Embed Size (px)
Citation preview

Machine Learning Machine Learning in the Study of in the Study of
Protein StructureProtein Structure
RuiRui KuangKuangColumbia UniversityColumbia UniversityCandidacy Exam Talk May 5th, 2004Candidacy Exam Talk May 5th, 2004
Committee: Christina S. Leslie (advisor), Committee: Christina S. Leslie (advisor), YoavYoav Freund, Tony Freund, Tony Jebara Jebara

Table of contentsTable of contents1. Introduction to protein structure and its
prediction2. HMM, SVM and string kernels3. Machine learning in the study of protein
structure• Protein ranking • Protein structural classification• Protein secondary structural and
conformational state prediction• Protein domain segmentation
4. Conclusion and Future work

Thanks to Carl-Ivar Branden and John Tooze
1. Introduction 2. HMM, SVM and string kernels3. Topics4. Conclusion and future work
Part 1: Introduction to Protein Structure and Its Prediction

Why study protein structureWhy study protein structure• Protein –
Derived from Greek word proteios meaning “of the first rank” in 1838 by Jöns J. Berzelius
• Crucial in all biological processes
• Function depends on structurestructure can help us to understand function

How to Describe Protein How to Describe Protein StructureStructure
• Primary structure: amino acid sequence• Secondary structure: local structure elements • Tertiary structure: packing and arrangement of
secondary structure, also called domain• Quaternary structure: arrangement of several
polypeptide chains
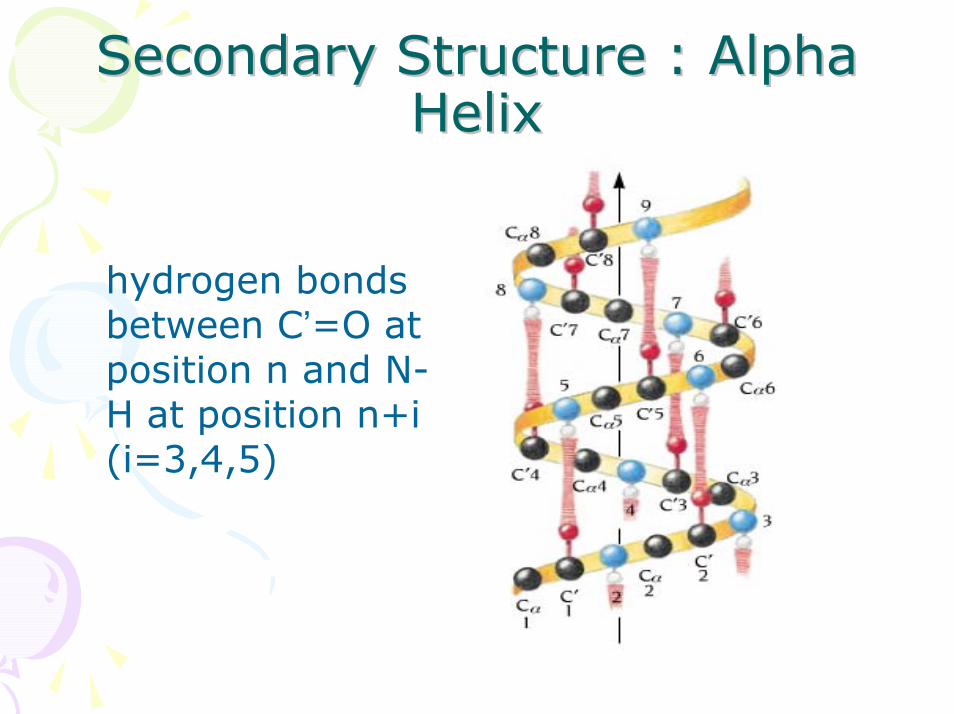
Secondary Structure : Alpha Secondary Structure : Alpha HelixHelix
hydrogen bonds between C’=O at position n and N-H at position n+i (i=3,4,5)

Secondary Structure : Beta Secondary Structure : Beta SheetSheet
Parallel Beta Sheet
Antiparallel Beta Sheet
We can also have a mix of both.
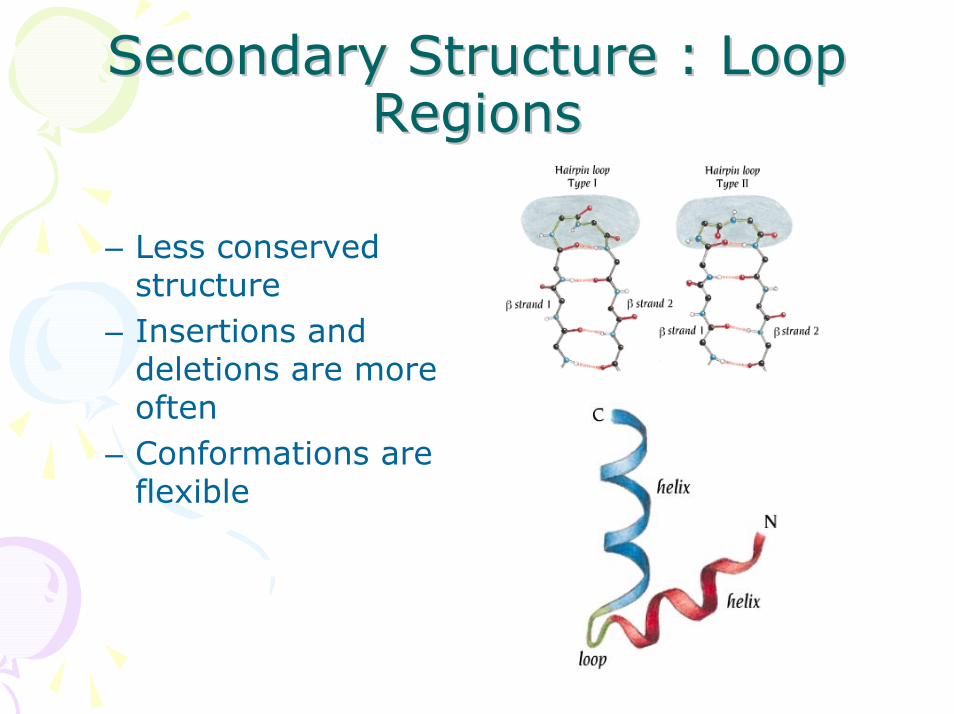
Secondary Structure : Loop Secondary Structure : Loop RegionsRegions
– Less conserved structure
– Insertions and deletions are more often
– Conformations are flexible

Phi – N - bondPsi – -C’ bond
Tertiary StructureTertiary Structure
αCαC

PhiPhi--PsiPsi angle distributionangle distribution

Protein DomainsProtein Domains
• A polypeptide chain or a part of a polypeptide chain that can fold independently into a stable tertiary structure.

Determination of Protein Determination of Protein StructuresStructures
• Experimental determination (time consuming and expensive)– X-ray crystallography – Nuclear magnetic resonance (NMR)
• Computational determination [Schonbrun2002 (B2)]
– Comparative modeling– Fold recognition ('threading')– Ab initio structure prediction (‘de novo’)

Picture due to Michal Linial
Sequence, Structure and Sequence, Structure and Function Function [[DominguesDomingues 2000 (B1)]2000 (B1)]
•>30% sequence similarity suggests strong structure similarity•Remote homologous proteins can also share similar structure
•Function associated with different structures•Super-family with the same fold can evolve into distinct functions.•66% of proteins having similar fold also have a similar function
Sequence (1,000,000)
Structure (24,000): discrete groups of folds with unclear boundaries
Function (Ill-defined)

Thanks to Nello Cristianini
1. Introduction 2. HMM, SVM and string kernels3. Topics4. Conclusion and future work
Part 2: Hidden Markov Model, Support Vector Machine and String Kernels
K( , )

Hidden Markov Models for Hidden Markov Models for Modeling Protein Modeling Protein [[KroghKrogh 19931993(B3)](B3)]
Alignment
MaximumLikelihood
OrMaximum a
posteriori
HMM
If we don’t know the alignment, use EM to train HMM.

Hidden Markov Models for Hidden Markov Models for Modeling Protein Modeling Protein [[KroghKrogh 19931993(B3)](B3)]
• Probability of sequence x through path q
• Viterbi algorithm for finding the best path
• Can be used for sequence clustering, database search…

Support Vector Machine Support Vector Machine [[BurgesBurges 1998(B4)]1998(B4)]
• Relate to structural risk minimization• Linear-separable case
– Primal qp problemMinimize subject to
– Dual convex problemMinimize
subject to &

Support Vector Machine Support Vector Machine [[BurgesBurges 1998(B4)]1998(B4)]
• Kernel: one nice property of dual qpproblem is that it only involves the inner product between feature vectors, we can define a kernel function to compute it more efficiently
• Example:

String Kernels for Text String Kernels for Text Classification Classification [[LodhiLodhi 20022002(M2)](M2)]
• String subsequence kernel –SSK :
• A recursive computation of SSK has the complexity of the computation O(n|s||t|). It is quadratic in terms of the length of input sequences. Not practical.
n
isui
iiu uu Σ∈= ∑
=
+− ,][:
11||λφ

1. Introduction 2. HMM, SVM and string kernels3. Topics4. Conclusion and future work
Part 3Part 3
Machine learning in the study of protein structure
3.1 Protein ranking 3.2 Protein structural classification3.3 Protein secondary structure and
conformational state prediction3.4 Protein domain segmentation

Part 3.1 Protein Ranking
Please!!!Stand in order
• Smith-Waterman • SAM-T98
• BLAST/PSI-BLAST• Rank Propagation

Thanks to Jean Philippe
Local alignment: Local alignment: SmithSmith--Waterman Waterman algorithmalgorithm
• For two string x and y, a local alignment with gaps is:
• The score is:
• Smith-Waterman score:

BLAST BLAST [[AltschulAltschul 1997 (R1)]1997 (R1)]: : a heuristic algorithm for matching DNA/Protein a heuristic algorithm for matching DNA/Protein
sequencessequences
• Idea: True matches are likely to contain a short stretch of identity
AKQKQDQDYDYYYYY…
AKQDYYYYE… cut SearchProtein
Database
match
Query: ………DYY………………Target: …ASDDYYQQEYY…
AKQ SKQ..KQD AQD..QDY ..DYY ..YYY…
Neighbor mapping
substitution score>T
Extend match Extend match

PSIPSI--BLAST: PositionBLAST: Position--specific specific Iterated BLAST Iterated BLAST [[AltschulAltschul 1997 (R1)]1997 (R1)]
• Only extend those double hits within a certain range.
• A gapped alignment uses dynamic programming to extend a central pair of aligned residues in both directions.
• PSI-BLAST can takes PSSM as input to search database

SAMSAM--T98 T98 [[KarplusKarplus 1999 (C3)]1999 (C3)]
Iterate 4
roundsNR Protein databaseQuery sequence Blast search
Buildalignment with hits
search
Profile/AlignmentHMM

• Affinity matrix
• D is a diagonal matrix of sum of i-th row of W
• Iterate
• F* is the limit of seuqnce{F(t)}
Local and Global Local and Global Consistency Consistency [[ZhouZhou 2003 (M1)]2003 (M1)]
22 2/)||||exp( δjiij xxW −−=
2/12/1 −−= WDDS
tSFtF )1()()1( αα −+=+
*maxarg ijcji Fy ≤=
Y

Rank propagation Rank propagation [Weston 2004 (R2)][Weston 2004 (R2)]
• Protein similarity network: – Graph nodes: protein sequences in the
database – Directed edges: a exponential function of
the PSI-BLAST e-value (destination node as query)
– Activation value at each node: the similarity to the query sequnce
• Exploit the structure of the protein similarity network
tqt KYKY α+=+ 1

Result Result [Weston 2004 (R2)][Weston 2004 (R2)]

Part 3.2 Protein structural classification
• Fisher Kernel• Mismatch Kernel• ISITE Kernel
• SVM-Pairwise• EMOTIF Kernel• Cluster Kernels
Where are my
relatives?

SCOPSCOP [[MurzinMurzin 1995 (C1)]1995 (C1)]
SCOP
Fold
Superfamily
Family
Positive Training Set
Positive Test Set
Negative Training Set
Negative Test Set
Family : Sequence identity > 30% or functions and structures are very similarSuperfamily : low sequence similarity but functional features suggest probable common evolutionary originCommon fold : same major secondary structures in the same arrangement with the same topological connections

CATH [[OrengoOrengo 1997 (C2)]CATH 1997 (C2)]• ClassSecondary structure composition
and contacts
• ArchitectureGross arrangement of secondary
structure
• TopologySimilar number and arrange of
secondary structure and same connectivity linking
• Homologous superfamily
• Sequence family

Fisher Kernel Fisher Kernel [[JaakkolaJaakkola 2000 (C4)]2000 (C4)]
• A HMM (or more than one) is built for each family
• Derive feature mapping from the Fisher scores of each sequence given a HMM H1:
),|(log 1 θθ HXPUX ∇=
∑−=k
jj
jij kE
ieiE
U )()()(

SVMSVM--pairwise pairwise [[LiaoLiao 2002 (C5)]2002 (C5)]
• Represent sequence P as a vector of pairwise similarity score with all training sequences
• The similarity score could be a Smith-Waterman score or PSI-BLAST e-value.
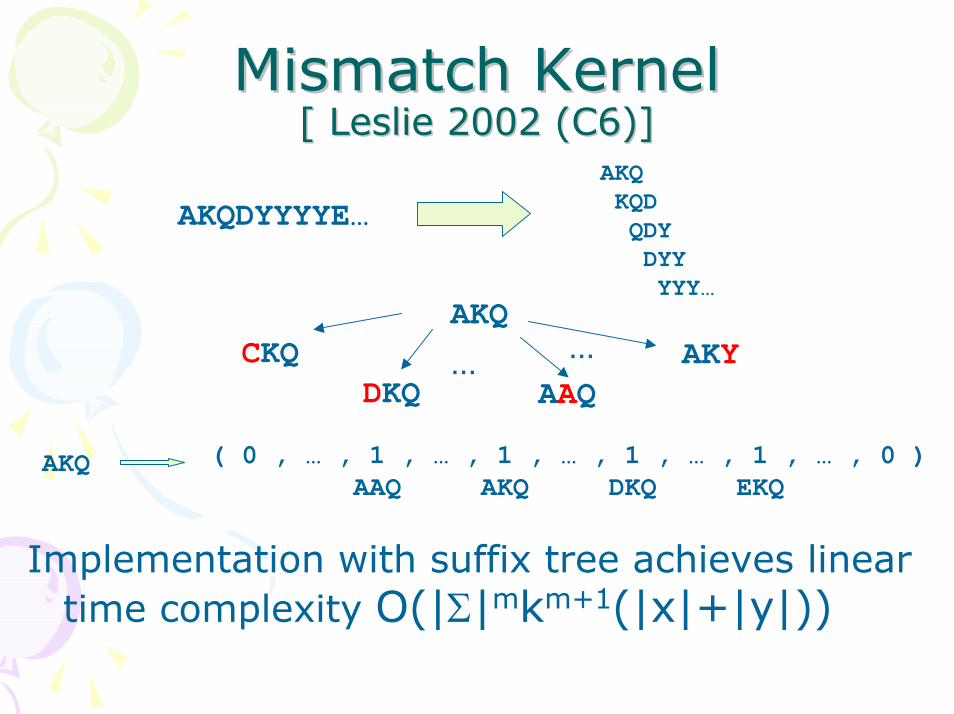
Mismatch Kernel Mismatch Kernel [ Leslie 2002 (C6)][ Leslie 2002 (C6)]
AKQKQDQDYDYYYYY…
AKQDYYYYE…
AKQCKQ
DKQ AAQ… AKY…
( 0 , … , 1 , … , 1 , … , 1 , … , 1 , … , 0 )AAQ AKQ DKQ EKQ
AKQ
Implementation with suffix tree achieves linear time complexity O(|Σ|mkm+1(|x|+|y|))

EMOTIF Kernel EMOTIF Kernel [Ben[Ben--HurHur 2003 (C8)]2003 (C8)]
• EMOTIF TRIE built from eBLOCKS [Nevill-manning 1998 (C7)]
• EMOTIF feature vector:where is the number of occurrences of the
motif m in x
Mmm xx ∈=Φ ))(()( φ)(xmφ

II--SITE Kernel SITE Kernel [[HouHou 2003 (C10)]2003 (C10)]
• Similar to EMOTIF kernel I-SITE kernel encodes protein sequences as a vector of the confidence level against structural motifs in the I-SITES library [Bystroff1998 (C9)]

Cluster kernels Cluster kernels [Weston 2004 (C11)][Weston 2004 (C11)]
• Neighborhood KernelsImplicitly average the feature vectors for sequences in the PSI-BLAST neighborhood of input sequence (dependent on the size of the neighborhood and total length of unlabeled sequences)
• Bagged KernelsRun bagged k-means to estimate p(x,y), the empirical probability that x and y are in the same cluster. The new kernel is the product of p(x,y) and base kernel K(x,y)

ResultsResults
Part 3.3: Protein secondary structure and conformational state prediction
• PrISM• HMMSTR
• PHD• PSI-PRED
Can we really do
that?

PHD: Profile network from PHD: Profile network from HeiDelbergHeiDelberg [[RostRost 1993 (P1)]1993 (P1)]
Accuracy: 70.8%
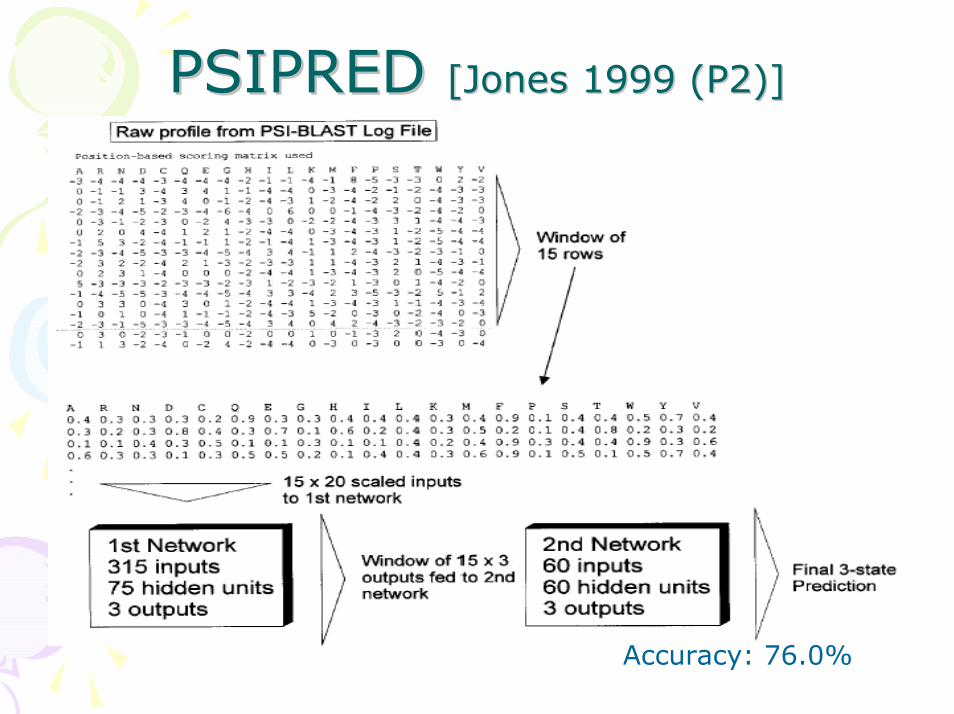
PSIPRED PSIPRED [Jones 1999 (P2)][Jones 1999 (P2)]
Accuracy: 76.0%

Conformational State Conformational State PredictionPrediction
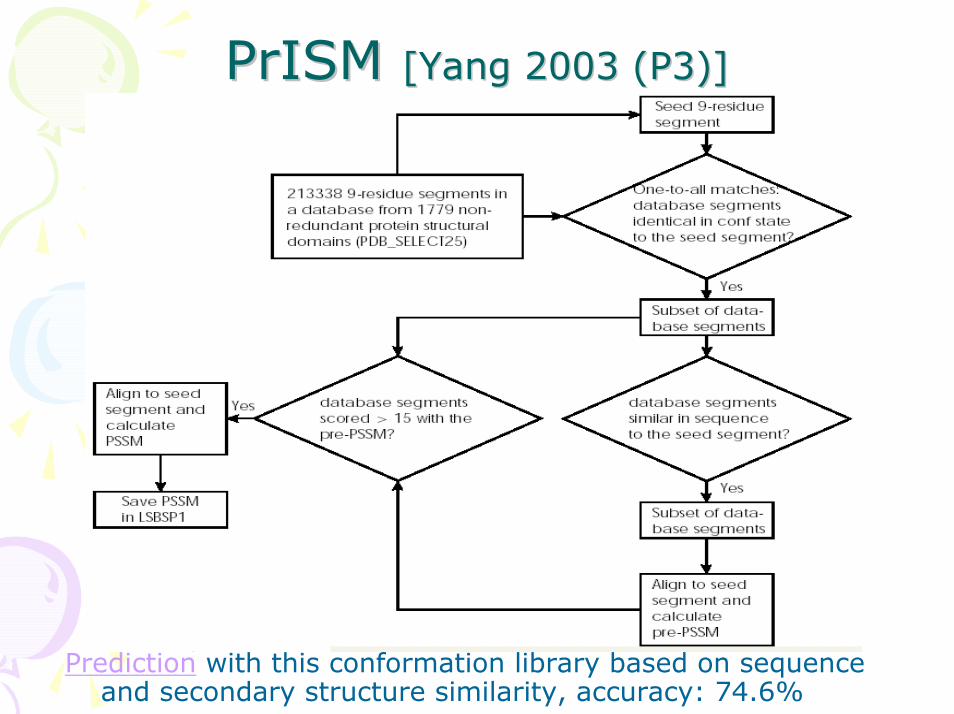
PrISMPrISM [Yang 2003 (P3)][Yang 2003 (P3)]
Prediction with this conformation library based on sequence and secondary structure similarity, accuracy: 74.6%
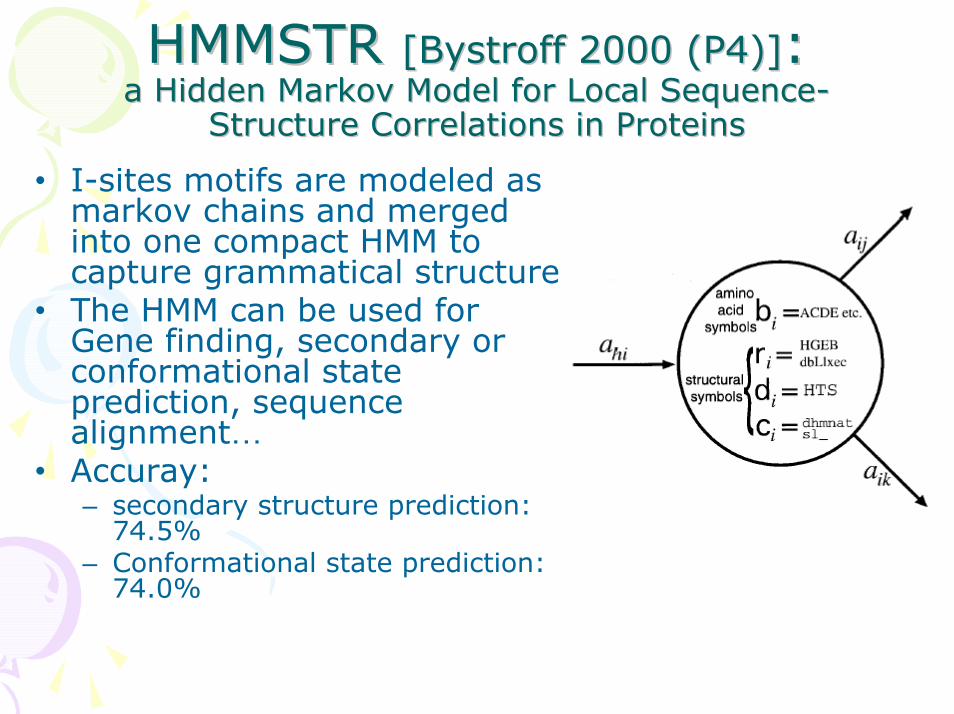
HMMSTR HMMSTR [[BystroffBystroff 2000 (P4)]2000 (P4)]: : a Hidden Markov Model for Local Sequencea Hidden Markov Model for Local Sequence--
Structure Correlations in ProteinsStructure Correlations in Proteins
• I-sites motifs are modeled as markov chains and merged into one compact HMM to capture grammatical structure
• The HMM can be used for Gene finding, secondary or conformational state prediction, sequence alignment…
• Accuray: – secondary structure prediction:
74.5%– Conformational state prediction:
74.0%

Part 3.4: Protein domain segmentation
• DOMAINATION • Pfam Database • Multi-experts
Cut? where???
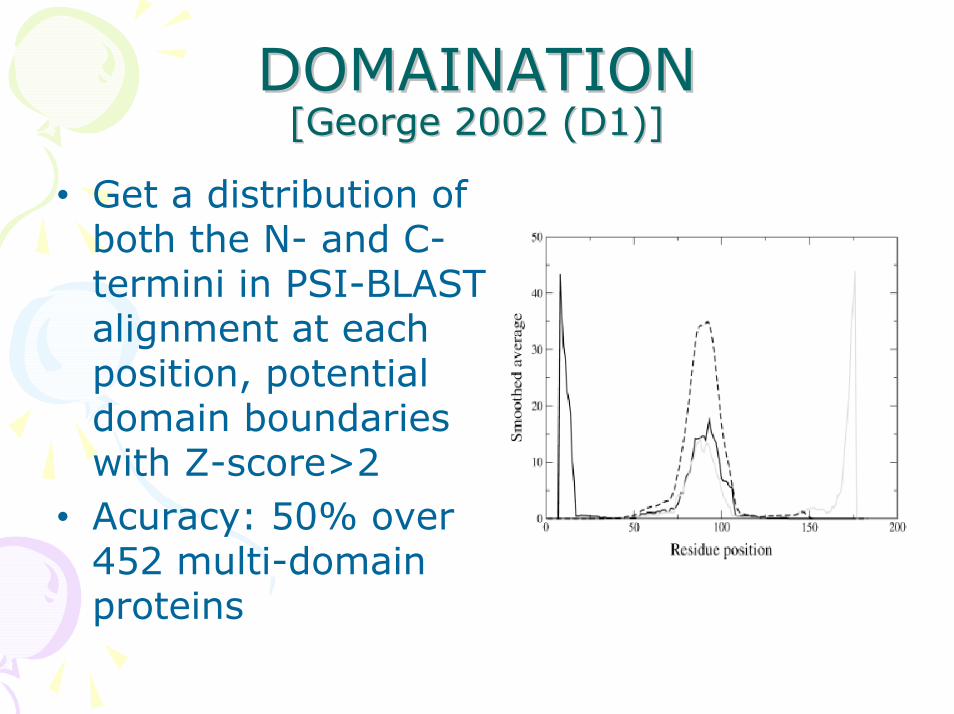
DOMAINATION DOMAINATION [George 2002 (D1)][George 2002 (D1)]
• Get a distribution of both the N- and C-termini in PSI-BLAST alignment at each position, potential domain boundaries with Z-score>2
• Acuracy: 50% over 452 multi-domain proteins

PfamPfam [[SonnhammerSonnhammer 1997 (D2)]1997 (D2)]
• A database of HMMs of domain families• Pfam A: high quality alignments and
HMMS built from known domains• Pfam B: domains built from Domainer
algorithm from the remaining protein sequences with removal of Pfam-A domains

A multiA multi--expert system from expert system from sequence information sequence information
[[NagarajanNagarajan 2003 (D3)]2003 (D3)]
Seed Sequence
Multiple Alignment
blast search
Neural Network
Correlation
Entropy
Sequence Participation
Contact Profile
Secondary Structure
Physio-Chemical PropertiesPutative Predictions
DNA DATA
Intron Boundaries
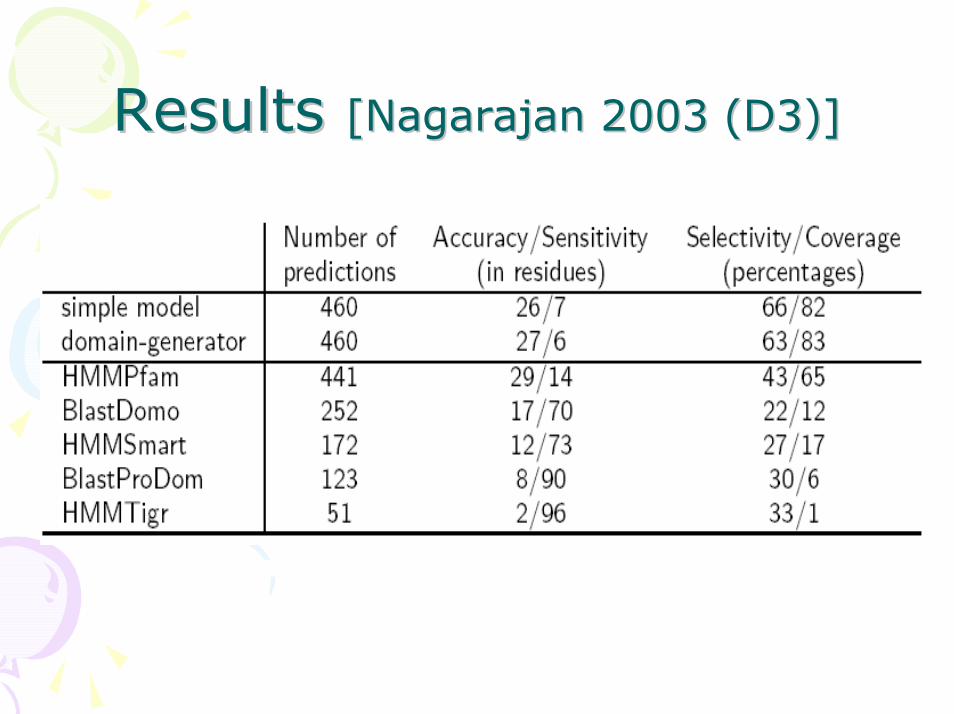
Results Results [[NagarajanNagarajan 2003 (D3)]2003 (D3)]

1. Introduction 2. HMM, SVM and string kernels3. Topics4. Conclusion and future work
Part 4: Conclusion and Future Work
Mars is not too far!?

ConclusionConclusion• Structural genomics plays important role
for understanding our life• Protein structure can be studied from
different perspectives with different methods
• Machine learning is one of the most important tools for understanding genome data
• Protein structure prediction is a challenging task given the data we have now

Future WorkFuture Work• Rank propagation with domain
activation regions• Profile kernel with secondary
structure information for protein classification
• Rank propagation for domain segmentation
• Specialist algorithm for protein conformational state prediction

The EndThe End

Determination of Protein Determination of Protein Structures Structures (back)(back)
• X-ray crystallography The interaction of x-rays with electrons arranged in a crystal can produce electron-density map, which can be interpreted to an atomic model. Crystal is very hard to grow.
• Nuclear magnetic resonance (NMR)Some atomic nuclei have a magnetic spin. Probed the molecule by radio frequency and get the distances between atoms. Only applicable to small molecules.

Hidden Markov Models for Hidden Markov Models for Modeling Protein Modeling Protein [[KroghKrogh 19931993(B3)] (B3)]
(back)(back)
Build HMM from sequences not alignedEM algorithm1. Choose initial length and parameters2. Iterate until the change of likelihood is
small– Calculate expected number of times
each transition or emission is used– Maximize the likelihood to get new
parameters

Thanks to Tony Jebara
Support Vector Machine Support Vector Machine [[BurgesBurges 1998(B4)] 1998(B4)] (back)(back)
• With probability 1-η the bound holds
– l is the number of data points– h is VC dimension
• Structural Risk Minimization– For each hi,
– Get bestα*=argmin Remp(α)
– Choose model with min J(α*,hi)
))4/log()1)/2(log(()()()(l
hlhRJR empηααα −+
+=≤

EMOTIF Database EMOTIF Database [[NevillNevill--manning 1998 (C7)]manning 1998 (C7)]
• A motif database of protein families• Substitution groups from separation score

EMOTIF Database EMOTIF Database [[NevillNevill--manning 1998 (C7)] manning 1998 (C7)] (back)(back)
•All possible motifs are enumerated from sequence alignments

II--SITE Motif Library SITE Motif Library [[BystroffBystroff 1998 (C9)] 1998 (C9)] (back)(back)
• Sequence segments (3-15 amino acids long) are clustered via K-means
• Within each cluster structure similarity is calculated in terms of dme and mda
• Only those clusters with good dme and mda are refined and considered motifs afterwords
Ndme
sji
L
i
i
ij
sji )( 2
1
5
5
1→
=
+
−=→∑ ∑ −
=αα ),(max)( 11,1 iiLiLmda ϕφ ∆∆= +−=

PrISMPrISM [Yang 2003 (P3)] [Yang 2003 (P3)] (back)(back)

PfamPfam [[SonnhammerSonnhammer 1997 (D2)] 1997 (D2)] (back)(back)
• Construction of Pfam A:– Pick seed sequences from several sources
and build seed alignment– Build HMM from seed alignment and use
to it pull in new members and align them to the HMM to get full alignment

Sonnhammer, 1997
PfamPfam [[SonnhammerSonnhammer 1997 (D2)] 1997 (D2)] (back)(back)
• Construction of Pfam B:– Domainer program merges homology segment
pairs into homologous segment sets together with links. This graph is partitioned into domains
– Use domainer program to build alignment from all protein segments not covered by Pfam-A
• Incremental updating– New sequence is added to the full alignment of
existing models if they score above a threshold– If the new sequence causes problems, the
seed alignment will be altered and Pfam-B will be regenerated afterwards.





























