Embed Size (px)
Citation preview

Métabolisme des lipoprotéines: de la physiologie à la maladie
Noël PerettiMD-PHD
UF de Nutrition PédiatriqueHôpital Édouard Herriot

PLAN
• Introduction:– Les lipides: structures et rôles– Besoins en lipides: spécificités de l’enfant
• Anomalies métaboliques par carence de lipides– Hypocholestérolémies familiales
• Anomalies métaboliques par excès de lipides:– Hypercholestérolémie– Hypertriglycéridémie
• Conclusion

Objectifs de cette formation• 1) rappel de la physiologie des lipides
– Réaliser que le cholestérol n’est pas que « mauvais » pour la santé
– Comprendre les mécanismes des maladies par excès ou carence de lipides
• 2) savoir évoquer une hypocholestérolémie familiale devant un syndrome de malabsorption intestinale du nourrisson– Savoir traiter les carences associées
• 3) se sensibiliser au problème des hypercholestérolémies chez l’enfant– Savoir quand et comment dépister

Structure et rôles des lipides
• Cholestérol :– CE– Rôles:
• structure et fonctions cellulaires, • précurseurs stéroïdes, vitD, acides biliaires
• Phospholipides– Diglycéride + acide phosphorique lié à molécule
(choline, éthanolamine, sérine, inositol)– Rôles:
• structure et fonctions cellulaires (SNC)

• Acides Gras : AG– Longueur chaîne carbonée:
• court<C8, moyen C8-C11, long>C12– Nombre et position des insaturations:
• oméga 3, oméga 6– Rôles:
• AGE,• structure et fonctions cellulaires (SNC), • précurseurs eicosanoïdes (inflammation, coagulation)
• Triglycérides : TG– 3 AG estérifiés sur les 3 fonctions alcool d’1 glycérol– Diglycérides et Monoglycérides– + abondants dans alimentation – Rôles:
• énergétique (stockage adipeux)• structure et fonctions cellulaires
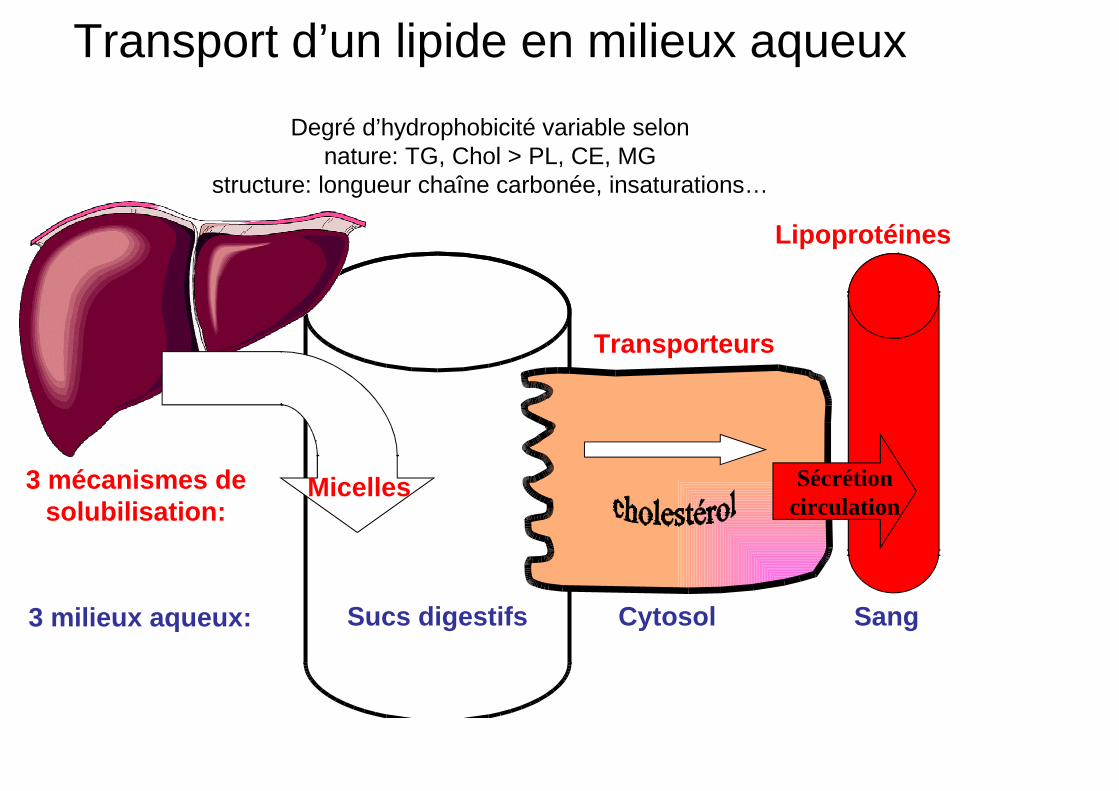
Transport d’un lipide en milieux aqueux
Degré d’hydrophobicité variable selon nature: TG, Chol > PL, CE, MG
structure: longueur chaîne carbonée, insaturations…
Sucs digestifs Cytosol Sang
Micelles Sécrétion circulation
Transporteurs
Lipoprotéines
3 milieux aqueux:
3 mécanismes de solubilisation:
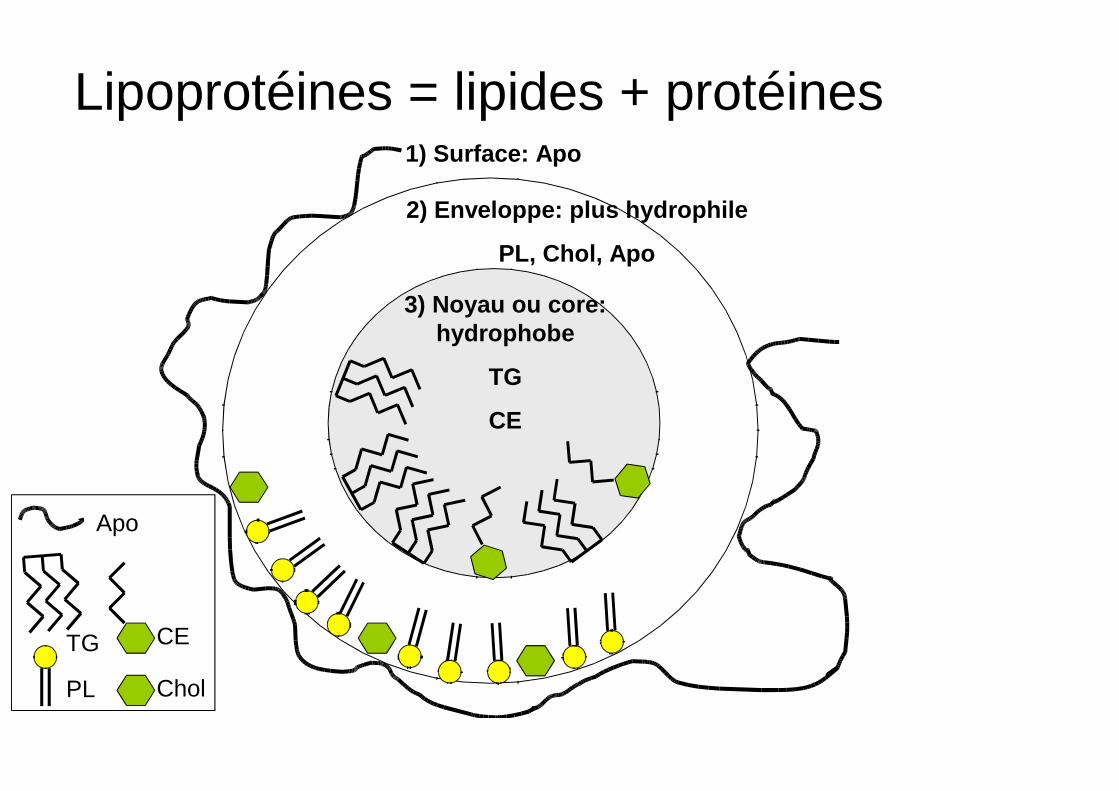
Lipoprotéines = lipides + protéines
3) Noyau ou core: hydrophobe
TG
CE
PL
TG
Chol
CE
2) Enveloppe: plus hydrophile
PL, Chol, Apo
1) Surface: Apo
Apo
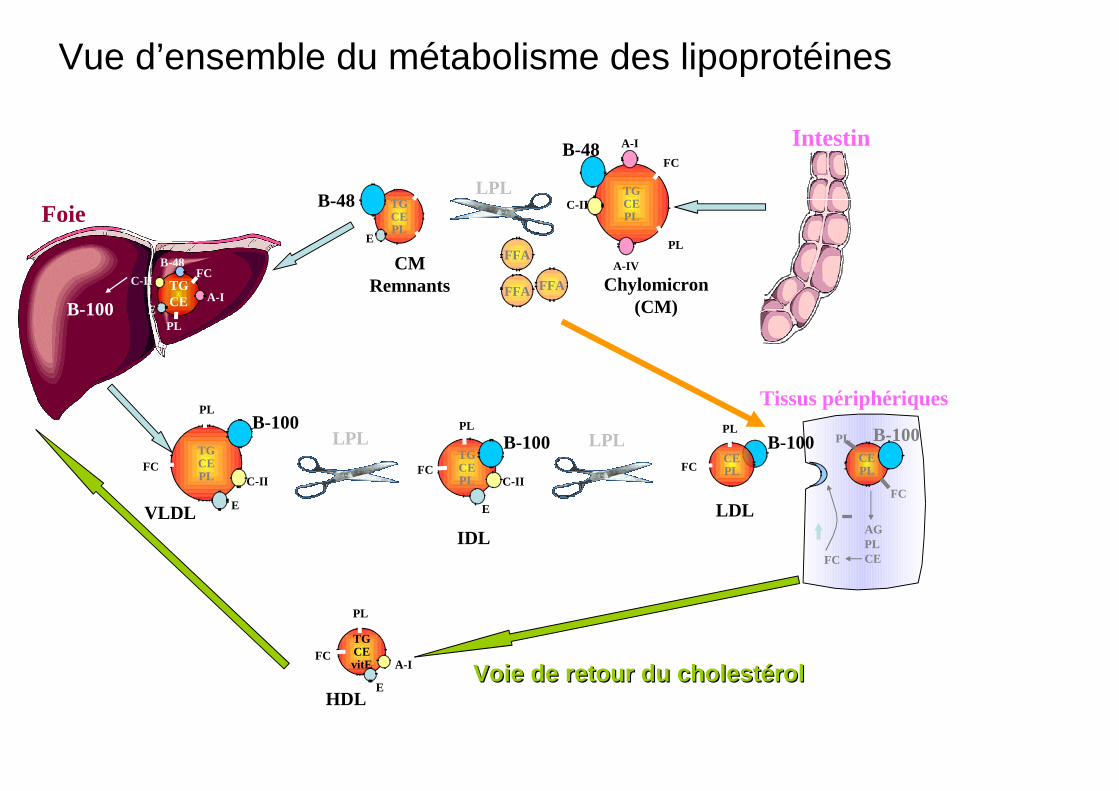
Foie
Tissus périphériques
FC
PL
CEPL
AGPLCEFC
Intestin
-
B-100
E
FC
PL
TGCEvitE
HDL
A-I
VLDL
C-IITGCEPL
B-48
PL
FC
A-I
Chylomicron (CM)
A-IV
Vue d’ensemble du métabolisme des lipoprotéines
Voie de retour du cholestérolVoie de retour du cholestérol
TGCEPL
E
CM Remnants
B-48LPL
FFA
FFA
FFA
B-48
E
FC
A-IC-II
PL
TGCEB-100
E
FC
PL
TGCEPL C-II
B-100B-100
E
FC
PL
TGCEPL C-II
IDL
FC
PL
CEPL
LDL
B-100LPL LPL

Apolipoprotéines: rôles
• Structure des lipoprotéines– Déficit en ApoB → absence de chylomicrons → malabsorption
• Solubilisation des lipideS
• Ligands aux récepteurs membranaires– Anomalie du récepteur de l’ApoB → hypercholestérolémie par
diminution de l’élimination des LDL
• Modulation d’activités enzymatiques– Anomalie de l’Apo C-II → défaut de stimulation de l’enzyme
responsable de la dégradation des TG (LPL) → accumulation TG

Principales apolipoprotéines
• Apo A-I:– > HDL– Transporteur de CETP: HDL2 VLDL– Activateur de LCAT: estérifier le Chol de la
surface des HDL pour l’internaliser et permettre la captation de nouvelles molécules de Chol en surface
– Rôle dans sortie du Chol de périphérie → foie
CE
TG

• Apo B
» Foie: Apo B100 → VLDL → LDL
• Gène» Intestin (et foie): Apo B48 → Chylomicrons
• Transport des TG, Chol et CE d’origine– Alimentaire pour l’Apo B48– Hépatiques pour l’Apo B100
• Capture des LDL par le foie (LDL-R ou ApoB/E-R)
Principales apolipoprotéines
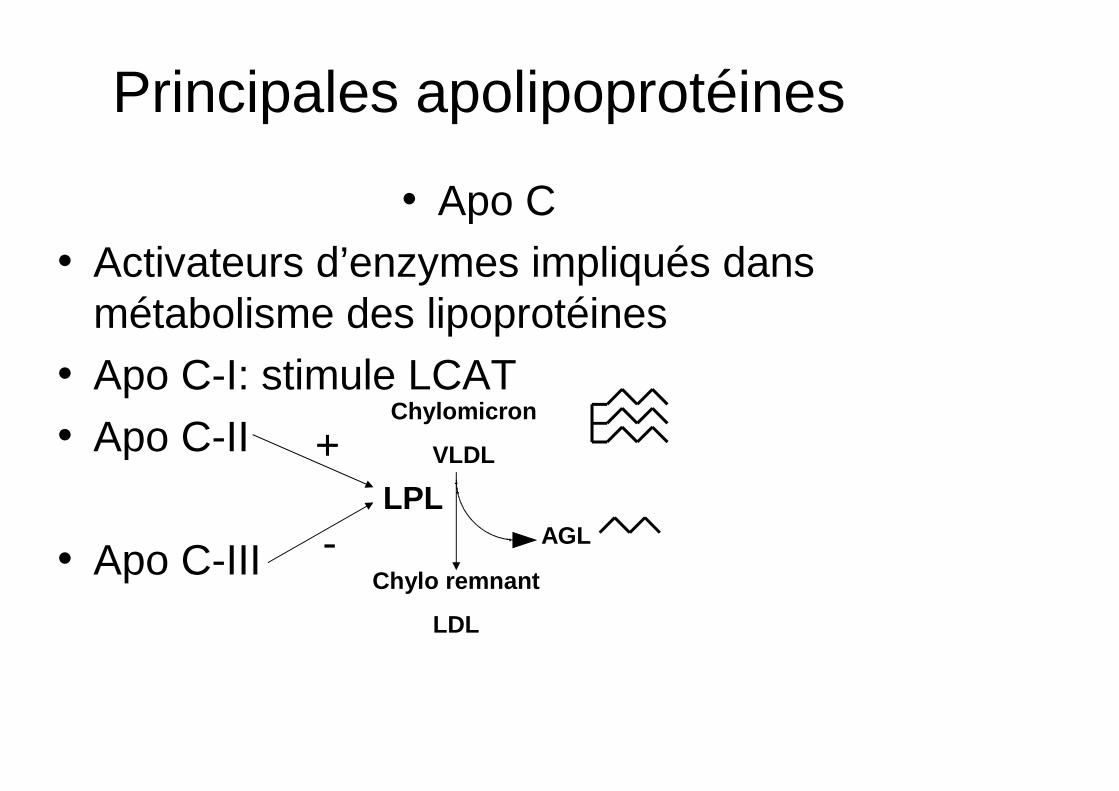
• Apo C• Activateurs d’enzymes impliqués dans
métabolisme des lipoprotéines• Apo C-I: stimule LCAT• Apo C-II
• Apo C-III
Principales apolipoprotéines
Chylomicron
VLDL
Chylo remnant
LDL
AGLLPL
+
-

• Apo E• Sécrétée par le foie• Associée VLDL, IDL, HDL et remnants
chylo (absente LDL)• Liaison hépatique aux:
– R Apo B/E (LDL-R) pour les VLDL, IDL– R Apo E pour remnants chylo
Principales apolipoprotéines
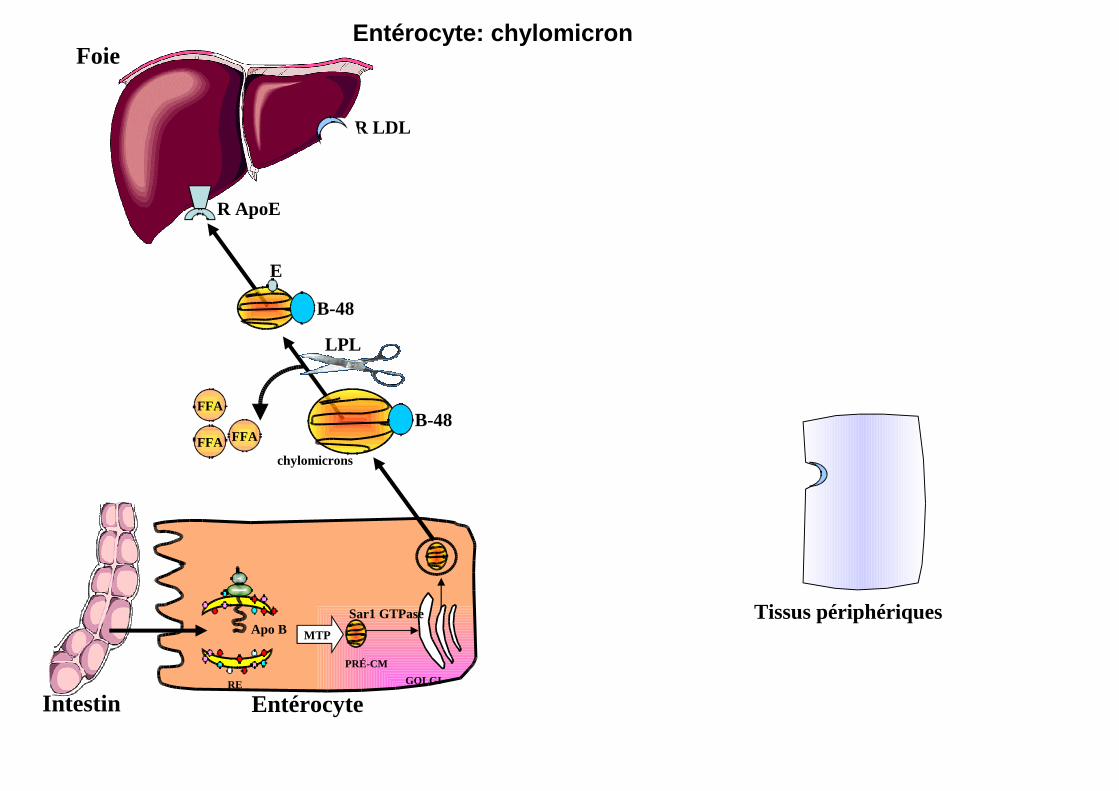
MTP
PRÉ-CM
RE
Apo B
GOLGI
Sar1 GTPase
E
Foie
Tissus périphériques
Intestin Entérocyte
Entérocyte: chylomicron
R ApoE
LPL
FFA
FFA
FFA
B-48
chylomicrons
B-48
R LDL
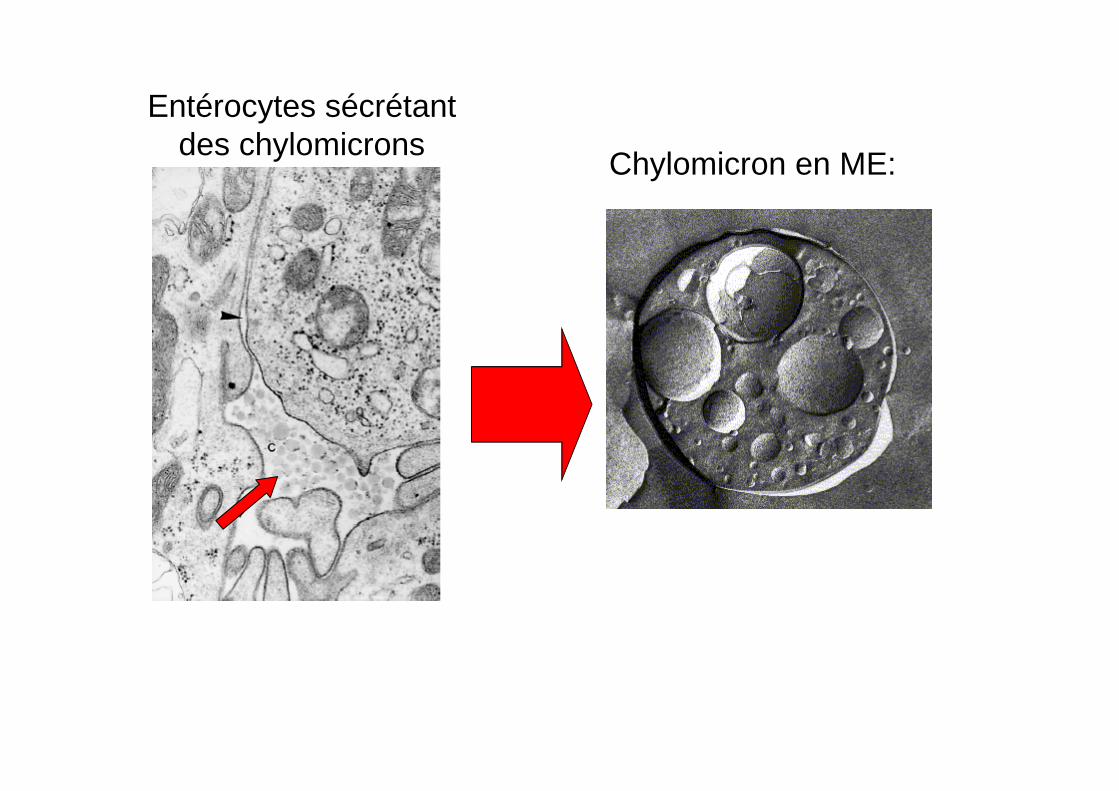
Chylomicron en ME:
Entérocytes sécrétant des chylomicrons
MTP
PRÉ-CM
RE
Apo B
GOLGI
Sar1 GTPase
chylomicron
LPL
FFA
FFA
FFA
B-48
B-48
E
R ApoE
Foie
Tissus périphériques
Intestin Entérocyte
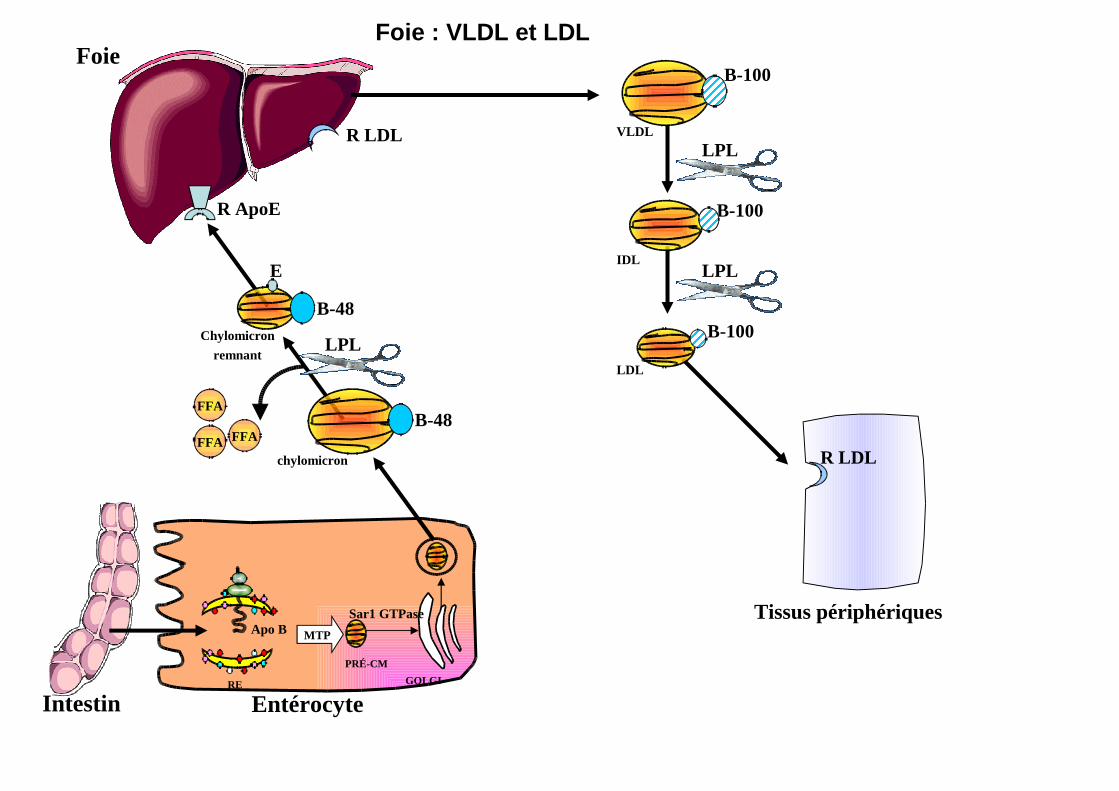
Chylomicron
remnant
R LDL
Foie : VLDL et LDL
B-100
B-100
VLDL
IDL
LDL
B-100
LPL
LPL
R LDL
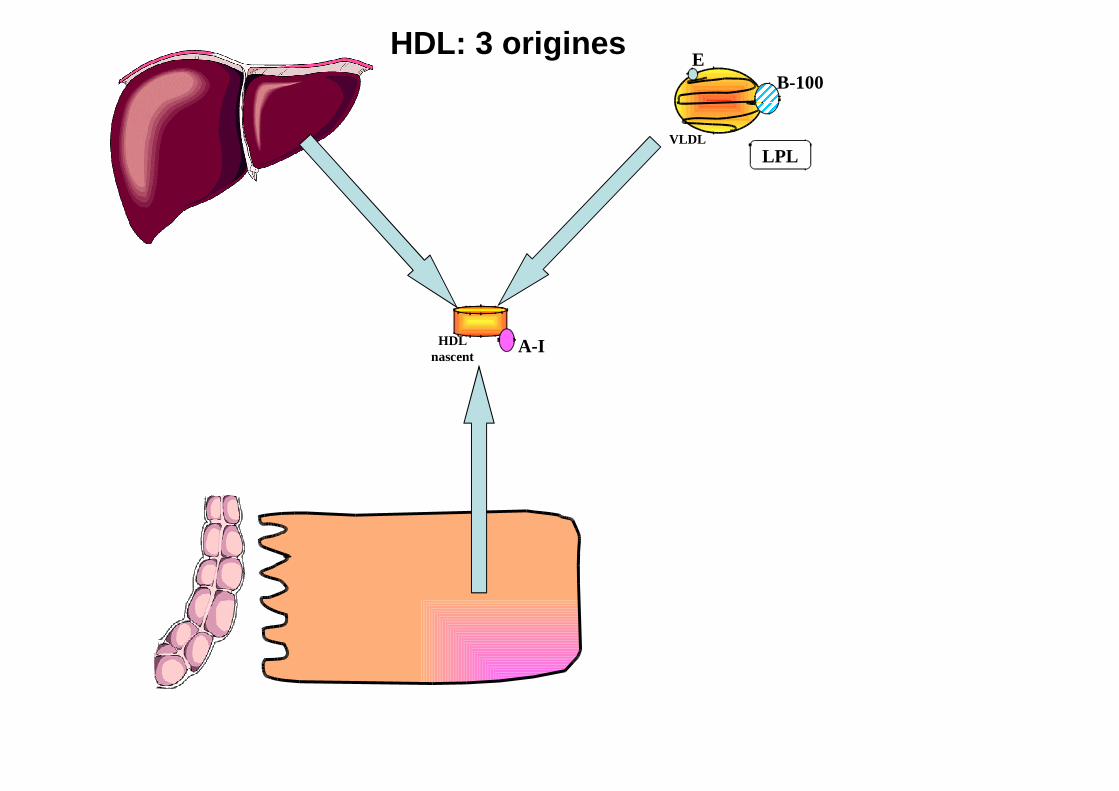
HDL nascent
A-I
HDL: 3 originesB-100
VLDLLPL
E
MTP
PRÉ-CM
RE
Apo B
GOLGI
Sar1 GTPase
chylomicron
LPL
FFA
FFA
FFA
B-48
B-48
E
R ApoE
Foie
Tissus périphériques
Intestin Entérocyte
B-100
Chylomicron
remnant
VLDL
B-100
IDL
LDL
B-100
R LDL
R LDL
E
E
LPL
HDL nascent
E
A-I
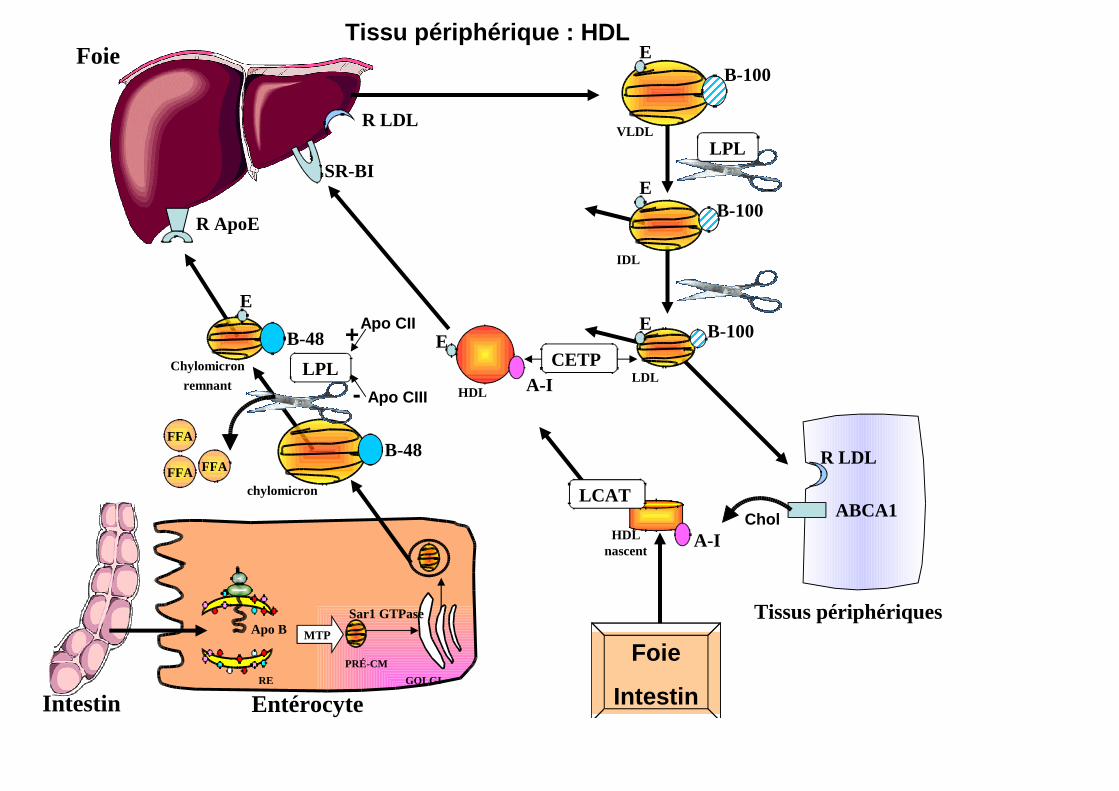
Tissu périphérique : HDL
Apo CII+
Apo CIII-
Foie
Intestin
LCAT
A-I
E
HDL
CETP
SR-BI
ABCA1Chol

Hypolipidémies: mécanismes
∀ ↓ entrée des lipide dans LP:↓ absorption intestinale:
• stéatose entérocytaire et↓ chylomicrons– Hypobéta, Abétalipoprotéinémie, Anderson
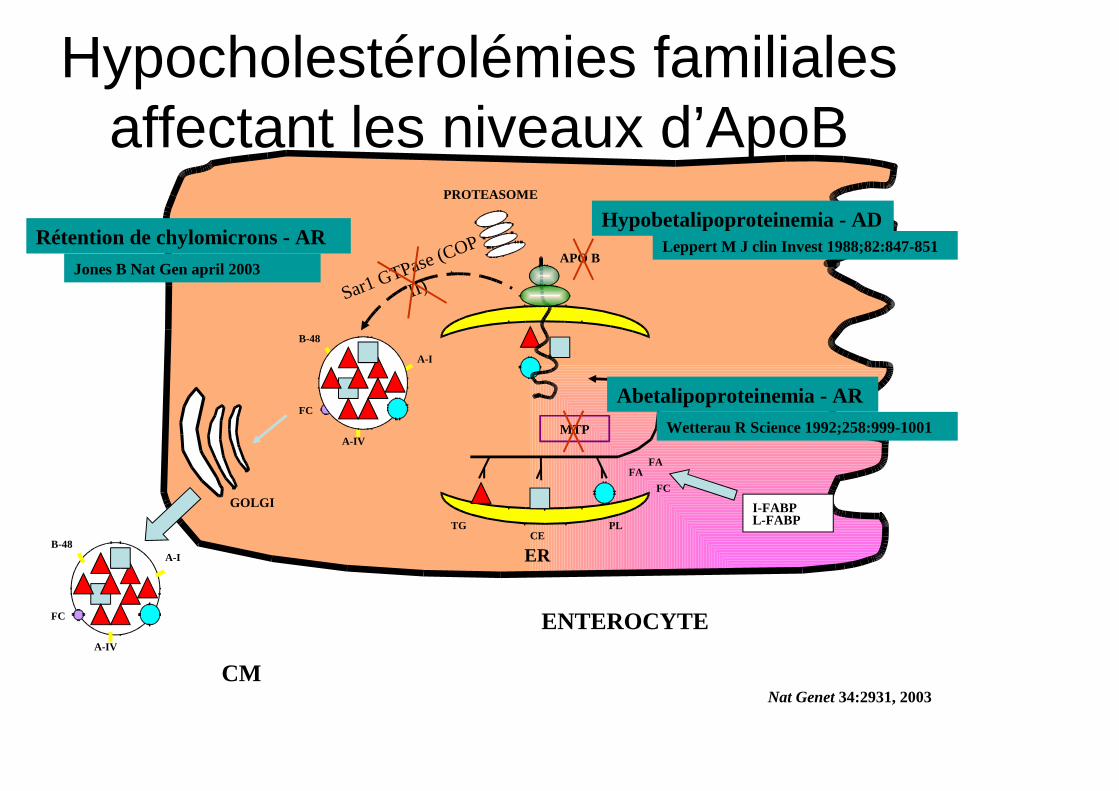
ENTEROCYTE
PROTEASOME
FAFA
FC
APO B
TGCE
PL
MTP
GOLGI
A-I
B-48
A-IV
FC
B-48
A-IV
FC
A-I
I-FABPL-FABP
ER
Sar1 GTPase (COP
II)
Nat Genet 34:2931, 2003CM
Abetalipoproteinemia - ARWetterau R Science 1992;258:999-1001
Hypobetalipoproteinemia - ADLeppert M J clin Invest 1988;82:847-851Rétention de chylomicrons - AR
Jones B Nat Gen april 2003
Hypocholestérolémies familiales affectant les niveaux d’ApoB
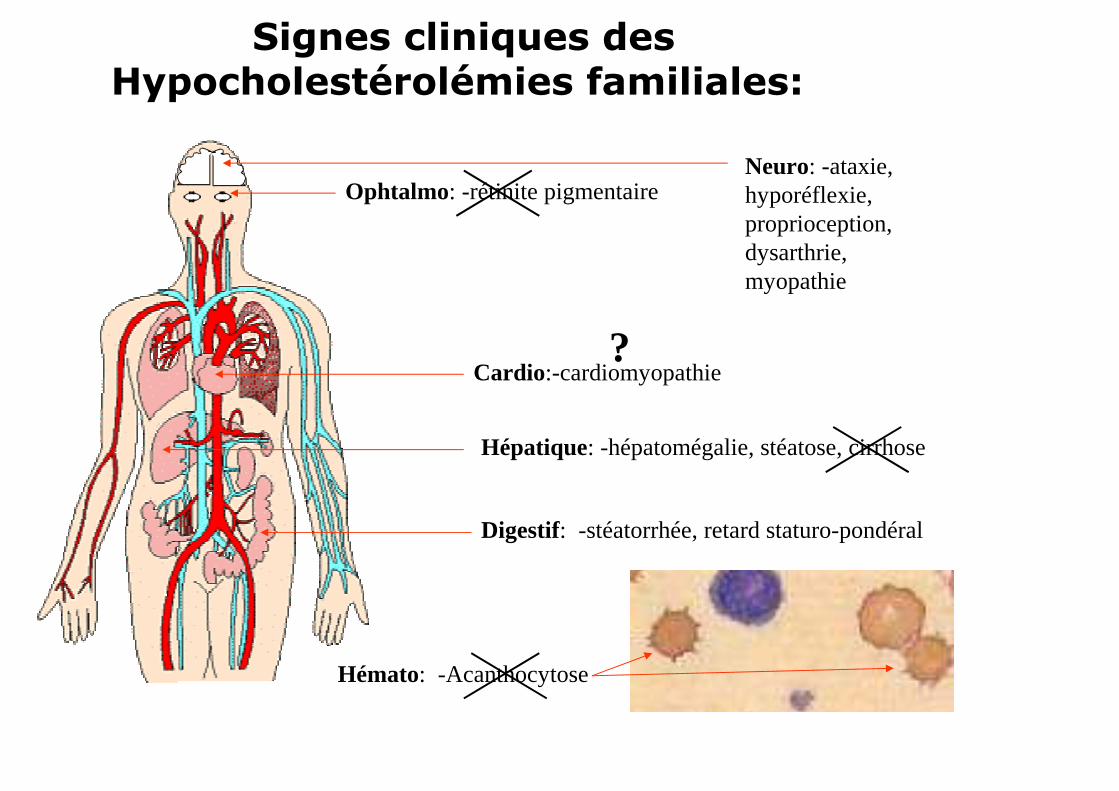
Signes cliniques des Hypocholestérolémies familiales:
Neuro: -ataxie, hyporéflexie, proprioception, dysarthrie, myopathie
Ophtalmo: -rétinite pigmentaire
Cardio:-cardiomyopathie
Hépatique: -hépatomégalie, stéatose, cirrhose
Digestif: -stéatorrhée, retard staturo-pondéral
Hémato: -Acanthocytose
?
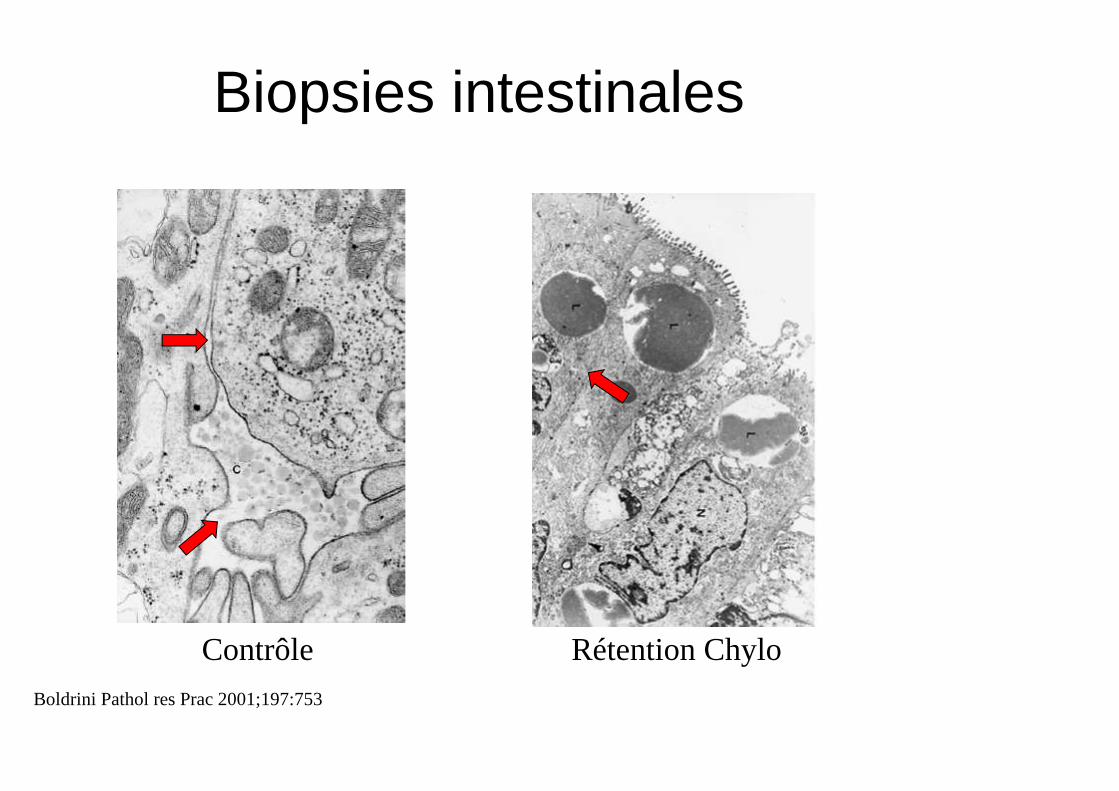
Biopsies intestinales
Contrôle Rétention Chylo
Boldrini Pathol res Prac 2001;197:753

Hypolipidémies: mécanismes
∀ ↓ entrée des lipide dans LP:↓ absorption intestinale:
• stéatose entérocytaire et↓ chylomicrons– Hypobéta, Abétalipoprotéinémie, Anderson
↓ efflux des cellules périphériques: • rétention dans cellules (macrophages) et ↓ HDL
– Anomalie ABCA1 (m. de Tangier): hypertrophie amygdales orangées, HSM, ophtalmo
– Déficit LCAT: protéinurie, IRC, anémie, ophtalmo
• ↓ transfert des lipides entre LP:↓ de Chol ester dans HDL
• Déficit CETP
athérogènes
MTP
PRÉ-CM
RE
Apo B
GOLGI
Sar1 GTPase
chylomicron
LPL
FFA
FFA
FFA
B-48
B-48
E
R ApoE
Foie
Intestin Entérocyte
B-100
Chylomicron
remnant
VLDL
B-100
IDL
LDL
B-100
R LDL
R LDL
E
E
LPL
HDL nascent
A-I
E
HDL
CETP
LCAT
E
A-I
SR-BI
ABCA1
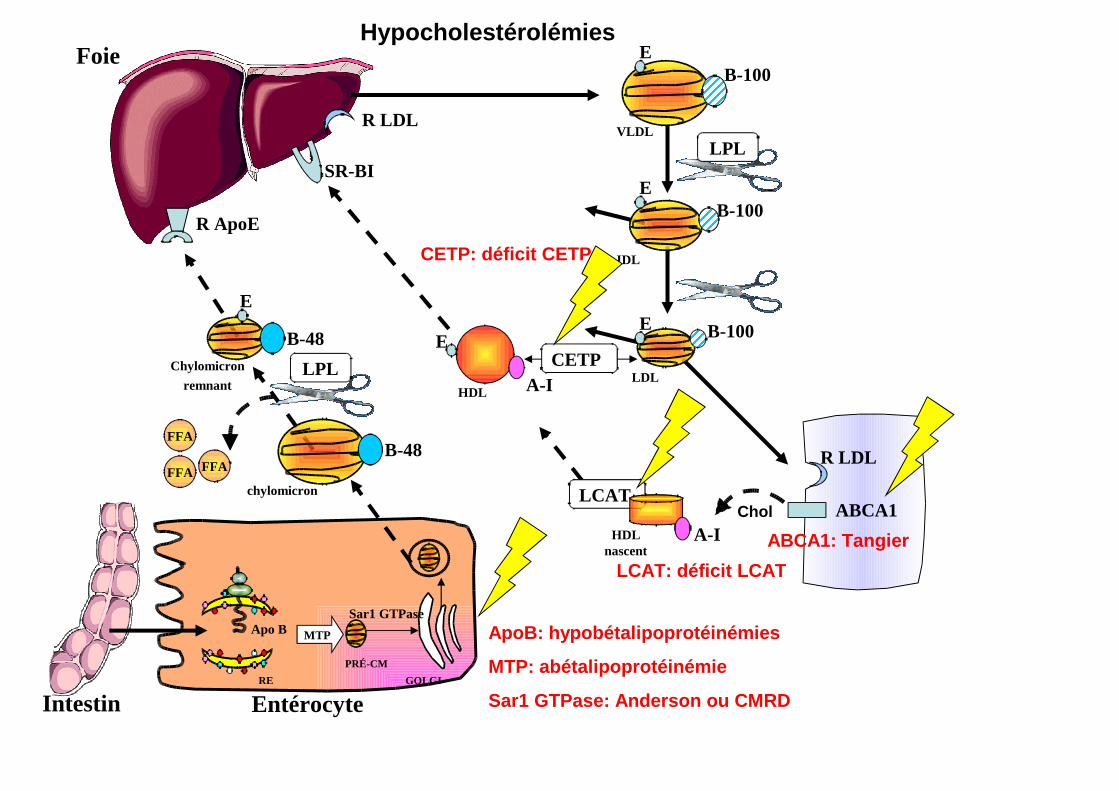
Hypocholestérolémies
ApoB: hypobétalipoprotéinémies
MTP: abétalipoprotéinémie
Sar1 GTPase: Anderson ou CMRD
ABCA1: Tangier
LCAT: déficit LCAT
CETP: déficit CETP
Chol

Hyperlipidémies: mécanismes
∀ ↑ LP “ pro " athérogènes:↑ production:
• Lp(a)
↓ dégradation:• R-LDL diminué ou anormal:
– hypercholestérolémie familiale hétérozygote (AD, fréquente 1/200-1/500, athérome précoce)
• Apo B100 anormale ne reconnaissant plus son récepteur• Anomalie Apo E ou R-Apo E: ↓ épuration chylo et VLDL∀ ↓ activité de LPL: ↓ hydrolyse TG∀ ↓ activité Apo C-II: ↓ stimulation LPL: ↓ hydrolyse TG
MTP
PRÉ-CM
RE
Apo B
GOLGI
Sar1 GTPase
chylomicron
LPL
FFA
FFA
FFA
B-48
B-48
E
R ApoE
Foie
Tissus périphériques
Intestin Entérocyte
B-100
Chylomicron
remnant
VLDL
B-100
IDL
LDL
B-100
R LDL=R ApoB/E
R LDL=R ApoB/E
E
LPL
HDL nascent
A-I
E
HDL
CETP
LCAT
E
A-I
SR-BI
ABCA1
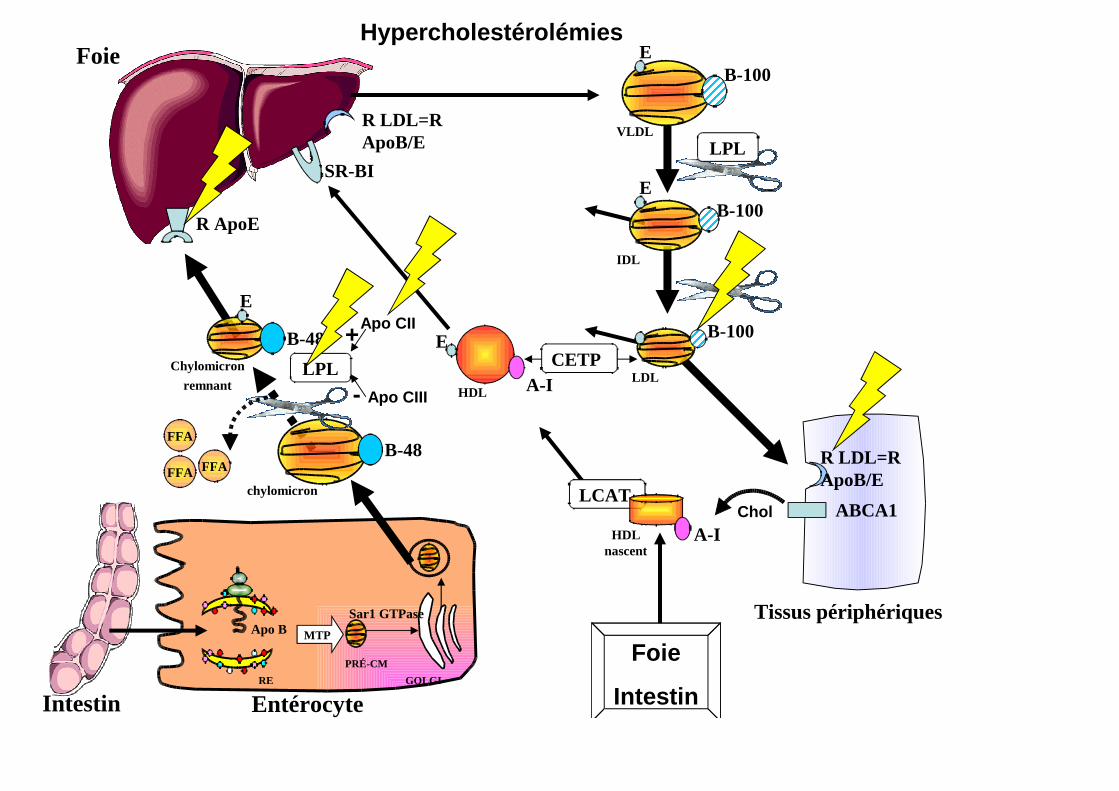
Hypercholestérolémies
Apo CII+
Apo CIII-
Chol
Foie
Intestin

Hyperlipidémies: classifications
• Primaires:
– Anomalie gène
• Secondaires:↑ des lipides circulants suite à une maladie
affectant leur élimination ou métabolisme• Syndromes cholestatiques• Syndromes néphrotiques• Diabète sucré• hypothyroïdie
Structure (Apo)
Métabolisme (Apo, R-Apo, Enzymes)

• 1) Electrophorèse lipides (Frederikson)• 2) Taux Chol et TG• 3) Physiopathologie
– Hypercholestérolémie pure: forme familiale ou polygénique
– Hyperlipidémie familiale combinée– Hypertriglycéridémie familiale (surpoids,
diète…)– Dysbétalipoprotéinémie: anomalie apo E
Hypercholestérolémies: classifications

Hyperlipidémies: résumé
Auto DominantFréquente 1/100-1/200
Clinique: variable, > 25 ans
HyperchylomicronémieExceptionnelle (1/100.000)↑ Chylo par ↓↓↓↓ activité LPL (anomalie LPL ou Apo CII)
Enfant: Xanthomes éruptifs, HSM, lipémie rétinienne,
pancréatites
Familiale HétérozygoteAuto Dominant →→→→ cf parents
↑ LDL par anomalie:R-LDL
Fréquente 1/200-1/500Déficience Familiale ApoBAD, ApoB100, 1/700-1/1000
Enfant: AsymptomatiqueAdulte: athérome précoce X50
Familiale HomozygoteExceptionnelle (1/1M)
Xanthomes cutanés <5 ansXanthomes tendineux 10 ans
Polygénique(non AD, bilan famille -)
+ communeClinique: idem Famil Hétéroz
Hyperlipoprotéinémie combinée familiale
HypertriglycéridémiesHypercholestérolémies
pures
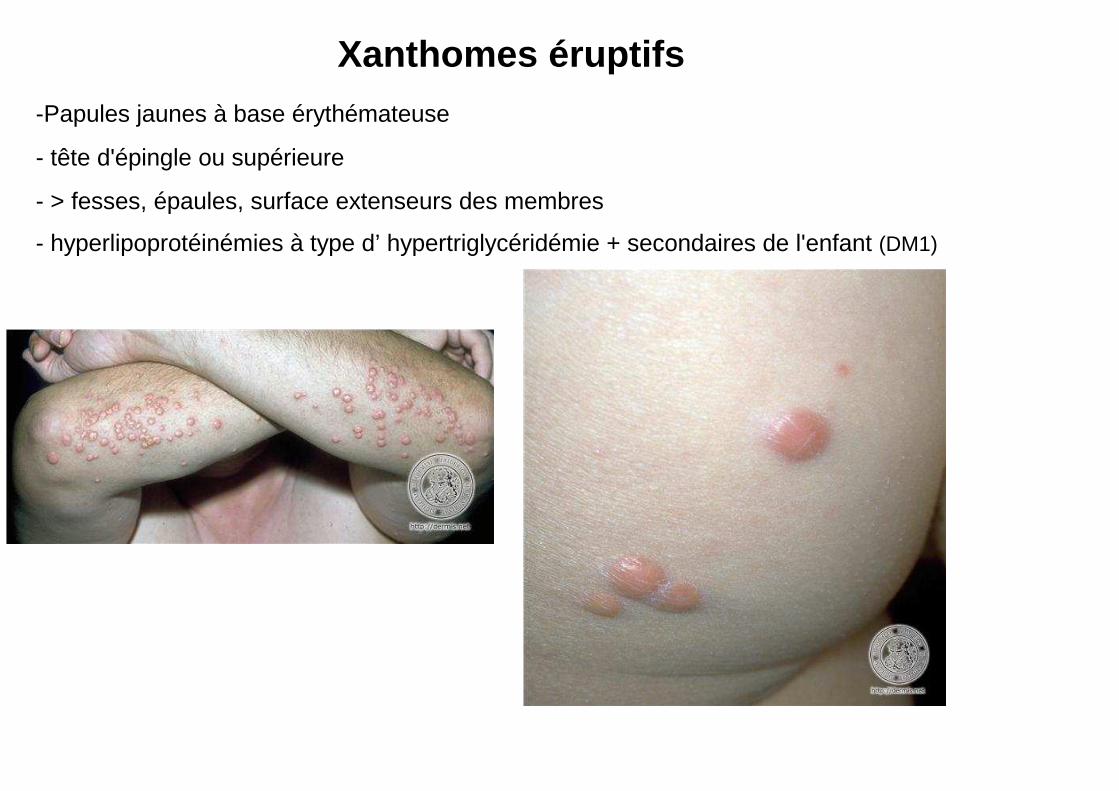
-Papules jaunes à base érythémateuse
- tête d'épingle ou supérieure
- > fesses, épaules, surface extenseurs des membres
- hyperlipoprotéinémies à type d’ hypertriglycéridémie + secondaires de l'enfant (DM1)
Xanthomes éruptifs

Arc cornéen
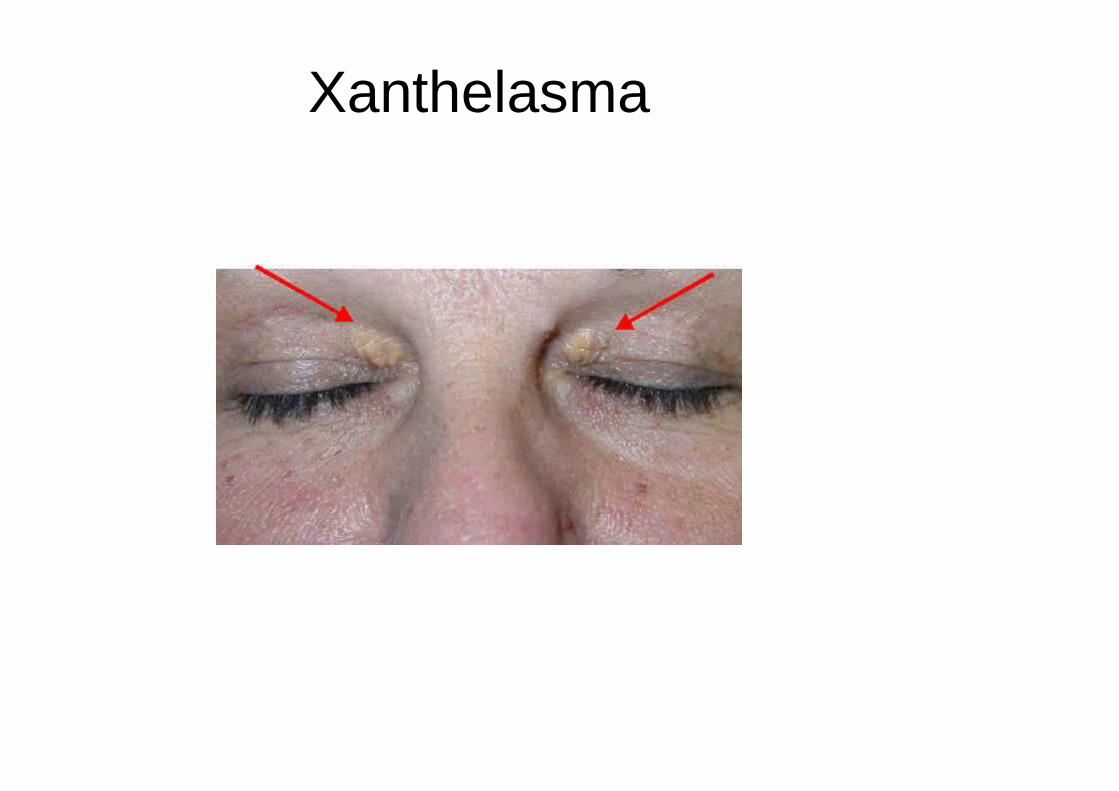
Xanthelasma

Hyperlipidémies: CAT
• 1) confirmer l’anomalie bio:– Redoser à 1m d’intervalle
< 1,5Apo B
< 1,14<1,48
> 100> 130
< 0,85< 1,02
< 75< 90
Triglycérides Avant 10 ans Après 10 ans
> 1,03> 40HDL Cholestérol
< 3,35> 130< 2,8< 110LDL Cholestérol
< 5,16> 200< 4,4< 170Cholestérol total
mmol/Lmg/dLmmol/Lmg/dL
ElevéSouhaitable

• 2) éliminer une forme secondaire:
– Métabolique: • hypothyroïdie, diabète, obésité, hypercalcémie idiopathique,
glycogénose, sphingolipidose.
– Viscéral: • Foie: cholestases, • Rein: syndrome néphrotique.
– Médicamenteuse: • corticoïdes, bétabloquants, contracetif oraux, trétinoïne,
alcool
– Divers: • Klinefelter, anorexie mentale

• 3) Evaluer risque cardiovasculaire:
• Enquête familiale: – 1 des 2 parents atteint– complications CV précoces (< 55 ans chez ♂ et <60 ans chez ♀) ?– traitement hypolipidémiant ?
• Faire bilan lipidique famille – 1er degré, en cas de naissance attendre l'âge de 2 ans
• Cliniquement : dépôts de cholestérol tardifs:
– xanthelasma, xanthome cutanés ou tendineux, arc cornéenHTA, souffles vasculaires (athérome), obésité.Pancréatites aiguës (hypertriglycéridémie type I)
• Profil lipidique: après 6 mois de traitement LDL toujours supérieur à 190mg/dL (4,89 mmol/L)
• Mesure EIM (épaisseur intima média)

• 4) traitement: Triple approche
– 1) mode de vie:• Sport, ↓ surpoids• Ado: stop tabac
– 2) diète:• ↓ graisses, ↑ légumes-fruits:
– graisse < 30 % des besoins énergétiques– cholestérol < 300 mg/j
• équilibrer: – ↓ AG saturés < 10% des apports énergétiques– ↑ AG polyinsaturés (surtout oméga 3) et monoinsaturés
– 3) médicaments:• ↓ absorption des graisses: Chélateur: Questran
– Compliance médiocre car mauvaise palatabilité– Carence vit liposoluble: surveillance et supplémentation
• ↓ synthèse endogène: statines (inhibiteurs HMGCoA R)• futur: inhibiteur spécifiques cholestérol: ézétimibe

Conclusion
• Hypocholestérolémie au cours d’une diarrhée chronique du nourrisson:– Pas forcément secondaire !
• Hypercholestérolémie:– Existe chez l’enfant– découverte enfant parent– Dépistage de la famille– Diète + hygiène vie + médicamenteux




























![Mosta Physiologie de La Synapse Neuroneuronale[1]](https://img.pdfslide.us/doc/110x75/55cf8f09550346703b9848aa/mosta-physiologie-de-la-synapse-neuroneuronale1.jpg)

