Embed Size (px)
Citation preview

Lysine Methylation Mapping of Crenarchaeal DNA-Directed RNAPolymerases by Collision-Induced and Electron-Transfer DissociationMass SpectrometryMikel Azkargorta,† Magdalena N. Wojtas,‡,¶ Nicola G. A. Abrescia,‡,§ and Felix Elortza*,†
†Proteomics Platform, CIC bioGUNE, ProteoRed-ISCIII, CIBERehd, 48160 Derio, Spain‡Structural Biology Unit, CIC bioGUNE, CIBERehd, 48160 Derio, Spain§Ikerbasque, Basque Foundation for Science, 48011 Bilbao, Spain
*S Supporting Information
ABSTRACT: Enzymatic machineries fundamental for infor-mation processing (e.g., transcription, replication, translation)in Archaea are simplified versions of their eukaryoticcounterparts. This is clearly noticeable in the conservation ofsequence and structure of corresponding enzymes (see forexample the archaeal DNA-directed RNA polymerase(RNAP)). In Eukarya, post-translational modifications(PTMs) often serve as functional regulatory factors for variousenzymes and complexes. Among the various PTMs, methyl-ation and acetylation have been recently attracting mostattention. Nevertheless, little is known about such PTMs inArchaea, and cross-methodological studies are scarce. We examined methylation and N-terminal acetylation of endogenouslypurified crenarchaeal RNA polymerase from Sulfolobus shibatae (Ssh) and Sulfolobus acidocaldarius (Sac). In-gel and in-solutionprotein digestion methods were combined with collision-induced dissociation (CID) and electron-transfer dissociation (ETD)mass spectrometry analysis. Overall, 20 and 26 methyl-lysines for S. shibatae and S. acidocaldarius were identified, respectively.Furthermore, two N-terminal acetylation sites for each of these organisms were assessed. As a result, we generated a high-confidence data set for the mapping of methylation and acetylation sites in both Sulfolobus species, allowing comparisons with thedata previously obtained for RNAP from Sulfolobus solfataricus (Sso). We confirmed that all observed methyl-lysines are on thesurface of the RNAP.
KEYWORDS: Sulfolobus, RNA polymerase, methylation, acetylation, ETD, CID, post-translational modification
■ INTRODUCTION
Phylogenetic classification based on rRNA sequences placesarchaeal organisms closer to eukaryotic than to bacterial cells.1
Accumulated biochemical, functional, and structural data haveshown that Eukarya and Archaea share essential structural andfunctional protein machinery features. For example, key playersin the eukaryotic genomic DNA replication apparatus arepresent, in a simplified form, in Archaea.2 Thus, these archaealsystems can be used as models for complex eukaryotic cellularprocesses. Recent structural studies of the complete DNA-directed RNA polymerase (RNAP) from Sulfolobus shibatae(Ssh) have shown its striking structural homology with theeukaryotic RNAP II.3,4 However, one of the major differencesbetween archaeal RNAP and eukaryotic RNAPs lies at the levelof regulatory activities. It is still an open question whether post-translational modifications (PTMs) can modulate the activity ofthese archaeal enzymes.Protein methylation is a common protein PTM where a
hydrogen atom is replaced by a methyl group, usually in lysineor arginine residues.5 Histone methylation is the best describedmethylation event; it regulates gene expression and affects
developmental and cellular response processes.6 Histones,however, are not the only eukaryotic proteins that are subjectto this modification.7 A number of non-histone proteinsundergo methylation, which modulates their stability, protein−protein interactions, or transactivational activity. p53, ribosomalprotein Rpl23ab, Dam1, TAF10, TFIID, RuBisCO, andcytochrome c can be lysine-methylated, among others.8
Recently, the non-histone methylome data for eukaryotes hasbeen significantly expanded by application of heavy methylSILAC approach, in combination with extensive fractionationand use of a battery of antibodies or affinity reagents.9,10
Nevertheless, our understanding of non-histone proteinmethylation and its physiological implications is still limited.In contrast with the wealth of information on methylation in
eukaryotes, little is known about this process in Archaea.Proteome-wide analysis in crenarchaeal Sulfolobus solfataricus(Sso) P2 has revealed the existence of a methylated peptide inSso7d protein,11 and analysis of Sso DNA-directed RNA
Received: January 27, 2014Published: March 13, 2014
Article
pubs.acs.org/jpr
© 2014 American Chemical Society 2637 dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−2648

polymerase has shown extensive methylation in this proteincomplex.12 Interestingly, the existence of a highly conserved,lysine methyltransferase with broad substrate specificity hasbeen recently confirmed in Sulfolobus islandicus.13 It isimportant to remember that archaeal organisms often grow invery hostile environments, e.g., at high temperatures (hyper-thermophiles) and/or under acidic and high ionic strengthconditions (acidophiles and halophiles), and thus theirbiochemistry is of extreme interest. Lysine methylation mightplay an important role in the heat stability of hyperthermophiliccrenarchaeal proteins through the modulation of their pKa,hydrophobicity, and solubility.12,14
Mass spectrometry (MS) has become the analytical tool parexcellence for the identification of proteins and discovery andcharacterization of PTMs.15 Collision-induced dissociation(CID) and electron-transfer dissociation (ETD) are comple-mentary methods for peptide fragmentation and subsequenttandem mass spectrometry analysis and identification. CID ismost effective when working with small, low-charge,unmodified peptides. Instead, ETD is considered to workbetter with high charge-states, generating more extensive seriesof ions than CID and leading to more comprehensive sequenceinformation. This is particularly useful in detecting labilemodifications, such as phosphorylations.16 Thus, using ETD inconjunction with CID can be a very successful approachbecause of differential dissociation characteristics and comple-mentary information these methods provide17−19
In this study, we analyzed methylation pattern ofendogenously purified RNAPs from two archaeal organisms,Ssh and Sulfolobus acidocaldarius (Sac), using CID- and ETD-based mass spectrometry. The sequence coverage for theidentified subunits ranged from 98.1% (for the Rpo3 subunit inSac) to 56.2% (for the Rpo12 subunit in Ssh). Usingconservative conditions for the preliminary screening andmanual validation of the obtained hits, we generated high-confidence methylation data sets for both Ssh and Sac RNAPs.We found 20 sites across 11 subunits for Ssh RNAP and 26
methyl-lysines distributed across 10 subunits for Sac RNAP,adding new information to a previous study of Sso RNAP andstrengthening the evidence and reliability of the methylationdata. We also found two subunits with N-terminal acetylation inboth analyzed species.
■ MATERIALS AND METHODS
Sulfolobus Biomass Production
Biomass production and RNAP purification of S. shibatae wascarried out using a protocol originally devised for the threearchaeal species Sulfolobus acidocaldarius, Sulfolobus shibatae,and Pyrococcus furiosus by Korkhin et al.20 and adapted to ourlaboratory infrastructure.For the present study archaeal (Sac and Ssh) cells were
asynchronously grown. S. shibatae growth media consisted ofSolution A (1.3 g (NH4)2 SO4, 0.28 g KH2PO4, 0.25 g MgSO4,0.07 g CaCl2, and 2 g Bactopeptone (Conda) in 1 L of media;pH 3, adjusted using sulfuric acid; the solution was autoclaved)and Solution B (5 g FeCl3, 0.45 g MnCl2, 1.13 g Na2B4O7,0.055 g ZnSO4, 0.013 g CuCl2, 0.008 g Na2MoO4, 0.008 gVOSO4, and 0.003 g CoSO4 in 100 mL of 1 M HCl filtratedthrough 0.22 μm filter). Complete media (A + B) wereprepared by mixing 1 L of solution A with 0.4 mL of solution Band 10 g of sucrose and prewarmed before adding theinoculum. To start the culture, 10 mL of media (A + B) in a 50
mL Schott bottle was inoculated with a piece of frozen cellpellet. The bottle, with the lid unscrewed, was placed within alarger beaker part-filled with water. The beaker was placed ontoa hot plate at 75 °C. The content of the Schott bottle wasgently stirred and aerated and used as further inoculum oflarger volumes. Once the inoculum reached 3 L, it was used as aseed for 18 L of media. The culture was put in six 5 LErlenmeyer flasks which were placed in a shaker (Innova, NewBrunswick) and agitated at 105 rpm, at 75 °C. The cells werecollected after 4 days and separated by centrifugation at 5500rpm for 20 min in a JLA 8.100 rotor. From these 18 L, weobtained ∼75 g of wet-cell pellet; this was flash-frozen in liquidnitrogen and stored at −80 °C. Our biomass yields werecomparable to those obtained originally by Korkhin et al.20
RNAP Purification
Ssh RNAP was purified to homogeneity, from 100 g of biomass,by ammonium precipitation, followed by anion exchangechromatography (QFF) on a heparin column (HiPrep FF16/10), and polished by gel filtration (HiLoad 16/60 Superdex200) as described previously.20 Highly purified Ssh RNAP (6mg/mL) in buffer containing 50 mM Tris pH 7.8, 25 mMMgCl2, 10% (v/v) glycerol, 10 mM β-mercaptoethanol, and500 mM KCl was aliquoted and flash-frozen in liquid nitrogenfor medium- and long-term storage.Purified Sac RNAP was a generous gift from Yakov Korkhin
and was stored at similar protein concentration as Ssh RNAP in50 mMTris/HC1, pH 7.8, 22 mM NH4CI, 10% (v/v) glycerol,10 mM β-mercaptoethanol, and 300 mM KCl.
Polyacrylamide Gel Electrophoresis and Staining
6X loading buffer was added to RNAP samples to a finalconcentration of 50 mM Tris pH 6.8, 5% glycerol, 1.67%mercaptoethanol, 1.67% SDS, and 0.0062% bromophenol blue.Protein samples were boiled for 5 min and resolved in 12.5%acrylamide gels using a Mini-Protean II electrophoresis cell(Bio-Rad). A constant voltage of 150 V was applied for 45 min.To avoid artifactual modification of acidic residues, the gel wasstained in SimplyBlue Safe Stain (Life Sciences), which doesnot require methanol or acetic acid fixatives or destains. The gelwas washed in Milli-Q water, and protein bands were cut intopieces and subjected to tryptic digestion followed by LC−MS/MS analysis.
Tryptic Digestion
In-Gel Tryptic Digestion. Gel bands were first cut intosmall pieces and washed in Milli-Q water. Reduction andalkylation was achieved by incubation with dithiothreitol (DTT,10 mM in 50 mM ammonium bicarbonate) at 56 °C for 20min, followed by an incubation in iodoacetamide (IA, 50 mMin 50 mM ammonium bicarbonate) for another 20 min, in thedark. Gel pieces were dried and incubated with trypsin (12.5μg/mL, in 50 mM ammonium bicarbonate) for 20 min on ice.After rehydration, the trypsin supernatant was discarded; gelbands were covered with 50 mM ammonium bicarbonate andincubated overnight at 37 °C. After digestion, acidic peptideswere further extracted with TFA 0.1% and dried in a RVC2 25SpeedVac concentrator (Christ). The peptides were resus-pended in 0.1% FA and sonicated for 5 min prior to theiranalysis.
In-Solution Tryptic Digestion. RNAP samples (30 μg)were dried in a RVC2 25 SpeedVac concentrator (Christ) andresuspended in 16 μL of 6 M urea. Protein was reduced andalkylated by incubation in 5 μL of DTT (200 mM, in 50 mM
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482638

ammonium bicarbonate) for 45 min, followed by incubation in4 μL of IA (1 M, in 50 mM ammonium bicarbonate) foranother 45 min, and finally in 20 μL of DTT for 45 min. Allincubations were carried out at 25 °C. Then, the sample wasdiluted to 100 μL by adding 50 mM ammonium bicarbonate,and trypsin (Trypsin Gold, Promega Corporation, Madison,WI, USA) was added to a final trypsin/protein ratio of 1:20.The sample was vortexed and incubated overnight at 37 °C.The peptides were dried in the SpeedVac concentrator,resuspended in 0.1% FA, and sonicated for 5 min. The sampleswere desalted in Zip Tip C18 pipet tips (Millipore) followingthe manufacturer’s protocol, dried in the SpeedVac concen-trator, resuspended in 0.1% FA, and sonicated for 5 min priorto analysis by MS.
NanoLC−MS/MS and Data Analysis
Peptide mixtures obtained from the digestions were separatedby online NanoLC (nLC) and analyzed using electrospraytandem mass spectrometry. Peptide separation was performedon a nanoACQUITY UPLC system (Waters) connected to anLTQ Orbitrap XL ETD mass spectrometer (Thermo Electron,Bremen, Germany). Approximately 500-ng samples wereloaded onto a Symmetry 300 C18 UPLC Trap column, 180μm × 20 mm, 5 μm (Waters). The precolumn was connectedto a BEH130 C18 column, 75 μm × 200 mm, 1.7 μm (Waters)equilibrated in 3% acetonitrile and 0.1% FA. The peptides wereeluted at 300 nL/min using a 30 min linear gradient of 3−50%acetonitrile for in-gel digested samples and a 60 min lineargradient of 3−50% acetonitrile for in-solution digested samples.Samples were loaded directly onto the nanoelectrospray ionsource (Proxeon Biosystems, Odense, Denmark).The mass spectrometer automatically switched between MS
and MS/MS acquisition in DDA mode. Full MS survey spectra(m/z 400−2000) were acquired in the orbitrap with 30000resolution at m/z 400. The five most intense ions weresubjected both to CID and ETD fragmentation in the linear iontrap, in an alternating fashion. Precursors with charge statesequal to or greater than 2 were specifically selected forfragmentation. Collision-energy applied to each peptide wasautomatically normalized as a function of the m/z and chargestate, and charge-state-dependent ETD time was applied.Analyzed peptides were excluded from further analysis during30 s using dynamic exclusion lists.Searches were performed using Mascot Search engine (www.
matrixscience.com, Matrix Science, London, U.K.) on Pro-teome Discoverer v.1.2. software (Thermo Electron, Bremen,Germany). Carbamidomethylation of cysteines was selected asfixed modification, and oxidation of methionines, methylationof lysine and arginine, and protein N-terminal acetylation asvariable modifications. Peptide mass tolerance of 5 ppm and 0.5Da fragment mass tolerance were adopted as search parameters,and 4 missed cleavages were allowed. Spectra were searchedagainst a mixed Sulfolobus + Saccharomyces cerevisiae databaseobtained from UniProt. Sac and Ssh databases were obtainedfrom UniProt/Swiss-Prot (version 2013_07, 540546 entries)and UniProt/TrEMBL (version 2013_07, 39870577 entries)and merged with yeast UniProt/Swiss-Prot database from thesame release. A decoy search was carried out in order toestimate the false discovery rate (FDR) for the samples.Proteins with at least 2 peptides passing the FDR < 5% filterwere considered for further analysis. Only unambiguouslyassigned, rank1 modified peptides with FDR < 5% wereselected in the initial screening. For the final filtering and
selection of the methylated hits, only manually validatedmodified sites present in both experimental replicates wereconsidered.
■ RESULTS
Identification of S. acidocaldarius and S. shibatae RNAPSubunits
RNAPs from both Sac and Ssh were analyzed following dualgel-based and gel-free strategies (Figure 1). Protein subunits
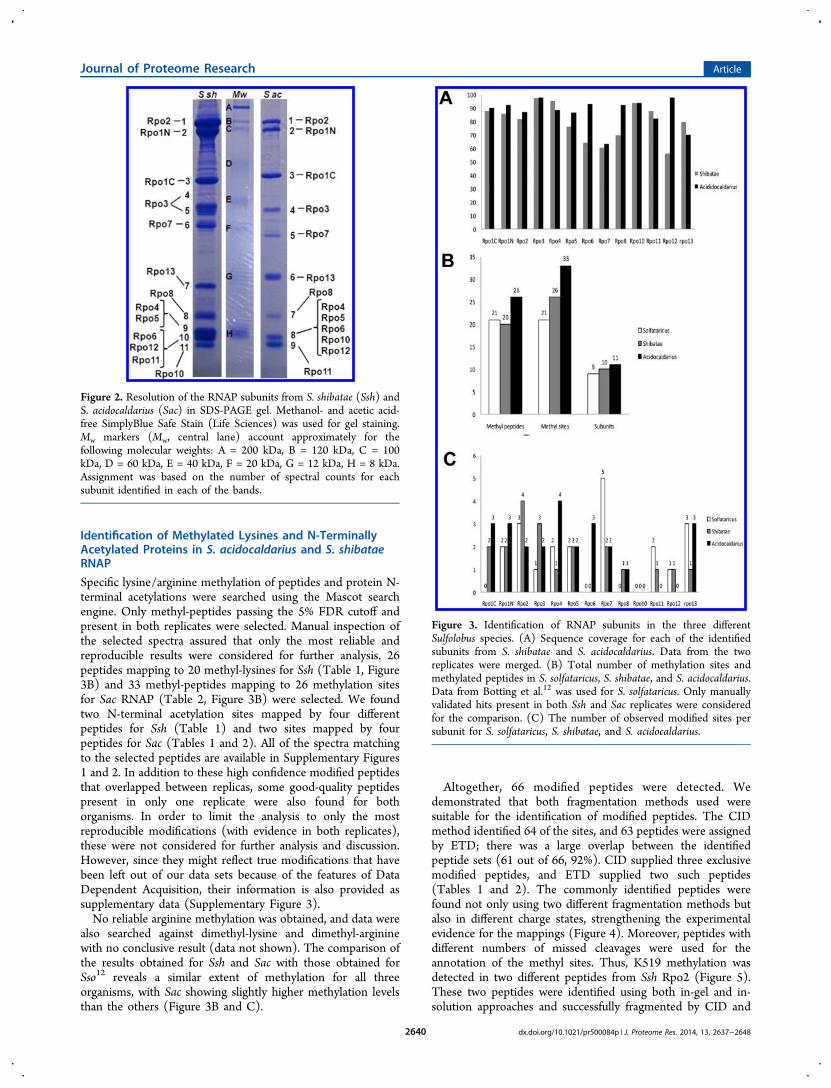
were resolved on an SDS-PAGE gel, and each observed bandwas in-gel digested and analyzed by nLC−MS/MS. Proteinextracts were also directly digested with trypsin in solution andanalyzed by mass spectrometry, using both CID and ETDfragmentation methods. Two independent replicates comingfrom different preparations were run for each procedure.Ssh RNAP yielded 11 prominent bands in the gel-based
approach, whereas RNAP from Sac yielded 9 different bands inthe gel (Figure 2). All 13 different RNAP subunits wereidentified following this approach for both organisms. Some ofthe subunits showed an anomalous electrophoretic mobility inthe gel. This was the case for the newly identified Rpo13,3,21
which resolved with higher than expected molecular weight.Interestingly, discrepancies in the electrophoretic mobility forsome Sac and Ssh subunits were detected (for example, Rpo7).Using the two approaches (gel-based and gel-free), we
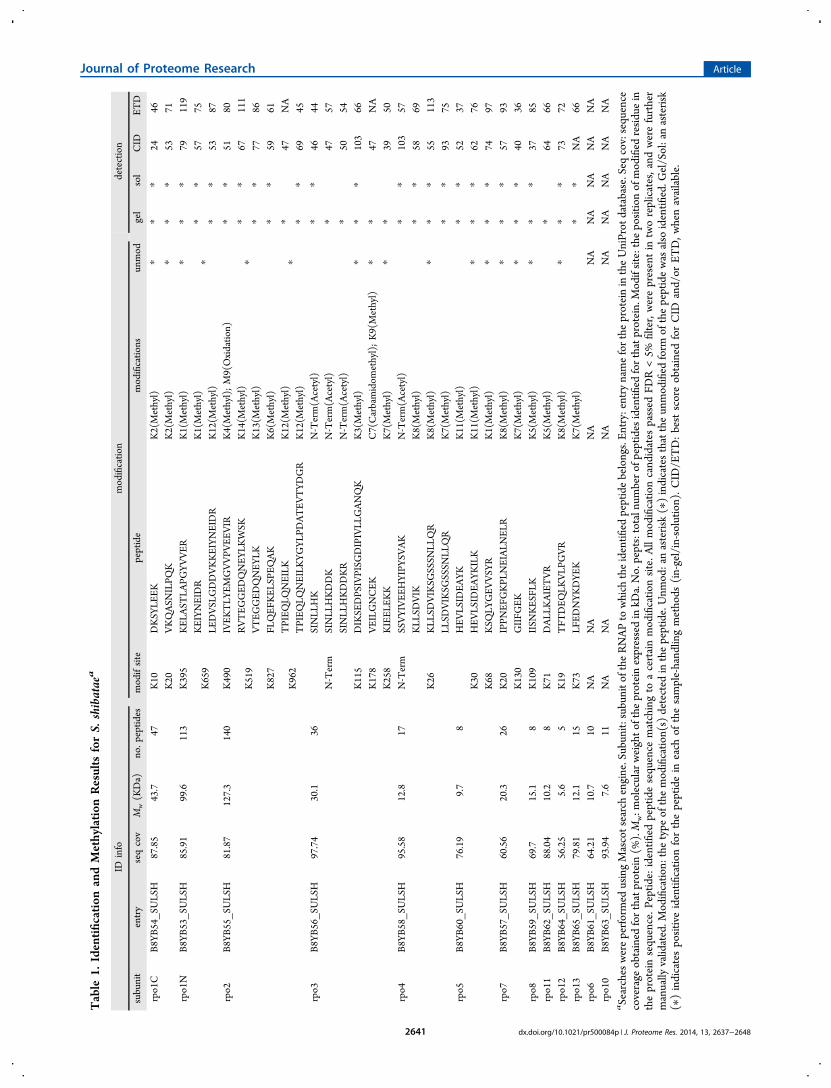
obtained an extensive mapping of all 13 subunits composingRNAP. Interestingly, over 90% sequence coverage was obtainedfor 7 of the RNAP subunits from Sac and 3 subunits from SshRNAP. More than 80% sequence coverage was obtained formost of the subunits from both organisms (11 out of 13 for Sac,7 out of 13 for Ssh), confirming that the samples wereextensively analyzed (Figure 3A). In addition to the 13 subunitsof the archaeal RNAPs, some other proteins were identified inboth Sac and Ssh preparations (Supplementary Tables 1 and 2).
Figure 1. Schematic representation of sample preparation and analysisworkflow. RNAP from S. shibatae and S. acidocaldarius was isolatedand processed using in-gel and in-solution approaches. Peptides wereanalyzed in the mass spectrometer using both CID and ETD asfragmentation methods.
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482639
Identification of Methylated Lysines and N-TerminallyAcetylated Proteins in S. acidocaldarius and S. shibataeRNAP
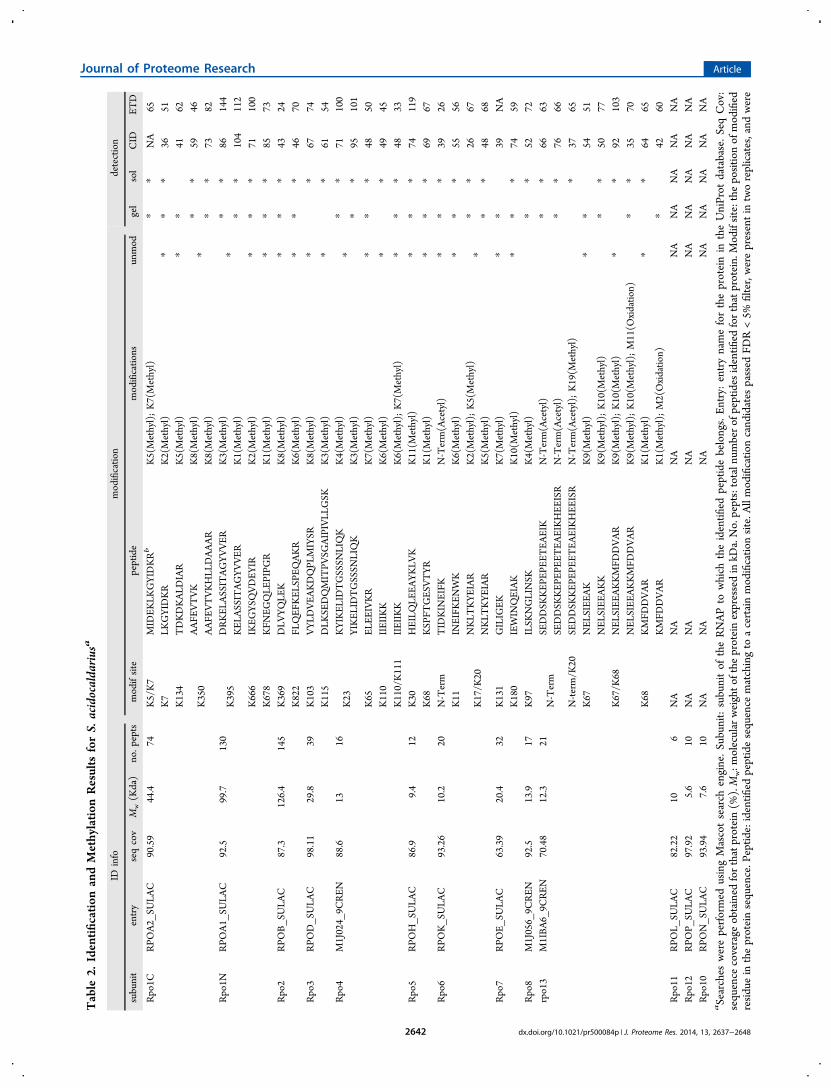
Specific lysine/arginine methylation of peptides and protein N-terminal acetylations were searched using the Mascot searchengine. Only methyl-peptides passing the 5% FDR cutoff andpresent in both replicates were selected. Manual inspection ofthe selected spectra assured that only the most reliable andreproducible results were considered for further analysis. 26peptides mapping to 20 methyl-lysines for Ssh (Table 1, Figure3B) and 33 methyl-peptides mapping to 26 methylation sitesfor Sac RNAP (Table 2, Figure 3B) were selected. We foundtwo N-terminal acetylation sites mapped by four differentpeptides for Ssh (Table 1) and two sites mapped by fourpeptides for Sac (Tables 1 and 2). All of the spectra matchingto the selected peptides are available in Supplementary Figures1 and 2. In addition to these high confidence modified peptidesthat overlapped between replicas, some good-quality peptidespresent in only one replicate were also found for bothorganisms. In order to limit the analysis to only the mostreproducible modifications (with evidence in both replicates),these were not considered for further analysis and discussion.However, since they might reflect true modifications that havebeen left out of our data sets because of the features of DataDependent Acquisition, their information is also provided assupplementary data (Supplementary Figure 3).No reliable arginine methylation was obtained, and data were
also searched against dimethyl-lysine and dimethyl-argininewith no conclusive result (data not shown). The comparison ofthe results obtained for Ssh and Sac with those obtained forSso12 reveals a similar extent of methylation for all threeorganisms, with Sac showing slightly higher methylation levelsthan the others (Figure 3B and C).
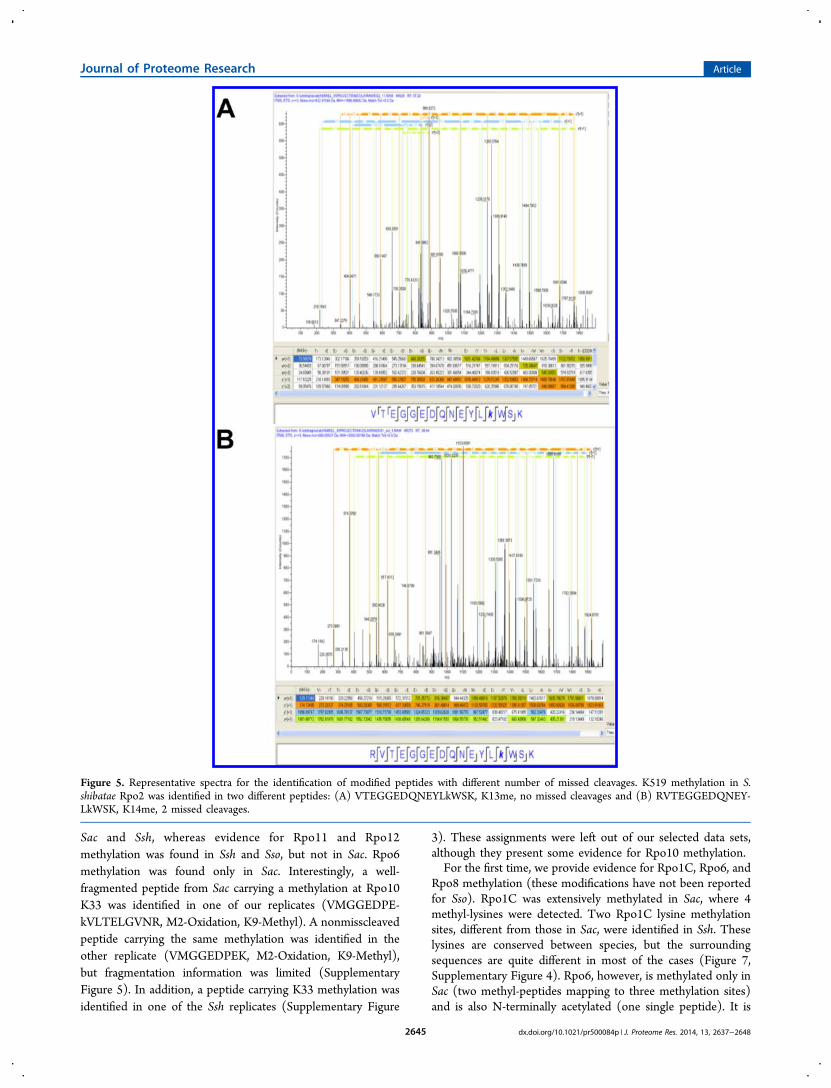
Altogether, 66 modified peptides were detected. Wedemonstrated that both fragmentation methods used weresuitable for the identification of modified peptides. The CIDmethod identified 64 of the sites, and 63 peptides were assignedby ETD; there was a large overlap between the identifiedpeptide sets (61 out of 66, 92%). CID supplied three exclusivemodified peptides, and ETD supplied two such peptides(Tables 1 and 2). The commonly identified peptides werefound not only using two different fragmentation methods butalso in different charge states, strengthening the experimentalevidence for the mappings (Figure 4). Moreover, peptides withdifferent numbers of missed cleavages were used for theannotation of the methyl sites. Thus, K519 methylation wasdetected in two different peptides from Ssh Rpo2 (Figure 5).These two peptides were identified using both in-gel and in-solution approaches and successfully fragmented by CID and
Figure 2. Resolution of the RNAP subunits from S. shibatae (Ssh) andS. acidocaldarius (Sac) in SDS-PAGE gel. Methanol- and acetic acid-free SimplyBlue Safe Stain (Life Sciences) was used for gel staining.Mw markers (Mw, central lane) account approximately for thefollowing molecular weights: A = 200 kDa, B = 120 kDa, C = 100kDa, D = 60 kDa, E = 40 kDa, F = 20 kDa, G = 12 kDa, H = 8 kDa.Assignment was based on the number of spectral counts for eachsubunit identified in each of the bands.
Figure 3. Identification of RNAP subunits in the three differentSulfolobus species. (A) Sequence coverage for each of the identifiedsubunits from S. shibatae and S. acidocaldarius. Data from the tworeplicates were merged. (B) Total number of methylation sites andmethylated peptides in S. solfataricus, S. shibatae, and S. acidocaldarius.Data from Botting et al.12 was used for S. solfataricus. Only manuallyvalidated hits present in both Ssh and Sac replicates were consideredfor the comparison. (C) The number of observed modified sites persubunit for S. solfataricus, S. shibatae, and S. acidocaldarius.
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482640
Table
1.Identification
andMethylation
ResultsforS.
shibataea
IDinfo
modificatio
ndetection
subunit
entry
seqcov
Mw(K
Da)
no.p
eptid
esmodifsite
peptide
modificatio
nsunmod
gel
sol
CID
ETD
rpo1C
B8Y
B54_S
ULS
H87.85
43.7
47K10
DKSY
LEEK
K2(Methyl)
**
*24
46K20
VKQASN
ILPQ
KK2(Methyl)
**
*53
71rpo1N
B8Y
B53_S
ULS
H85.91
99.6
113
K395
KEL
AST
LAPG
YVVER
K1(Methyl)
**
*79
119
K659
KEIYNEIDR
K1(Methyl)
**
*57
75LE
DVSL
GDDVKKEIYNEIDR
K12(M
ethyl)
**
5387
rpo2
B8Y
B55_S
ULS
H81.87
127.3
140
K490
IVEK
TLY
EMGVVPV
EEVIR
K4(Methyl);M9(Oxidatio
n)*
*51
80
K519
RVTEG
GED
QNEY
LKWSK
K14(M
ethyl)
**
*67
111
VTEG
GED
QNEY
LKK13(M
ethyl)
**
7786
K827
FLQEF
KEL
SPEQ
AK
K6(Methyl)
**
5961
K962
TPIEQ
LQNEILK
K12(M
ethyl)
**
47NA
TPIEQ
LQNEILK
YGYLP
DATEV
TYDGR
K12(M
ethyl)
**
6945
rpo3
B8Y
B56_S
ULS
H97.74
30.1
36N-Term
SINLL
HK
N-Term(Acetyl)
**
4644
SINLL
HKDDK
N-Term(Acetyl)
*47
57SINLL
HKDDKR
N-Term(Acetyl)
*50
54K115
DIKSE
DPS
IVPISG
DIPIVLL
GANQK
K3(Methyl)
**
*103
66K178
VEILG
NCEK
C7(Carbamidom
ethyl);K9(Methyl)
**
47NA
K258
KIEEL
EKK
K7(Methyl)
**
3950
rpo4
B8Y
B58_S
ULS
H95.58
12.8
17N-Term
SSVYIVEE
HYIPYSV
AK
N-Term(Acetyl)
**
103
57
K26
KLL
SDVIK
K8(Methyl)
**
*58
69KLL
SDVIKSG
SSSN
LLQR
K8(Methyl)
**
55113
LLSD
VIKSG
SSSN
LLQR
K7(Methyl)
**
9375
rpo5
B8Y
B60_S
ULS
H76.19
9.7
8HEV
LSID
EAYK
K11(M
ethyl)
**
5237
K30
HEV
LSID
EAYKILK
K11(M
ethyl)
**
*62
76K68
KSQ
LYGEV
VSY
RK1(Methyl)
**
*74
97rpo7
B8Y
B57_S
ULS
H60.56
20.3
26K20
IPPN
EFGKPL
NEIALN
ELR
K8(Methyl)
**
*57
93K130
GIIFG
EKK7(Methyl)
**
*40
36rpo8
B8Y
B59_S
ULS
H69.7
15.1
8K109
IISN
KES
FLK
K5(Methyl)
**
*37
85rpo11
B8Y
B62_S
ULS
H88.04
10.2
8K71
DALL
KAIETVR
K5(Methyl)
*64
66rpo12
B8Y
B64_S
ULS
H56.25
5.6
5K19
TFT
DEQ
LKVLP
GVR
K8(Methyl)
**
*73
72rpo13
B8Y
B65_S
ULS
H79.81
12.1
15K73
LFED
NYKDYEK
K7(Methyl)
**
NA
66rpo6
B8Y
B61_S
ULS
H64.21
10.7
10NA
NA
NA
NA
NA
NA
NA
NA
rpo10
B8Y
B63_S
ULS
H93.94
7.6
11NA
NA
NA
NA
NA
NA
NA
NA
aSearches
wereperformed
usingMascotsearchengine.Subunit:
subunito
fthe
RNAPto
which
theidentified
peptidebelongs.En
try:entrynamefortheproteinintheUniProt
database.Seq
cov:sequence
coverage
obtained
forthatprotein(%
).M
w:m
olecularweighto
fthe
proteinexpressedinkD
a.No.pepts:totalnum
berofpeptides
identified
forthatprotein.Modifsite:the
positio
nofmodified
residuein
theproteinsequence.P
eptid
e:identifi
edpeptidesequence
matchingto
acertainmodificatio
nsite.A
llmodificatio
ncandidates
passed
FDR<5%
filter,werepresentin
tworeplicates,and
werefurther
manually
validated.M
odificatio
n:thetype
ofthemodificatio
n(s)detected
inthepeptide.Unm
od:anasterisk(∗)indicatesthattheunmodified
form
ofthepeptidewasalso
identifi
ed.G
el/Sol:anasterisk
(∗)indicatespositiveidentifi
catio
nforthepeptidein
each
ofthesample-handlingmethods
(in-gel/in-solution).C
ID/ETD:bestscoreobtained
forCID
and/or
ETD,w
henavailable.
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482641
Table
2.Identification
andMethylation
ResultsforS.
acidocaldariusa
IDinfo
modificatio
ndetection
subunit
entry
seqcov
Mw(K
da)
no.p
epts
modifsite
peptide
modificatio
nsunmod
gel
sol
CID
ETD
Rpo1C
RPO
A2_
SULA
C90.59
44.4
74K5/K7
MID
EKLK
GYID
KRb
K5(Methyl);K7(Methyl)
**
NA
65K7
LKGYID
KR
K2(Methyl)
**
*36
51K134
TDKDKALD
IAR
K5(Methyl)
**
4162
K350
AAFE
VTVK
K8(Methyl)
**
*59
46AAFE
VTVKHLL
DAAAR
K8(Methyl)
**
7382
Rpo1N
RPO
A1_
SULA
C92.5
99.7
130
K395
DRKEL
ASSITAGYVVER
K3(Methyl)
**
*86
144
KEL
ASSITAGYVVER
K1(Methyl)
**
104
112
K666
IKEG
YSQ
VDEY
IRK2(Methyl)
**
*71
100
K678
KFN
EGQLE
PIPG
RK1(Methyl)
**
*85
73Rpo2
RPO
B_S
ULA
C87.3
126.4
145
K369
DLV
YQLE
KK8(Methyl)
**
*43
24K822
FLQEF
KEL
SPEQ
AKR
K6(Methyl)
**
*46
70Rpo3
RPO
D_S
ULA
C98.11
29.8
39K103
VYLD
VEA
KDQPL
MIYSR
K8(Methyl)
**
6774
K115
DLK
SEDQMITPV
SGAIPIVLL
GSK
K3(Methyl)
**
6154
Rpo4
M1J024_
9CREN
88.6
1316
K23
KYIKEL
IDTGSSSN
LIQK
K4(Methyl)
**
*71
100
YIKEL
IDTGSSSN
LIQK
K3(Methyl)
**
95101
K65
ELEE
IVKR
K7(Methyl)
**
*48
50K110
IIEIIKK
K6(Methyl)
**
4945
K110/K111
IIEIIKK
K6(Methyl);K7(Methyl)
**
*48
33Rpo5
RPO
H_S
ULA
C86.9
9.4
12K30
HEILQ
LEEA
YKLV
KK11(M
ethyl)
**
*74
119
K68
KSP
FTGES
VTYR
K1(Methyl)
**
*69
67Rpo6
RPO
K_S
ULA
C93.26
10.2
20N-Term
TID
KIN
EIFK
N-Term(Acetyl)
**
*39
26K11
INEIFK
ENWK
K6(Methyl)
**
*55
56
K17/K
20NKLT
KYEIAR
K2(Methyl);K5(Methyl)
**
*26
67NKLT
KYEIAR
K5(Methyl)
**
4868
Rpo7
RPO
E_SU
LAC
63.39
20.4
32K131
GILIG
EKK7(Methyl)
**
39NA
K180
IEWIN
QEIAK
K10(M
ethyl)
**
*74
59Rpo8
M1J056_
9CREN
92.5
13.9
17K97
ILSK
NGLINSK
K4(Methyl)
**
5272
rpo13
M1IBA6_
9CREN
70.48
12.3
21N-Term
SEDDSK
KEP
EPEE
TEA
EIK
N-Term(Acetyl)
**
6663
SEDDSK
KEP
EPEE
TEA
EIKHEE
ISR
N-Term(Acetyl)
**
7666
N-term/K
20SE
DDSK
KEP
EPEE
TEA
EIKHEE
ISR
N-Term(Acetyl);K19(M
ethyl)
*37
65K67
NEL
SIEE
AK
K9(Methyl)
**
5451
K67/K
68NEL
SIEE
AKK
K9(Methyl);K10(M
ethyl)
**
*50
77NEL
SIEE
AKKMFD
DVAR
K9(Methyl);K10(M
ethyl)
*92
103
NEL
SIEE
AKKMFD
DVAR
K9(Methyl);K10(M
ethyl);M11(O
xidatio
n)*
*35
70K68
KMFD
DVAR
K1(Methyl)
**
6465
KMFD
DVAR
K1(Methyl);M2(Oxidatio
n)*
4260
Rpo11
RPO
L_SU
LAC
82.22
106
NA
NA
NA
NA
NA
NA
NA
NA
Rpo12
RPO
P_SU
LAC
97.92
5.6
10NA
NA
NA
NA
NA
NA
NA
NA
Rpo10
RPO
N_S
ULA
C93.94
7.6
10NA
NA
NA
NA
NA
NA
NA
NA
aSearches
wereperformed
usingMascotsearch
engine.Subunit:subunitof
theRNAPto
which
theidentifi
edpeptidebelongs.En
try:
entrynamefortheproteinin
theUniProt
database.SeqCov:
sequence
coverage
obtained
forthatprotein(%
).M
w:m
olecularweighto
fthe
proteinexpressedinkD
a.No.pepts:totalnum
berof
peptides
identified
forthatprotein.Modifsite:the
positio
nofmodified
residuein
theproteinsequence.P
eptid
e:identifi
edpeptidesequence
matchingto
acertainmodificatio
nsite.A
llmodificatio
ncandidates
passed
FDR<5%
filter,werepresentin
tworeplicates,and
were
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482642

ETD, further strengthening the evidence for this methylationsite.
Comparison of Lysine Methylation in RNAPs from theThree Sulfolobus Species
Overall results for RNAP methylation revealed a similarnumber of methylation sites for each organism, with SacRNAP being slightly more methylated than others (Figure 3B).RNAP subunits of these three Sulfolobus species have a highlevel of sequence identity, with Sac being the least similar. Inparticular, subunits Ssh Rpo8 and Rpo13 have only 43% and39% sequence similarity with Sac (see Supplementary Table 3).The proportion of lysine residues is mostly conserved acrossspecies in subunits Rpo1N, Rpo1C, Rpo2, Rpo3, Rpo7, Rpo10,Rpo12, and Rpo13. In the remaining subunits (Rpo4, Rpo6,Rpo8, and Rpo11), lysine residues are more abundant in Sacthan in the other two species (Supplementary Table 4). Thiseffect is most noticeable for Rpo6 (9% lysines in Sac against the4.2% in Ssh and 2.1% in Sso). Interestingly, this subunit hasonly been reported to be methylated in Sac.Our data on Ssh and Sac RNAPs show that methylation
occurs predominantly on α-helices and loops and only in onecase on an incoming β-strand (Supplementary Figure 4). Thissuggests a structural specificity as it appears that ordered andaccessible parts of the protein, mainly in an α-helical secondarystructure, are more likely to be methylated. The methylationsites in Ssh RNAP were mapped on the available RNAP-DNAbinary complex (PDB ID 4B1O), confirming that the methyl-lysines are located on the surface of this 13 multisubunitenzyme and away from the DNA fragment (Figure 6). Finally,the ratio between methylated and nonmethylated lysinesexposed on the surface is relatively low (∼12%; Figure 6),supporting a selective methylation pattern. These observationsare in agreement with the data reported by Botting et al.,12
suggesting a structure-dependent methylation pattern, andstrengthening the likelihood that the methyltransferase actsafter protein folding.
■ DISCUSSION
We discovered some novel methylation sites in Ssh and SacRNAPs, supporting and expanding the results of previousstudies of archaeal protein methylation. Botting et al. havecharacterized RNAP methylation in Sso12 and found 21 methyl-lysines across 9 subunits of the complex. Our data show theexistence of 20 sites in 11 subunits for Ssh and 26 methyl-lysines distributed across 10 subunits for Sac RNAP (Figure3B). In contrast to the results of large-scale experimentsperformed on mammalian cells,9 there are no reliable reports ofarginine methylation in Archaea, suggesting that lysine is theprevalent residue for archaeal RNAP methylation. We alsofound N-terminal acetylation of Rpo3 and Rpo4 in Ssh andRpo6 and Rpo13 in Sac. N-terminal acetylation of Rpo1N,Rpo2, and Rpo4 has been reported for Sso.12
Use of parallel approaches improves the results of the RNAPmethylation analysis. We used both in-gel and in-solutiondigestion procedures and combined CID and ETD fragmenta-tion methods. This strategy allowed extensive sample analysis(Figure 3A), providing a large number of data for methylationmapping. We found that CID and ETD were equally useful inthe discovery of modified sites. The majority of modifiedpeptides (61 out of 66) were found using both fragmentationmethods. CID contributed three exclusive methyl-peptides, andtwo such peptides were discovered using ETD (Tables 1 andT
able
2.continued
furthermanually
validated.M
odificatio
n:thetype
ofthemodificatio
n(s)detected
inthepeptide.Unm
od:anasterisk(∗)indicatesthattheunmodified
form
ofthepeptidewasalso
identified.G
el/Sol:an
asterisk(∗)indicatespositiveidentificatio
nforthepeptideineach
ofthesample-handlingmethods
(in-gel/in-solution).C
ID/ETD:bestscoreobtained
forCID
and/or
ETD,w
henavailable.bThe
peptide
MID
EKLK
GYID
KRwas
identified
inboth
replicates,b
utwith
anoxidized
methioninein
oneof
thereplicates.T
hebestscorewas
selected
forthetable,correspondingto
thenon-oxidized
form
.
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482643
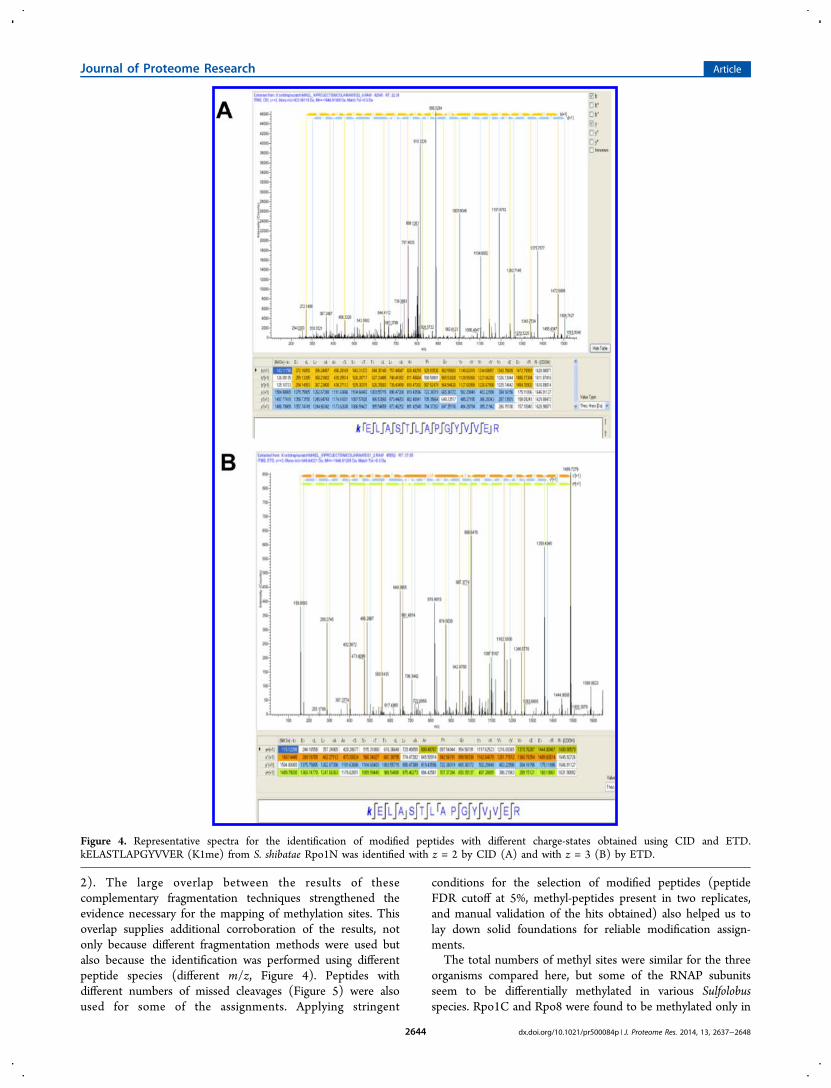
2). The large overlap between the results of thesecomplementary fragmentation techniques strengthened theevidence necessary for the mapping of methylation sites. Thisoverlap supplies additional corroboration of the results, notonly because different fragmentation methods were used butalso because the identification was performed using differentpeptide species (different m/z, Figure 4). Peptides withdifferent numbers of missed cleavages (Figure 5) were alsoused for some of the assignments. Applying stringent
conditions for the selection of modified peptides (peptideFDR cutoff at 5%, methyl-peptides present in two replicates,and manual validation of the hits obtained) also helped us tolay down solid foundations for reliable modification assign-ments.The total numbers of methyl sites were similar for the three
organisms compared here, but some of the RNAP subunitsseem to be differentially methylated in various Sulfolobusspecies. Rpo1C and Rpo8 were found to be methylated only in
Figure 4. Representative spectra for the identification of modified peptides with different charge-states obtained using CID and ETD.kELASTLAPGYVVER (K1me) from S. shibatae Rpo1N was identified with z = 2 by CID (A) and with z = 3 (B) by ETD.
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482644
Sac and Ssh, whereas evidence for Rpo11 and Rpo12methylation was found in Ssh and Sso, but not in Sac. Rpo6methylation was found only in Sac. Interestingly, a well-fragmented peptide from Sac carrying a methylation at Rpo10K33 was identified in one of our replicates (VMGGEDPE-kVLTELGVNR, M2-Oxidation, K9-Methyl). A nonmisscleavedpeptide carrying the same methylation was identified in theother replicate (VMGGEDPEK, M2-Oxidation, K9-Methyl),but fragmentation information was limited (SupplementaryFigure 5). In addition, a peptide carrying K33 methylation wasidentified in one of the Ssh replicates (Supplementary Figure
3). These assignments were left out of our selected data sets,although they present some evidence for Rpo10 methylation.For the first time, we provide evidence for Rpo1C, Rpo6, and
Rpo8 methylation (these modifications have not been reportedfor Sso). Rpo1C was extensively methylated in Sac, where 4methyl-lysines were detected. Two Rpo1C lysine methylationsites, different from those in Sac, were identified in Ssh. Theselysines are conserved between species, but the surroundingsequences are quite different in most of the cases (Figure 7,Supplementary Figure 4). Rpo6, however, is methylated only inSac (two methyl-peptides mapping to three methylation sites)and is also N-terminally acetylated (one single peptide). It is
Figure 5. Representative spectra for the identification of modified peptides with different number of missed cleavages. K519 methylation in S.shibatae Rpo2 was identified in two different peptides: (A) VTEGGEDQNEYLkWSK, K13me, no missed cleavages and (B) RVTEGGEDQNEY-LkWSK, K14me, 2 missed cleavages.
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482645
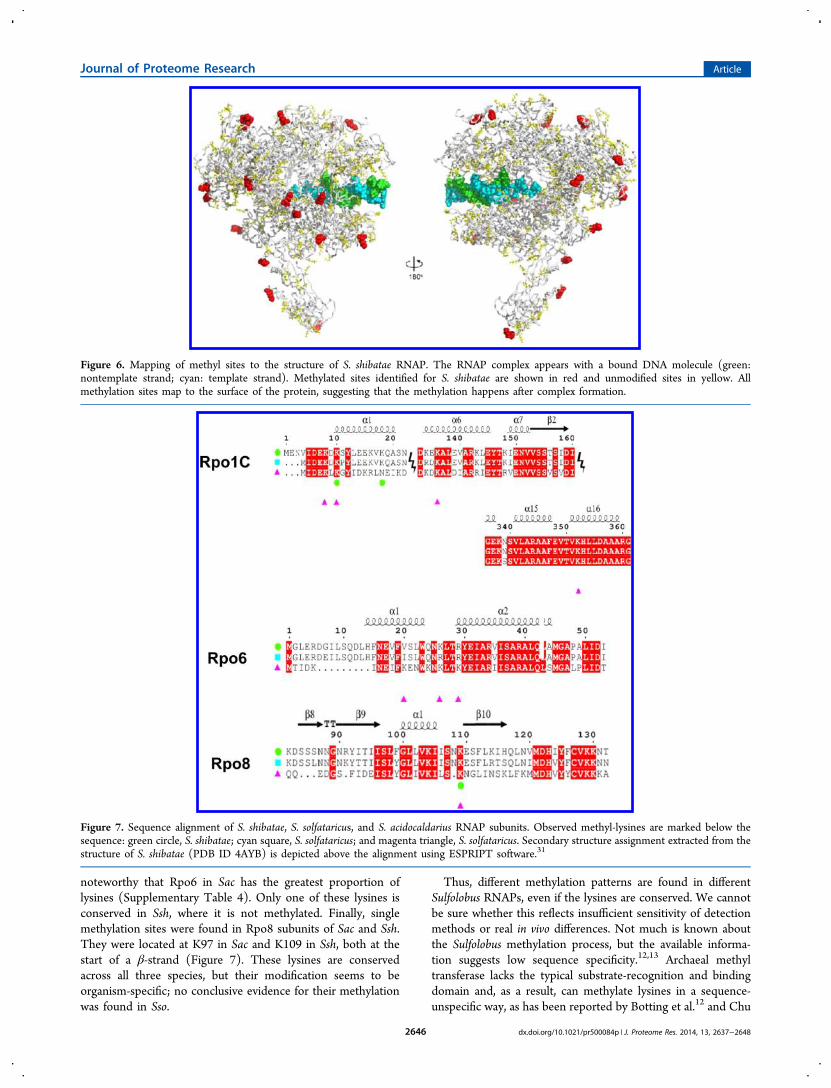
noteworthy that Rpo6 in Sac has the greatest proportion oflysines (Supplementary Table 4). Only one of these lysines isconserved in Ssh, where it is not methylated. Finally, singlemethylation sites were found in Rpo8 subunits of Sac and Ssh.They were located at K97 in Sac and K109 in Ssh, both at thestart of a β-strand (Figure 7). These lysines are conservedacross all three species, but their modification seems to beorganism-specific; no conclusive evidence for their methylationwas found in Sso.
Thus, different methylation patterns are found in differentSulfolobus RNAPs, even if the lysines are conserved. We cannotbe sure whether this reflects insufficient sensitivity of detectionmethods or real in vivo differences. Not much is known aboutthe Sulfolobus methylation process, but the available informa-tion suggests low sequence specificity.12,13 Archaeal methyltransferase lacks the typical substrate-recognition and bindingdomain and, as a result, can methylate lysines in a sequence-unspecific way, as has been reported by Botting et al.12 and Chu
Figure 6. Mapping of methyl sites to the structure of S. shibatae RNAP. The RNAP complex appears with a bound DNA molecule (green:nontemplate strand; cyan: template strand). Methylated sites identified for S. shibatae are shown in red and unmodified sites in yellow. Allmethylation sites map to the surface of the protein, suggesting that the methylation happens after complex formation.
Figure 7. Sequence alignment of S. shibatae, S. solfataricus, and S. acidocaldarius RNAP subunits. Observed methyl-lysines are marked below thesequence: green circle, S. shibatae; cyan square, S. solfataricus; and magenta triangle, S. solfataricus. Secondary structure assignment extracted from thestructure of S. shibatae (PDB ID 4AYB) is depicted above the alignment using ESPRIPT software.31
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482646

et al.13 Currently available data suggest that the specificity ofmethylation pattern, rather than being determined by aminoacid sequence, might be guided by the local secondary structureand accessibility of the residue to be modified. This supportsthe idea of a certain structural specificity that may helpunderstand the differences in the methylation patternsdescribed.Lysine methylation in archaeal organisms has been directly
linked to protein stabilization.14,22,23 Methylation of lysinesincreases the pKa of the side chains, changing the hydropathy ofproteins and strengthening ionic interactions, and thereforecontributing to the side-chain stability.12 Lysine methylationmaps onto the surfaces of the three RNAP complexes,suggesting that the modifications happen after the assemblyof subunits into a full-complement RNAP enzyme. Bremang etal. have found that in mammalian HeLaS3 cells methylatedresidues are over-represented in proteins that form complexes.9
Their results suggest that methylation may play a role in theformation or stabilization of complexes. This modificationmight also be involved in the formation or stabilization ofarchaeal protein complexes. Methylation of certain sites mightstrengthen the protein structure through chemical modifica-tions; it might also improve the protein stability by competingfor that site with modifications that compromise proteinsurvival, such as ubiquitination. This mechanism has beenproposed for the stabilization of some eukaryan proteins,23 anda similar relationship between methylation and SAMPylationcould be proposed for archaeal organisms.24
Recent studies of methylation of other molecular machinessuch as ribosomes from Saccharomyces cerevisiae have revealedthe existence of both surface and buried methylation sites, thelatter being involved in the regulation of the interactionsbetween ribosomal proteins and rRNA.25 However, since noburied methyl-lysines were found in our study, this does notseem to apply to archaeal organisms. This observation impliesthat these PTMs are not involved in the interactions with theDNA or RNA bound by the complex. Similarly, one could alsospeculate that, in addition to its probable stabilizing role, lysinemethylation on the RNAP surface might serve as a rudimentalregulatory mechanism modulating RNA binding during its exitfrom the enzyme. Biophysical studies have shown that nascentmRNA binds to Rpo4/7;19 methylation sites on the stalkRpo4/7 domain might guide the exit of mRNA from theRNAP. In Eukarya, this putative crude regulatory mechanismwould have been superseded by the more sophisticatedregulatory system.We also identified some functionally related proteins,
purified together with the archaeal RNA subunits (seeSupplementary Tables 1 and 2). Heat stabilization-relatedthermosome subunits alpha (P46219), beta (P28488), andgamma (Q9HH21)26,27 were among the most abundant co-purified proteins in Ssh RNAP extracts. DNA/RNA-bindingproteins involved in transcription regulation, such as Alba(Q4J973 and P60848 UniProt entries)28,29 and TBP (TATAbox-binding protein, inferred from homology in UniProtM1IYI2),30 were also identified together with RNAP subunits.We also found some uncharacterized proteins such as single-stranded DNA-binding protein (ORF Name SacRo-n12I_04725, predicted protein, M1J131), uncharacterizedprotein M1IBK9, and the probable exosome complexexonuclease 1 (ECX1, Q4JB27), which should be highlightedas putative interactors with RNAP complexes. Interestingly,modified peptides for some of these putative interactors were
also identified in our approach (Supplementary Figure 3),showing that not only RNAP proteins may be modified andsuggesting additional regulation levels for their interaction.Further experiments should be performed to assess thesignificance of the proteins co-purified with the RNAP. Thepossibility that methylation might be a signal for therecruitment of specific proteins and cofactors, modulatingprotein degradation or protein activity, should not beoverlooked.
■ CONCLUSIONTo summarize, we performed in-solution and in-gel digestion ofRNAP from Ssh and Sac and combined CID and ETDfragmentation methods in mass spectrometry analysis tocharacterize methylation and N-terminal acetylation of theRNAP component subunits. We identified 20 methyl-lysines forSsh and 26 for Sac RNAP. Moreover, two N-terminalacetylation sites were found for each of the analyzed species.When we mapped the obtained data to the three-dimensionalstructure of the RNAP-DNA complex from Ssh, it becameapparent that all of the methyl-lysines were on the surface, farfrom the bound DNA. We hypothesize that these particularPTMs in archaeal RNAP would fulfill additional roles to that ofincreasing the stability of the enzyme in harsh environments,such as the modulation of the RNAP binding to othercomponents of the transcriptional machinery; however, furtherstudies are needed to test this hypothesis.
■ ASSOCIATED CONTENT*S Supporting Information
Supplementary tables and figures as described in the text. Thismaterial is available free of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author
*Tel: +34 94 406 13 15. Fax: +34 94 657 25 02. E-mail:[email protected] Address¶M.N.W.: EMBL-Grenoble, France.Notes
The authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe thank Yakov Korkhin for the gift of purified S.acidocaldarius RNAP and Stephen D. Bell for S. shibatae cells.Marina Ondiviela and Pietro Roversi are also thanked for helpduring S. shibatae biomass production, and Ibon Iloro andIraide Escobes for sample preparation and useful discussion.This work is supported by the CIC bioGUNE (to MA andMNW) and the Spanish Ministerio de Economia yCompetitividad BFU2012-33947 (to NGAA).
■ REFERENCES(1) Woese, C. R.; Kandler, O.; Wheelis, M. L. Towards a naturalsystem of organisms: proposal for the domains Archaea, Bacteria, andEucarya. Proc. Natl. Acad. Sci. U.S.A. 1990, 87 (12), 4576−9.(2) Beattie, T. R.; Bell, S. D. Molecular machines in archaeal DNAreplication. Curr. Opin. Chem. Biol. 2011, 15 (5), 614−9.(3) Korkhin, Y.; Unligil, U. M.; Littlefield, O.; Nelson, P. J.; Stuart, D.I.; Sigler, P. B.; Bell, S. D.; Abrescia, N. G. Evolution of complex RNA
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482647

polymerases: the complete archaeal RNA polymerase structure. PLoSBiol. 2009, 7 (5), e1000102.(4) Wojtas, M. N.; Mogni, M.; Millet, O.; Bell, S. D.; Abrescia, N. G.Structural and functional analyses of the interaction of archaeal RNApolymerase with DNA. Nucleic Acids Res. 2012, 40 (19), 9941−52.(5) Paik, W. K.; Paik, D. C.; Kim, S. Historical review: the field ofprotein methylation. Trends Biochem. Sci. 2007, 32 (3), 146−52.(6) Lee, D. Y.; Teyssier, C.; Strahl, B. D.; Stallcup, M. R. Role ofprotein methylation in regulation of transcription. Endocr. Rev. 2005,26 (2), 147−70.(7) Zhang, X.; Wen, H.; Shi, X. Lysine methylation: beyond histones.Acta Biochim. Biophys. Sin. (Shanghai) 2012, 44 (1), 14−27.(8) Huang, J.; Berger, S. L. The emerging field of dynamic lysinemethylation of non-histone proteins. Curr. Opin. Genet. Dev. 2008, 18(2), 152−8.(9) Bremang, M.; Cuomo, A.; Agresta, A. M.; Stugiewicz, M.;Spadotto, V.; Bonaldi, T. Mass spectrometry-based identification andcharacterisation of lysine and arginine methylation in the humanproteome. Mol. Biosyst. 2013, 9 (9), 2231−47.(10) Moore, K. E.; Carlson, S. M.; Camp, N. D.; Cheung, P.; James,R. G.; Chua, K. F.; Wolf-Yadlin, A.; Gozani, O. A general molecularaffinity strategy for global detection and proteomic analysis of lysinemethylation. Mol. Cell 2013, 50 (3), 444−56.(11) Assiddiq, B. F.; Snijders, A. P.; Chong, P. K.; Wright, P. C.;Dickman, M. J. Identification and characterization of sulfolobussolfataricus P2 proteome using multidimensional liquid phase proteinseparations. J. Proteome Res. 2008, 7 (6), 2253−61.(12) Botting, C. H.; Talbot, P.; Paytubi, S.; White, M. F. Extensivelysine methylation in hyperthermophilic crenarchaea: potentialimplications for protein stability and recombinant enzymes. Archaea2010, 2010.(13) Chu, Y.; Zhang, Z.; Wang, Q.; Luo, Y.; Huang, L. Identificationand characterization of a highly conserved crenarchaeal protein lysinemethyltransferase with broad substrate specificity. J. Bacteriol. 2012,194 (24), 6917−26.(14) Febbraio, F.; Andolfo, A.; Tanfani, F.; Briante, R.; Gentile, F.;Formisano, S.; Vaccaro, C.; Scire, A.; Bertoli, E.; Pucci, P.; Nucci, R.Thermal stability and aggregation of sulfolobus solfataricus beta-glycosidase are dependent upon the N-epsilon-methylation of specificlysyl residues: critical role of in vivo post-translational modifications. J.Biol. Chem. 2004, 279 (11), 10185−94.(15) Jensen, O. N. Interpreting the protein language usingproteomics. Nat. Rev. Mol. Cell Biol. 2006, 7 (6), 391−403.(16) Mikesh, L. M.; Ueberheide, B.; Chi, A.; Coon, J. J.; Syka, J. E.;Shabanowitz, J.; Hunt, D. F. The utility of ETD mass spectrometry inproteomic analysis. Biochim. Biophys. Acta 2006, 1764 (12), 1811−22.(17) Fisk, J. C.; Li, J.; Wang, H.; Aletta, J. M.; Qu, J.; Read, L. K.Proteomic analysis reveals diverse classes of arginine methylproteins inmitochondria of trypanosomes. Mol. Cell. Proteomics 2013, 12 (2),302−11.(18) Sobott, F.; Watt, S. J.; Smith, J.; Edelmann, M. J.; Kramer, H. B.;Kessler, B. M. Comparison of CID versus ETD based MS/MSfragmentation for the analysis of protein ubiquitination. J. Am. Soc.Mass Spectrom. 2009, 20 (9), 1652−9.(19) Zubarev, R. A.; Zubarev, A. R.; Savitski, M. M. Electron capture/transfer versus collisionally activated/induced dissociations: solo orduet? J. Am. Soc. Mass Spectrom. 2008, 19 (6), 753−61.(20) Korkhin, Y.; Littlefield, O.; Nelson, P. J.; Bell, S. D.; Sigler, P. B.Preparation of components of archaeal transcription preinitiationcomplex. Methods Enzymol. 2001, 334, 227−39.(21) Wojtas, M.; Peralta, B.; Ondiviela, M.; Mogni, M.; Bell, S. D.;Abrescia, N. G. Archaeal RNA polymerase: the influence of theprotruding stalk in crystal packing and preliminary biophysical analysisof the Rpo13 subunit. Biochem. Soc. Trans. 2011, 39 (1), 25−30.(22) Sabo, T. M.; Bakhtiari, D.; Walter, K. F.; McFeeters, R. L.;Giller, K.; Becker, S.; Griesinger, C.; Lee, D. Thermal coefficients ofthe methyl groups within ubiquitin. Protein Sci. 2012, 21 (4), 562−70.(23) Yang, X. D.; Lamb, A.; Chen, L. F. Methylation, a newepigenetic mark for protein stability. Epigenetics 2009, 4 (7), 429−33.
(24) Humbard, M. A.; Miranda, H. V.; Lim, J. M.; Krause, D. J.; Pritz,J. R.; Zhou, G.; Chen, S.; Wells, L.; Maupin-Furlow, J. A. Ubiquitin-like small archaeal modifier proteins (SAMPs) in Haloferax volcanii.Nature 2010, 463 (7277), 54−60.(25) Sainsbury, S.; Niesser, J.; Cramer, P. Structure and function ofthe initially transcribing RNA polymerase II-TFIIB complex. Nature2013, 493 (7432), 437−40.(26) Kagawa, H. K.; Osipiuk, J.; Maltsev, N.; Overbeek, R.; Quaite-Randall, E.; Joachimiak, A.; Trent, J. D. The 60 kDa heat shockproteins in the hyperthermophilic archaeon Sulfolobus shibatae. J. Mol.Biol. 1995, 253 (5), 712−25.(27) Trent, J. D.; Nimmesgern, E.; Wall, J. S.; Hartl, F. U.; Horwich,A. L. A molecular chaperone from a thermophilic archaebacterium isrelated to the eukaryotic protein t-complex polypeptide-1. Nature1991, 354 (6353), 490−3.(28) Aravind, L.; Iyer, L. M.; Anantharaman, V. The two faces ofAlba: the evolutionary connection between proteins participating inchromatin structure and RNA metabolism. Genome Biol. 2003, 4 (10),R64.(29) Mani, J.; Guttinger, A.; Schimanski, B.; Heller, M.; Acosta-Serrano, A.; Pescher, P.; Spath, G.; Roditi, I. Alba-domain proteins ofTrypanosoma brucei are cytoplasmic RNA-binding proteins thatinteract with the translation machinery. PLoS One 2011, 6 (7), e22463.(30) De Carlo, S.; Lin, S. C.; Taatjes, D. J.; Hoenger, A. Molecularbasis of transcription initiation in Archaea. Transcription 2010, 1 (2),103−11.(31) Gouet, P.; Courcelle, E.; Stuart, D. I.; Metoz, F. ESPript:analysis of multiple sequence alignments in PostScript. Bioinformatics1999, 15 (4), 305−8.
Journal of Proteome Research Article
dx.doi.org/10.1021/pr500084p | J. Proteome Res. 2014, 13, 2637−26482648





























