Embed Size (px)
DESCRIPTION
This is the report from a mini-project at Stocholms University in the course - Applied Bioinformatics. During the project I also came over the interesting theme of 'The minor spliceosome'. This is a second spliceosome system which acts outside the nucleus and is known to have an effect on cell proliferation which was interesting for the All-In-G1 project.
Citation preview

Logos for Ribosome Binding Sites and First Introns in the Human Genome
Authors: Bjorn Sponberg, Ganapathy Varma, Ramya Seetharaman Masters Students, Bioinformatics Program, Stockholm University
Abstract
The handling and analysis of biological sequences remains one of the prime tasks in the field of
Bioinformatics. Logos are a useful and easy to use tool in genome analysis as they provide an instant
graphic representation of evolutionary conserved elements in sequence datasets. Sequence motifs are
short patterns in biological sequences that are biologically active. Motifs are therefore evolutionary
conserved in sequences that are active in the same biologic process.
The Kozak sequence are located in the start codons context in mRNA and is known to play an
important role in the recognition by ribosomes. Kozak sequence is therefore crucial for regulating
translation.
The first intron in every gene differs from the other introns as it has typical features such as generally
longer sequence length and unique sequence motifs. If unique motifs are detected they can aid
tremendously in gene prediction.
Four DNA datasets from the human genome was collected. One dataset representing the start codon
sequence context in mRNAs and a suitable control dataset. A second dataset of first intron sequences
and a suitable control dataset. For creating logos, the appropriate sequences (start codon sequence
context and first introns respectively) were mined from ensembl 60 using Biomart and RSAT. Python
scripts were then executed to extract the pieces of sequence bits from the datasets that later was used as
input for creating logos. The logo building tool, Weblogo, is a built in function in python and was used
to construct the logos.
Characteristic patterns of Ribosome Binding Sites was observed in weblogo. The Ribosome Binding
Site logo showed a conserved ATG nucleotide start codon along with the characteristic -3 purines and
the +4 guanine flanking bases. In the first intron dataset weblogo displayed characteristic intron motifs.
In addition a unique motif was detected at the 5' end wich was suspected to be a unique motif to first
introns.
Keywords: Bioinformatics, Genome Analysis, Motif, Logo, Pattern, Ribosome Binding Site, mRNA,
Ribosome, Translation, Coding Sequence, Gene, Kozak Sequence, Open Reading Frame, Codon, Exon,
Intron, First Intron, Splice Sites, Python, Biomart, RSAT, ensembl60, Weblogo.

Introduction
Much of the non-coding sequence regions in DNA was earlier termed “Junk-DNA”. However, in the
last two decades many important discoveries have been done in the field which makes the term
misleading.
In genes, non coding elements in the proximal regions close to transcription start site (TSS) is known
to be important for regulation of translation. The non coding first introns in eukaryotic genes contain
motifs that are believed to regulate gene transcription [1].
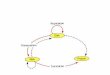
The central dogma in cell biology describes the events that take place from gene transcription to
translation. Gene transcription is a key biologic process that produces a blueprint of the sequence
information stored in a gene. The blueprint of the gene comes in the form of a physically independent
unspliced transcript called pre-mRNA. After the removal of non-coding sequences within the genes
blueprint (Called introns), the coding sequences (Called exons) is coupled together to produce a
messenger RNA nucleotide string (mRNA).
The resulting mRNA is ready to be transported to ribosomes in the cytosol for translation to a new
sequence form – a string of amino acids which are called proteins (Figure 1). These events just
described are really one biologic process that transfer information from genes to functional proteins –
and is the core of the central dogma [2].
In the world of biological sequences (Including nucleotide sequences) regulatory motifs are very
important. Regulatory motifs communicates between the biological sequences they are a part of and
their outside world. Usually this means, to communicate with a motif binding biologic component(s)
( Usually proteins or RNA's) that are present in the sequences nearby surroundings.
Together, a motif binding component and a regulatory sequence motif triggers a biological process
when they find each other. Since regulatory sequence motifs are a very important element of a
evolutionary conserved biological process, they are themselves conserved.
However, protein binding motifs are often only semi conserved, not fully conserved (Base by base) –
as is the case for example with enzyme restriction DNA binding sites. Restriction enzymes does not
need dynamic regulation due to their all-or-none activity (They destroy invading DNA elements) [3].
However, most motif binding components do trigger and regulate a certain biologic process.
Consequently, a motif-type that regulates needs to show some kind of variability that discriminates

between different output levels of the biological process it are meant to regulate.
In addition, evolutionary robustness could also play a role in the variable nature of sequence motifs.
Since, the more mutations a motif can handle without harming the host – the more biologic fit becomes
the host.
Figure 1. Illustration of the central dogma, including the splicing event of the pre-mRNA [4].

Start codon context
As with most of biologic molecular discoveries, the discovery of an m-RNA motif to be involved in
translation, was made in prokaryotes in the 1970's. This first ribosome binding site motif was called a
Shine-Dalgarno sequence. A variety in this motif (Among different genes) was believed to affect
ribosome binding affinity. How strong the ribosome bound to mRNA was assumed to be important to
the translation output (Protein production).
Later, the theory became that the secondary structure of the 5' end mRNA strand also was important to
ribosome binding affinity and not only the motif-type [4, 5].
After this the same kind of m-RNA motifs (Ribosome binding motif) was found in Eukaryotes as well.
However, in eukaryotes the motif is referred to as the Kozak sequence (Figure 2).
Figure 2. An Weblogo example of a Kozak sequence that surrounds the start codon (ATG) [6].
Often the literature talks about the “AUG nucleotide context” to describe how well the Kozak
sequence promotes mRNA translation. The most crucial elements of the AUG context is believed to be
the -3 position and +4 position (See figure 2). For the start codon to be in an optimal context the -3
position must be a purine base (A or G) and the +4 position must be a guanine base (G) . If not, the start
codon is in a suboptimal contex. MRNA's with a start codon in a suboptimal context is relatively
abundant [7].

Figure 3. mRNA structure (Right) is also important for ribosome binding activity, not only motif-type
[8].
First introns
Splicing in eukaryotic mRNA was discovered in the late 1970s and the intensive focus on exon-intron
structure in eukaryotic genes was born. Before splicing the mRNA exist in a form called pre-mRNA
(Figure 4 under). It was later found that a RNA-Protein complex called spliceosome was responsible
for the removal of introns and ligation of the introns flanking exons. The spliceosome is a protein
complex that also consists of small snRNA's. The function to the small snRNA's is probably to
recognize motifs in the intron/exon boundaries, or within the intron. This motif recognition is believed
to help the spliceosome to correctly cut in the 5' end and 3' end splice points (GT and AG respectively).
Thus, conserved motifs that are complementary to these splicesome snRNA's should be found in the
intron or intron/exon boundary regions.

Figure 4. Pre-mRNA. The first intron (Intron 1) is generally longer than the other introns [9].
These conserved sequence elements includes the 5' end splice, the branch point sequence, the
polypyrimidine tract and the 3' end splice site (Figure 5 under) [10]. The branch point is expected to be
located 18-40 nt upstream from the polypyrimidine tract at the 3' end [11].
Figure 5. The four main conserved motifs in introns: GU, Branchpoint(A), Polypyrimidine and AG
[12].
On average, the first introns are known to have longer sequence regions than other introns. The extra
length in first introns can stem from an intron-mediated enhancement or silencer element(s) (Motifs).
However, exactly how the first introns regulates gene transcription is still not known [12].

Methods
Start codon context: Downloading sequence data (See also figure 6 under)
RSAT (http://rsat.ulb.ac.be/rsat/retrieve-seq_form.cgi) was used to download human CDS sequences.
1.The human gene sequences was downloaded as CDS. Flanking regions relative to start codon was
set to -10 and +5 (+200 and +215 for the control dataset).
2.Downloaded all genes by marking “all” in the Genes section.
3.Prevented overlap with neighboring genes. Admitted imprecise positions. No Mask repeats.
4.The output was shown on the server. Copied and pasted the results into a gedit page. The dataset
was truncated because all genes was too much for weblogo to handle. Truncated until Weblogo
accepted 27168 sequences (See also Result).
Figure 6. Screenshot from the parameter settings in RSAT [13].

The script
We used a Python script to find the ATG start codon within the 15 base long dataset sequences.
We used the re module by importing it with the command: import re
The motif created in the prog variable: prog = re.compile('.{5}.ATG..')
The motif included nine bases upstream of the A in ATG, and two downstream of the G in ATG.
These motifs was sent to the weblogo functions imported from the motif module. The module was
imported with the command:
from Bio.AlignAce.Motif import Motif
See also the Python script in the bin directory named:
prog_ATG_context.py
First introns
Downloading sequence data (See also figure 7 under):
•RSAT was used to obtain data from ensembl for the Homo sapiens dataset.
•Ensembl Gene Id list was pasted into the window. 'First intron' option was selected.
•The result , which had complete sequences of all the first introns of the genes in the list specified
was saved as a text file.
•This data file was corrected for small errors such as warning messages and small length sequences
were removed and the data was ready for use in the python script.

Figure 7. Screenshot from the parameter settings in RSAT [14].
The script
Since the dataset was already extracted as first intron in RSAT it was not necessary to discriminate
first introns from exons with programming. However, it was necessary to cut slices from both ends and
join them to make one weblogo representation of both ends. The script cut 20 bases long slices from
both ends and joined them before sending to weblogo for conservation pattern display.
See also the script in the bin directory named:
prog_first_intron.py
System development
Personal notebooks, filesharing etc was shared at code.google

Results
Start codon context
Logos for 744 and 27168 random human genes respectively are shown in the two figures under
(Figures 8 and 9). The logos flanks the start codon -6 upstream and +1 downstream which was set in
the Python script (See Methods).
Sequence counting begins at the A in the start codon ATG. A is annotated as +1 (Position 7 in the
weblogo figures). Position 6 in the start codon context weblogo figures would be annotated as -1 and so
forth.
The most crucial elements for ribosome recognition in the ATG sequence context is present in the
result (See also introduction). At position -3: A and G (Or the purines) are most conserved. At position
+4: G is the most conserved base.
Figure 8. Human CDS flanked -6 and +1 in weblogo (Based on 744 sequences).
The weblogo for the 27168 random human gene dataset is shown in figure 9. The same ATG context
can be seen as in the 744 sequence dataset logo in figure 8.

Figure 9. Human CDS flanked -6 and +1 displayed in weblogo (Based on 27168 sequences).
A control dataset tested the script. A web logo for 4822 random human genes flanking ATG
downstream +200 and +215 in the same CDS dataset was created (Figure 10).
Figure 10. Human CDS +200 and +215 (based on 4822 sequences).
In this case all human CDS was searched for ATG in the specified region (Downstream +200 +215).
Any ATG triplet found here would most probably not function as a start codon and would be fewer in
number. The number of ATG motifs found in this region was 4822 relative to 27168 in the -6 and +1
flanking dataset (Figure 9 over). As expected, no conserved bases in this ATG context could be noticed
(Figure 10).

First introns
When cutting 20 nt downstream of 5' end and 20 nt upstream of 3' first intron ends only the two end
splice points and the polypyrimidine tract was observed according to the literature. In addition to these
known features, a 4 base long AG rich region adjacent downstream to the 5' end splicing point was
observed (Figure 11).
Figure 11. A weblogo display of a dataset based on 1082 first intron sequences.
Since the branch point motif was expected to be located ca. 18-40 nt upstream of the polypyrimidine
tract the 3' end slicing length was increased to 60 nt. However, no branch point was observed (Figure
12).
Figure 12. This logo shows an increased 60 base slice from the 3' end for the first intron dataset.
The control dataset was obtained from an first exon dataset. No intron-conserved elements should be
observed as was the case in figure 11 and 12 over. This control confirmed that the results over was not
a case of randomly luck relative to the information found in the literature.

Figure 13. Weblogo based on a 1247 exon sequence control dataset.

Discussion
Start codon context
From the literature it is known that the most crucial conserved bases in the ATG context are a purine at
position -3 (Position 4 in weblogo result) and a guanine at position +4 (Position 10 in weblogo) [7].
Figures 8 and 9 in Results shows that both of these elements was conserved in the input dataset. The -3
position is almost solely dominated by the purines. However, position +4 also have some adenine
present, but guanine dominates - as expected.
In addition, the results suggests that cytosine is most dominant in position -1 over guanine and adenine
respectively. Cytosine also show some slightly dominances at position -2. These two extra element
features can be seen in the 744 sequence dataset as well as in the 27168 sequence dataset and are
probably biological relevant to the ATG context.
However, unique to the 27168 sequence dataset is a dominating cytosine conserved over guanine and
adenine -4 upstream of ATG. Since this dataset is over 30 times larger as the 744 sequence dataset, this
unique feature is probably more reliable. That is, this slight cytosine dominance over guanine and
adenine in position -4 is probably a better reflection of the biological truth.
The control dataset is extracted from position +200 to +215 downstream of start codon in the same
CDS dataset. Any ATG's found in this region would not be expected to have any similar context as the
ATG's found in the -10 to +5 region. Figure 13 also confirm this. The figure shows no conserved
elements in the ATG context at all.
Since ATG is not a common triplet in this downstream region [6], only 4822 sequences was obtained
from the CDS dataset that generated 27168 in the -6 +1 region. This also reflects its non-conserved
function in this region of the CDS.

First introns
The 5' GU and 3'AG splicing points are the only two universally conserved elements in the intron.
These two motifs was fully conserved in the resulting weblogos in figure 11 (See results).
The polypyrimidine tract on the 3' end slice was also a distinct pattern that emerged in the weblogo
result.
What the result did lack was the branching point A and its surrounding elements. In Yeast, a branching
point context is very well conserved: UACUAAC (The A in bold is the branching point). This
sequence is complementary to the internal region in U2 snRNA. Mutation experiments in the branch
point region (18 to 40 bases upstream of the polypyrimidine tract) have shown this sequence to be
critical for the splicing events in Yeast. However, the branching point sequence context is known not to
be as conserved in higher eukaryotes, as in Yeast. Thus, this fact could very well be the reason for the
branch point motif to not show up in the weblogo in figures 11 and 12. Further, It is reported that the
polypyrimidine tract assists in binding the U2 snRNA correctly to the branching point in higher
eukaryotes. This biological process can indirectly wipe out the branching point motif as a conserved
motif in the human genome [16]. The branching point motif would not be necessary for correct U2
snRNA binding in higher eukaryotes.
The four conserved elements seen in figure 11 (See Results) downstream of the 5' end splicing point
was not expected. Could these conserved elements be unique to the first introns? Yes, they could. It is
known that the 5'end GT motif is encompassed within a larger less conserved motif [16]. This is
probably what is seen in figure 11. That is, within this GT context region lies unique information to the
intron in question e.g. silencers and/or enhancers to each respective intron/exon overlap region.

Conclusion
Regulatory motifs in biological sequences are known to be semi conserved. Their variable base
conservation makes them hard to detect without informatics methods. Weblogo is an application that
can display conserved regions in sequence datasets.
A dataset of 27168 sequences -6 and +1 downstream of start codon (ATG) from the human genome
was displayed in weblogo. The result showed that the expected conservation of purines in the -3
position and a guanine in the +4 position did emerge.
A dataset of 1082 sequences of first introns from the human genome was displayed in weblogo.
Weblogo displayed two types of result. In one case the 5' end was cut 20 downstream and the 3' end cut
60 bases upstream. This to look for a conserved branching point motif that was known to exist in lower
eukaryotes. The result showed that the branching point was not detected in the human first intron
dataset. In the second case the 5' and 3' end was both cut by 20 bases downstream and upstream
respectively. The result showed that the two splicing points in the 3' and 5'end (GT and AG
respectively) was fully conserved. The polypyrimidine tract elements in the 3'end region was also
conserved.
The lack of finding the branching point in the human first intron dataset could be that the
complementary U2 snRNA found in Yeast binds to the branching point motif differently in humans. In
humans the U2 snRNA binds to the branching point (A) is mediated by proteins already bound to the
polypyrimidine tract – which was found in the human dataset. This observation could explain a
evolutionary process that did not need a conserved branching point motif. Thus, the lack of this motif
in the human genome could be explained.
In the human first intron dataset Weblogo also detected a four base long conserved region, rich in
purines (A and G). These conserved elements could serve a local intron specific regulatory function. It
could either serve to enhance or to silence splicing, which would later in the central dogma pathway
affect the rate of protein production.

REFERENCES
1. A brief history of the status of transposable elements: from junk DNA to major players in evolution.
Biémont C.
Genetics. 2010 Dec;186(4):1085-93.
2. Thieffry D, Sarkar S. Forty years under the central dogma.
Trends Biochem Sci. 1998 Aug;23(8):312-6.
3. Stormo GD.DNA binding sites: representation and discovery. Bioinformatics. 2000 Jan;16(1):16-23.
4. http://compbio.pbworks.com/w/page/16252897/Introduction-and-Basic-Molecular-Biology5. Smit,M.H. and van Duin,J. (1990) Secondary structure ofthe ribosome binding site determines translational efficiency: aquantitative analysis. Proc. Natl. Acad. Sci. USA, 87, 7668–7672.6. Harhay et al. Kozak consensus sequence surrounding bovine start methonine using WebLogo [29]. BMC Genomics 2005 6:166 doi:10.1186/1471-2164-6-166
7. Kochetov AV. Bioinformatics. AUG codons at the beginning of protein coding sequences are frequent in eukaryotic mRNAs with a suboptimal start codon context. 2005 Apr 1;21(7):837-40. Epub 2004 Nov 5.
8. http://www.ask.com/wiki/Primary_structure?qsrc=3044
9. http://library.thinkquest.org/C006188/basics/pictures/introns.gi
10. http://en.wikipedia.org/wiki/Spliceosome
11. http://www.natur e.com/nrg/journal/v5/n5/full/nrg1327.html
12. Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers.
Nat Rev Genet. 2004 May;5(5):389-96.
13. http://rsat.ulb.ac.be/rsat/retrieve-seq_form.cgi
14. http://rsat.ulb.ac.be/rsat/
15. http://oregonstate.edu/instruction/bb492/lectures/EuTranscriptIII.html
16. Douglas L. Black MECHANISMS OF ALTERNATIVE PRE-MESSENGER RNA SPLICING
Annual Review of Biochemistry Vol. 72: 291-336
17. Marketa Zvelebil and Jeremy O. Baum Understanding Bioinformatics 18. A. Malcom Campbell and Laurie J. Heyer Discovering Genomics, Proteomics and Bioinformatics
19. Jeremy J. Ramsden Bioinformatics- An Introduction

20. Jonathan Pevsner Bioinformatics and Functional Genomics











![Ribosome Stoichiometry: From Form to Function · Ribosome abundance: A major model, also termed the ribosome concentration hypothesis [3], that explains how ribosomes could exert](https://img.pdfslide.us/doc/110x75/60de31e56d30fc4fb30719b8/ribosome-stoichiometry-from-form-to-function-ribosome-abundance-a-major-model.jpg)