Embed Size (px)
Citation preview

LEBER'S DISEASE
REPORT OF FOUR CASES IN ONE FAMILY*
JOHN E. RAAF, M.D.t AND HuGO L. BAIR, M.D.Rochester, Minnesota
In 1871 Theodore Leber- published hispaper, "Ueber hereditare und congenitalangelegte Sehnervenleiden," and, eventhough this was by no means the first account of the disease, his description wasso widely acknowledged that the conditionhas since frequently been termed "Leber'sdisease." Leber's paper provided an impetus to the study of hereditary opticatrophy, and since his time there havebeen many papers on the subject.
Julia Bell, in 1931, made an exhaustivestudy of the disease." The material onwhich she worked had been collected frompublished reports of cases which provided1,182 affected individuals for study: 1,018were Europeans, the remaining 164 wereJapanese. From her study Bell pointed outthe main features of the disease as follows: 1. There is a predilection for themale sex (84.8 percent males amongEuropeans and 59.1 percent males amongJapanese). 2. The average age at the onsetof the disease among Europeans is 23years for males and 25 years for females,whereas among Japanese the average ageat the onset of the disease is 21 years formales, 20 years for females. 3. The impairment in vision advances relativelyrapidly, probably in most cases providing the maximal disability within twomonths from the time of the onset; rarelydo symptoms advance after six months.4. In the majority of cases the diseasecomes on while patients are in goodhealth, and there is no other disease char-
*From the Department of Surgery, TheMayo Foundation, and the Section on Ophthalmology, The Mayo Clinic.
t Formerly Fellow in Surgery, now residingin Portland, Oregon.
acteristically associated with hereditaryoptic atrophy. 5. Usually the patient's solecomplaint is diminution of vision; thevisual defect most common is centralscotoma although peripheral contractionof the visual fields is also frequently seen.6. Color blindness is usually the only othersymptom shown by patients with hereditary optic atrophy. 7. The appearance ofthe fundus in the early stages of hereditary optic atrophy may vary from complete normality to the picture of opticneuritis, which is characterized by reddening of the nerve head, blurring of thedisc margins, tortuosity and dilatation ofthe vessels, and in some instances, smallhemorrhages. When present, the inflammatory signs are temporary and are soonfollowed by atrophy of the nerve whichprogresses and may advance after diminution of vision has become stationary. Eventhough the sight may subsequently markedly improve, the disc never regains itsnormal appearance. 8. Estimates of thenumber of patients suffering from hereditary optic atrophy whose vision subsequently improves range from 10 to 29percent. Points which seem to bear on theprognosis for the individual are: (a) onset at an early age appears to indicate arelatively favorable outlook, and (b) notonly is liability to the disease inherited,but the tendency to improvement is alsoinherited, improvement occurring morereadily in some families than in others.9. Transmission of the disease throughthe father is relatively rare; in 93.6 percent of the cases in which Europeans wereaffected the mother transmitted the disease.
The first patient in our series was a girl,
384

LEBER'S DISEASE 385
aged 18 years. She presented herself at theclinic on April 29, 1936, complaining ofblurring and loss of vision of four months'duration. She had had no serious illnessesand was unaware of any hereditary dis-
Fig. 1 (Raaf and Bair). Visual fields ofdaughter (May 2, 1936), showing large, bilateral cecocentral scotomas.
ease in her family. On January 1, 1936,she had suddenly noticed blurring ofvision while reading. Within four or fivedays her vision had become so poor thatshe had had to stop school. She consulted
hour. Roentgenograms of the thorax andhead, and also of the sella turcica andoptic foramina, were negative.
Inasmuch as no toxic factor could befound to account for the patient's amblyopia, she was questioned further regardingthe onset of her condition. There wasfound to be a conflict at home and this,it was thought, might be the basis of anhysterical amblyopia. The results of psychiatric examination, however, did notdefinitely confirm the diagnosis of hysterical blindness.
Vision in the left eye was 1/60, in theright eye 2/60; the ocular fundi werenormal. On examination of the visualfields, large bilateral cecocentral scotomaswere found (fig. 1). It was then thoughtthat the patient had some obscure type ofretrobulbar neuritis rather than functional amblyopia. Spinal puncture gave negative results. Examination of the ocularfundi and visual fields was again made on
Fig. 2 (Raaf and Bair). Subsequent appearance of visual fields of daughter (May 6th andMay 8th), showing tendency of the scotomas to assume a right homonymous position.
several physicians, and a diagnosis ofoptic neuritis in the right eye was made.Her tonsils were removed, but loss ofvision gradually increased.
On general examination the patient appeared to be well developed and wellnourished. Examination of the teeth, nose,throat, and pelvis did not reveal any fociof infection. The urine and blood appeared to be normal; no lead or arsenicwas found in the urine. The sedimentationrate of erythrocytes was 5 mm. in one
May 6th, and then again on May 8th; theresults of these examinations, however,did not differ greatly from those of theoriginal examination (fig. 2). The scotomas tended to assume a right homonymous position, and it was thought that theconstancy of the scotomas spoke for anorganic lesion.
That the condition might be Leber'sdisease was suspected when the patient'smother, aged 42 years, was referred to theSection on Ophthalmology 15 days after
386 JOHN E. RAAF AND HUGO L. BAIR
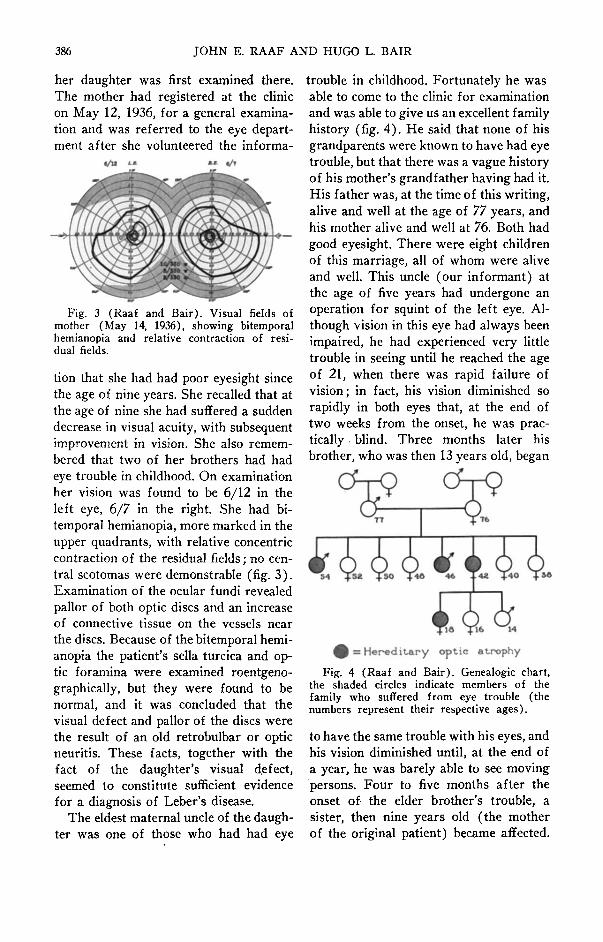
her daughter was first examined there.The mother had registered at the clinicon May 12, 1936, for a general examination and was referred to the eye department after she volunteered the informa-
Fig. 3 (Raaf and Bair). Visual fields ofmother (May 14, 1936), showing bitemporalhemianopia and relative contraction of residual fields.
tion that she had had poor eyesight sincethe age of nine years. She recalled that atthe age of nine she had suffered a suddendecrease in visual acuity, with subsequentimprovement in vision. She also remembered that two of her brothers had hadeye trouble in childhood. On examinationher vision was found to be 6/12 in theleft eye, 6/7 in the right. She had bitemporal hemianopia, more marked in theupper quadrants, with relative concentriccontraction of the residual fields; no central scotomas were demonstrable (fig. 3).Examination of the ocular fundi revealedpallor of both optic discs and an increaseof connective tissue on the vessels nearthe discs. Because of the bitemporal hemianopia the patient's sella turcica and optic foramina were examined roentgenographically, but they were found to benormal, and it was concluded that thevisual defect and pallor of the discs werethe result of an old retrobulbar or opticneuritis. These facts, together with thefact of the daughter's visual defect,seemed to constitute sufficient evidencefor a diagnosis of Leber's disease.
The eldest maternal uncle of the daughter was one of those who had had eye
trouble in childhood. Fortunately he wasable to come to the clinic for examinationand was able to give us an excellent familyhistory (fig. 4). He said that none of hisgrandparents were known to have had eyetrouble, but that there was a vague historyof his mother's grandfather having had it.His father was, at the time of this writing,alive and well at the age of 77 years, andhis mother alive and well at 76. Both hadgood eyesight. There were eight childrenof this marriage, all of whom were aliveand well. This uncle (our informant) atthe age of five years had undergone anoperation for squint of the left eye. Although vision in this eye had always beenimpaired, he had experienced very littletrouble in seeing until he reached the ageof 21, when there was rapid failure ofvision; in fact, his vision diminished sorapidly in both eyes that, at the end oftwo weeks from the onset, he was practically . blind. Three months later hisbrother, who was then 13 years old, began
• =Hereditary optic atrophy
Fig. 4 (Raaf and Bair). Genealogic chart,the shaded circles indicate members of thefamily who suffered from eye trouble (thenumbers represent their respective ages).
to have the same trouble with his eyes, andhis vision diminished until, at the end ofa year, he was barely able to see movingpersons. Four to five months after theonset of the elder brother's trouble, asister, then nine years old (the motherof the original patient) became affected.

LEBER'S DISEASE 387
Because these three children were affectedwithin a few months of each other it wasthought that they were suffering fromsome contagious eye disease. Each hadthen had some form of treatment: the twobrothers received local applications anddrops; the sister, 97 chiropractic treatments. All three experienced improvementin vision within seven months to a year.
The vision of this maternal uncle onexamination at the clinic was found to be6/15 in the right eye and 6/60 in the left.The squint for which he had been operated on at the age of five was no doubtpartially responsible for the poor vision in his left eye. Examination of hisvisual fields revealed in the right eye anupper-temporal-quadrant defect continuous with the physiologic blind spot, andin the left eye a cecocentral scotoma continuous with a defect in the upper temporal quadrant. There was concentric contraction of the residual fields in both eyes(fig. 5).
The other uncle with eye trouble has. not been able to come to the clinic for
examination, but he wrote that his eyesight had begun to improve about a yearafter the onset of the disease. He estimated that he had regained approximately80 percent of his original vision, beingable to read fine print without glasses.There was some evidence that he still hadslight residual central scotomas, for hesaid that at times he had difficulty locatingtraffic signals but that, after locating them,he could see them quite clearly.
The other five children in the family,uncles and aunts of the original patient,have never had any trouble with theireyes. Neither of the two brothers whosuffered from the disease has married,and, so far as we have been able to findout, none of the children of the otherbrothers and sisters except the originalpatient has been affected. The patient hasa sister, aged 16 years, and a brother,
aged 14 years, but neither has had any eyetrouble.
A point to which attention should becalled is that the residual visual fields ofthe patient's mother (fig. 3) and uncle
Fig. 5 (Raaf and Bair). Visual fields ofuncle (May 25, 1936), showing upper-temporalquadrant defect in right eye, cecocentralscotoma in left eye, and concentric contractionof residual fields in both eyes.
(fig. 5) are both suggestive of a bitemporal type of hemianopia. Numerous authors have considered the possibility ofan intracranial lesion, particularly in theregion of the sella turcica, being the etiologic factor in Leber's disease. Bruner"reported that roentgenographic examination of the patient in his case, as well asof the patient's sister and nephew, all ofwhom had hereditary optic atrophy, revealed enlarged sphenoidal cells. Themother and another sister of the patientwho did not have optic atrophy had smaller sphenoidal cells. Bruner quoted Berger"as saying, "A number of cases of hereditary optic atrophy are due to irregularityin the growth of the sphenoid bone." Indiscussing Bruner's paper, Bedell" reported four cases of hereditary opticatrophy, three brothers and a cousin beingaffected. All of these three brothers hadvery prominent clinoid processes, withdeep sella turcicas ; the cousin's sellaturcica did not give evidence of this abnormality.
Fisher" suggested that Leber's disease isprimarily attributable to an inherited temporary disorder of the pituitary gland.

388 JOHN E. RAAF AND HUGO L. BAIR
Fig. 6 (Raaf and Bair). Central visualfields of daughter on July 13, 1937, showingpartial resolution of central scotomas.
He pointed out that the pituitary glandmight hypertrophy at puberty or at themenopause, and thus implicate the visualpathways, producing the field defects seenin hereditary optic atrophy. He made
roentgenograms of the sella turcica in twocases of typical hereditary optic atrophy;in one the sella turcica was normal but,in the other, it was filled with a cellular,honeycomblike shadow. He interpretedthe latter as being an abnormal sellaturcica. Fisher" later reported three casesof two brothers and an uncle who hadLeber's disease. The roentgenogram ofthe sella turcica of one of the brothersrevealed slight expansion, undermining anirregularity of the postclinoid process;Fisher thought that this supported hisview that Leber's disease results from anexaggerated function of the pituitarygland.
Roentgenograms were made in 60 ofthe cases collected by JuliaBell in orderto determine whether there was any abnormality in the region of the sella turcica.In 34 of these cases the sella turcica wasfound to be normal, and the variationfrom normal in the remaining 26 caseswas so slight or inconstant as to enable noconclusions to be drawn regarding theassociation of an intrasellar lesion withLeber's disease.
A diagnosis of pituitary tumor wasconsidered in the case of our patient's
0.5. 6/15 0.0. 6/lZ
mother since her visual fields showed abitemporal-hemianopic type of defect.Roentgenographic examination of thesella turcica of both mother and daughterwere, however, as has been said, normal.The patient's uncle, whose visual fields'also suggested a bitemporal hemianopicdefect, was not examined roentgenographically. It must therefore be concluded that even though two of the patients in this series did have a bitemporaltype of defect in the visual fields, no relationship can be considered to have beenestablished between an intrasellar neoplastic lesion and Leber's disease.
Treatment in the daughter's case consisted of the intravenous administrationof typhoid vaccine. She received a courseof four injections, following which anexamination of the visual fields and central vision on May 19th, showed practically no change from the time of the original examination. On examination of theocular fundi there was thought to be aslight pallor in the temporal part of thediscs. Another series of four injections ofintravenous typhoid vaccine was administered, but again examination of the visual fields and fundi on June 2d revealedno change, although subjectively the patient thought that her sight had improved.
The patient again returned on March 8,1937, at which time her vision was foundto be about the same as previously, andthe optic discs showed a marked pallorwith some evidence of loss of substance.She was given four intravenous injectionsof typhoid vaccine and, on March 18th,her visual fields showed beginning resolution of the central scotomas, consisting ina tongue of relatively improved visionentering the scotoma from each uppertemporal quadrant. There was also subjective improvement in the patient'svision. She was again seen on July 13,1937, and stated that her vision had begunto improve rapidly about 1~ months pre-

LEBER'S DISEASE 389
viouslyand had steadily increased until,at this time, she was able to read ordinaryprint, although slowly. Her visual acuitywas found to be 6/12 in the right eye and6/15 in the left eye. The visual fieldsshowed a quite definite partial resolutionof the scotomas, although the island ofcentral vision for a I-mm. object at onemeter's distance was very small, being lessthan one-half degree in the left eye andabout three degrees in diameter in theright eye (fig. 6). Another series of intravenous typhoid injections was given, butthere was no subjective improvement invision. The visual acuity was then recorded as 6/10 in the right eye and 6/15in the left eye. The visual fields showedthe scotomas to be very slightly smallerbut otherwise to be essentially the same.The patient was able to recognize a 2-mm.red color and 5-mm. red and blue colorsat one meter's distance. There was noapparent change in the ocular fundi.
The prognosis as to the complete returnof vision is, of course, indeterminate.Bell's statement that not only a liabilityto the disease is inherited but also a tend-
cncy toward improvement, suggested thatthe outlook for this patient should befavorable inasmuch as her mother andtwo uncles had had a good return ofvision. Her course so far has supportedthis, although the presence of markedatrophy of the optic discs makes it probable that much residual defect will bepresent.
SUMMARY
In this series of four cases of Leber'sdisease in one family, the bitemporal character of the residual field defect in the twoolder patients and the homonymous position of the scotomas in the patient withthe acute form of the disease suggested alesion in the region of the optic chiasmrather than in the optic nerve proper.However, roentgenologic examination ofthe sellar region in the last-named caseand in one of the former cases yielded nodemonstrable evidence of a causativelesion of the pituitary or adjacent structures. There were no other associatedsigns of pituitary dysfunction in thesecases.
REFERENCES
t Leber, von Th. Ueber hereditare und congenital-angelegte Sehnervenleiden. Arch. f. Ophth.,1871, v, 17 (Pt. 2), p. 249. Abstr. Schmidt's Jahrb., 1872, v. 153,p. 204.
'Bell, Julia. Hereditary optic atrophy (Leber's disease). Cambridge University Press, 1931.(Pearson, Karl., ed. Anomalies and diseases of the eye. Nettleship Memorial Volume. v. 2,pt. 4).
I Bruner, W. E. Hereditary optic atrophy with X-ray findings. Trans. Amer, Ophth. Soc., 1912,v, 13 (pt. 1), p. 162.
• Berger. Quoted by Bruner, W. E.'Bedell, A. J. Discussion. Trans. Amer, Ophth, Soc., 1912, v. 13 (pt.l), p. 174.• Fisher, J. H. Leber's disease (hereditary optic atrophy) ; a suggestion as to its cause. Trans.
Ophth, Soc. U. Kingdom, 1916,v, 36, p. 298.A further case of Leber's disease, and two allied cases, associated with changes
in the sella turcica. Trans. Ophth. Soc. U. Kingdom, 1917, v, 37, p. 251.





























