were shown to result instructural alterations of select hippocampal andamygdala neurons (9). These changes in cellularcytoskeleton, leading to af®nity of neurons to silverstains (argyrophilia) were not induced by hyper-thermia alone: they were not found in animalssubjected to the same magnitude and duration ofhyperthermia, but in whom seizures were preventedby a short-acting barbiturate. The same studyshowed that although neurons in hippocampusand amygdala, in a distribution consistent with theinjury found in TLE, were altered for at least 2weeks, neuronal death was negligible. First, a time-course of in situ end labeling, performed 1, 4, 8.5, 20or 48 hours after the seizures, did not revealappreciable numbers of dying neurons. In addition,cell counts in highly involved limbic regions weresimilar in animals one month after prolongedhyperthermic seizures, hyperthermia alone or con-trol conditions (9).
However, several key questions remained. First,are the apparently transient alterations of neur-onal structure induced by hyperthermic seizuresassociated with functional disruption suf®cient toalter the excitation-inhibition balance in theinvolved circuits, promoting the development ofTLE? Second, are there other, more subtleneuroanatomical/structural alterations of hippo-campal neurons, short of overt death, that mayin¯uence the hippocampal circuit to promoteexcitability?
Methods and Results
Addressing the ®rst question, Chen et al. (10),demonstrated the presence of persistent functionalmodulation of hippocampal circuitry in this imma-ture rat model of febrile seizures. Speci®cally,hyperthermia-induced seizures (but not hyper-
thermia alone) caused a selective presynapticincrease of inhibitory synaptic transmission inhippocampus, that lasted into adulthood. Thispaper thus documented, using controlled, prospec-tive methods, that in a previously normal immaturebrain, experimental prolonged febrile seizurescaused long-lasting modi®cations of the balance ofexcitation and inhibition in neuronal microcircuitswithin the limbic system. The changes found,however, seemed to imply increased activity ofinhibitory interneurons, and the relationshipbetween these alterations of synaptic communica-tion and the development of limbic epilepsy was notresolved. In addition, these dramatic changes wereshown in vitro, and their relevance to the wholeorganism required further study.
The consequences of prolonged febrile seizuresin the immature rat model on the development ofspontaneous seizures were the focus of a secondstudy (7). Using in vivo and in vitro approaches inthis model, it was determined that prolongedhyperthermia-induced seizures in immature ratcaused long-term enhanced susceptibility tolimbic convulsants that lasted to adulthood.After induction of prolonged (20 minutes)hyperthermic seizures, animals were allowed togrow to adulthood, then underwent extensivehippocampal EEGs and behavioral monitoring.Both EEGs and behavioral measures failed todemonstrate spontaneous seizures in these adultrats who had experienced hyperthermic seizuresduring infancy. However, 100% of animals devel-oped hippocampal seizures upon systemic admin-istration of a threshold dose of kainic acid, anactivator of a glutamate receptor subtype. Thus,whereas this dose of the excitatory trigger did notcause seizures in most adult rats that did notexperience prolonged febrile seizures in ``infancy'',i.e., both normothermic controls and those under-going hyperthermia with seizure blockade, themajority of adult animals who had experiencedprolonged febrile seizures early in life progressedto status epilepticus (SE). These ®ndings, of aprofound increase in vulnerability to pro-convul-sant provocation, were con®rmed in vitro:Spontaneous epileptiform discharges were notobserved in hippocampal-entorhinal cortex slicesderived from either control or experimentalgroups. However, Schaeffer collateral stimulationinduced prolonged, self-sustaining, SE-like dis-charges exclusively in slices from experimentalrats. These data indicate that hyperthermic seizuresin the immature rat model of prolonged febrileseizures do not cause spontaneous limbic seizuresduring adulthood. However, they reduce thresh-olds to chemical convulsants in vivo and electrical
39

stimulation in vitro, indicating persistent enhance-ment of limbic excitability that may facilitate thedevelopment of epilepsy.
What are the underlying mechanisms for thisprofound and persistent enhancement of hippo-campal excitability? In the absence of overt loss ofhippocampal principal cells, functional changes inkey components of the hippocampal circuit areunder investigation. The possibility of subtle loss ofdiscrete, vulnerable neuronal populations is beingexamined; recruitment of newly born neurons andre-wiring of dentate gyrus circuitry (e.g., granulecell axonal ``sprouting'') after febrile seizures arebeing considered. In addition, modulation ofprograms of gene expression in speci®c neuronalsubtypes, that should govern their functionalproperties, are being pursued, to provide a betterunderstanding of the molecular-cellular mechan-isms by which prolonged febrile seizures in theimmature rat model may promote a seizure-pronestate.
Conclusion
Prolonged febrile seizures in the immature ratmodel modulate hippocampal excitability long-term. The molecular and electrophysiologicalmechanisms underlying this enhanced excitabilitymay be unique- and thus amenable to therapeutictargeting. The precise mechanisms and conse-quences of these seizures, and the implication ofthese new data for the human situation requirefurther investigations.
Acknowledgement
Supported by NIH NS 35439.
References
1. SHINNAR S. Febrile Seizures. In: JOHNSON RT, eds. CurrentTherapy in Neurological Disease. Philadelphia: Decker,1990; 29±32.
2. VERITY CM, GOLDING J. Risk of epilepsy after febrileconvulsions: a national cohort study. Brit Med J1991;303:1373±1376.
3. HAUSER WA. The prevalence and incidence of convulsivedisorders in children. Epilepsia 1994;35 (Suppl 2):S1±S6.
4. SLOVITER RS, PEDLEY TA. Subtle hippocampal malforma-tion: importance in febrile seizures and development ofepilepsy. Neurology 1998;50:846±849.
5. SHINNAR S. Prolonged febrile seizures and mesial temporalsclerosis. Ann Neurol 1998;43:411±412.
6. LEWIS DV. Febrile convulsions and mesial temporalsclerosis. Curr Opin Neurol 1999;12:197±201.
7. DUBE C, CHEN K, EGHBAL-AHMADI M, BRUNSON K, SOLTESZ I,BARAM TZ. Prolonged febrile seizures in the immature ratmodel enhance hippocampal excitability long-term. AnnNeurol 2000;47:336±344.
8. BARAM TZ, GERTH A, SCHULTZ L. Febrile seizures: anappropriate-aged model suitable for long-term studies.Brain Res Dev Brain Res 1997;98:265±70.
9. TOTH Z, YAN XX, HEFTOGLU S, RIBAK CE, BARAM TZ.Seizure-induced neuronal injury: vulnerability to febrileseizures in an immature rat model. J Neurosci1998;18:4285±4294.
10. CHEN K, BARAM TZ, SOLTESZ I. Febrile seizures in thedeveloping brain result in persistent modi®cation ofneuronal excitability in limbic circuits. Nat Med1999;5:888±894.
The basis for the use of antiepileptic drugs in psychiatric disorders
U. F. MaltDepartment of Psychosomatic and Behavioural Medicine, The National Hospital, University of Oslo, N-0027 Oslo, Norway.
Objectives
The aims of the present presentation are twofold.First to provide an overview of the clinical andresearch basis for the increased interest in and use ofantiepileptic drugs in the treatment of bipolardisorders. Secondly to present clinical case storiesto elucidate possible new indications for antic-onvulsant drugs, and diagnostic and therapeuticchallenges in the interface between neurology andpsychiatry.
Methods
Methods include computer based Medline,PubMed and manual journal search for relevant
studies supplemented by single case reports ofdif®cult to treat psychiatric disorders referred to atertiary University centre.
Results
Valproate and carbamazepine were suggested tobe possible options for the treatment of severeaffective disorders in 1966 and 1971 respectively.In 1978 the ®rst double-blind study occurreddemonstrating ef®cacy of carbamazepine in manicpatients and subsequently in bipolar I andschizoaffective disorders (1). During the twodecades to follow, randomised double-blind pla-cebo trials (RCT) and clinical experience suggest
40

ef®cacy of valproate and carbamazepine asprophylactic treatment for bipolar I disorders(i.e. the patient has a history of both depressiveand manic episodes). Both drugs also have anestablished position as effective treatments ofhypomanic and manic episodes Currently, valpro-ate has taken over for lithium as the mostlyprescribed drug for the prophylactic treatment ofbipolar I disorders in the United States. Ef®cacydata are less convincing regarding treatment andprophylaxis of bipolar II prophylaxis (depressiveand hypomanic episodes), however. In the pres-ence of a family history of bipolar disorder,classical clinical features and no EEG pathology®ndings, lithium still remains the best provenprophylactic drug for bipolar II disorder.
Recent studies suggest that the antimanic effectof lamotrigine is less striking compared to thatseen with valproate and carbamazepine. However,lamotrigine may be of particular value asprophylactic treatment of depressive episodes inbipolar I patients (2). Clinical experience alsosuggests that this is valid for bipolar II patients.The main problems of bipolar II patients arerecurrent depressive episodes which often areassociated with functional impairment. The diag-nosis of bipolar II disorder is often overlooked,however, mainly due to failure to identify thehypomanic episodes in their history. Accordingly,bipolar II patients often are misdiagnosed asrecurrent depression or completely overlookeddue to their episodic nature, often normal pre-episode personality and normalising attributionalstyle of the patient (e.g. ``episodic overwork'';``situational stress''. Nevertheless, bipolar II dis-orders represent the majority of bipolar spectrumpatients seen in psychiatric outpatient services andin general hospital and primary care settings aswell. No studies have compared the effect oflamotrigine with that of lithium, however.Considering the high prevalence of bipolar IIdisorders, and the relative lack of effectiveprophylactic treatment with few side effects,future studies are clearly needed to substantiatethe promising ®ndings from the ®rst RCTs.
Outside neurology, gabapentin has been used totreat chronic pain. Recent RCTs do not suggestthat gabapentin is very effective as isolatedtreatment for bipolar I disorders (3). Some anti-anxiety effects have been observed, however.Comorbid anxiety is rather common in patientswith bipolar disorders, suggesting a therapeuticrole for gabapentin and pregabalin as add-ontreatment of comorbid bipolar disorders.
No controlled studies have addressed theef®cacy of topiramateas single treatment for
psychiatric disorders, but studies do suggest thattopiramate may be of value as add-on treatmentto other drugs in dif®cult to treat manic episodes(4). In contrast to lamotrigin and gabapentin,cognitive impairment has been reported as a side-effect of topiramate. If such side effects also areseen when used as add-on treatment where thedoses commonly are lower than those used in thetreatment of epilepsy, remain to be established.Currently, there is no good evidence suggesting arole for felbamate, remacemide, vigabatin ortiagabin as therapeutic or prophylactic agents inthe mental disorders.
Own clinical experiences; the prophylactic andpossible therapeutic effects of lamotrigine ondepression in bipolar disorders, may suggest arole for this type of drugs in the treatment ofother depressive disorders, in particular rapidcycling forms of depression (ICD-10 F38.1: briefrecurrent depression). Regular antidepressants donot seem to work very well in these patients, andno effective treatment is currently availablealthough small dose neuroleptics have beenrecommended.
The remarkable antidepressant properties oflamotrigine lead us to try this drug in dif®cult totreat patients with brief recurrent depression.Prior to lamotrigine treatment the patients neitherhad responded to valproate nor carbamazepine orlithium (one patients). After four years treatmentwith lamotrigine, striking long-term ef®cacy hasbeen observed in these patients. In one caserelapse occurred after stopping the drug (due topregnancy) with completely normalisation whenthe drug was reinitiated. These observations havelead us to believe that the effect of lamotrigine inbrief recurrent depression should be explored incontrolled studies.
All responders had minor EEG dysfunction(episodic theta or theta-delta activity), however.Although neurological assessments did not sup-port the presence of a neurological disorder orepilepsy (including absence of spike-waves onEEG), the relationship between episodic thetaactivity and behavioural dysfunction is poorlyunderstood. This is an area of research whichshould be explored further in close collaborationbetween neurologist, clinical psychiatrists andneuropsychologists.
Interestingly, some recent controlled studieshave reported that gabapentin may be effectivetreatment of social phobia and generalised anxietydisorder. These ®ndings are con®rmed in studieson adult Wistar rats (5). In anxiety models,increased number of open entries and time spentin open arms have been observed in gabapentin
41

treated animals, in higher doses even with effectssimilar to that seen when the rats are treated withbenzodiazepines. Considering the minor differ-ences between gabapentin and its successor ±pregabalin ± it is likely that this antianxietyproperty also characterises pregabalin.
The ®ndings rise the question to which extentnew drugs originally developed to treat epilepsymay have a role in the treatment of chronicanxiety disorders. Chronic anxiety disorders arerather prevalent in the population, but currentlyavailable treatment options are few and of limitedef®cacy. Effects of valproate, carbamazepine andlamotrigine have been reported in open studies ofdif®cult to treat psychiatric disorders like border-line personality and post-traumatic stress disorder,but controlled studies are lacking. However, if orto which extent the therapeutic effects are relatedto the presence of EEG-dysrythmia or otherneurological soft signs are currently not settled.
Conclusion
The clinical effects of anticonvulsants observed inpatients with different types of psychiatric andbehavioural disorders without epilepsy suggestthat the term ``antiepileptic'' is too narrow andmisleading. Terms like ``neurostabiliser''may bemore appropriate. This points to a much wider
range of indications for these drugs in the future.The ®ndings also suggest that important psycho-pharmacological break through in the treatmentof major psychiatric disorders may be expectedfrom further development of ``neurostabilisers''.This assumption stresses the need for a muchcloser collaboration between neurologists andpsychiatrists in the study of main-stream psychia-tric disorders like depression and anxiety dis-orders in the future.
References
1. POST RM, DENICOFF KD, FRYE MA et al. A history of the useof anticonvulsants as mood stabilisers in the last two decadesof the 20th Century. Neuropsychobiology 1998;38:152±166.
2. CALABRESE JR, BOWDEN CL, SACHS GS et al. A double-blindplacebo-controlled study of lamotrigine monotherapy inoutpatients with bipolar I depression. J Clin Psychiatry1999;60:79±88.
3. GHAEMI SN, KATZOW JJ, DESAI SP, GOODWIN FK. Gabapentintreatment of mood disorders: a preliminary study. J ClinPsychiatry 1998;59:426±429.
4. ROY CHENGAPPA KN, RATHORE D, LEVINE J et al. Topiramateas add-on treatment for patients with bipolar mania. BipolarDisorders 1999;42±53.
5. DE-PARIS F, BUSNELLO JV, VIANNA MRM et al. The antic-onvulstant compound gabapentin possesses anxiolytic butnot amnesic effects in rats. Behaviour Pharmacol2000;11:169±173.
Drug development in the genomic era
D. A. Hosford12
1Divisions of Clinical Genetics and Biological Sciences, Glaxo Wellcome R&D, Gunnels Wood Road, Stevenage Hertfordshire SG1 2NY, United Kingdom, 2 Departments of
Medicine (Neurology) and Neurobiology, Duke University Medical Center, Durham North Carolina 27710, USA
Objectives
To compare and contrast strategies and technolo-gies of drug development during the era before andafter completion of the mapping of the humangenome.
Pre-genomic era
Drug development before the human genome hasbeen mapped (termed pre-genomic era) may becharacterised as a mixture of serendipity and ofrational drug design to alleviate disease symptoms.In the ®eld of epilepsy, phenobarbital exempli®esthe role of serendipity, having been found by chanceto have anticonvulsant properties; whereas antic-onvulsant compounds such as phenytoin, car-bamazepine and lamotrigine were developedrationally using animal models and targeted
chemical synthetic strategies (1). In the ®eld ofpsychiatry, chlorpromazine and imipramine werefound serendipitously to have antipsychotic andantidepressant properties, respectively; whereas theselective serotonin reuptake inhibitors (SSRIs) werethe result of rational antidepressant drug develop-ment (2).
Rational drug development in the pre-genomicera uses many of the strategies and technologieswhich will be employed in the genomic era. Theprocess by which drug development operates isgenerally as follows. First, biological targets of theeventual drug's intended action are chosen: forexample, type II sodium channels for an intendedantiepileptic drug; or serotonin transporters for anantidepressant compound. Next, chemical classesthat have hypothetical or precedented action at the
42

biological target are chosen, and high-throughputscreens are conducted using chemical librariescontaining hundreds of thousands of compoundsto choose a particular chemical structure (termed alead series) that best possesses desired activity at thetarget. Compounds from the lead series are furthertested using in vitro assays or animal models toensure that these compounds have biologicalactivity at the target, and pharmacokinetic andtoxicological properties of the compounds aremeasured. The optimal compound (termed leadcompound) that is both safe and ef®cacious inanimal models is then tested in human volunteers totest safety and to help determine an appropriatedose. The lead compound is then tested in arelatively small number of patients who exhibitthe disease of interest, for proof of the conceptunderlying the compound's intended action.Compounds which demonstrate proof of conceptthen undergo larger clinical trials to con®rm thatthey are ef®cacious and well-tolerated. Only aftersuccessful testing of this type may drugs besubmitted for the ®nal hurdle of drug registrationand approval for marketing. This process ofrational drug development may take up to 15years, and the successfully marketed drug may bethe sole survivor of a process that has eliminated10,000 compounds (3).
Drug development in the genomic era
The Human Genome Project (HGP) and a privateproject being undertaken by Celera have nearlycompleted their goal of a high-quality sequence ofthe human genome. HGP will also identifyapproximately 100,000 common variants, includingsingle nucleotide polymorphisms (SNPs), that canserve as genetic markers (4). The SNP Consortium(TSC) will add at least an additional 750,000 SNPsthat will be available as genetic markers (http://snp.cshl.org). Together, these efforts will deliver avast amount of information that can be used fromeither a genetics or genomics perspective to aid drugdiscovery and development. There are 4 principalways in which this new genomics information willimpact drug discovery and development. Each willbe described and illustrated separately.
First, it will become more feasible to conductrapid case-and-control association studies, usingthe high density SNP supplied by HGP and TSCto perform high-resolution scans of the completegenome. Hence diseases with unmet therapeuticneed can be screened, and any genes that areassociated with each disease population can beconsidered either as targets for drug development,or as candidates for further experimental research
to identify a tractable target (5). Depending onthe population of patients chosen as cases, thisexercise offers promise in elucidating susceptibilitygenes which alter the risk of partial epilepsies, aswell as those which alter the risk of unipolardepression. However, even if the resulting asso-ciated genes are themselves plausible targets orprovide plausible targets for drug action, it maybe dif®cult to ascertain the precise manner inwhich a drug must interact with the target to havethe desired therapeutic action in patients with thedisease. Consider patients with familial hemiplegicmigraine as an example. Although the diseasegene product has been identi®ed as the pore-forming subunit of P/Q-type voltage-dependentcalcium channels, other mutations within thissubunit can cause profoundly different phenotypeswithin patients who exhibit no migraine; andmutations of this subunit in several geneticmodels cause seizures and neither overt hemi-plegia nor migraine (6). This example of theunclear relationship between mutation and phe-notype will undoubtedly apply to many patientswith epilepsies or with psychiatric disease, andwill necessitate additional experimental workbefore therapeutic molecules can be tailored togene targets. However, this example also illus-trates that diseases which appear unrelatedphenotypically, may have similar underlyingcauses. Hence, there may be additional opportu-nities to discover and develop similar medicinesfor patients with different diseases.
A second manner in which the genomic era willenhance drug development will be through insilico data-mining, to uncover genes that can beviewed as tractable targets (5). For example, newgenes with homology to catecholamine transpor-ters will be candidates for psychiatric diseasessuch as schizophrenia, bipolar disorder, or uni-polar depression. Likewise, novel ion channelgenes may be plausible targets for certainepilepsies. Molecules which act at these targetscan then be tested in models and in man toascertain if the target is relevant to the disease.Hence, genomics information of this type willspur further discovery research and thereby maylead to new drug development.
A third way in which new genomics informa-tion will add value to drug development is via apharmacogenetics approach, to identify drug-speci®c response pro®les for drug ef®cacy andfor drug-related adverse events. This will beaccomplished as follows. SNPs spanning thegenome will be identi®ed in genotyping conductedin clinical trials or in post-marketing surveillanceof new drugs. Patients will be grouped by ef®cacy
43

and by adverse events, and SNP haplotypepro®les that are signi®cantly associated witheither drug ef®cacy or adverse events will besought. Identi®cation of SNP pro®les for ef®cacyor adverse events will permit subsequent use ofthese haplotype pro®les as predictive tools inpatients being considered for these drugs byhealth-care staff (5). For example, the SNP pro®leof a patient being considered for a new anti-psychotic compound would be compared toresponse pro®les of prior tested patients. If thispatient had a SNP pro®le suggesting lack ofef®cacy or likelihood of adverse event, then thisparticular antipsychotic drug would not beprescribed. Moreover, this also provides signi®-cant opportunities to streamline drug developmentby only including in clinical trials those patientspredicted to respond.
A fourth manner in which genomics informa-tion will impact drug development is to enablecoupling of a disease diagnostic with a therapeuticagent (5). Polymorphisms that are present indisease genes and susceptibility genes can be usedas diagnostics, not only to improve the likelihoodthat a patient harbouring a particular polymorph-ism actually has the disease in question (assuringthe diagnosis), but also to predict that a particulardrug acting through that genetic mechanisms islikely to offer therapeutic bene®t. For example,suppose that polymorphisms in a particularsusceptibility gene (termed gene XYZ456 forthis discussion) are associated with a syndromeof partial epilepsy. If a patient with newlydiagnosed seizures is genotyped and shown tohave polymorphisms in gene XYZ456, that willnot only help con®rm the diagnosis of a particularsyndrome of epilepsy, but also suggest theparticular drug which is associated with successfultreatment of that epilepsy. Moreover, the like-lihood of an ef®cacious response and lack ofadverse events can also be predicted by comparingthe patient's SNP pro®le with drug responsepro®les for that drug. (as per example three,
paragraph above). Hence, the end result will betreatment tailored to the patient based on thepatient's pharmacogenetic pro®le.
Conclusions
In the genomic era, drug development will bemore complex but more ef®cient in selecting theright targets, in bringing ef®cacious medicines topatients who are likely to bene®t, and in reducingadverse events associated with current empiricmethods of treatment. Current strategies andtechnologies of drug development, typical of ourpre-genomic era, will still be employed, but thesestrategies will be used to evaluate gene-basedtargets that are much more likely to yieldmarketed drugs than the current 10,000:1 failurerate. Moreover, in addition to facilitating phar-macogenetic approaches, the new genomics infor-mation will improve diagnostic accuracy of somediseases. These advances will offer a world inwhich the right medicine is truly tailored to theright, and rightly diagnosed, patient (5).
References
1. MCNAMARA JO. Drugs effective in the therapy of the epilepsies.In: HARDMAN JG, LIMBIRD LE, eds. Goodman and Gilman'sThe Pharmacological Basis of Therapeutics, 9th edition. NewYork: McGraw-Hill, 1996; 461±86.
2. KAPLAN HI, SADDOCK BJ. Synopsis of Psychiatry, 8th edition.Baltimore: Williams and Wilkins, 1998;932±54.
3. PAYTON M. Drug discovery and development process in thepharmaceutical industry. Presentation to Genetics ofAsthma International Network, Runymede, UK, 2000.
4. COLLINS FS, PATRINOS A, JORDAN E, CHAKRAVARTI A,GESTELAND R, WALTERS L. New goals for the U.S.Human Genome Project: 1998±2003. Science1998;282:682±9.
5. ROSES AD. Pharmacogenetics and the practice of medicine.Nature 2000;405:857±65.
6. COOPER EC and JAN LY (1999) Ion channel genes and humanneurological disease: recent progress, prospects, and chal-lenges. PNAS 96:4759±4766.
Adaptive electric ®eld control of epileptic seizures
B. J. Gluckman12, H. Nguyen1, S. J. Schiff13
1Krasnow Institute for Advanced Studies, 2Department of Physics and Astronomy, 3Department of Psychology. Mail Stop 2A1, George Mason University, Fairfax, VA, 22030, USA
Objectives
Epilepsy is a dynamical disease ± symptoms areproduced by aberrant dynamics of neuronal net-works. Although numerous attempts have beenmade to suppress epileptic seizures in human
patients by stimulating sites remote from theepileptic focus, the ef®cacy of doing this withouttaking the dynamics of the seizures into accounthave been unimpressive.
44

Surprisingly, there has been little effort focusedon the direct dynamical control of seizures. Recentexperiments have explored electrical current injec-tion directly into epileptiform networks to achievecontrol using periodic pacing, nonlinear controlschemes, and linear feedback. However, thesemethods require insertion of electrodes into theparenchyma of the brain.
Since the experiments of Rushton in 1927, it hasbeen recognized that electrical ®elds in¯uence thethreshold of excitability of neurons in an orienta-tion speci®c manner. An electric ®eld orientedparallel to the somatic-dendritic axis of neuronswhose spike initiation zone is asymmetrically placedwith respect to the centroid of the neurons isoptimal for modulating neuronal ®ring.
Recently DC electric ®elds applied external tobrain tissue were shown to suppress spontaneousepileptiform activity (1). However, neuronal adap-tation as well as electrode and tissue polarizationrender the effects of DC ®elds transient.
We here report the ®rst evidence that adaptiveelectric ®eld control seizures of seizures is feasible.Transient adaptation appears to be eliminated withthis approach. Although our experiments utilizeepileptiform activity as the dynamics that wemodulate, applications of our methodology arenot limited to seizures.
Methods
Longitudinally or transversely cut hippocampalslices were placed in the ®eld produced by parallelAg-AgCl electrode plates. The neural layers of theslice are oriented with respect to the mapped ®eld.Elevated [K+] in the perfusate leads to seizure-likeevents. An adaptive feedback control algorithmcontinually calculates the strength of the required®eld based on measured neuronal signal powerwithin frequency bands characteristic of seizures.
Results
We replicated sustained seizure suppression duringcontrol in 10 separate experiments with slices from10 different rats. In the CA1, there is a characteristicDC depolarization of the tissue associated withthese seizure-like events similar to In Vivo seizures.This DC shift was completely eliminated duringsuppression for some slices, while it was partiallyretained for others. In both cases the high frequencyactivity, towards which the suppression algorithm is
directed, is signi®cantly attenuated. Control canoften be maintained for arbitrary periods of time,and we have maintained control for as long as 6minutes in slices exhibiting seizure events every5±100 seconds. We have not identi®ed a time periodbeyond which this control system loses its ability tosuppress seizure dynamics in this network. Uponsudden release of control, an immediate andexcessively large seizure is often generated.
Suppression is achieved by using negative feed-back. By reversing the gain on the algorithm toprovide positive feedback, which reverses theapplied ®eld polarity, seizures can be eitherenhanced or even created where no seizures wereobserved beforehand. With positive feedback con-trol, the adaptively applied ®eld can enhance briefnetwork bursts with small positive DC potentialshifts, into large seizure-like events with thesubstantial DC negative shifts characteristic ofseizures.
Non-adaptive DC ®elds of comparable amplitudeare not similarly effective in suppressing seizures.
Conclusion
These results demonstrate that a novel approach toadaptive seizure control using externally appliedelectric ®elds is feasible. By employing ®elds it is notnecessary to impale brain tissue with stimulatingelectrodes, and this has obvious advantages forminimizing the invasiveness of devices employingsuch technology.
On the other hand, by using positive feedback,the methodology presented here forms a novelapproach to imposing function prosthetically. Bysensing the ongoing background activity of neur-onal circuitry, adaptive electric ®elds may permitthe modulation of network behavior in a morephysiological manner than previously possible.
A full length manuscript detailing these resultswill appear as: Gluckman BJ, Nguyen H, Schiff SJ.Adaptive electric ®eld suppression of epilepticseizures. J Neurosci, in press, 2001.
Acknowledgement
Support: NIH 7K02MH01493, 2R01MH50006.
Reference
1. GLUCKMAN BJ, NEEL EJ, NETOFF TI, DITTO WL, SPANO ML,SCHIFF SJ. Electric ®eld suppression of epileptiform activity inhippocampal slices. J Neurophysiol 1996;76:4202±4205.
45

Cell grafts in epilepsy: therapeutic prospects and problems
B. S. Meldrum1, A. G. Chapman1, E. Tang2, K. Keaney1, S. Patel2, A. Chadwick2, H. Hodges3
Departments of 1Neurology, 3Psychology, and 2Reneuron Ltd., Institute of Psychiatry, Kings College London, Denmark Hill, London SE5 8AF, UK
Objectives
To evaluate the potential therapeutic use ofconditionally immortalised neuroepithelial stemcells in rodent models of epilepsy.
Introduction
Previous attempts to use brain cell grafts as atherapeutic approach have used embryo-derivedcells that have (partially) differentiated into eitherGABAergic or monoaminergic neurons (1±3).These have produced limited bene®t when injectedinto epileptic foci or nodal control points (4) inrodent models of epilepsy. The clearest effects tendto be seen where the graft is correcting a previouslyestablished de®cit (5). Neuroepithelial stem cellsretain the capacity to differentiate into neurons orglia. A conditionally immortalised clonal line frommice, MHP36, has been shown to produce improve-ment in a range of behavioural tests in rats withvarious neurodegenerative syndromes includinghippocampal CA1 cell loss following global ischae-mia (6), focal cortical and striatal degenerationfollowing middle cerebral artery occlusion (7), andcortical cholinergic insuf®ciency (8). We havechosen to test this cell line in three rodent modelsof epilepsy, 1) sound-induced generalised motorseizures in genetically-epilepsy prone rats(GEPrats), 2) amygdala-kindled limbic seizuresand 3) spontaneous limbic seizures followingpilocarpine-induced status epilepticus.
Methods
Rat models of chronic epilepsy:(i) GEP rats were bred at the Institute of
Psychiatry and tested by exposure to mixed tonesat 96dB for 60 sec. Those showing a maximalseizure response (stage 9) were selected. Age 10±12weeks the GEPrats were implanted bilaterally in thecentral nucleus of the inferior colliculus either withstem cells or stem cell medium.(®rst series) orMHP36 cells, ®broblasts or medium (second series).Animals were tested for sound-induced seizuresover the following 12 weeks (®rst series) or 23 weeks(second series).
(ii) Electrical kindling of the amygdala wasperformed in male Wistar rats (weighing200±220 g, at start). A left basolateral amygdalastimulating electrode, hippocampal guide cannulae,and cortical recording electrodes were implanted
under general anaesthesia. Kindling stimulation wasinitiated 5-7 days later and continued daily until 3successive ``stage 5'' Racine responses were elicited.Rats subsequently received into each dorsal hippo-campus MHP36 cells, MHP 15 cells, ®broblasts orculture medium. In each group after-dischargeduration, seizure duration and clinical seizure stagewere assessed each week 8±16 weeks post implant.
(iii) Male Wistar rats received pilocarpine380 mg/kg i.p. and status epilepticus was moni-tored; after 3h of continuous status it wasterminated by diazepam (10mg/kg i.p.) and pheno-barbital (25mg/kg i.p.). Video monitoring (12h/day,7 days/week) for spontaneous limbic seizures wasinitiated after 100 days. After 3 weeks of baselinevideo monitoring, rats received bilateral implants ofMHP36 cells or cell-free medium at 4 sites extendingfrom anterior to posterior of the piriform cortex.Following an interval of 9 weeks from implantationsurgery, video-monitoring was resumed for a threeweek period.
Brains were perfused with 4% paraformaldehyde,frozen and serially sectioned at 50 mm and stainedwith cresyl fast violet to visualise lesions and b-galactosidase antibodies for MHP36 cells.
All experiments were carried out in accordancewith the UK Scienti®c Procedures Act,1986 and theEthical Review Process of the Institute ofPsychiatry.
Results
(i) Sound-induced seizures in GEP rats
A macrophage response to the implantation wasprominent within the inferior colliculus at the site ofMHP36 cell implantation. b-galactosidase positivecells were visible around the needle tract directlyabove the inferior colliculus. The mean latency totonic clonic seizures was prolonged 8 and 12 weeksafter MHP36 cell implantation in the ®rst series. Inboth series a proportion of rats implanted withMHP36 cells showed reduction in the seizure scorefor sound-induced seizures. Seizure response ceasedor was reduced in 10 out of 27 MHP36 grafted ratsand in 3 out of 23 control rats, a marginallysigni®cant difference.
(ii) Limbic seizures in amygdala-kindled rats
No differences were found between the 4 groupsgrafted with MHP36, MHP15, ®broblasts or
46

medium in terms of current threshold for inducing afully-kindled seizure, after-discharge or seizureduration and Racine seizure score.
(iii) Limbic seizures in rats subject to pilocarpine-induced status epilepticus ± Mean seizurefrequency was identical in MHP36 and controlrats during the 3 weeks before implantation (1.76and 1.59 seizures/day). Monitoring 9-12 weeksafter implantation showed that seizure frequencywas increased in both groups:- MHP36 cell groupto 6.44 seizures/day and control rats to 3.00seizures/day. Statistical analysis revealed that theseizure frequency was signi®cantly increased post-implantation in the MHP36 group (P<0.01).
Discussion
We chose 3 rodent models to provide informationabout two potential clinical targets, drug refractorycomplex partial seizures (which are commonlytreated with anterior temporal lobe resection) andprimary generalised seizures, where evidence for ananatomical focus is often absent. Hippocampalsclerosis is commonly seen in the ®rst syndrome, butproof that the syndrome is dependent on theselective loss of a particular cell-type is missing.There is massive cell loss in the piriform cortexfollowing pilocarpine-induced status epilepticus,but little evidence for cell loss in the other twosyndromes.
The sites chosen for stem cell implants wererelated to our understanding of epileptogenesis inthese models. We also chose to perform the implantonly when the seizure pattern is fully established i.e.an effect on the process of epileptogenesis has notbeen studied.
No convincing evidence for long-term improve-ment in seizure responses following MHP36 cellimplantation has been found. The slight improve-ments found in rats with sound-induced seizuresmay be related to local damage within the inferiorcolliculus, despite our attempt to control for thisusing implants of ®broblasts. An increase in seizurefrequency following control or MHP36 cellimplantation in status epilepticus rats is probablyattributable to cortical damage produced by the 8
injections into tissue primed for seizure activity.Possibilities that neuroepithelial stem cells fromother brain regions, or grafting into different sitesmight be more effective remain to be tested.
Summary/Conclusions
A conditionally-immortalised neuroepithelial stemcell line which has been shown to produce markedfunctional improvement in rats with ischaemic andother lesions has been implanted in the inferiorcolliculus in Genetically-epilepsy prone rats, in thehippocampus in amygdala-kindled rats and in thepiriform cortex in rats with limbic seizures followingstatus epilepticus. Grafted cells showed poorsurvival in the inferior colliculus but did grow inthe hippocampus and piriform cortex. We ®nd noconvincing evidence of seizure reduction that can beattributed to the MHP36 cells.
References
1. FINE A, MELDRUM BS, PATEL S. Modulation of experimentallyinduced epilepsy by intracerebral grafts of fetal GABAergicneurons. Neuropsychologia 1990;28:627±634.
2. HOLMES G, THOMPSON JT, HUH K, STUART JD, CARL GF.Effects of neural transplantation on seizures in the immatureGenetically Epilepsy Prone rat. Exp Neurol 1992;116:52±63.
3. BENGZON J, KOKAIA Z, LINDVALL O. Speci®c functions ofgrafted locus coeruleus neurons in the kindling model ofepilepsy. Exp Neurol 1993;122:143±154.
4. LOÈ SCHER W, EBERT U, LEHMANN H, ROSENTHAL C, NIKKHAH G.1998. Seizure suppression in kindling epilepsy by grafts offetal GABAergic neurons in rat substantia nigra. J NeurosciRes 1998;51:196±209.
5. CLOUGH R, STATNICK M, MARING-SMITH M et al. Fetal raphetransplants reduce seizure severity in serotonin-depletedGEPRs. NeuroReport 1996;8:341±346.
6. SINDEN JD, RASHID-DOUBELL F, KERSHAW TR et al. Recoveryof spatial learning by grafts of a conditionally immortalizedhippocampal neuroepithelial cell line into the ischaemia-lesioned hippocampus. Neurosci 1997;81:599±608.
7. VEIZOVIC T, STROEMER P, BEECH J, SOWINSKI P, HODGES H.Stem cell grafts resolve sensory dysfunction followingMCAO in the rat. J Cereb Blood Flow Metab 1999;19(Suppl.1):616.
8. GRAY JA, GRIGORYAN G, VIRLEY D, PATEL S, SINDEN JD,HODGES H. Conditionally immortalized, multipotential andmultifunctional neural stem cell lines an approach to clinicaltransplantation. Cell Transplantation 2000;9:153±168.
47

The impact of the Human Genome Project and new genetic strategies onthe diagnosis and treatment of epilepsy and psychiatric disease
A. Wynshaw-BorisUCSD School of Medicine, La Jolla, CA, USA 92093-0627
It is quite clear that the revolution in geneticanalysis of disease will continue to have atremendous impact on the understanding of thefunction of the brain. The imminent completion ofthe total sequence of the human genome, computeralgorithms to identify attractive candidate genes foranalysis, and powerful strategies for mappingcomplex traits will allow for the rapid identi®cationof chromosomal regions and candidate genesresponsible for neurologic diseases. The naturalvariation of these genes will be determined in largepopulations, which will allow for the rapid identi-®cation of variations associated with disease.
Genetic animal models, particularly geneticallymanipulated mice, can be used to determine the invivo function of these genes in terms of phenotypeas well as by expression pro®ling using cDNA andprotein chip arrays. Such studies will provideopportunities for the functional classi®cation ofneurologic diseases. It is anticipated that thesestudies will lead to more accurate diagnoses andprognoses, and will provide novel targets forpotential therapies for devastating neurologic dis-eases, including epilepsy and psychiatric disorders.Examples of the uses of these strategies andtechnologies will be discussed.
48

POSTER ABSTRACTS
1.
Serum magnesium is reduced in women with lifetime depressive episodes andis associated with cognitive impairment
E. Albertsen Malt1, S. Olafsson2
1Dept. of Psychiatry, Haukeland University Hospital, N-5022 Bergen, Norway, 2Dept. of Internal medicine, Haukeland University Hospital, N-5022 Bergen, Norway
Objectives
To explore magnesium (Mg) as trait marker andexplore the association between Mg and cognitiveimpairment in women with mood disorders andcontrols.
Methods
Morning serum Mg was measured in 31 femalepatients with past or current depressive episodes(ICD-10 diagnoses F32 or F33) and 55 femalecontrols. The Schedeules for Clinical Assessment inNeuropsychiatry (SCAN) was used for diagnosticassessment. Cognitive impairment was assessed bythe computerized Gottschalk-Gleser content analy-sis method.
Results
Mg was signi®cantly lower in patients with lifetimemood disorder than in controls (P=0.036). Nodifference in Mg was observed according to currentdepression (n=12) or remission (n= 19).
Mg correlated signi®cantly with cognitive impair-ment in the patient group (0=0.48, P=0.013) andexplained 20% of variance in cognitive impairmentscores. No such association was seen in controls.
Comments
Low Mg may be a trait marker in mood disorders,and Mg and other medications showing N-methyl-D-aspartate (NMDA)-antagonistic and gamma-aminobutyric (GABA)A-agonistic effects may beused therapeutically. Low Mg is associated withdecreased oxidative stress tolerance, increased focalin¯ammation, alterations in light sensitivity andcircadian rythms, neurogenic pain, migraine andincreased liability to bone fractures, and mayunderlie the associations seen between depressionand cardiovascular disorders, hip fractures, painand migraine. Increased NMDA-receptor antagon-ism by Mg may reduce long-term potentiation(LPT) in hippocampus and might lead to increase incognitive impairment in susceptible individuals withmood disorders.
2.
A novel postsynaptic density protein: the monocarboxylate transporter MCT2is colocalized with glutamate receptors in postsynaptic densities of parallel®ber-Purkinje cell synapses
L. Bergersen1,4, O. Wñrhaug1,4, J. Helm1, M. Thomas1, P. Laake3, A. P. Halestrap2, O. P. Ottersen1
1Department of Anatomy, Institute of Basic Medical Sciences, University of Oslo, POB 1105 Blindern, N-0317 Oslo, Norway, 2Department of Biochemistry, School of Medical
Sciences, University of Bristol, Bristol BS8 1TD, United Kingdom, 3Section of Medical Statistics, University of Oslo, POB 1122 Blindern, N-0317 Oslo, Norway, 4The Norwegian
University of Sport and Physical Education, POB 4014 UllevaÊl Hageby, 0806 N-Oslo, Norway
Confocal immuno¯uorescence microscopy showedstrong MCT2 labeling of Purkinje cell bodies andpunctate labeling in the molecular layer. Byimmunogold cytochemistry it could be demon-strated that the MCT2 immunosignal was concen-trated at postsynaptic densities of parallel ®ber-
Purkinje cell synapses. The distribution of MCT2transporters within the individual postsynapticdensities mimicked that of the d2 glutamatereceptor, as shown by use of two different goldparticle sizes. The MCT2 distribution was alsocompared with the distributions of other mono-
49

carboxylate carriers (MCT1 and MCT4). TheMCT1 immunolabeling was localized in the endo-thelial cells while MCT4 immunogold particles wereassociated with glial pro®les, including thoseabutting the synaptic cleft of the parallel®ber- spine synapses. The postsynaptic density(PSD) molecules identi®ed so far can be dividedin ®ve classes: receptors, their anchoring molecules,molecules involved in signal transduction, ionchannels, and attachment proteins. Here we provide
evidence that this list of molecules must now beextended to comprise an organic molecule trans-porter: the monocarboxylate transporter MCT2.The present data suggest that MCT2 has speci®ctransport functions related to the synaptic cleft andthat this transporter may allow an in¯ux of lactatederived from perisynaptic glial processes. Theexpression of MCT2 in synaptic membranes mayallow energy supply to be tuned to the excitatorydrive.
3.
Redistribution of neuroactive amino acids in rat hippocampus and striatumduring hypoglycemia: a quantitative immunogold study
V. Gundersen1, F. Fonnum2, O. P. Ottersen1, J. Storm-Mathisen1
1Anatomical Institute, University of Oslo, P.O.Box 1105 Blindern, N-0317 Oslo, Norway, 2Norwegian Research Establishment, Division for Environmental Toxicology, Kjeller,
Norway
Objectives
We wanted to investigate the redistribution ofneuroactive amino acids in the brain duringhypoglycemia.
Methods
Rats were given insulin or saline intraperitoneallybefore perfusion ®xation with glutaraldehyde/for-maldehyde. Aspartate, glutamate, GABA andglutamine were localized in hippocampus andstriatum by light and electron microscopic immuno-cytochemistry using antibodies speci®cally recog-nizing the aldehyde ®xed amino acids.
Results
In hippocampus light microscopy showed thatduring hypoglycemia, aspartate-like immuno-reactivity became more intense, particularly in theterminal ®elds of excitatory afferents and in GABAcontaining neuronal cell bodies. In both hippocam-pus and striatum quantitative electron microscopydemonstrated a strong hypoglycemia inducedincrease in the density of gold particles signallingaspartate in excitatory and inhibitory nerve term-inals and myelinated axons. Aspartate was co-localized with glutamate in excitatory type nerveterminals (i.e., forming asymmetric synapses on
spines) and with GABA in inhibitory type terminals(i.e., forming symmetric synapses on cell bodies).The densities of glutamate-like and glutamine-likeimmunoreactivities were markedly reduced in alltissue elements, the reductions were less in excita-tory terminals than in other parts of the excitatoryneurons (i.e., hippocampal pyramidal and granulecells). In glial cells the density of glutamineimmunogold particles showed only slight attenua-tion. The level of GABA immunogold particles didnot change signi®cantly in any tissue pro®les duringhypoglycemia.
Conclusion
The results suggest that when the brain is short ofglucose for energy production, glutamate andglutamine are utilized as energy substrates.Excitatory terminals have the capacity to keeprelatively high levels of these amino acids, probablydue to replenishment by glia-derived glutamine.Under hypoglycemic conditions both excitatory andinhibitory terminals build up a higher level ofaspartate, which may be released from these nerveendings. This phenomenon may be related to thepattern of neuronal death characteristic of hypo-glycemia.
50

4.
Genetic association studies of idiopathic generalized epilepsy
L. Nashef1,2, J. Blower1, B. A. Chioza3, H. J. Goodwin3, D. McCormick1, P. Asherson4, A. J. Makoff3
1Department of Neurology, Kent & Canterbury Hospital, Canterbury CT1 3NG, 2King's College Hospital, Denmark Hill, London SE5 9RS, 3Department of Psychological Medicine,
Institute of Psychiatry, Denmark Hill, London SE5 8AF, 4Social Genetic and Developmental Psychiatry Research Centre, Institute of Psychiatry, Denmark Hill, London SE5 8AF
Familial segregation in common forms of idio-pathic generalized epilepsy (IGE) is complex.These disorders are therefore less amenable totraditional linkage studies, which have successfullyidenti®ed disease-causing mutations within ionchannel genes in human Mendelian idiopathicepilepsies, and in animal models. Susceptibilitygenes in idiopathic generalized epilepsy (IGE)may be shared between subtypes and/or besubtype speci®c. We are performing associationstudies of common IGEs, focusing initially onshared susceptibility genes. We have so farcollected DNA from a) 230 unselected IGEprobands with generalized spike wave on EEGand a compatible clinical presentation; b) age andsex matched controls, and c) available parents.
Negative associations to date include the a4nicotinic acetylcholine receptor subunit and meta-
botropic glutamate receptors 7 and 8. Positiveassociations with the unselected IGE samplepresented so far include a) two markers in thea1a calcium channel subunit gene, one of which iscon®rmed by family studies and b) a positiveassociation with two markers at 15q13-q14 (whichincludes the a7 nicotinic acetylcholine receptorsubunit gene), although within family analysissuggests a possible population artifact with onemarker. Preliminary results also appear to con®rman association with the human m-opioid receptorpreviously reported in idiopathic absence epilepsypatients.
Our experience supports the use of associationstudies in the investigation of the genetics ofidiopathic epilepsies.
5.
Molecular dating of senile plaques in the brains of individuals withDown syndrome.
R. Torp1, B. Y. Azizeh2, E. Head2, C. Cotman2
1Dept. of Anatomy, Inst. for Basic Medical Sciences, University of Oslo, Norway., 2Inst. For Brain Aging and Dementia, University of California at Irvine, USA
The brains of individuals with Down syndrome(DS) are characterized by the development of allthe neuropathological hallmarks of Alzheimerksdisease (AD) by the age of 40 years. An early andpredominant pathological feature is the formationof senile plaques in several cortical and subcor-tical brain regions. b-Amyloid (Ab) is a constitu-ent of senile plaques found with increasing age inindividuals with DS. In an attempt to search for amolecular means of determining the initial corticalsites of Ab deposition in DS we used an af®nity-puri®ed polyclonal antibody recognizing race-mized, chronobiologically older, senile plaques.The immunostaining characteristics of the anti-body (d7C16) indicate that it is present in allplaque subtypes. The youngest DS case exhibit
weak positive staining but the extent of immuno-labelling increased with age. In addition, chron-obiologially older plaques were initially found inclusters in the outer layers of the frontal andenthorinal cortex but, as the disease progressed,d7C16-positive plaques spread into deeper corticallayers, suggesting a progression of Ab-plaquesfrom the super®cial to the deeper layers.Morphologically the racemized ®brils consists ofmore highly interwoven ®brils than non-racemizedAb. However, the use of antibodies to modi®edforms of the Ab protein leads to the interestingconcept of actually ``dating'' senile plaques andreaching some conclusion about when the diseasestarts in DS, which would then have implicationsfor disease onset in AD.
51

6.
Effects of long-term lamotrigine and valproate treatment on brain aminoacid levels and enzyme activities in male Wistar rats
B. Hassel1, E. Taubùll2, L. Gjerstad2, L. S. Rùste2
1Norwegian Defence Research Establishment, Kjeller, Norway, 2Dept. of Neurology, Rikshospitalet, University of Oslo, Norway
Background/methods
The mechanism of action of valproate and lamo-trigine has previously been studied in acute experi-ments or in vitro. We treated Wistar rats for 90 dayswith lamotrigine, 5mg/kg, or valproate, 200mg/kgor 400mg/kg, twice daily, and we compared theeffects with those of acute valproate treatment,400mg/kg or 800mg/kg for 1 hour, on amino acidlevels and enzyme activities in rat brain.
Results
Chronic lamotrigine treatment selectively elevatedhippocampal GABA levels (25%) and increased themaximal activities of enzymes of the GABA shunt.Taurine levels increased by 16±27% in hippocam-pus, frontal and parietal cortices, but not incerebellum. The levels of glutamate and aspartateas well as the activity of hexokinase and a-ketoglutarate dehydrogenase remained at controllevels. Serum lamotrigine 4 hours after last dose was41.7t1.5 mM (meantSEM).
Chronic valproate treatment led to a 17% decreasein GABA in frontal cortex, which paralleled adecrease in a-ketoglutarate dehydrogenase activityand glutamate levels. No changes in GABA were seen
in hippocampus, cerebellum or parietal cortex, buthippocampal taurine increased by 12%. Interest-ingly, the activity of glutaminase, an enzyme involvedin transmitter glutamate formation, was reduced25±40% in hippocampus. Serum valproate 4 hoursafter last dose was 143t39 mM and 355t66 mM, forthe two doses, respectively.
Acute valproate treatment led to a 24% increasein hippocampal GABA levels, as reported earlier byseveral groups. Both chronic and acute valproatetreatment reduced the levels of aspartate in the threeforebrain areas, but not in cerebellum.
Conclusion
Chronic lamotrigine treatment increases hippocam-pal GABA levels and GABA shunt activity; this®nding suggests increased GABAergic activityduring chronic lamotrigine treatment. Long-termvalproate treatment, on the other hand, does notappear to increase cerebral GABA levels, althoughacute treatment does. The reduction in hippocam-pal glutaminase activity may suggest a down-regulation of hippocampal glutamatergic neuro-transmission during chronic valproate treatment.
52