Embed Size (px)
Citation preview

GPP statement: This Non-Interventional Study will be conducted in compliance with
the Clinical Study Protocol, Good Pharmacoepidemiology Practices
and applicable regulatory requirement(s).
Sponsoring LEO entity:
LEO Pharma GmbH
Frankfurter Str. 233
D-63263 Neu-Isenburg
Protocol Code Number
LEO Study ID:
NIS-KYNTHEUM-
1400
Date: 8- NOV-2017
Version: 2.0
KEY STUDY PERSONNEL
STUDY MANAGER
PHARMACOVIGILANCE SCIENTIST
STATISTICIAN
National Coordinating Investigator
Non-Interventional Study Protocol
Management of moderate to severe plaque psoriasis
with Kyntheum® in Daily Practice
Prospective, observational, non-interventionaL, multicenter real-world evIdence (RWE)
study of Brodalumab 210mg (Kyntheum®) to manage patients with modERate-to-severe
plaque psOriasis in daily practice (LIBERO)
LIBERO

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 2 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
APPROVERS
The following persons within LEO Pharma (LEO) have approved this Study Protocol by
signing the Non-Interventional Study Protocol Approval Form adjoined as a separate page to
this document:
The following persons outside LEO have approved this Clinical Study Protocol by signing the
Clinical Study Protocol Approval Form adjoined as a separate page to this document:

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 3 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
1. Table of Contents
Contents
1. Table of Contents.......................................................................................................... 3
2. List of Abbreviations ......................................................................................................... 5
3. Responsible Parties ............................................................................................................ 7
4. Abstract ............................................................................................................................. 8
5. Amendments and Updates ............................................................................................... 13
6. Milestones ....................................................................................................................... 13
7. Rationale and Background .............................................................................................. 14
8. Research Question and Objectives .................................................................................. 15
9. Research Methods ........................................................................................................... 16
9.1 Study Design ............................................................................................................... 16
9.2 Setting .......................................................................................................................... 18
9.3 Variables ...................................................................................................................... 22
9.4 Data Source ................................................................................................................. 24
9.5 Study Size .................................................................................................................... 24
9.6 Data Management ........................................................................................................ 24
9.7 Data Analysis ............................................................................................................... 25
9.7.1 Interim Analyses .......................................................................................................... 27
9.8 Quality Control ............................................................................................................ 27
9.9 Limitations of Research Methods ................................................................................ 27
9.10 Other Aspects .............................................................................................................. 28
10 Protection of human subjects .............................................................................................. 29
11 Management and Reporting of Adverse Events and Other Experiences ............................. 32
12 Plans for Disseminating and Communicating Study Results .............................................. 36
Annex 1. List of Stand Alone Documents ................................................................................ 38
Annex 2: Signature Page .......................................................................................................... 39
Annex 3: Contact details .......................................................................................................... 40
References ................................................................................................................................ 42

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 4 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
APPENDICES
1. List of Stand Alone Documents (08. NOV 2017, 1 page)
2. Approver´s Signature Page
3. Contact Details of all key study personell (08. NOV 2017, 2 pages)

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 5 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
2. List of Abbreviations
ADR Adverse Drug Reaction
AE Adverse Event
AMG Arzneimittelgesetz (German Medicine Law)
ANFOMED Anfomed GmbH (CRO)
BfArM Bundesinstitut für Arzneimittel und Medizinprodukte
BSA Body Surface Area
CHMP Committee for Medicinal Products for Human Use
CI Confidence Interval
CRF Case Report Form
CRO Contract Research Organization
DLQI Dermatology Quality of Life Index
EC Ethics Committee
EEA European Economic Area
EMA European Medicines Agency
FPI First Patient In
Ig Immunglobulin
IL Interleukin
ITT Intention To Treat
LEO LEO Pharma GmbH (Sponsor)
LOCF Last Observation Carried Forward
LPI Last Patient In
LPO Last Patient Out
MedDRA Medical Dictionary for Regulatory Activities
NIS Non Interventional Study
OE Other Event
PaGA Patient Global Assessment
PASI Psoriasis Area and Severity Index
PBI Patient Benefit Index
PEI Paul Ehrlich Institute
PGA Physician Global Assessment

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 6 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Q2W Every other week
QA Quality Assurance (Qualitätssicherung)
QW Weekly
RCT Randomized Clinical Trial
RWE Real World Evidence
SAE Serious Adverse Event
SAP Statistical Analysis Plan
SOC System Organ Class
SOP Standard Operating Procedure
sPGA Static Physician Global Assessment
SmPC Summary of Product Characteristics
Tab Table
TSQM Treatment Satisfaction Questionnaire for Medication
V Visit
W Week
WHO World Health Organization

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 7 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
3. Responsible Parties
LEO Pharma GmbH (LEO) is the sponsor of the NIS and Anfomed GmbH (ANFOMED) is
the Contract Research Organisations (CRO) authorised by LEO to act on behalf of LEO.
LEO Pharma A/S, Industriparken 55, DK-2750 Ballerup, Denmark will be the Data
Controller.
(LEO) is the study manager and responsible for overall project
coordination.
is the principal investigator of the study and
will provide ongoing expert input into the conduct of the study and evaluation of study
results, including review of safety data to enable early and ongoing risk management during
the conduct of the Study. He will provide guidance on development and review of study
materials, documents (e.g. protocol, Case Report Forms, disease-specific questionnaires) and
procedures, will support the successful roll-out of the Registry and encourage active
participation from the selected investigators and sites.
is responsible for writing the protocol
and the final publication with scientific support and under the supervision of the national
coordinating investigator.
is responsible for statistical analysis and for writing the
biometric clinical study report.
Contact and responsibilities of all parties contributing to the study, including all investigators,
are listed in Annex 3. For key contributors LEO will keep CVs and documentation regarding
Conflicts of Interest and make such documentation available upon request from relevant
parties, e.g. authorities, journals. LEO will keep a record of all relevant sponsor personnel.
The CRO will keep a record of all involved CRO personnel.
NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 8 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
4. Abstract
Title Prospective, observationaL, non-interventional, multicenter, real-world
evIdence (RWE) study with Brodalumab 210mg (Kyntheum®) to manage
patients with modERate-to-severe plaque psOriasis in daily practice
(LIBERO)
Version 2.0, 08. NOV 2017
Rationale
and
background
Kyntheum® is the first IL 17- Receptor A Blocker, which has shown in clinical
trials superior efficacy and a comparable safety profile vs. Ustekinumab
(Lebwohl et al, NEJM 2016). In July 2017 Kyntheum® obtained approval in
the EEA (European Eonomic Area) for the treatment of patients with
moderate-to-severe plaque psoriasis in adult patients, who are candidates for
systemic treatment. To-date there is no evidence, how the management of
moderate-to-severe psoriasis patients with Kyntheum® results in daily practice
and how different patient profiles are managed with Kyntheum® in the
diversity of a real world setting.
Research
questions
and
objectives
This NIS aims
to assess the short and long-term management outcome in patients with
moderate-to-severe psoriasis starting with Kyntheum® under daily
practice conditions
to describe different patient profiles managed with Kyntheum® in the
diversity of a real world setting
The NIS will deliver real world evidence in the German healthcare setting
regarding patient profiles, physician´s decision algorithm (e.g. more biologic
naive, after failure of anti-TNF-biologics or ustekinumab etc.) and brodalumab
effectiveness.
Patient reported outcomes will create substantial insights regarding care with a
modern biologic outside of randomized clinical trials (RCT).
Study
design
Open-label, multicenter, real-world, 12 and 52 weeks, prospective, non-
interventional, non-controlled (single-arm), observational study
Population Inclusion criteria
Adult patients (≥ 18 years)
Patients must be a candidate for systemic therapy with Kyntheum®
according to SmPC and local guidelines.
Decision to start treatment with Kyntheum® in a patient was taken
NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 9 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
before enrolment in the study.
Patients must have understood and voluntarily signed the ICF.
Exclusion criteria
Patients who are enrolled in any interventional clinical trial in parallel.
Patients who are already treated or have been previously treated with
Kyntheum® before enrolment.
Patients, who did not sign the informed consent or incapacitated
patients, who are not able to provide informed consent and/ or are under
institutionalized care.
Variables Primary endpoints
Proportion of patients achieving an absolute PASI value of 3 at week
12
Proportion of patients who continued with Kyntheum® after 12 weeks
of treatment achieving sPGA success (defined as clear = 0 and almost
clear = 1) at week 52
Secondary endpoints
Short term effectiveness as measured by the proportion of patients
achieving
o PASI 75/ 90/ 100 at week 12
o sPGA-success (clear = 0 or almost clear = 1) at week 12
o sPGA = 0 at week 12
o Time to response (absolute PASI ≤ 3, PASI 75/90, PGA 0/1,
PGA 0)
Long term effectiveness of continued Kyntheum® treatment as
measured by the proportion of patients over time on a quarterly basis
and at week 52 measured by
o mean PASI
o Proportion of patients achieving a PASI ≤ 3
o PASI 75/ 90/ 100
o sPGA = 0/1 und sPGA = 0
Description of the frequency of adverse events as per MedDRA
preferred term and systemic organ class (SOC) during induction phase
(week 0-12) and maintenance phase (week 13-52).
NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 10 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Description of patient profiles with regard to
o demographics (e.g. gender, age, health insurance status,
socioeconomic status, disease duration),
o disease severity (sIGA, sPaGA, BSA, PASI),
o previous treatment regimens,
o satisfaction with previous treatment and
o reasons for choosing Kyntheum®
Patient Reported Outcomes (PROs) at week 12 and week 52 as
measured by
o Mean change in Patient Global Assessment (PaGA) and
proportion of patients with sPaGA of 0 clear or 1 almost clear.
o Mean change in overall PBI and subscales on psoriasis
symptoms, social life and emotional well-being and proportion
of scales/ subscales with a score of 4 or 5
o Mean proportion of patients, who were satisfied/ very satisfied
with treatment (TSQM-9)
o Mean change in DLQI Score and proportion of patients, who
achieved a DLQI Score of 0-1 (= no impact on QoL)
Physician and patient reported outcome variables will be collected in the first
four weeks on a biweekly basis followed by quarterly follow-ups (W12, W24,
W36, W48) and a close-out visit at W52.
Data
sources
Data will be collected by the physicians from the standard medical records of
the participating out-patient clinics and private practices.
At their regular visits patients will be asked to complete a questionnaire on
assessing severity of their psoriasis (PaGA) during the 52 weeks treatment
period. They will be asked to answer a short series of questionnaires about their
quality of life (DLQI), general and emotional well-being (PBI) and their
satisfaction with therapy (TSQM-9).
Study size The study aims to include 500 patients in about 100-150 sites all over
Germany. A participating site should be willing to enrole at least 3-6 patients to
the study.
The primary purpose of this NIS is descriptive and the number of 500 patients
is a representative patient clientele for Germany aspired by epidemiological
data. With this sample size it is expected to see the same or similar outcome
NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 11 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
compared to randomized clinical trials (RCT) with Kyntheum®. The results
could bridge the RCT data with the real-world experience.
With 500 documented cases:
In case of dichotomous variables, a 95% confidence interval for the
underlying binomial probabilities will have a maximum length of 8.94
percentage points.
95% confidence intervals for the underlying mean values of
quantitative variables will have a length of 0.176 standard deviations.
Rare events with an incidence down to 0.006 (1/167) will appear at
least once in the sample with 95% probability.
Patient groups formed by anamnestic or other characteristics will
usually be large enough to allow reliable subgroup analyses.
Data
analysis
Populations to be analyzed:
The ITT (Intention-To-Treat) population being all patients who have given
written informed consent to use of their data for the short and longer term
effectiveness.
The safety population being the subset of the ITT- patients who have either
confirmed in patient reported data that they have administered the initially
prescribed medication at least once, or have confirmed to site staff at a later
contact that they have administered the initially prescribed medication at least
once.
The short-term effectiveness population being the subset of the safety
population who has data on at least one of the effectiveness outcome variables
available until week 12 (LOCF).
The long-term effectiveness population being the subset of the effectiveness
population, who agreed to continue with Kyntheum® after week 12, has
received at least once the study medication in maintenance phase and has at
least one of the effectiveness outcome variables available after week 12.
Statistical analysis
Taking into account the non-interventional character of the study, statistical
analysis will be performed in a descriptive and explorative way after 12 and 52
weeks of treatment. Continuous variables will be described by number of valid
(evaluable) cases, mean, standard deviation, minimum, lower quartile, median,
upper quartile and maximum. Categorical variables will be described by
NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 12 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
number of cases, frequency, and percentage. 95%-confidence intervals will be
computed where suitable.
A statistical analysis plan will provide details of analyses, data tabulation, and
listings (see Annex 4)
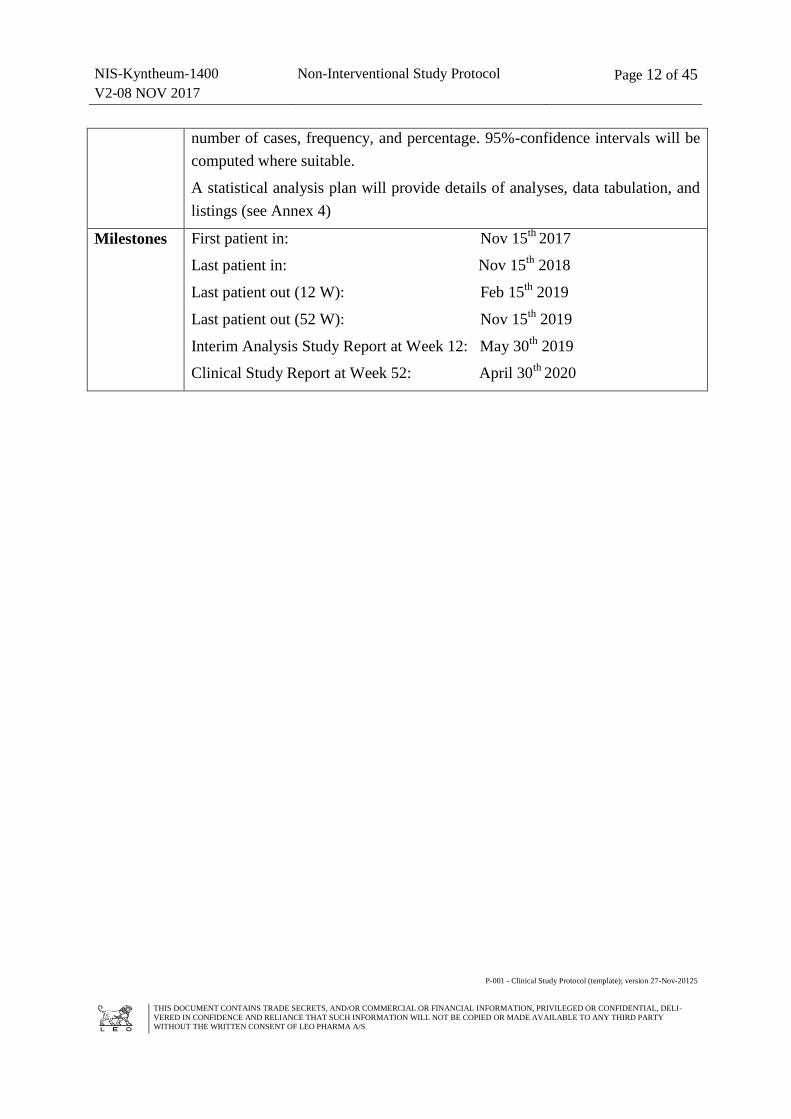
Milestones First patient in: Nov 15th
2017
Last patient in: Nov 15th
2018
Last patient out (12 W): Feb 15th
2019
Last patient out (52 W): Nov 15th
2019
Interim Analysis Study Report at Week 12: May 30th
2019
Clinical Study Report at Week 52: April 30th
2020

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 13 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
5. Amendments and Updates
None
6. Milestones
Milestone Planned Date
Start of data collection November 15th
2017
End of data collection November 15th
2019
Interim report (12 Weeks) May 30th
2019
Final study report (52 Weeks) April 30th
2020
The start of the study is defined as the date on which the first information on a study patient is
recorded in the study dataset. LEO will ensure that End-of-Data Collection (End of Study)
notification is submitted to the concerned authorities and ECs.
Based on upcoming knowledge, LEO might choose to terminate the study prematurely. In
such case the study sites, ECs and authorities will be informed promptly.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 14 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
7. Rationale and Background
Psoriasis is a chronic relapsing immunological skin disease characterized by proliferation of
keratinocytes and accumulation of immune cells in the affected skin, that occurs in 2 - 3% of
the Caucasian population.1,2,3 Despite the availability of several therapies, many patients
remain untreated, do not have an adequate response or have treatment- related toxic effects.4
The Interleukin (IL)-17 pathway has shown to play a key role in the immunopathogenesis of
psoriasis.5 Genomewide association studies have linked IL-17 pathway-related genes with
psoriasis6,7 and IL-17 mRNA levels are higher in psoriatic lesions than in normal skin.
8,9
Numbers of T-helper (h) 17 cells are increased in psoriatic lesions and are stimulated by IL-23
to release IL-17 cytokines.10
IL-17 cytokines, which include IL-17A, IL-17C, IL- 17F, and IL-
17A/F, can induce expression of psoriasis-related proinflammatory molecules in
keratinocytes, leading to the recruitment and accumulation of neutrophils, T cells, and
dendritic cells. 11
,12
,13
Kyntheum® (Brodalumab 210mg) is the first human monoclonal IgG2 antibody that
selectively binds to the human IL-17 A receptor and thereby blocks its interactions with a
number of cytokines of the IL-17 family (IL-17-A, - C, -E, -F). 14
,15
,16
In July 2017, the European Medicines Agency's (EMA's) Committee for Medicinal Products
for Human Use (CHMP) recommended the granting of a marketing authorisation for
Kyntheum® for the treatment of moderate to severe plaque psoriasis in adults who are
candidates for systemic therapy.17
The approval of Kyntheum® was based on three randomized, double-blind, placebo-controlled
phase 3 trials including a 12 week induction phase followed by withdrawal/ retreatment or a
maintenance phase.
AMAGINE-1 (n=661) evaluated the efficacy, safety, and withdrawal and retreatment
effect of brodalumab compared with placebo.18
AMAGINE-2 (n=1831) and AMAGINE-3 (n=1881) were 12-week induction trials
followed by re-randomization at week 12 and evaluated the efficacy and safety of
induction and maintenance of brodalumab compared with ustekinumab and placebo.19
Results showed that Kyntheum® 210mg offered more patients complete skin clearance
measured by Improvement of the Psoriasis Area Severity Index by 100% (PASI 100) at 12
weeks compared to patients treated with ustekinumab [AMAGINE-2: 44% (n=272) versus
22% (n=65), p<0.001; AMAGINE-3: 37% (n=229) versus 19% (n=58), p<0.001].18,19

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 15 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
In AMAGINE-1 83% of patients on Kyntheum® 210mg achieved PASI 75 compared to 3% of
patients treated with placebo at 12 weeks [83.3% (n=185) versus 2.7% (n=6), p<0.001] and
76% of patients achieved static Physcian Global Assessment (PGA) success versus 1% of
patients treated with placebo [75.7% (n=168) versus 1.4% (n=3), p<0.001].18
In the AMAGINE trials more than half (53-56%) of patients on continuous Kyntheum®
treatment achieved PASI 100 at week 52.18,19
Patients also reported experiencing improved health-related quality of life after four weeks of
treatment with Kyntheum®. After 12 weeks of treatment, seven of ten patients (72%, n=29/40,
p<0.0001) reported psoriasis no longer impaired their health-related quality of life, (0/1
DLQI) compared with placebo (5%, n=2/37).20
Data from the three large randomised, controlled AMAGINE clinical trials, found
Kyntheum® to be well tolerated, with an acceptable safety profile.
21 The most common
adverse events were arthralgia, headache, fatigue, diarrhoea and oropharyngeal pain.18
The approval of a new pharmaceutical product is based on data from randomized, controlled
clinical trials in patient populations that are based on stringent inclusion and exclusion
criteria. Even when the studies are designed to be representative of the patients described in
the indication, clinical trials often do not cover the diversity of the real-world setting.
To-date there is no evidence, how the management of moderate-to-severe psoriasis patients
with Kyntheum® results in the daily practice and how different patient profiles are managed
with Kyntheum® in the diversity of a real world setting in Germany.
8. Research Question and Objectives
The primary objective of this non-interventional observational study is to assess the short-
and long-term management outcome of patients with moderate-to-severe psoriasis
starting with Kyntheum® under daily practice conditions by assessing the proportion of
patients achieving an absolute PASI 3 at week 12 and – in patients, who continued
Kyntheum® after 12 weeks of treatment - the proportion of patients achieving sPGA success
(defined as clear/ almost clear) at week 52.
Secondary objective is to describe different patient profiles managed with Kyntheum® in
the diversity of a real world setting with regard to demographics, disease severity, previous
treatment regimen and satisfaction with it. LIBERO will deliver real world evidence on
physician´s decision algorithm (e.g. more biologic naive? After failure of anti-TNF-biologics
or ustekinumab? ) in the German healthcare system and effectiveness in the respective groups.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 16 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Patient reported outcomes will create substantial insights regarding the patient´s benefit as
well as satisfaction with the first IL-17-Receptor A Blocker outside of randomized clinical
trials (RCTs).
9. Research Methods
9.1 Study Design
DESIGN
This study is an open-label, multicenter, real-world, 12 and 52 weeks, prospective, non-
controlled (single-arm) observational study.
The study is a ‘non-interventional study’ as defined for drug studies in AMG §4 (23) and in
Directive 2001/20/EC22
with primary data collection and will follow the guidelines for Good
Pharmacoepidemiology Practices.23
This means that:
The assignment of a patient to Kyntheum® is not decided in advance by the study protocol
but falls within current practice.
The prescription of Kyntheum® is clearly separated from the decision to include the
patient in the study.
Kyntheum® is prescribed in accordance with the terms of the marketing authorization.
17
Kyntheum® is obtained through the usual distribution channels.
No additional diagnostic or monitoring procedures shall be applied to the patients.
Epidemiological methods shall be used for the analysis of collected data.
ENDPOINTS
To meet the primary and secondary objectives of this study the following endpoints have been
defined:
Primary endpoints
Proportion of patients achieving an absolute PASI value of 3 at week 12.
Proportion of patients who continued with Kyntheum® after 12 weeks of treatment.
achieving sPGA success (defined as clear = 0 and almost clear = 1) at week 52.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 17 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Secondary endpoints
Short-term effectiveness as measured by the proportion of patients achieving
o PASI 75/ 90/ 100 at week 12
o sPGA-success (clear = 0 or almost clear = 1) at week 12
o sPGA = 0 at week 12
o Time to response (absolute PASI ≤ 3, PASI 75/90, PGA 0/1, PGA 0)
Long-term effectiveness of continued Kyntheum® treatment as measured by the
proportion of patients (LOCF) over time on a quarterly basis and at week 52 measured
by
o mean PASI
o Proportion of patients achieving a PASI ≤ 3
o PASI 75/ 90/ 100
o sPGA = 0/1 und sPGA = 0
Description of the frequency of adverse events as per MEDRA24
preferred term and
systemic organ class (SOC) during induction phase (week 0-12) and maintenance
phase (week 13-52).
Description of patient profiles with regard to
o demographics (e.g. gender, age, health insurance status, socioeconomic status,
disease duration),
o disease severity (sPGA, sPaGA, BSA, PASI),
o previous treatment regimens,
o satisfaction with previous treatment and
o reasons for choosing Kyntheum®
Patient Reported Outcomes (PROs) at week 12 and week 52 as measured by
o Mean change in Patient Global Assessment (PaGA) and proportion of patients
with a PaGA of 0 = clear or 1 = almost clear.
o Mean change in overall Patient Benefit Index (PBI) and subscales on psoriasis
symptoms, social life and emotional well-being and proportion of scales/
subscales with a score of 4 or 5

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 18 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
o Mean proportion of patients, who were satisfied/ very satisfied with treatment
(TSQM-9)
o Mean change in DLQI Score and proportion of patients, who achieved a DLQI
Score of 0-1 (= no impact on QoL)
9.2 Setting
POPULATION
The study aims to include 500 moderate-to-severe plaque psoriasis patients in about 100 - 150
dermatological sites (private practices, out-patient clinics) all over Germany over a 12- and
52-weeks period.
Each participating site is expected to include between 3 and 6 patients. Patients should be
included in the study only once.
Inclusion requires that Kyntheum® is prescribed in accordance with the approved terms of the
EU marketing authorization and that none of the stated contraindications apply.
Caution should be taken by the treating physician concerning any precautions, warnings and
potential drug interactions stated in the terms of the marketing authorization (SmPC).
SELECTION CRITERIA
Inclusion criteria
Adult patients (≥ 18 years)
Patients must be a candidate for systemic therapy with Kyntheum® according to
SmPC17
and local guidelines.25,26
Decision to start treatment with Kyntheum® in a patient was taken before enrolment in
the study.
Patients must have understood and voluntarily signed the ICF.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 19 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Exclusion criteria
Patients who are enrolled in any interventional clinical trial in parallel.
Patients who are already treated or have been previously treated with Kyntheum®
before enrolment.
Patients, who did not sign the informed consent or incapacitated patients, who are not
able to provide informed consent and/ or are under institutionalized care.
PARTICIPATING SITES
Office based dermatologists or out-patients clinics, who are experienced for the clinical care
and follow-up of psoriasis patients, are invited to participate in the NIS.
DATA COLLECTION
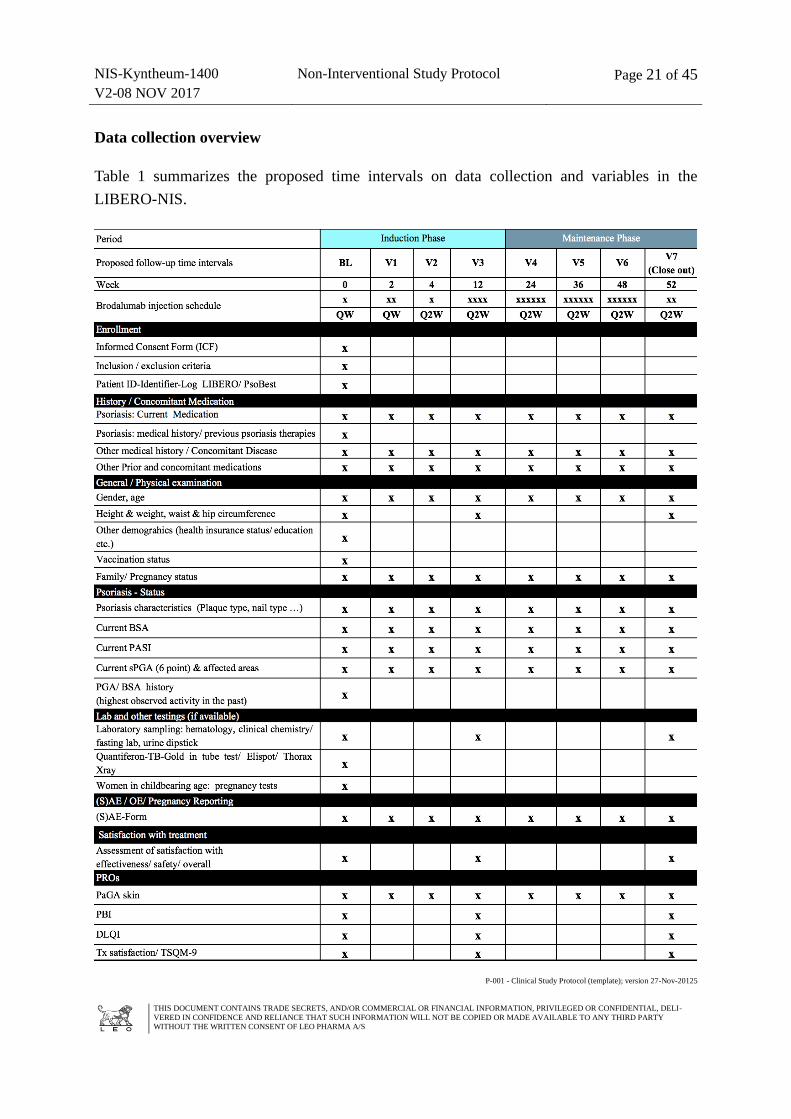
Prospective data collection from consented patients will be performed by the participating
dermatologists during the course of routine patient care at the time of first injection of
Kyntheum® (Baseline) and at the following time intervals: week (W)2, W4, W12, quarterly in
the subsequent months (W24, W36, W48) until W52 (close-out visit). See further details in
Table 1 and in section 9.3. on variables.
As this is an observational study, participation will not change the patient/physician
relationship, nor influence the physician’s drug prescription or therapy choices or other
management of the patient’s disease.
Data collected by physicians
Data will be collected by the physicians from the standard medical records of the participating
out-patient clinics and private practices.
At baseline physicians will complete a questionnaire on the patient´s demographics, physical
examination, vaccination status, psoriasis and medical history, prior and current psoriasis
treatment and concomitant medication and – in women in childbearing age – pregnancy
status/ pregnancy prevention control method, respectively. If available, relevant lab testing
results are collected.
At all visits the clinical psoriasis status is assessed by describing psoriasis type and
characteristics, affected areas, BSA, PASI and sPGA (6-point scale, ranging from 0 = clear
skin to 5 = very severe disease; a score of 3 indicates moderate disease). Date of
administration of recent Kyntheum® injections and any change in psoriasis or concomitant

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 20 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
medication, concomitant diseases and – in woman in childbearing age – pregnancy status is
recorded. If available, relevant lab testing results are collected.
Adverse Events and Other Experiences which during the study are brought or come to the
attention of a healthcare professional involved in the study will be documented in separate
forms and processed as described in section 11.
Physician´s satisfaction with efficacy and safety of psoriasis treatment regimen is assessed at
Baseline visit, at the end of the induction phase (W12) and at the close-out visit on W52
during the maintenance phase.
Data collected by patients
At each visit patients are also asked to assess their psoriasis via a static Patient Global
Assessment (PaGA) mirroring the sPGA 6-point scale (0 = clear skin to 5 = very severe
disease; a score of 3 indicates moderate disease).
At Baseline, at the end of the induction phase (W12) and during the close-out visit on W52
they will be asked to answer a short series of questionnaires about their quality of life
(DLQI)27
, general and emotional well-being (PBI)28
and their satisfaction with previous and
current therapy (TSQM-9).29,30
NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 21 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Data collection overview
Table 1 summarizes the proposed time intervals on data collection and variables in the
LIBERO-NIS.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 22 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
PATIENT DISCONTINUATION
Patients will be considered as withdrawals (data until moment of withdrawal included in the
analyses) if they indicate they do not want to provide any further follow-up data after having
completed at least the first visit in its entirety.
Patients will be considered as lost to follow-up (included in the analyses) if they prove to be
non-contactable by a participating physician before the end of the follow-up.
Missing questionnaires at one or several time points, or changing of participating physician, is
not a criterion for patient exclusion from the analyses.
Definition of the analyses datasets will be further detailed in the Statistical Analysis Plan.
Subsequent management of missing data will also be described in the Data Management Plan.
9.3 Variables
VARIABLES COLLECTED BY THE PHYSICIAN
Baseline Visit (at inclusion)
Date of the signed informed consent
Inclusion/ exclusion criteria
Pseudonymous patient ID identifier Log (to ensure alignment of patient IDs if a
patient is enrolled in parallel in PsoBEST)
General patient characteristics (demographics, health insurance status, socio-
economic data, concomitant diseases, height, weight, waist and hip circumference)
Vaccination status (date of last vaccinations and last tuberculosis testing incl.
Quantiferon – TB Gold/ ELISPOT test result)
Pregnancy status/ pregnancy prevention method incl. date and result of last
pregnancy test(s) if available (in women of childbearing age only)
Lab-testings (last available results on clinical chemistry, hematology and urine
dipstick, fasting glucose and lipid panel)

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 23 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Psoriasis history (disease duration, psoriasis characteristics e.g. plaque-type, nail-
type, PGA/ BSA highest observed activity in the past, previous psoriasis therapies)
Current psoriasis status (BSA, PASI, sPGA (6-point scale)1 and affected area)
Psoriasis management (reasons for choosing Kyntheum®, date of first Kyntheum
®
administration, other psoriasis medication and other concomitant medication)
At all Follow-up visits:
Date of visit and date of previous Kyntheum® injections since last visit
Current psoriasis status (BSA, PASI, sPGA (6-point scale) and location of lesions)
Any change in psoriasis management
Any change in concomitant disease and medication
Any (S)AE, (S)ADR as defined in section 11
Any change in pregnancy status (in woman of childbearing age only)
At W12 and W52 Follow up in addition:
Any change in Lab – testing results (if of clinical significance, AE form to be
completed)
Height and weight, waist and hip circumference
Overall satisfaction with efficacy and tolerability
VARIABLES COLLECTED BY THE PATIENT
At all Visits
Current psoriasis status assessed by patients via sPGA
1 Scores on the static physician’s global assessment (sPGA) range from 0 (clear skin) to 5 (very severe disease);
a score of 3 indicates moderate disease.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 24 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
At Baseline, W12 and W52
Satisfaction questionnaire with previous treatment (Baseline) and abbreviated
Treatment Satisfaction Questionnaire for Medication (TSQM-9) at W12 and W52.
Disease and Life Quality Index (DLQI) – Questionnaire
Patient Benefit Index (PBI)
9.4 Data Source
Study data to be collected from the site will be transcribed from the routine medical records of
the participating patients. In addition, participating patients will be asked to provide data
through patient questionnaires, as described in section 9.3.
9.5 Study Size
The study aims to include 500 patients in about 100-150 sites all over Germany. The primary
purpose of this NIS is descriptive and the number of 500 patients is a representative patient
clientele for Germany aspired by epidemiological data. With this sample size it is expected to
see the same or similar outcome compared to randomized clinical trials (RCT) with
Kyntheum®. The results could bridge the RCT data with the real world experience.
With 500 documented cases:
In case of dichotomous variables a 95% confidence interval for the underlying
binomial probabilities will have a maximum length of 8.94 percentage points,
95% confidence intervals for the underlying mean values of quantitative variables will
have a length of 0.176 standard deviations,
Rare events with an incidence down to 0.006 (1/167) will appear at least once in the
sample with 95% probability.
Patient groups formed by anamnestic or other characteristics will usually be large
enough to allow reliable subgroup analyses.
9.6 Data Management
The Study Site will receive from ANFOMED data collection tools (one or two study binders)
containing paper based CRFs and PRO questionnaires for three patients as well as the

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 25 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
protocol, Kyntheum®
SmPC, the patient identifier log, patient informed consent forms and
(S)AE-/ pregnancy reporting forms.
Text field entries and any data collected on paper should be made in legible German.
The completed questionnaires need to be sent by the study site to ANFOMED after visit 3
(W12), visit 5 (W36), and visit 7 (W52).
The Study Site Responsible must sign off the complete data set for each patient, confirming
that a signed informed consent of the patient is available and the accuracy and completeness
of the collected data. Only signed data sets will be included in the database.
Any Adverse Events, other experiences or pregnancies should be reported according to
section 11 and should be signed off separately by a physician who may or may not be
involved in the study.
Data Management will be carried out according to a Data Management Plan, which must be
written and approved before the design of the study database is finalized. The data
management provider should approve all data formats before the data collection tools are
made available to the sites.
If the written informed consent of a patient is known not to be available in spite of it being
required, data for this patient is not entered into or is deleted from the database.
If a patient is erroneously included in the study more than once only the data relating to the
first inclusion will be kept in the database and be available for analysis. Data from later
inclusions will be transferred to the first dataset when relevant, i.e. if collected within the time
frame of the first follow-up period.
If a patient is included in the study in spite of not being treated according to the approved
terms of the marketing authorization, data is kept in the database and analyzed separately and
as part of the overall analyses as described in the Statistical Analysis Plan.
Coding of medical history, concomitant illness and adverse events is performed per MedDRA
Codes and concomitant medication per WHO-Drug classification
9.7 Data Analysis
Statistical Methods
This study is observational and epidemiological methods will be employed for data analyses.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 26 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Taking into account the non-interventional character of the study, statistical analysis will be
performed in a descriptive and explorative way after 12 and 52 weeks of treatment.
Descriptive analysis will be performed of all collected data except data collected only for the
purpose of data cleaning, i.e. all data listed in section 9.3. Continuous variables will be
described by number of valid (evaluable) cases, mean, standard deviation, minimum, lower
quartile, median, upper quartile and maximum. Categorical variables will be described by
number of cases, frequency, and percentage. 95%-confidence intervals will be computed
where suitable.
The Statistical Analysis Plan describes the statistical analyses as foreseen at the time of
planning the study. Any known deviations from the planned analyses, the reason for such
deviations and all alternative/additional statistical analyses that may be performed as well as
the final statistical analysis must be described in a revised Statistical Analysis Plan (SAP)
before completion of data collection. All later deviations and/or alterations will be
summarised in the Clinical Study Report.
Populations
The following populations will be analyzed:
The ITT (Intention-To-Treat) population being all patients who have given written
informed consent to use of their data for the short and longer term effectiveness.
The safety population being the subset of the ITT- patients who have either confirmed in
patient reported data that they have administered the initially prescribed medication at least
once, or have confirmed to site staff at a later contact that they have administered the initially
prescribed medication at least once.
The short-term effectiveness population being the subset of the safety population who has
data on at least one of the effectiveness outcome variables available until week 12 (LOCF).
The long-term effectiveness population being the subset of the effectiveness population,
who agreed to continue with Kyntheum® after week 12, has received at least once the study
medication in maintenance phase and has at least one of the effectiveness outcome variables
available after week 12.
Safety analysis
All AEs reported in Kyntheum® treated patients reported to LEO Safety Contact Person or
directly to competent authorities according to section 11 will be all captured in the study
database and listed or tabulated in the final report. AEs will be classified by system organ
class (SOC), preferred terms (PT) in accordance with the current version of the Medical
dictionary for Regulatory Activities (MedDRA), and by causality and seriousness.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 27 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
9.7.1 Interim Analyses
To assess short term effectiveness during induction phase with Kyntheum®
an interim analysis
is planned after week 12.
To assess long term effectiveness during maintenance phase final analysis is planned after the
week 52 visit is completed in patients, who agreed to continue with Kyntheum® after week
12.
9.8 Quality Control
Monitoring
For quality control monitoring visits can be performed at the participating sites. During a
monitoring visit it will be verified, if the signed informed patient consent is available and if
the documented data are identical with the source data from the patient charts (source data
verification).
Safety data reconciliation
A monthly reconciliation of the events and experiences received from the sites will be
performed between LEO GmbH and GPV according to standard LEO processes. Before
closure of the LIBERO database for interim- or final analysis after week 12 and week 52,
LEO DE and Anfomed will perform reconciliation of reported events between the LEO Safety
Database and the LIBERO database. Reconciliation is further specified in a separate Safety
Plan.
9.9 Limitations of Research Methods
Generally, two types of bias are distinguished in epidemiology and should be considered:
selection and information bias.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 28 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Selection bias
Selection bias is a entails the selective recruitment into the study of subjects that are not
representative of the exposure or outcome pattern in the source population. The selection bias
in this study may result from selectively prescribing of a medication to more severe patients.
As a result of a higher background risk, the reported event rate may be higher and give the
appearance of an elevated risk related to the use of the primary medication. The collection of
the Baseline information will help to identify the presence of elevated background risk so that
event rates can be compared through sub-group analysis (those with or without prior events)
to determine if the elevated risk is likely to be related to background risk or use of
Kyntheum®
Information bias
Information bias arises when incorrect information about either exposure or outcome or any
covariates is collected in the study. This could occur in this study if physicians do not report
certain types of events of potential interest because they feel they are not related to the safety
of the medication. In order to address this, a data quality audit will be conducted with
physicians by the CRO to determine how well their medical records data matches the
information provided in the NIS.
9.10 Other Aspects
Audits and Inspections
The Quality Assurance (QA) unit at LEO may audit the study to ensure that study procedures
comply with the protocol and LEO standard operating procedures, and that collected data is
correct and complete. Representatives from IEC or Competent Authority may in rare cases
wish to inspect the study on site. Upon receiving notification of such inspection, the Study
Site Responsible must immediately contact LEO and must make the records available as
requested.
Archiving of Study Data and Documentation
During the course of the study the Site Responsible must as a minimum file the essential
documents (Section 3), the protocol (all used versions), the list of participating patients, the
written informed consents, the CRFs and the progress reports in the Study Site File. After
final database lock the Site Responsible must as a minimum store the list of participating
patients and the signed Informed Consent Forms on site for 10 years.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 29 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
The Site Responsible should store additional study documentation for a longer period of time
as required by any local regulations and/or hospital/clinic requirement.
10 Protection of human subjects
Ethical conduct of the study
This study is a non-interventional study where the existence of the study has no impact on the
patient. The treatment of the participating patients will not be any different from patients not
participating in the study, except for collection of informed consent to use of the patient’s data
and collection of the following non-standard Patient Reported Outcomes: PaGA, DLQI, PBI,
TSQM-9
This study will be conducted in accordance with the current version of the
Declaration of Helsinki (Fortalza, 2013)31
Good Pharmacoepidemiology Practices23
VFA Empfehlungen zur Verbesserung der Qualität und Transparenz von nicht-
interventionellen Studien (NIS)32
Gemeinsame Empfehlung des BfArM und des PEI zur Planung, Durchführung und
Auswertung von Anwendungsbeobachtungen33
AKG-Kodex 34
Fundamental principles on the lawful processing of personal data provided by the EU
directive on data protection (95/46/EC) must be respected.35
LEO Pharma A/S, Industriparken 55, Ballerup will be the data holder.
Information and Patient Consent/ Assent
The Site Study Responsible must give the patient oral and written information about the study
in a form that the patient can understand, and obtain the patient’s assent written consent
before collection by LEO of identifiable patient information (hereinafter referred to as
personal data).
Before consenting, the patient must be given sufficient time to consider and to pose questions.
Since the study is observational the consent only concerns the data collection per se and is not
consent to any interventional procedure or treatment.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 30 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
The patient must agree that his/her pseudononymized, key-coded data will be processed,
stored and may be transferred to third parties, e.g. other companies or authorities, that may be
located in other countries with potentially different regulations for data. Written informed
consent to use of collected data must always be obtained before collection of any personal,
sensitive data.
Data erroneously collected from patients for which written consent is not available, will not
be included in or will be deleted from the database, as feasible.
The patient has the right to withdraw his/her consent at any time without prejudice. In the
Informed Consent Form it is stated, that if consent is withdrawn, any data collected before
withdrawal of consent will be kept. The original, signed Informed Consent Forms must be
kept on the Site.
For details, see the Patient Information Sheet and Informed Consent Form.
Submissions
ANFOMED will ensure that the protocol and any amendments and the Patient Information
Sheet/Informed Consent Form and Questionnaires are submitted to the relevant Independent
Ethics Committees (IECs)/Institutional Review Boards (IRBs) and/or competent authorities
and/or other national or regional authorities according to German requirements. According to
applicable regulations, LEO, the appointed CRO or the Site Study Responsible will submit
required documents to the IEC / IRB, such as:
periodic updates on the progress of the study
notification of the end-of-study
a summary of the study results
LEO will keep a copy of all documents submitted and an updated list of all submission and
approval dates of all documents submitted to the IEC / IRB and/or authorities and will
provide the Site Responsible with a copy of this list. Copies of the documents will be
distributed upon request.
Remuneration of the participating sites is calculated based on the German “Gebührenordnung
für Ärzte (GOÄ)”. The study will be registered in a public accessable database.
According to AMG §67(6) the study will be notified to the “Bundesoberbehörde”, the
“kassenärztliche Bundesvereinigung” and the “Spitzenverbände der Krankenkassen”.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 31 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Data Protection
The patient personal data and treating physician personal data, which may be included in the
NIS database held by ANFOMED during study, but owned by LEO, shall be treated at all
times in compliance with all local applicable privacy laws and regulations
Patient´s personal information will remain pseudonymous and key-coded at all times.
The key-coded data will be stored in a secure database at ANFOMED/ LEO. They will not be
made available to the public. “Key-coded” means that at the time of patient enrolment in the
registry, he/ she will be assigned a code that will be used instead of his/her name to identify
him/her in any NIS-related documentation. The patient´s personal data are therefore
considered as “pseudonymous” as they will be identified by the assigned code rather than the
patient´s name.
A patient identification list will be kept by the Principal Investigator on site during the course
of the NIS and up to 10 years thereafter. The list will be kept confidential and will be
accessible only to the medical personnel at Principal Investigator’s office, monitors who will
visit the site to verify the data that are entered into the registry database, regulatory and health
authorities and ANFOMED or LEO auditors.
Regulatory and health authorities and other study personnel representing ANFOMED/ LEO
or its agents, may be granted direct access to the patient´s medical records to verify the
registry procedures or the accuracy of the data. The information will be treated strictly
confidentially and reviewed with the permission of the Principal Investigator.
LEO may disclose the key-coded data to its affiliated companies and subsidiaries, as well as
to its service providers and contractors, including, but not limited to ANFOMED and its
agents, and regulatory and health authorities. The key-coded data will be used by these parties
for performing the NIS, including review and scientific analysis.
The collected data will be combined with data from other participants in the Study and the
results may be used for scientific reasons including but not limited to research, publications or
presented at congresses. When the study results are published or reported, patient´s identity
will not be disclosed.
When archiving or processing personal data pertaining to the treating physician and/or to the
patients, the Sponsor shall take all appropriate measures to safeguard and prevent access to
this data by any unauthorized (third) party.
The use of key-coded or indirect identifiable data is essential for the following reasons:
Patients are followed up for 52 weeks, with collection of longitudinal information.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 32 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Quality control of the data will be done with the treating physicians in order to guarantee
the data quality, with possible correction requests for missing or inconsistent data as
regards key data.
The patient will be informed of his/her right of access, objection and correction of the data
recorded during this study, and that this right may be exercised at any time through his/her
physician.
11 Management and Reporting of Adverse Events and Other Experiences
DEFINITIONS
Adverse Event (AE)
An adverse event (AE) is any untoward medical occurrence in a patient administered a
medicinal product and which does not necessarily have to have a causal relationship with this
treatment. An AE can therefore be any unfavourable and unintended sign (e.g. an abnormal
laboratory finding), symptom, or disease temporally associated with the use of a medicinal
product, whether or not considered related to the medicinal product.
Adverse Drug Reaction (ADR)
An Adverse Drug Reaction (ADR) is a response to a medicinal product which is noxious and
unintended. Response in this context means that a causal relationship between a medicinal
product and an adverse event is at least a reasonable possibility. ADRs may arise from use of
the product within or outside the terms of the marketing authorisation or from occupational
exposure. Conditions of use outside the marketing authorisation include off-label use,
overdose, misuse, abuse and medication errors.
Other Experiences (OE)
Other Experiences (OE) are considered ‘non events’ which describe the circumstances around
the use of a drug which potentially could cause drug related problems or which could
potentially give positive knowledge of a drug.
These may or may not be associated with adverse events.
These cases include but are not limited to:
unintended beneficial effects,

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 33 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
drug exposure in utero,
drug exposure before and/or during pregnancy,
drug exposure via breast milk,
paternal drug exposure before and/or during pregnancy,
occupational exposure,
drug overdose or abuse,
drug misuse,
medication error (including potential and intercepted),
off label use/unapproved use,
lack of efficacy,
drug and food interaction,
any suspicion of counterfeit product
any suspected transmission of an infectious agent by a medicinal product
CLASSIFICATION
Seriousness
A serious ADR or Adverse Event is any ADR or Adverse Event which results in death, is life
threatening, requires inpatient hospitalisation or prolongation of existing hospitalisation,
results in persistent or significant disability or incapacity, or is a congenital anomaly or birth
defect.
Life-threatening in this context refers to a reaction in which the patient was at risk of death a
the time of the event; it does not refer to an event which hypothetically might have caused
death if it were more severe.
Medical and scientific judgement should be exercised in deciding whether other situations
should be considered serious, such as important medical events that might not be immediately
life-threatening or result in death or hospitalisation but might jeopardise the patient or might
require intervention to prevent one of the other outcomes listed above.
Any suspected transmission of an infectious agent via a medicinal product is considered a
serious ADR.
Severity

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 34 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Severity is a clinical observation and describes the intensity of the event.
Mild: Transient symptoms, no interference with the patient’s daily activities
Moderate: Marked symptoms, moderate interference with the patient’s daily activities
Severe: Considerable interference with the patient’s daily activities.
Causality
Related: A reasonable temporal relationship between the medicinal product
administration and the event where there is no other obvious explanation for the
occurrence of the event
Not related: There is evidence for (an) alternative explanation(s) for the event (e.g. the
event is explained by one or more of the following: a) the patient’s medical condition, b) a
concomitant medication for which the event is labelled, or c) the event occurred prior to
the introduction of the medicinal product)
RESPONSIBILITIES
Physician’s responsibility for reporting of Adverse Drug Reactions, Adverse Events and
Other Experiences
Physicians and other healthcare professionals involved in the study should report to LEO any
SAE, AE, OE, ADR or pregnancy in patients who are or were treated with Kyntheum® during
the study within 24 hours after becoming aware of it.
The provided LEO Adverse Event Form – Marketed Products or in case of pregnancies – the
Pregnancy Form (Part 1) should be used for the reports and completed carefully, including the
section on causality. A copy of the completed form must be sent within 24 hours to the LEO
Safety Contact Person by email or fax (contact details below) .
Email: [email protected]
Fax: + 49 6102 201 125

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 35 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
LEO will notify the competent authorities of all serious and non-serious ADRs related to LEO
product(s) and pregnancies according to the currently applicable pharmacovigilance
guidelines for non-interventional studies.
If the patient is enrolled in parallel in the PsoBest Registry both study IDs (PsoBest ID and
LIBERO ID) must be included in the forms sent to LEO and to PsoBest. In order to reduce
duplicate reporting and as LEO is reporting the cases to the competent authorities the
physician does not need to report to the Paul Ehrlich Institute (PEI) if the patient participates
in LIBERO. If - for one reason or another – the physician has also reported the event to PEI,
this should be clearly marked on the form (please tick in tick box on the form) to facilitate
identification of duplicate reports.
The original “LEO Adverse Events Form - Marketed Products” and any follow-up
documentation submitted by email or fax to [email protected] remain in the
study documentation folder and will be sent together with the other forms to ANFOMED after
visit 3 (week 12), visit 5 (week 36), and visit 7 (week 52).
In addition, the physician should notify local competent authorities (PEI for biological
medicinal products and BfArM for the rest) or the concerning manufacturer/ marketing
authorization holder, but not both, of AEs at least possibly related to non-LEO products
including a reference to the Study ID or study title.
Important additional information obtained later, e.g. a report of diagnostic procedures or a
hospital discharge summary should be provided in pseudonymous, key-coded form to LEO as
follow-up report by the same route as the initial AE/OE report.
The physician should record a precise medical term for an AEs preferably a diagnosis. Please
observe that death or surgery are considered as outcome, not as an event, so it is the event
leading to death or surgery which should be recorded.
Only one AE or OE should be recorded per form. If no diagnosis is available, the physician
should record each sign and symptom in separate form. For each AE or OE the causality,
severity and outcome must be included.
LEO may request further information from the physician in order to fully assess the safety
reports.
LEO responsibilities for reporting of safety-related data

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 36 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
LEO will notify the competent authorities of all serious and non-serious ADRs related to LEO
product(s) and pregnancies according to currently applicable pharmacovigilance guidelines
for non-interventional studies.
All safety data collected in the study database or reported to LEO pharmacovigilance will be
summarized in the Non-Interventional Study Report.
12 Plans for Disseminating and Communicating Study Results
LEO will prepare a Non-Interventional Study Report based on the results obtained. The Final
Study Report shall be reviewed and approved by the Coordinating Investigator and be
available within one year from collection of the last data point. In case the Coordinating
Investigator changes during study conduct, all investigators will be informed and asked for
their consent to delegate this responsibility to the new Coordinating Investigator. The sites
that have enrolled patients in the study will be informed about the results when the report is
finalized. Summary results will be posted on a publicly accessible database.
LEO aims to have the results of this study published and acknowledges the right of the
participating sites to publish results from this study.
The principles guiding publications from this NIS dictate that usable scientifically or
medically significant information, hypotheses or analysis are widely disseminated, that good
publication practice is adhered to (and in particular the GPP2 guidelines (http://www.gpp-
guidelines.org), that redundant publication is avoided, that publications are of high scientific
and linguistic standard, that an ethical approach is applied at all times, that the reputations of
the NIS, the (financial) Sponsors and their stakeholders and the participating investigators
should be up held as well as all relevant regulatory requirements.
A primary publication based on all study data and following the STROBE guidelines36
in an
international peer reviewed journal is foreseen with the Coordinating Investigator as the first
or last author. Any manuscripts for secondary publications or abstracts may not be submitted
until after the primary publication manuscript has been accepted for publication. Any
manuscript or abstract must be sent to LEO for commenting at least 30 calendar days prior to
submission.
LEO has the right to use the data and results for regulatory and reimbursement purposes, and
for internal presentation within the company and to partners. In addition, LEO may use the
data for analysis of pooled data from various studies.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 37 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
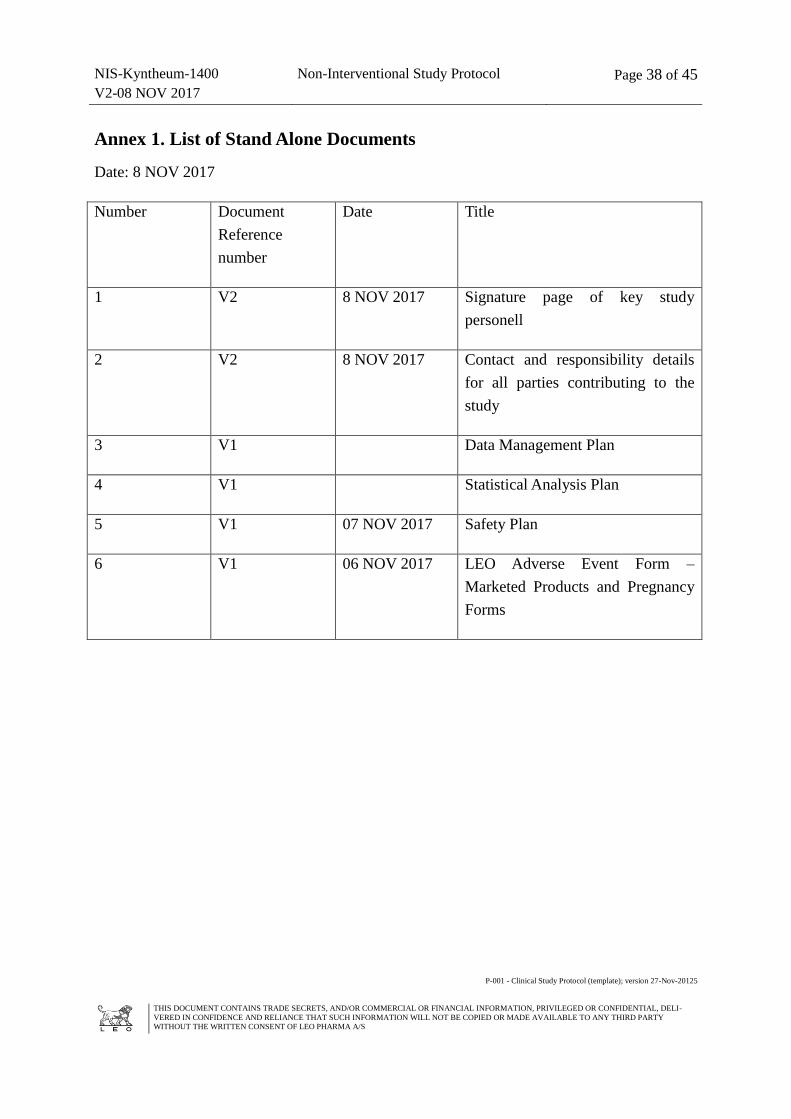
NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 38 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Annex 1. List of Stand Alone Documents
Date: 8 NOV 2017
Number Document
Reference
number
Date Title
1 V2 8 NOV 2017 Signature page of key study
personell
2 V2 8 NOV 2017 Contact and responsibility details
for all parties contributing to the
study
3 V1 Data Management Plan
4 V1 Statistical Analysis Plan
5 V1 07 NOV 2017 Safety Plan
6 V1 06 NOV 2017 LEO Adverse Event Form –
Marketed Products and Pregnancy
Forms

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 39 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Annex 2: Signature Page
The following persons approve the Study Protocol:
“Prospective, observational, non-interventionaL, multicenter real-world evIdence (RWE)
study of Brodalumab 210mg (Kyntheum®) to manage patients with modERate-to-severe
plaque psOriasis in daily practice (LIBERO)”
Version 2.0 – 8 NOV 2017
Place, Date
Place, Date
Place, Date
Place, Date
Place, Date
Place, Date

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 40 of 45
Anfomed GmbH
Röttenbacher Str. 17
91096 Möhrendorf
Study Manager
LEO Pharma GmbH
Frankfurter Str. 233
D-63263 Neu-Isenburg
Protocol author
Biostatistics
Anfomed GmbH
Röttenbacher Str. 17
91096 Möhrendorf

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 41 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
Pharmacovigilance
LEO Pharma GmbH
Frankfurter Str. 233
D-63263 Neu-Isenburg
Ethic committee
Freiburger Ethik-Kommission
GmbH international
Mozartstraße 21
D-79104 Freiburg
(Germany)

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 42 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
References
1 Goff KL, Karimkhani C, Boyers LN, et al. The global burden of psoriatic skin disease. Br J
Dermatol 2015; 172: 1665-8.
2 Gupta R, Debbaneh MG, Liao W. Genetic epidemiology of psoriasis. Curr Dermatol Rep
2014; 3: 61-78.
3 World Health Organization (WHO). Global Report on Psoriasis. Available
from: http://apps.who.int/iris/bitstream/10665/204417/1/9789241565189_eng.pdf (Accessed
July 2017)
4 Lebwohl MG, Bachelez H, Barker J, et al. Patient perspectives in the management of
psoriasis: results from the population- based Multinational Assessment of Psoriasis and
Psoriatic Arthritis Survey. J Am Acad Dermatol 2014; 70(5): 871–881.
5 Beringer A, Noack M, Miossec P. IL-17 in Chronic Inflammation: From Discovery to
Targeting. Trends Mol Med. 2016 Mar;22(3): 230-41. 6 Elder JT. Genome-wide association scan yields new insights into the immunopathogenesis of
psoriasis. Genes Immun 2009; 10: 201-9. 7 Ellinghaus E, Ellinghaus D, Stuart PE, et al. Genome-wide association study identifies a
psoriasis susceptibility locus at TRAF3IP2. Nat Genet 2010; 42: 991-5. 8 Li J, Li D, Tan Z. The expression of interleukin-17, interferon-gamma, and macrophage
inflammatory protein-3 alpha mRNA in patients with psoriasis vulgaris. J Huazhong Univ Sci
Technolog Med Sci 2004; 24: 294-6. 9 Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-
17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine
production by human keratinocytes. J Invest Dermatol 1998; 111: 645-9. 10
Lowes MA, Kikuchi T, Fuentes-Duculan J, et al. Psoriasis vulgaris lesions contain discrete
populations of Th1 and Th17 T cells. J Invest Dermatol 2008; 128: 1207-11. 11
Johnston A, Fritz Y, Dawes SM, et al. Keratinocyte overexpression of IL-17C promotes
psoriasiform skin inflammation. J Immunol 2013; 190: 2252-62. 12
Nograles KE, Zaba LC, Guttman-Yassky E, et al. Th17 cytokines interleukin
(IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J
Dermatol 2008; 159: 1092-102. 13
Ramirez-Carrozzi V, Sambandam A, Luis E, et al. IL-17C regulates the innate immune
function of epithelial cells in an autocrine manner. Nat Immunol 2011; 12: 1159-66. 14
Campa M, Mansouri B, Warren R, Menter A. A Review of Biologic Therapies Targeting IL-
23 and IL-17 for Use in Moderate-to-Severe Plaque Psoriasis. Dermatol Ther (Heidelb). 2016
Mar;6(1): 1-12.

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 43 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
15
Coimbra S, Figueiredo A, Santos-Silva A. Brodalumab: an evidence-based review of its
potential in the treatment of moderate-to-severe psoriasis. Core Evid. 2014 Jul 21;9:89-97.
16 Russell CB, Rand H, Bigler J, Kerkof K, Timour M, Bautista E, Krueger JG,
Salinger DH, Welcher AA, Martin DA. Gene expression profiles normalized in
psoriatic skin by treatment with brodalumab, a human anti-IL-17 receptor
monoclonal antibody. J Immunol. 2014 Apr 15;192(8):3828-36. 17
European Commission, Community register of medicinal products for human use,
Kyntheum® (brodalumab). Available from: http://ec.europa.eu/health/documents/community-
register/html/h1155.htm (Accessed July 2017)
18 Papp KA, Reich K, Paul C, Blauvelt A, Baran W, Bolduc C, Toth D, Langley RG,Cather J,
Gottlieb AB, Thaçi D, Krueger JG, Russell CB, Milmont CE, Li J, Klekotka PA, Kricorian G,
Nirula A. A prospective phase III, randomized, double-blind, placebo-controlled study of
brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016
Aug;175(2):273-86. 19 Lebwohl M, Strober B, Menter A, Gordon K, Weglowska J, Puig L, Papp K, Spelman L,
Toth D, Kerdel F, Armstrong AW, Stingl G, Kimball AB, Bachelez H, Wu JJ, Crowley J,
Langley RG, Blicharski T, Paul C, Lacour JP, Tyring S, Kircik L, Chimenti S, Callis Duffin K,
Bagel J, Koo J, Aras G, Li J, Song W, Milmont CE, Shi Y, Erondu N, Klekotka P, Kotzin B,
Nirula A. Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. N Engl J
Med. 2015 Oct;373(14):1318-28 and Supplement. 20
Gordon KB, Kimball AB, Chau D, Viswanathan HN, Li J, Revicki DA, Kricorian G,
Ortmeier BG. Impact of brodalumab treatment on psoriasis symptoms and health-related
quality of life: use of a novel patient-reported outcome measure, the Psoriasis Symptom
Inventory. Br J Dermatol. 2014 Mar;170(3):705-15. 21
Attia A, Abushouk AI, Ahmed H, Gadelkarim M, Elgebaly A, Hassan Z, Abdel-Daim MM,
Negida A. Safety and Efficacy of Brodalumab for Moderate-to-Severe Plaque Psoriasis: A
Systematic Review and Meta-Analysis. Clin Drug Investig. 2017 May;37(5):439-451. 22
Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the
approximation of the laws, regulations and administrative provisions of the Member States
relating to the implementation of good clinical practice in the conduct of clinical trials on
medicinal products for human use. Official Journal of the European Communities L/21/34
1.5.2001. 23
Epstein, M on behalf of ISPE. Guidelines for Good Pharmacoepidemiology Practices
(GPP). Pharmacoepidemiology and Drug Safety 2016; 25:2-10. 24
Maintenance and Support Services Organization. Medical dictionary for regulatory
activities, accessed on Oct 6th
2017 (https://www.meddra.org/basics)

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 44 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
25 Mrowietz U, de Jong EM, Kragballe K, Langley R, Nast A, Puig L, Reich K, Schmitt J,
Warren RB. A consensus report on appropriate treatment optimization and transitioning in the
management of moderate-to-severe plaque psoriasis. J Eur Acad Dermatol Venereol. 2014
Apr;28(4):438-53. 26
Von Kiedrowski R, Dirschka T, Kurzen H, Ostendorf R, Petering H, Reinhold U, Sebastian
M, Termeer C: Aktualisierte Empfehlungen für die ambulante Versorgung von Psoriasis
Patienten: Praxisnaher Behandlungspfad Psoriasis vulgaris 2016. Onkoderm e.V. 2016
accessed at Oct 6th
2017 via https://www.praxis-
kiedrowski.de/files/2016_behandlungspfad_psoriasis.pdf 27 Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)--a simple practical measure
for routine clinical use. Clin Exp Dermatol. 1994 May;19(3):210-6. 28
Feuerhahn J, Blome C, Radtke M, Augustin M. Validation of the patient benefit index for
the assessment of patient-relevant benefit in the treatment of psoriasis. Arch Dermatol Res.
2012 Aug;304(6):433-41. 29
Bharmal M, Payne K, Atkinson MJ, Desrosiers MP, Morisky DE, Gemmen E. Validation of
an abbreviated Treatment Satisfaction Questionnaire for Medication (TSQM-9) among
patients on antihypertensive medications. Health Qual Life Outcomes. 2009 Apr 27;7: 36. 30
Bagel J, Levi E, Tyring S, Knuckles ML. Real-life treatment profile of calcipotriene and
betamethasone dipropionate topical suspension in patients with psoriasis vulgaris. J Drugs
Dermatol. 2014 Nov;13(11):1374-9. 31
World Medical Association Declaration of Helsinki. Ethical principles for Medical Research
Involving Human Subjects, Helsinki 1964, amended in Tokyo 1975, Venice 1983, Hong Kong
1989, South Africa 1996, Edinburgh 2000, Seoul 2008 and Fortaleza 2013. 32
Verband Forschender Arzneimittelhersteller e.V: VFA Empfehlungen nicht interventionelle
Studien. Stand 2014, eingesehen am 6. Okt 2017, abrufbar unter dem Link:
https://www.vfa.de/de/arzneimittel-forschung/datenbanken-zu-arzneimitteln/nisdb/nis-
empfehlungen
33
Empfehlungen des Bundesinstituts für Arzneimittel und Medizinprodukte und des Paul-
Ehrlich-Instituts zur Planung, Durchführung und Auswertung von Anwendungs-
beobachtungen vom 7. Juli 2010; eingesehen am 6. Okt. 2017, abrufbar unter dem Link:
http://www.pei.de/SharedDocs/Downloads/pu/klinische-pruefung/100707-awb-
kommentierung-fachkreise.pdf?__blob=publicationFile&v=1
34
Verhaltenskodex der Mitglieder des „Arzneimittel und Kooperation im Gesundheitswesen
e.V.“ AKG e.V. in der Fassung vom 07.04.2008, zuletzt geändert am 22.04.2015 (durch das
Bundeskartellamt anerkannt mit Beschluss vom 25. September 2014), eingesehen am 6.Okt.
2017, abrufbar unter dem Link: http://www.ak-gesundheitswesen.de/verhaltenskodex/

NIS-Kyntheum-1400
V2-08 NOV 2017
Non-Interventional Study Protocol Page 45 of 45
P-001 - Clinical Study Protocol (template); version 27-Nov-20125
THIS DOCUMENT CONTAINS TRADE SECRETS, AND/OR COMMERCIAL OR FINANCIAL INFORMATION, PRIVILEGED OR CONFIDENTIAL, DELI-
VERED IN CONFIDENCE AND RELIANCE THAT SUCH INFORMATION WILL NOT BE COPIED OR MADE AVAILABLE TO ANY THIRD PARTY
WITHOUT THE WRITTEN CONSENT OF LEO PHARMA A/S
35
EU Directive 95/46/EC of the European Parliament and of the Council of 24 October 1995
on the Protection of Individuals with Regard to the Processing of Personal Data and on the
Free Movement of Such Data. Official Journal of the European Communities L281/31
23.11.1995. Directive on data protection (95/46/EC)
36
Von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP, for the
STROBE initiative. Strengthening the Reporting of Observational Studies in Epidemiology
(STROBE) Statement: guidelines for reporting observational studies. Lancet 2007; 370:1453-
7





























