Embed Size (px)
Citation preview

Master Bioinformatica y Biologıa Computacional
Manejo, visualizacion y calculostopologicos en redes biologicas
Daniel Lopez y David Ochoa
Modulo de Redes y Biologıa de Sistemas

Indice general
1. Introduccion 4Redes Biologicas a estudiar . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2. Manejo y Visualizacion de Redes en Cytoscape 6Importando redes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
Importar red galFitered . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6Importar atributos de la red . . . . . . . . . . . . . . . . . . . . . . . . . 7
Visualizacion de las redes y Analisis de expresion . . . . . . . . . . . . . . . . 10Layouts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10Visualizando Datos de Expresion (VizMapper) . . . . . . . . . . . . . . . 11Filtrado . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14Interpretar la Red . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
Analisis funcional . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16Generar clusters de coexpresion . . . . . . . . . . . . . . . . . . . . . . . 16
Enriquecimiento de terminos GO . . . . . . . . . . . . . . . . . . . . . . . . . 18
3. Analisis topologico en R 20Cargando distintos tipos de redes . . . . . . . . . . . . . . . . . . . . . . . . . 20Parametros topologicos mas generales . . . . . . . . . . . . . . . . . . . . . . . 21
Caminos mınimos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21Diametro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22Componente conexo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
Cualidades de las Redes Biologicas . . . . . . . . . . . . . . . . . . . . . . . . 25Coeficiente de clustering . . . . . . . . . . . . . . . . . . . . . . . . . . . 25Redes de mundo pequeno (Small-World Networks) . . . . . . . . . . . . 26Redes libres de escala (Scale-free Networks) . . . . . . . . . . . . . . . . 28
4. Conclusiones 33Agradecimientos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
Apendices 33Instalacion de Cytoscape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
Ejecutar Cytoscape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35Instalar plugins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
Instalacion de igraph en R . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37Navegando por Cytoscape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
Interface Cytoscape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
2

Menus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38Plugins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
3

Capıtulo 1
Introduccion
El objetivo de este tutorial es familiarizarse con el manejo y visualizacion de redesbiologicas ası como aprender a calcular algunos de sus parametros topologicos mas rele-vantes. Aunque existen multitud de herramientas para trabajar con redes, en esta practicasolo se utilizaran Cytoscape y R por motivos didacticos. El fin no es crear un manual dedichos programas, ni ahondar entre los cientos de opciones y plug-ins que existen, sino masbien hacerse con una vision general del analisis de redes y sobre todo en las posibilidadesque dichos analisis ofrecen.
Es importante recordar que para la realizacion de la practica es imprescindible tenerpreviamente instalado Cytoscape con todos los plug-ins que utilizaremos (Instalacion deCytoscape) ası como R con la librerıa igraph (Instalacion de igraph en R).
La practica esta encuadrada dentro del modulo de Redes Biologicas y Biologıa deSistemas y pretende servir ademas como complemento a la clase de Teorıa de Grafosvista previamente ası como a las clases venideras en las que se trataran algunas de lasredes biologicas mas relevantes en la actualidad.
Redes Biologicas a estudiar
En esta practica vamos a utilizar dos redes biologicas. Por un lado, utilizaremos losdatos procedentes del siguiente artıculo publicado en Science en 2001:
Ideker et al. Integrated genomic and proteomic analyses of a systematically per-turbed metabolic network. Science (2001) vol. 292 (5518) pp. 929-34
Es importante entender el articulo del que proceden los datos. A grandes rasgos, Idekeret al. llevan a cabo un estudio sistemico en levadura de la respuesta del metabolismo dela galactosa a distintas perturbaciones como delecion o sobrexpresion de genes, o cambiosde temperatura o de las condiciones del medio. Ante tales perturbaciones, cuantificaronel mRNA y la expresion de proteınas a escala genomica y los integraron con redes deinteraccion entre proteınas y proteına-DNA disponibles en aquel momento.
4

Asimismo, en otra parte de la practica trabajaremos con la red de interacciones fısicasentre proteınas de Saccharomyces cerevisiae, obtenida desde numerosas fuentes y que sepublico en:
Han et al. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature (2004) vol. 430 (6995) pp. 88-93
No es necesario descargar las redes del material suplementario del articulo ya que losdatos se encuentran accesibles dentro del directorio Cytoscape_v2.8.0/sampleData dela carpeta de Cytoscape.
5

Capıtulo 2
Manejo y Visualizacion de Redes enCytoscape
Importando redes
Para empezar a trabajar con Cytoscape lo primero que necesitamos es crear o cargaruna red. Cytoscape reconoce numerosos formatos de archivos (.sif, .nnf, .gml, .xls, . . . ),permite la importacion desde un archivo tabular (.txt, .xls) y ofrece la posibilidad decargar una red a traves de un webservice. Si nos interesa, tambien podemos crear omodificar nuestra propia red, anadiendo los nodos y los enlaces pertinentes. Por ultimo,Cytoscape tiene la posibilidad de cargar una sesion previamente guardada (.cys). Lasesion, ademas de guardar la red, tambien guarda los atributos modificados (color, forma,layout,. . . ) ası como la posicion de las ventanas y algunas otras preferencias.
Importar red galFitered
Uno de los archivos con los que trabajaremos en la practica es galFiltered.sif quese encuentra en el directorio sampleData de la carpeta de instalacion de Cytoscape (verseccion Redes Biologicas a estudiar). Antes de cargarlo conviene echarle un vistazo conun editor de texto. El formato SIF (Simple Interaction File) es el mas sencillo. Constaunicamente de 3 columnas que representan el nodo de origen, el tipo de interaccion yel nodo de destino en este orden. Generalmente, mientras los nodos suelen ser identifi-cadores de genes o proteınas, los tipos de interaccion suelen hacer alusion a la relacionque mantienen. Para importar esta red seguiremos los pasos siguientes:
Ir al menu File → Import → Network (multiple file types).
Seleccionar la opcion Local y darle a Select.
Abrir el directorio sampleData que se encuentra dentro de la carpeta de instalacionde Cytoscape
Seleccionar galFiltered.sif y hacer click primero en Open y luego en Import.
6

Tras esto veras algo como la Figura 2.1.
Figura 2.1: Importacion correcta de la red galFiltered.sif
Importar atributos de la red
En ocasiones nos puede interesar anadir informacion complementaria de los nodoscomo anotacion funcional, ubicacion celular, etc. En esta ocasion anadiremos datos deexpresion genica. Para ello:
Ve al menu File → Import → Attribute from Table (Text/MS Excel)
En nuestro caso queremos importar informacion de los nodos por lo que deberemosasegurarnos de que este seleccionado el boton Node de la seccion Data Sources
Haz click en Select File(s) y selecciona el fichero galExpData.csv
En la seccion Advanced haz click en Show Text File Import Options y seleccionala coma como separador de campos
Haz click tambien en Transfer first line attribute names de la seccion AttributeNames
Finalmente selecciona la opcion Import Everything (Key is always ID) paraasegurarse que se importan todas las columnas y no solo aquellas que coinciden conalguno de los parametros que ya tienes en la red
7

Figura 2.2: Import Annotation File
Si todo ha ido bien deberıas ver algo similar a la Figura 2.2. Para terminar haz clicken Import .
Detente un momento y trata de observar lo que estamos haciendo. Hemos importadoun fichero con datos de expresion para los distintos genes que tenemos en la red obtenidosa partir de los experimentos de Ideker et al. que ya vimos en el apartado Redes Biologicasa estudiar. En este fichero (galExpData.csv) tenemos 8 columnas. La primera (GENE )hace referencia al identificador del gen que estamos anotando. La segunda COMMON serefiere al nombre comun que recibe dicho gen. Esta es una informacion anadida que noestaba en nuestra red original por lo que es un ejemplo de como cambiar los identificadoresde los nodos anadiendo atributos a las redes. Las 6 columnas siguientes, representan3 condiciones experimentales diferentes (gal1R, gal4R, gal80R), en cada una de ellasreprimiendo la expresion de un gen en particular gal1, gal4 o gal80 respectivamente.Para cada una de las condiciones tenemos ademas 2 columnas: una que hace referenciaal propio valor de expresion (exp) y otra en la que se especifica el valor de significancia(sig) o p-value del mismo.
En la parte inferior de Cytoscape se encuentra el panel de datos. Dicho panel es utilpara mostrar los atributos de los nodos y enlaces que el propio usuario selecciona sobrela red. Para ver los nuevos atributos que has importado, selecciona un nodo de la red yhaz click en el boton Select All Attributes. De esta manera, podremos ver detalladamente,todos los atributos que hayamos importado o incluso calculado en el propio Cytoscape.
8

Problema
Ahora que hemos importado los atributos de la red, tenemos todos los datos deexpresion de los nodos en nuestro Data Panel. ¿Serıas capaz de encontrar aquellosgenes que estan diferencialmente expresados cuando reprimimos el gen gal80 usandoel Data Panel?¿Alguno de estos genes puede estar relacionado con el metabolismo dela galactosa?
9

Visualizacion de las redes y Analisis de expresion
El hecho de visualizar una red de una manera u otra puede parecer un asunto trivial,pero nada mas lejos de la realidad. En muchas ocasiones, la representacion de la rednos puede ayudar a observar propiedades o extraer conclusiones que quiza de otro modonunca hubieramos apreciado. Hagamos una pequena demostracion:
Selecciona en el menu Layout → Cytoscape Layouts → Force-Directed Lay-out.
A continuacion en el tab VizMapper selecciona la opcion Sample1 en la cajaCurrent Visual Style.
Tras aplicar este estilo, podemos ver como, ademas de aplicar distintos cambios no in-formativos como el color de los nodos, tambien hemos alterado la etiqueta, el color yel tipo de las conexiones en funcion de si la interaccion es de tipo proteına-proteına oproteına-DNA. Como ya hablamos en la seccion Redes Biologicas a estudiar, Ideker etal. emplearon ambos tipos de evidencias en la reconstruccion de la red por lo que ahorapodemos visualizar de una manera clara y sencilla los dos tipos de interacciones. Aho-ra comprenderas que una visualizacion apropiada ayuda mucho a la comprension de lasredes. Veamos como podemos personalizar la visualizacion a nuestro gusto.
Layouts
La mejor forma de entender los Layouts es probandolos. Ası que aplica unos cuantosdel menu Layout. Algunos de ellos son pesados (weighted) en funcion de algun atributoy otros en cambio, simplemente ordenan la red en funcion de la topologıa. Otra opcioninteresante es que puedes aplicar un layout solo a los nodos que tienes seleccionados.Esta opcion es muy practica especialmente en determinados redes con topologıas muyparticulares.
Una vez aplicado un layout, es frecuente que te interese mover algunos nodos para quela visualizacion quede a tu gusto. Obviamente, puedes mover los nodos uno a uno peropara mejorar la visualizacion es mas interesante el panel Align and Distribute que puedesencontrar en el menu Layout → Align and Distribute. Con este panel es mucho massencillo obtener una visualizacion de la red personalizada. De igual manera, el panel Scale(Layout → Scale) puede ser de gran utilidad.
Problema
De entre todos los nodos de la red existe uno que no tiene identificador. ¿Serıascapaz de encontrarlo?
10

Visualizando Datos de Expresion (VizMapper)
Los datos de expresion se suelen utilizar en Cytoscape para cambiar los atributosvisuales de la red. La herramienta VizMapper, integrada en el propio nucleo del programa,permite cambiar la representacion grafica en base a la informacion disponible. Esta opcionofrece una gran potencia a la hora de integrar e interpretar los resultados en un contextobiologico.
Etiquetado de los nodos
Para cambiar los atributos de los nodos recurriremos por tanto a VizMapper :
En primer lugar abre el VizMapper, bien seleccionando el tab en el panel de laizquierda o bien haciendo click en el icono de la barra de herramientas.
En el desplegable Current Visual Style seleccionamos default y el aspecto de la redvolvera al original. Para no modificar este estilo crearemos otro estilo como copia deeste haciendo click en el icono, seleccionando Copy existing visual style... y dandoun nombre al estilo personalizado.
Haz Zoom hasta que las etiquetas de los nodos sean visibles.
Haz click en la segunda columna de la fila Node Label in Visual Mapping Brows-er. Esto abrira un desplegable en el que elegiremos la opcion COMMON.
Este cambio provocara que en lugar de mostrar el identificador del gen como etique-ta de los nodos, mostrara el nombre comun (COMMON ) que hemos proporcionado alimportar los datos de expresion de la red en la seccion Importar atributos de la red.
Coloreando los nodos
Una forma habitual de colorear los nodos cuando se tienen datos de expresion esmediante la gama rojo/verde para representar represion o sobrexpresion respectivamente.Para ello:
Haz doble-click sobre la lınea Node Color en la seccion Unused Visual Proper-ties del Visual Mapping Browser. Con esto la lınea de Node Color se moveraa las primeras opciones de Visual Mapping Browser.
Selecciona la celda Please select a value de la lınea Node Color. Esto produciraun desplegable con los atributos disponibles para colorear. Selecciona ”gal80Rexp”(datos de expresion del experimento de represion de gal80 ).
Selecciona ahora en el desplegable la opcion Continuous Mapping para que la grad-uacion de color sea de manera continua. Tras esto, la red cambiara a una escala degrises.
11

Para cambiar los colores de la escala, haz click sobre el gradiente para editarlo. Hazdoble click sobre el triangulo que se encuentra mas a la izquierda en color negro.Este es el color que representara el color mas bajo de expresion por lo que asocialocon un color rojo brillante.
Repite la misma accion con el segundo triangulo negro. Esto cambiara el gradientede rojo a blanco.
A continuacion, desliza el siguiente triangulo (blanco) hacia un valor proximo a 0para representar aquellos genes que no estan ni sobrexpresados ni reprimidos.
Haz click sobre Add para anadir un nuevo triangulo a la escala. Haz click sobre ely selecciona un color verde brillante.
Cambia tambien el color del ultimo triangulo al mismo verde brillante.
Cierra la ventana y comprueba que la red se asemeja al gradiente que se muestraen la Figura 2.3
Figura 2.3: Gradiente de expresion de VizMapper
Un problema al que nos enfrentamos es que algunos de los nodos no tienen datos deexpresion y por tanto no estan siendo coloreados en base a nuestra escala y mantienenel color rosa original que se puede confundir dentro de nuestro gradiente. Para corregireste comportamiento bastarıa con cambiar el color por defecto de los nodos a uno fuerade nuestro espectro:
Haz click en la red que aparece dentro de Defaults en el panel de VizMapper.
Selecciona Node Fill Color y sustituyelo por un color gris. Haz click en Apply.
Disminuye el zoom para visualizar los nodos sin datos de expresion.
12
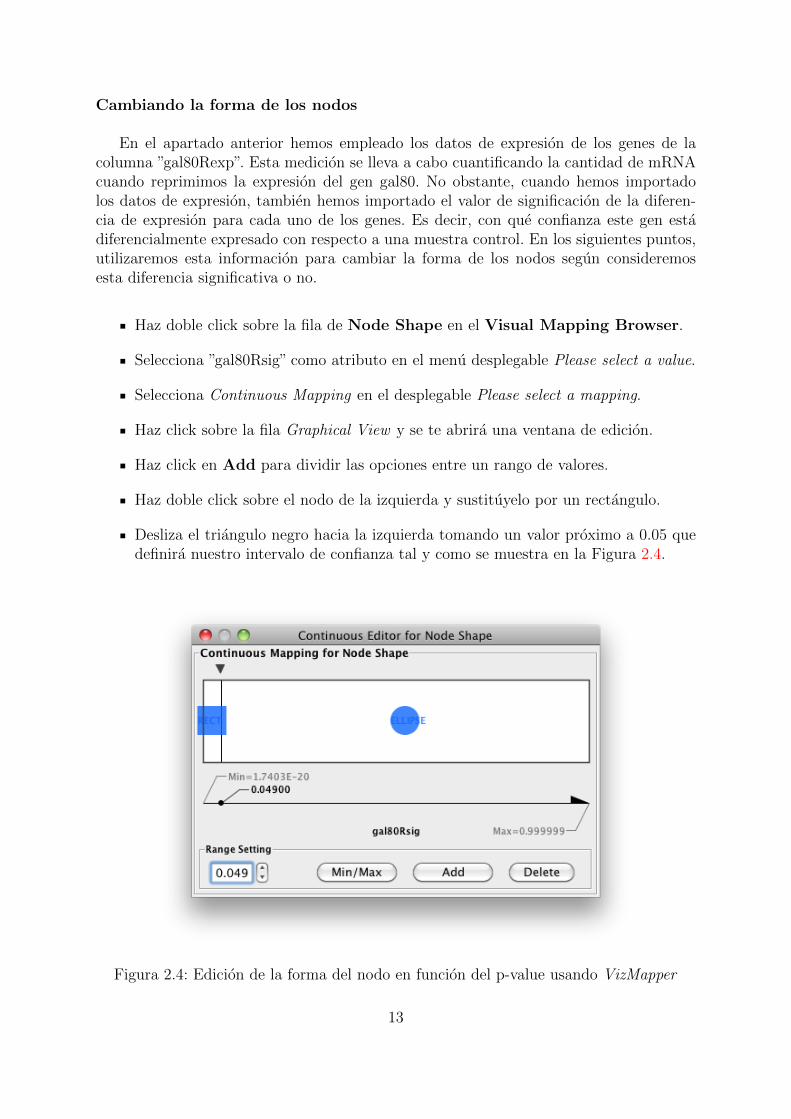
Cambiando la forma de los nodos
En el apartado anterior hemos empleado los datos de expresion de los genes de lacolumna ”gal80Rexp”. Esta medicion se lleva a cabo cuantificando la cantidad de mRNAcuando reprimimos la expresion del gen gal80. No obstante, cuando hemos importadolos datos de expresion, tambien hemos importado el valor de significacion de la diferen-cia de expresion para cada uno de los genes. Es decir, con que confianza este gen estadiferencialmente expresado con respecto a una muestra control. En los siguientes puntos,utilizaremos esta informacion para cambiar la forma de los nodos segun consideremosesta diferencia significativa o no.
Haz doble click sobre la fila de Node Shape en el Visual Mapping Browser.
Selecciona ”gal80Rsig” como atributo en el menu desplegable Please select a value.
Selecciona Continuous Mapping en el desplegable Please select a mapping.
Haz click sobre la fila Graphical View y se te abrira una ventana de edicion.
Haz click en Add para dividir las opciones entre un rango de valores.
Haz doble click sobre el nodo de la izquierda y sustituyelo por un rectangulo.
Desliza el triangulo negro hacia la izquierda tomando un valor proximo a 0.05 quedefinira nuestro intervalo de confianza tal y como se muestra en la Figura 2.4.
Figura 2.4: Edicion de la forma del nodo en funcion del p-value usando VizMapper
13

Ahora podemos comparar de un simple vistazo, aquellos nodos cuya expresion estadiferencialmente alterada por el hecho de reprimir la expresion del gen ”gal80”.
De igual manera, puedes aplicar todo lo aprendido con VizMapper para cambiarcualquier otro parametro visual a tu gusto. Puede ser un buen momento para guardartodos los avances que has realizado hasta ahora.
Filtrado
Como vimos al inicio de la seccion Visualizacion de las redes y Analisis de expresion,en esta red estamos representando una combinacion de interacciones proteına-proteına(pp) y proteına-DNA (pd). A continuacion, vamos a filtrar las interacciones ”pp” paraquedarnos unicamente con las interacciones proteına-DNA.
Haz click sobre el tab Filters del Control Panel.
En el desplegable Attribute/Filters selecciona ”edge.interaction” y anadelo me-diante el boton Add.
Escribe las letras ”pp” en la caja de texto para especificar que quieres todas aquellasinteracciones que coinciden con esta expresion.
Haz click en el boton Apply Filters para aplicar el filtro que hemos creado y verascomo se seleccionan algunas de las interacciones.
Seleccionamos en el menu Edit → Delete Selected Nodes and Edges puestoque estas son las interacciones que no nos interesan.
Aplicamos un layout como Force-Directed Layout para conseguir una visualiza-cion mas amigable.
Interpretar la Red
Podemos ver que hay 3 genes brillantes (altamente sobrexpresados) en la misma regiondel grafo y que hay 2 nodos que interaccionan con los 3: GAL4 (YPL248C) y GAL11(YOL051W). Vamos a crear una subred a partir de esta para facilitar la interpretacion:
Seleccionamos GAL4 y GAL11 y seleccionamos sus vecinos mas cercanos (Select→ Nodes → First Neighbors of Selected Nodes).
Creamos la subred mediante el menu File→ New→ Network→ From SelectedNodes, All Edges.
Aplicamos un layout para obtener figuras como las que se muestran en la Figura2.5.
14

GAL80 GAL4
GAL1MIG1
GAL7
GAL2GCY1CYC1
GAL10
GAL11
GAL1
GAL11
CYC1
GAL10
GAL4
GCY1
GAL80
MIG1
GAL7
GAL2
GAL1
GAL11
CYC1
GAL10
GAL4
GCY1
GAL80
MIG1
GAL7
GAL2
Gal4RGal1R Gal80R
Figura 2.5: Expresion de la subred de regulacion de GAL4 y GAL11 en los distintos casosexperimentales analizados tras reprimir Gal1, Gal4 y Gal80 respectivamente. El color delos nodos representa los niveles de expresion y la forma si el p-value es menor (cuadrado)o mayor (cırculo) de 0.05.
Problema
Hasta ahora has analizado la subred de GAL4 y GAL11 a partir de la expresion delos genes tras reprimir GAL80. No obstante, podrıa ser interesante ver lo que sucedesi reprimimos GAL1 o GAL4 ya que poseemos esos datos de expresion y los podemosmostrar simplemente cambiando el mapping del VizMapper tal y como se muestra enla Figura 2.5.
Teniendo en cuenta que no tenemos datos de direccionalidad (que proteına regulaa que gen), ¿serıas capaz de plantear un modelo de regulacion? En el articulo deIdeker et al. se plantea un posible modelo. ¿Estas de acuerdo con el?
Te puede servir de ayuda obtener mas informacion de los nodos. Prueba la opcionLinkOut → Entrez → Gene haciendo click con el boton derecho sobre cualquierade ellos.
15
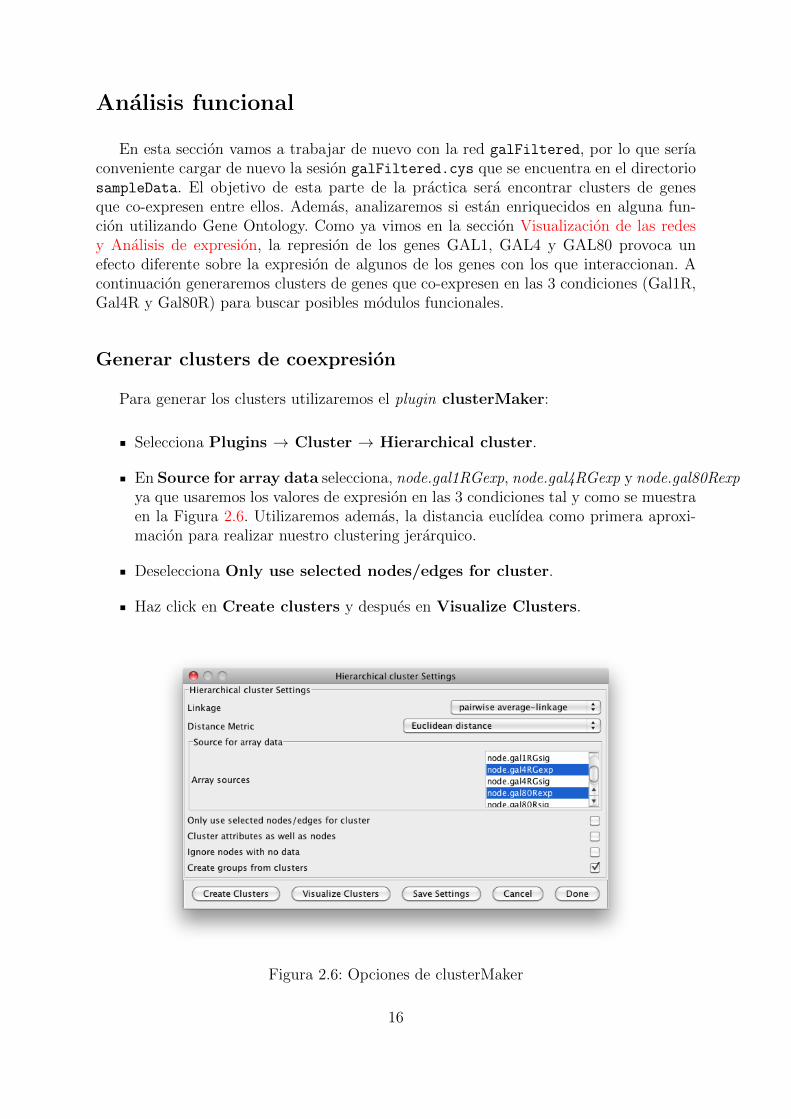
Analisis funcional
En esta seccion vamos a trabajar de nuevo con la red galFiltered, por lo que serıaconveniente cargar de nuevo la sesion galFiltered.cys que se encuentra en el directoriosampleData. El objetivo de esta parte de la practica sera encontrar clusters de genesque co-expresen entre ellos. Ademas, analizaremos si estan enriquecidos en alguna fun-cion utilizando Gene Ontology. Como ya vimos en la seccion Visualizacion de las redesy Analisis de expresion, la represion de los genes GAL1, GAL4 y GAL80 provoca unefecto diferente sobre la expresion de algunos de los genes con los que interaccionan. Acontinuacion generaremos clusters de genes que co-expresen en las 3 condiciones (Gal1R,Gal4R y Gal80R) para buscar posibles modulos funcionales.
Generar clusters de coexpresion
Para generar los clusters utilizaremos el plugin clusterMaker:
Selecciona Plugins → Cluster → Hierarchical cluster.
En Source for array data selecciona, node.gal1RGexp, node.gal4RGexp y node.gal80Rexpya que usaremos los valores de expresion en las 3 condiciones tal y como se muestraen la Figura 2.6. Utilizaremos ademas, la distancia euclıdea como primera aproxi-macion para realizar nuestro clustering jerarquico.
Deselecciona Only use selected nodes/edges for cluster.
Haz click en Create clusters y despues en Visualize Clusters.
Figura 2.6: Opciones de clusterMaker
16

Si todo ha ido bien observaras un resultado como el que se muestra en la Figura 2.7.En ella puedes observar un heatmap con los datos de expresion en las 3 condiciones asıcomo un dendrograma en el que se observa la clusterizacion de los valores. De esta forma,puedes hacer click en cualquier clado del dendrograma y de esta forma seleccionar losgenes agrupados en el.
Figura 2.7: Resultado de clusterMaker
En el clustering podemos observar como en la parte superior nos ha agrupado algunosgenes cuya expresion se ve especialmente afectada ya que los vemos en colores brillantes.Vamos a tratar de analizar exhaustivamente estos casos. Para ello:
Selecciona en el dendrograma el clado superior en el que aparecen una serie degenes diferencialmente expresados. Veras como la expresion de dichos genes aparecerepresentada en otro heatmap en la parte central de la ventana, ası como los iden-tificadores de los mismos.
Oculta la ventana para volver a la red y veras los genes que has seleccionado enclusterMaker tambien seleccionados en la red.
Problema
Ahora que has identificado los nodos que estaban presentes en este cluster decoexpresion, ¿son estos resultados consistentes con los resultados que obtuviste enel analisis de expresion (Figura 2.5)? ¿Que crees que sucederıa si el clustering lorealizaramos solo con las muestras Gal4R y Gal80R?
17

Enriquecimiento de terminos GO
A continuacion vamos a emplear el plugin BiNGO para encontrar terminos GeneOntology sobrerrepresentados en un grupo de genes. Gene Ontology es, como su propionombre indica, una ontologıa. Esto es, un lenguaje controlado que representa conceptosen 3 ambitos de la Biologıa Celular: proceso biologico, componente celular y funcionmolecular. Es por tanto un vocabulario de conceptos para etiquetar genes o sus productosde una forma ordenada y jerarquica.
Trabajando con redes es muy frecuente tener un grupo de genes o proteınas con unaserie de terminos funcionales asociados a cada uno. Por ello puede ser interesante realizarun test hipergeometrico para ver que funciones estan sobrerrepresentadas. Es precisa-mente esto lo que hace BiNGO y lo que trataremos de realizar en esta seccion. Puedesanalizar cualquier subseleccion de nodos pero ahora trataremos de ver el enriquecimientoen terminos GO de los genes que hemos encontrado coexpresados en clusterMaker (GAL1,GAL7 y GAL10).
Figura 2.8: Opciones del plugin BINGO.
Asegurate que los nodos a analizar estan seleccionados.
Selecciona ahora en el menu Plugins → Start BiNGO 2.4.4.
En el menu de BiNGO introduce un nombre para el cluster y revisa el resto deopciones. Algunos parametros corresponden al test estadıstico que vamos a ejecutaro a la correccion por multiple testing. Otro parametro importante es que ontologıade Gene Ontology vamos a emplear. En este caso, analizaremos al nivel de procesobiologico. Si necesitas ayuda haz click en el boton Help.
18
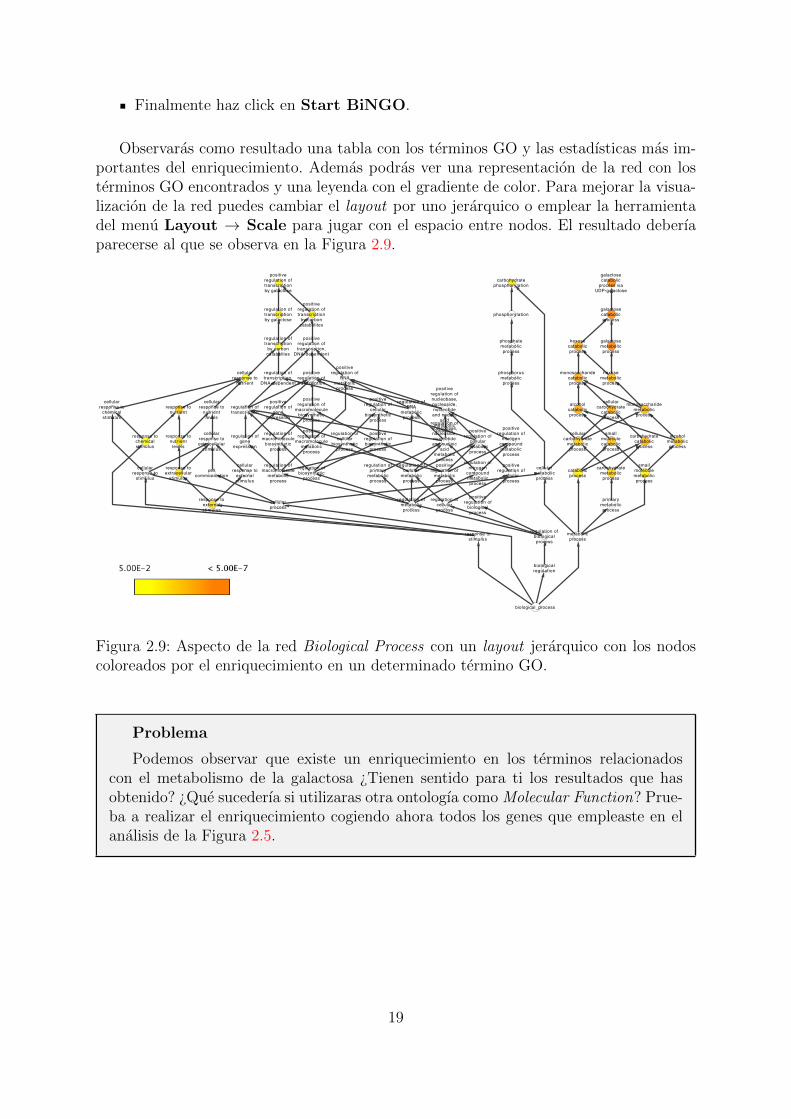
Finalmente haz click en Start BiNGO.
Observaras como resultado una tabla con los terminos GO y las estadısticas mas im-portantes del enriquecimiento. Ademas podras ver una representacion de la red con losterminos GO encontrados y una leyenda con el gradiente de color. Para mejorar la visua-lizacion de la red puedes cambiar el layout por uno jerarquico o emplear la herramientadel menu Layout → Scale para jugar con el espacio entre nodos. El resultado deberıaparecerse al que se observa en la Figura 2.9.
carbohydrate catabolic process
positive regulation of
macromolecule metabolic process
cellular carbohydrate
metabolic process
regulation of nucleobase, nucleoside, nucleotide and nucleic
acid metabolic process
positive regulation of biosynthetic
process
small molecule catabolic process
positive regulation of
cellular metabolic process
regulation of cellular
biosynthetic process
alcohol metabolic process
positive regulation of
nitrogen compound metabolic process
small molecule metabolic process
carbohydrate metabolic process
catabolic process
regulation of cellular
metabolic process
regulation of primary
metabolic process
regulation of nitrogen
compound metabolic process
positive regulation of
metabolic process
positive regulation of
cellular process
regulation of biosynthetic
process
cellular metabolic process
phosphate metabolic process
galactose catabolic
process via UDP-galactose
galactose catabolic process
galactose metabolic process
carbohydrate phosphorylation
hexose catabolic process
hexose metabolic process
monosaccharide catabolic process
phosphorus metabolic process
phosphorylation
positive regulation of
RNA metabolic process
regulation of transcription by galactose
positive regulation of transcription by galactose
regulation of transcription
by carbon catabolites
positive regulation of transcription
by carbon catabolites
positive regulation of transcription,
DNA-dependent
positive regulation of transcription
regulation of transcription
cellular response to
chemical stimulus
response to nutrient
positive regulation of
gene expression
positive regulation of
macromolecule biosynthetic
process
cellular response to
nutrient levels
primary metabolic process
metabolic process
response to stimulus
positive regulation of
biological process
regulation of biological process
regulation of metabolic process
biological_process
biological regulation
regulation of cellular process
response to nutrient
levels
cell communication
response to chemical stimulus
response to extracellular
stimulus
response to external stimulus
cellular response to
stimulus
cellular response to extracellular
stimulus
regulation of RNA
metabolic process
alcohol catabolic process
positive regulation of
cellular biosynthetic
process
monosaccharide metabolic process
cellular carbohydrate
catabolic process
positive regulation of nucleobase, nucleoside, nucleotide and nucleic
acid metabolic process
regulation of macromolecule
metabolic process
cellular response to
nutrient
regulation of transcription,
DNA-dependent
regulation of macromolecule
biosynthetic process
regulation of gene
expression
cellular response to
external stimulus
cellular process
Figura 2.9: Aspecto de la red Biological Process con un layout jerarquico con los nodoscoloreados por el enriquecimiento en un determinado termino GO.
Problema
Podemos observar que existe un enriquecimiento en los terminos relacionadoscon el metabolismo de la galactosa ¿Tienen sentido para ti los resultados que hasobtenido? ¿Que sucederıa si utilizaras otra ontologıa como Molecular Function? Prue-ba a realizar el enriquecimiento cogiendo ahora todos los genes que empleaste en elanalisis de la Figura 2.5.
19

Capıtulo 3
Analisis topologico en R
La lista de parametros que se usan para describir la topologıa de una red es enorme-mente larga y depende en gran medida de la observacion que se quiera realizar. En estaseccion, aprenderemos a calcular algunos de los parametros topologicos mas importantespara el estudio y analisis de redes biologicas, y los compararemos con los obtenidos pararedes artificiales. Para ello nos valdremos del entorno R y de su paquete de analisis degrafos igraph. Si bien hemos visto como Cytoscape nos ofrecece multitud de opciones a lahora de interactuar con nuestras redes, R es una herramienta especialmente interesantecuando queremos trabajar con una mayor cantidad de datos o cuando queremos realizaranalisis de mayor complejidad.
Para realizar los siguientes pasos necesitaremos tener instalado tanto R como el pa-quete igraph. Si aun no lo tienes instalado, puedes recurrir al apartado anexo Instalacionde igraph en R. Una vez instalado, bastara con abrir una consola de R y cargar la librerıaigraph mediante el siguiente comando:
> library(igraph)
Puesto que esta puede ser nuestra primera experiencia con el paquete, conviene echarun vistazo a la ayuda del mismo. Nos valdremos del comando help.start(). Esto nosabrira un navegador donde encontraremos la ayuda de R. Bastara con hacer click en Pack-ages y posteriormente en igraph. Echa un vistazo por encima y reconoceras muchas delas funcionalidades de las que hemos hecho uso en Cytoscape. Del mismo modo, encon-traras funciones para calcular parametros topologicos como el diametro de la red o elcamino mınimo entre dos nodos que implementan algunos de los algoritmos vistos en laclase de Teorıa de grafos.
Cargando distintos tipos de redes
En primer lugar, importaremos la red de interacciones de proteınas fyi de Han et al.(ver seccion Redes Biologicas a estudiar). Este fichero es basicamente una lista de lospares de nodos que interaccionan. Para importarlo utilizaremos la funcion read.graph()
20

que, ademas de permitir cargar ficheros locales, permite importar redes situadas en unservidor remoto. Una vez cargada, podemos utilizar la funcion summary() para mostrarun resumen de la misma:
> fyi <- read.graph("http://csbg.cnb.csic.es/master2012/www/data/fyi.net",
+ directed=FALSE)
> summary(fyi)
Vertices: 1379
Edges: 2493
Directed: FALSE
No graph attributes.
No vertex attributes.
No edge attributes.
A continuacion, crearemos una red aleatoria equivalente a fyi respecto al numero denodos y conexiones. Existen dos modelos matematicos para generar redes aleatorias: elModelo de Erdos-Renyi (1959) y el Modelo de Gilbert (1959), ambos implementados enla funcion erdos.renyi.game(). El primero genera una red aleatoria dado el numerode nodos y la probabilidad de que dos nodos esten conectados (G(n,p)), mientras queel segundo genera una red aleatoria con un numero concreto de nodos y conexiones(G(n,m)). Este ultimo sera el que utilicemos en nuestro caso, ya que nos interesa generaruna red aleatoria con el mismo numero de nodos y conexiones que fyi. Recuerda quepuedes consultar la ayuda de cualquier funcion con el comando help() o empleando eloperador ? (ej. ?erdos.renyi.game).
> erdosgraph <- erdos.renyi.game(length(V(fyi)), length(E(fyi)),
+ type="gnm")
Problema
Ahora que has sido capaz de crear redes aleatorias mediante el Modelo de Gilbert,¿serıas capaz de generar una red usando el Modelo de Erdos-Renyi de 100 nodos yuna probabilidadd de 0.5? ¿Has obtenido el numero de nodos y conexiones esperado?
Parametros topologicos mas generales
Caminos mınimos
La librerıa igraph nos permite calcular facilmente los caminos mınimos entre nodos.Utilizando por ejemplo la funcion shortest.paths() podemos calcular la distancia mı-nima entre un conjunto de nodos y el resto de la red. La distancia mınima media de todala red nos da una idea de como de alejados estan los nodos
21

> average.path.length(erdosgraph)
[1] 5.672712
> average.path.length(fyi)
[1] 9.410451
Diametro
El diametro de una red se define como el mayor camino mınimo en una red, o dichode otro modo, el camino mınimo entre los dos nodos mas alejados de la red. La Figura 3.1muestra el diametro de la red fyi
> diameter(erdosgraph)
[1] 12
> diameter(fyi)
[1] 25
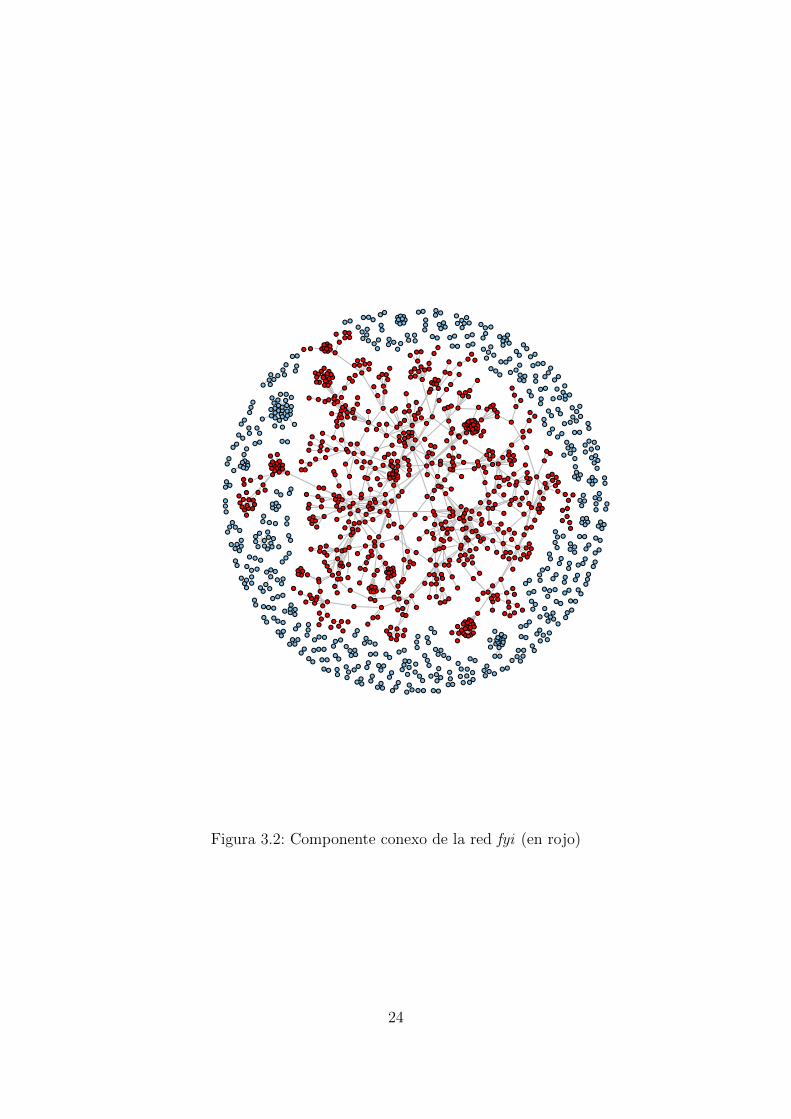
Componente conexo
Podemos igualmente analizar el tamano de los componentes conexos de la red. Lafuncion cluster() calcula todas las subredes cuyos nodos estan conectados entre sı.
> c <- clusters(fyi) #calculamos los clusters
> c$no #¿cuantos componentes distintos hay?
[1] 162
> table(c$csize) #¿Como de grandes son cada uno de ellos?
2 3 4 5 6 7 8 9 10 11 13 16 30 778
81 31 11 14 4 4 3 5 3 2 1 1 1 1
Problema
Como puedes observar, en la red fyi hay 162 componentes conexos, de los cuales81 estan formados solo por dos nodos. ¿A que crees que se debe el hecho de que noexistan nodos aislados?
Calcula los componentes conexos para nuestra red aleatoria equivalente segun elModelo de Gilbert. ¿Que diferencias hay? ¿A que crees que se deben?
22

●
●
●
●
●
●
●
●
●
●
●
● ●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
●
●
●●
●
●●
●●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
● ●●
●
●
●●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
● ●
●
●●
●
●●
●
●
●●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
● ●●
●●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●●●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●●●●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●●
●
●
●
●
●
●●
●●● ●
● ●
●
●●●
●●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●●
●●
●
●●● ●
●
● ●●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●●●●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
● ●
●
●
●
●
●●
●
●
●
●
●
●●
●●
●●● ●
●●
●●
●●●●●●
●●
●
●●
●●
●
●
●
●● ●
●
●●
●
●●
●●●●
●
●
● ●
●●●
● ●● ●
●●
●●
●
●
●
●
●
●●
●
●
● ●●●
●
●
●
●●
● ●●●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●● ●●
●
●●
●●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
● ●●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
● ●●●
●●
●
●
●
●
●
●●
●
●●
●●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●●
●●
●
●
●● ●●●●
●
●
● ●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●●
●●●
●
●
●
● ●
●
●
●
●
●
●
● ●
●
●
●
●●
●
●
●
● ●●●●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●
●
●
● ●
●●●
●
●
●
●
●
●
●●
●●
●
●
● ●●
●●●●●
● ●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●●
●●
●
●
●
●
● ●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●●
●
●
●
●●
●
●
●●
●●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●●
●●●
●
● ●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●● ●
●
●
●
●●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●●●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
Figura 3.1: Diametro de la red fyi (en rojo)
23
●
●
●
●
●
●
●
●
●
●
●
● ●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
●
●
●●
●
●●
●●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
● ●●
●
●
●●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
● ●
●
●●
●
●●
●
●
●●
●
●
●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
● ●●
●●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●●●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●●●●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●●
●
●
●
●
●
●●
●●● ●
● ●
●
●●●
●●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●●
●●
●
●●● ●
●
● ●●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●●●●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●●
● ●
●
●
●
●
●●
●
●
●
●
●
●●
●●
●●● ●
●●
●●
●●●●●●
●●
●
●●
●●
●
●
●
●● ●
●
●●
●
●●
●●●●
●
●
● ●
●●●
● ●● ●
●●
●●
●
●
●
●
●
●●
●
●
● ●●●
●
●
●
●●
● ●●●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●● ●●
●
●●
●●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
● ●●
●
●
●
●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
● ●●●
●●
●
●
●
●
●
●●
●
●●
●●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●●
●●
●
●
●● ●●●●
●
●
● ●
●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●●
●●●
●
●
●
● ●
●
●
●
●
●
●
● ●
●
●
●
●●
●
●
●
● ●●●●
●
●
●
● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●●
●
●
●
●
● ●
●●●
●
●
●
●
●
●
●●
●●
●
●
● ●●
●●●●●
● ●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●●
●●
●
●
●
●
● ●
●●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●●
●
●
●
●●
●
●
●●
●●
●
●● ●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●●
●
●●●
●●●
●
● ●
●
●
●
●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●●
●
●
●●●
●
●
●
●
●
●
●
●
●
●
●● ●
●
●
●
●●
●
●●
●●
●
●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●●
●
●
●
●
●●●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
Figura 3.2: Componente conexo de la red fyi (en rojo)
24

Cualidades de las Redes Biologicas
Coeficiente de clustering
El coeficiente de clustering o transitividad cuantifica la probabilidad de que los nodosadyacentes a un nodo esten conectados. En un grafo, la transitividad de un nodo es uno sitodos sus nodos vecinos estan conectados entre sı, y cero si no hay ninguna conexion entreellos. Para entenderlo mejor, crearemos un pequeno grafo t1 y calcularemos el coeficientede clustering para uno de los nodos:
> t1 <- graph.formula(A-B:C:D:E) #creamos un grafo con 5 nodos
> t1$layout <- layout.circle #aplicamos un layout en cırculo
> V(t1)$color <- "white" #definimos el color de los nodos
> V(t1)[name=="A"]$color <- "orange" #asignamos un color especıfico al nodo A
> V(t1)$size <- 40 #tama~no de los nodos
> V(t1)$label.cex <- 3 #tama~no de la etiqueta de los nodos
> V(t1)$label <- V(t1)$name #la etiqueta de cada nodo sera su nombre
> E(t1)$color <- "black" #color de la conexion entre dos nodos
> E(t1)$width <- 3 #grosor de la conexion entre dos nodos
> tr <- transitivity(t1, type="local", vids="A") #transitividad del nodo A
> plot(t1, main=paste("Transitividad de 'A':", tr))
Transitividad de 'A': 0
A
BC
DE
Cuando se anaden conexiones entre los nodos adyacentes el coeficiente de clusteringaumenta.
> t2 <- add.edges(t1, V(t1)[name %in% c("C","D")], color="red", width=3)
> tr <- transitivity(t2, type="local", vids="A")
> plot(t2, main=paste("Transitividad de 'A':", round(tr,4)))
25

Transitividad de 'A': 0.1667
A
BC
DE
La funcion transitivity() tambien se puede utilizar para calcular el coeficiente declustering global en un grafo. Para calcular dicho parametro topologico para las redes deerdos y fyi bastarıa con escribir lo siguiente:
> transitivity(erdosgraph)
[1] 0.003635963
> transitivity(fyi)
[1] 0.5430382
Problema
Hemos observado como el coeficiente de clustering de la red erdos es 0.0036 mien-tras que para fyi es 0.543. Si recuerdas la definicion de coeficiente de clustering,¿consideras que son numeros razonables para redes biologicas o sociales?
Por otro lado, ¿crees que el hecho de tener mayor diametro y caminos mınimos maslargos es compatible con el hecho de tener un coeficiente de clustering global mayor?¿Como dibujarıas dos redes pequenas para que se cumplieran dichas condiciones?
Redes de mundo pequeno (Small-World Networks)
Hasta ahora hemos visto como nuestra red erdos presenta ciertas diferencias y seme-janzas con la red fyi. Mientras que el diametro y el camino mınimo medio tienen valores
26

parecidos, el coeficiente de clustering es varios ordenes de magnitud inferior. El primerintento para modelar una red que, a diferencia del Modelo de Erdos-Renyi, cumpliera elprincipio de transitividad caracterıstico de las redes biologicas, fue propuesto en un ar-ticulo en Nature (1998) y se conoce como Modelo de Watts y Strogatz. Las redes creadassegun este modelo tienen una estructura ordenada en forma de anillo, en la que cada nodoesta ademas conectado a los nodos vecinos situados a una determinada distancia.
> strogatzgraph <- watts.strogatz.game(1, 10, 2, 0)
> transitivity(strogatzgraph)
[1] 0.5
> average.path.length(strogatzgraph)
[1] 1.666667
> plot(strogatzgraph, layout=layout.circle)
●
●
●●
●
●
●
● ●
●
0
1
23
4
5
6
7 8
9
Sin embargo, veamos que sucede si creamos una red con tantos nodos como nuestrared fyi :
> bigstrogatzgraph <- watts.strogatz.game(1, length(V(fyi)), 2, 0)
> transitivity(bigstrogatzgraph)
[1] 0.5
27

> average.path.length(bigstrogatzgraph)
[1] 172.7504
Las redes creadas segun el Modelo de Watts y Strogatz tienen un alto coeficientede clustering, sin embargo, a medida que aumenta el numero de nodos crece tambien elcamino mınimo medio. Estas redes cumplen por tanto solo parcialmente la propiedad deRedes de Mundo Pequeno tıpica de las redes biologicas.
Los propios autores del Modelo de Watts y Strogatz observaron como barajandoaleatoriamente algunas de las conexiones (rewiring), eran capaces de reducir drastica-mente el camino mınimo medio sin afectar al coeficiente de clustering. Esto cumple los 2requisitos vistos hasta ahora en nuestra red biologica fyi.
> strogatzgraph <- watts.strogatz.game(1, length(V(fyi)), 2, 0.2)
> transitivity(strogatzgraph)
[1] 0.1181512
> average.path.length(strogatzgraph)
[1] 6.197887
Redes libres de escala (Scale-free Networks)
Como hemos visto, el Modelo de Watts y Strogatz con rewiring es interesante yaque ofrece un compromiso entre las redes aleatorias de Erdos-Renyi, donde los grafosgenerados son de mundo pequeno pero no son transitivos y el modelo original de Watts yStrogatz. No obstante hay una caracterıstica tıpica de las redes biologicas que no cumplen:no son Redes Libres de Escala.
Como recordareis de la clase de Teorıa de Grafos, el Grado (Degree) de un nodose define como el numero de conexiones que entran a dicho nodo. En las Redes Libresde Escala, existe un gran numero de nodos poco conectados, mientras que los nodosaltamente conectados (hubs) son muy escasos. Se dice que la distribucion de nodos de lasredes libres de escala sigue una ley de potencia P (k) ∼ ck−γ:
> deg.dist.fyi <- degree.distribution(fyi)
> plot(deg.dist.fyi, xlab="k", ylab="P(k)", main="Scale-free network")
28
●
●
●
●
●
●
● ●
● ●●
● ●●
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
0 5 10 15 20 25 30
0.0
0.1
0.2
0.3
Scale−free network
k
P(k
)
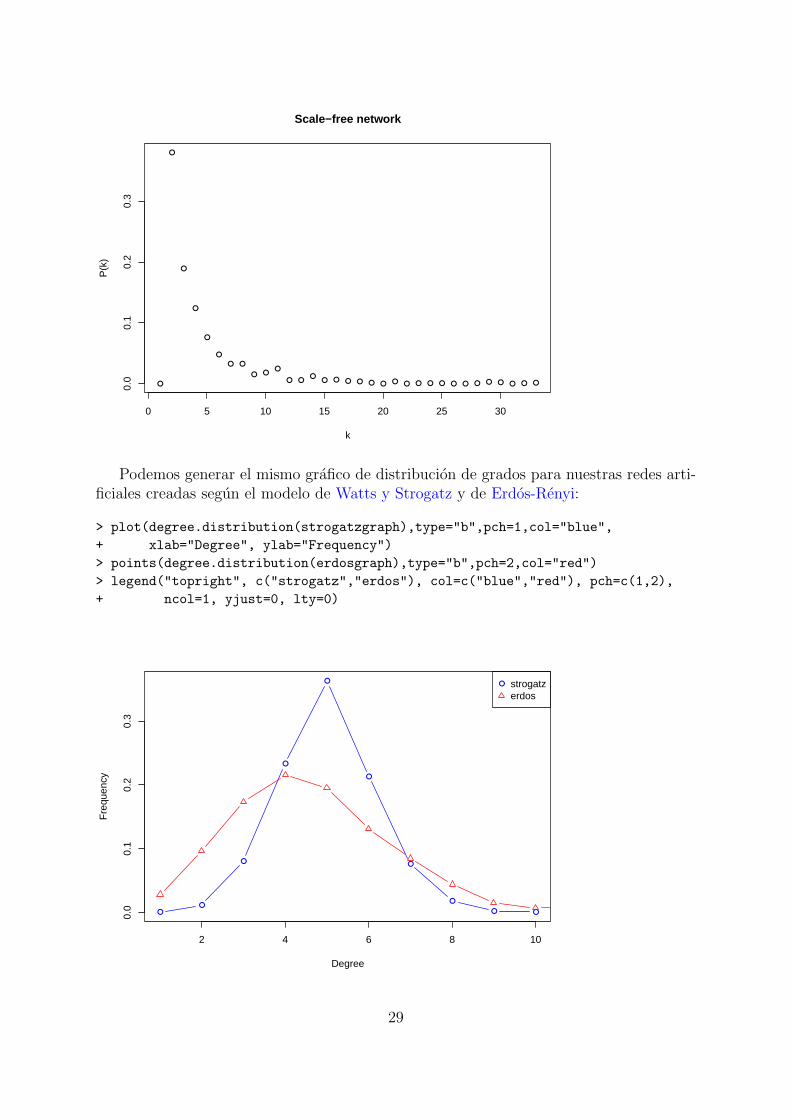
Podemos generar el mismo grafico de distribucion de grados para nuestras redes arti-ficiales creadas segun el modelo de Watts y Strogatz y de Erdos-Renyi:
> plot(degree.distribution(strogatzgraph),type="b",pch=1,col="blue",
+ xlab="Degree", ylab="Frequency")
> points(degree.distribution(erdosgraph),type="b",pch=2,col="red")
> legend("topright", c("strogatz","erdos"), col=c("blue","red"), pch=c(1,2),
+ ncol=1, yjust=0, lty=0)
●●
●
●
●
●
●
●
● ●
2 4 6 8 10
0.0
0.1
0.2
0.3
Degree
Fre
quen
cy
● strogatzerdos
29

El termino scale free fue acunado y publicado por Albert-Laszlo Barabasi y RekaAlbert en un articulo en Science (1999) gracias a sus estudios sobre la World Wide Web.Barabasi y Albert definieron un modelo para generar este tipo de redes siguiendo unmecanismo denominado union preferencial (preferential attachment) que cumple lassiguientes condiciones:
Los nodos se van anadiendo a la red en un proceso iterativo.
Cada nodo se unira a los nodos existentes con un numero fijo de conexiones.
La probabilidad de unirse a un nodo concreto, es directamente proporcional al gradode dicho nodo.
Los nodos con un grado mayor atraeran mas conexiones, mientras que los nodos menosconectados, permaneceran poco conectados cumpliendo la ley de potencia que caracterizaa las redes scale free. A este efecto de ”los ricos seran mas ricos y los pobres sera maspobres” se le conoce como efecto Matthew.
> barabasigraph <- barabasi.game(100, m = 1, directed=FALSE, out.pref = TRUE)
> plot(barabasigraph, layout=layout.fruchterman.reingold, vertex.size=4,
+ vertex.label=NA)
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●
●
●●
●
●
●●
●
● ●
●●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●●
●
●
●●
●
●
●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●●
30
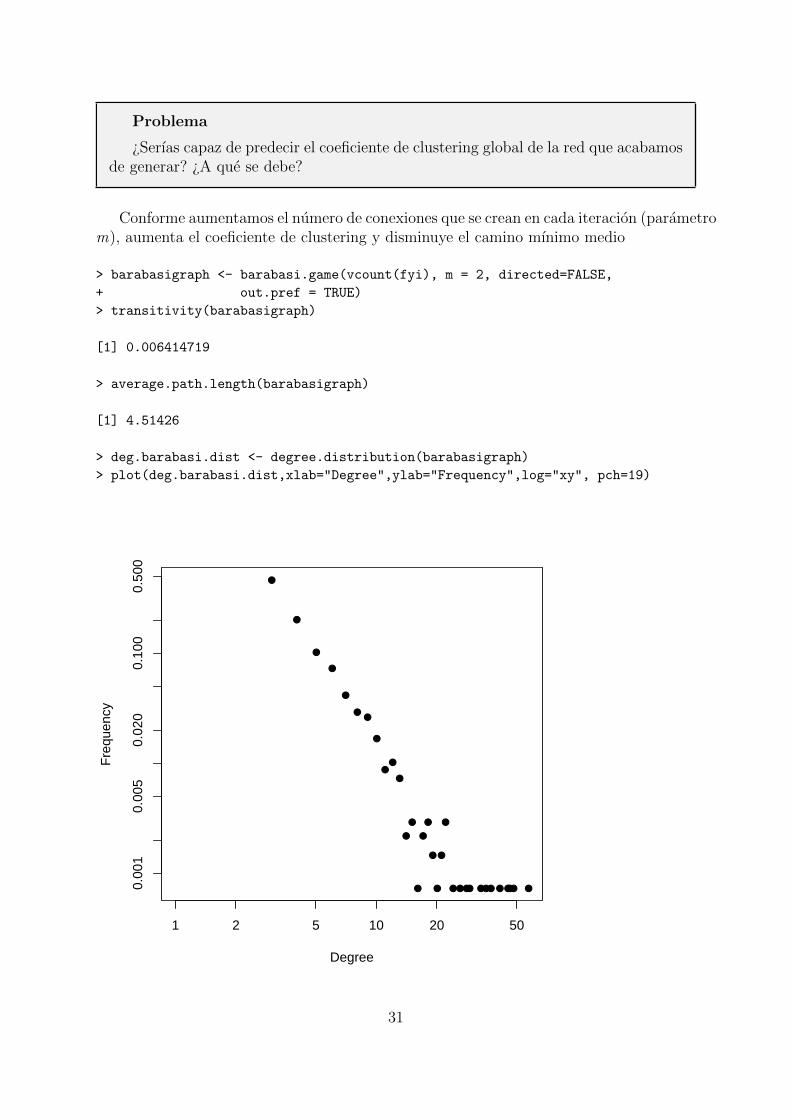
Problema
¿Serıas capaz de predecir el coeficiente de clustering global de la red que acabamosde generar? ¿A que se debe?
Conforme aumentamos el numero de conexiones que se crean en cada iteracion (parametrom), aumenta el coeficiente de clustering y disminuye el camino mınimo medio
> barabasigraph <- barabasi.game(vcount(fyi), m = 2, directed=FALSE,
+ out.pref = TRUE)
> transitivity(barabasigraph)
[1] 0.006414719
> average.path.length(barabasigraph)
[1] 4.51426
> deg.barabasi.dist <- degree.distribution(barabasigraph)
> plot(deg.barabasi.dist,xlab="Degree",ylab="Frequency",log="xy", pch=19)
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●●●● ●●● ● ●●● ●
1 2 5 10 20 50
0.00
10.
005
0.02
00.
100
0.50
0
Degree
Fre
quen
cy
31
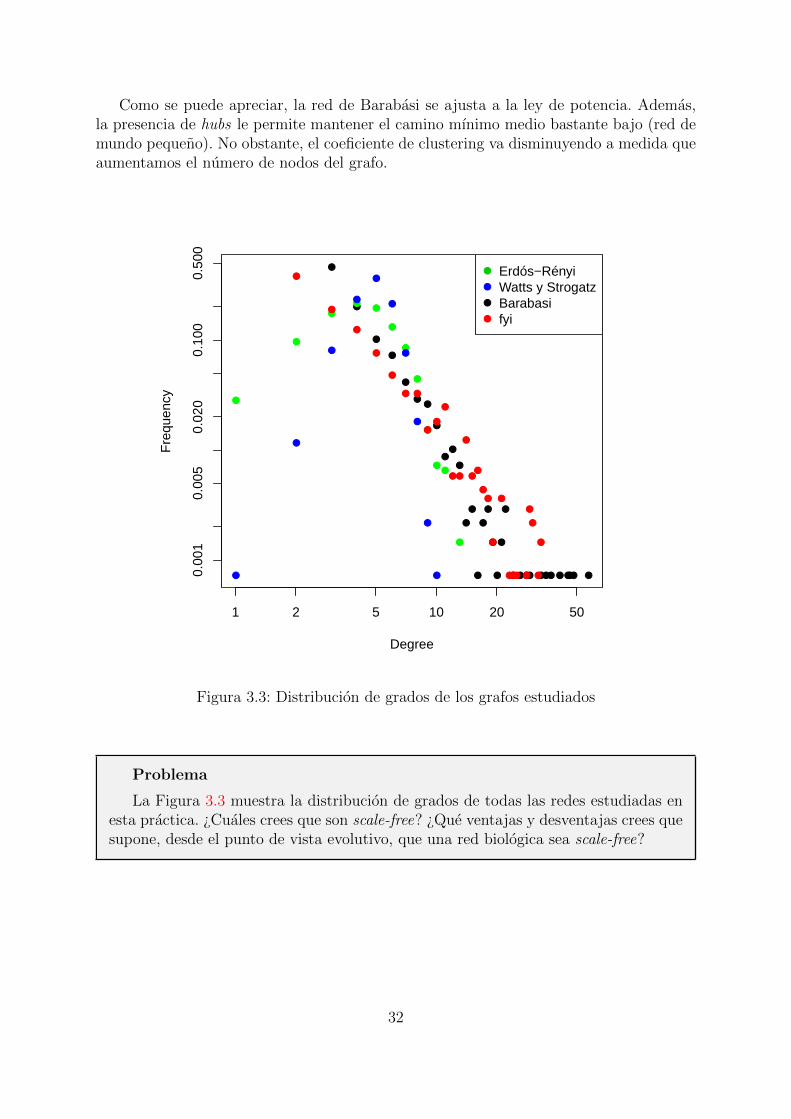
Como se puede apreciar, la red de Barabasi se ajusta a la ley de potencia. Ademas,la presencia de hubs le permite mantener el camino mınimo medio bastante bajo (red demundo pequeno). No obstante, el coeficiente de clustering va disminuyendo a medida queaumentamos el numero de nodos del grafo.
●
●
●
●
●
●●
●
●●
●
●
●
●
●
●
●
●
●
●
●●●● ●●● ● ●●● ●
1 2 5 10 20 50
0.00
10.
005
0.02
00.
100
0.50
0
Degree
Fre
quen
cy ●
●
●
●●
●
●
●
●
●●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
●
● ●
●●
●
●●
●
●●
●●
●
●
●●● ●
●
●
●
●
●
●
●
●
Erdós−RényiWatts y StrogatzBarabasifyi
Figura 3.3: Distribucion de grados de los grafos estudiados
Problema
La Figura 3.3 muestra la distribucion de grados de todas las redes estudiadas enesta practica. ¿Cuales crees que son scale-free? ¿Que ventajas y desventajas crees quesupone, desde el punto de vista evolutivo, que una red biologica sea scale-free?
32

Capıtulo 4
Conclusiones
A lo largo de este tutorial hemos visto los conceptos mas basicos para trabajar conCytoscape. Ahora deberıas ser capaz de instalar tanto el programa como sus plugins,crear e importar tus propias redes ası como aquellas procedentes de artıculos cientıficos,cambiar la visualizacion de las redes a tu antojo y trabajar con algunos plugins.
Evidentemente esto es solo un repaso general. Si aun te has quedado con ganas de masecha un vistazo a la pagina http://cytoscape.wodaklab.org/wiki/Welcome donde podrasencontrar material muy interesante como tutoriales, presentaciones. . . . Ademas tambienpuedes encontrar artıculos interesantes que pretenden servir de guıa para llevar a caboanalisis dentro de Cytoscape. Un buen ejemplo es el siguiente:
Cline et al. Integration of biological networks and gene expression data using Cy-toscape. Nat Protoc (2007) vol. 2 (10) pp. 2366-82
En la segunda parte de la practica hemos visto como trabajar con redes valiendose delentorno de trabajo R y la librerıa igraph. Hemos aprendido a cargar redes biologicas, agenerar redes artificiales segun diferentes modelos y a calcular los parametros topologicosmas comunes. En la web de igraph (http://igraph.sourceforge.net/) puedes encontrarmanuales y la documentacion de la librerıa, ası como la librerıa en otros lenguajes deprogramacion como C, Python o Ruby.
Agradecimientos
Agradecemos a Mike Smoot, David de Juan y Florencio Pazos por su ayuda y materialproporcionado para llevar a cabo este tutorial ya que sin ellos no hubiera sido posible.
33

Apendices
34

Instalacion de Cytoscape
En esta practica vamos a trabajar con la version de Cytoscape 2.8. La version sobrela que se trabaja es algo especialmente crıtico en Cytoscape ya que los plugins estandisenados para una version especıfica y no necesariamente trabajan en las demas. Por estarazon, y para no perder demasiado tiempo en la instalacion hemos creado una carpetatanto con Cytoscape 2.8 como con todos los plugins que se utilizaran. Puesto que norequiere privilegios de administracion, bastara con descargar el fichero comprimido desdela web de la practica (link), descomprimirlo y ejecutar el siguiente comando desde eldirectorio en cuestion:
1 . / cytoscape . sh
Para instalar Cystoscape desde cero existen diferentes opciones:
Instalar automaticamente los paquetes especıficos para Windows, Mac y Linux.
Instalar Cytoscape desde un paquete comprimido distribuido.
Compilar Cytoscape desde el codigo fuente.
Instalar desde el repositorio de Subversion.
Lo mas habitual es recurrir directamente a la seccion de descargas de la pagina webde Cytoscape (http://www.cytoscape.org/). Para descargarlo os pedira vuestro nombre ee-mail. La ultima version cuando se escribio este manual era la 2.8.1 y los desarrolladoresse encuentran inmersos en las versiones 2.9 y 3.0. El problema actual es que muchos delos plugins tienen que ser rehechos para cada una de las versiones. Por esta razon, enocasiones, es mas recomendable bajar a una version anterior en la que todos los pluginsfuncionen correctamente. Para ello, basta con ir a la seccion All releases de la pagina deCytoscape y elegir la apropiada. Como la mayorıa de aplicaciones en Java no requierenprivilegios de administrador, Cytoscape se puede instalar en un directorio a nivel deusuario.
Ejecutar Cytoscape
Para abrir la aplicacion desde Linux o Mac OSX basta con hacer doble click en elicono creado por el instalador o bien ejecutar el fichero cytoscape.sh. En Windows encambio hay que abrir el fichero cytoscape.bat. Tambien es posible ejecutar directamenteel programa desde la consola con el siguiente comando (desde el directorio que contienela aplicacion):
1 java −Xmx512M −j a r cytoscape . j a r −p p lug in s
La opcion -Xmx512M especifica la memoria que Java destinara a Cytoscape. Esteparametro puede interesar alterarlo para trabajar en ordenadores con memoria suficientey redes grandes. Para nuestro caso deberıa ser suficiente con 512Mb. La opcion -p plugins
35
especifica en que directorio se encuentran los plugins. Por tanto, es recomendable noabrir directamente el fichero .jar mediante doble-click porque a pesar de que tenga uncomportamiento aparentemente normal, puede que no tengas algunas funcionalidades queestan en la carpeta plugins.
Instalar plugins
Una de las grandes cualidades de Cytoscape es que es de codigo abierto, por lo quecualquier usuario puede aportar nuevas herramientas creando un plugin y distribuyen-dolo. Esto ha convertido a Cytoscape en una aplicacion muy poderosa con multitud deutilidades para resolver, de los problemas mas generales, a los mas especıficas.
Figura 1: Ventana Manage Plugins
En esta practica vamos a necesitar distintos plugins por lo que antes de nada convienesaber como instalarlos. Para ello, una vez abierta la ventana principal, seleccionamos enel menu Plugins la opcion Manage Plugins. Esto nos abrira una ventana con los queestan disponibles en el servidor. Puesto que los plugins son dependientes de la versionde Cytoscape, puede que algunos no esten adaptados a la version actual. Si habeis hechocaso a mi recomendacion y habeis instalado la version 2.8 de Cytoscape, este problema noos afectara para los plugins de esta practica. Si no fuera el caso, existen 2 posibilidades:instalar una version anterior de Cytoscape e instalar los plugins adecuados; o instalar elplugin desactualizado y confiar en que no pierdas ninguna funcionalidad. Si te decantaspor esta ultima opcion tienes que hacer click en la caja con el rotulo Show outdated
36

Plugins como se ve en la Figura 1. De una u otra forma conviene asegurarse de tenertodas las extensiones instaladas.
En esta practica vamos a trabajar con los siguientes plugins :
Network Analyzer
ClusterMaker
AllegroMCODE
BINGO
Una vez instalados, los encontraremos en el menu Plugins de la ventana principal deCytoscape. Algunos plugins pueden requerir reiniciar Cytoscape para que funcionen.
Instalacion de igraph en R
En primer lugar hay que instalar R. Los pasos detallados para instalar R en linux,MacOS X y Windows se pueden encontrar en la propia web de R (link). En caso de usarUbuntu, la mejor opcion es anadir el repositorio de R al archivo sources.list y usar aptpara completar la instalacion. Para instalar igraph basta con entrar en R y escribir:
1 i n s t a l l . packages ( ”igraph ”)
Una vez elegido el servidor mas cercano, la instalacion se completara de forma au-tomatica. Para empezar a trabajar con igraph en el entorno de R simplemente carga lalibrerıa con la siguiente funcion:
1 l i b r a r y ( ”igraph ”)
37

Navegando por Cytoscape
Esta seccion tiene como objetivo familiarizarse con el interfaz de Cytoscape: los menus,ventanas, opciones,. . . Conviene ser consciente de las opciones que hay y las posibilidadesque ofrece el programa para cuando se pretenda hacer algo saber a que menu, boton opestana hay que recurrir.
Interface Cytoscape
Abre Cytoscape y observa las distintas partes de la ventana principal:
En la parte superior del escritorio de Cytoscape encontraras la barra de herramientascon los botones mas importantes. Pasa el raton sobre ellos para ver para que sirvecada uno.
En el extremo superior derecho esta la ventana Main Network View donde semostrara la informacion sobre la red. Esta region esta originalmente en blanco.
A la izquierda se encuentra el Control Panel. Es una lista con las redes que tienesabiertas. Contiene su nombre ası como el numero de nodos y conexiones. La redaparecera en verde si has creado una vista de ella o en rojo si no hay una visualiza-cion de la misma. Para crearla o destruir la vista basta con hacer click con el botonderecho sobre el nombre de la red.
Inmediatamente debajo del Control Panel encontraras el Network Overview Panelque contiene una visualizacion general de la representacion de la red.
En el extremo inferior derecho puedes encontrar el Data Panel que contiene lainformacion mas importante sobre los nodos, las conexiones o los atributos de lared.
Estos dos ultimos paneles pueden ser extraıdos y hacerse flotantes sobre el escritorio parafacilitar la visualizacion, simplemente haciendo click sobre el icono que se encuentra enel extremo superior derecho.
Menus
Vamos a ver muy por encima los menus mas importantes de Cytoscape.
File
El menu File contiene las principales funciones para trabajar con ficheros:
Open para abrir ficheros de sesion de Cytoscape.
New para crear una nueva red.
38

Save para guardar una nueva red.
Import para importar datos como redes o atributos.
Export para exportar datos o imagenes.
Print para imprimir.
Quit cierra todas las ventanas de Cytoscape y sale del programa.
Edit
El menu Edit contiene:
Undo y Redo para deshacer y rehacer respectivamente cambios hechos en AttributeBrowser, Network Editor y en el Layout.
Opciones para crear y destruir las representaciones graficas de las redes y las propiasredes.
Opciones para eliminar los nodos y conexiones seleccionadas en la red actual.
Preferences → Properties contiene todas las propiedades del Cytoscape y de susplugins.
View
Este menu es para mostrar u ocultar las ventanas:
Control Panel
Data Panel
Results Panel
VizMapper
Select
El menu Select contiene:
Opciones para seleccionar nodos o conexiones.
La opcion Select → Use Filters permite crear filtros para la seleccion automaticade porciones de la red cuyos atributos de nodos o conexiones cumplan un determi-nado criterio.
39

Layout
El menu Layout es una lista de opciones para cambiar el aspecto de la representacionvisual de las redes:
Rotate, Align, Scale y Distribute permiten trabajar con la visualizacion general.
La seccion que se encuentra en la parte inferior contiene una gran variedad de algo-ritmos que automaticamente alteran la visualizacion de las redes. Los hay pesadospor algun parametro o no pesados.
Plugins
Contiene las opciones mas importantes para administrar o usar las extensiones. Estemenu puede variar notablemente a medida que instalemos nuevos plugins con nuevasfuncionalidades.
Help
El menu de ayuda te permite lanzar el visualizador de la ayuda para navegar por loscontenidos del manual de Cytoscape.
40





























