Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
126 (2008) 139–148www.elsevier.com/locate/jconrel
Journal of Controlled Release
Long-term in vitro study of paclitaxel-eluting bioresorbablecore/shell fiber structures
Amir Kraitzer, Lia Ofek, Reut Schreiber, Meital Zilberman ⁎
Department of Biomedical Engineering, Faculty of Engineering, Tel-Aviv University, Tel-Aviv 69978, Israel
Received 10 October 2007; accepted 16 November 2007Available online 28 November 2007
Abstract
Paclitaxel-eluting bioresorbable core/shell fiber structures for stent applications and local cancer treatment were developed and studied. Thesestructures were composed of a polyglyconate core and a porous PDLGA shell loaded with the anti-proliferative agent paclitaxel, prepared usingfreeze drying of inverted emulsions. The investigation of these new composite fibers focused on the effects of the emulsion's composition(formulation) and process kinetics on the long-term drug release from the fibers, in light of the shell's morphology and degradation profile.Paclitaxel release from the porous shell was relatively slow due to its extremely hydrophobic nature. It exhibited three phases of release, whichcorresponded to the degradation profile of the host PDLGA. We found that the effect of the emulsion formulation on the release profile is moresignificant than the effect of the process kinetics. The copolymer composition had the most dominant effect on the drug release profile from thecomposite fibers. The polymer content also affected the release profile, whereas the drug content and the organic:aqueous phase ratio resulted inminor effects. Emulsions with a less hydrophobic nature are favorable for effective controlled release of the hydrophobic paclitaxel from theporous shell.© 2007 Elsevier B.V. All rights reserved.
Keywords: Paclitaxel; Drug-eluting fibers; Controlled release; Poly(DL-lactic-co-glycolic acid); Degradation
1. Introduction
Drug-loaded fibers have a high potential for various bio-medical applications. Their advantages include ease of fabrica-tion and a high surface area for controlled release and localizeddelivery of bioactive agents to their target. Only few controlledrelease fiber systems based on polymers have been investigatedto date [1–8]. The two basic types of drug-loaded fibers thathave been reported are monolithic fibers and reservoir fibers. Insystems that use monolithic fibers, the drug is dissolved ordispersed throughout the polymer fiber. For example, curcumin,paclitaxel and dexamethasone have been melt spun with poly(L-lactic acid) (PLLA) to generate drug-loaded fibers [1] andaqueous drugs have been solution spun with PLLA [2]. Varioussteroid-loaded fiber systems have demonstrated expected first-order release kinetics [3–5]. In systems that use hollow re-
⁎ Corresponding author. Tel.: +972 3 6405842; fax: + 972 3 6407939.E-mail address: [email protected] (M. Zilberman).
0168-3659/$ - see front matter © 2007 Elsevier B.V. All rights reserved.doi:10.1016/j.jconrel.2007.11.011
servoir fibers, drugs such as dexamethasone and methotrexanehave been added to the internal section of the fiber [6–8]. Thedisadvantages of these systems include poor mechanical prop-erties due to drug incorporation and limitations in drug loading.Furthermore, many drugs and all proteins do not tolerate meltprocessing and organic solvents.
In one of our recent studies we presented a new concept ofcore/shell fiber structures which successfully meet these chal-lenges [9]. These composite fibers combine a dense polymercore fiber and a drug-loaded porous shell structure, i.e. the drugor protein is located in a separate compartment (a “shell”)around a melt spun “core” fiber. The shell is prepared usingmild processing conditions. This can result in good mechanicalproperties as well as in the desired drug release profile. Our newfibers are ideal for forming thin, delicate, biomedically im-portant structures for various applications, such as fiber-basedbioresorbable endovascular stents that mechanically supportblood vessels while delivering drugs for preventing restenosisdirectly to the blood vessel wall, or drug delivery systems for

140 A. Kraitzer et al. / Journal of Controlled Release 126 (2008) 139–148
cancer treatment. The next two paragraphs focus on these twoapplications.
Restenosis is the re-narrowing of a blood vessel which causesa reduction in the diameter of the lumen, consequently restrictingblood flow after an intravascular procedure. In-stent restenosis ismainly characterized by an abnormal increase in tissue growth(neointimal hyperplasia) of the tissue composing the bloodvessel [10,11]. Re-narrowing is the result of a complex series ofbiological events in response to the initial injury of the vesselcaused by balloon expansion and the presence of the permanentstent implant [12]. Further progress in the treatment of coronaryartery disease has been the development of drug-eluting stentsthat dramatically reduce the incidence of in-stent restenosisto less than 5% [13]. Several drug-coated metal stents havereceived FDA approval to date. The most popular ones areTAXUS™ (Boston Scientific) — a paclitaxel-eluting stent, andCypher™ (J&J) — a sirolimus eluting stent. These are metalstents coated with a thin stable dense polymer layer in which thedrug is located. Bioresorbable stents are designed to support thevascular wall during the vessel healing process, graduallytransferring the mechanical load to the vessel wall while the stentmass and strength decrease over time. Restenosis usually occurswithin 3 to 6 months after coronary intervention, and rarelythereafter. Interest in bioresorbable vascular stents, with orwithout drug delivery, is therefore increasing. These stents areusually made of fibers. For example, Tamai et al. [14,15]described the Igaki/Tamai stent, a bioresorbable balloon ex-pandable zigzag coil design based on a PLLAmonofilament.Wehave developed and studied a fiber-based expandable stentdesign [16,17], which was prepared using a linear continuouscoil array principle, by which four furled lobes convert into asingle large lobe upon balloon expansion. This expandable stentdesign is based on melt-spun bioresorbable fibers that weremade from a relatively high molecular weight PLLA. Itdemonstrated excellent initial radial compression strength andgood in vitro degradation resistance for 20 weeks.
Cancer may be defined as a disease characterized by uncon-trolled proliferation and the spread of abnormal forms of thebody's own cells in the body [18]. In cancer therapy, surgicaltreatment is usually carried out for patients with a resectablecarcinoma. However, treatment failure due to local recurrenceof primary tumors or metastatic spread often occurs. Surgicaladjuvant chemotherapy employing an anticancer agent duringor immediately after surgery would therefore be beneficial [18].A delivery system loaded with paclitaxel at the tumor resectionsite will provide a high local concentration of the drug, which isdetrimental to malignant cells that may have survived surgery,thus preventing tumor re-growth and metastasis. In glioblas-toma, localized prolonged release of anti-proliferative drugs,which induces fewer side effects, can be applied to regionswhere systemic administration is not effective. Drug deliv-ery systems explored for localized paclitaxel delivery to dateinclude microspheres, surgical pastes and implants [18]. Anexample for a biodegradable implant is the Gliadel® wafer,a biodegradable wafer used to deliver the anticancer drugcarmustine directly into a brain tumor site after the surgicalremoval of the tumor. It has been FDA approved since 1996.
Paclitaxel is a potent cell proliferation inhibitor and is knownto be very effective in the treatment of cancer [18] as well as inpreventing restenosis [18,19]. However, it is potentially cy-totoxic, and the paclitaxel dose that can be delivered is relativelysmall [20,21]. Paclitaxel has been shown to markedly attenuatestent-induced intimal thickening of the lumen [22,23]. Paclitax-el's anti-proliferative effect is reversible [24]. Its short cellularresidence time (1 h), along with the reversible anti-proliferativeactivity, suggest that it should be formulated in sustained-release dosage form [25]. The TAXUS trials revealed significantinhibition of coronary stenosis by paclitaxel [26–28]. Drug-eluting polymer-coated stents have thus moved into the lime-light as vehicles for local drug administration [29,30]. In ad-dition to usage in bioresorbable drug-eluting stents, our novelcoating technique can be used for stable stents, where only thedrug-loaded coat degrades with time. Studies on paclitaxel-eluting bioresorbable microspheres for cancer treatment [31,32]have shown that its release in an aqueous medium is relativelyslow due to paclitaxel's high hydrophobicity. Since paclitaxelhas demonstrated effectiveness in both drug-eluting stents andlocal cancer treatment applications, its controlled release in adesired manner is highly important. Highly porous structuresthat encapsulate the drug, such as our fiber's “shell”, maytherefore be advantageous for both applications.
The current study focuses on composite core/shell fiberstructures loaded with paclitaxel. The porous shell (drug-con-taining section) was prepared using the technique of freezedrying an inverted emulsion. The effect of the emulsion'scomposition (formulation) and process kinetics on the emul-sion's stability and on the resulting shell's microstructure havealready been reported [9]. The current article focuses on thelong-term (9 months, relevant for cancer treatment) in vitrorelease of paclitaxel and its mechanisms. It describes the effectsof the emulsion's formulation and process kinetics on thepaclitaxel release profile from the fibers, in light of the shell'smorphology and degradation profile. It should be noted thatthese paclitaxel-eluting composite fiber structures can alsoserve as a good model for composite fibers loaded with otherhydrophobic bioactive agents for a wide range of biomedicalapplications.
2. Materials and methods
2.1. Materials
Maxon™ polyglyconate monofilament (3–0) sutures, with adiameter of 0.20–0.25 mm, Syneture, USA were used as corefibers.
Bioresorbable porous structures (the shell coating) weremade of the following polymers:
• 75/25 poly(DL-lactic-co-glycolic acid), inherent viscosity(i.v.) =0.65 dL/g (in CHCl3 at 30 °C, approximately97,100 g/mol), Absorbable Polymer Technologies Inc.,USA. This polymer is termed 75/25 PDLGA.
• 50/50 poly(DL-lactic-co-glycolic acid), inherent viscosi-ty (i.v.) =0.56 dL/g (in CHCl3 at 30 °C, approximately

141A. Kraitzer et al. / Journal of Controlled Release 126 (2008) 139–148
80,000 g/mol), Absorbable Polymer Technologies Inc.,USA. This polymer is termed 50/50 PDLGA.
Drug: Paclitaxel (Genexol™) was purchased from Sam YangCorp. (Seoul, Korea).
2.1.1. Reagents1,1,1,3,3,3-Hexafluoro-2-propanol (H1008) was purchased
from Spectrum Chemical Mfg. Corp.Chloroform (CHCl3) HPLC grade and methylene chloride
(CH2Cl2) HPLC grade were purchased from Frutarom, Israel.Acetonitrile (CH3CN) HPLC grade was purchased from J.T.
Baker.
2.2. Preparation of core/shell fiber structures
2.2.1. Fiber surface treatmentThe sutures were surface-treated in order to enhance the
adhesion between the core fiber and the coating. The poly-glyconate fibers were slightly stretched on special holders anddipped in 1,1,1,3,3,3-hexafluoro-2-propanol (hexafluorisopro-panol) for 40 s. The fibers were then washed with ethanol anddried at room temperature.
2.2.2. Emulsion formationA known amount of 75/25 PDLGA was dissolved in chlo-
roform to form an organic solution and paclitaxel was addedto the solution. Double-distilled water was then poured intothe organic phase (in a test tube) and homogenization ofthe emulsion was performed using a homogenizer (PolytronPT3100 Kinematica, 12 mm rotor) operating at 16,500 rpm(medium rate) for 3 min, for most investigated samples. As areference sample we chose an emulsion formulation containing17.5% w/v polymer in the organic solution, 1.43% w/w pac-litaxel (relative to the polymer load), and an organic to aqueous(O:A) phase ratio of 2:1 v/v. Other formulations included 22.5%w/v polymer, 2.9% and 4.3% w/w paclitaxel, O:A phase ratiosof 4:1 and copolymer composition of 50/50 PDLGA. Certainsamples were prepared using homogenization rates of 5500 rpm(low rate) or 25,000 rpm (high rate) in order to investigate theeffect of processing kinetics on the porous shell structure.
2.2.3. Core/shell fiber structure formationThe treated core polyglyconate fibers were dip-coated (while
placed on holders) in fresh emulsions and then frozen imme-diately in a liquid nitrogen bath. The holders+samples werethen placed in a pre-cooled (−105 °C) freeze dryer (Virtis 101equipped with a nitrogen trap) capable of working with organicsolvents (freezing temperature of the condenser was approx-imately −105 °C) and freeze dried in order to preserve themicrostructure of the emulsion-based core/shell fiber structures.Drying was performed in two stages:
1. The freeze dryer chamber pressure was reduced to 100 mTorrwhile the temperature remained at −105 °C.
2. The condenser was turned off and its plate temperatureslowly rose to room temperature while the pressure was
monitored between 100 mTorr and 700 mTorr. During thisstep the liquid nitrogen trap condensed the excess water andsolvent vapors.
The samples were stored in desiccators until use.
2.2.4. Morphological characterizationThe morphology of the composite core/shell fiber structures
(cryogenically fractured surfaces) was observed using a JeolJSM-6300 scanning electron microscope (SEM) at an accel-erating voltage of 5 kV. The SEM samples were Au sputteredprior to observation. The mean pore diameter of the observedmorphologies was analyzed using the Sigma Scan Pro softwareand statistics were drawn using SPSS. Statistical significancewas determined using the ANOVA (Tukey–Kramer) method.Statistical significance was determined at pb0.05.
2.3. In vitro paclitaxel release studies
The composite core/shell fiber structures (triplicates) wereimmersed in phosphate buffered saline (PBS) at 37 °C andpH=7.4 for 37 weeks in order to determine paclitaxel's releasekinetics from these structures. The release studies were con-ducted in closed glass vessels containing 3 ml PBS medium,using a horizontal bath shaker operated at a constant rate of130 rpm. The medium was removed (completely) periodically,at certain sampling times, and fresh medium was introduced.This experiment enabled simulating conditions similar to thosewhich exist in a blood vessel.
2.3.1. Extraction procedurePaclitaxel extraction from the medium was performed as
follows: the 3 ml medium was completely removed at each timepoint and placed in a scintillation vial. 3 ml acetonitrile and 1 mlmethylene chloride were added. Methylene chloride evapora-tion was performed under a nitrogen stream and the paclitaxelconcentration was then estimated using HPLC. An extractionfactor was used for correction. Known weights of paclitaxelwere dissolved in 1 ml of methylene chloride and subjected tothe same extraction procedure in order to determine the re-covery efficiency of the extraction procedure. Recoveries werealways greater than 75% and the value of the measured drugwas corrected accordingly.
The paclitaxel content of the medium samples was determinedusing Jasco High Performance Liquid Chromatography (HPLC)with a UV2075 plus detector and a reverse phase column (ZorbaxODS 5 µm, inner diameter d=4.6 mm, length=150 mm), andwas kept at 25 °C. The mobile phase consisted of a mixture ofacetonitrile and double-distilled water (55/45, v/v) at a flow rateof 1 ml/min with a quaternary gradient pump (PU 2089 plus)without gradient. 100 µl samples were injected with an auto-sampler (AS 2057 Plus). The column effluent was eluted for15 min and detected at 227 nm. The area of each eluted peak wasintegrated using the EZstart software version 3.1.7.
The cumulative release profiles were determined relative tothe initial amount of paclitaxel in the composite fibers (quantityreleased during the incubation period+ the residue remaining in

Fig. 1. SEM fractographs showing cross sections of the reference compositefiber.
142 A. Kraitzer et al. / Journal of Controlled Release 126 (2008) 139–148
the fibers). All experiments were performed in triplicates. Re-sults are presented as means±standard errors. The effects of theemulsion's formulation on the release profile were studied byexamining the following parameters: polymer content in theorganic phase (%w/v), drug content relative to polymer content(%w/w), organic:aqueous (O:A) phase ratio and copolymercomposition. The effect of the process kinetics (homogenizationrate) on the release profile was also studied.
2.4. Residual drug recovery from composite fibers
Residual paclitaxel recovery from the composite fibers wasmeasured as follows: the fibers were placed in 1 ml methylenechloride and 3 ml of a 50/50 acetonitrile/water solution wasadded. Methylene chloride evaporation was performed under anitrogen (99.999%) stream and the paclitaxel concentration wasthen estimated using HPLC. An extraction factor was used forcorrection. Five solutions with known concentrations ofpaclitaxel were subjected to the same extraction procedure inorder to determine the recovery efficiency of the extractionprocedure. Recoveries were always greater than 75% and thevalue of the residual drug was corrected accordingly.
2.5. Paclitaxel's chemical stability
Twomilligrams of paclitaxel were placed in phosphate bufferedsaline (PBS) at 37 °C for 160 days in order to determine paclitaxel'sstability throughout the release experiment. Closed scintillationvials containing 3ml PBSmediumwere placed in a horizontal bathshaker operated at a constant rate of 130 rpm. The paclitaxel contentof a single vial was measured at each point of time.
Paclitaxel was extracted as follows: 3 ml methylene chloridewere added to the 3 ml PBS/paclitaxel mixture and stirred for10 min in order to dissolve the drug. One milliliter was carefullyremoved from the bottom of the vial (the organic phase) using apipette and placed in a new vial. Five milliliters of acetonitrile/double-distilled water (the medium) were added to the vial andthen evaporated under nitrogen conditions. The content of eachvial was transferred to a test tube and diluted to 20 ml. Thepaclitaxel content of the medium samples was determined usingHPLC, as described above.
2.6. In vitro degradation study of the porous PDLGA structure
Porous 75/25 PDLGA film structures especially prepared fordegradation trial were based on the reference emulsion formulation.The inverted emulsion was prepared as described earlier in theparagraph about emulsion formation (in theMaterials and methodssection describing preparation of core/shell fiber structures), pouredinto an aluminum plate and freeze dried in liquid nitrogen. Eachdried film sample was then incubated in 20 ml phosphate bufferedsaline (PBS) containing 0.05% (v/v) sodium azide (as preservative)at 37 °C and pH=7.4 for 17 weeks, under static conditions. Eachsample was taken out at weekly intervals and dried using a vacuumoven (35 °C for 1 h), and stored in a dessicator.
The weight-average molecular weight and number-averagemolecular weight of the samples were determined by gel
permeation chromatography (GPC). The GPC (Waters 21515isocratic pump, operating temperature 40 °C using a columnoven) was equipped with a refractive index detector (Waters2414, operating temperature 40 °C) and calibrated with poly-L-lactic acid MW kit standards (Polysciences Inc., USA). Datawere analyzed using the Breeze version 3.3 software. Thesamples were dissolved in methylene chloride, filtered, andeluted through 4 Styragel columns (model WAT044234 HR1THF, WAT044237 HR2, WAT044225 HR4, WAT054460 HR5,300×7.8 mm, 5 µm particle diameter) at a flow rate of 1 ml/min. The elution medium was Baker analyzed HPLC grademethylene chloride.
3. Results and discussion
The freeze drying technique is unique in being able to pre-serve the liquid structure in solids. We used this technique inorder to produce inverted emulsions in which the continuousphase contained polymer and drug dissolved in a solvent, withwater being the dispersed phase. SEM fractographs showing thebulk morphology of the reference specimen are presented inFig. 1. The diameter of the treated core fiber was in the range of200–250 µm and a shell thickness of 30–70 µm was obtainedfor most emulsion formulations. Relatively high contents ofhydrophobic components (such as PDLGA and paclitaxel) re-sulted in an increase in shell thickness, due to the emulsion's
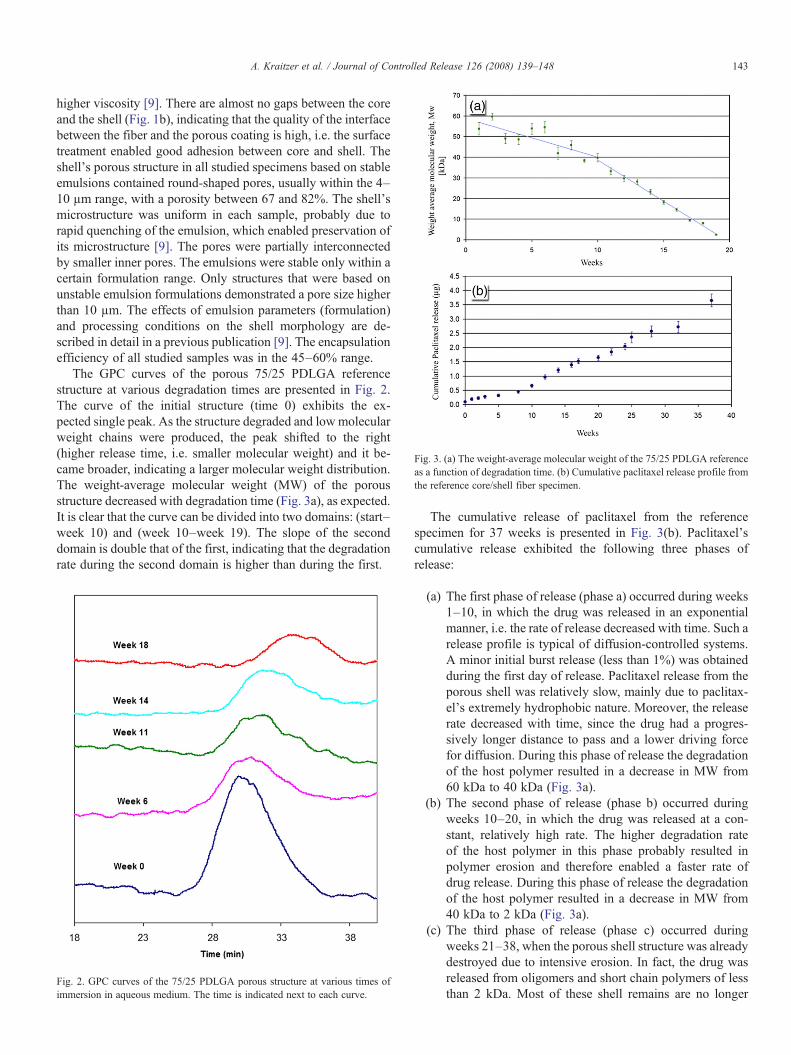
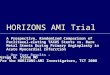
Fig. 3. (a) The weight-average molecular weight of the 75/25 PDLGA referenceas a function of degradation time. (b) Cumulative paclitaxel release profile fromthe reference core/shell fiber specimen.
143A. Kraitzer et al. / Journal of Controlled Release 126 (2008) 139–148
higher viscosity [9]. There are almost no gaps between the coreand the shell (Fig. 1b), indicating that the quality of the interfacebetween the fiber and the porous coating is high, i.e. the surfacetreatment enabled good adhesion between core and shell. Theshell's porous structure in all studied specimens based on stableemulsions contained round-shaped pores, usually within the 4–10 µm range, with a porosity between 67 and 82%. The shell'smicrostructure was uniform in each sample, probably due torapid quenching of the emulsion, which enabled preservation ofits microstructure [9]. The pores were partially interconnectedby smaller inner pores. The emulsions were stable only within acertain formulation range. Only structures that were based onunstable emulsion formulations demonstrated a pore size higherthan 10 µm. The effects of emulsion parameters (formulation)and processing conditions on the shell morphology are de-scribed in detail in a previous publication [9]. The encapsulationefficiency of all studied samples was in the 45–60% range.
The GPC curves of the porous 75/25 PDLGA referencestructure at various degradation times are presented in Fig. 2.The curve of the initial structure (time 0) exhibits the ex-pected single peak. As the structure degraded and lowmolecularweight chains were produced, the peak shifted to the right(higher release time, i.e. smaller molecular weight) and it be-came broader, indicating a larger molecular weight distribution.The weight-average molecular weight (MW) of the porousstructure decreased with degradation time (Fig. 3a), as expected.It is clear that the curve can be divided into two domains: (start–week 10) and (week 10–week 19). The slope of the seconddomain is double that of the first, indicating that the degradationrate during the second domain is higher than during the first.
Fig. 2. GPC curves of the 75/25 PDLGA porous structure at various times ofimmersion in aqueous medium. The time is indicated next to each curve.
The cumulative release of paclitaxel from the referencespecimen for 37 weeks is presented in Fig. 3(b). Paclitaxel'scumulative release exhibited the following three phases ofrelease:
(a) The first phase of release (phase a) occurred during weeks1–10, in which the drug was released in an exponentialmanner, i.e. the rate of release decreased with time. Such arelease profile is typical of diffusion-controlled systems.A minor initial burst release (less than 1%) was obtainedduring the first day of release. Paclitaxel release from theporous shell was relatively slow, mainly due to paclitax-el's extremely hydrophobic nature. Moreover, the releaserate decreased with time, since the drug had a progres-sively longer distance to pass and a lower driving forcefor diffusion. During this phase of release the degradationof the host polymer resulted in a decrease in MW from60 kDa to 40 kDa (Fig. 3a).
(b) The second phase of release (phase b) occurred duringweeks 10–20, in which the drug was released at a con-stant, relatively high rate. The higher degradation rateof the host polymer in this phase probably resulted inpolymer erosion and therefore enabled a faster rate ofdrug release. During this phase of release the degradationof the host polymer resulted in a decrease in MW from40 kDa to 2 kDa (Fig. 3a).
(c) The third phase of release (phase c) occurred duringweeks 21–38, when the porous shell structure was alreadydestroyed due to intensive erosion. In fact, the drug wasreleased from oligomers and short chain polymers of lessthan 2 kDa. Most of these shell remains are no longer
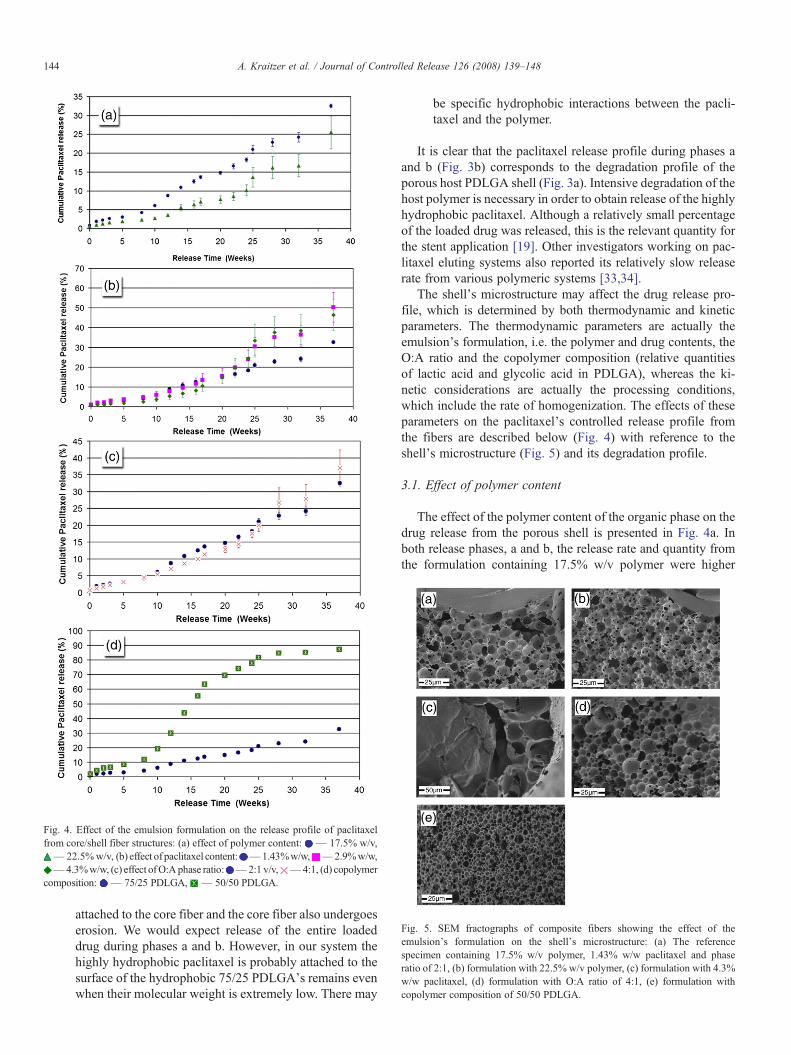
Fig. 4. Effect of the emulsion formulation on the release profile of paclitaxelfrom core/shell fiber structures: (a) effect of polymer content: — 17.5% w/v,— 22.5%w/v, (b) effect of paclitaxel content: — 1.43%w/w, — 2.9%w/w,— 4.3%w/w, (c) effect ofO:Aphase ratio: — 2:1 v/v, — 4:1, (d) copolymer
composition: — 75/25 PDLGA, — 50/50 PDLGA.
Fig. 5. SEM fractographs of composite fibers showing the effect of theemulsion's formulation on the shell's microstructure: (a) The referencespecimen containing 17.5% w/v polymer, 1.43% w/w paclitaxel and phaseratio of 2:1, (b) formulation with 22.5% w/v polymer, (c) formulation with 4.3%w/w paclitaxel, (d) formulation with O:A ratio of 4:1, (e) formulation withcopolymer composition of 50/50 PDLGA.
144 A. Kraitzer et al. / Journal of Controlled Release 126 (2008) 139–148
attached to the core fiber and the core fiber also undergoeserosion. We would expect release of the entire loadeddrug during phases a and b. However, in our system thehighly hydrophobic paclitaxel is probably attached to thesurface of the hydrophobic 75/25 PDLGA's remains evenwhen their molecular weight is extremely low. There may
be specific hydrophobic interactions between the pacli-taxel and the polymer.
It is clear that the paclitaxel release profile during phases aand b (Fig. 3b) corresponds to the degradation profile of theporous host PDLGA shell (Fig. 3a). Intensive degradation of thehost polymer is necessary in order to obtain release of the highlyhydrophobic paclitaxel. Although a relatively small percentageof the loaded drug was released, this is the relevant quantity forthe stent application [19]. Other investigators working on pac-litaxel eluting systems also reported its relatively slow releaserate from various polymeric systems [33,34].
The shell's microstructure may affect the drug release pro-file, which is determined by both thermodynamic and kineticparameters. The thermodynamic parameters are actually theemulsion's formulation, i.e. the polymer and drug contents, theO:A ratio and the copolymer composition (relative quantitiesof lactic acid and glycolic acid in PDLGA), whereas the ki-netic considerations are actually the processing conditions,which include the rate of homogenization. The effects of theseparameters on the paclitaxel's controlled release profile fromthe fibers are described below (Fig. 4) with reference to theshell's microstructure (Fig. 5) and its degradation profile.
3.1. Effect of polymer content
The effect of the polymer content of the organic phase on thedrug release from the porous shell is presented in Fig. 4a. Inboth release phases, a and b, the release rate and quantity fromthe formulation containing 17.5% w/v polymer were higher

145A. Kraitzer et al. / Journal of Controlled Release 126 (2008) 139–148
than the release rate and quantity from the formulation con-taining 22.5% w/v polymer. Since larger drug quantities arereleased during phase b, the differences between these twoformulations are more significant during this phase of releasethan during phase a. It is suggested that since the pore size andporosity remained almost unchanged with the emulsion's poly-mer content (Fig. 5a,b and Table 1) but denser “polymericwalls” were probably created between adjacent pores of theformulation with 22.5% w/v polymer, a relatively high polymercontent increases the interactions between the matrix and pac-litaxel, resulting in a lower diffusion coefficient which results inslower drug release. During the third phase of release, in whichthe drug was released from shell remains with a very lowmolecular weight, the polymer content had almost no effect onthe rate of drug release, as expected.
3.2. Effect of drug content
The effect of the paclitaxel content on its release profile ispresented in Fig. 4b. Since the drug content strongly affects theemulsion's stability [9], a relatively low range of drug contents(1.4% w/w–4.3% w/w) was used. The highly hydrophobicpaclitaxel diffuses out very slowly from the shell. Therefore, thedrug content did not show a significant effect on the releaseprofile during the first phase of release. During the second phaseof release the release rate and the amount of released drugdecreased slightly with the increase in the drug content from2.8% w/w to 4.3% w/w (Fig. 4b). We would expect an increasein the release rate with the increase in drug content, due to ahigher driving force for diffusion. However, paclitaxel is ahydrophobic drug and a higher paclitaxel content in the organicphase of the emulsion therefore results in a higher interfacialtension (difference between the surface tension of the organicand aqueous phases), leading to a less stable emulsion witha larger pore size (Fig. 5a,c and Table 1). Larger pores areexpected to reduce the release rate when the porosity andinterconnectivity do not change too much. It can thereforebe concluded that in this specific range of drug contents, themorphological changes do not enable the driving force for
Table 1The examined specimens and the structural characteristics of their shells
Process parameters Mean pore size±SD [µm]
Mean porosity±SD [%]
Reference a 6.42±2.32 69±6Polymer content [% w/v] 22.5 5.34±1.64 ( b) 72±8Paclitaxel content [% w/w] 2.86 9.22±4.54 ( b) 73±3
4.286 29.96±3.1 ( b) 82±9Organic to aqueous phase
ratio [v/v]4:1 5.96±2.41 ( b) 80±2
Homogenization rate [rpm] 5500 8.23±3.15 ( b) 77±825,000 5.98±1.6 (P=0.053) 75±10
PDLGA copolymercomposition [w/w]
50/50 4.1±1.35 ( b) 67±6
a The reference sample was prepared using the following conditions:emulsion-based on 75/25 PDLGA, 17.5% w/v polymer, 1.43% w/w paclitaxel,O:A=2:1 v/v, homogenization rate=16,500 rpm.b Significant compared to the reference.
diffusion to affect paclitaxel's release profile. During the thirdphase of release (20–38 weeks) the release rate from the re-maining short polymers and oligomers of the formulations with2.9% w/w and 4.3% w/w paclitaxel is significantly higher thanthat obtained from the formulation with 1.4% w/w paclitaxel(Fig. 5a). The driving force for diffusion plays amajor role at thisstage, after intensive polymer erosion when the morphologicalfeatures of the initial microstructure no longer exist.
3.3. Effect of O:A phase ratio
The effect of the O:A phase ratio on paclitaxel's releaseprofile from the composite fibers and on the shell's micro-structure are presented in Figs. 4c and 5a,d, respectively. Therelease profile during release phases a and b (20 weeks) as wellas the pore size (Table 1) exhibited almost no change in the O:Arange of 2:1 to 4:1. It should be mentioned that the relativelynarrow O:A range chosen for this study resulted from theemulsion's stability considerations. The porosity of samplesderived from emulsions with O:A ratios higher than 4:1 may notbe high enough to enable effective release of water-insolubleagents such as paclitaxel. On the other hand, samples derivedfrom emulsions with O:A ratios less than 2:1 are not stable.During the third phase of release (20–40 weeks) the release ratefrom the 4:1 phase ratio was higher than from the 2:1 phaseratio. A relatively high organic phase content provides morebinding sites for the hydrophobic paclitaxel. This higher drugcontent enables a higher release rate only when the drug diffusesout from very low molecular weight components which remainin the third phase of release.
3.4. Effect of copolymer composition
The effect of the copolymer composition, i.e. the relativequantities of lactic acid (LA) and glycolic acid (GA) in thecopolymer, on the drug release profile and on the shell'smicrostructure is presented in Figs. 4d and 5a,e, respectively.The rate of release from the 50/50 PDLGA formulation is higherthan that obtained from the 75/25 PDLGA formulation(reference) during phases a and b (Fig. 4d). It is important tonote that the differences in the release rate and quantity are lowduring the first phase of release but very high during the secondphase of release. After 20 weeks of degradation the 50/50formulation released 70% of the drug, whereas the 75/25 for-mulation released only 13%. These differences are attributedmainly to differences in the degradation rate of these two co-polymers and differences in their hydrophilic/hydrophobic na-ture. The 50/50 copolymer degrades faster than the 75/25copolymer and therefore during intensive erosion (phase b) itreleases the drug at a faster rate. Furthermore, the less hy-drophobic nature of the 50/50 copolymer relative to the 75/25copolymer results in less drug attachment to its surface andtherefore also contributes to a higher paclitaxel release rate.
The mean pore diameter increased with the increase in thecopolymer's LA content. The 50/50 composition exhibited amean pore diameter of 4.1 µm, whereas the 75/25 composition(reference) exhibited a mean pore diameter of 6.4 µm (Table 1).
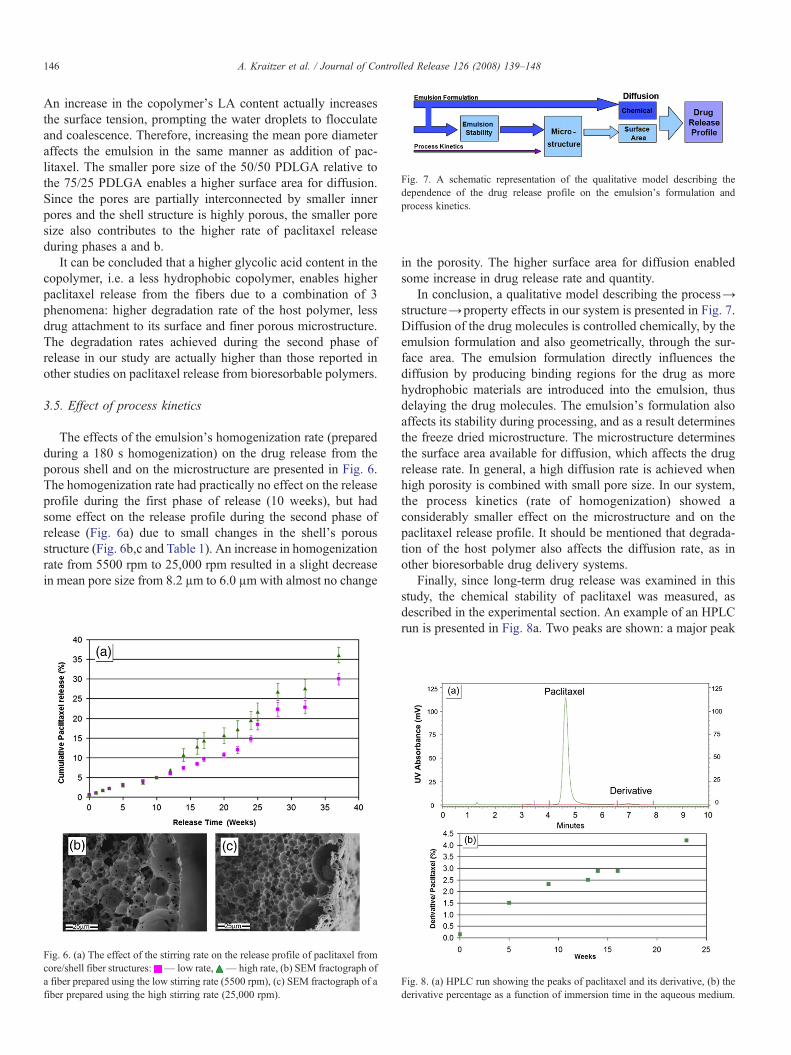
Fig. 7. A schematic representation of the qualitative model describing thedependence of the drug release profile on the emulsion's formulation andprocess kinetics.
146 A. Kraitzer et al. / Journal of Controlled Release 126 (2008) 139–148
An increase in the copolymer's LA content actually increasesthe surface tension, prompting the water droplets to flocculateand coalescence. Therefore, increasing the mean pore diameteraffects the emulsion in the same manner as addition of pac-litaxel. The smaller pore size of the 50/50 PDLGA relative tothe 75/25 PDLGA enables a higher surface area for diffusion.Since the pores are partially interconnected by smaller innerpores and the shell structure is highly porous, the smaller poresize also contributes to the higher rate of paclitaxel releaseduring phases a and b.
It can be concluded that a higher glycolic acid content in thecopolymer, i.e. a less hydrophobic copolymer, enables higherpaclitaxel release from the fibers due to a combination of 3phenomena: higher degradation rate of the host polymer, lessdrug attachment to its surface and finer porous microstructure.The degradation rates achieved during the second phase ofrelease in our study are actually higher than those reported inother studies on paclitaxel release from bioresorbable polymers.
3.5. Effect of process kinetics
The effects of the emulsion's homogenization rate (preparedduring a 180 s homogenization) on the drug release from theporous shell and on the microstructure are presented in Fig. 6.The homogenization rate had practically no effect on the releaseprofile during the first phase of release (10 weeks), but hadsome effect on the release profile during the second phase ofrelease (Fig. 6a) due to small changes in the shell's porousstructure (Fig. 6b,c and Table 1). An increase in homogenizationrate from 5500 rpm to 25,000 rpm resulted in a slight decreasein mean pore size from 8.2 µm to 6.0 µm with almost no change
Fig. 6. (a) The effect of the stirring rate on the release profile of paclitaxel fromcore/shell fiber structures: — low rate, — high rate, (b) SEM fractograph ofa fiber prepared using the low stirring rate (5500 rpm), (c) SEM fractograph of afiber prepared using the high stirring rate (25,000 rpm).
in the porosity. The higher surface area for diffusion enabledsome increase in drug release rate and quantity.
In conclusion, a qualitative model describing the process→structure→property effects in our system is presented in Fig. 7.Diffusion of the drug molecules is controlled chemically, by theemulsion formulation and also geometrically, through the sur-face area. The emulsion formulation directly influences thediffusion by producing binding regions for the drug as morehydrophobic materials are introduced into the emulsion, thusdelaying the drug molecules. The emulsion's formulation alsoaffects its stability during processing, and as a result determinesthe freeze dried microstructure. The microstructure determinesthe surface area available for diffusion, which affects the drugrelease rate. In general, a high diffusion rate is achieved whenhigh porosity is combined with small pore size. In our system,the process kinetics (rate of homogenization) showed aconsiderably smaller effect on the microstructure and on thepaclitaxel release profile. It should be mentioned that degrada-tion of the host polymer also affects the diffusion rate, as inother bioresorbable drug delivery systems.
Finally, since long-term drug release was examined in thisstudy, the chemical stability of paclitaxel was measured, asdescribed in the experimental section. An example of an HPLCrun is presented in Fig. 8a. Two peaks are shown: a major peak
Fig. 8. (a) HPLC run showing the peaks of paclitaxel and its derivative, (b) thederivative percentage as a function of immersion time in the aqueous medium.

147A. Kraitzer et al. / Journal of Controlled Release 126 (2008) 139–148
of paclitaxel after an elution time of 4–5 min, and a minorpeak of its derivative after an elution time of 7 min. Thederivative percentage (derivative content divided by paclitax-el content) as a function of immersion time in the aqueousmedium is presented in Fig. 8b. As expected, the derivativepercentage increases with time, but even after 23 weeks it islower than 5%, thus demonstrating that 95% of the paclitaxelremained stable.
4. Summary and conclusions
Bioresorbable core/shell fiber structures for biomedical ap-plications were developed and studied. These structures werecomposed of a polyglyconate core and a porous PDLGA shellloaded with the anti-proliferative agent paclitaxel, preparedusing freeze drying of inverted emulsions. A long-term in vitrorelease study was performed. The investigation of these newcomposite fibers focused on the effects of the emulsion's com-position (formulation) and process kinetics on the drug releaseprofile from the fibers, in light of the shell's morphology anddegradation profile. These unique fiber structures can be used asbasic elements of vascular stents and also for local cancertreatment.
In general, porous “shell” structures (porosity of 67–82%and pore size of 4–10 µm) were obtained with good adhesion tothe core fiber. Paclitaxel release from the porous shell wasrelatively slow due to its extremely hydrophobic nature. Itexhibited three phases of release. It is clear that the first phase(1–10 weeks) was governed by diffusion, whereas the secondphase (10–20 weeks) was governed by degradation. The releaseprofile of paclitaxel from the porous shell corresponds to thedegradation profile of the host 75/25 PDLGA.
The copolymer composition had the most dominant effect onthe drug release profile from the composite fibers. An increasein the glycolic acid content (or decrease in lactic acid content)resulted in a tremendous increase in the release rate during thesecond phase, which was attributed mainly to the increaseddegradation rate and decreased drug attachment to the hostpolymer. A decrease in polymer content also resulted in anincrease in the release rate during the second phase of release,whereas the drug content and the O:A phase ratio in the studiedrange affected the release profile only during the third phase ofrelease.
The effect of the emulsion formulation on the release profileis more significant than the effect of the process kinetics. Fur-thermore, emulsions with a less hydrophobic nature are fa-vorable for effective controlled release of the hydrophobicpaclitaxel from the porous shell, since they do not provide manybinding regions for the drug and their higher stability enablesobtaining highly porous shell structures with a large surface areafor diffusion after freeze drying.
Acknowledgements
The authors are grateful to the RAMOT (Horowitz) Foun-dation and to the Slezak Foundation, Tel-Aviv University, forsupporting this research.
References
[1] S.H. Su,C.L. Landau, R.Y. Chao, R.B. Timmons, R.S.Meidell, L. Tang, R.C.Eberhart, Expandable bioresorbable endovascular stent with anti-platelet andanti-inflammation treatments, Circulation 104 (II) (2001) 500–507.
[2] N. Alikacem, T. Yoshizawa, C. Wilson, K.D. Nelson, Quantitative MRImaging study of intravitreal sustained release of VEGF in rabbits, Invest.Ophthalmol. Vis. Sci. 41 (2000) 1561–1569.
[3] R.L. Dunn, D.H. Lewis, J.M. Goodson, Monolithic fibers for controlleddelivery of tetracycline, Proc. Int. Symp. Control. Rel. Bioact. Mater. 9(1982) 157–163.
[4] R.L. Dunn, J.P. English, W.C. Stoner, A.G. Potter, B.H. Perkins,Biodegradable fibers for the controlled release of tetracycline in treatmentof peridontal disease, Proc. Int. Symp. Control. Rel. Bioact. Mater. 14(1987) 289–294.
[5] R.L. Dunn, D.H. Lewis, L.R. Beck, Fibrous polymer for the delivery ofcontraceptive steroids to the female reproductive track, in: D.H. Lewis(Ed.), Controlled Release of Pesticides and Pharmaceuticals, PlenumPress, New York, 1981, pp. 125–146.
[6] M.D.J. Eenink, J. Feijen, J. Oligslanger, J.H.M. Albers, J.C. Rieke, P.J.Greidonus, Biodegradable hollow fibers for the controlled release ofhormones, J. Control. Rel. 6 (1987) 225–237.
[7] G. Polacco, M.G. Cascone, L. Lazzeri, S. Ferrara, P. Giusti, Biodegradablehollow fibers containing drug-loaded nanoparticles as controlled releasesystems, Polym. Int. 51 (12) (2002) 1464–1472.
[8] L. Lazzeri, M.G. Cascone, S. Quiriconi, L. Morabito, P. Giusti,Biodegradable hollow microfibers to produce bioactive scaffolds,Polym. Int. 54 (2005) 101–107.
[9] A. Kraitzer, M. Zilberman, Paclitaxel-loaded composite fibers: micro-structure and emulsion stability, J. Biomed. Mater. Res. A. 81 (2) (2007)427–436.
[10] M.N. Babapulle, M.J. Eisenberg, Coated stents for prevention ofrestenosis: part I, Circulation 106 (2002) 2734–2740.
[11] S.H. Duda, T.C. Poerner, B. Wiesinger, J.H. Rundback, G. Tepe, J.Wiskirchen, K.K. Haase, Drug-eluting stents: potential applications forperipheral occlusive disease, J. Vasc. Interv. Radiol. 14 (2003) 291–301.
[12] A.L. Lewis, L.A. Tolhurst, P.W. Stratford, Analysis of a phosphorylcho-line-based polymer coating on a coronary stent pre- and post-implantation,Biomaterials 23 (2002) 1697–1706.
[13] T.C. Woods, A.R. Marks, Drug-eluting stents, Ann. Rev. Med. 55 (2004)169–178.
[14] H. Tamai, K. Igaki, E. Kyo, K. Kosuga, A. Kawashima, S. Matsui, H.Komori, T. Tsuji, S. Motohara, H. Uehata, Initial and 6-month results ofbiodegradable poly-L-lactic acid coronary stents in humans, Circulation102 (2000) 399–404.
[15] H. Tamai, K. Igaki, T. Tsuji, E. Kyo, K. Kosuga, A. Kawashima, S. Matsui,H. Komori, S. Motohara, H. Uehata, E. Takeuchi, A biodegradable poly-L-lactic acid coronary stent in porcine coronary artery, J. Interv. Cardiol. 12(1999) 443–449.
[16] M. Zilberman, N.D. Schwade, R.C. Eberhart, Protein-loaded bioresorbablefibers and expandable stents: mechanical properties and protein release, J.Biomed. Mater. Res. B. 69 (2004) 1–10.
[17] M. Zilberman, R.C. Eberhart, Drug-eluting bioresorbable stents for variousapplications, Ann. Rev. Biomed. Eng. 8 (2006) 153–180.
[18] A.B. Dhanikula, R. Panchagnula, Localized paclitaxel delivery, Int. J.Pharm. 183 (2) (2000) 85–100.
[19] S. Silber, E. Grube, The Boston scientific antiproliferative paclitaxeleluting stent (TAXUS), in: P.W. Serruys (Ed.), Handbook of CoronaryStents, Martin Dunitz, London, 2001, pp. 311–319.
[20] AHFS Drug Information, American Society of Health System Pharmacists,Bethseda, 1989, pp. 1075–1086.
[21] E.K. Rowinsky, R.C. Donehower, Drug therapy: paclitaxel (Taxol), N.Engl. J. Med. 332 (1995) 1002–1014.
[22] T. Yamawaki, H. Shimokawa, T. Kozai, K. Miyata, T. Higo, E. Tanaka, K.Egashira, T. Shiraishi, H. Tamai, K. Igaki, Intramural delivery of a specifictyrosine kinase inhibitor with biodegradable stent suppresses the restenoticchanges of the coronary artery in pigs in vivo, J. Am. Coll. Cardiol. 32(1998) 780–786.

148 A. Kraitzer et al. / Journal of Controlled Release 126 (2008) 139–148
[23] D.E.Drachman, E.R. Edelman, P. Seifert,A.R.Groothuis,D.A.Bornstein,K.R.Kamath,M. Palasis, D. Yang, S.H. Nott, C. Rogers, Neointimal thickening afterstent delivery of paclitaxel: change in composition and arrest of growth oversix months, J. Am. Coll. Cardiol. 36 (2000) 2325–2332.
[24] H. Suh, B. Jeong, R. Rathi, S.W. Kim, Regulation of smooth muscle cellproliferation using paclitaxel-loaded poly(ethylene oxide)-poly(lactide/glycolide) nanospheres, J. Biomed. Mater. Res. 42 (1998) 331–338.
[25] F. Alexis, S.S. Venkatraman, S.K. Rath, F. Boey, In vitro study of releasemechanisms of paclitaxel and rapamycin from drug-incorporated biode-gradable stent matrices, J. Cont. Rel. 98 (2004) 67–74.
[26] E. Grube, S. Silber, K.E. Hauptmann, R. Mueller, L. Buellesfeld, U.Gerckens, M.E. Russel, TAXUS I: six- and twelve-month results from arandomized, double-blind trial on a slow-release paclitaxel-eluting stentfor de novo coronary lesions, Circulation 107 (2003) 38–42.
[27] K. Tanabe, P.W. Serruys, M. Degertekin, G. Guagliumi, E. Grube, C.Chan, T. Munzel, J. Belardi, W. Ruzyllo, L. Bilodeau, H. Kelbaek, J.Ormiston, K. Dawkins, L. Roy, B.H. Strauss, C. Disco, J. Koglin, M.E.Russell, A. Colombo, Chronic arterial responses to polymer-controlledpaclitaxel-eluting stents: comparison with bare metal stents by serialintravascular ultrasound analysis: data from the randomized TAXUS-IItrial, Circulation 109 (2004) 196–200.
[28] K. Tanabe, P.W. Serruys, E. Grube, P.C. Smits, G. Selbach, W.J. van derGiessen, M. Staberock, P. de Feyter, R. Müller, E. Regar, M. Degertekin,J.M.R. Ligthart, C. Disco, B. Backx, M.E. Russell, TAXUS III trial: in-
stent restenosis treated with stent-based delivery of paclitaxel incorpo-rated in slow-release polymer formulation, Circulation 107 (2003)559–564.
[29] G.W. Stone, S.G. Ellis, D.A. Cox, J. Hermiller, C. O'Shaughnessy, J.T.Mann, M. Turco, R. Caputo, P. Bergin, J. Greenberg, J.J. Popma, M.E.Russell, A polymer-based, paclitaxel-eluting stent in patients withcoronary artery disease, N. Engl. J. Med. 350 (2004) 221–231.
[30] T. Tsuji, H. Tamai, K. Igaki, E. Kyo, K. Kosuga, T. Hata, T. Nakamura, S.Fujita, S. Takeda, S. Motohara, H. Uehata, Biodegradable stents as aplatform to drug loading, Int. J. Cardiovasc, Interv. 5 (2003) 13–16.
[31] L. Mu, S.S. Feng, A novel controlled release formulation for the anticancerdrug Paclitaxel (Taxol): PLGA nanoparticles containing vitamin E TPGS,J. Cont. Rel. 86 (2003) 33–48.
[32] T.L. Richard, H.L. Burt, Paclitaxel loaded poly (L-lactic acid) (PLLA)microspheres II. The effect of processing parameters on microspheremorphology and drug release kinetics, Int. J. Pharm. 281 (2004) 103–106.
[33] D. Chitkara, A. Shikanov, N. Kumar, A.J. Domb, Biodegradable injectablein situ depot-forming drug delivery systems, Macromol. Biosci. 6 (2006)977–990.
[34] A. Shikanov, B. Vaisman, M.Y. Krasko, A. Nyska, A.J. Domb, Poly(sebatic acid-co-ricinoleic acid) biodegradable carrier for paclitaxel: in-vitro release and in-vivo toxicity, J. Biomed. Mater. Res. A. 69 (1) (2004)47–54.