Embed Size (px)
DESCRIPTION
Study guide for genetics disorders
Citation preview

Disease Name
Hemophilia (classic)
Ornithine Transcarbamylase Deficiency
Fragile X
Glucose Phosphate Dehydrogenase Deficiency
(Favism)
Lesch Nyhan Syndrome
Diseases - X linked
Duchenne Muscular Dystrophy
Androgen Insensitivity Syndrome
Rett Syndrome
MELAS
Diseases - X linked
Diseases - Mitochondrial

MERRF
Cystic Fibrosis
Sickle Cell Anemia
Beta-Thalassemia
Alpha-Thalassemia
Diseases - Mitochondrial
Diseases - Autosomal recessive with
heterozygote advantage

Phenylketonuria
Ornithine Transcarbamylase Deficiency
Tay Sachs Disease
Familial Hypercholesterolemia
Diseases - Inborn errors of metabolism

Marfan Disease
Ehlers-Danlos
Diseases - Autosomal dominant
(negative)

Osteogenesis Imperfecta
Huntington's Disease
Achondroplasia
Diseases - Autosomal dominant
(negative)
Familial Hypercholesterolemia
Limb-Girdle Muscular Dystrophy
Praeder Willi
Angelman Syndrome
Diseases - Examples of imprinting
Diseases - Autosomal dominant
(negative)

Patau Syndrome
Edwards Syndrome
Down Syndrome
Diseases - Chromosomal

Turner Syndrome
Triple X Syndrome
Kleinfelter's Syndrome
XYY
Diseases - Chromosomal
DiGeorge Syndrome
Alzheimer's Disease
Autism
Gastroschisis
Diseases - Chromosomal
Diseases - Complex genetic
Diseases - Errors in development

Treacher Collins Syndrome
Diaphragmatic Hernia
Pulmonary Hypoplasia
Diseases - Errors in development
VACTERL
Esophageal Atresia
Diseases - Errors in development

Tracheal-esophageal Fistula
Polydactyly
Holoprosencephaly
Diseases - Errors in development

Exencephaly/anencephaly and Spina Bifida
Diseases - Errors in development

Diseases - Errors in development
ASD (Atrial Septal Defect)

VSD (Ventricular Septal Defect)
Diseases - Errors in development
ASD (Atrial Septal Defect)

Persistent Atrioventricular Canal
Persistent Truncus Arteriosus
Transposition of the Great Vessels
Diseases - Errors in development

Tetralogy of Fallot
Syndactyly
Diseases - Errors in development

Synostosis
Hand Foot Genital Syndrome
DiGeorge Syndrome
Diseases - Errors in development
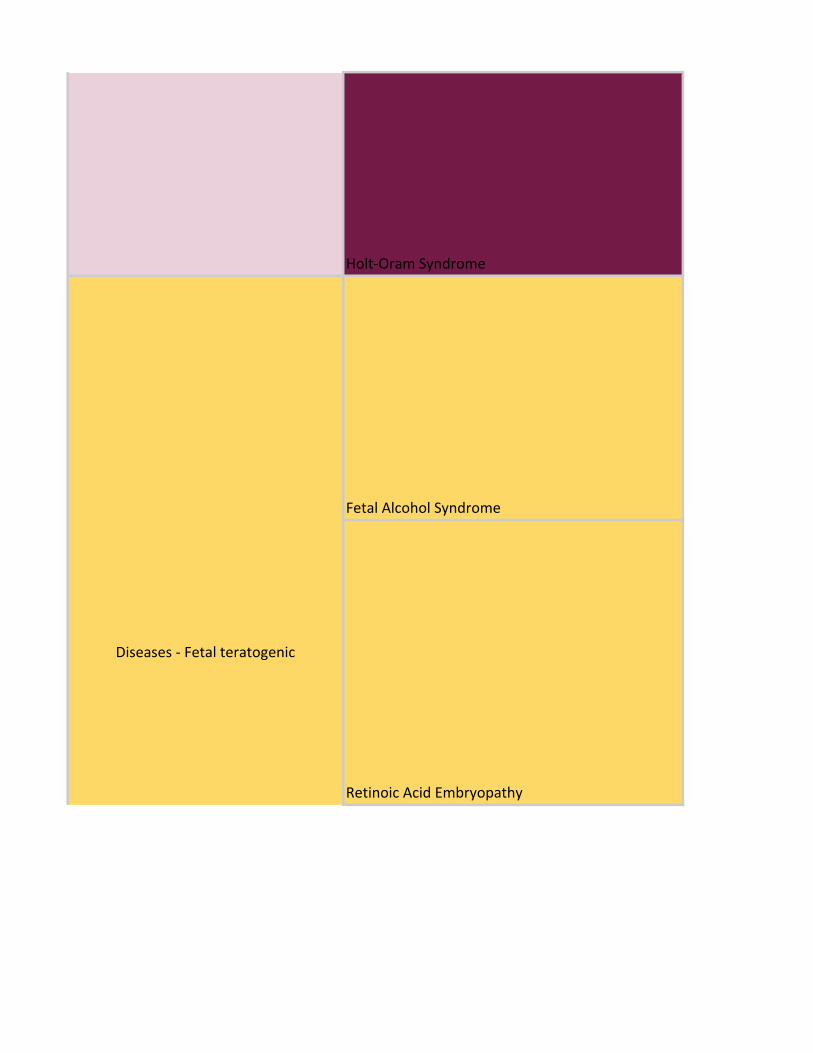
Holt-Oram Syndrome
Fetal Alcohol Syndrome
Retinoic Acid Embryopathy
Diseases - Fetal teratogenic
Diseases - Errors in development
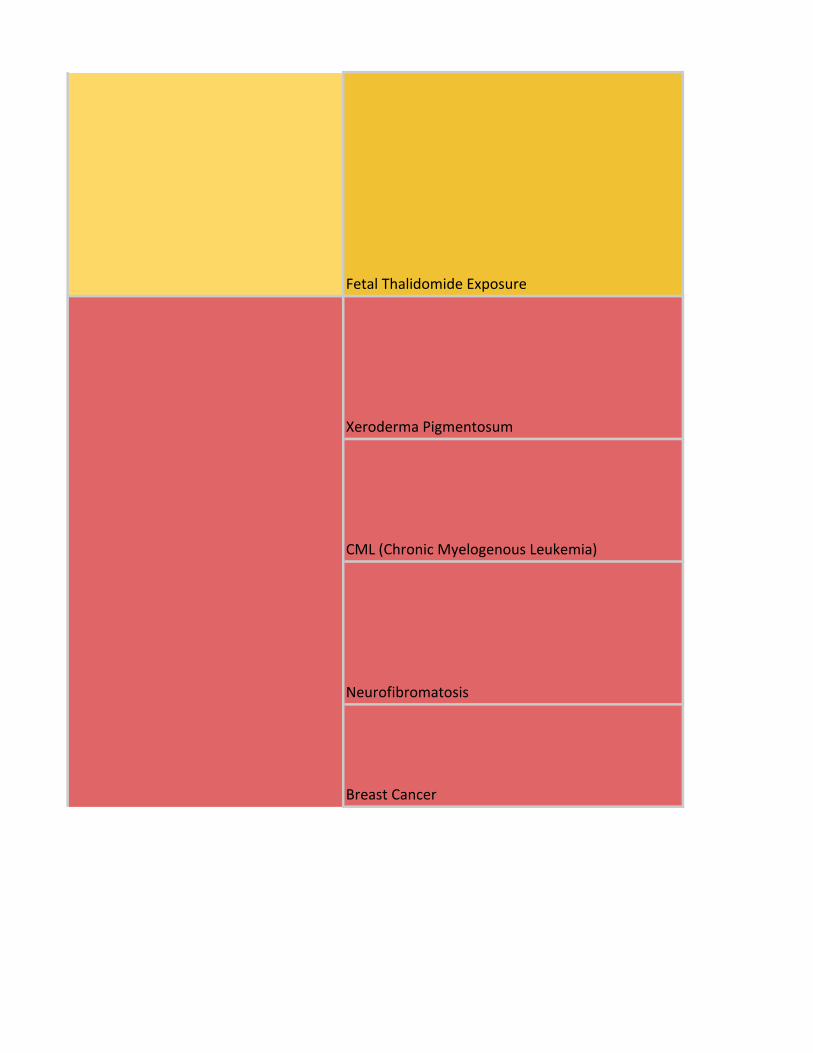
Fetal Thalidomide Exposure
Xeroderma Pigmentosum
CML (Chronic Myelogenous Leukemia)
Neurofibromatosis
Breast Cancer
Diseases - Fetal teratogenic
Diseases - Cancer related mutations

HNPCC (Hereditary Non-Polyposis Colorectal
Cancer)
FAP (Familial Adenomatous Polyposis)
Retinoblastoma
Li-Fraumeni
Disease Name
Wolf-Hirschhorn Syndrome
Cri du chat Syndrome
Miller-Dieker syndrome
WAGR syndrome
Williams syndrome
Diseases - Cancer related mutations

Ataxia-telangiectasia
Fanconi anemia
Bloom syndrome
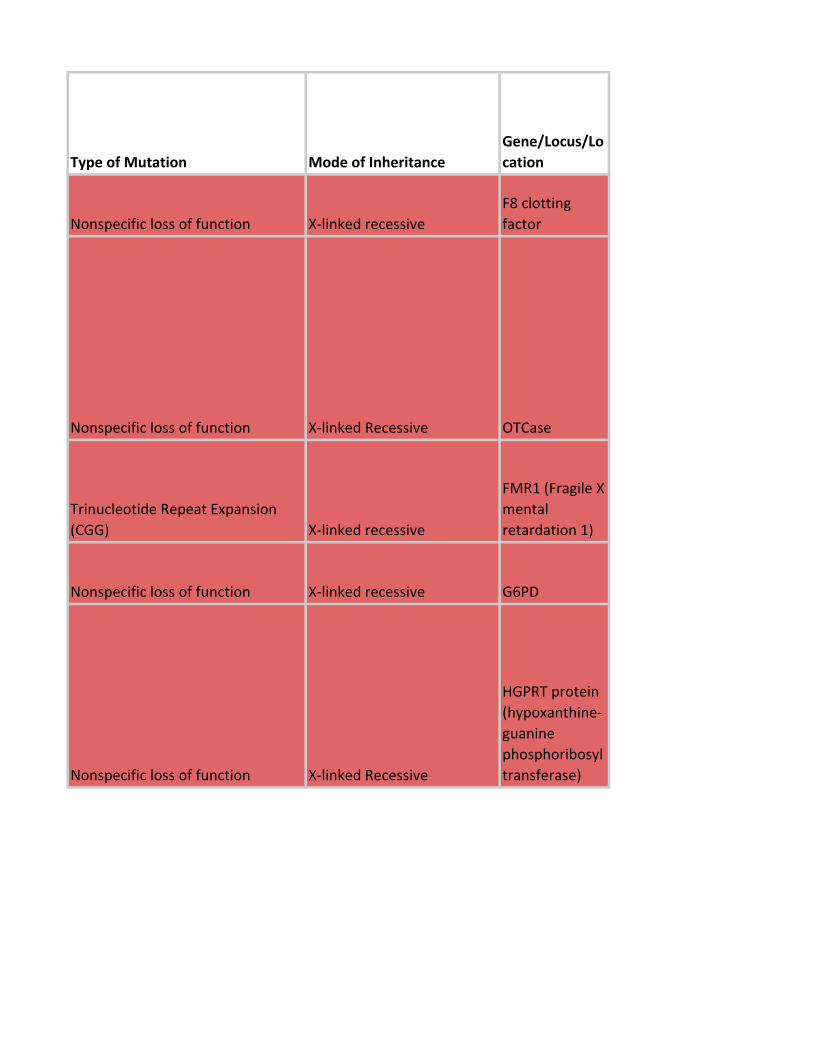
Type of Mutation Mode of Inheritance
Gene/Locus/Lo
cation
Nonspecific loss of function X-linked recessive
F8 clotting
factor
Nonspecific loss of function X-linked Recessive OTCase
Trinucleotide Repeat Expansion
(CGG) X-linked recessive
FMR1 (Fragile X
mental
retardation 1)
Nonspecific loss of function X-linked recessive G6PD
Nonspecific loss of function X-linked Recessive
HGPRT protein
(hypoxanthine-
guanine
phosphoribosyl
transferase)
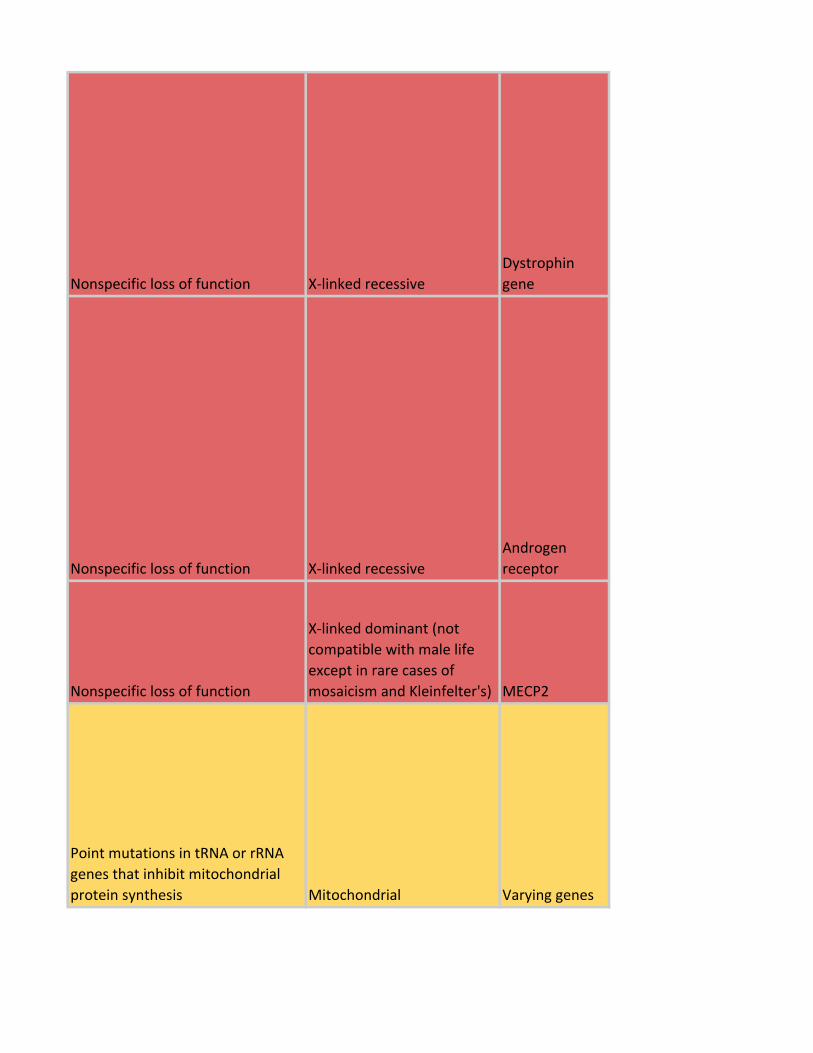
Nonspecific loss of function X-linked recessive
Dystrophin
gene
Nonspecific loss of function X-linked recessive
Androgen
receptor
Nonspecific loss of function
X-linked dominant (not
compatible with male life
except in rare cases of
mosaicism and Kleinfelter's) MECP2
Point mutations in tRNA or rRNA
genes that inhibit mitochondrial
protein synthesis Mitochondrial Varying genes
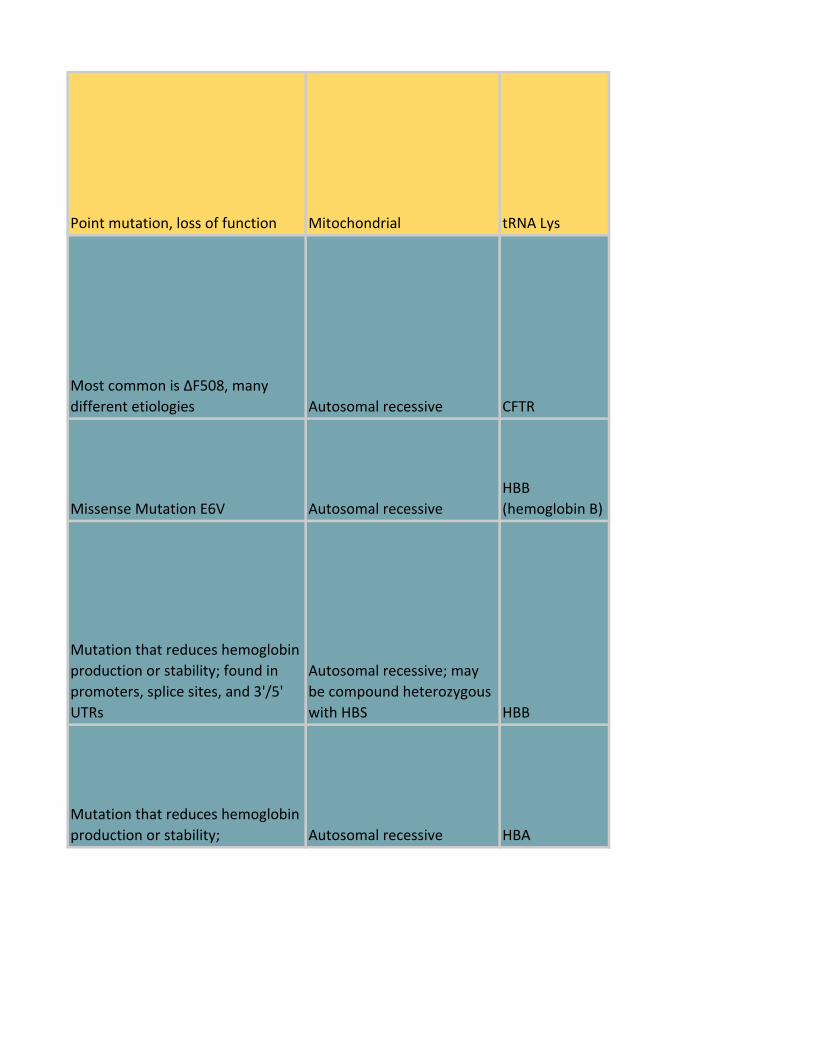
Point mutation, loss of function Mitochondrial tRNA Lys
Most common is ΔF508, many
different etiologies Autosomal recessive CFTR
Missense Mutation E6V Autosomal recessive
HBB
(hemoglobin B)
Mutation that reduces hemoglobin
production or stability; found in
promoters, splice sites, and 3'/5'
UTRs
Autosomal recessive; may
be compound heterozygous
with HBS HBB
Mutation that reduces hemoglobin
production or stability; Autosomal recessive HBA
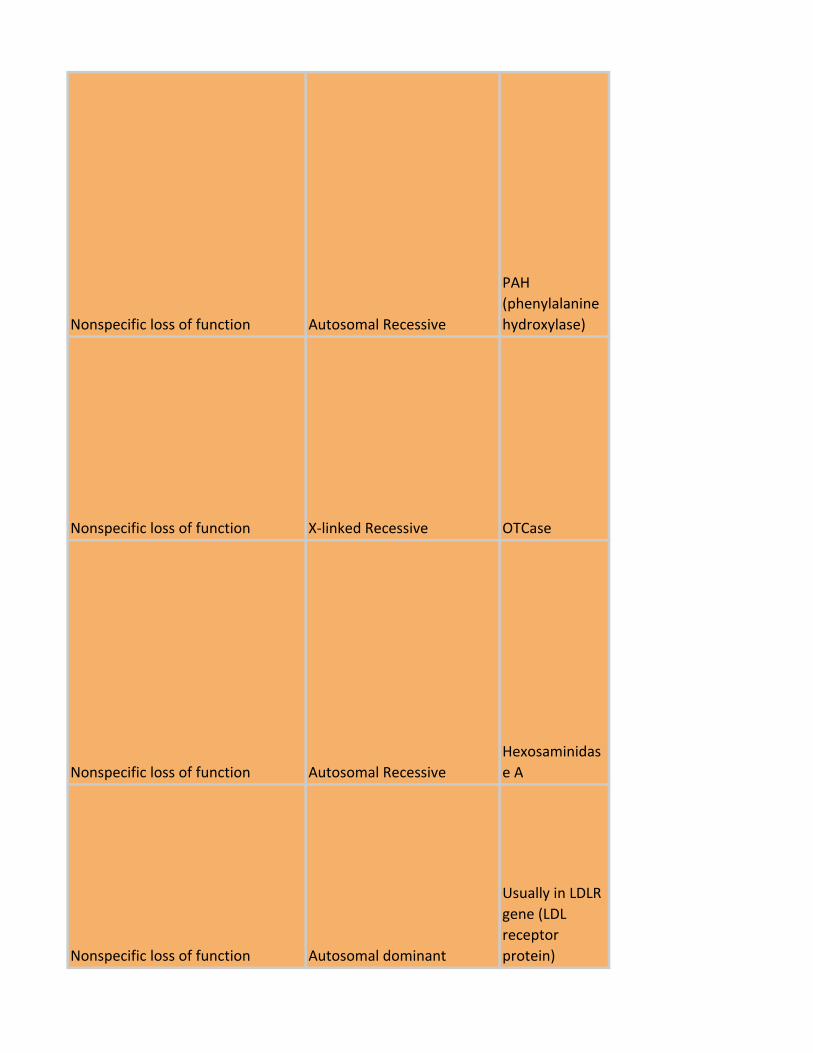
Nonspecific loss of function Autosomal Recessive
PAH
(phenylalanine
hydroxylase)
Nonspecific loss of function X-linked Recessive OTCase
Nonspecific loss of function Autosomal Recessive
Hexosaminidas
e A
Nonspecific loss of function Autosomal dominant
Usually in LDLR
gene (LDL
receptor
protein)

Loss of function that does not
impede expression
Autosomal Dominant
Negative
Fibrillin protein
FBN1
Loss of function that does not
impede expression
Autosomal Dominant
Negative
Collagen
proteins
(COL;1/3/5;A1/
2
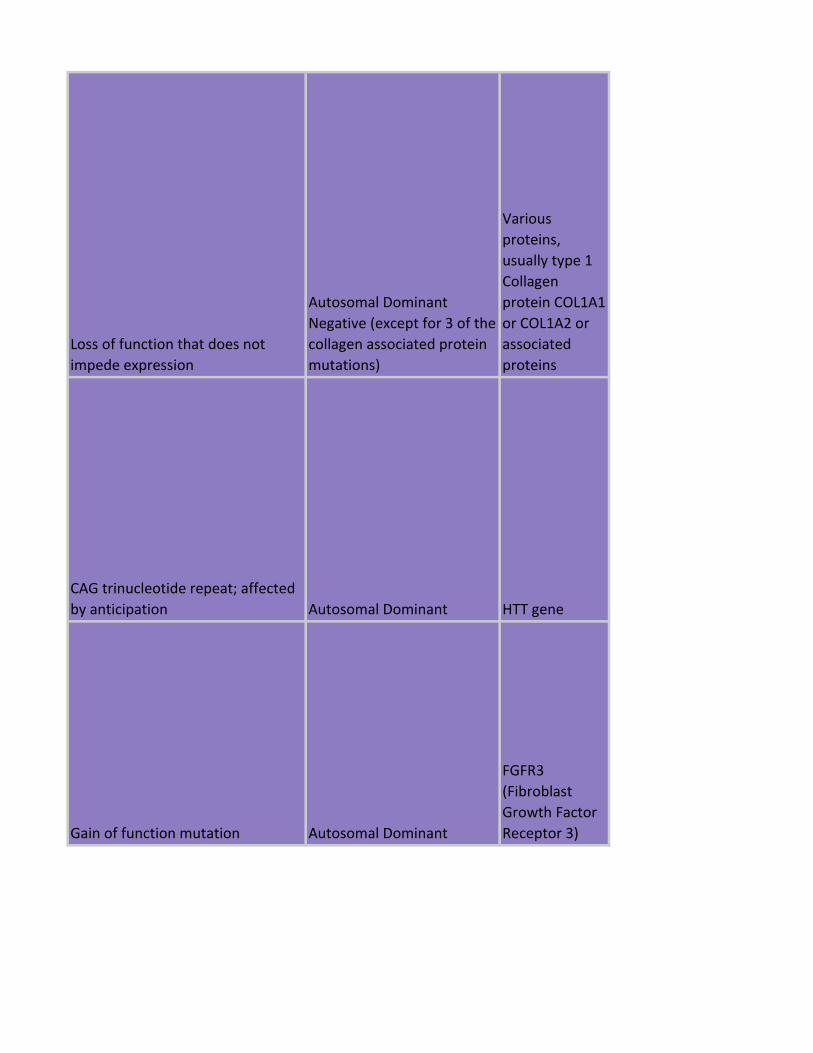
Loss of function that does not
impede expression
Autosomal Dominant
Negative (except for 3 of the
collagen associated protein
mutations)
Various
proteins,
usually type 1
Collagen
protein COL1A1
or COL1A2 or
associated
proteins
CAG trinucleotide repeat; affected
by anticipation Autosomal Dominant HTT gene
Gain of function mutation Autosomal Dominant
FGFR3
(Fibroblast
Growth Factor
Receptor 3)

Nonspecific loss of function Autosomal dominant
Usually in LDLR
gene (LDL
receptor
protein)
Activation of a cryptic splice donor
site resulting in frame shift
Autosomal dominant or
recessive Calpain 3 gene
Deletion of paternal PWS segment
of chromosome 15 or maternal
uniparental disomy
Maternally imprinted;
Paternally
inherited/expressed
More than one
gene is
expressed in
this region;
chromosomal
abnormality
Deletion of maternal AS segment
of chromosome 15 or paternal
uniparental disomy
Paternally imprinted;
Maternally
inherited/expressed
More than one
gene is
expressed in
this region;
chromosomal
abnormality
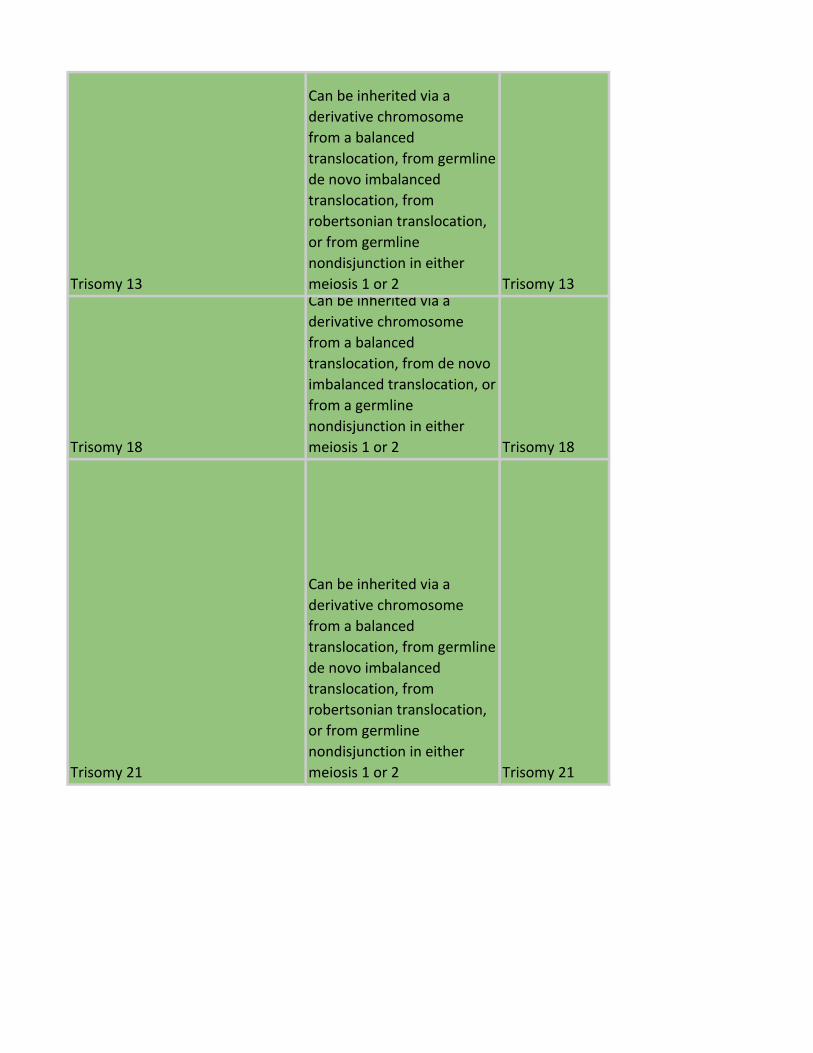
Trisomy 13
Can be inherited via a
derivative chromosome
from a balanced
translocation, from germline
de novo imbalanced
translocation, from
robertsonian translocation,
or from germline
nondisjunction in either
meiosis 1 or 2 Trisomy 13
Trisomy 18
Can be inherited via a
derivative chromosome
from a balanced
translocation, from de novo
imbalanced translocation, or
from a germline
nondisjunction in either
meiosis 1 or 2 Trisomy 18
Trisomy 21
Can be inherited via a
derivative chromosome
from a balanced
translocation, from germline
de novo imbalanced
translocation, from
robertsonian translocation,
or from germline
nondisjunction in either
meiosis 1 or 2 Trisomy 21
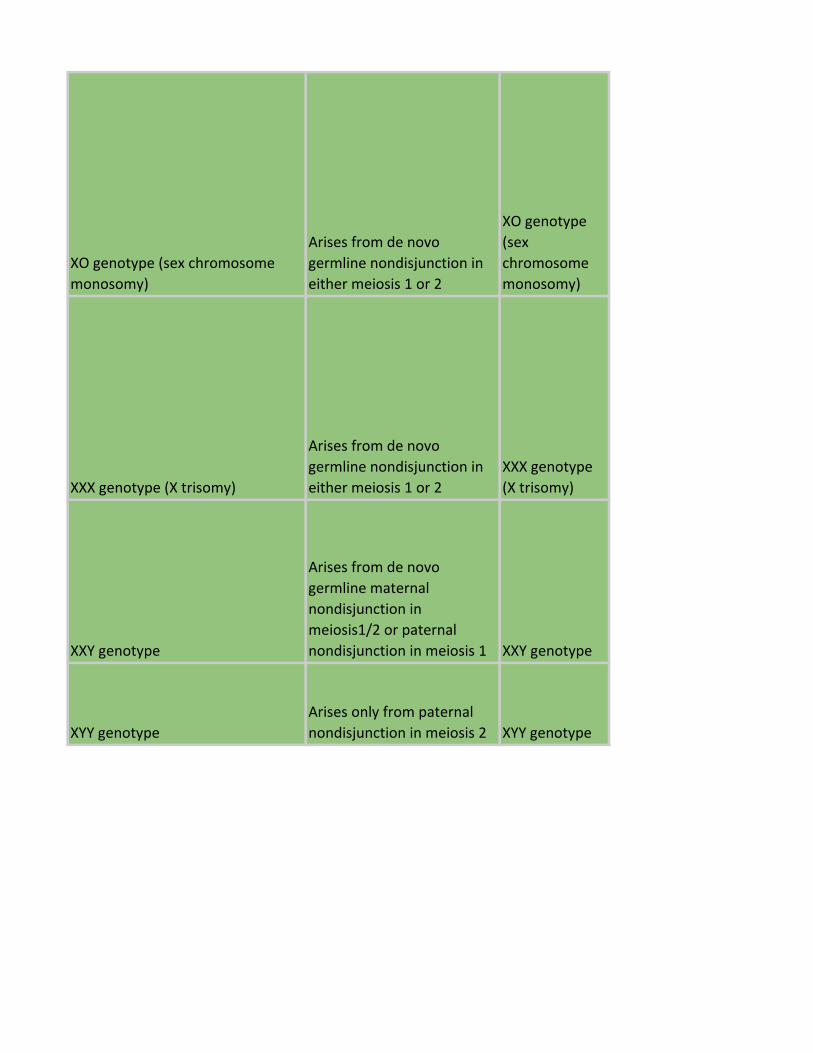
XO genotype (sex chromosome
monosomy)
Arises from de novo
germline nondisjunction in
either meiosis 1 or 2
XO genotype
(sex
chromosome
monosomy)
XXX genotype (X trisomy)
Arises from de novo
germline nondisjunction in
either meiosis 1 or 2
XXX genotype
(X trisomy)
XXY genotype
Arises from de novo
germline maternal
nondisjunction in
meiosis1/2 or paternal
nondisjunction in meiosis 1 XXY genotype
XYY genotype
Arises only from paternal
nondisjunction in meiosis 2 XYY genotype
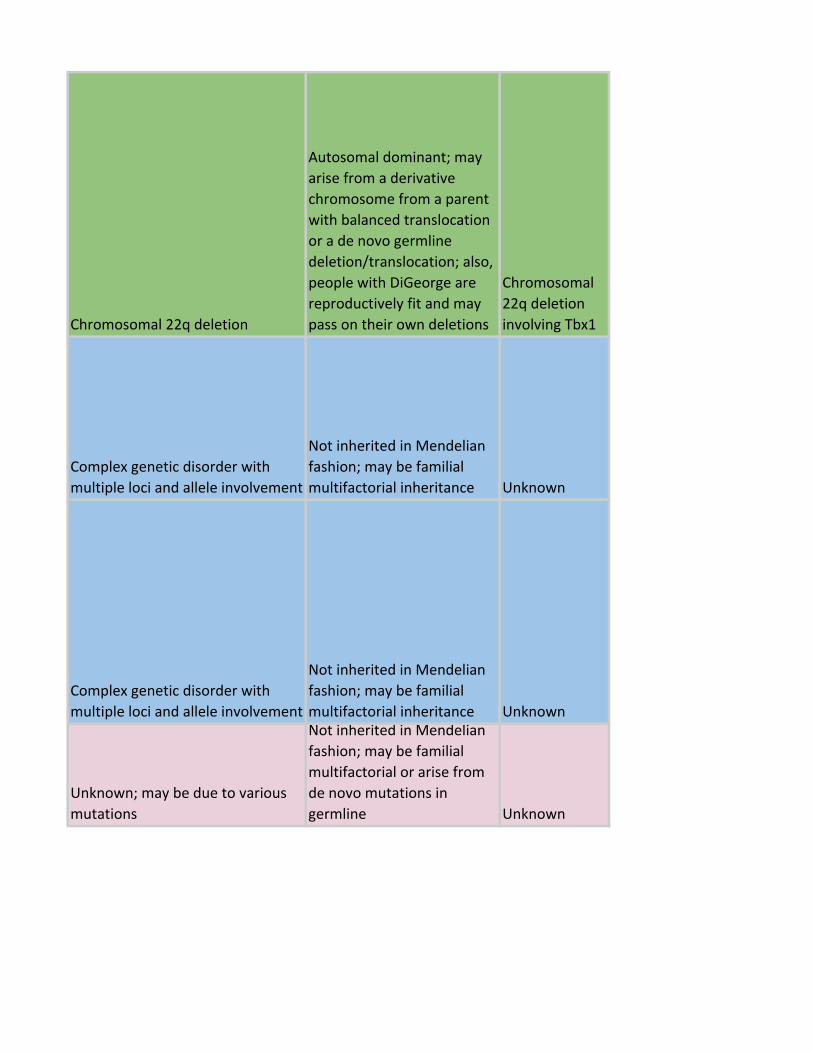
Chromosomal 22q deletion
Autosomal dominant; may
arise from a derivative
chromosome from a parent
with balanced translocation
or a de novo germline
deletion/translocation; also,
people with DiGeorge are
reproductively fit and may
pass on their own deletions
Chromosomal
22q deletion
involving Tbx1
Complex genetic disorder with
multiple loci and allele involvement
Not inherited in Mendelian
fashion; may be familial
multifactorial inheritance Unknown
Complex genetic disorder with
multiple loci and allele involvement
Not inherited in Mendelian
fashion; may be familial
multifactorial inheritance Unknown
Unknown; may be due to various
mutations
Not inherited in Mendelian
fashion; may be familial
multifactorial or arise from
de novo mutations in
germline Unknown
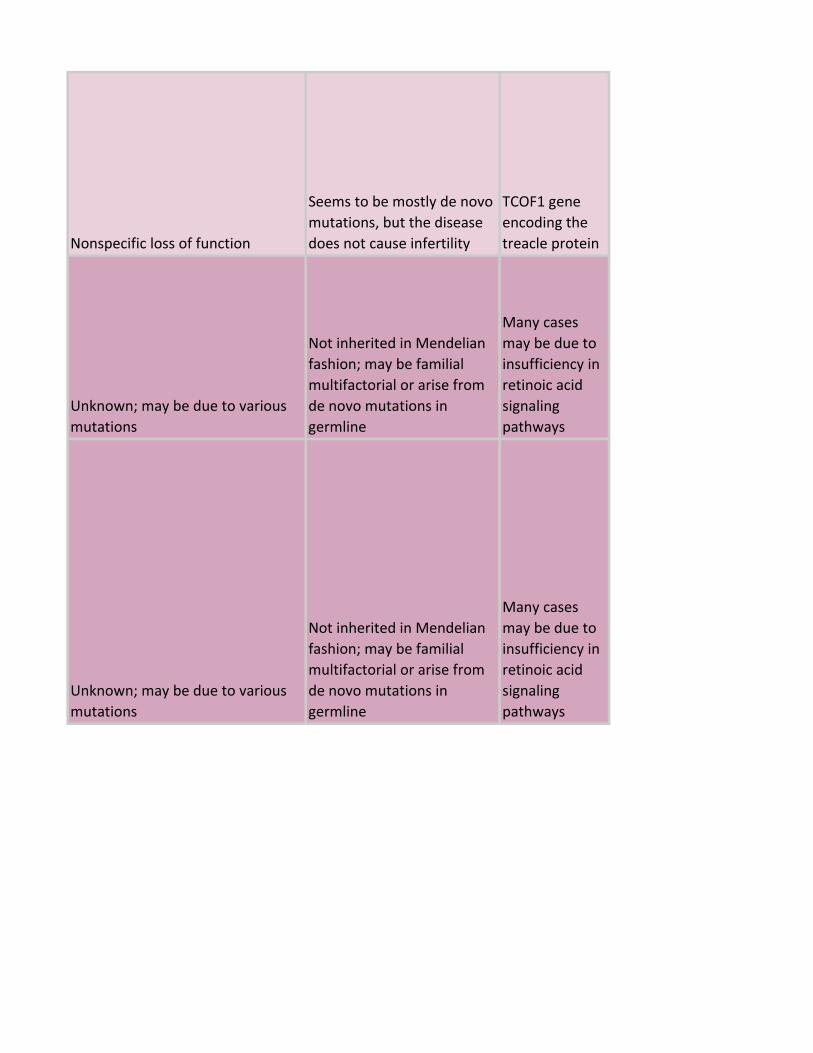
Nonspecific loss of function
Seems to be mostly de novo
mutations, but the disease
does not cause infertility
TCOF1 gene
encoding the
treacle protein
Unknown; may be due to various
mutations
Not inherited in Mendelian
fashion; may be familial
multifactorial or arise from
de novo mutations in
germline
Many cases
may be due to
insufficiency in
retinoic acid
signaling
pathways
Unknown; may be due to various
mutations
Not inherited in Mendelian
fashion; may be familial
multifactorial or arise from
de novo mutations in
germline
Many cases
may be due to
insufficiency in
retinoic acid
signaling
pathways

Various mutations in the Shh
signaling pathway Unknown
Suspected to
be Shh
signaling
pathway
Unknown; may be due to various
mutations
Not inherited in Mendelian
fashion; may be familial
multifactorial or arise from
de novo mutations in
germline
It is one of the
possible effects
of a disrupted
Shh signaling
pathway
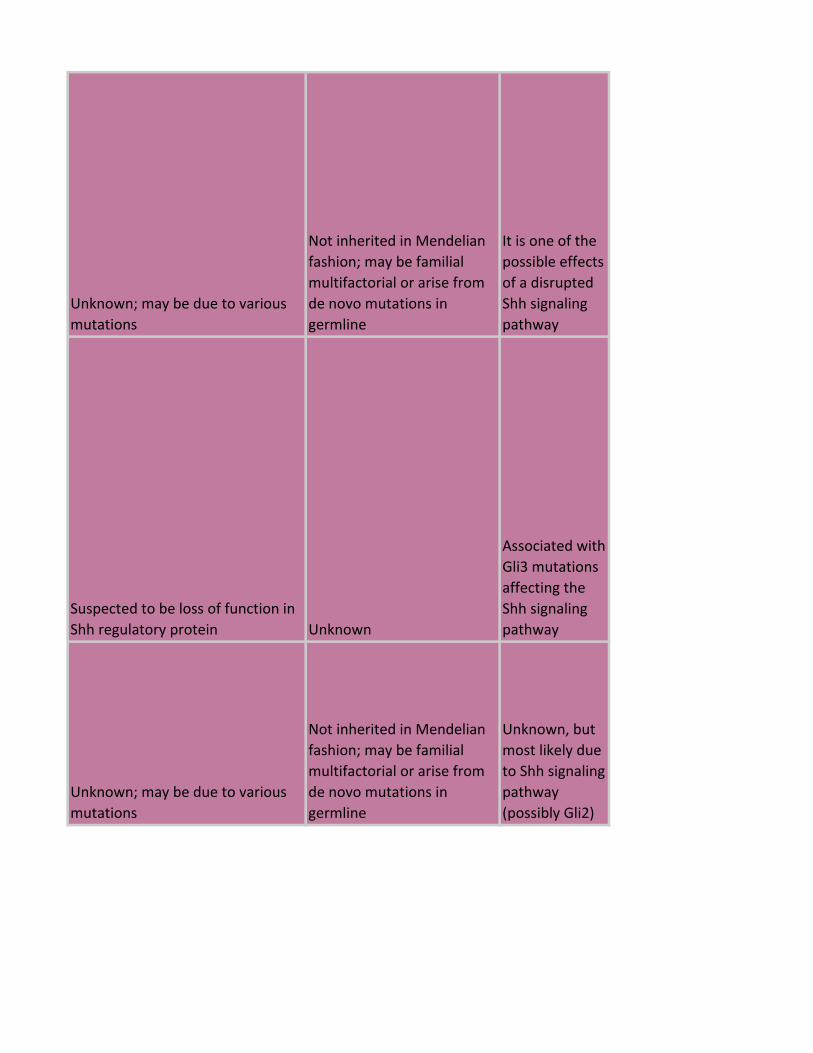
Unknown; may be due to various
mutations
Not inherited in Mendelian
fashion; may be familial
multifactorial or arise from
de novo mutations in
germline
It is one of the
possible effects
of a disrupted
Shh signaling
pathway
Suspected to be loss of function in
Shh regulatory protein Unknown
Associated with
Gli3 mutations
affecting the
Shh signaling
pathway
Unknown; may be due to various
mutations
Not inherited in Mendelian
fashion; may be familial
multifactorial or arise from
de novo mutations in
germline
Unknown, but
most likely due
to Shh signaling
pathway
(possibly Gli2)

Complex genetic disorder with
multiple alleles
Not inherited in Mendelian
fashion; may be familial
multifactorial inheritance
Unknown; it
may be related
to the Shh
pathway
(VACTERL)

Tbx1 and Shh
signaling
pathway
components or
regulators, also
present in
retinoic acid
embryopathies
Some causes are inherited in
an autosomal dominant
manner
Known causes include
chromosomal deletions and
genetic mutations that cause loss
of function
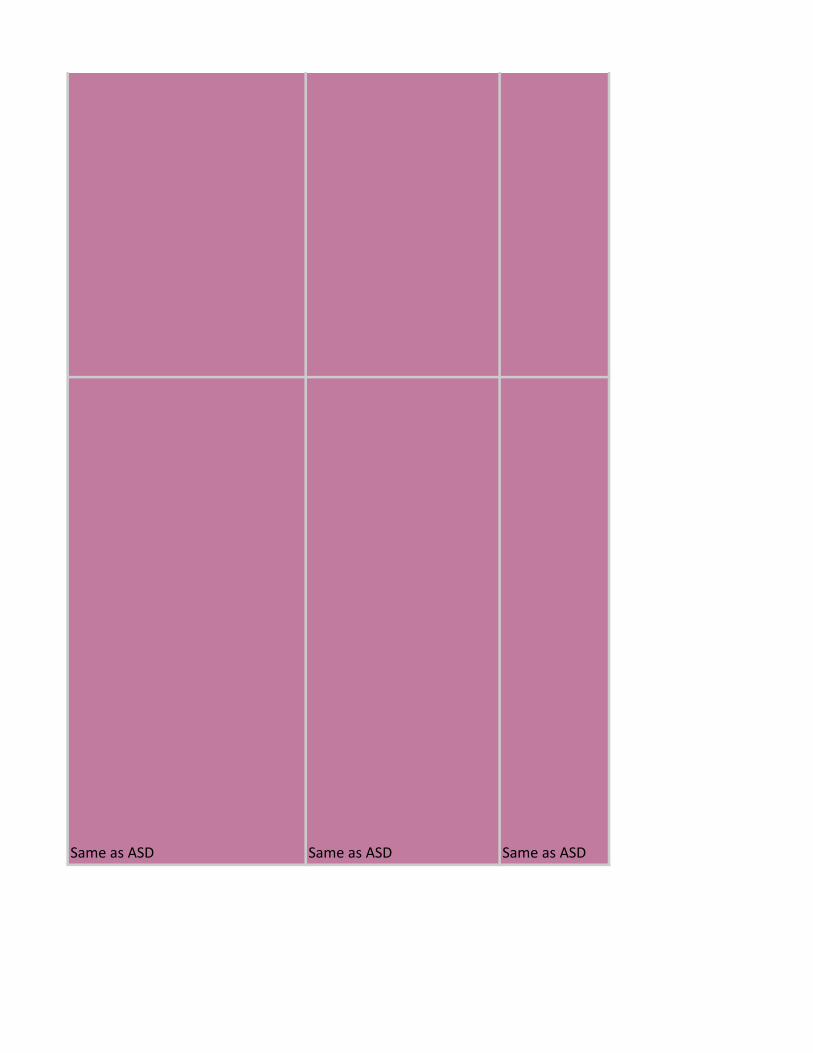
Same as ASD Same as ASD Same as ASD
Tbx1 and Shh
signaling
pathway
components or
regulators, also
present in
retinoic acid
embryopathies
Some causes are inherited in
an autosomal dominant
manner
Known causes include
chromosomal deletions and
genetic mutations that cause loss
of function
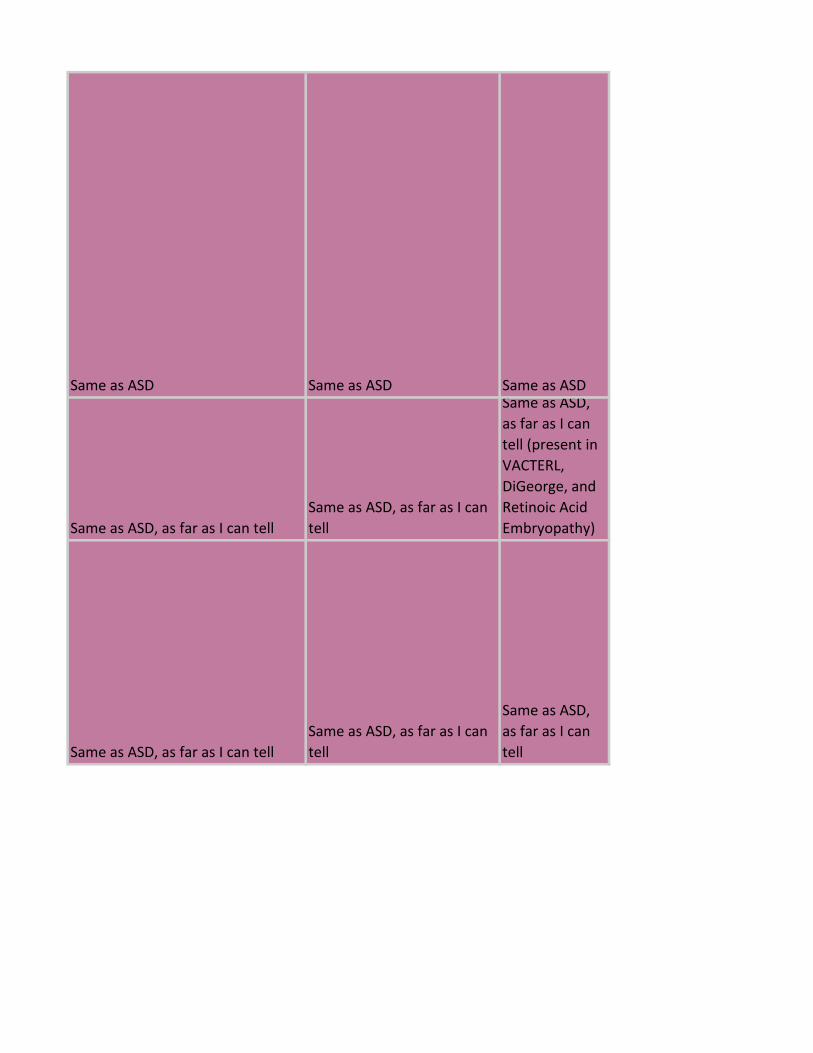
Same as ASD Same as ASD Same as ASD
Same as ASD, as far as I can tell
Same as ASD, as far as I can
tell
Same as ASD,
as far as I can
tell (present in
VACTERL,
DiGeorge, and
Retinoic Acid
Embryopathy)
Same as ASD, as far as I can tell
Same as ASD, as far as I can
tell
Same as ASD,
as far as I can
tell

Trisomy 21, DiGeorge deletion,
mutations related to Shh Varies
Trisomy 21,
Tbx1 deletion,
mutations
related to Shh
May be due to various kinds of
mutations, both loss of function
and gain of function
Autosomal recessive or
dominant
Associated with
gain of function
mutations in
FGFReceptors;
disruptions in
Hoxd13
patterning; and
mutations in
BMP
antagonists
(e.g. Noggin)
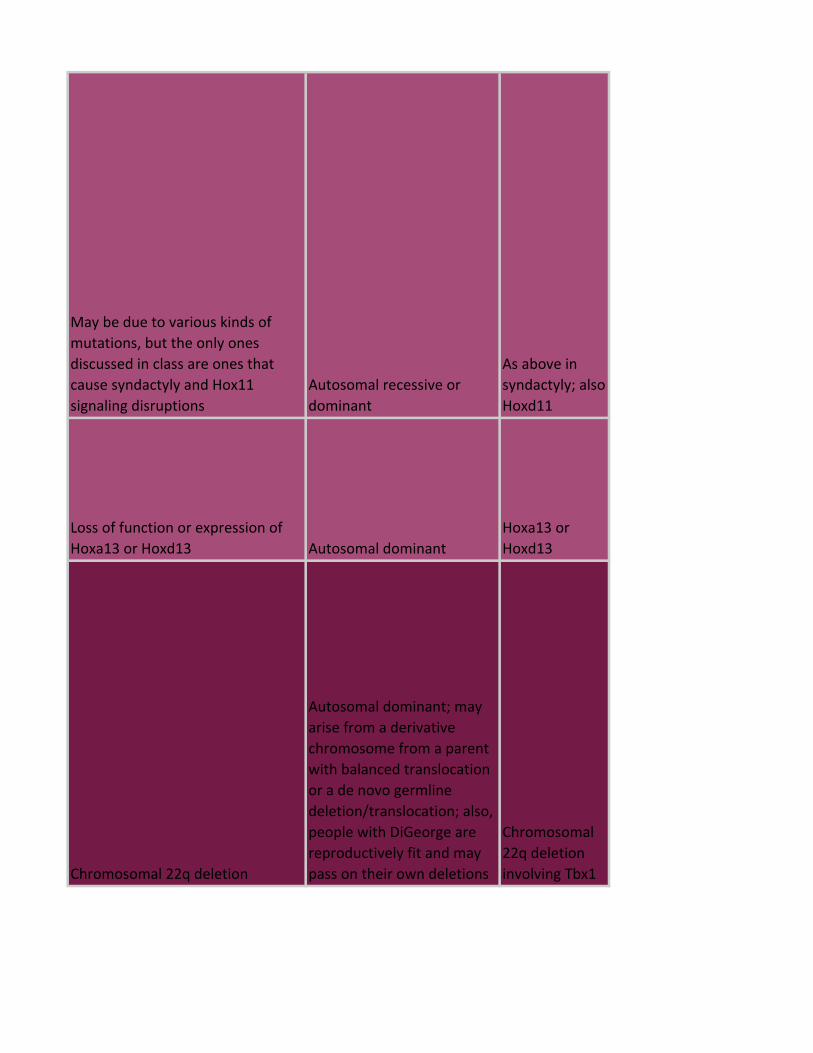
May be due to various kinds of
mutations, but the only ones
discussed in class are ones that
cause syndactyly and Hox11
signaling disruptions
Autosomal recessive or
dominant
As above in
syndactyly; also
Hoxd11
Loss of function or expression of
Hoxa13 or Hoxd13 Autosomal dominant
Hoxa13 or
Hoxd13
Chromosomal 22q deletion
Autosomal dominant; may
arise from a derivative
chromosome from a parent
with balanced translocation
or a de novo germline
deletion/translocation; also,
people with DiGeorge are
reproductively fit and may
pass on their own deletions
Chromosomal
22q deletion
involving Tbx1

Nonspecific loss of function Autosomal dominant Tbx5
Not genetic; caused by etoh
consumption in pregnancy
Not genetic; caused by etoh
consumption in pregnancy
Causes
downregulation
of Shh and
retinoic acid
Not genetic; caused by excess
retinoid intake during pregnancy,
notably from acutane
Not genetic; caused by
excess retinoid intake during
pregnancy, notably from
acutane
Retinoids are
converted into
retinoic acid,
mimicking
upregulation of
RALDH genes
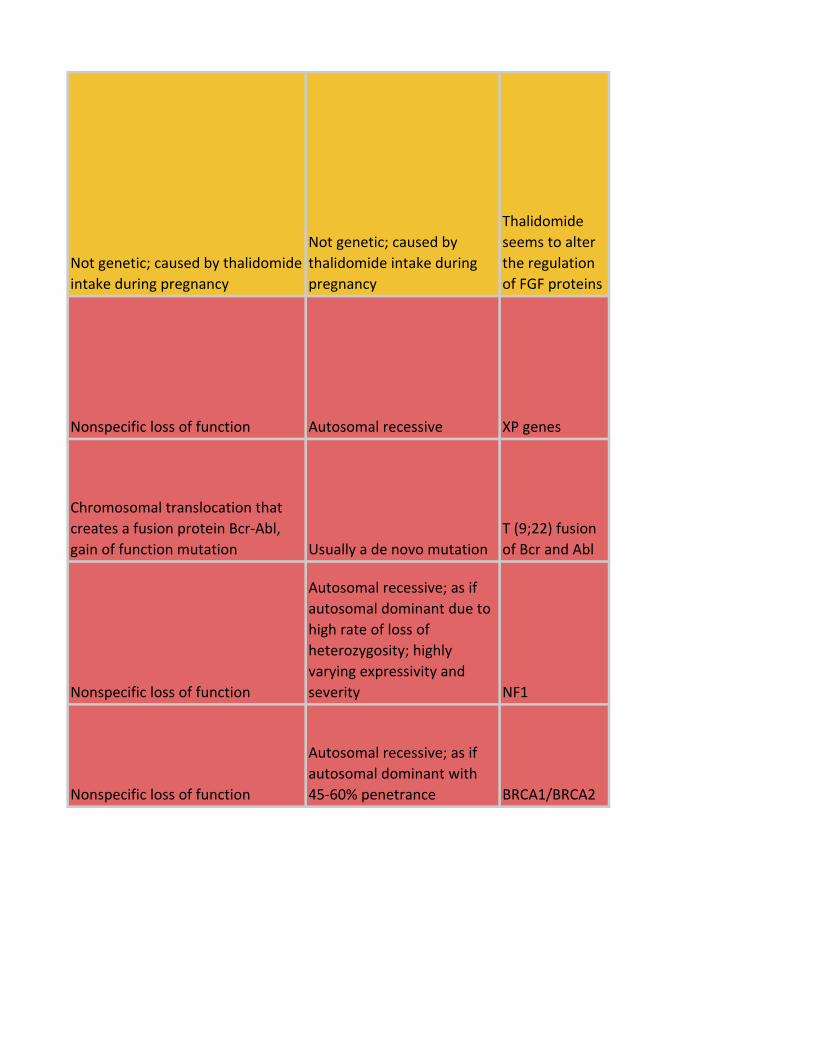
Not genetic; caused by thalidomide
intake during pregnancy
Not genetic; caused by
thalidomide intake during
pregnancy
Thalidomide
seems to alter
the regulation
of FGF proteins
Nonspecific loss of function Autosomal recessive XP genes
Chromosomal translocation that
creates a fusion protein Bcr-Abl,
gain of function mutation Usually a de novo mutation
T (9;22) fusion
of Bcr and Abl
Nonspecific loss of function
Autosomal recessive; as if
autosomal dominant due to
high rate of loss of
heterozygosity; highly
varying expressivity and
severity NF1
Nonspecific loss of function
Autosomal recessive; as if
autosomal dominant with
45-60% penetrance BRCA1/BRCA2
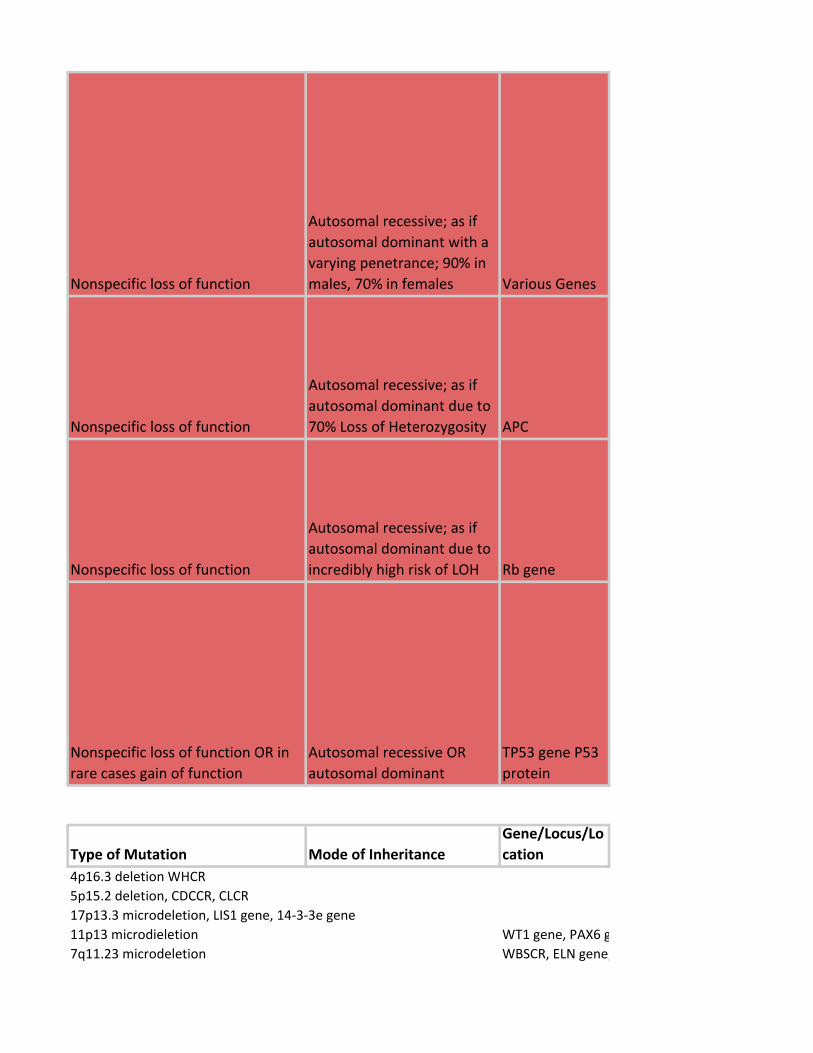
Nonspecific loss of function
Autosomal recessive; as if
autosomal dominant with a
varying penetrance; 90% in
males, 70% in females Various Genes
Nonspecific loss of function
Autosomal recessive; as if
autosomal dominant due to
70% Loss of Heterozygosity APC
Nonspecific loss of function
Autosomal recessive; as if
autosomal dominant due to
incredibly high risk of LOH Rb gene
Nonspecific loss of function OR in
rare cases gain of function
Autosomal recessive OR
autosomal dominant
TP53 gene P53
protein
Type of Mutation Mode of Inheritance
Gene/Locus/Lo
cation
4p16.3 deletion WHCR
5p15.2 deletion, CDCCR, CLCR
17p13.3 microdeletion, LIS1 gene, 14-3-3e gene
11p13 microdieletion WT1 gene, PAX6 gene
7q11.23 microdeletion WBSCR, ELN gene, LIMK1 gene

DNA recomb repair enzymes, 11q22 ATM gene
DNA recomb repair enzymes, 16q24, FA-A gene
DNA repair enzymes, 15q26, BLM gene
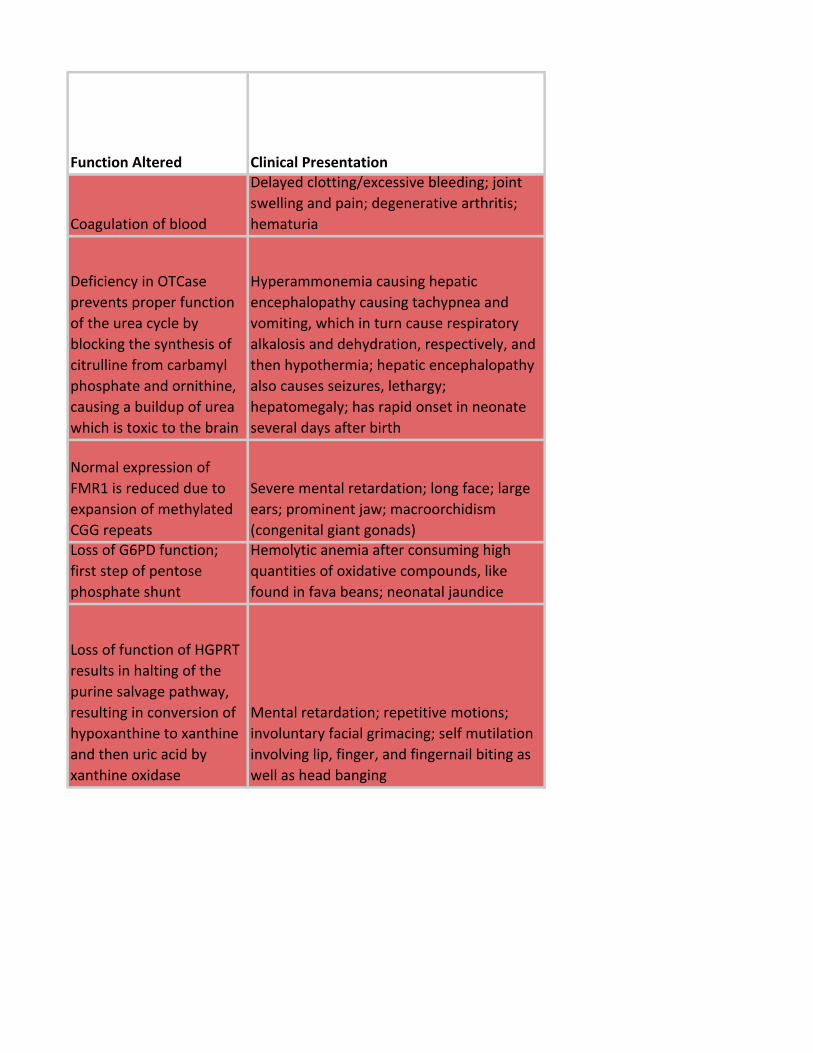
Function Altered Clinical Presentation
Coagulation of blood
Delayed clotting/excessive bleeding; joint
swelling and pain; degenerative arthritis;
hematuria
Deficiency in OTCase
prevents proper function
of the urea cycle by
blocking the synthesis of
citrulline from carbamyl
phosphate and ornithine,
causing a buildup of urea
which is toxic to the brain
Hyperammonemia causing hepatic
encephalopathy causing tachypnea and
vomiting, which in turn cause respiratory
alkalosis and dehydration, respectively, and
then hypothermia; hepatic encephalopathy
also causes seizures, lethargy;
hepatomegaly; has rapid onset in neonate
several days after birth
Normal expression of
FMR1 is reduced due to
expansion of methylated
CGG repeats
Severe mental retardation; long face; large
ears; prominent jaw; macroorchidism
(congenital giant gonads)Loss of G6PD function;
first step of pentose
phosphate shunt
Hemolytic anemia after consuming high
quantities of oxidative compounds, like
found in fava beans; neonatal jaundice
Loss of function of HGPRT
results in halting of the
purine salvage pathway,
resulting in conversion of
hypoxanthine to xanthine
and then uric acid by
xanthine oxidase
Mental retardation; repetitive motions;
involuntary facial grimacing; self mutilation
involving lip, finger, and fingernail biting as
well as head banging
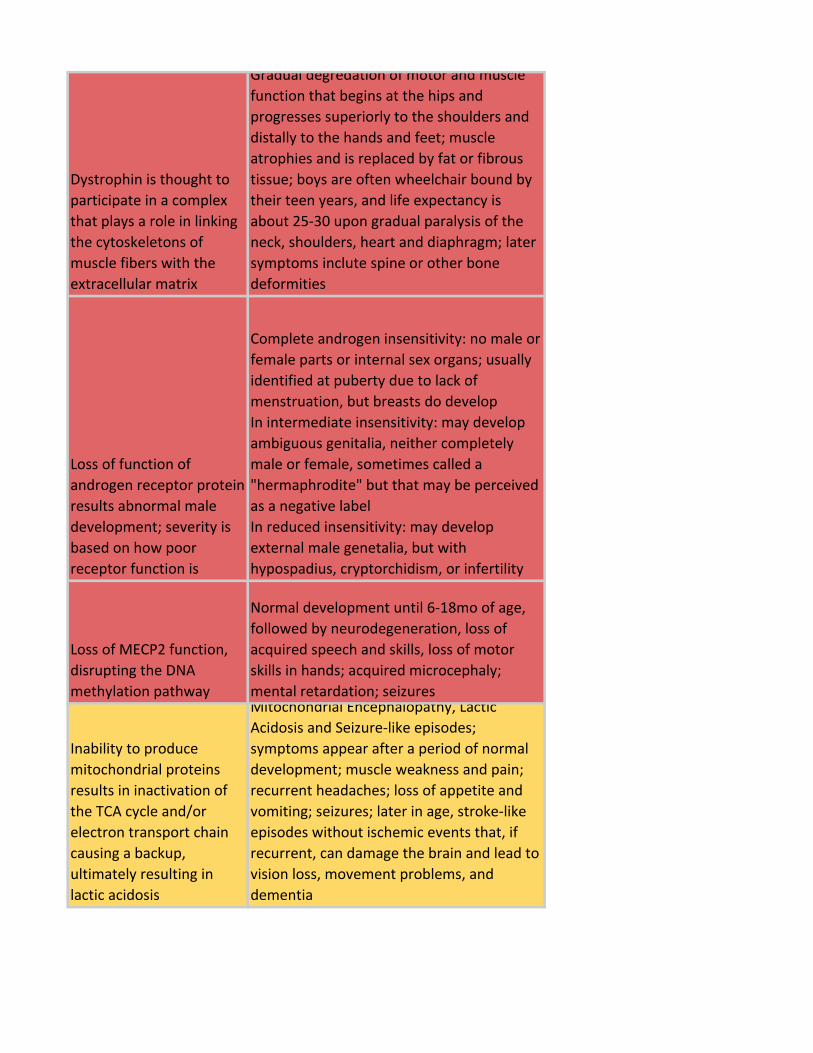
Dystrophin is thought to
participate in a complex
that plays a role in linking
the cytoskeletons of
muscle fibers with the
extracellular matrix
Gradual degredation of motor and muscle
function that begins at the hips and
progresses superiorly to the shoulders and
distally to the hands and feet; muscle
atrophies and is replaced by fat or fibrous
tissue; boys are often wheelchair bound by
their teen years, and life expectancy is
about 25-30 upon gradual paralysis of the
neck, shoulders, heart and diaphragm; later
symptoms inclute spine or other bone
deformities
Loss of function of
androgen receptor protein
results abnormal male
development; severity is
based on how poor
receptor function is
Complete androgen insensitivity: no male or
female parts or internal sex organs; usually
identified at puberty due to lack of
menstruation, but breasts do develop
In intermediate insensitivity: may develop
ambiguous genitalia, neither completely
male or female, sometimes called a
"hermaphrodite" but that may be perceived
as a negative label
In reduced insensitivity: may develop
external male genetalia, but with
hypospadius, cryptorchidism, or infertility
Loss of MECP2 function,
disrupting the DNA
methylation pathway
Normal development until 6-18mo of age,
followed by neurodegeneration, loss of
acquired speech and skills, loss of motor
skills in hands; acquired microcephaly;
mental retardation; seizures
Inability to produce
mitochondrial proteins
results in inactivation of
the TCA cycle and/or
electron transport chain
causing a backup,
ultimately resulting in
lactic acidosis
Mitochondrial Encephalopathy, Lactic
Acidosis and Seizure-like episodes;
symptoms appear after a period of normal
development; muscle weakness and pain;
recurrent headaches; loss of appetite and
vomiting; seizures; later in age, stroke-like
episodes without ischemic events that, if
recurrent, can damage the brain and lead to
vision loss, movement problems, and
dementia

Nonfunctioning tRNA Lys
results in the inability to
produce mitochondrial
proteins necessary for TCA
and Ox-Phos
Myoclonic Epilepsy with Ragged Red Fibers;
Progressive myoclonic (muscular) epilepsy;
ragged red fibers - aggregations of diseased
mitochondria in subsarcolemmal region of
muscle fibers; short stature; hearing loss;
lactic acidosis; exercise intolerance; poor
night vision
Poor Cl transporter
activity; insufficient
transcription; poor
trafficking of Cl trasporter;
functional protein but
rapid degredation
Commonly, pancreatic insufficiency;
gastrointestinal/nutritional insufficiency;
high salt loss through sweat; chronic
bronchopulmonary infection; sometimes
infertility; frequency varying by population -
1/29 in Ashkenazi Jews, 1/28 in N.
Americans of N. European descent, and
1/61 in African Americans
Hypoxic state causes HBSS
to partially denature and
form an aggregate
Low oxygen states cause hypertension,
anemia, stroke, and ischemic pain; ischemia
of the spleen requiring splenectomy is
common at a young age; septicemia risk in
the first 3 years of life
Loss of HBB chains results
in poor functioning or
nonfunctioning globin
tetramers; if it is
compound heterozygous
with sickle cell HBB (HBS),
the patient presents as
sickle cell homozygous
Compound heterozygotes as sickle cell
anemia homozygotes but not as severe;
beta-thalassemia homozygotes have
transfusion dependent anemia with facial,
skull, and other bone abnormalities due to
expansion of the bone marrow
Loss of HBA chain results
in HBB tetramers which
are unstable and lead to
hemolysis; called HB H
disease
Hemolytic anemia, edema, and heart
conditions

Deficiency in PAH
prevents conversion of
phenylalanine to tyrosine,
which at increased
concentrations is toxic to
the brain
Difficulty in executive function, social
difficulties, eczema, irritability,
psychological or behavioral issues, loss of
IQ; when untreated, mental retardation,
seizures, and tremors; PKU can be
completely controlled with diet alone, but it
is INCREDIBLY difficult - must completely cut
out all meat, eggs, legumes, dairy, and most
grains, can mostly only eat fruits and
vegetables with awful tasting protein
supplements; a single day of poor eating can
result in a permanent loss of IQ
Deficiency in OTCase
prevents proper function
of the urea cycle by
blocking the synthesis of
citrulline from carbamyl
phosphate and ornithine,
causing a buildup of urea
which is toxic to the brain
Hyperammonemia causing hepatic
encephalopathy causing tachypnea and
vomiting, which in turn cause respiratory
alkalosis and dehydration, respectively, and
then hypothermia; hepatic encephalopathy
also causes seizures, lethargy;
hepatomegaly; has rapid onset in neonate
several days after birthA sphingolipidosis, a
subset of lysosomal
storage diseases that
prevents metabolism of
complex fatty acids; an
accumulation of
sphingolipids in cell
membranes and myelin
sheaths results in
neuronal distension
followed by CNS and
systemic defects
Mental retardation; blindness; cherry red
spot on retina; rapid disease progression of
mental and physical deterioration of other
senses, movements, and the ability to
swallow; in infantile Tay-Sachs, death
usually occurs before age 4
LDL receptor is important
in the uptake of
chylomicron, VLDL, and
IDL remnants, as well as
LDL lipoproteins (via ApoB
interaction); causes an
overall increase in bodily
cholesterol
Hypertension due to occlusive arterial
disease and atherosclerosis; accelerated
heart disease and angina; heart attacks,
transient ischemic attacks, and strokes;
visible deposits of cholesterol on the skin in
yellow patches, particularly above the eyes;
milky blood

Expression of bad fibrillin
gene interferes with
normal protein function,
has widespread systemic
effects
High variability of expression requires
complex diagnostic criteria (Ghent):
With contributing family history, one major
organ system involvement and minor
involvement of another system. (major in
brackets, minor not)
Without contributing family history, two
major organ system involvements and
minor involvement of a third system.
Skeletal: [≥4 pectus carinatum, severe
pectus excavatum, long limbs, scoliosis,
wrist and thumb signs, medial displacement
of medial malleoli causing pes planus (flat
feet) and protrusio acetabulae (hip socket is
too deep into the pelvis)]; 2 major or one
major and two of the following - moderate
pectus excavatum, joint hypermobility,
highly arched palate with teeth crowding,
abnormal facies
Ocular: [2 of the following minor conditions]
- flat cornea, increased axial length of the
eye, decreased pupillary miosis (constriction
response)
Cardiovascular: [dilation of the ascending
aorta involving the sinuses of valsalva,
dissection of ascending aorta]; mitral valve
Expression of bad collagen
protein interferes with
normal collagen function,
can affect many parts of
the body
Joint hypermobility, frequent dislocations or
joint subluxations; skin is hyperextensible,
soft, easy to tear and bruise; poor wound
healing; mitral and aortic valve problems;
premature arthritis; the most severe and
rarest vascular type has less joint
involvement but more smooth muscle
issues including arterial dissections and
intestinal rupture, abnormal facies with
large protruding eyes and thin face,
transluscent skin, and prone to collapsed
lungs

Expression of bad collagen
protein interferes with
normal collagen function,
can affect many parts of
the body
Eight different types depending on the
mutations involved, all but one are any
combination of 1) generally lethal, 2)
deforming, or 3) mutations not in collagen
but in collagen associated protein; the one
that is none of these is type I osteogenesis
imperfecta, a mild, null COL1A1 mutant that
is autosomal dominant negative and due to
de novo mutation in the germ line ~60% of
the time; symptoms include loose joints,
spinal curvature, poor muscle tone, slight
protrusion of the eyes with grey blue sclera,
easily fractured bones, and possibly early
hearing loss
CAG (polyglutamine)
repeat causes some HTT
to misfold; misfolded
proteins aggregate within
neurons, preventing them
from being destroyed;
aggregation leads to
neuronal death and
plaque formation
Risk determined by number of repeats: <26,
normal; 27-35, risk for offspring; 36-39, at
risk, reduced penetrance; 40-55, adult
onset; >55, juvenile onset(prior to age 20);
in rare cases, may occur in children;
symptoms include changes in memory and
behavior, particularly an increase in ager or
violence, depression, mania, and OCD;
inability to perform job duties; gradual loss
of motor control, muscle movement,
speech, and cognitive ability; muscle spasms
Regularly, FGFs act to
initiate limb bud
outgrowth and then
perhaps have a negative
regulatory effect on bone
growth; studies show this
FGFR3 mutation is
constitutively active,
resulting in very short
bones
Improportionate dwarfism; shortening of
the proximal limbs, fingers and toes; large
prominent forehead with small midface and
flattened nasal bridge; spines and knees can
be bent one way or another (kyphosis or
lordosis and bowleg or knock knee)

LDL receptor is important
in the uptake of
chylomicron, VLDL, and
IDL remnants, as well as
LDL lipoproteins (via ApoB
interaction); causes an
overall increase in bodily
cholesterol
Hypertension due to occlusive arterial
disease and atherosclerosis; accelerated
heart disease and angina; heart attacks,
transient ischemic attacks, and strokes;
visible deposits of cholesterol on the skin in
yellow patches, particularly above the eyes;
milky blood
As muscular dystrophy
Like Duchenne Muscular Dystrophy, but
with varying expressivity and severity; it is
often not fatal, and sometimes distal motor
function is completely unaffected; however,
heart and respiratory function may still be
weakened; the most affected muscles are
those of the shoulder and hip girdles; age of
onset varies from 10-30 years; some
patients with mild forms live near normal
lives with little muscle weakness
Multigene syndromes
largely affecting the brain;
PWS genes and AS genes
are geographically very
close in the chromosome,
so it is common for both
to be deleted together
Hypotonia, poor development, short
stature, small hands and feet, mental
retardation, polyphagia, obesity is common
Multigene syndromes
largely affecting the brain;
PWS genes and AS genes
are geographically very
close in the chromosome,
so it is common for both
to be deleted together
Extremely happy demeanor, waving of
hands and forearms with limp wrists and
bent elbows like marionettes

Extra copy of
chromosome 13 causes
disease state due to gene
dosage effects
Phenotypic findings are similar to those of
the embryonic signaling pathway
developmental disorders and include:
mental retardation, microcephaly,
holoprosencephaly, eye defects, and
meningomyelocele (mild spina bifida);
polydactyly, rocker-bottom feet, cleft
palate, low set ears, missing skin or hair;
abnormal genitalia and kidney defects;
heart defects
~80% of patients die by age 1
Extra copy of
chromosome 18 causes
disease state due to gene
dosage effects
Microcephaly; low set, malformed ears;
micrognathia and cleft lip and/or palate
(classical cleft or Pierre-Robin sequence);
ocular hypertelorism with narrow palpebral
fissures and ptosis; cryptorchidism
Extra copy of
chromosome 21 causes
disease state due to gene
dosage effects
Making a diagnosis: hypotonia; slanted
palpebral fissures, midface hypoplasia;
brushfield spots (white flecks in the iris); ear
anomolies and skin tags; high arched palate;
single palmar crease; wide spread large toe;
clinodactyly (inward bending of the fifth
finger)
Also, Down Syndrome patients often have
heart defects, obstructice bowel issues,
narrowing of developmental facial tubes
resulting in frequent ear and sinus
infections, altered leukemia risks,
hypothyroidism, increases risk of seizures
and Alzheimer's, pointed cornea, and
standing eversion of the feet

Monosomy of the
pseudoautosomal regions
of the X chromosome
results in
haploinsufficiency
Often identified by phenotype at birth:
edema of the feet and hand, abnormal
facies, webbed neck, broad, shield shaped
chest, widely spread nipples
In development, they have short stature,
limited breast development, lack of
menstruation
Other symptoms include possible coarcation
of the aorta, infertility due to premature
degeneration of ovaries, and kidney
abnormalities
Trisomy of the
pseudoautosomal regions
of the X chromosome
causes only minor, usually
unnoticeable phenotypic
changes and few health
complications; often
undiagnosed
Usually tall statured women with mild
developmental, learning, or speech
disabilities; no significant phenotypical
changes; usually fertile, but some may
become sterile; histology shows 2 Barr
bodies within cellsTwo copies of X
chromosome in males
results in androgen
deficiency, affecting
develompent of
secondary sex traits;
typically not diagnosed
until puberty
Tall with long legs; sexual immaturity and
hypogonadism; infertility; decreased muscle
tone; poor libido; decreased bone density;
gynecomastia with increased risk of breast
cancer; social dysfunction, poor verbal skills,
or social dysfunction
Like XXX; very minor
phenotypical changes,
often going undiagnosed
Tall stature; increased risk of behavioral
problems such as ADD/ADHD,
impulsiveness, and aggression; ~50% have
reduced IQ
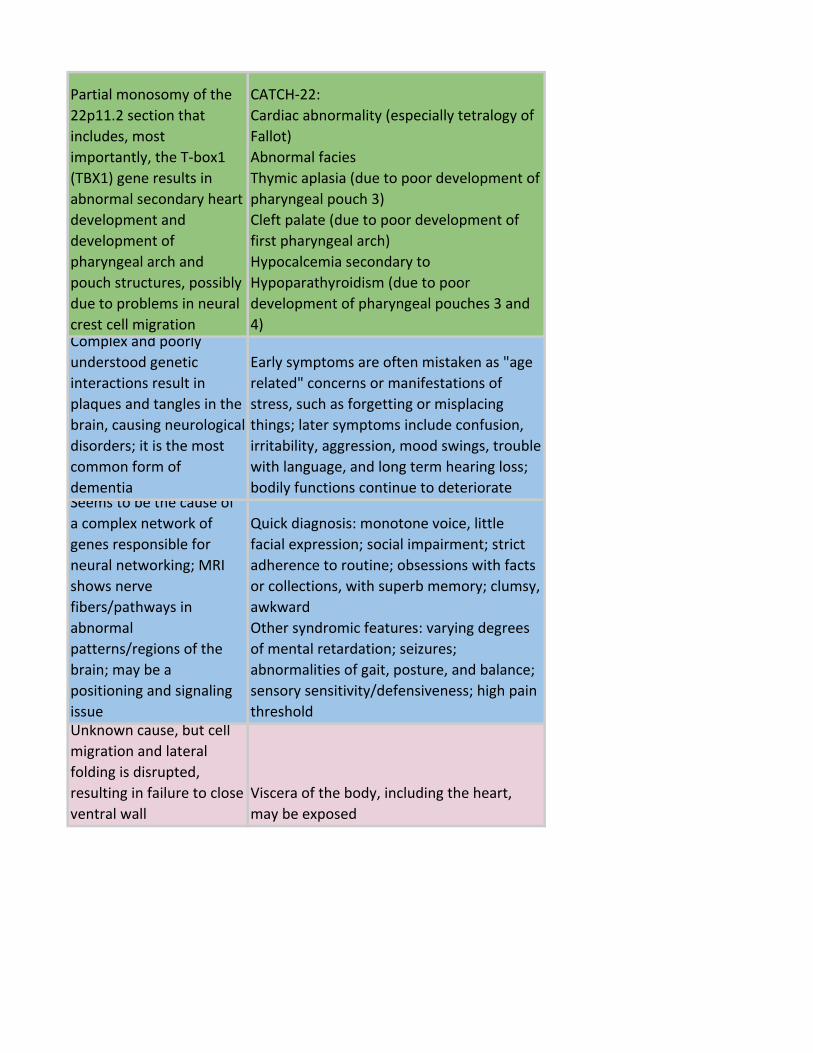
Partial monosomy of the
22p11.2 section that
includes, most
importantly, the T-box1
(TBX1) gene results in
abnormal secondary heart
development and
development of
pharyngeal arch and
pouch structures, possibly
due to problems in neural
crest cell migration
CATCH-22:
Cardiac abnormality (especially tetralogy of
Fallot)
Abnormal facies
Thymic aplasia (due to poor development of
pharyngeal pouch 3)
Cleft palate (due to poor development of
first pharyngeal arch)
Hypocalcemia secondary to
Hypoparathyroidism (due to poor
development of pharyngeal pouches 3 and
4)Complex and poorly
understood genetic
interactions result in
plaques and tangles in the
brain, causing neurological
disorders; it is the most
common form of
dementia
Early symptoms are often mistaken as "age
related" concerns or manifestations of
stress, such as forgetting or misplacing
things; later symptoms include confusion,
irritability, aggression, mood swings, trouble
with language, and long term hearing loss;
bodily functions continue to deteriorateSeems to be the cause of
a complex network of
genes responsible for
neural networking; MRI
shows nerve
fibers/pathways in
abnormal
patterns/regions of the
brain; may be a
positioning and signaling
issue
Quick diagnosis: monotone voice, little
facial expression; social impairment; strict
adherence to routine; obsessions with facts
or collections, with superb memory; clumsy,
awkward
Other syndromic features: varying degrees
of mental retardation; seizures;
abnormalities of gait, posture, and balance;
sensory sensitivity/defensiveness; high pain
thresholdUnknown cause, but cell
migration and lateral
folding is disrupted,
resulting in failure to close
ventral wall
Viscera of the body, including the heart,
may be exposed

Disruption of TCOF1
causes massive apoptosis
of neural crest cells in the
first branchial arch via an
unknown mechanism
This mutation results in hypoplasia of the
mandible and facial bones resulting in
narrow face and no zygomatic processes;
low, malformed external and middle ears;
very small chin that may lead to high or cleft
palate and teeth probems; sagging or
drooping lower eyelids due to lack of
underlying structuresUnknown mechanism, but
cell migration of
pleuroperitoneal folds
from the the body wall to
form the diaphragm is
disrupted, resulting in a
connected
pericardioperitoneal
cavity
Lung and heart development may be
impaired due to herniation of abdominal
viscera into the thoracic cavity; presence of
diaphragmatic hernia and pulmonary
hyperplasia may be indicative of retinoic
acid insufficiency
Retinoic acid is not only
responsible for anterior-
posterior patterning of
vertebrae and hindbrain,
but also the foregut; lack
of RA signaling is
suspected to prevent the
activation of appropriate
Hox genes that determine
where the lung buds will
form, resulting in
improper development of
lungs
Children with pulmonary hypoplasia have
small lungs, and may be missing lobes or
entire lungs; pulmonary hypoplasia is a
common cause of neonatal death and a
common finding in stillbirths; while it is
largely fatal, it is not incompatible with life

Shh has a widespread
effect on multiple bodily
systems:
Expression in the
notochord has an effect
on somite migration to
form vertebral structures
Shh plays a role in
separating the cloaca into
an anal canal and a
urogenital sinus (kind of
like the lungs and
esophagus at the other
end)
Shh is necessary for
secondary heart field cell
migration and separation
of the great vessels
Shh plays a role in
separation of lung buds
from the esophagus
It forms a gradient in AP
limb patterning
VACTERL is an acronym for associated
symptoms, diagnosis is made if 3 or more
(according to lecture, not notes) are
present:
Vertebral defects including spina bifida
occulta, tethered cord, and abnormalities in
vertebrae and ribs
Anorectal malformations from imperforate
anus to fistula of the urinary tract and
digestive system
Cardiovascular defects including ASD,
persistent truncus, and tetralogy of Fallot
Tracheal-Esophageal fistula or esophageal
atresia
Renal dysplasia
Limb malformations
Knockouts in Shh in mice
models show that Shh
plays a role in separating
the lung buds from the
esophagus; in esophageal
atresia, the proximal and
distal segments of the
esophagus are not
continuous, and the
proximal end may
terminate in a blind sac
Esophageal atresia with proximal blind sac
results in persistent regurgitation of food
after feeding; esophageal atresia may
occure with or without the distal esophagus
opening into the trachea; if it does, air can
be forced into the stomach through the
fistula while coughing; while a patient in this
case cannot feed, if for any reason they
have stomach contents this may result in
their regurgitation into the trachea

Knockouts in Shh in mice
models show that Shh
plays a role in separating
the lung buds from the
esophagus; in tracheal-
esophageal fistula without
atresia, the esophagus is
continuous but has an
abnormal opening into the
trachea distal to the
epiglottis
Tracheal-esophageal fistula may cause air to
be forced into the stomach through the
fistula while coughing, which may result in
the regurgitation of food and other stomach
contents into the trachea
Gli3 restricts Shh signaling
to the zone of polarizing
activity and the ulnar or
caudal aspect of the limb
bud and developing arm,
from where it creates a
gradient for
anterior/posterior
patterning; in Gli3
mutants, Shh is allowed to
be expressed in other
cells, creating multiple
overlapping gradients and
extra fingers
Polydactyly is typically preaxial or postaxial,
but it can also be central; preaxial
polydactyly means to have an extra thumb,
postaxial means to have an extra fifth
finger, while central polydactyly means to
have a duplicate index, middle, or ring
finger, in order of prevalence
Unknown cause, but
suspected to be disrupted
Shh signaling causing
midline neural and
craniofacial patterning to
be disrupted
Midline brain and craniofacial defects that
usually result in the absence of normal
structures and vary in severity from very
mild brain malformations and normal facies
to severe: cyclopea; absence of nasal
septum; single nostril; single incisor; cleft
lip; single small cerebral hemisphere due to
incomplete division of the forebrain

Exencephaly/anencephaly
and spina bifida are
caused by incomplete
closure of the neural tube
during development at
the head and most
commonly the lumbar
spine, respectively
Exencephaly/anencephaly results in a still
birth of a child without a brain - the brain
develops normally but is degraded by
exposure to the amniotic fluid; as a result of
not having the brain and skull to influence
migration of other derivatives, these
children also tend to have abnormally low,
compacted, and proportinately broad facial
features
Spina bifida most commonly occurs at the
lumbar spine and varies in severity - some
defects may be as severe as an open
vertebral canal which exposes the spine to
amniotic fluid and begins degredation;
others may have exposed dural sacs that
have herniated out of lesions, or they may
even be too small for that to occur (spina
bifida occulta); in severe cases, the children
are paralyzed, in less severe cases they are
normal with surgical repair, in the most mild
there is no intervention necessary

Atrial septal defects can
arise from abnormalities
in Shh signaling (VACTERL)
causing improper
migration to the heart to
form septal structures
They may also arise from
abnormalities in
secondary heart field cells
(DiGeorge Tbx1 deletion)
that may result in poor
migration, differentiation,
or survival (unknown
mechanism)
The septum primum and septum secundum
are like two overlapping sheets of paper
with holes cut out of them, with the septum
primum on the left and the septum
secundum on the right; the septum
secundum is held in place while the septum
primum is allowed to flap away from it;
before birth, the right atrium is at higher
pressure which causes the septum primum
to flap, allowing blood to travel through
both holes to the other side
An atrial septal defect can be caused by
absence of either the septum primum or
septum secundum, or by excessive
apoptosis, each creating an open passage
for the heart to flow through
An atrial septal defect is a local defect in
atrial septation that causes mixing of
oxygenated and deoxygenated blood; after
birth, the left side of the heart becomes
higher in pressure than the right, causing
blood to flow from left atrium to right
atrium
A similar, but more severe defect in
development is a common atrium where
the entire atrial septum is missing; this is
caused by absolute absence of the septa
primum and secundum
A similar but less severe defect is a patent
foramen ovale, present in about 20% of
people: these are present when the septa
primum and secundum fail to fuse after
birth; they are generally asymptomatic
because the higher pressure on the left
after birth pushes the two together and, as
they have holes cut out at different spots,
prevent blood from flowing into the right
atrium

Retinoic acid
embryopathy also causes
ASD as retinoic acid plays
a role in neural crest
migration to the AV
cushions that form the
bottom of the atrial
septum- these neural
crest cells apoptose and
do not contribute to
septal structures, but it is
suspected they play some
role in directing septation
In VSD, it seems that the
same defects that cause
malformation of atrial
septal structures also
prevents formation of the
ventricular septum; this is
most commonly due to
poor migration of
secondary heart field cells
to the outflow tract of the
primary heart tube; these
cells are major
contributors to the conal
cushions, which grow
down the heart to meet
the muscular ventricular
septum and the
endocardial cushions to
form the thin,
membranous portion of
the interventricular
septum (pars
membranaceae)
VSD causes shunting of oxygenated blood
from the left ventricle to the right ventricle
The septum primum and septum secundum
are like two overlapping sheets of paper
with holes cut out of them, with the septum
primum on the left and the septum
secundum on the right; the septum
secundum is held in place while the septum
primum is allowed to flap away from it;
before birth, the right atrium is at higher
pressure which causes the septum primum
to flap, allowing blood to travel through
both holes to the other side
An atrial septal defect can be caused by
absence of either the septum primum or
septum secundum, or by excessive
apoptosis, each creating an open passage
for the heart to flow through
An atrial septal defect is a local defect in
atrial septation that causes mixing of
oxygenated and deoxygenated blood; after
birth, the left side of the heart becomes
higher in pressure than the right, causing
blood to flow from left atrium to right
atrium
A similar, but more severe defect in
development is a common atrium where
the entire atrial septum is missing; this is
caused by absolute absence of the septa
primum and secundum
A similar but less severe defect is a patent
foramen ovale, present in about 20% of
people: these are present when the septa
primum and secundum fail to fuse after
birth; they are generally asymptomatic
because the higher pressure on the left
after birth pushes the two together and, as
they have holes cut out at different spots,
prevent blood from flowing into the right
atrium
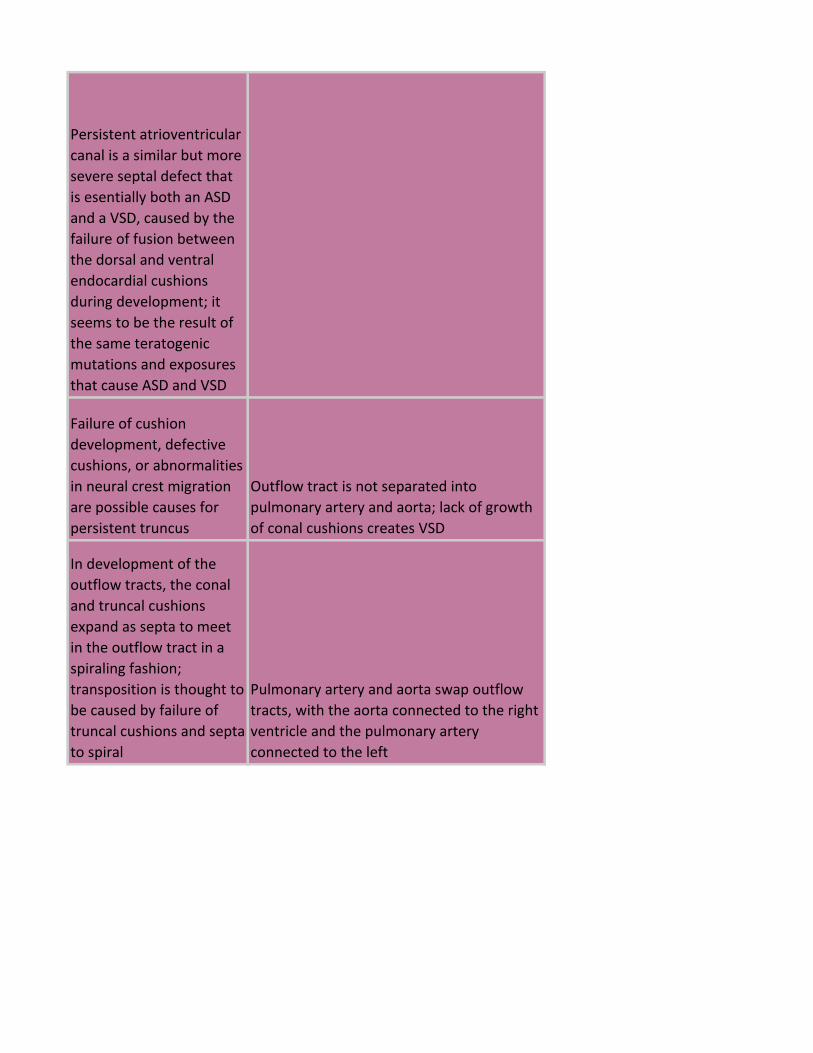
Persistent atrioventricular
canal is a similar but more
severe septal defect that
is esentially both an ASD
and a VSD, caused by the
failure of fusion between
the dorsal and ventral
endocardial cushions
during development; it
seems to be the result of
the same teratogenic
mutations and exposures
that cause ASD and VSD
Failure of cushion
development, defective
cushions, or abnormalities
in neural crest migration
are possible causes for
persistent truncus
Outflow tract is not separated into
pulmonary artery and aorta; lack of growth
of conal cushions creates VSD
In development of the
outflow tracts, the conal
and truncal cushions
expand as septa to meet
in the outflow tract in a
spiraling fashion;
transposition is thought to
be caused by failure of
truncal cushions and septa
to spiral
Pulmonary artery and aorta swap outflow
tracts, with the aorta connected to the right
ventricle and the pulmonary artery
connected to the left

Cause of tetralogy of
Fallot in Down syndrome
patients was never
discussed, but the cardiac
disruption speaker
mentioned their
association; causes for
other mutations are
similar to those for ASD
and VSD
Tetralogy of Fallot consists of 4
malformations:
1) Overriding aorta
2) Ventricular septal defect
3) Pulmonary stenosis
4) Right ventricle hypertrophy, which I
believe is secondary to narrowed pulmonary
artery and resultant pulmonary
hypertension
In tetralogy of Fallot, the conal septum is
deviated, creating all four characteristic
malformations
Mutations' effects on the
BMP pathway in limbs are
difficult to understand
because it not only results
in patterning apoptosis of
the digits but also the
condensation of cartilage
into bone; constitutively
active FGFRs signal cells to
resist patterning
apoptosis, preventing
separation of the digits;
mutations in BMP
antagonists may cause
syndactyly via improper
apoptosis or excessive
bone growth from
cartilage blueprints;
Hoxd13 mutations are
independent from BMP
pathways and result in
syndactyly due to
disrupted proximal-distal
patterning;
Any number of digits may be fused with
varying severity; in extreme cases, all five
digits may be fused together

Synostosis is the fusion of
bones, with syndactyly
being a specific case; the
other case is caused by
disruption of Hoxd11
expression that results in
fusion more proximal to
disruption of Hoxd13
(numbers get smaller as
we approach the head);
mutations in FGFRs that
cause syndactyly may also
result in craniosynostosis
(premature fusion of the
skull)
Abnormal absence of Hox11 in the presence
of Hox9 and Hox10 drives humeral fate in
forearm, causing fusion of the radius and
ulna; craniosynostosis phenotype depends
on which sutures fuse, but are always
indicated by abnormal cranial proportions -
for example, if the sagittal suture fuses
early, the brain will continue ot expand
posteriorly, resulting in an enlarged
occipitus and long, boat shaped headHox13 is expressed
caudally and distally on
appendages; loss of
appropriate expression or
function results in
improper development of
these regions
Hand-foot-genital syndrome includes
extremely rare defects including
hypospadius (closure of urethra that leaves
opening on ventral aspect of penis); uterine
fusion defects; brachydactyly and
syndactyly or synostosis
Partial monosomy of the
22p11.2 section that
includes, most
importantly, the T-box1
(TBX1) gene results in
abnormal secondary heart
development due to lack
of Tbx1 expression by
secondary heart field cells
and development of
pharyngeal arch and
pouch structures, possibly
due to problems in neural
crest cell migration
CATCH-22:
Cardiac abnormality (especially tetralogy of
Fallot due to poor migration of secondary
heart field cells)
Abnormal facies
Thymic aplasia (due to poor development of
pharyngeal pouch 3)
Cleft palate (due to poor development of
first pharyngeal arch)
Hypocalcemia secondary to
Hypoparathyroidism (due to poor
development of pharyngeal pouches 3 and
4)
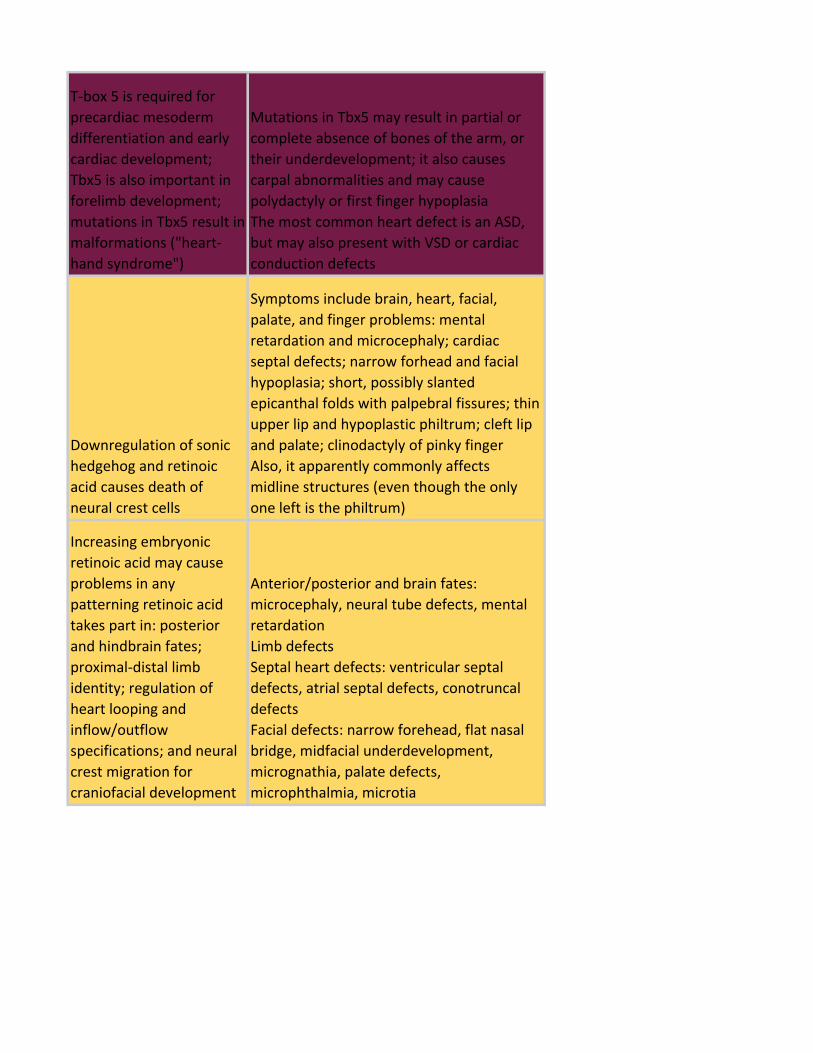
T-box 5 is required for
precardiac mesoderm
differentiation and early
cardiac development;
Tbx5 is also important in
forelimb development;
mutations in Tbx5 result in
malformations ("heart-
hand syndrome")
Mutations in Tbx5 may result in partial or
complete absence of bones of the arm, or
their underdevelopment; it also causes
carpal abnormalities and may cause
polydactyly or first finger hypoplasia
The most common heart defect is an ASD,
but may also present with VSD or cardiac
conduction defects
Downregulation of sonic
hedgehog and retinoic
acid causes death of
neural crest cells
Symptoms include brain, heart, facial,
palate, and finger problems: mental
retardation and microcephaly; cardiac
septal defects; narrow forhead and facial
hypoplasia; short, possibly slanted
epicanthal folds with palpebral fissures; thin
upper lip and hypoplastic philtrum; cleft lip
and palate; clinodactyly of pinky finger
Also, it apparently commonly affects
midline structures (even though the only
one left is the philtrum)
Increasing embryonic
retinoic acid may cause
problems in any
patterning retinoic acid
takes part in: posterior
and hindbrain fates;
proximal-distal limb
identity; regulation of
heart looping and
inflow/outflow
specifications; and neural
crest migration for
craniofacial development
Anterior/posterior and brain fates:
microcephaly, neural tube defects, mental
retardation
Limb defects
Septal heart defects: ventricular septal
defects, atrial septal defects, conotruncal
defects
Facial defects: narrow forehead, flat nasal
bridge, midfacial underdevelopment,
micrognathia, palate defects,
microphthalmia, microtia

Downregulation of FGF10
and FGF8 proteins causes
death of cells in limb bud
mesenchyme and the
apical ectodermal ridge;
thalidomide also acts as
an antiangiogenic factor,
preventing blood cells
prom perfusing the
developing limbs
Thalidomide poisoning in the embryo
results most notably in truncation of the
limbs; additional deformities include those
of the eyes, hearts, gut, and urogenital
tractsLoss of function of XP
results in reduced
efficiency of nucleotide
excision DNA repair
mechanism in response to
UV light induced
pyrimidine dimers
Sensitivity to sunlight, unusually high
number of freckles, irregular dark spots on
the skin, sensitive eyes
Fusion of the two proteins
creates a constitutively
active tyrosine kinase that
activates many proteins in
the cell
Leukemias are sometimes asymptomatic
and have rapid onset; often, they will
present as colds: malaise, body/joint pain,
low grade fever; other symptoms include
enlarged spleen, anemia, and easy bruising
Loss of neurofibromin
protein results in
unchecked neuronal cell
growth
Cafe au lait spots; proliferation of usually
small, benign tumors along the PNS,
although these may get quite large in some
patients; often the patients body will be
covered in little nodules where the tumors
are within the skin; some types include
hearing loss
dsDNA breaks are
repaired less efficiently
Dramatically increased risk for breast cancer
bilaterally and at an earlier age of onset;
increased risk for ovarian cancer; increased
risk of prostate cancer for males with the
BRCA2 mutation
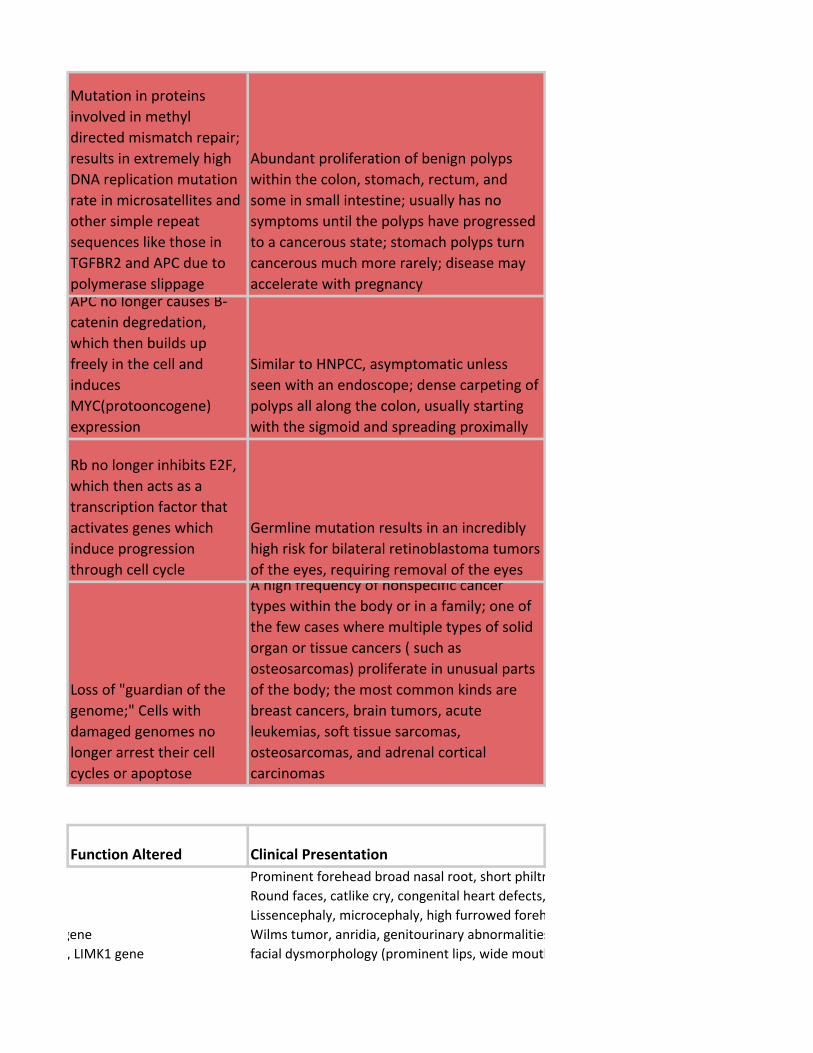
Mutation in proteins
involved in methyl
directed mismatch repair;
results in extremely high
DNA replication mutation
rate in microsatellites and
other simple repeat
sequences like those in
TGFBR2 and APC due to
polymerase slippage
Abundant proliferation of benign polyps
within the colon, stomach, rectum, and
some in small intestine; usually has no
symptoms until the polyps have progressed
to a cancerous state; stomach polyps turn
cancerous much more rarely; disease may
accelerate with pregnancyAPC no longer causes B-
catenin degredation,
which then builds up
freely in the cell and
induces
MYC(protooncogene)
expression
Similar to HNPCC, asymptomatic unless
seen with an endoscope; dense carpeting of
polyps all along the colon, usually starting
with the sigmoid and spreading proximally
Rb no longer inhibits E2F,
which then acts as a
transcription factor that
activates genes which
induce progression
through cell cycle
Germline mutation results in an incredibly
high risk for bilateral retinoblastoma tumors
of the eyes, requiring removal of the eyes
Loss of "guardian of the
genome;" Cells with
damaged genomes no
longer arrest their cell
cycles or apoptose
A high frequency of nonspecific cancer
types within the body or in a family; one of
the few cases where multiple types of solid
organ or tissue cancers ( such as
osteosarcomas) proliferate in unusual parts
of the body; the most common kinds are
breast cancers, brain tumors, acute
leukemias, soft tissue sarcomas,
osteosarcomas, and adrenal cortical
carcinomas
Function Altered Clinical Presentation
Prominent forehead broad nasal root, short philtrum, down-turned mouth, congenital heart defects, growth retardation and severe mental retardation
Round faces, catlike cry, congenital heart defects, microcephaly, mental retardation
Lissencephaly, microcephaly, high furrowed forehead, death early.
WT1 gene, PAX6 gene Wilms tumor, anridia, genitourinary abnormalities, (mental) retardation. Wilms most common.
WBSCR, ELN gene, LIMK1 gene facial dysmorphology (prominent lips, wide mouth, short palpebral tissues, short upturned nose, long philtrum) CV disease, endocrine abnormalities, prenatal growth deficiency, failure to thrive in infancy, connective tissue ab, mild mental deficiency

DNA recomb repair enzymes, 11q22 ATM gene ionizing radiation sensitivity, cerebellar ataxia depletion of Purkinje, prog nystagmus, slurred speech, oculocutaneous telangiectasia, immunodeficiency, death in second decade of life
DNA recomb repair enzymes, 16q24, FA-A gene crosslinking agent sensitivity, café-au-lait spots, hypogonadism, microcephaly, hypoplastic or aplastic thumbs, renal malformation, acute leukemia, progressive aplastic anemia, head and neck tumors
DNA repair enzymes, 15q26, BLM gene hypersensitivity to damaging agents, long narrow face, erythema with telangiectasia in butterfly distribution over nose and cheeks, high pitched voice, small stature, small mandible, protuberant ears, absence of upper lateral incisors, hypopigmentation and hyperpigmentation, immunodeficiency with decreased IgA, IgM, IgG levels, predisposition to several types of cancer

Treatments discussed in class
Citrulline supplementation, protein
restriction, arginine supplementation,
benzoic acid, and phenylacetate; liver
transplant
Avoid exposure to broad beans and
other foods high in oxidative
compounds
Allopurinol, a suicide inhibitor of
xanthine oxidase, to reduce bodily
levels of uric acid

Hydroxycarbamide or Hydroxyurea
induces transcription of gamma
hemoglobin to compensate for lack of
HBB
Hydroxycarbamide or Hydroxyurea
induces transcription of gamma
hemoglobin to compensate for lack of
HBB

Diet management, BH4 cofactor
supplementation
Citrulline supplementation, protein
restriction, arginine supplementation,
benzoic acid, and phenylacetate; liver
transplant
Statins, hypolipidemic agents, and
dietary management

Limit contact sports and other activities
that could exacerbate the aorta
Limit activities that could result in joint
dislocation, as repeated events will
accelerate the arthritic degeneration

Statins, hypolipidemic agents, and
dietary management

Folic acid supplementation while trying
to get pregnant; supplementation must
occur BEFORE you know you are
pregnant, because by that time defects
have already been created

Children with craniosynostosis must
have the sutures surgically reopened
and possibly their skull reconstructed
to avoid risk of brain damage

Limit sunlight exposure
Gleevec (imatinib) is a targeted
tyrosine kinase inhibitor that stops the
rapid proliferation of CML cells,
preventing its progression to "blast
crisis," the terminal phase
Prophylactic salpingoophorectomy,
mastectomy, tamoxifen, herceptin (if
HER2 positive)

Close monitoring, endoscopic polyp
removal, or colectomy if the polyps are
too abundant
Too many polyps to remove
individually; requires colon resection
Treatments discussed in class
Prominent forehead broad nasal root, short philtrum, down-turned mouth, congenital heart defects, growth retardation and severe mental retardation
Round faces, catlike cry, congenital heart defects, microcephaly, mental retardation
Lissencephaly, microcephaly, high furrowed forehead, death early.
Wilms tumor, anridia, genitourinary abnormalities, (mental) retardation. Wilms most common.
facial dysmorphology (prominent lips, wide mouth, short palpebral tissues, short upturned nose, long philtrum) CV disease, endocrine abnormalities, prenatal growth deficiency, failure to thrive in infancy, connective tissue ab, mild mental deficiency

ionizing radiation sensitivity, cerebellar ataxia depletion of Purkinje, prog nystagmus, slurred speech, oculocutaneous telangiectasia, immunodeficiency, death in second decade of life
crosslinking agent sensitivity, café-au-lait spots, hypogonadism, microcephaly, hypoplastic or aplastic thumbs, renal malformation, acute leukemia, progressive aplastic anemia, head and neck tumors
hypersensitivity to damaging agents, long narrow face, erythema with telangiectasia in butterfly distribution over nose and cheeks, high pitched voice, small stature, small mandible, protuberant ears, absence of upper lateral incisors, hypopigmentation and hyperpigmentation, immunodeficiency with decreased IgA, IgM, IgG levels, predisposition to several types of cancer






















facial dysmorphology (prominent lips, wide mouth, short palpebral tissues, short upturned nose, long philtrum) CV disease, endocrine abnormalities, prenatal growth deficiency, failure to thrive in infancy, connective tissue ab, mild mental deficiency

ionizing radiation sensitivity, cerebellar ataxia depletion of Purkinje, prog nystagmus, slurred speech, oculocutaneous telangiectasia, immunodeficiency, death in second decade of life
crosslinking agent sensitivity, café-au-lait spots, hypogonadism, microcephaly, hypoplastic or aplastic thumbs, renal malformation, acute leukemia, progressive aplastic anemia, head and neck tumors
hypersensitivity to damaging agents, long narrow face, erythema with telangiectasia in butterfly distribution over nose and cheeks, high pitched voice, small stature, small mandible, protuberant ears, absence of upper lateral incisors, hypopigmentation and hyperpigmentation, immunodeficiency with decreased IgA, IgM, IgG levels, predisposition to several types of cancer























hypersensitivity to damaging agents, long narrow face, erythema with telangiectasia in butterfly distribution over nose and cheeks, high pitched voice, small stature, small mandible, protuberant ears, absence of upper lateral incisors, hypopigmentation and hyperpigmentation, immunodeficiency with decreased IgA, IgM, IgG levels, predisposition to several types of cancer























hypersensitivity to damaging agents, long narrow face, erythema with telangiectasia in butterfly distribution over nose and cheeks, high pitched voice, small stature, small mandible, protuberant ears, absence of upper lateral incisors, hypopigmentation and hyperpigmentation, immunodeficiency with decreased IgA, IgM, IgG levels, predisposition to several types of cancer





























