Embed Size (px)
Citation preview

MEET THE PLATELET
K. Krishnan MD Department of Internal
Medicine
Acknowledgements My teachers at PGI,
Chandigarh, Hammersmith Hospitals, UK and U of Michigan, Ann Arbor
American Society of Hematology for images

LEARNING OBJECTIVES
Understand platelet development and function
Understand the classification of platelet disorders
Understand the clinical manifestations of platelet disorders
Understand the methods available to diagnose platelet disorders
Understand the pharmacological agents used to treat platelet disorders

PLATELET HEMATOLOGY
Platelet development and kinetics Platelet tests Clinical aspects of platelet disorders Qualitative platelet disorders
Platelet function disorders Congenital Acquired
Quantitative platelet disorders Thrombocytopenia Thrombocytosis
Platelet therapeutics

PLATELET DEVELOPMENT Small anucleate fragments formed from the
megakaryocyte cytoplasm Characteristic discoid shape Hematopoeitic stem cells are converted into MGKs by
exposure to the specific growth factor, thrombopoietin Tpo initiates a maturation program
Amplifies the megakaryocyte DNA Synthesis of platelet-specific proteins Cytosketal elements, membrane systems and receptor proteins
are bulk produced Platelet production begins when microtubules aggregate in the
cell cortex, elaborate pseudopodia These pseudopodia develop into proplatelets Platelets are assembled at the end of proplatelets Microtubules deliver intracellular organelles into these
proplatelets Platelets are released from the ends of proplatelets

PLATELET KINETICS
Platelets are produced in bone marrow by megakaryocytes
MGKs produce platelets by cytoplasmic shedding into bone marrow sinusoids
1000-5000 platelets per MGK 35k to 50k platelets per microl of whole
blood per day Platelet life span 8-10 days Removed from circulation by monocyte-
macrophage system

Determinants of megakaryocytopoiesis and thrombopoiesis.
Battinelli E et al. PNAS 2001;98:14458-14463
©2001 by National Academy of Sciences

EARLY MEGAKARYOCYTE
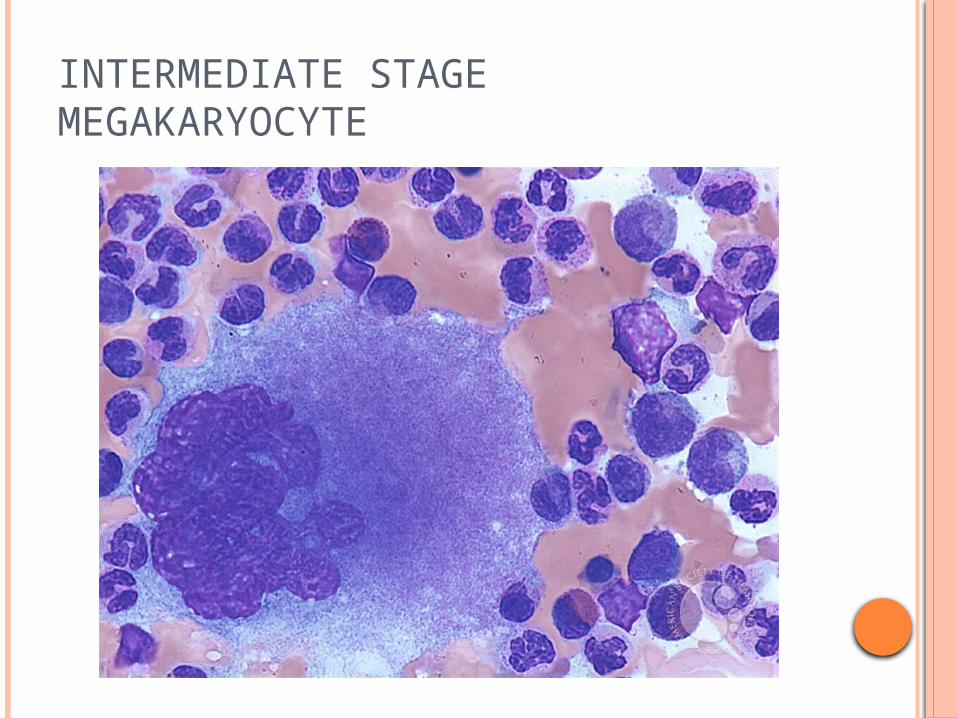
INTERMEDIATE STAGE MEGAKARYOCYTE
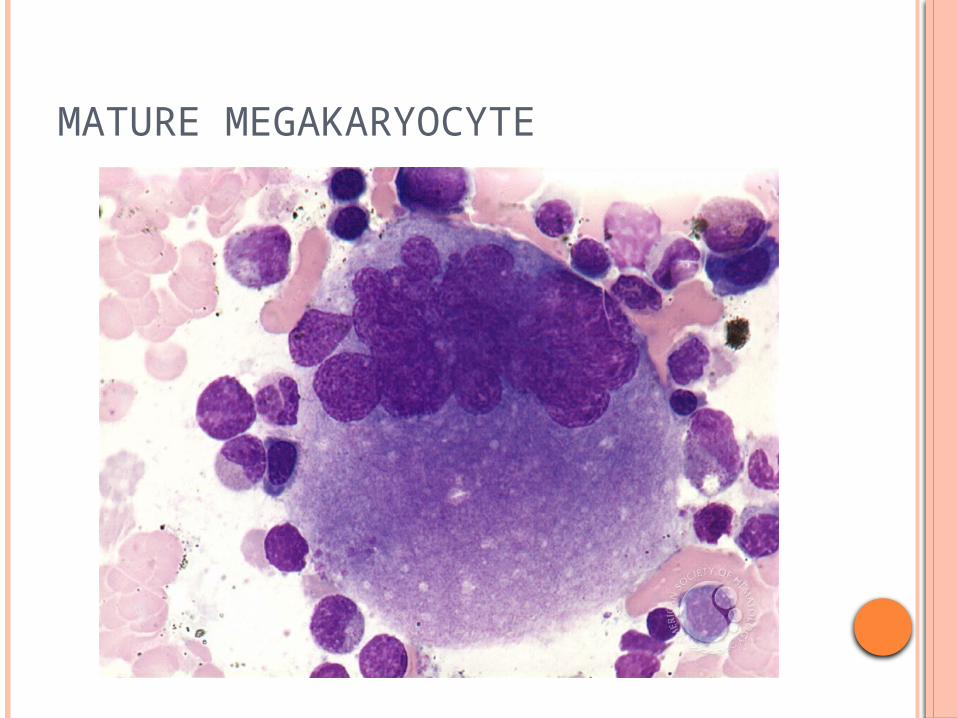
MATURE MEGAKARYOCYTE

PLATELET FUNCTIONS
Adhere to sites of vascular injury Generate biological mediators Secrete granule contents Form multicellular aggregates Serve as a nidus for plasma coagulation
reactions

PLATELET FUNCTIONS
For these platelet functions, Structural rearrangements Utilize multiple membrane receptors
Bind small molecule mediators Bind adhesive glycoproteins and constituents of
vascular endothelium Activate a network of complex signaling pathways

HOW TO ASSESS PLATELETS
Automatic/Manual Platelet count Peripheral smear Bone marrow examination and specialised
tests Platelet function testing
PFA test/screening test Specific tests using platelet aggregometry (many
methods/instruments) Thrombin, Collagen, ADP, Arachidonic acid, Ristocetin
Antibody assays


CLINICAL FEATURES IN PLATELET DISORDERS
Splenomegaly/Chronic liver disease Petechiae or dry purpura
Begins in the dependent portions of the body due to venous pressure-ankles and feet in an ambulatory patient
Occurs when platelet count decreases; not seen in disorders of platelet function
Differentiate dry non-palpable purpura from palpable purpura seen in vasculitis eg. Henoch-Schonlein purpura
Wet purpura- look in mouth, oral mucosa Sign of severe thrombocytopenia Denotes risk for significant hemorrhage
Excessive bruising Seen in disorders of platelet function and number

CLINICAL FEATURES OF PLATELET DISORDERSHIGH PLATELET COUNT
Thrombocytosis Symptoms due to high platelet count
Easy bruising Bleeding due to platelet dysfunction Thrombotic tendencies TIAs Erythromelalgia Mild splenomegaly

BRUISING
PU
RP
UR
A
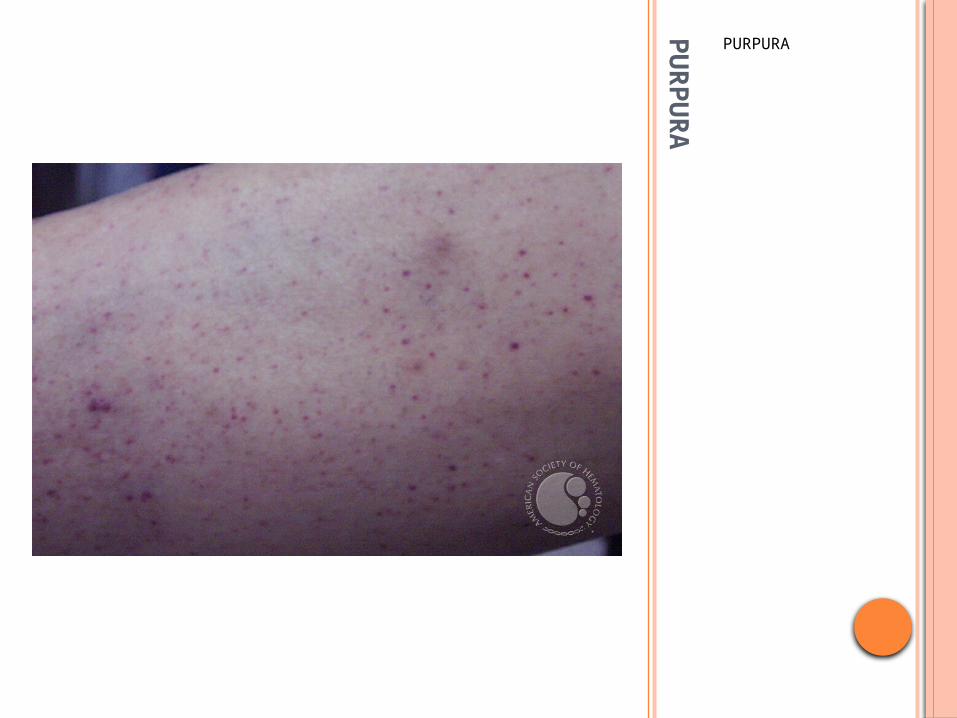
PURPURA

PU
RP
UR
A
Seen in dependent areas of the body

Palpable purpura: Henoch-Schonlein Purpura

Date of download: 6/10/2012Copyright © 2012 American Medical Association.
All rights reserved.
SCURVY
Arch Dermatol. 2010;146(8):938-938. doi:10.1001/archdermatol.2010.162

PLATELET FUNCTION DISORDERS Defects of platelet-vessel wall interaction (disorders of
adhesion) Von Willebrand disease Bernard Soulier syndrome
Defects in platelet- platelet interaction (disorders of aggregation) Congenital afibrinogenemia Glanzman’s thrombasthenia
Disorders of platelet secretion and abnormalities of granules Storage pool deficiency Quebec platelet disorders
Disorders of platelet secretion and signal transduction Defects in platelet- agonist interaction (TXA2, COX, Collagen, ADP)
Defects in cytoskeletal regulation Wiskott- Aldrich syndrome
Disorders of platelet coagulant-protein interaction (membrane phospholipid defects) Scott syndrome



INHERITED PLATELET DISORDERS
Rare, heterogenous group Not often seen in clinical practice Yet fascinating abnormalities that provide
insight into normal platelet biochemistry and physiology

INHERITED PLATELET DISORDERS
Disorders of Platelet membrane Platelet granule packaging Hereditary macrothrombocytopenias Platelet signaling disorders Platelet coagulant function disorders

PLATELET MEMBRANE DISORDERSGLANZMAN’S THROMBASTHENIA
“Weak platelets” Platelets carry out most of the functions Platelet count is normal Platelet morphology is normal Platelets adhere normally to vascular
endothelium Platelets secrete granules and perform
normal signalling functions Platelets DO NOT AGGREGATE due to loss
of GpIIb/IIIa receptor Normally this complex binds fibrinogen linked
into multicellular aggregates

PLATELET MEMBRANE DISORDERSGLANZMAN’S THROMBASTHENIA
Inherited Most are compound heterozygotes Life long mucosal bleeding Life long platelet transfusions Recombinant Factor VII
Acquired Rare, autoantibodies that bind to GpIIb/IIIa
epitopes Seen in ITP and in patients with normal counts Steroids may not work Immunotherapy/Rituxan may work

BERNARD SOULIER SYNDROME
Autosomal recessive Gp1b deficiency or defect Gp1b is the principal receptor for vWF No functioning Vwf receptor Platelets cannot adhere to vascular endothelium Giant platelets and thrombocytopenia
Large size due to lack of interaction between actin binding proteins in platelet cytoskeleton and cytoplasmic domain of gp1b
Lack of gp1b bound sialic acid residues causes shortening of platelet survival leading to thrombocytopenia
Platelet transfusions, DDAVP and fibrinolytic inhibitors like EACA

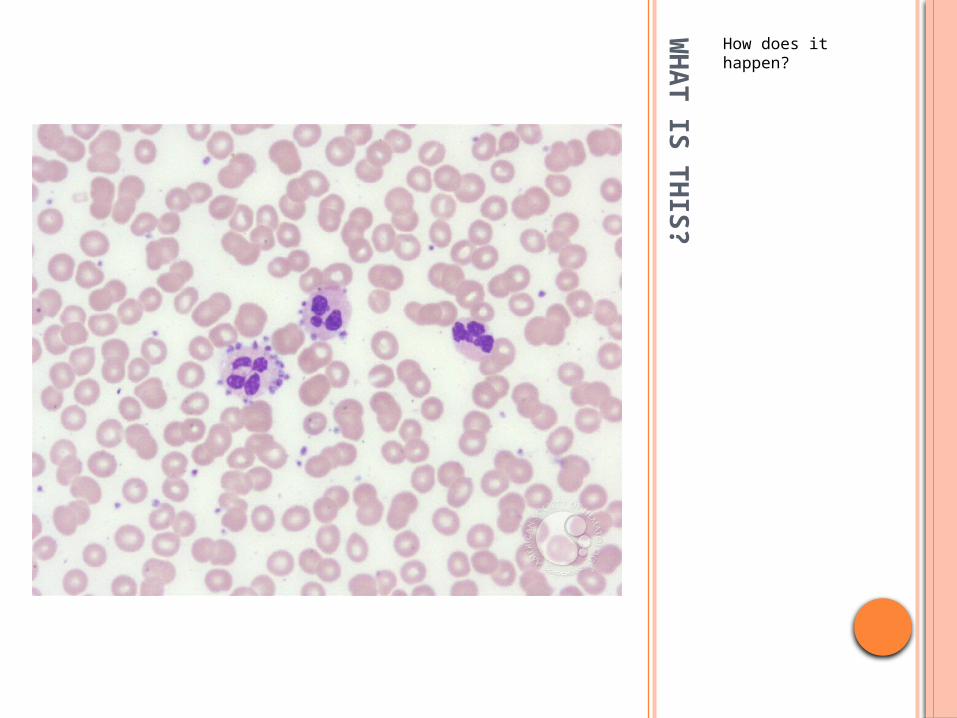
WHAT IS THIS?

ACQUIRED QUALITATIVE PLATELET DISORDERS
Drugs Aspirin
Treat with platelet transfusions for severe bleeding NSAIDs Glycoprotein inhibitors like Abciximab ADP receptor antagonists like Clopidrogel
Uremia Toxic effects of uremia plasma, impaired platelet-
vessel wall adhesion and increased production of NO Platelet transfusions ineffective Treat with dialysis, DDAVP, conjugated estrogens
Myeloproliferative disorders Myelodysplastic disorders
WH
AT IS
TH
IS?
How does it happen?
PS
EU
DO
-TH
RO
MB
OC
YTO
PEN
IA
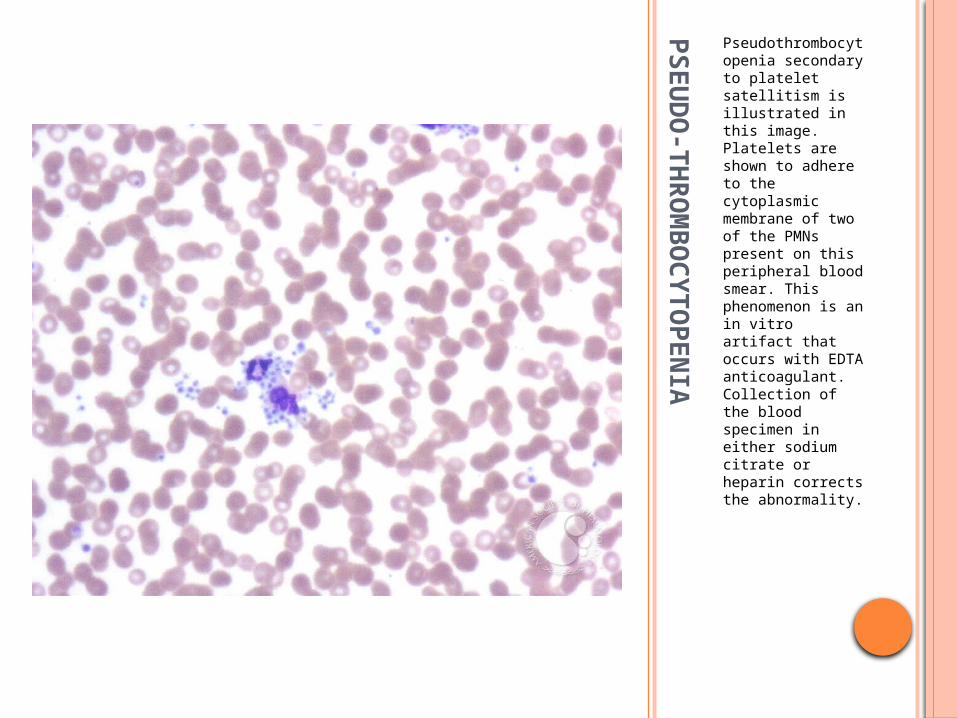
Pseudothrombocytopenia secondary to platelet satellitism is illustrated in this image. Platelets are shown to adhere to the cytoplasmic membrane of two of the PMNs present on this peripheral blood smear. This phenomenon is an in vitro artifact that occurs with EDTA anticoagulant. Collection of the blood specimen in either sodium citrate or heparin corrects the abnormality.

CLASSIFICATION OF THROMBOCYTOPENIA Impaired or decreased production
Congenital May –Hegglin anomaly Bernard- Soulier syndrome Wiskott- Aldrich syndrome TAR Congenital amegakaryocytic thrombocytopenia
Neonatal Infective/viral Drug induced Acquired
Increased platelet destruction Immune
ITP Drug induced HIT
Non-immune Thrombocytopenia in pregnancy and pre-eclampsia HIV TTP DIC HUS Drugs
Disorders related to distribution or dilution Splenic sequestration Kasabach-Merritt syndrome Hypothermia Loss of platelets- massive blood transfusion, extracorporeal circulation

THROMBOCYTOPENIA
Impaired or decreased platelet production Megakaryocyte hypoplasia
Usually congenital and include Fanconi anemia, thrombocytopenia with absent radii
(TAR syndrome), Wiskott- Aldrich syndrome, Bernard- Soulier syndrome, May Heglin anomaly, congenital amegakaryocytic thromobocytopenia
Ineffective thrombopoeisis Megaloblastic anemia
Miscellaneous Viral Marrow infiltration by malignancy, myelofibrosis
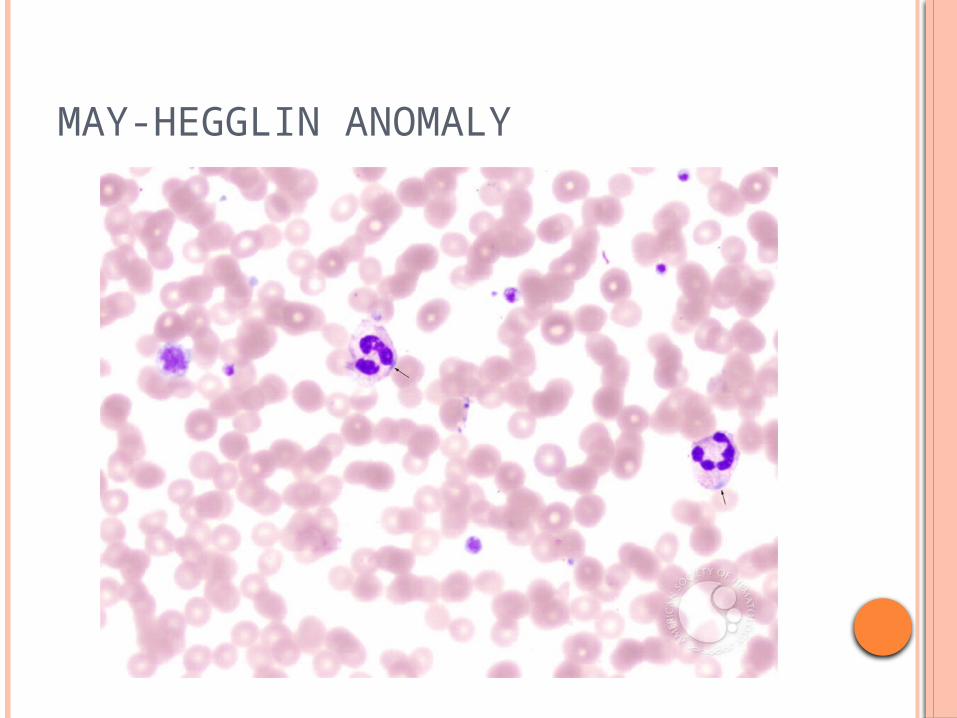
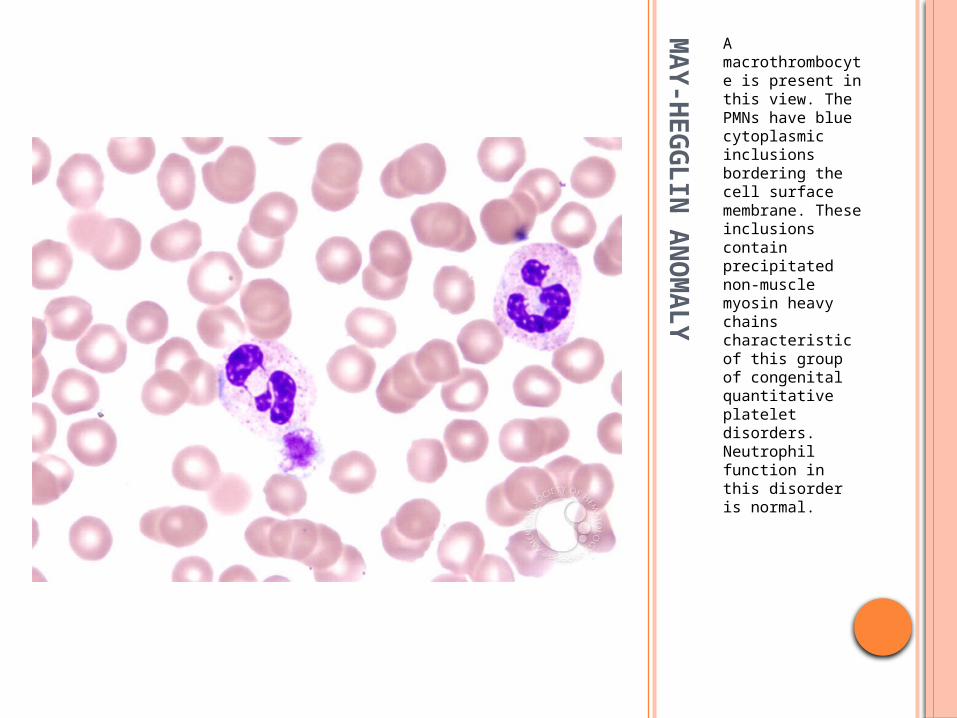
MAY-HEGGLIN ANOMALY
MAY-H
EG
GLIN
AN
OM
ALY
A macrothrombocyte is present in this view. The PMNs have blue cytoplasmic inclusions bordering the cell surface membrane. These inclusions contain precipitated non-muscle myosin heavy chains characteristic of this group of congenital quantitative platelet disorders. Neutrophil function in this disorder is normal.

CONGENITAL AMEGAKARYOCYTIC THROMBOCYTOPENIA
AR disorder causing bone marrow failure Seen in infancy Platelet count <20 Petechiae and physical anomalies Develop aplastic anemia, MDS and leukemia Stem cell transplantation is curative Mutations in the c-mpl gene leading to loss of
the thrombopoietin receptor function Loss of TPO receptor function causes
reduction in MGK progenitors and high TPO levels

ACQUIRED HYPOPLASIA
Drugs Chemotherapy drugs Zidovudine Ethanol Interferon therapy Anticonvulsants Antibacterial agents like chloramphenicol

INFECTION INDUCED THROMBOCYTOPENIA
Many viral and bacterial infections without DIC
Infections affect platelet survival and production; immune mechanisms can also be at work (Infectious mononucleosis, early HIV)
At times, bone marrow exam may be required for occult infections

THROMBOCYTOPENIAINCREASED PLATELET DESTRUCTION
Immune thrombocytopenic purpura Acute
Disorder of children Abrupt onset Follows an infection usually nonspecific respiratory or
GI virus Diagnosis is clinical Most patients recover without treatment within 3
weeks Severe cases can be treated with IVIG, platelet
transfusions and splenectomy Occasionally seen in adults

THROMBOCYTOPENIAINCREASED PLATELET DESTRUCTION
Chronic ITP 20-50 yrs of age Females:males 2:1 Mucocutaneous bleeding, menorrhagia, recurrent
epistaxis or easy bruising Immune mediated destruction of platelets Autoantibodies against platelet glycoproteins

CLINICAL PICTURE OF ACUTE AND CHRONIC ITP
Characteristics Acute Chronic
Age at onset 2-6 yrs 20-50 yrs
Sex predilection None Female over male 3:1
Prior infection Common Unusual
Onset of bleeding Sudden Gradual
Platelet count <20 30-80
Duration 2-6 wk Months to years
Spontaneous remission
90% Uncommon
Seasonal pattern High in winter/spring
None
Therapy 70% steroid responsive
30% steroid responsive
Splenectomy rare Splenectomy<45 yr 90% response >45 yr 40% response

BONE MARROW IN ITPMEGAKARYOCYTIC HYPERPLASIA

PhlWHWWegmasia Cerulea Dolens
Bawrham K, Shah T. N Engl J Med 2007;356:e3.WHA

HEPARIN-INDUCED THROMBOCYTOPENIA (HIT)
Differs from other drug induced thrombocytopenias Thrombocytopenia never severe ie <20k Not associated with bleeding but with thrombosis
Antibody to a complex of platelet specific PF4 and heparin (anti-PF4/heparin)
Antibody activates platelets through the FcYR II a receptor; also activates endothelial cells
Many patients exposed to heparin develop this antibody though not all develop HIT and even less develop HITT

HIT
Both standard heparin and LMWH can cause HIT-former more common
Heparin exposure 5-10 days Rarely HIT can develop several days after
heparin discontinued called delayed onset HIT
Diagnostic algorithm 4Ts Thrombocytopenia Timing of platelet drop Thrombosis oTher cause of thrombocytopenia not evident

CLINICAL TEACHING POINTS ABOUT HIT
Early recognition; HIT remains a clinical diagnosis
Thrombosis can be arterial and/or venous When HIT suspected, doppler legs Anticoagulate when HIT suspected even in the
absence of thrombosis because of higher rate of thrombosis (alternate AC followed by 3-6 months of warfarin)
Risk of thrombosis persists for about 1 month after diagnosis of HIT
Do not introduce warfarin alone in setting of HIT or HITT as it may precipitate thrombosis especially venous gangrene. Start after several days of alternate anticoagulation

ALTERNATE ANTICOAGULANTS IN HIT/HITT
Direct thrombin inhibitors Argatroban Lepirudin
Both approved in the US Bivalirudin
Effective but not FDA approved
Antithrombin-binding polysaccharide Fondaparinux Effective but not FDA approved in the US
Anti-Xa Danaproid
No longer available in the US

PREGNANCY AND THROMBOCYTOPENIA
You are asked to see a pregnant patient with thrombocytopenia.
What is the differential diagnosis?

Differential diagnosis of thrombocytopenia in pregnancy
MAHAThrombocytopenia
Coagulopathy
HTNLiver disease
Renal disease
CNS Time of onset
ITP ------ Mild to severe ------- -------- --------- --------- ---------Anytime common in first tri
Gestational -------- Mild ------- --------- --------- --------- --------- 2nd-3rd tri
Preeclampsia
MildMild to moderate
Absent to mild
Mod- to severe
------- Protein Seizures 3rd trim
HELLPModerate to severe
Mod to severe MildAbsent to severe
Mod to severeAbsent to moderate
Absent to moderate
3rd trim
HUS Mod to severe Mod to severe AbsentAbsent to mild
Absent Mod to severeAbsent to mild
Post partum
TTP Mod to severe Severe Absent Absent AbsentAbsent to moderate
Absent to severe
2nd- 3rd
trim
AFLP Mild Mild to mod SevereAbsent to mild
SevereAbsent to mild
Absent to mild
3rd tri

NON-IMMUNE MECHANISMS OF PLATELET DESTRUCTION
Thrombocytopenia in pregnancy and preeclampsia Gestational thrombocytopenia
Commonest cause Usually mild Healthy with no prior history of thrombocytopenia Mechanism unknown Return to normal a few weeks after delivery

NON IMMUNE CAUSES OF PLATELET DESTRUCTION
Thrombocytopenia in preeclampsia and hypertensive states in pregnancy Thrombocytopenia occurs in about 15- 20% of
preeclampsia Some have microangiopathic hemolysis,
elevated liver enzymes, and low platelet count-HELLP syndrome
Thrombocytopenia is due to platelet destruction Perhaps an underlying low grade DIC or ?
Immune process Delivery is the treatment for this condition-
thrombocytopenia will resolve in a few days post delivery

MICROANGIOPATHIC HEMOLYTIC ANEMIA (MAHA)

NON IMMUNE CAUSES OF PLATELET DESTRUCTION
Thrombotic thrombocytopenic purpura Triad of microangiopathic hemolytic anemia,
thrombocytopenia, neurological abnormalities Sometimes the pentad- fever + renal dysfunction Four types
Single acute episode Recurrent episodes Drug induced Chronic relapsing-rare form, starts in infancy

TTP
Hyaline thrombi in end arterioles and capillaries
Hyaline thrombi are composed of platelets and von Willebrand factor with little or no fibrin or fibrinogen
Deposition of these platelet-vWf thrombi leads to thrombocytopenia
Degree of thrombocytopenia is related to extent of microvascular platelet aggregation
RBCs flowing under arterial pressure fragment when they have to manouever these thrombi in the microvessels

TTP
Thrombotic lesions give rise to other manifestations Organ ischemia
Neurological Visual Abdominal-pain due to mesenteric ischemia, bleeding
due to thrombocytopenia Renal
Overwhelming renal damage is not usual; if so, consider HUS

TTP
Hemolysis can be severe Smear shows marked decrease in platelets,
RBC polychromasia and RBC fragmentation (microspherocytes, shistocytes) called MICROANGIOPATHIC HEMOLYTIC ANEMIA
Coagulation tests remain normal

TTP
Accumulation of unusually large von Willebrand factor (ULVWF)
In the plasma, ULVWF is rapidly cleaved by a VWF cleaving metalloprotease also called “ a disintegrin-like and metalloprotease domain with thrombospondin type 1 motifs” (ADAMTS 13)

SO WHAT HAPPENS IN TTP?
Familial chronic relapsing TTP Deficiency or absence of the Vwf cleaving
protease Sporadic
Autoantibody against the protease causing deficiency or loss of function
Measurement of the vWF protease enzyme (not rapid enough for clinical use)

THROMBOCYTOPENIA IN THE ICU Sepsis is commonest Often multifactorial, exact cause may be difficult to
pinpoint Infection, sepsis, shock Heparin Other drugs DIC Massive blood transfusion Post transfusion purpura CPR Cardiopulmonary bypass ARDS Pulmonary emboli Intravascular catheters

DRUG INDUCED THROMBOCYTOPENIA
Drug dependent antibodies specific for the drug structure and bind tightly to the platelets by the Fab region in the presence of the drug
Platelets seem to be the favorite target of these drug dependent antibodies
When should DIT be suspected? Unexpected occurrence of thrombocytopenia Recurrent episodes of thrombocytopenia with quick
recovery Misdiagnosis of ITP Beware of quinine containing agents like tonic water,
bittter lemon; foods such as tahini containing sesame seeds, herbal remedies like Jui herbal tea
List of drugs from www.ouhsc.edu/platelets

ANTITHROMBOTIC AGENTS AND THROMBOCYTOPENIA
Presents as acute ITP 0.1% - 2% of patients have severe
thrombocytopenia within several hours of exposure to Abiciximab, Tirobifan or Eptifibatide
About 12% can become acutely thrombocytopenic after second exposure to Abiciximab
Immediate reactions are due to presence of naturally occurring antibodies against structural elements of abiciximab or due to structural changes to GpIIb/IIIa induced by binding of Tirobifan and Eptifitabide.

Immune-Mediated Thrombocytopenia.
Warkentin TE. N Engl J Med 2007;356:891-893.

THROMBOCYTOPENIA
Dysplastic megakaryocytes Myelodysplastic syndromes Chemotherapy effects
Failure of function of megakaryocytes due to defects in DNA synthesis
B12 deficiency Folate deficiency

DYSPLASTIC MEGAKARYOCYTE

DYSPLASTIC MEGAKARYOCYTE

APPROACH TO THROMBOCYTOPENIA
Plt <150
Hb and WBC count
Normal
Smear
Fragmented red cells
DIC/TTP
Normal RBC, platele
ts norma
l
Consider Drug
s, Infection, ITP, Congenital
Abnormal
Bone marrow exam

THROMBOCYTOSIS
Reactive thrombocytosis Associated with blood loss and surgery Post splenectomy Iron deficiency anemia Inflammation and disease Stress or exercise
Clonal thrombocytosis Polycythemia vera CML Myelofibrosis Primary or Essential thrombocythemia MDS associated

THROMBOCYTOSIS IN CML
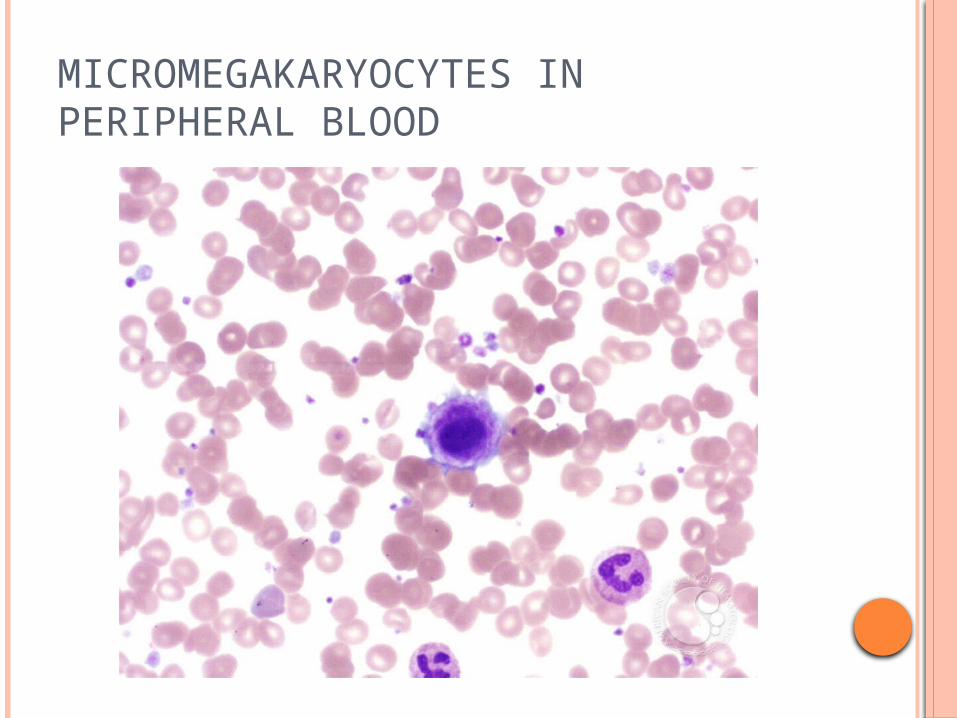
MICROMEGAKARYOCYTES IN PERIPHERAL BLOOD

PLATELET THERAPEUTICS
Platelet transfusions Platelet pheresis Manipulation of the immune system
IVIG, Steroids, Rituxan, Splenectomy, immunosuppressives Prevention of complications Reduction of platelet number
Hydrea Suppression of megakaryocyte platelet production
Anagrelide Stimulation of megakaryocyte production
Thrombopoeitin mimetics or TPO mimetics Romiplostim Eltromobag
Inhibitors of platelet aggregation Aspirin, Clopidrogel, NSAIDs Gp IIb/IIIa inhibitors Dipyridamole

THROMBOPOEITIN MIMETICS Romiplostim Trade name is Nplate TPO receptor agonist Route: subcutaneous Mechanism: Like
endogenous TPO- increases platelet production by binding and activating TPO receptor
Indications: Chronic ITP Dose titration based on
platelet count
Eltromobag Trade name is
Promacta TPO receptor agonist Route: oral Mechanism: similar
to Nplate Indications: Chronic
ITP Dose titration based
on platelet count