Embed Size (px)
Citation preview

Localization of O-Glycosylation Sites on Glycopeptide Fragmentsfrom Lactation-associated MUC1ALL PUTATIVE SITES WITHIN THE TANDEM REPEAT ARE GLYCOSYLATION TARGETS IN VIVO*
(Received for publication, April 1, 1997, and in revised form, July 21, 1997)
Stefan Muller‡, Steffen Goletz§, Nicolle Packer¶, Andrew Gooley¶, Alexander M. Lawsoni,and Franz-Georg Hanisch‡**
From the ‡Institute of Biochemistry, Medical Faculty of the University, 50931 Cologne, the §Max Delbruck Center ofMolecular Medicine, 13125 Berlin, Federal Republic of Germany, the ¶Macquarie University Center for AnalyticalBiotechnology, Sydney, New South Wales 2109, Australia, and the iMRC Glycosciences Laboratory, Imperial CollegeSchool of Medicine, Northwick Park Hospital, Harrow HA13UJ, United Kingdom
Since there is no consensus sequence directing theinitial GalNAc incorporation into mucin peptides, O-glycosylation sites are not reliably predictable. We havedeveloped a mass spectrometric sequencing strategythat allows the identification of in vivo O-glycosylationsites on mucin-derived glycopeptides. Lactation-associ-ated MUC1 was isolated from human milk and partiallydeglycosylated by trifluoromethanesulfonic acid to thelevel of core GalNAc residues. The product was frag-mented by the Arg-C-specific endopeptidase clostripainto yield tandem repeat icosapeptides starting with thePAP motif. PAP20 glycopeptides were subjected to se-quencing by post-source decay matrix-assisted laser de-sorption ionization mass spectrometry or by solid phaseEdman degradation to localize the glycosylation sites.The masses of C- or N-terminal fragments registered forthe mono- to pentasubstituted PAP20 indicated thatGalNAc was linked to the peptide at Ser5,Thr6 (GSTA)and Thr14 (VTSA) but contrary to previous in vitro gly-cosylation studies also at Thr19 and Ser15 located withinthe PDTR or VTSA motifs, respectively. Quantitativedata from solid phase Edman sequencing revealed nopreferential glycosylation of the threonines. These dis-crepancies between in vivo and in vitro glycosylationpatterns may be explained by assuming that O-glycosyl-ation of adjacent peptide positions is a dynamically reg-ulated process that depends on changes of the substratequalities induced by glycosylation at vicinal sites.
Post-translational modification of proteins by glycosylationhas extensively been studied in the case of N-linked glycans,and accordingly, rules for the substitution of asparagine resi-dues by dolichol phosphate-linked glycans are well established(1). While N-glycosylation is directed by the consensus peptidemotif Asn-X-(Ser/Thr), no strict sequence dependence is knownfor the initiation of O-glycosylation. Currently, there are sev-eral approaches to the identification of O-glycosylation sites:studies on in vitro O-glycosylation of synthetic peptides (2–4)or studies on in vivo processed mucin-type glycoproteins (5–7).
The relative merits of both approaches have been discussed (8).The results obtained so far agree with the statement that thereare no clear-cut motifs for the addition of GalNAc at Ser or Thrresidues; however, the non-random patterns of O-glycosylationsuggest influences of flanking sequences (2). Wang et al. (3)proposed different motifs for threonine and serine glycosyla-tion, and the studies of O‘Connell et al. (2) revealed criticalpositions in the vicinity of putative O-glycosylation sites. Theobviously less specific GalNAc addition to Ser/Thr residuescompared with N-glycosylation is reflected also in the existenceof at least four distinct species of UDP-GalNAc/peptide N-acetylgalactosaminyltransferase(s) (GalNAc-transferase) forwhich different substrate specificities have been established(9). Accordingly, the differentiation and organ localization of acell should determine its characteristic equipment of activeGalNAc-transferases (T1–T4) and, hence, the site-specific O-glycosylation of proteins processed within this cell.
Site specificity of O-glycosylation strongly affects the antige-nicity of a protein as has been revealed for the highly immu-nogenic peptide motif PDTR within MUC1 tandem repeats(10). Most antibodies reactive to the PDTR sequence are moreor less influenced by GalNAc incorporation into threonine res-idues of the flanking motifs VTSA and GSTA by showing asignificant enhancement or decrease of their binding activities(10, 11). In vitro studies have established that preferentiallyThr residues within VTSA and GSTA are glycosylated (12–15),whereas only Ser within GSTA serves as a substrate to a minordegree (14, 15).
To establish the actual sites of O-glycosylation on the in vivoprocessed mucin, we have applied a mass spectrometric se-quencing strategy (15) using the MUC1 glycoform in humanmilk as a model. A fragmentation of the highly glycosylatedmucin by specific endopeptidases becomes possible only after alimited deglycosylation with trifluoromethanesulfonic acid(TFMSA).1 This treatment is reported to result in the removalof complex carbohydrates, but to leave during short reactiontimes a considerable portion of the core GalNAc residues intact(16, 17). This partially deglycosylated mucin (GalNAc-MUC1)can be effectively fragmented by clostripain cleaving at Arg-C(18). Clostripain fragmentation of the tandem repeat peptideresults in a series of differentially glycosylated icosapeptides* This study was supported by Grant Ha 2092/4-1 of the Deutsche
Forschungsgemeinschaft and by a Max Delbruck Center fellowship (toS. G.). The costs of publication of this article were defrayed in part bythe payment of page charges. This article must therefore be herebymarked “advertisement” in accordance with 18 U.S.C. Section 1734solely to indicate this fact.
** To whom correspondence and reprint requests should be ad-dressed: Institut fur Biochemie II, Medizinische Fakultat der Univer-sitat, Joseph-Stelzmann-Str. 52, 50931 Koln, Germany. Tel.: 49 2214784493; Fax: 49 221 4786977.
1 The abbreviations used are: TFMSA, trifluoromethanesulfonic acid;HexNAc, N-acetylhexosamine; HMFGM, human milk fat globule mem-brane; PSD-MALDI-(TOF)-MS, post-source decay matrix-assisted laserdesorption ionization mass spectrometry; RP-HPLC, reversed phase-high performance liquid chromatography; PTH, phenylthiohydantoin;PBS, phosphate-buffered saline; mAb, monoclonal antibody; BSA, bo-vine serum albumin; ACN, acetonitrile.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 272, No. 40, Issue of October 3, pp. 24780–24793, 1997© 1997 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org24780
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

PAP20 as shown in Sequence 1:
PDTRuC
PAPGSTAPPAHGVTSAPDTRPAP20
uC
PAPGSTAPPA
SEQUENCE 1
As demonstrated previously for a series of in vitro glycosylatedpeptides corresponding to the MUC1 tandem repeats (TAP25)(15), the sequencing of PAP20 peptides was possible by massspectrometric analysis of post-source decay fragments in aMALDI-TOF instrument. Independent evidence for the local-ization of O-glycosylation sites and, moreover, quantitativeestimations of the degree of glycosylation at each site wereobtained from solid phase Edman sequencing. The results ob-tained suggest that in vitro data should be carefully reevalu-ated by the analysis of in vivo processed glycoprotein and thata prediction of O-glycosylation sites cannot be based solely onpeptide sequence.
MATERIALS AND METHODS
Isolation of Mucin—Human milk was obtained from healthy womenduring days 2–5 of lactation and immediately frozen at 220 °C. Milksamples from multiple donors were thawed at 4 °C and combined, andmilk fat was separated from skim milk by centrifugation at 3,000 3 g(4 °C) for 1 h (Fig. 1). Secretory MUC1 from skim milk was isolated byextraction with hot phenol and subsequent gel filtration on SephacrylS300 as described previously (19) (Fig. 1). This preparation is desig-nated MUC1(skim milk). Milk fat was used for the preparation ofhuman milk fat globule membranes (HMFGM) according to the proce-dure of Shimidzu and Yamauchi (20). For the isolation of membrane-
associated MUC1, the HMFGM were homogenized and sonicated in0.45 mM sodium phosphate, 125 mM sodium chloride, pH 7.2 (PBS),containing 4 M urea, before they were extracted with an equal volume of90% phenol for 30 min at 65 °C. The extract was allowed to cool to roomtemperature; 0.5 volumes of chloroform were added, and the phaseswere separated by centrifugation at 30,000 3 g (10 °C) for 30 min. Theaqueous phase was dialyzed against several changes of demineralizedwater, concentrated by ultrafiltration, and applied to a column (2.5 3100 cm) of Sephacryl S300 equilibrated in PBS. Fractions were ana-lyzed by colorimetric microassays for hexoses (21) and sialic acid (22)and in an enzyme-linked immunosorbent assay with monoclonal anti-body BW835. The combined void fractions, containing the mucin, wereextensively dialyzed against demineralized water, concentrated byultrafiltration, and freeze-dried. This preparation is designatedMUC1(HMFGM).
SDS-Polyacrylamide Gel Electrophoresis and Immunoblot Analy-sis—SDS-polyacrylamide gel electrophoretic separations of mucin sam-ples were carried out in the Mini Protean II gel electrophoresis appa-ratus (Bio-Rad, Munchen, Germany) according to the method ofLaemmli. Gradient gels (3–15%), overlaid with a 3% stacking gel, wererun at 200-V constant voltage for 50 min and stained with CoomassieBrilliant Blue R-250. In addition, gels were blotted onto polyvinylidenedifluoride membranes using a semi-dry blotting apparatus and thebuffer system of Towbin et al. (23). Remaining protein binding siteswere blocked with 3% BSA in PBS (1 h, room temperature), and mucinwas detected by an overnight incubation with mAb BW835 (Behring-werke, Marburg, Germany, 2 mg/ml in 0.5% BSA in PBS) followed by1-h incubations with biotinylated goat anti-mouse immunoglobulin se-rum (E 413, Dako, 1:5,000 in 0.5% BSA in PBS) and avidin-conjugatedalkaline phosphatase (Boehringer Mannheim, 1:2,500 in 0.5% BSA inPBS) using nitro blue tetrazolium/5-bromo-4-chloro-3-indoyl phosphateas substrate. Alternatively, blotted glycoproteins were detected afteroxidation with sodium periodate according to the method ofO’Shannessy et al. (24).
Partial Deglycosylation of Mucin with TFMSA—MUC1(skim milk) orMUC1(HMFGM) were deglycosylated by treatment with anhydrousTFMSA (Sigma, Unterhaching, Germany) in the absence (16) or pres-ence of anisole (17) (Fig. 2). Lyophilized mucin was resuspended in 0.1M hydrochloric acid at 4 mg/ml and heated at 70 °C for 60 min tohydrolyze sialic acids. Hydrochloric acid was removed by dialysisagainst demineralized water. Following the method of Sojar and Bahlaliquots corresponding to 0.1–8 mg of native mucin were lyophilized inTeflon-lined screw-cap vials, flushed with argon, and precooled on ice,before 0.2–0.4 ml of TFMSA (220 °C) were added for each milligram ofmucin. The tubes were flushed with argon once more and then imme-diately closed and incubated on ice for the appropriate time. The reac-tion was stopped by cooling the tubes on dry ice in methanol and slowlyadding 3 volumes of ice-cold 60% pyridine/water. Finally, the deglyco-sylated samples were dialyzed against several changes of buffer, appro-priate for the following manipulations, and stored frozen at 220 °C. Themethod of Gerken et al. (17) was followed as described.
The time course of N-acetylgalactosamine (GalNAc) exposure duringremoval of complex glycans by TFMSA treatment was followed bykinetic analysis (Fig. 2). 0.1 mg of MUC1(HMFGM) was desialylatedand treated with TFMSA for 0, 15, 30, and 60 min in the absence ofanisole or for 0, 1, 2, and 6 h in the presence of anisole. After neutral-ization the samples were diluted with 50 mM NH4HCO3, pH 8.0, andchromatographed on Superdex 75pg (1 3 20 cm) using the same bufferas eluant and a flow rate of 0.4 ml/min. The samples were analyzed byquantitative monosaccharide composition analysis (see below) or byHelix pomatia agglutinin binding to follow the time course of the sugarremoval and the immunochemical accessibility of exposed GalNAc.Samples were diluted 1:16 in 0.1 M sodium carbonate, pH 9.6, and 50 mlof a serial 1:2 dilution in the same buffer were bound to 96-well poly-styrene plates by drying in a desiccator. Remaining protein bindingsites were blocked with 3% BSA in PBS at 37 °C for 1 h, and the platewas incubated with 50 ml of a 2 mg/ml solution of biotinylated H.pomatia agglutinin (Sigma) in 0.5% BSA in PBS at 37 °C for 1 h. Boundlectin was detected with avidin-alkaline phosphatase (BoehringerMannheim, Germany, 1:2,500 in 0.5% BSA in PBS) using p-nitrophenylphosphate as substrate.
Chemical Analysis of Glycans—Amino sugars in the deglycosylatedsamples were analyzed as peracetylated alditols using gas chromatog-raphy/electron impact mass spectrometry (Fison MD800) in the scanmode. 100 ml (20%) of the dialyzed samples and 2 mg of meso-erythritolwere lyophilized in micro-reaction vials and hydrolyzed with 4 N hydro-chloric acid for 18 h at 100 °C. Subsequent reduction and peracetylationof monosaccharides was performed as described elsewhere (25). To
FIG. 1. Preparative routes followed during generation and iso-lation of MUC1-derived glycopeptides.
In Vivo O-Glycosylation on MUC1 Tandem Repeats 24781
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
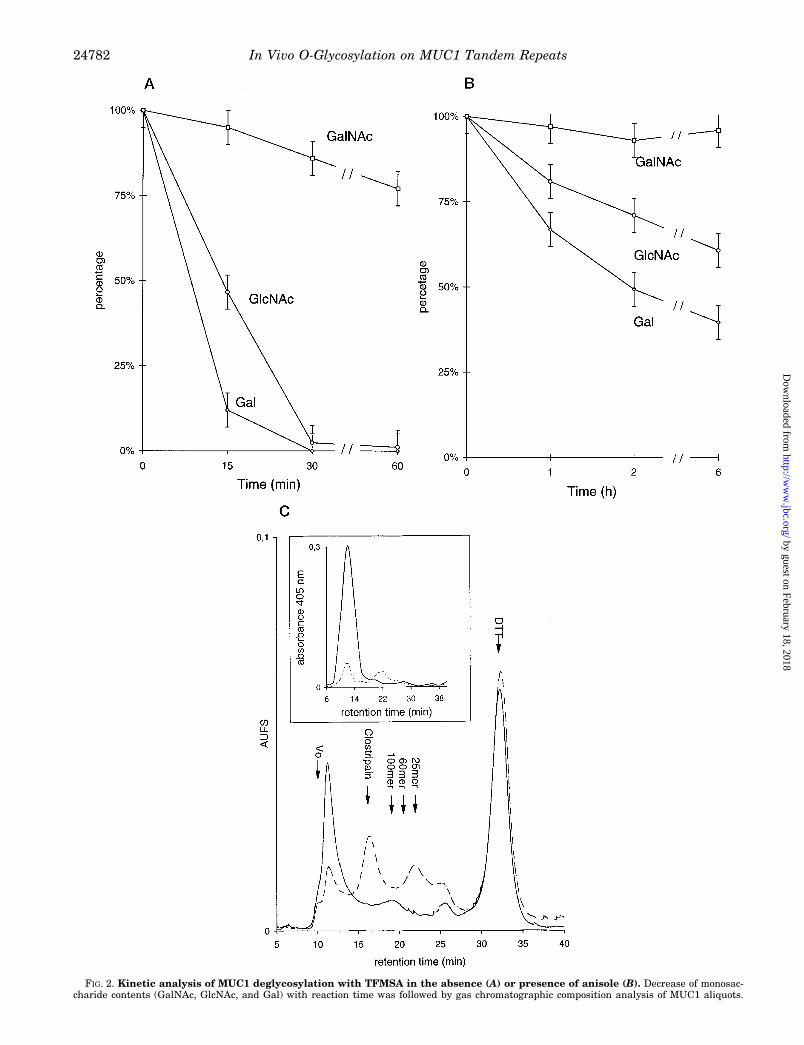
FIG. 2. Kinetic analysis of MUC1 deglycosylation with TFMSA in the absence (A) or presence of anisole (B). Decrease of monosac-charide contents (GalNAc, GlcNAc, and Gal) with reaction time was followed by gas chromatographic composition analysis of MUC1 aliquots.
In Vivo O-Glycosylation on MUC1 Tandem Repeats24782
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
analyze total monosaccharides in the deglycosylated and gel-filtratedsamples per-O-trimethylsilyl,1-O-methyl esters were prepared accord-ing to the method of Chaplin and Kennedy (26). Gas chromatography ofperacetylated alditols was performed on a DB5ms capillary (J & WScientific, 0.25 mm 3 15 m). Splitless sample injection (200 °C) wasused. The capillary temperature was held constant at 50 °C for 1 min,raised to 120 °C with 30 °C/min, and then to 260 °C with 7.5 °C/min.The same capillary was used for the analysis of per-O-trimethylsilyl,1-O-methyl esters. The temperature was kept at 100 °C for 1 min andthen raised to 280 °C with 7.5 °C/min. On-line mass spectrometry wasperformed with electron impact ionization at 70 eV in the positive mode.Mass spectra were recorded in the range from 50 to 500 mass units.
A synthetic glycopeptide GPT-(GlcNAcb1–6GalNAca)-TTPITT(kindly provided by Prof. H. Paulsen, Institute of Organic Chemistry,University of Hamburg) or partially deglycosylated MUC1 were treatedwith 50 mM NaOH, 1 M NaBH4 for 16 h at 50 °C, and after destructionof the excess borohydride with dilute acetic acid the reductively b-elim-inated alditols were desalted on Dowex 50WX8, followed by removal ofborate as its methyl ester, extensive drying and permethylation asdescribed (19). Gas-chromatography/mass spectrometry of methylateddihexosamine alditols was performed on an MD800 (Fisons). The sam-ples were solubilized in chloroform and injected onto a DB5 capillarycolumn (15 m), which was heated from 100 to 300 °C at 10 °C/minfollowed by isothermic elution at 300 °C for 10 min. Mass spectra wererecorded in cyclic scans from m/z 100 to 800.
Preparation of Partially Deglycosylated MUC1 Glycopeptides—Threealiquots corresponding to 8 mg of native mucin were desialylated andtreated with TFMSA according to Sojar and Bahl (16) for 20 min (P1),30 min (P3), or 60 min (P2) (Fig. 1). The neutralized samples weredialyzed into 25 mM sodium phosphate, pH 7.5; 0.2 mM calcium acetateand concentrated by ultrafiltration. 0.5 ml (50%) of the concentratedsamples (8 mg/ml) were supplemented with 2.5 mM dithiothreitol, andproteolytic digestion was performed by adding 120 mg of clostripain (1mg/ml in 2.5 mM dithiothreitol, 2 mM calcium acetate, activated at roomtemperature for 3 h) and incubating at 37 °C for 24 h. An aliquot of thedigest was chromatographed on Superdex 75pg (1 3 20 cm) in 50 mM
sodium phosphate, 500 mM NaCl, pH 7.0. Elution of mucin/glycopep-tides was registered at 214 nm, and aliquots of the fractionated eluatewere measured for MUC1 peptide epitopes (see below). The column hadbeen calibrated with a mixture of synthetic MUC1 tandem repeatoligomers (kindly provided by Dr. J. Hilgers, Free University Hospital,Amsterdam).
Isolation of Partially Deglycosylated MUC1 Glycopeptides by RP-HPLC—Digested partially deglycosylated mucin samples were clearedby centrifugation for 5 min at 15,000 3 g and separated by reversedphase-HPLC on a 250 3 4.6 mm PLRP-S column (Polymer Laborato-ries) (Fig. 1 and Fig. 3). The samples were injected as 5 3 100-mlaliquots, and the column was eluted at a flow rate of 1 ml/min using alinear gradient from 2% (v/v) acetonitrile (ACN) in 0.1%(v/v) trifluoro-acetic acid (solvent A) to 80% (v/v) ACN in 0.09% (v/v) (solvent B) during80 min. Peaks were detected at 214 nm, and 1-ml fractions were col-lected. 10 ml of each fraction were used for immunochemical detectionwith a mixture of the MUC1-specific mAbs BC3 and VA1 (see “Immu-nochemical Procedures”). Immunoreactive fractions (19–25 and 26–30min) were combined, lyophilized in a speed vac, and rechromatographedon a 250 3 2.1 mm C18 column (Vydac) using a flow rate of 0.25 ml/minand a gradient of 0–100% solvent B during 100 min (P1, P2). Rechro-matography of P3 glycopeptides was performed with a flow rate of 0.3ml/min and a gradient from 0 to 100% during 80 min. 0.3-ml fractionswere collected, and 5 ml were used for immunochemical detection asdescribed above. Immunoreactive fractions were lyophilized in a speedvac and used for matrix-assisted laser desorption ionization-mass spec-trometry (MALDI-MS) or Edman sequencing (Fig. 1).
Immunochemical Procedures—The monoclonal antibodies BC3 andVA1 are directed against peptide epitopes within the MUC1 tandemrepeat sequence and were kindly provided by Dr. Ian McKenzie (AustinResearch Institute, Heidelberg, Australia). BW835 (Behringwerke,Marburg, Germany) is specific for exposed core 1 disaccharide within a
peptide motif of the MUC1 tandem repeat sequence (27).Samples were immobilized onto 96-well polystyrene plates (Nunc,
Wiesbaden, Germany), and bound antibodies were quantified by alter-nating incubations with polyclonal anti-mouse immunoglobulin (Z 259,Dako, Hamburg, Germany) and a complex of alkaline phosphatase/anti-alkaline phosphatase, as described previously (27).
Matrix-assisted Laser-desorption Mass Spectrometry (MALDI-MS)—The matrix CCA (a-cyano-4-hydroxycinnamic acid; saturated solutionin 2/3 aqueous 0.1% trifluoroacetic acid, 1/3 acetonitrile) was mixed 1:1(v/v) with the probe in 50% methanol, placed (0.7–0.8 ml) onto a pol-ished stainless steel target, and air-dried. Linear, reflectron, and PSDmass spectrometric analyses were performed with a MALDI-TOF in-strument (Bruker-Reflex, Bruker-Franzen Analytic, Bremen, Ger-many) (15) using a pulsed UV laser beam (nitrogen laser, l 5 337 nm).
Amino sugars were measured as peracetylated alditols after hydrolysis in 4 N HCl (23), galactose as trimethylsilyl derivative of its 1-O-methylglycoside after methanolysis (26). According to the quantitative monosaccharide analyses, the untreated MUC1(HMFGM) contained Gal,GlcNAc, and GalNAc in the molar proportions of 3.5:3.2:1.0. C, gel exclusion chromatography of proteolytically digested GalNAc-MUC1 onSuperdex 75pg. The column (1 3 20 cm) which had been calibrated with a series of MUC1 icosapeptide oligomers (25-, 60-, and 100-mer) was runwith 50 mM sodium phosphate, 500 mM NaCl, pH 7.0, at a flow rate of 0.4 ml/min. The eluate was registered spectrophotometrically at 214 nm.The inset shows the corresponding immunoreactivity profiles of the eluted fractions using MUC1 tandem repeat peptide-specific antibodies BC3and VA1. The immunoreactivity of MUC1-derived glycopeptides eluting at 22 min is expectedly lower by orders of magnitude compared with thepolyvalent mucin in the void volume. Profile without addition of clostripain (O) or after digestion with the enzyme (- - -).
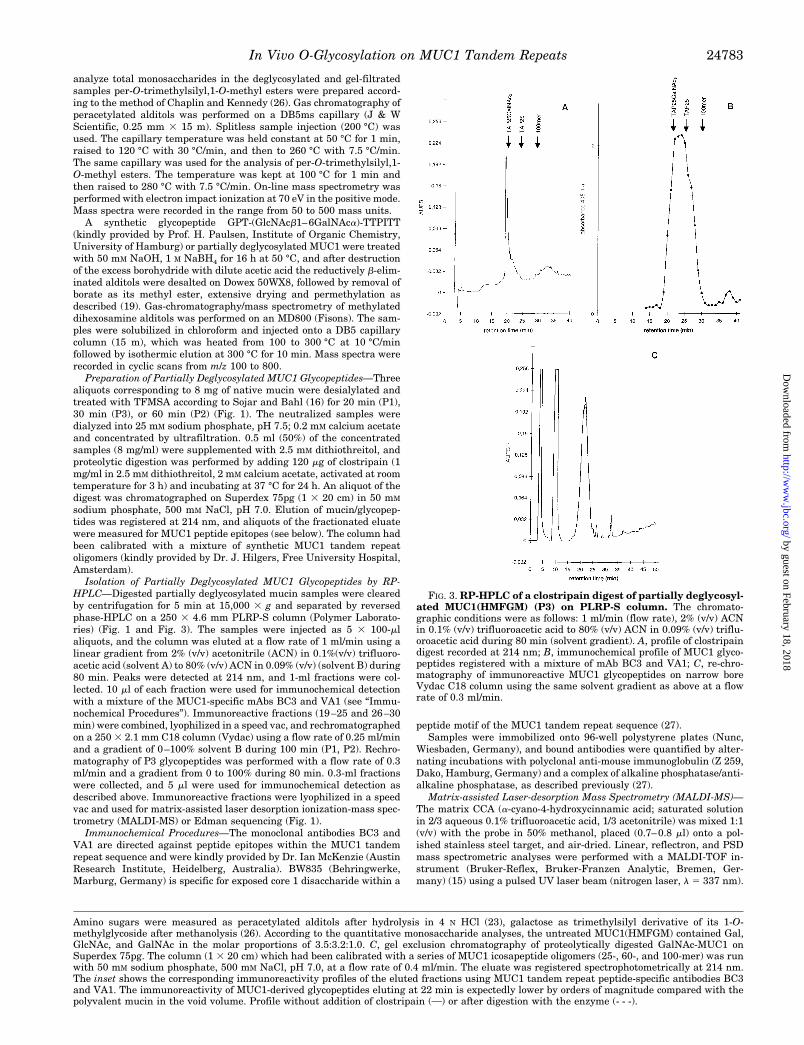
FIG. 3. RP-HPLC of a clostripain digest of partially deglycosyl-ated MUC1(HMFGM) (P3) on PLRP-S column. The chromato-graphic conditions were as follows: 1 ml/min (flow rate), 2% (v/v) ACNin 0.1% (v/v) trifluoroacetic acid to 80% (v/v) ACN in 0.09% (v/v) triflu-oroacetic acid during 80 min (solvent gradient). A, profile of clostripaindigest recorded at 214 nm; B, immunochemical profile of MUC1 glyco-peptides registered with a mixture of mAb BC3 and VA1; C, re-chro-matography of immunoreactive MUC1 glycopeptides on narrow boreVydac C18 column using the same solvent gradient as above at a flowrate of 0.3 ml/min.
In Vivo O-Glycosylation on MUC1 Tandem Repeats 24783
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
Irradiance was slightly above the threshold of ion detection. Residualgas pressure was at 0.8 3 1027 3 1027 mbar. Ion spectra were obtainedin the positive ion mode. Acceleration and reflector voltages were set to28.5 and 30 kV, respectively. The molecular parent ion was isolatedwith a pulsed field by deflection of all other ions (precursor selection).Complete PSD fragment ion spectra were obtained by stepwise reduc-ing the reflector voltage to produce overlapping mass ranges, combina-tion of the part spectra, and calibration according to the manufacturer’srecommendations. No smoothing or filtering was applied to the shownspectra.
Edman Degradation—Glycopeptides were sequenced on a HPJ000Gwith 3.1 chemistry and a biphasic sequencing column or after covalentattachment to Sequelon AATM membranes on a Beckman GlycoSitesequenator according to previously published procedures (6, 28). Quan-titation of the serine/threonine HexNAc substitution was calculatedusing a glycopeptide derived from the Dictyostelium discoideum recom-binant expressed glycoprotein PsA, which contains a 100% substitutedGlcNAcb-Thr in position 4. The corrected yield for this cycle (repetitiveyield for the glycopeptide ITATPAPT) was 95%. The migration behaviorof GlcNAcb-Thr is almost identical to the corresponding GalNAca de-rivative on reversed phase-HPLC (see also Refs. 6 and 28).
RESULTS
Isolation of Mucin
A detailed description of the purification procedure and thechemical characterization of MUC1 from human skim milk isgiven in Refs. 19 and 29. The yield of MUC1(skim milk) weobtained in the present study was 50 mg/1 liter of defattedmilk. For MUC1(HMFGM) the total yield was 7 mg calculatedfor 100 g of wet milk fat. Quality and homogeneity of bothpreparations were checked by SDS-polyacrylamide gel electro-phoresis with 10 mg of mucin/lane and subsequent electro-blotting, followed by the glycoprotein staining procedure ofO’Shannessy or immunochemical detection with the MUC1-specific mAb BW835. For both mucin preparations the glyco-protein stain revealed a single, broad band that hardly enteredthe 3–15% gel and that was isographic with the single bandobtained by staining Western blots with mAb BW835. Thesedata confirm that MUC1 is the major component in both mucinpreparations and demonstrate the absence of detectableamounts of contaminating glycoproteins. In addition, therewere no visible bands after staining the gels (100 mg mucin/lane) with Coomassie R-250, indicating that MUC1(skim milk)as well as MUC1(HMFGM) were almost free of non-glycosyl-ated protein.
Kinetics of MUC1 Deglycosylation with TFMSA
Although treatment with anhydrous TFMSA is generallyregarded as an efficient method for the deglycosylation of pro-teins (16, 17), it leaves a significant amount of the peptide-linked GalNAc uncleaved, when used for limited incubationtimes at 0 °C (16, 17). We took advantage of this fact by usingresidual core GalNAc as a marker for O-glycosylation sites.Kinetic studies were performed to find conditions where re-moval of peripheral saccharides was quantitative and the lossof peptide-linked GalNAc could be neglected. Because the pres-ence of GalNAc in MUC1-linked O-glycans is restricted topeptide-linked positions in core 1 (minor) or core 2 (major)structures (19),2, the amount of GalNAc residues and the ratioof GlcNAc/GalNAc, as measured by quantitative monosaccha-ride analysis, should give a good estimation of the proceedingdeglycosylation reaction. In the absence of anisole the removalof complex glycans and the chain truncation to the level of coreGalNAc proceeded within 60 min (Fig. 2A). Extension of theincubation times over 60 min resulted in substantial losses ofthe core GalNAc residues which was also reflected in a de-creased H. pomatia agglutinin-binding rate (data not shown).
2 W. Chai, A. M. Lawson, and F.-G. Hanisch, unpublished results.
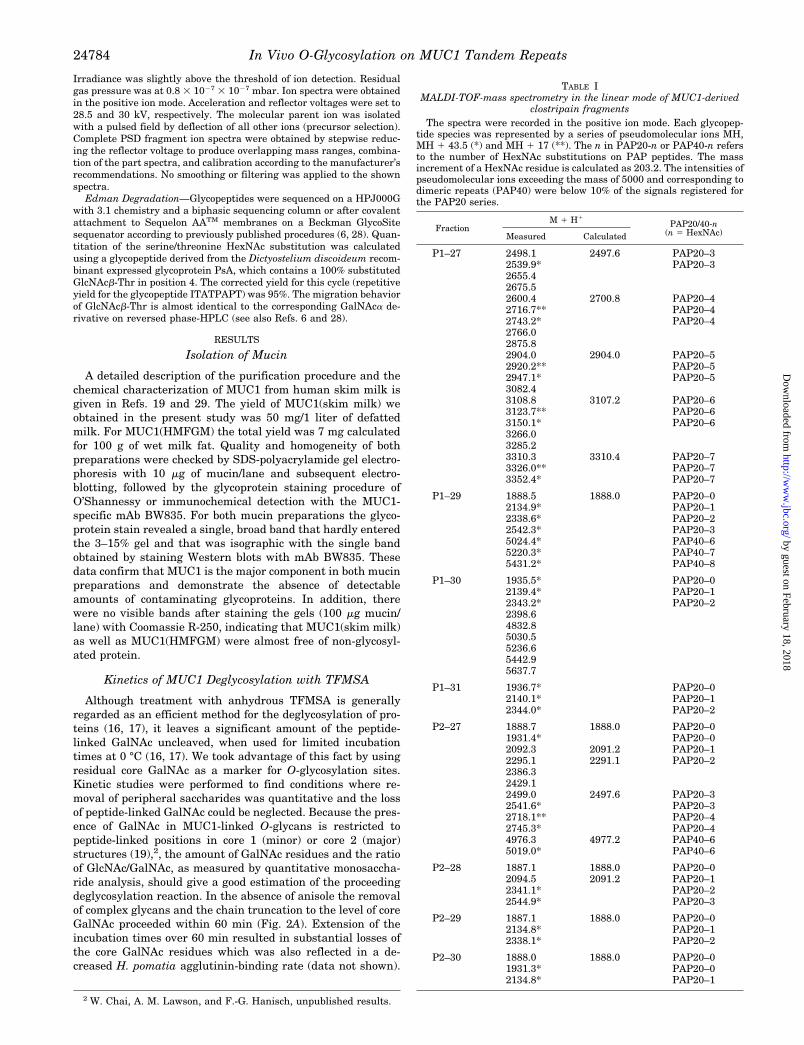
TABLE IMALDI-TOF-mass spectrometry in the linear mode of MUC1-derived
clostripain fragmentsThe spectra were recorded in the positive ion mode. Each glycopep-
tide species was represented by a series of pseudomolecular ions MH,MH 1 43.5 (*) and MH 1 17 (**). The n in PAP20-n or PAP40-n refersto the number of HexNAc substitutions on PAP peptides. The massincrement of a HexNAc residue is calculated as 203.2. The intensities ofpseudomolecular ions exceeding the mass of 5000 and corresponding todimeric repeats (PAP40) were below 10% of the signals registered forthe PAP20 series.
FractionM 1 H1
PAP20/40-n(n 5 HexNAc)Measured Calculated
P1–27 2498.1 2497.6 PAP20–32539.9* PAP20–32655.42675.52600.4 2700.8 PAP20–42716.7** PAP20–42743.2* PAP20–42766.02875.82904.0 2904.0 PAP20–52920.2** PAP20–52947.1* PAP20–53082.43108.8 3107.2 PAP20–63123.7** PAP20–63150.1* PAP20–63266.03285.23310.3 3310.4 PAP20–73326.0** PAP20–73352.4* PAP20–7
P1–29 1888.5 1888.0 PAP20–02134.9* PAP20–12338.6* PAP20–22542.3* PAP20–35024.4* PAP40–65220.3* PAP40–75431.2* PAP40–8
P1–30 1935.5* PAP20–02139.4* PAP20–12343.2* PAP20–22398.64832.85030.55236.65442.95637.7
P1–31 1936.7* PAP20–02140.1* PAP20–12344.0* PAP20–2
P2–27 1888.7 1888.0 PAP20–01931.4* PAP20–02092.3 2091.2 PAP20–12295.1 2291.1 PAP20–22386.32429.12499.0 2497.6 PAP20–32541.6* PAP20–32718.1** PAP20–42745.3* PAP20–44976.3 4977.2 PAP40–65019.0* PAP40–6
P2–28 1887.1 1888.0 PAP20–02094.5 2091.2 PAP20–12341.1* PAP20–22544.9* PAP20–3
P2–29 1887.1 1888.0 PAP20–02134.8* PAP20–12338.1* PAP20–2
P2–30 1888.0 1888.0 PAP20–01931.3* PAP20–02134.8* PAP20–1
In Vivo O-Glycosylation on MUC1 Tandem Repeats24784
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
On the contrary, deglycosylation in the presence of anisoleresulted in decreased reaction rates, and even after prolongedincubation times up to 6 h the residual GlcNAc was detected at60% relative to the untreated sample (Fig. 2B). The length ofthe polylactosamine-type chains on lactation-associated MUC1and evidence that indicates a stepwise deglycosylation processfrom the non-reducing terminus of the glycans (17) could ex-plain this observation. On the basis of these quantitative ki-
netic data the method of Sojar and Bahl (16) was preferred forthe preparation of MUC1-derived glycopeptides. As measuredby quantitative hexosamine analysis, the content of GalNAcduring TFMSA treatments up to 30 min decreased only to 86%,while the GlcNAc content during the same period droppedbelow 2% (Fig. 2A). The low amounts of GlcNAc relative toGalNAc detected after 30 min of deglycosylation indicate thatresidual dihexosamine GlcNAcb1–6GalNAca corresponding to
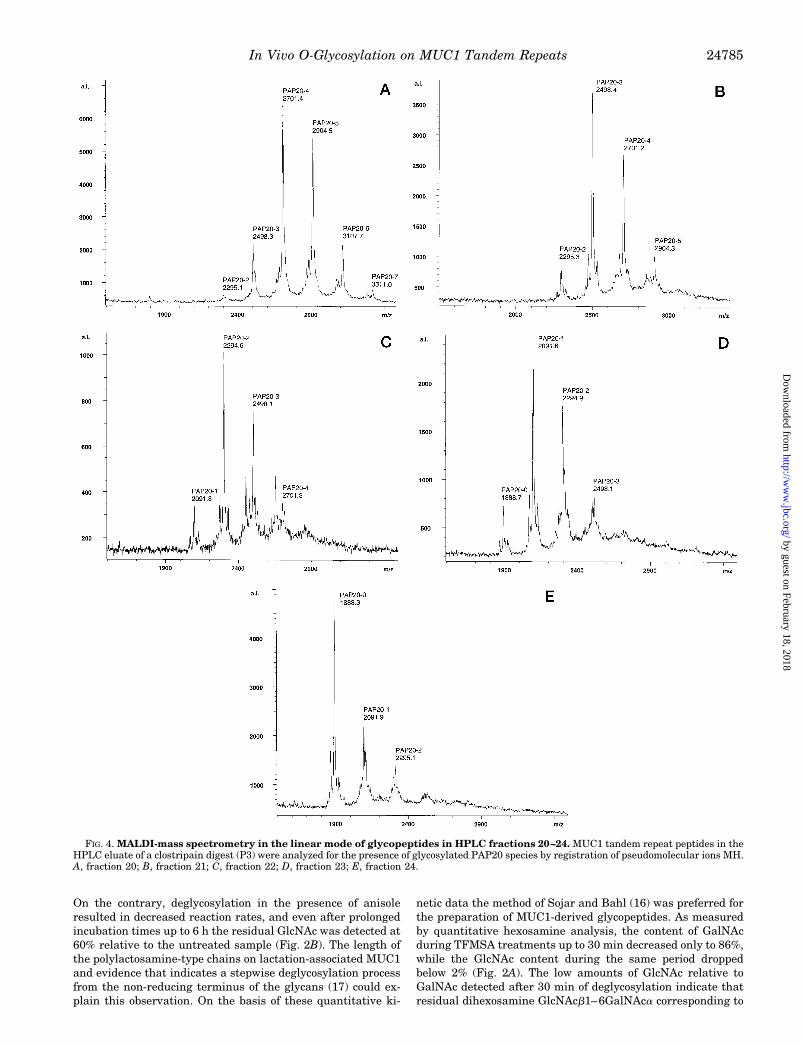
FIG. 4. MALDI-mass spectrometry in the linear mode of glycopeptides in HPLC fractions 20–24. MUC1 tandem repeat peptides in theHPLC eluate of a clostripain digest (P3) were analyzed for the presence of glycosylated PAP20 species by registration of pseudomolecular ions MH.A, fraction 20; B, fraction 21; C, fraction 22; D, fraction 23; E, fraction 24.
In Vivo O-Glycosylation on MUC1 Tandem Repeats 24785
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
a partial structure of the abundant core 2 glycans on MUC1from skim milk should be present only in trace amounts. Thiswas confirmed by GC/MS analysis of the permethylated aldi-tols derived by reductive b-elimination from the partially de-glycosylated mucin. Using a synthetic glycopeptide GPT-(Glc-NAcb1–6GalNAca)-TTPITT the dihexosamine alditol could beregistered in the scan mode at detection limits of 25 fmol perinjection. On the basis of this detection limit no GlcNAcb1–6GalNAc-ol was found on analysis of MUC1 that had beendeglycosylated by TFMSA treatment for 30 min. Also accordingto quantitative Edman degradation data (see below), theamount of GlcNAcb1–6GalNAc present should be less than 5%of the more common GalNAc substitution, since the corre-sponding PTH derivatives of serine or threonine wereundetectable.
In conclusion, these data demonstrate that 1) TFMSA cleav-age of peripheral sugars is rapid, 2) that 86% of GalNAc areretained for up to 30 min, and 3) that residual glycosylationconsists of GalNAc and only trace amounts of GlcNAc.
Preparation of Partially Deglycosylated Glycopeptides
MUC1 is a large and complex molecule, and the preparationof defined fragments is a prerequisite for sequencing experi-ments. Because there are several sites for specific proteolyticcleavage within the MUC1 tandem repeat sequence, we triedthe specific enzymatic fragmentation of native MUC1(skimmilk) or MUC1(HMFGM) with a variety of proteolytic enzymes(trypsin, staphyloccocus V8 proteinase, clostripain, papaya pro-teinase IV, Asp-N). The results obtained with these enzymeswere unsatisfactory (data not shown), and it turned out thatthe fully glycosylated mucin expressed in the lactating mam-mary gland and its desialylated derivative are almost resistantto commercially available specific proteinases. Since non-gly-cosylated as well as mono-, di-, or tri-GalNAc-substituted syn-thetic MUC1 peptides (TAPPAHGVTSAPDTRPAPGSTAPPA,designated TAP25) were good substrates (.90% cleavage asdetermined by RP-HPLC, data not shown) for clostripain andpapaya proteinase IV, it is obvious that, at least for theseenzymes, proteolytic resistance should be due to extensive gly-cosylation of the mucin. In addition these data indicate thatproteolytic efficiency can be increased by reducing glycosyla-tion to the level of core GalNAc, and hence, preparativeamounts (8 mg each) of MUC1 were partially deglycosylated bytreatment with TFMSA for 20 min (P1), 60 min (P2) (skim milkpreparations), and 30 min (P3, HMFGM preparation), and thedeglycosylated samples were digested with the Arg-C-specificproteinase clostripain. Aliquots of the digest that had been runover Superdex 75pg revealed that according to the profilesregistered at 214 nm, about 80% of the mucin had been de-graded by the enzyme (Fig. 2C). Only weak immunoreactivityto MUC1 peptide-specific antibodies corresponding to undi-gested MUC1 was detected in the excluded fraction (Fig. 2C).Clostripain-catalyzed fragmentation of MUC1 proceededmainly to the level of 20-mers (corresponding to one repeat) asrevealed by column calibration using a series of repeat oli-gomers (25-, 60-, and 100-mer). The pool of PAP20 glycopep-tides was isolated from crude digests by RP-HPLC on a PLRP-Scolumn. The HPLC profile registered at 214 nm for preparationP3 is shown in Fig. 3A, and the corresponding immunochemicalprofile, registered by enzyme-linked immunosorbent assaywith a mixture of MUC1-specific mAbs (BC3, VA1), is given inFig. 3B. The main peak in the UV profile eluted between 19 and25 min and represented the total glycopeptide fraction derivedfrom GalNAc-MUC1. Immunoreactivity eluted between 20 and30 min and several shoulders reflect the partial separation ofindividual glycoforms. Dithiothreitol, which was a component
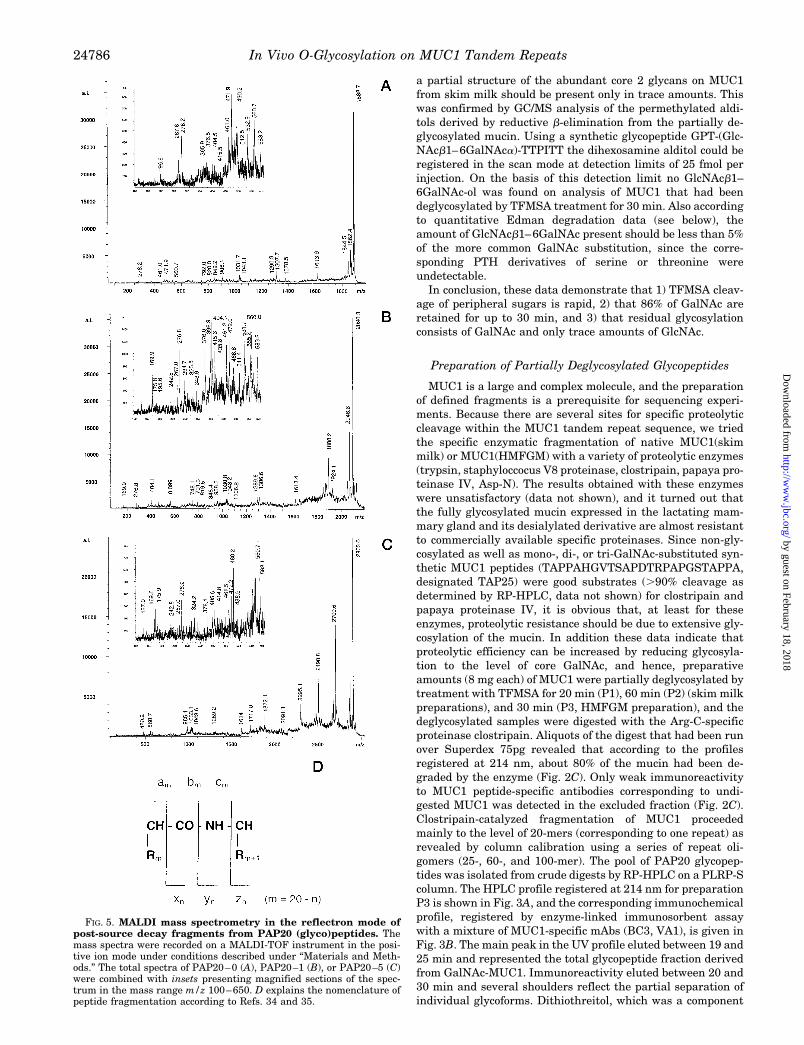
FIG. 5. MALDI mass spectrometry in the reflectron mode ofpost-source decay fragments from PAP20 (glyco)peptides. Themass spectra were recorded on a MALDI-TOF instrument in the posi-tive ion mode under conditions described under “Materials and Meth-ods.” The total spectra of PAP20–0 (A), PAP20–1 (B), or PAP20–5 (C)were combined with insets presenting magnified sections of the spec-trum in the mass range m/z 100–650. D explains the nomenclature ofpeptide fragmentation according to Refs. 34 and 35.
In Vivo O-Glycosylation on MUC1 Tandem Repeats24786
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

of the digestion buffer, coeluted with the PAP 20 glycopeptidesin the main peak at 20 min.
The retention times were in good agreement with thosemeasured for a series of synthetic MUC1 peptides. Non- totriglycosylated TAP25 peptides eluted between 20 and 25 min,and non-glycosylated repeat oligomers, containing three to fivetandem repeats, eluted between 26 and 30 min (Fig. 3C). Foreach preparation (P1–P3) the fractions between 19–25 min and26–30 min (Fig. 3A) were pooled, dried in a speed vac, andrechromatographed on a narrow-bore Vydac C18 column. Afterrechromatography the latter fraction was demonstrated by lin-ear MALDI MS not to contain any detectable MUC1-derivedglycopeptides. PAP20 glycopeptides (fractions 19–25 min) wereonly partially resolved on rechromatography and eluted indifferent solvent gradients as a broad peak at about 28 min (P1,P2) or 21 min (P3), respectively. Subfractions corresponding tothis peak and representing glycopeptides in P1-(27–32), P2-(27–32), and P3-(19–25) were collected and analyzed in thelinear mode MALDI mass spectrometry or for sequencing ex-periments (P3-(19–25)). As demonstrated by gel filtration onSuperdex 75pg and by linear MALDI mass spectrometry (seebelow), the subfractions from rechromatography containedmainly the monomeric tandem repeat peptides.
Glycopeptide Analysis by Linear Mode MALDI MassSpectrometry
Glycopeptides resulting from clostripain-catalyzed fragmen-tation of partially deglycosylated MUC1 mucin were collectedfrom the HPLC eluates, and the various fractions obtainedfrom preparations 1 to 3 (P1 to P3) were subjected to MALDI-mass spectrometry in the linear mode. The positively chargedpseudomolecular ions MH (MH 1 17, MH 1 43.5) registered forPAP20 glycopeptides from preparations 1 and 2 are listed inTable I, and the corresponding data obtained for preparation 3are presented in Fig. 4. In summary, only HexNAc carryingPAP20 peptides were detectable in the various fractions on thebasis of their pseudomolecular ions and the mass increment ofthe monosaccharide substituents (203.2 units) (Fig. 4 and Ta-ble I) Besides unglycosylated PAP20 (MH 1888.0) glycoforms ofPAP20 containing mainly 1–5 sugar residues and trace
amounts of higher glycosylated species were identified in frac-tion P1–27 (Table I) corresponding to the calculated MH at m/z2091.2, 2294.4, 2497.6, 2700.8, 2904.0, 3107.2, and 3310.4. Onthe other hand, the corresponding fraction P2–27 derived frommore extensively deglycosylated MUC1 contained onlyPAP20–0 to PAP20–4 glycoforms. The more polar, higher gly-cosylated species (PAP20–3, PAP20–4, PAP20–5, etc.) wereexpectedly eluted in the early fractions of the registered pro-files (Fig. 3 and Fig. 4), whereas the late fractions containedprimarily unglycosylated PAP20 or the correspondingPAP20–1 and PAP20–2 species. Moreover, the fractions elut-ing at higher acetonitrile concentrations were to a minor degreecomposed of larger glycopeptides that correspond to dimericrepeats PAP40 (less than 10% of the PAP20 peptides accordingto the signal intensities in the semi-quantitative MALDI MS).In general, the degree of peptide glycosylation as indicated bythe pseudomolecular ions of the polar species contained infractions P1–27 or P2–27 or in fraction P3–20 was expectedlydependent on the duration of TFMSA treatment that had beenemployed for the different preparations (P1, P2, P3).
Glycopeptide Sequencing by Post-source Decay MALDIMass Spectrometry
Analysis of post-source decay fragments for the sequencing ofglycopeptides was performed on PAP20–0 to PAP20–5 isolatedin P3.
PAP20–0—PSD-MALDI-MS analysis of the non-glycosyl-ated PAP20 (Fig. 5A) registered at m/z 1888.0 gave a nearlycomplete series of intense C-terminal yn and (yn 218) fragmentions, with a main fragment y2 (Fig. 5D). The main fragmentresults from cleavage of the Asp-Thr bond that had been iden-tified also in the previous study on synthetic MUC1 tandemrepeat peptides as a preferred cleavage site. The N-terminalfragment ions of the a or b series (am, bm) together with their217 companions that result from the loss of NH3 supported theevidence from C-terminal fragmentation, however, were lesscomplete than the y-fragment series (Fig. 5D). In conjunctionwith unidirectional subfragment ion series (internal sequenceions) derived from the various proline fragments the peptidesequence of PAP20 could unequivocally be determined as iden-
TABLE IIFragment masses measured for HexNAc-substituted PAP20–1 peptides
The relative masses m/z measured for the PSD fragments from the C-terminal end (yn ions and their companions) or N-terminal end ofPAP-20–1 (bm ions and their companions) were listed in accordance to the peptide sequence. Masses given in brackets were of low intensity,whereas for ions, indicated by 1, accurate mass assignment was impossible. The masses are accompanied by assignments of the fragment speciesgiven in brackets, followed by supporting ions (yn or bm companions, x, or a series ions) which provide additional evidence for the cleavage at distinctsites.
ynAmino acidsequence
Masses of C-terminal fragments, HexNAc Masses of N-terminal fragments, HexNAcbm
0 1 0 1
20 Pro 1885.2 (y) 2090.3 (y) 119 Ala 169.9 (b) 218 Pro 1924.1 (y) 267.0 (b) 317 Gly 1 416 Ser 1564.6 (y) 1794.4 (y), x 411.0 (b) 515 Thr 1478.6 (y) 511.4 (b) 614 Ala 1378.3 (y) 1580.2 (y), y-17 583.2 (b) 713 Pro 1306.6 (y) 1510.0 (y), y-17 679.8 884.5 (b) 812 Pro 1209.6 (y) 1439.4 (x) 980.1 (b), b-17 911 Ala 1113.8 (y) 848.4 (b) 1052.3 (b), a 1010 His 1 1245.0 (y), y-18 1 1171.4 (b-17) 11
9 Gly 903.7 (y) 1106.8 (y), y-18 1 128 Val 848.4 (y) 1076.7 (x) 1139.9 (b) 1343.7 (b) 137 Thr 748.1 (y) 951.1 (y), y-17 1242.4 (b) 1444.9 (b), a 146 Ser 647.0 (y) 1329.9 (b) 1516.6 (b-17) 155 Ala 560.0 (y) 164 Pro 488.8 (y) 173 Asp 392.5 (y) 1613.4 (b) 1816.3 (b) 182 Thr 276.8 (y) 1901.6 (b-17) 191 Arg 175.8 (y) 2046.6 (a) 20
In Vivo O-Glycosylation on MUC1 Tandem Repeats 24787
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

tical to the MUC1 tandem repeat sequence. On sequencing ofthe various PAP20 glycoforms by PSD-MALDI-MS, the sameseries of fragment ions and their companions were registered.However, fragment ions that are potentially glycosylated gen-erally occur at lower intensities. This can be explained, partic-ularly for peptides with a low degree of glycosylation, by thedispersity of each glycoform resulting from a substitutionisomery as revealed by the more complex fragment spectra.
PAP20–1 to PAP20–3—In the PSD spectrum of PAP20–1(Fig. 5B), the monoglycosylated peptide, the HexNAc substitu-tion could be localized at two to three different positions (Thr14
and Ser5 and/or Thr6) based on glycosylated C- or N-terminalfragments (Table II). In the y fragment series ions at m/z 951.1(y7), 1106.8 (y9), 1245.0 (y10), 1510.0 (y13), or 1580.2 (y14) inconjunction with x ions and yn217 or yn218 companions indi-cate glycosylation at one of the positions Thr14, Ser15, or Thr19.Since no glycosylated fragment ions corresponding to y2 to y6
were detectable, the GalNAc substitution can be tentativelyassigned to Thr14. Also PAP20–2, the glycoform carrying twoGalNAc residues, was demonstrated to be a mixture of severalposition isomers. Unequivocal evidence for a HexNAc substitu-tion was obtained with reference to the position Thr19 at m/z480.9 (y2) representing the ion mass of a monoglycosylatedfragment (Table III). The higher glycosylated PAP20 species,PAP20–3 to PAP20–5, turned out to be less heterogeneouswith regard to the substitution of single sites. Accordingly, theintensity of the signals indicating glycosylation increased andGalNAc substitution could be more specifically localized. Thetriglycosylated peptide, PAP20–3, revealed unequivocal evi-dence for a monoglycosylation of Thr19 by C-terminal fragmentions at m/z 480.3 (y2) and 692.0 (y4) (Table IV). HexNAcsubstitution of Ser5 and Thr6 was revealed by the triglycosyl-ated fragment ions at m/z 1121.4 (b6), 1192.7 (b7), 1165.3 (a7),1290.0 (b8), 1261.4 (a8), 1105.0 (b6217), and 1175.2 (b7217).With regard to the positions Thr14 and Ser15 at least one of theputative glycosylation sites carries one or two HexNAc residuesas evidenced by the triglycosylated fragment ions at m/z1384.6 (x7), 1845.9 (x12), 1634.6 (y10217) and 1899.6 (y13217).A minor subfraction of PAP20–3 should be substituted at Ser5
with a dihexosamine which is indicated by the fragment ion atm/z 818.0 (b5) and its companion b5217.
PAP20–4—The ambiguities remaining after analysis of thePAP20 species with a lower degree of glycosylation (i.e. glyco-
sylation of the Thr14 and Ser15 positions) could be eliminatedon PSD fragment analysis of the tetraglycosylated PAP20–4.Again a HexNAc substitution of Thr19 was revealed by thefragment ions at m/z 480.3 (y2), 595.6 (y3), 692.1 (y4), and theircompanions (Table V). The intensity of the glycosylated y2
relative to the non-glycosylated y2 supports the assumptionthat an increasing portion of the PAP20–4 fraction was glyco-sylated at that position, when compared with peptides inPAP20–1 to PAP20–3 (see also Fig. 5C). The most highlyglycosylated C-terminal fragments y7 (m/z 1562.0), y9 (m/z1744.5), y11217 (m/z 1909.4) which are substituted with fourHexNAc residues demonstrate that both positions, Ser15 andThr14, carry at least one sugar residue (Table V). The same lineof evidence holds true for the analysis of Ser5 and Thr6 glyco-sylation. The triglycosylated, N-terminal fragments b6 (m/z1121.9) and b8 (m/z 1290.9) which comprise both positionsshould, accordingly, reflect their substitution with HexNAc.Thus, fraction PAP20–4 reveals the first unequivocal evidencethat all putative sites within the MUC1 tandem repeat can betargets for O-glycosylation.
PAP20–5—On analysis of post-source decay fragments de-rived from PAP20–5, each of the five putative glycosylationsites could be revealed as HexNAc-substituted. In detail, theC-terminal series of fragments at m/z 480.2 (y2), 692.0 (y3),and 764.0 (y4) indicated glycosylation of Thr19 with a HexNAcresidue (Fig. 5C and Table VI). Comparing the signal intensi-ties of the main fragment ion at m/z 480.2 (y2) relative to thoseat m/z 276.9 (y2) an increasing proportion of Thr19-glycosyl-ated species is obvious (Fig. 5, A–C). The fragment massescorresponding to y6 and x6 (containing the mass increments oftwo HexNAc residues), accordingly, prove that Ser15 is alsoglycosylated. Unequivocal evidence for a HexNAc substitutionof Thr14 comes from the y fragment series at m/z 1920.2 (y9),2127.0 (y11), 2225.8 (y12), or their companions at m/z 1846.2(y8-17), 2041.6 (y10-17), and 2208 (y12-17) corresponding toHexNAc5-substituted C-terminal fragments (Table VI). N-ter-minal fragmentation of the bm and am series demonstrated thatSer5 and Thr6 are substituted with one or two HexNAc resi-dues. In particular, the ion masses corresponding to b5 and itscompanion b5217 indicate (at least for an enriched subpopula-tion of glycopeptides in PAP20–5) the presence of a dihex-osamine in position Ser5. Position Thr6 obviously carries one ortwo HexNAc residues according to the fragment masses at m/z
TABLE IIIFragment masses measured for HexNAc-substituted PAP20–2 peptides
The relative masses m/z measured for the PSD fragments from the C-terminal end (yn ions and their companions) or N-terminal end of PAP20–2(bm ions and their companions) were listed in accordance to the peptide sequence. For further details refer to Table II.
ynAmino acidsequence
Masses of C-terminal fragments, HexNAc Masses of N-terminal fragments, HexNAcbm
0 1 2 0 1 2
20 Pro 1887.6 (y) 2091.7 (y), y-18 2293.6 (y) 119 Ala 1788.0 (y) 169.8 (b) 218 Pro 1718.5 (y) 1 2123.9 (y) 267.0 (b) 317 Gly 2028.8 (y) 416 Ser 1564.0 (y) 1972.9 (y), y-18 597.4 (b-17) 515 Thr 1478.8 (y) 1681.9 (y) 512.2 (b) 919.0 (b), b-17 614 Ala 1 1580.8 (y), y-17 1784.3 (y), y-18 587.7 (b) 713 Pro 1306.3 (y) 1510.8 (y) 681.5 (b) 1086.9 (b) 812 Pro 1642.7 (x) 911 Ala 1113.5 (y) 1544.8 (x), y-18 1010 His 1041.5 (y) 1447.1 (y), y-17 985.6 (b) 1392.7 (b), a 119 Gly 1108.0 (y) 1041.5 (b) 1246.6 (b) 1431.8 (b-17) 128 Val 1050.2 (y), y-18 1252.9 (y), y-17 1142.3 (b) 1317.5 (a) 137 Thr 747.4 (y) 934.2 (y-17) 1243.8 (b) 1446.6 (b), b-17 146 Ser 646.7 (y) 1516.8 (b-17) 1708.8 (a) 155 Ala 559.4 (y) 762.8 (y) 164 Pro 489.3 (y) 691.8 (y) 1498.3 (b) 1904.6 (b), b-17 173 Asp 393.5 (y) 1613.4 (b) 2021.2 (b), b-17 182 Thr 277.0 (y) 480.9 (y), y-18 1916.8 (b) 2105.1 (b-17) 191 Arg 175.7 (y) 2073.4 (b) 2249.9 (a) 20
In Vivo O-Glycosylation on MUC1 Tandem Repeats24788
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
1192.5 (b7) or 1492.8 (b8) which correspond to tri- or tetragly-cosylated ion species, respectively (Table VI).
In summary, all five putative sites are glycosylated withHexNAc residues. The detection of dihexosamines by PSDMALDI MS was restricted to the higher glycosylated species,where this structural component should be enriched. The pres-ence of minor amounts of dihexosamines did not affect the massspecrometric sequencing.
Identification of Glycosylation Sites by EdmanDegradation
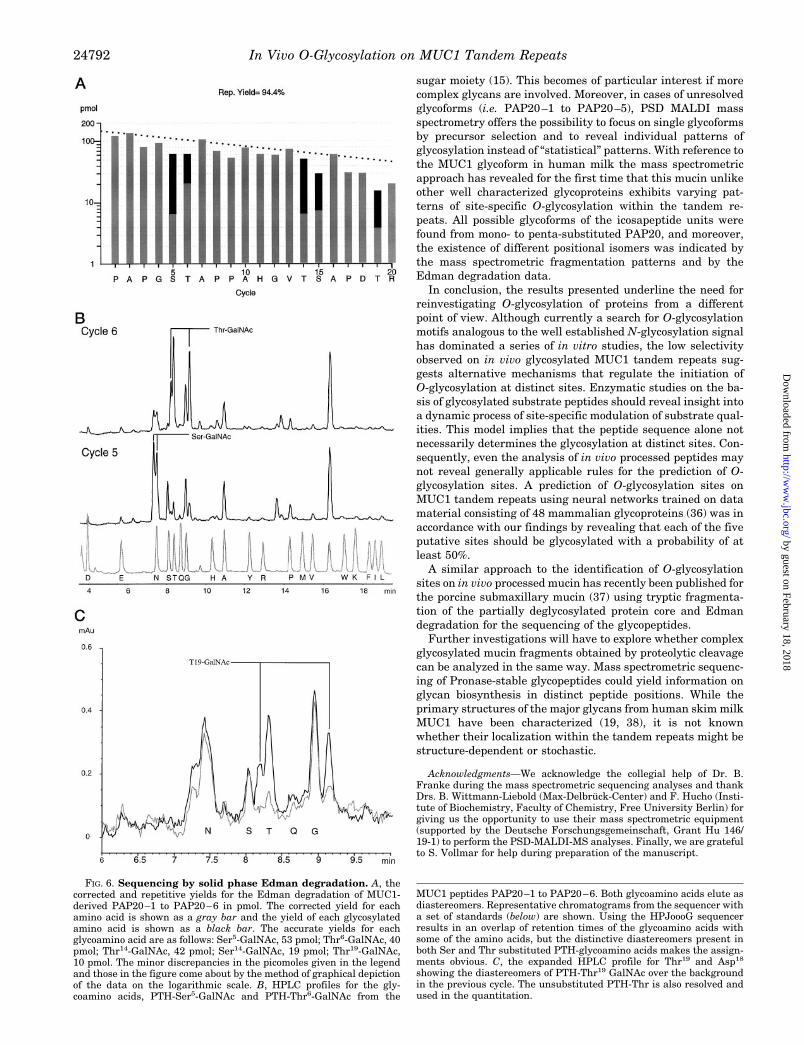
Amino acids that are substituted with a monosaccharide arereadily recovered in the Edman sequenator. Edman degrada-tion of MUC1-derived glycopeptides was performed on a repre-sentative mixture of the Vydac C18 HPLC fractions 20–24 (P3)and on the highly glycosylated species (mainly PAP20–4 andPAP20–5) contained in HPLC fraction 20 (P3) of the same run.Fig. 6 shows the corrected and repetitive yields (pmol) of theglycosylated or non-glycosylated PTH-amino acids obtained forthe whole PAP20 peptide(s). No indications for the presence ofminor sequences were found, and there was not any readablesequence after cycle 20.
Confirming the above reported mass spectrometric findings,all serine and threonine positions within PAP20 were demon-strated to be glycosylated. The relative molar quantities ofglycosylated or non-glycosylated Ser or Thr derivatives weredetermined by correcting the observed yield for the phenylthio-hydantoin (PTH)-Ser(HexNAc) or Thr(HexNAc) by a factor of1.28 which we have shown elsewhere is the correction factor fora fully substituted Thr or Ser (28). Since the unsubstituted Serand Thr undergo partial dehydration in the sequenator, theyare recovered in much lower yields than their glycosylatedcounterparts. Hence, only the glycosylated Ser and Thr can bequantitated with any accuracy. Evidence for the presence of aSer or Thr substituted with a dihexosamine was tested usingthe GlycoSite, a solid phase Edman sequenator (Beckman In-struments Inc.). GlycoSite analysis of the HPLC fraction 20(P3) did not indicate the dihexosamine was present, at least atthe level of 1 pmol (equivalent to #5% glycosylation at any oneparticular site, data not shown). There was no obvious prefer-
ence of threonine over serine positions regarding the degree ofGalNAc substitution. The most highly glycosylated positionwas Thr14 showing 79.6 or 93.9% substitution in the represent-ative mixture or in HPLC fraction 20, respectively. Unexpect-edly, the degree of glycosylation at Ser5 was higher than at thevicinal Thr6 position, a finding that contrasts with the previousin vitro pattern (14). At least one-third of the Ser15 and Thr19
positions was revealed as glycosylated, when analyzing therepresentative mixture of glycopeptides. On average 2.7 of the5 putative sites were calculated to carry a GalNAc substitution.The substitution pattern of individual repeats obviously showsfluctuations and may reflect a random mechanism of GalNAcaddition to the peptide core or a limited random deglycosyla-tion by TFMSA.
DISCUSSION
The present contribution reports on the localization of O-glycosylation sites on MUC1 tandem repeat peptides that hadbeen processed in vivo by mammary glandular epithelial cellsduring lactation. By using controlled conditions for the limiteddeglycosylation of MUC1 leaving core GalNAc residues intactand by enzymatic fragmentation with clostripain to the level ofmonomeric repeats, a representative mixture of glycopeptidescould be obtained that reflects the site-specific glycosylationstatus of the lactation-associated mucin. We have demon-strated by mass spectrometric sequencing using PSD-MALDI-MS and by solid phase Edman sequencing that all fiveputative glycosylation sites within the 20 amino acid repeatsserve as substrates for GalNAc-transferase(s). Although in pre-vious in vitro glycosylation studies only the threonine residueswithin the VTSA and GSTA sequences were identified as themain sites of initial O-glycosylation (12–14), and the serinewithin GSTA was found to be glycosylated to a lower degree(14), we here present evidence that also threonine within thePDTR motif and serine within VTSA are glycosylated in vivo byglandular epithelial cells of the breast. This discrepancy is notmerely explained by taking cell-specific enzyme repertoiresinto account, since GalNAc-transferase(s) in human milk hadbeen used in previous in vitro studies (14, 15). The restrictedpattern of O-glycosylation observed under in vitro conditions
TABLE IVFragment masses measured for HexNAc-substituted PAP20–3 peptides
The relative masses m/z measured for the PSD fragments from the C-terminal end (yn ions and their companions) or N-terminal end of PAP20–3(bm ions and their companions) were listed in accordance to the peptide sequence. For further details refer to Table II.
ynAmino acidsequence
Masses of C-terminal fragments, HexNAc Masses of N-terminal fragments, HexNAcbm
0 1 2 3 0 1 2 3
20 Pro 1888.4 (y) 2091.5 (y) 2293.8 (y), y-18 2496.3 (y) 119 Ala 1789.9 (y) 1994.7 (y) 169.7 (b) 218 Pro 1924.3 (y),
y-172123.5 (y) 266.8 (b) 3
17 Gly 1826.3 (y) 2232.2 (y) 323.5 (b) 416 Ser 2175.9 (y), y-17 818.0 (b), b-17 515 Thr 512.2 (b) 918.5 (b), a 1121.4 (b), b-17 614 Ala 1 1580.4 (y) 1783.1 (y) 582.8 (b) 973.5 (b-17) 1192.7 (b), b-17 713 Pro 1306.4 (y) 1509.3 (y),
y-171712.2 (y) 1899.6 (y-17) 680.6 (b) 1086.7 (b), a 1290.0 (b), a 8
12 Pro 1616.6 (y) 1845.9 (x) 776.9 (b) 911 Ala 1010 His 1244.9 1446.2 1634.6 (y-17) 119 Gly 1108.0 (y), x 1244.9 (b) 128 Val 1050.1 137 Thr 749.0 (y) 951.6 (y),
y-171384.6 (x) 1446.2 (b) 1648.1 (b) 14
6 Ser 646.6 (y) 155 Ala 560.2 (y) 1400.0 (b) 1576.2 (a) 1789.9 (b-17) 2010.4 (b) 164 Pro 489.6 (y) 692.0 1698.3 (b), a 173 Asp 1613.4 (b) 1814.7 (b) 2019.9 (b) 2222.8 (b) 182 Thr 276.9 (y) 480.3 1712.2 (b) 2324.9 (b), b-17 191 Arg 2074.4 (b) 2275.8 (b) 20
In Vivo O-Glycosylation on MUC1 Tandem Repeats 24789
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
does not result from short reaction times, since the PDTRPmotif was not glycosylated even after incubation periods in theorder of days (14, 15). Furthermore, the observed pattern of invitro glycosylation has been demonstrated to be independent ofthe repeat number and, hence, of the substrate peptide length(12). In conclusion, it has to be assumed that mucin peptideglycosylation under in vivo conditions follows different mecha-nisms than in vitro and, accordingly, distinct rules. The moreselective pattern of O-glycosylation observed in a non-cellular,stochastic system is ruled by flanking peptide sequences or bythe corresponding substrate specificities of the various Gal-NAc-transferases. This holds particularly true if O-glycosyla-tion of synthetic peptides is investigated on the level of mono-saccharide incorporation. Previous studies did not address thepossibility that glycosylation of Thr or Ser may favorably alterthe substrate qualities at vicinal sites that otherwise would nothave been used by GalNAc-transferase(s). A dynamic model ofinitial O-glycosylation includes the possibility that GalNAcaddition to poor substrate positions may be mediated by thecooperative action of glycosyltransferases on adjacent sites andthe formation of complex glycans that may be necessary for theinitiation of glycosylation at these sites due to the carbohy-drate-induced conformational changes of the peptide. In sup-port of this hypothesis a recent paper by Brockhausen et al. (30)provided evidence that a negative regulatory influence of vici-nal glycosylation is exerted on distinct substrate positions of apeptide.
Glycosylation of the PDTRP motif has a significant impact onthe immunogenicity and antigenicity of MUC1 (31). The nakedpeptide motif is known to be highly immunogenic and plays acrucial role in the non-MHC-restricted cytotoxic T-cell responsein cancer patients (32). Murine monoclonal antibodies reactive
to this motif have been demonstrated to bind to MUC1 in aglycosylation-dependent manner (10, 11). Most of these anti-bodies are non-reactive to the glycoform of MUC1 expressed bythe lactating mammary gland but are strongly binding to thecarcinoma-associated glycoform of the mucin. As previouslyshown, this effect partly results from glycosylation at vicinalsites (10, 14). However, a direct effect by glycosylation of thePDTR motif cannot be ruled out according to the results of thisstudy. Further investigations on the carcinoma-associated mu-cin have to be performed to reveal whether the altered antige-nicity of MUC1 could be caused by underglycosylation of thePDTR peptide motif.
The reflectron analysis of post-source decay fragments hasbeen established by Kaufmann et al. (33) for the sequencing ofpeptides and recently applied on the sequencing of glycopep-tides by Goletz et al. (15). The present paper describes for thefirst time the site-specific glycosylation pattern of a humanmucin by using this variant of MALDI-mass spectrometry forthe sequencing of proteolytic glycopeptide fragments. Thisnovel approach has previously been tested for a series of syn-thetic glycopeptides corresponding to the MUC1 tandem repeat(15). As revealed in this previous study, a sensitive and reliablelocalization of O-glycosylation sites is feasible by the analysis ofpost-source decay fragments. The most prominent fragments,the y ion series (34, 35) (Fig. 5D), represented a nearly completepattern of ionized scission products from the C-terminal end ofPAP20 peptides and comprised a large proportion of the glyco-sylated fragments. The b series of fragments (34, 35) (Fig. 5D)from the N-terminal end was less complete and dominated bynon-glycosylated species. The preferred cleavage sites withinthe PAP20 peptide sequence were at the various X-Pro linkagesand the Asp-Thr linkage. The major proline fragments by
TABLE VFragment masses measured for HexNAc-substituted PAP20–4 peptides
The relative masses m/z measured for the PSD fragments from the C-terminal end (yn ions and their companions) or N-terminal end of PAP20–4(bm ions and their companions) were listed in accordance to the peptide sequence. For further details refer to Table II.
ynAmino acidsequence
Masses of C-terminal fragments, HexNAc Masses of N-terminal fragments, HexNAcbm
0 1 2 3 4 0 1 2 3 4
20 Pro 2094.0 (y),y-17
2297.9 (y) 2499.7 (y),y-18
2701.8 (y) 1
19 Ala 169.8 (b) 218 Pro 2129.5 (y) 2331.9 (y) 2533.5 (y) 267.2 (b) 317 Gly 1 416 Ser 2179.6 (y) 2382.5 (y),
y-18411.5 (b) 801.8
(b), a5
15 Thr 890.7 (a) 1121.9(b)
6
14 Ala (1784.3)(y)
584.3 (b) 7
13 Pro 1307.6 (y) 1510.3 (y) (1715.0)(y)
1916.8 (y) 680.0 (b) 848.2 (b) 1290.9 (b) 8
12 Pro 1413.4 (y) (1614.4)(y), y-17
1817.7(y),x
9
11 Ala 1518.0(y),y-18
1904.4(y-17)
549.3 (b) 1025.0 (a) 10
10 His 1245.5 (y),y-18
1448.3 (y) 1 986.2 (b) 1162.0 (a) 11
9 Gly 903.5 (y) 1106.9 (y), x 1744.5 (y) 1245.5 (b) 128 Val 1032.4
(y-18)1142.5 (b) 1735.4
(b-17)13
7 Thr 1356.8(y),x
1562.0 (y) 14
6 Ser 647.4 (y) 349.3 (y) (1735.4)(b)
15
5 Ala 559.3 (y) 1400.5 (b) 1 164 Pro 489.3 (y) 692.1 (y) 1 1 2311.0 (b) 173 Asp 393.4 (y) 595.6 (y),
y-18(1) 1817.7 (b) 18
2 Thr 277.3 (y) 480.3 (y),y-17
(1) 1916.8 (b) 19
1 Arg 175.7 (y) (1) 2076.8 (b) 1 2482.0 (b) 20
In Vivo O-Glycosylation on MUC1 Tandem Repeats24790
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

themselves yielded a series of secondary ions correspondingmainly to the y and b series ions. Since the respective primaryand secondary fragments were registered as a series of variablyglycosylated species, it has to be assumed that each of theglycoforms, PAP20–1 to PAP20–5, represented a mixture ofpositional isomers. The complexity of the PSD mass spectra isfurther increased by the formation of am, and to a minor extendof cm, xn, and zn fragments (33, 34), and their identification maysometimes become difficult by overlapping fragment massesand a restricted mass accuracy. However, within a limitedmass range only small deviations from the calculated masseswere observed which showed, moreover, the same tendency andsimilar extents. In support of the reliability of the mass spec-trometric sequencing approach, fragment identification wasbased on a series of accompanying ions like yn 2 17, yn 2 18originating from the loss of NH3 or water, respectively, whichare sometimes of higher intensity than the y or b series ions.Considering the various diagnostic fragment ion series, theglycosylation status at individual sites could be unequivocallydetermined on the basis of several overlapping fragments. Theamounts of dihexosamines may have been overestimated onthe basis of the mass spectrometric data due to the enrichmentof this component in the highly glycosylated PAP20 species.GC-MS analysis of permethylated alditols and solid phase Ed-man degradation have revealed that GlcNAcb1–6GalNAcshould be present only in trace amounts. More complex glycans
containing additional hexose were not detectable in the glyco-peptide fractions of partially deglycosylated mucin according tothe pseudomolecular ions registered in linear MALDI-massspectrometry. It has to be emphasized that the glycopeptidesunder study had been derived from MUC1 deglycosylated inthe absence of anisole. In the presence of the radical scavengerthe efficiency of deglycosylation decreased strongly and evenafter hours the polylactosamine-type chains were only partiallycleaved (approximately 50%). Accordingly, the conditions ofdeglycosylation with TFMSA have to be optimized for eachglycoprotein, since the structure of the glycans and their chainlengths seem to influence the kinetics of the reaction.
The results of mass spectrometric sequencing were con-firmed by an independent method using a solid phase variant ofautomated Edman sequencing (8). This technique has the ad-vantages that the glycosylated amino acid derivatives can beidentified and quantified, which gives the opportunity to esti-mate the degree of glycosylation at each site. In the presentstudy it was possible to estimate the percentage of glycosyla-tion at the five putative sites within a peptide repeat. Thequantitative data revealed that there is no preference of thre-onine substitution over serine in vivo and that the pattern oflactation-associated MUC1 deviates also in this respect fromthe previously analyzed in vitro pattern (14). The mass spec-trometric strategy, on the other hand, yields more structuralinformation by allowing also the concomitant sequencing of the
TABLE VIFragment masses measured for HexNAc-substituted PAP20–5 peptides
The relative masses m/z measured for the PSD fragments from the C-terminal end (yn ions and their companions) or N-terminal end of PAP20–5(bm ions and their companions) were listed in accordance to the peptide sequence. For further details refer to Table II.
ynAmino acidsequence
Masses of C-terminal fragments, HexNAc Masses of N-terminal fragments, HexNAcbm
0 1 2 3 4 5 0 1 2 3 4
20 Pro 1886.7 (y) 2091.1(y),y-17
2295.1 (y) 2498.8(y),y-17
2700.6(y),y-18
2903.0 98.5 (b) 1
19 Ala 1 2402.0(y),x,y-18
(2605.2)(y)
2
18 Pro (2127.0)(y)
2328.3(y),y-18,x
2532.1 (y) 267.0 (b) 3
17 Gly (2028.5)(y)
2231.2 (y) 2639.1 324.2 (b) 4
16 Ser 1971.6(y),y-15
2158.2(y-17)
2364.0(y-15)
411.2 (b) 1(b),b-17 5
15 Thr 2276.5(y-15)
2480.4(y-15)
512.0 (b) 6
14 Ala (1563.5)(y-17)
1784.3 (y) 583.1 (b) 1192.5(b),a
7
13 Pro 1305.8 (y) 1510.6(y),y-17
1 2102.9(y-17)
680.8 (b) 1290.5 (b) 1492.8(b),a,a-17
8
12 Pro 1192.5 (y) 1413.5 (y) 1,y-17,x 1820.0(y),x
(2225.8)(y),y-17
776.5 (b) 1184.4 (b) 1589.2 (b) 9
11 Ala 1518.7 (y) 1 (2127.0)(y)
915.6 (b) 1458.0 (b) 1 (b-17) 10
10 His 1 1448.0(y),y-15
(1650.9(y),x
(1854.4)(y)
2041.6(y-17)
985.1 (b) 1392.0(b),b-17,c
1577.4(b-17)
11
9 Gly 903.9 (y) 1108.3 (y) 1 1515.5 (y) 1717.0(y),y-15
1920.2 (y) 1217.8 (a) 1(b),a-17 1650.9 (b) 1854.4(b),c,a
12
8 Val 1033.1(y-17)
1458.0 (y) 1642.1(y-17)
1846.2(y-17)
1141.7 (b) (1548.5)(b),b-17
1749.7(b),b-17
1936.7(b-17)
13
7 Thr 1 933.4(y-18)
1341.0(y),y-17
1749.7(y-15)
2037.6(b-17)
14
6 Ser 647.4 (y) 850.4 (y) 1 (y),x 1515.5(b-17)
2158.8 (c) 15
5 Ala 560.7 (y) 764.0(y),y-17
16
4 Pro 488.9 (y) 692.0 (y) 1702.0(b)
2091.1(b-17)
2310.8(b)
17
3 Asp 1833.1(c)
1991.5(a), c
2194.3(a), a-17
2380.4(a-17)
18
2 Thr 276.9 (y) 480.2(y), y-18
1934.0(c)
(2102.9)(b-17)
2498.8(a)
19
1 Arg 175.9 (y) 2074.2(b), a, a-17
(2276.5)(b), a, a-17
2480.4(b)
2682.2(b)
20
In Vivo O-Glycosylation on MUC1 Tandem Repeats 24791
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from
sugar moiety (15). This becomes of particular interest if morecomplex glycans are involved. Moreover, in cases of unresolvedglycoforms (i.e. PAP20–1 to PAP20–5), PSD MALDI massspectrometry offers the possibility to focus on single glycoformsby precursor selection and to reveal individual patterns ofglycosylation instead of “statistical” patterns. With reference tothe MUC1 glycoform in human milk the mass spectrometricapproach has revealed for the first time that this mucin unlikeother well characterized glycoproteins exhibits varying pat-terns of site-specific O-glycosylation within the tandem re-peats. All possible glycoforms of the icosapeptide units werefound from mono- to penta-substituted PAP20, and moreover,the existence of different positional isomers was indicated bythe mass spectrometric fragmentation patterns and by theEdman degradation data.
In conclusion, the results presented underline the need forreinvestigating O-glycosylation of proteins from a differentpoint of view. Although currently a search for O-glycosylationmotifs analogous to the well established N-glycosylation signalhas dominated a series of in vitro studies, the low selectivityobserved on in vivo glycosylated MUC1 tandem repeats sug-gests alternative mechanisms that regulate the initiation ofO-glycosylation at distinct sites. Enzymatic studies on the ba-sis of glycosylated substrate peptides should reveal insight intoa dynamic process of site-specific modulation of substrate qual-ities. This model implies that the peptide sequence alone notnecessarily determines the glycosylation at distinct sites. Con-sequently, even the analysis of in vivo processed peptides maynot reveal generally applicable rules for the prediction of O-glycosylation sites. A prediction of O-glycosylation sites onMUC1 tandem repeats using neural networks trained on datamaterial consisting of 48 mammalian glycoproteins (36) was inaccordance with our findings by revealing that each of the fiveputative sites should be glycosylated with a probability of atleast 50%.
A similar approach to the identification of O-glycosylationsites on in vivo processed mucin has recently been published forthe porcine submaxillary mucin (37) using tryptic fragmenta-tion of the partially deglycosylated protein core and Edmandegradation for the sequencing of the glycopeptides.
Further investigations will have to explore whether complexglycosylated mucin fragments obtained by proteolytic cleavagecan be analyzed in the same way. Mass spectrometric sequenc-ing of Pronase-stable glycopeptides could yield information onglycan biosynthesis in distinct peptide positions. While theprimary structures of the major glycans from human skim milkMUC1 have been characterized (19, 38), it is not knownwhether their localization within the tandem repeats might bestructure-dependent or stochastic.
Acknowledgments—We acknowledge the collegial help of Dr. B.Franke during the mass spectrometric sequencing analyses and thankDrs. B. Wittmann-Liebold (Max-Delbruck-Center) and F. Hucho (Insti-tute of Biochemistry, Faculty of Chemistry, Free University Berlin) forgiving us the opportunity to use their mass spectrometric equipment(supported by the Deutsche Forschungsgemeinschaft, Grant Hu 146/19-1) to perform the PSD-MALDI-MS analyses. Finally, we are gratefulto S. Vollmar for help during preparation of the manuscript.
FIG. 6. Sequencing by solid phase Edman degradation. A, thecorrected and repetitive yields for the Edman degradation of MUC1-derived PAP20–1 to PAP20–6 in pmol. The corrected yield for eachamino acid is shown as a gray bar and the yield of each glycosylatedamino acid is shown as a black bar. The accurate yields for eachglycoamino acid are as follows: Ser5-GalNAc, 53 pmol; Thr6-GalNAc, 40pmol; Thr14-GalNAc, 42 pmol; Ser14-GalNAc, 19 pmol; Thr19-GalNAc,10 pmol. The minor discrepancies in the picomoles given in the legendand those in the figure come about by the method of graphical depictionof the data on the logarithmic scale. B, HPLC profiles for the gly-coamino acids, PTH-Ser5-GalNAc and PTH-Thr6-GalNAc from the
MUC1 peptides PAP20–1 to PAP20–6. Both glycoamino acids elute asdiastereomers. Representative chromatograms from the sequencer witha set of standards (below) are shown. Using the HPJoooG sequencerresults in an overlap of retention times of the glycoamino acids withsome of the amino acids, but the distinctive diastereomers present inboth Ser and Thr substituted PTH-glycoamino acids makes the assign-ments obvious. C, the expanded HPLC profile for Thr19 and Asp18
showing the diastereomers of PTH-Thr19 GalNAc over the backgroundin the previous cycle. The unsubstituted PTH-Thr is also resolved andused in the quantitation.
In Vivo O-Glycosylation on MUC1 Tandem Repeats24792
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

REFERENCES
1. Bause, E. (1983) Biochem. J. 209, 331–3362. O’Connell, B. C., Tabak, L. A., and Ramusubbu, N. (1991) Biochem. Biophys.
Res. Commun. 180, 1024–10303. Wang, Y., Abernethy, J. L., Eckhardt, A. E., and Hill, R. L. (1992) J. Biol.
Chem. 267, 12709–127164. Wang, Y., Agrwal, N., Eckhardt, A. E., Stevens, R. D., and Hill, R. L. (1993)
J. Biol. Chem. 268, 22979–229835. Pisano, A., Redmond, J. W., Williams, K. L., and Gooley, A. A. (1993)
Glycobiology 3, 429–4356. Pisano, A., Packer, N. H., Redmond, J. W., Williams, K. L., and Gooley, A. A.
(1995) Glycobiology 4, 837–8447. Nehrke, K., Hagen, F. K., Tabak, L. A. (1996) J. Biol. Chem. 271, 7061–70658. Gooley, A. A., and Williams, K. L. (1994) Glycobiology 4, 413–4179. Clausen, H., and Bennett, E. P. (1996) Glycobiology 6, 635–646
10. Spencer, D. I. R., Price, M. R., Tendler, S. J. B., DeMatteis, C. I., Stadie, T., andHanisch, F.-G. (1996) Cancer Lett. 100, 11–15
11. Burchell, J., and Taylor-Papadimitriou, J. (1993) Epithelial Cell Biol. 2,155–162
12. Nishimori, I., Johnson, N. R., Sanderson, S. D., Perini, F., Mountjoy, K., Cerny,R. L., Gross, M. L., and Hollingsworth, M. A. (1994) J. Biol. Chem. 269,16123–16130
13. Nishimori, I., Perini, F., Mountjoy, K., Sanderson, S. D., Johnson, N., Cernay,R. L., Gross, M. L., Fontenot, J. D., and Hollingsworth, M. A. (1994) CancerRes. 54, 3738–3744
14. Stadie, T. R. E., Chai, W., Lawson, A. M., Byfield, P. G. H., and Hanisch, F.-G.(1995) Eur. J. Biochem. 229, 140–147
15. Goletz, S., Thiede, B., Hanisch, F.-G., Peter-Katalinic, J., Schultz, M., Muller,S., Seitz, O., and Karsten, U. (1997) Glycobiology, in press
16. Sojar, H. T., and Bahl, O. M. P. (1987) Arch. Biochem. Biophys. 259, 52–5717. Gerken, T. A., Gupta, R., and Jentoft, N. (1992) Biochemistry 31, 639–64818. Aitken, A., Geisow, M. J., Findley, J. B. C., Holmes, C., and Yarwood, A. (1989)
Protein Sequencing: A Practical Approach (Findley, J. B. C., and Geisolo, M.J., eds) pp. 43–68, IRL Press at Oxford University Press, Oxford
19. Hanisch, F.-G., Uhlenbruck, G., Peter-Katalinic, J., Egge, H., Dabrowski, J.,and Dabrowski, U. (1989) J. Biol. Chem. 264, 872–883
20. Shimidzu, M., and Yamauchi, K. (1982) J. Biochem. (Tokyo) 91, 515–52421. Monsigny, M., Petit, C., and Roche, A.-C. (1988) Anal. Biochem. 175, 525–53022. Bhavanandan, V. P., and Sheykhnazari, M. (1993) Anal. Biochem. 213,
438–44023. Towbin, H., Staehelin, T., and Gordon, T. (1979) Proc. Natl. Acad. Sci. U. S. A.
76, 4350–435424. O‘Shannassy, D. J., Voorstad, P. L., and Quarles, R. H. (1987) Anal. Biochem.
163, 204–20925. Merkle, R. K., and Poppe, I. (1994) Methods Enzymol. 230, 1–1526. Chaplin, M. F., and Kennedy, J. F. (1986) Carbohydrate Analysis: A Practical
Approach, IRL Press at Oxford University Press, Oxford27. Hanisch, F.-G., Stadie, T., and Boblet, K. (1995) Cancer Res. 55, 4036–404028. Pisano, A., Jadine, D. R., Packer, N. H., Redmond, J. W., Williams, K. L.,
Gooley, A. A., Farnsworth, V., Carson, W., and Cartier, P. K. (1997) inTechniques in Glycobiology (Townsend, R., and Hotchkiss, A., eds) pp.299–320, Marcel Dekker, Inc., New York
29. Hanisch, F.-G., Uhlenbruck, G., Dienst, C., Stottrop, M., and Hippauf, E.(1985) Eur. J. Biochem 149, 323–330
30. Brockhausen, I., Toki, D., Brockhausen, J., Peters, S., Bielfeldt, T., Kleen, A.,Paulsen, H., Meldal, M., Hagen, F., and Tabak, L. A. (1996) Glycoconj. J. 13,849–856
31. Taylor-Papadimitriou, J., and Epenetos, A. A. (1994) Trends Biotechnol. 12,227–233
32. Jerome, K. R., Barnd, D. L., Bendt, K. M., Boyer, C. M., Taylor-Papadimitriou,J., McKenzie, I. F. C., Bast, R. C., and Finn, O. J. (1991) Cancer Res. 51,2908–2916
33. Kaufmann, R., Kirsch, D., and Spengler, B. (1994) Int. J. Mass Spectrom. IonProcesses 131, 355–385
34. Biemann, K. (1988) Biomed. Environ. Mass Spectrom. 16, 99–11135. Roepstorff, P., and Fohlmann, J. (1984) Biomed. Mass Spectrom. 11, 60136. Hansen, J. E., Lund, O., Engelbrecht, J., Bohr, H., Nielsen, J. O., Hansen,
J.-E. S., and Brunak, S. (1995) Biochem. J. 308, 801–81337. Gerken, T. A., Owens, C. L., and Pasumarthy, M. (1997) J. Biol. Chem. 272,
9709–971938. Hanisch, F.-G., Peter-Katalinic, J., Egge, H., Dabrowski, U., and Uhlenbruck,
G. (1990) Glycoconj. J. 7, 525–543
In Vivo O-Glycosylation on MUC1 Tandem Repeats 24793
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from

Franz-Georg HanischStefan Müller, Steffen Goletz, Nicolle Packer, Andrew Gooley, Alexander M. Lawson and
REPEAT ARE GLYCOSYLATION TARGETS IN VIVO Lactation-associated MUC1: ALL PUTATIVE SITES WITHIN THE TANDEM
-Glycosylation Sites on Glycopeptide Fragments fromOLocalization of
doi: 10.1074/jbc.272.40.247801997, 272:24780-24793.J. Biol. Chem.
http://www.jbc.org/content/272/40/24780Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/272/40/24780.full.html#ref-list-1
This article cites 34 references, 12 of which can be accessed free at
by guest on February 18, 2018http://w
ww
.jbc.org/D
ownloaded from





























