Embed Size (px)
Citation preview
Paul J. McMurdie w/ contribution from Prof Susan Holmes, Stanford University
Lecture 3: Mixture Models for Microbiome data
1
Lecture 3: Mixture Models for Microbiome data
Outline: - Hypothesis Test Intro (t, wilcoxan) - Multiple Testing (FDR) - Mixture Models (Negative Binomial) - DESeq2 / Don’t Rarefy. Ever.
2
Hypothesis Tests - review
• A hypothesis is a precise disprovable statement.
• “Null hypothesis” - the default position. “Nothing special”
• Alternative/Rejection: Evidence disagrees with the Null
• Null hypothesis cannot be confirmed by the data.
3
Hypothesis Tests - P-value• If the Null Hypothesis is true, then a statistic, used to
perform the test, will follow the Null distribution.
• P-value is the tail probability under the Null distribution
4

Hypothesis Tests - P-value
P-value restated: “The probability of observing an event as extreme, or more extreme, by chance.”
This is “Type-1 Error”, or “False Positives”
5
Hypothesis Tests - some examples
test R functiont-test t.testMann-Whitney U-test wilcox.testcorrelation test cor.testChi-Square test chisq.testNeg-Binom Wald test DESeq2::nbinomWaldTest
6
Hypothesis Tests - t-test
• one-sample t-test: mean is different from zero • two-sample t-test: means are different from each other assume variances are the same • Welch’s t-test: means are different from each other estimates variance of each class separately • paired t-test: each value in one class has a “pair” in other
!Wikipedia: Student’s t-test !All of the above in R: t.test
7
Mann-Whitney U-statistic calculation:
– Convert all observations to ranks
– Compute the sum of ranks in each class, R1 and R2
– U1 =R1 –n1(n1 +1)/2
– U2 =R2 –n2(n2 +1)/2
– U = min(U1, U2)
Mann-Whitney U-test Wilcoxon rank sum test
Hypothesis Tests - Wilcoxan test
8
• Assumptions:
• Independent observations
• Observations can be ordered with respect to each other
• Null hypothesis: Two groups come from same distribution
• A random draw from each sample class A, B:
• P(A>B) = P(B>A)
• Two-sided alternative hypothesis: P(A>B) = P(B>A)
• Interpretation: rejection implies different medians
• More robust to outliers, more generally applicable than t-test
• This is 2-class special case of Kruskal-Wallis Rank Sum Test
Mann-Whitney U-test Wilcoxon rank sum test
Hypothesis Tests - Wilcoxan test
kruskal.test
9
Mann-Whitney U-test Wilcoxon rank sum test
Hypothesis Tests - Wilcoxan test
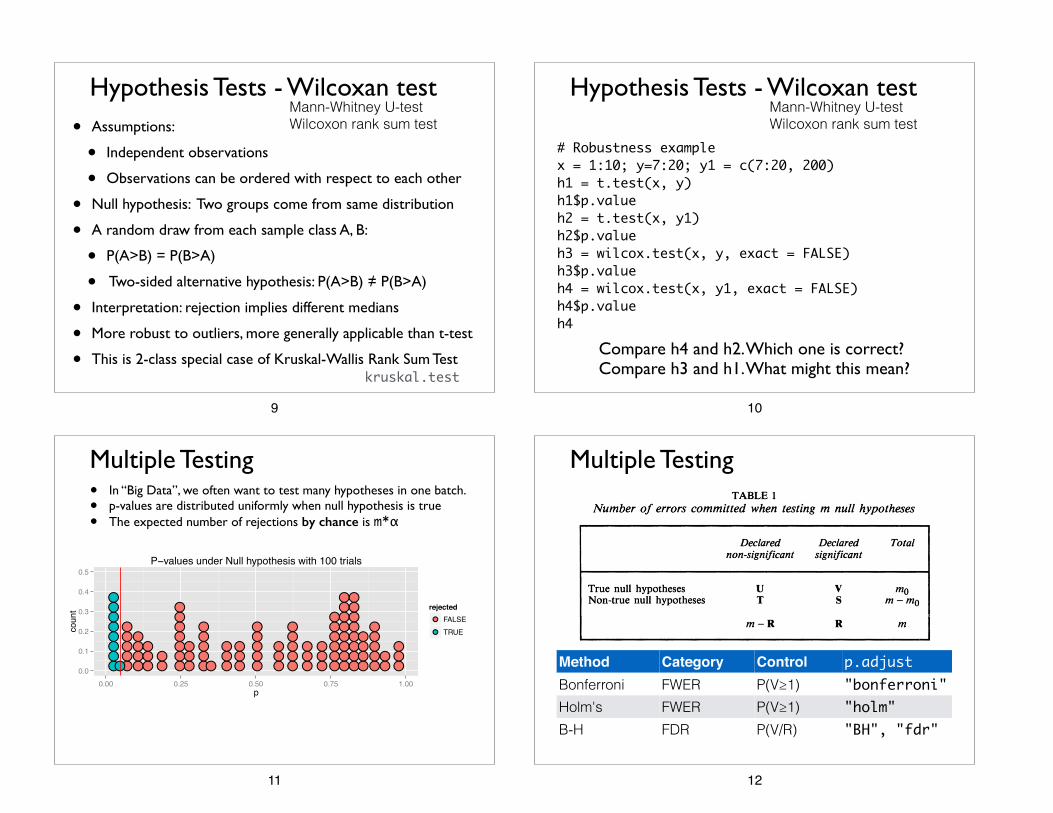
# Robustness examplex = 1:10; y=7:20; y1 = c(7:20, 200)h1 = t.test(x, y)h1$p.valueh2 = t.test(x, y1)h2$p.valueh3 = wilcox.test(x, y, exact = FALSE)h3$p.valueh4 = wilcox.test(x, y1, exact = FALSE)h4$p.valueh4
Compare h4 and h2. Which one is correct? Compare h3 and h1. What might this mean?
10
Multiple Testing• In “Big Data”, we often want to test many hypotheses in one batch. • p-values are distributed uniformly when null hypothesis is true • The expected number of rejections by chance is m*α
●●●●●
●●●●
●●●
●●
●●●●●●●
●●●
●●●●●
● ●●●
●●●
●●●●●
●●●
●●●●●
●●●
●●●
●●●●●●
●●●●●●●●
●●●●●●●●
●●●●
●●●●●
●●
●●●
●●●●●●●●
●0.0
0.1
0.2
0.3
0.4
0.5
0.00 0.25 0.50 0.75 1.00p
coun
t rejected●
●
FALSETRUE
P−values under Null hypothesis with 100 trials
11
Multiple Testing
Method Category Control p.adjustBonferroni FWER P(V≥1) "bonferroni"Holm's FWER P(V≥1) "holm"B-H FDR P(V/R) "BH", "fdr"
12

• To ensure overall significance at a given α, one performs each individual test at α’ = α/m • Useful when need to correct for just a few hypotheses • Very stringent, results in “loss of power” - increase in Type II error, decreases sensitivity
Multiple Testing - Bonferroni
13
• Rather than control probability of any errors, FDR instead controls the proportion of False Positives in the set of positives.
• Input: p-values for a set of univariate tests
• Output: p-values that are adjusted to FDR: “q-values”
• e.g. A collection of tests rejected at PFDR<=0.05 will have 5% or fewer false positives
• This is what is meant be “controlling” the false positive rate
Multiple Testing - Benjamini-Hochberg
Benjamini & Hochberg (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological), 57(1), 289–300.
14
Multiple Testing - Independent Filtering
•Is a general approach that can substantially increase the [statistical] efficiency of experiments
•Uses filter/ test pairs that are independent under the null hypothesis
•but correlated under the alternative
Bourgon, Gentleman, & Huber (2010) Independent filtering increases detection power for high-throughput experiments. PNAS 107(21) 9546-9551
Independent filtering
e.g. remove features with very low mean abundance
15
Poisson sampling simulation “shot noise” !
See: `shot-noise-simulation.Rmd/HTML` !
Non-mixture generative model
16
Transition: Mixture Models
Technical details in: mixture-model-Holmes-mathy-details.pdf
17
Example: Finite mixture of two normals
Flip a fair coin.
If it comes up heads
Generate a random number from a Normal with mean 1 and variance 0.25. R: `rnorm` function.
If it comes up tails
Generate a random number from a Normal with mean 2 and variance 0.25.
This is what the resulting histogram would look like if we did this 10,000 times.
Finite Mixture Model
18
Example: Finite mixture of two normals
However in many cases the separation is not so clear.
Challenge: Here is a histogram generated by two Normals with the same variances.
Can you guess the two parameters for these two Normals?
Finite Mixture Model
19
Here we knew the answer
(the source every data point)
!In practice, this information is usually missing, and we call it a latent variable
!Discovering the hidden class: EM
For simple parametric components, can use EM (Expectation-Maximization) algorithm to infer the value of the hidden variable.
Finite Mixture Model
20

Very popular iterative procedure
Lots of implementations. E.g. FlexMix
http://cran.r-project.org/web/views/Cluster.html
http://cran.r-project.org/web/packages/flexmix/index.html
Expectation Maximization (EM)
http://en.wikipedia.org/wiki/Expectation–maximization_algorithm
1. First, initialize θ to some random values. 2.Compute best value for U. 3. Use the just-computed values of U
to compute a better estimate for θ. Parameters associated with a particular value of U only use data points whose associated latent variable has that value. 4. Iterate steps 2 and 3 until convergence
21
Infinite Mixture ModelSometimes mixtures can be useful without us having to find who came from which distribution.
This is especially the case when we have (almost) as many different distributions as observations.
In some cases the total distribution can still be studied, even if we don’t know the source of each component distribution.
e.g. Gamma-Poisson a.k.a. Negative Binomial
22
Infinite Mixture Model - N.B.Generative Description:
Negative Binomial is useful for modeling: • Overdispersion (in Ecology) • Simplest Mixture Model for Counts • Different evolutionary mutation rates • Throughout Bioinformatics and Bayesian Statistics • Abundance data
Summarized Mathematically:
variance:Poisson Overdispersion
23
Finite Mixture Models
Mixture of Normals with different means and variances.
Mixtures of multivariate Normals with different means and covariance matrices
Decomposing the mixtures using the EM algorithm.
Common Infinite Mixture Models
Gamma-Poisson for read countsDirichlet-Multinomial (Birthday problem and the Bayesian setting).
Summary of Mixture Models
24
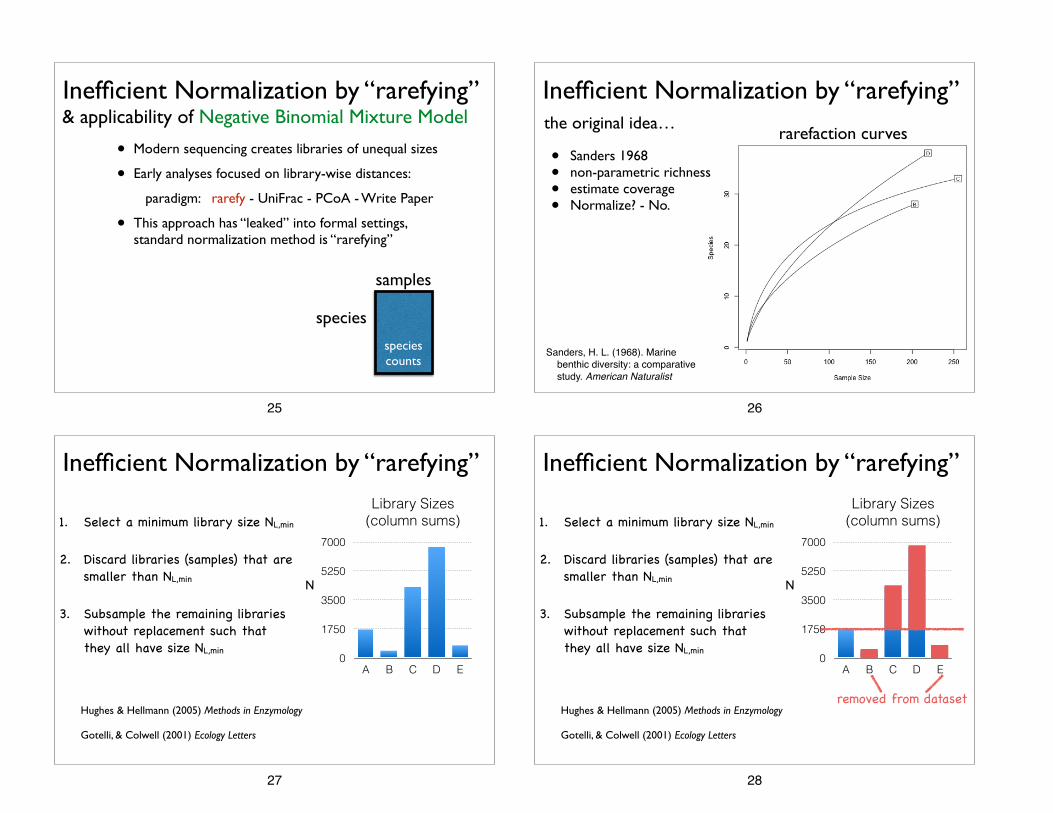
• Modern sequencing creates libraries of unequal sizes
• Early analyses focused on library-wise distances:
paradigm: rarefy - UniFrac - PCoA - Write Paper
• This approach has “leaked” into formal settings, standard normalization method is “rarefying”
Inefficient Normalization by “rarefying”
species
samples
species counts
& applicability of Negative Binomial Mixture Model
25
the original idea…rarefaction curves
• Sanders 1968 • non-parametric richness • estimate coverage • Normalize? - No.
Sanders, H. L. (1968). Marine benthic diversity: a comparative study. American Naturalist
Inefficient Normalization by “rarefying”
26
Gotelli, & Colwell (2001) Ecology Letters
Hughes & Hellmann (2005) Methods in Enzymology
1. Select a minimum library size NL,min
2. Discard libraries (samples) that are smaller than NL,min
3. Subsample the remaining libraries without replacement such that they all have size NL,min
Library Sizes (column sums)
0
1750
3500
5250
7000
A B C D E
N
Inefficient Normalization by “rarefying”
27
Gotelli, & Colwell (2001) Ecology Letters
Hughes & Hellmann (2005) Methods in Enzymology
Library Sizes (column sums)
0
1750
3500
5250
7000
A B C D E
N
removed from dataset
1. Select a minimum library size NL,min
2. Discard libraries (samples) that are smaller than NL,min
3. Subsample the remaining libraries without replacement such that they all have size NL,min
Inefficient Normalization by “rarefying”
28
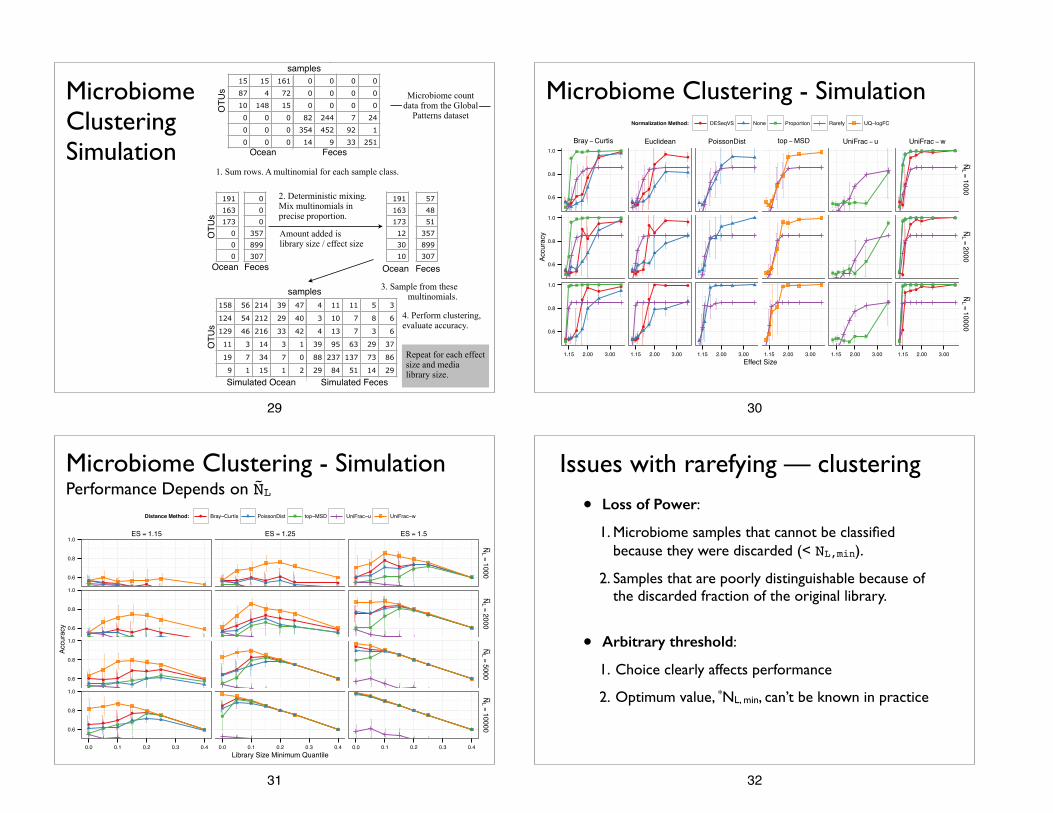
Microbiome Clustering Simulation
samples
OTU
s
test null
OTU
s
191163173123010
574851357899307
191163173000
000
357899307
15 15 161 0 0 0 0
87 4 72 0 0 0 0
10 148 15 0 0 0 0
0 0 0 82 244 7 24
0 0 0 354 452 92 1
0 0 0 14 9 33 251
samples
OTU
s
Ocean Feces
OTU
s
Ocean Feces
1. Sum rows. A multinomial for each sample class.
2. Deterministic mixing. Mix multinomials in precise proportion.
Ocean Feces
Microbiome Clustering Simulationsamples
OTU
s
Environment
OTU
s
1. Sum rows for each environment.
2. Sample from multinomial.
Differential Abundance Simulation
3. Multiply randomly
selected OTUs within test class by
effect size.
4. Perform differential abundance tests,
evaluate performance.
158 56 214 39 47 4 11 11 5 3
124 54 212 29 40 3 10 7 8 6
129 46 216 33 42 4 13 7 3 6
11 3 14 3 1 39 95 63 29 37
19 7 34 7 0 88 237 137 73 86
9 1 15 1 2 29 84 51 14 29
OTU
s
samples
Simulated Ocean Simulated Feces
3. Sample from these multinomials.
4. Perform clustering, evaluate accuracy.
BA
Microbiome count data from the Global
Patterns dataset
Repeat for each effect size and media library size.
Repeat for each environ-ment, number of sam-ples, effect size, and median library size.
Amount added is library size / effect size
29
Microbiome Clustering - Simulation
Bray −Curtis Euclidean PoissonDist top −MSD UniFrac − u UniFrac −w
●●●
●●
●
●
●
●●●
●
●
●
●●
●●●
●
●
●●
●
●●●
●●
●
●●
●●●
●
●
●
● ●
●●●
●
●
● ● ●
●●
●
●●
● ● ●
●●
●
●
● ●● ●
●
●
● ● ●● ●
●
0.6
0.8
1.0
0.6
0.8
1.0
0.6
0.8
1.0
N ~L=
1000N ~
L=
2000N ~
L=
10000
1.15 2.00 3.00 1.15 2.00 3.00 1.15 2.00 3.00 1.15 2.00 3.00 1.15 2.00 3.00 1.15 2.00 3.00Effect Size
Accu
racy
Normalization Method: ● DESeqVS None Proportion Rarefy UQ−logFC
30
Performance Depends on ÑLMicrobiome Clustering - Simulation
ES = 1.15 ES = 1.25 ES = 1.5
●● ●
●●
●●
● ●●
●●
●
●
● ● ●
● ● ●
●
● ●●
● ●●
●
● ●●
●●
● ●
●
●
●●
●●
●
●●
● ●●
●
●
●
●●
●●
●
●
●
●
●
● ● ●
●
● ●
● ●●
●
●
●● ●
●
●
●
●
●●
●●
●
●
●
0.6
0.8
1.0
0.6
0.8
1.0
0.6
0.8
1.0
0.6
0.8
1.0
N ~L=
1000N ~
L=
2000N ~
L=
5000N ~
L=
10000
0.0 0.1 0.2 0.3 0.4 0.0 0.1 0.2 0.3 0.4 0.0 0.1 0.2 0.3 0.4Library Size Minimum Quantile
Accu
racy
Distance Method: ● Bray−Curtis PoissonDist top−MSD UniFrac−u UniFrac−w
31
Issues with rarefying — clustering
• Loss of Power:
1. Microbiome samples that cannot be classified because they were discarded (< NL,min).
2. Samples that are poorly distinguishable because of the discarded fraction of the original library.
• Arbitrary threshold:
1. Choice clearly affects performance
2. Optimum value, *NL, min, can’t be known in practice
32
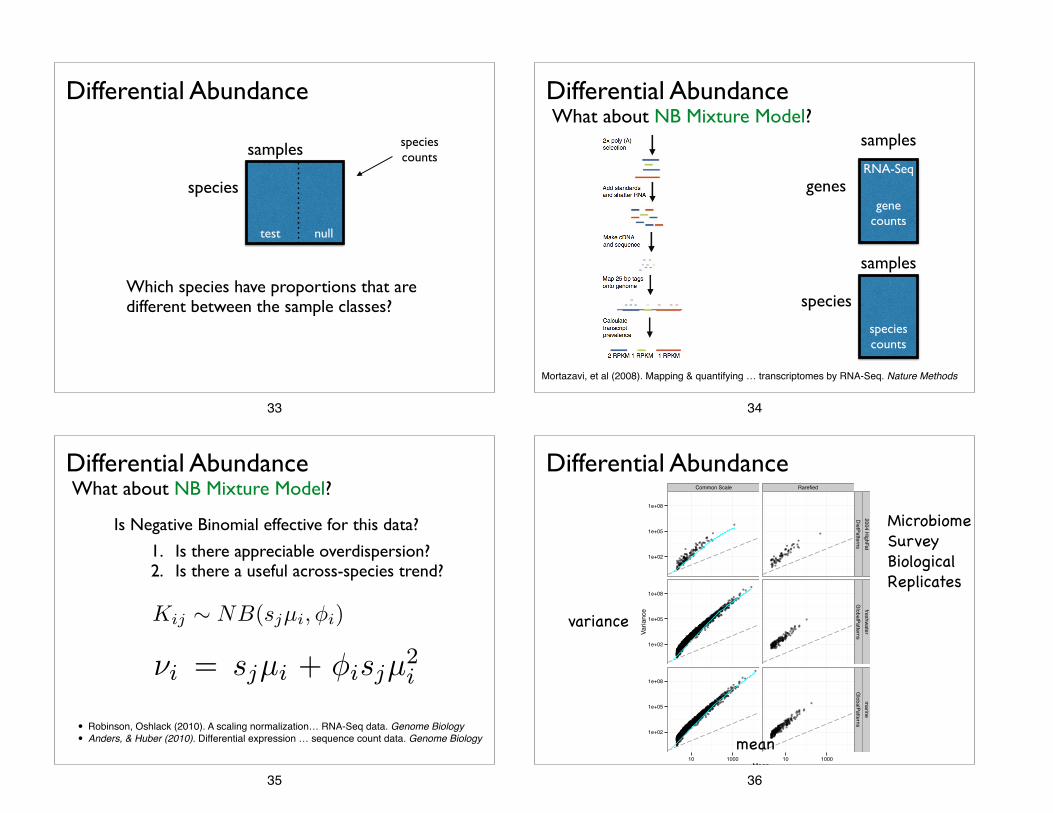
Differential Abundance
species
samples species counts
test null
Which species have proportions that are different between the sample classes?
33
Differential Abundance
Mortazavi, et al (2008). Mapping & quantifying … transcriptomes by RNA-Seq. Nature Methods
genes
samples
species
samples
RNA-Seq
species counts
gene counts
What about NB Mixture Model?
34
1. Is there appreciable overdispersion? 2. Is there a useful across-species trend?
2
a Poisson random variable in most cases [5]. How-ever, we are usually interested in understanding vari-ation among biological replicates, wherein a mixturemodel is necessary to account for the added uncer-tainty [6]. Taking a hierarchical model approach withthe Gamma-Poisson has provided a satisfactory fit toRNA-Seq data [7], as well as a valid regression frame-work that uses generalized linear models [8]. A Gammamixture of Poisson variables gives the negative binomial(NB) distribution [6, 7] and several RNA-Seq analysispackages now model the counts, K, for gene i, in sam-ple j according to:
Kij ⇠ NB(sjµi,�i) (1)
where sj is a linear scaling factor for sample j thataccounts for its library size, µi is the mean proportionfor gene i, and �i is the dispersion parameter for genei. The variance is ⌫i = sjµi + �isjµ
2i , with the NB
distribution becoming Poisson when � = 0. Recogniz-ing that � > 0 and estimating its value is important ingene-level tests. This reduces false positive genes thatappear significant under a Poisson distribution, but notafter accounting for non-zero dispersion.
The uncertainty in estimating �i for every gene whenthere is a small number of samples — or a small num-ber of biological replicates — can be mitigated by shar-ing information across the thousands of genes in an ex-periment, leveraging a systematic trend in the mean-dispersion relationship [7]. This approach substantiallyincreases the power to detect differences in proportions(differential expression) while still adequately control-ling for false positives [9]. Many R packages imple-menting this model of RNA-Seq data are now available,differing mainly in their approach to modeling disper-sion across genes [10].
Analogous to the development of gene expression re-search, culture independent [11] microbial ecology re-search has migrated away from detection of species (orOperational Taxonomic Units, OTUs) through microar-ray hybridization of rRNA gene PCR amplicons [12]to direct sequencing of highly-variable regions of theseamplicons [13], or even direct shotgun sequencing ofmicrobiome metagenomic DNA [14]. Although theselatter microbiome investigations use the same DNAsequencing platforms and represent the processed se-quence data in the same manner — a feature-by-samplecontingency table where the features are OTUs insteadof genes — the modeling and normalization methodsjust described for RNA-Seq analysis have not been
transferred to microbiome research [15, 16].Standard microbiome analysis workflows begin with
an ad hoc library size normalization by random subsam-pling without replacement, or so-called rarefying [17].Rarefying is most often defined by the following steps.
1. Select a minimum library size, NL.
2. Discard libraries (samples) that are smaller thanNL in size.
3. Subsample the remaining libraries without replace-ment such that they all have size NL.
Often NL is chosen to be equal to the size of the small-est library that is not considered an artifact, though inexperiments with large variation in library size iden-tifying artifact samples can be subjective. In manycases researchers have also failed to repeat the randomsubsampling step or record the pseudorandom num-ber generation seed/process – both of which are es-sential for reproducibility. To our knowledge, rare-fying was first recommended for microbiome countsin order to moderate the sensitivity of the UniFracdistance [18] to library size, especially differencesin the presence of rare OTUs attributable to librarysize [19]. In these and similar studies the principalobjective is to compare microbiome samples from dif-ferent sources, a research task that is increasingly ac-cessible with declining sequencing costs and the abilityto sequence many samples in parallel using barcodedprimers [20,21]. Rarefying is now an exceedingly com-mon precursor to microbiome multivariate workflowsthat seek to relate sample covariates to sample-wisedistance matrices [17, 22, 23]; for example, integratedas a recommended option in QIIME’s [24] beta_-diversity_through_plots.py workflow, inSub.sample in the mothur software library [25], andin daisychopper.pl [26]. This perception in themicrobiome literature of “rarefying to even samplingdepth” as a standard normalization procedure appearsto explain why rarefied counts are also used in studiesthat attempt to detect differential abundance of OTUsbetween predefined classes of samples [27–31], in addi-tion to studies that use proportions directly [32].
Statistical motivationUnfortunately, rarefying biological count data is unjus-tified despite its current ubiquity in microbiome anal-yses. The following is a minimal example to explain
2
a Poisson random variable in most cases [5]. How-ever, we are usually interested in understanding vari-ation among biological replicates, wherein a mixturemodel is necessary to account for the added uncer-tainty [6]. Taking a hierarchical model approach withthe Gamma-Poisson has provided a satisfactory fit toRNA-Seq data [7], as well as a valid regression frame-work that uses generalized linear models [8]. A Gammamixture of Poisson variables gives the negative binomial(NB) distribution [6, 7] and several RNA-Seq analysispackages now model the counts, K, for gene i, in sam-ple j according to:
Kij ⇠ NB(sjµi,�i) (1)
where sj is a linear scaling factor for sample j thataccounts for its library size, µi is the mean proportionfor gene i, and �i is the dispersion parameter for genei. The variance is ⌫i = sjµi + �isjµ
2i , with the NB
distribution becoming Poisson when � = 0. Recogniz-ing that � > 0 and estimating its value is important ingene-level tests. This reduces false positive genes thatappear significant under a Poisson distribution, but notafter accounting for non-zero dispersion.
The uncertainty in estimating �i for every gene whenthere is a small number of samples — or a small num-ber of biological replicates — can be mitigated by shar-ing information across the thousands of genes in an ex-periment, leveraging a systematic trend in the mean-dispersion relationship [7]. This approach substantiallyincreases the power to detect differences in proportions(differential expression) while still adequately control-ling for false positives [9]. Many R packages imple-menting this model of RNA-Seq data are now available,differing mainly in their approach to modeling disper-sion across genes [10].
Analogous to the development of gene expression re-search, culture independent [11] microbial ecology re-search has migrated away from detection of species (orOperational Taxonomic Units, OTUs) through microar-ray hybridization of rRNA gene PCR amplicons [12]to direct sequencing of highly-variable regions of theseamplicons [13], or even direct shotgun sequencing ofmicrobiome metagenomic DNA [14]. Although theselatter microbiome investigations use the same DNAsequencing platforms and represent the processed se-quence data in the same manner — a feature-by-samplecontingency table where the features are OTUs insteadof genes — the modeling and normalization methodsjust described for RNA-Seq analysis have not been
transferred to microbiome research [15, 16].Standard microbiome analysis workflows begin with
an ad hoc library size normalization by random subsam-pling without replacement, or so-called rarefying [17].Rarefying is most often defined by the following steps.
1. Select a minimum library size, NL.
2. Discard libraries (samples) that are smaller thanNL in size.
3. Subsample the remaining libraries without replace-ment such that they all have size NL.
Often NL is chosen to be equal to the size of the small-est library that is not considered an artifact, though inexperiments with large variation in library size iden-tifying artifact samples can be subjective. In manycases researchers have also failed to repeat the randomsubsampling step or record the pseudorandom num-ber generation seed/process – both of which are es-sential for reproducibility. To our knowledge, rare-fying was first recommended for microbiome countsin order to moderate the sensitivity of the UniFracdistance [18] to library size, especially differencesin the presence of rare OTUs attributable to librarysize [19]. In these and similar studies the principalobjective is to compare microbiome samples from dif-ferent sources, a research task that is increasingly ac-cessible with declining sequencing costs and the abilityto sequence many samples in parallel using barcodedprimers [20,21]. Rarefying is now an exceedingly com-mon precursor to microbiome multivariate workflowsthat seek to relate sample covariates to sample-wisedistance matrices [17, 22, 23]; for example, integratedas a recommended option in QIIME’s [24] beta_-diversity_through_plots.py workflow, inSub.sample in the mothur software library [25], andin daisychopper.pl [26]. This perception in themicrobiome literature of “rarefying to even samplingdepth” as a standard normalization procedure appearsto explain why rarefied counts are also used in studiesthat attempt to detect differential abundance of OTUsbetween predefined classes of samples [27–31], in addi-tion to studies that use proportions directly [32].
Statistical motivationUnfortunately, rarefying biological count data is unjus-tified despite its current ubiquity in microbiome anal-yses. The following is a minimal example to explain
Is Negative Binomial effective for this data?
Differential AbundanceWhat about NB Mixture Model?
• Robinson, Oshlack (2010). A scaling normalization… RNA-Seq data. Genome Biology!• Anders, & Huber (2010). Differential expression … sequence count data. Genome Biology
35
Common Scale Rarefied
1e+02
1e+05
1e+08
1e+02
1e+05
1e+08
1e+02
1e+05
1e+08
DietPatterns
GlobalPatterns
GlobalPatterns
2004 HighFat
freshwaterm
arine
10 1000 10 1000Mean
Varia
nce
variance
mean
Microbiome Survey Biological Replicates
Differential Abundance
36
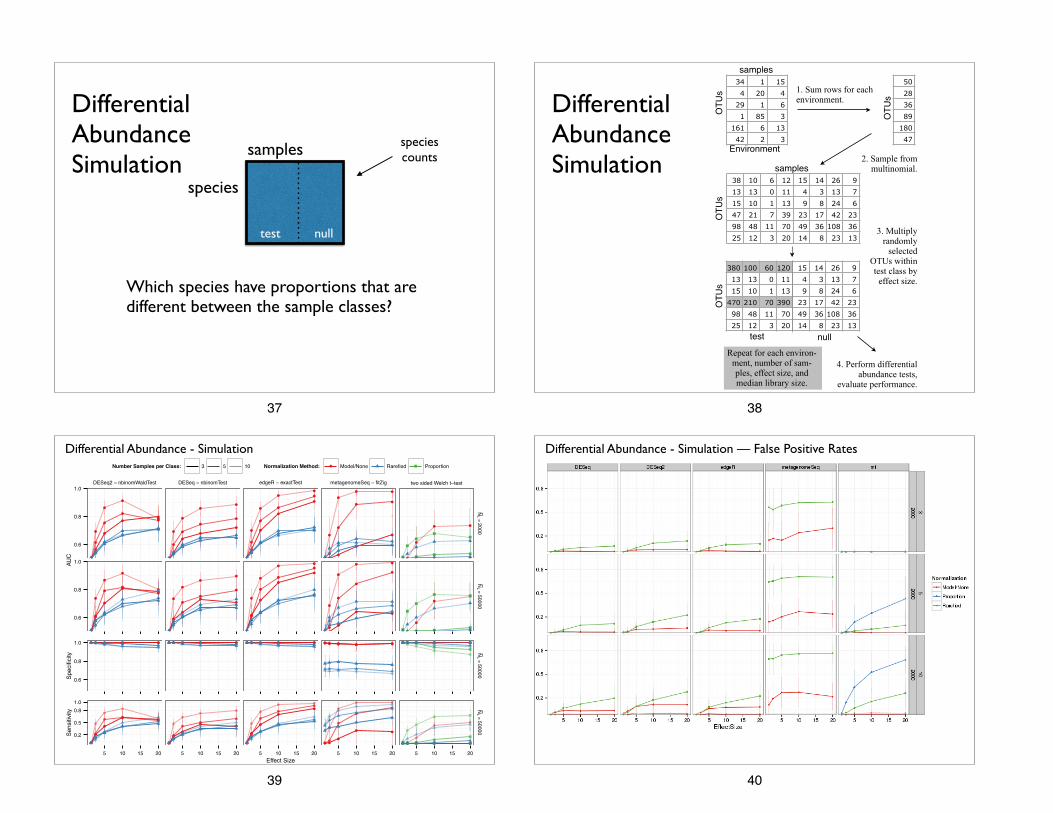
Differential Abundance Simulation
species
samples species counts
test null
Which species have proportions that are different between the sample classes?
37
Differential Abundance Simulation
38 10 6 12 15 14 26 913 13 0 11 4 3 13 715 10 1 13 9 8 24 647 21 7 39 23 17 42 2398 48 11 70 49 36 108 3625 12 3 20 14 8 23 13
380 100 60 120 15 14 26 913 13 0 11 4 3 13 715 10 1 13 9 8 24 6470 210 70 390 23 17 42 2398 48 11 70 49 36 108 3625 12 3 20 14 8 23 13
samples
OTU
s
test null
OTU
s
5028368918047
34 1 154 20 429 1 61 85 3
161 6 1342 2 3
samples
OTU
s
Ocean Feces
OTU
s
Ocean Feces
1. Sum rows. A multinomial for each sample class.
2. Deterministic mixing. Mix multinomials in precise proportion.
Ocean Feces
Microbiome Clustering Simulationsamples
OTU
s
Environment
OTU
s
1. Sum rows for each environment.
2. Sample from multinomial.
Differential Abundance Simulation
3. Multiply randomly
selected OTUs within test class by
effect size.
4. Perform differential abundance tests,
evaluate performance.
OTU
s
samples
Simulated Ocean Simulated Feces
3. Sample from these multinomials.
4. Perform clustering, evaluate accuracy.
BA
Microbiome count data from the Global
Patterns dataset
Repeat for each effect size and media library size.
Repeat for each environ-ment, number of sam-ples, effect size, and median library size.
Amount added is library size / effect size
38
Differential Abundance - Simulation
DESeq2 − nbinomWaldTest DESeq − nbinomTest edgeR − exactTest metagenomeSeq − fitZig two sided Welch t−test
●●●
●
●
●●
●
●
●
●
●
●●●
●●●
●●
●
●
●
●
●●
●
●●●
●●●
●
●
●
●
●
●
●
●
●
●
●
●
●●●
●●
●
●
●
●
●●
●
●
●
●
●●●
●
●
● ●
●
●
●
●
●
●
●
●
●●●
●●
● ●
●
●●
●
●
●
●●
●●● ●
●
●
●
●
●
●
●
●
●
●
●
●●● ●●
●
●
●
●
●
●
●
●
●
●●●● ●●● ●●
●
●●
●
●●
●
●●● ●●● ●●
●
●●
●
●●
●
0.6
0.8
1.0
0.6
0.8
1.0
N ~L=
2000N ~
L=
50000
AUC
Number Samples per Class: 3 5 10 Normalization Method: ● Model/None Rarefied Proportion
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
0.6
0.8
1.0
N ~L=
50000Spec
ifici
ty
●
●
●
●●
●
●●
●●
●
●
●
●
●
● ●●
● ●
● ● ● ● ●
0.2
0.5
0.81.0
N ~L=
50000
5 10 15 20 5 10 15 20 5 10 15 20 5 10 15 20 5 10 15 20Effect Size
Sens
itivi
ty
39
Differential Abundance - Simulation — False Positive Rates
40

1. Rarefied counts worse sensitivity in every analysis method we attempted.
2. Rarefied counts also worse specificity (high FPs)
• No accounting for overdispersion
• Added noise from subsampling step
Issues with rarefying — Differential Abundance
41
!
Transition: Lab 3 !
Negative Binomial mixture model for differential abundance multiple testing
42





























