Embed Size (px)
Citation preview

GENERAL APPROACH TO DIAGNOSIS OF HEMOLYTIC ANEMIA
Dr jp,asst prof,ich,mch,kottayam
LEARNING OBJECTIVES
To identify hemolytic anemia
To list 3 common causes of hemolytic anemia
Name 4 specific tests for diagnosis of hemolytic anemia
How to identify and treat thalassemia,hereditary spherocytosis and sickle cell
anemia?
What is hemolytic anemia?
An essential feature of hemolytic anemia is a reduction in the normal red cell
survival of 120 days. Premature destruction of red cells may result from
orpuscular abnormalities (within the red cell corpuscle), that is,
abnormalities of membrane,enzymes, or hemoglobin; or from
extracorpuscular abnormalities, that is, immune or nonimmune mechanisms.
Case scenarios
2wks old child has unconjugated hyperbilirubinemia. He has no blood loss.
His and mother’s blood group is o+. his father had jaundice in childhood .
he had splenectomy later
Wha t is your comment ?
6 month old child has anemia. She had swelling fingers two times so far. His
anemia has not responded with hematinics.what are the possibilities?
3 yr old child needs monthly transfusion to treat anemia. He has hemolytic
facies hsm,pigmentation He is on defersirox .what is the dx?

What is the approach to hemolytic anemia?
The approach to the diagnosis of hemolytic anemia should include:
• Consideration of the clinical features suggesting hemolytic disease
• Laboratory demonstration of the presence of a hemolytic process
• Determination of the precise cause of the hemolytic anemia by special
hematologic
investigations.
History- key points
Jaundice in newborn period/later
Recurrent transfusion
Ftt
Abnormal facies
Drug induced hemolysis
What are the Clinical Features?
The following clinical features suggest a hemolytic process:
• Ethnic factors: Incidence of sickle gene carrier in the African-American
population (8%), high incidence of thalassemia trait in people of Mediterranean
ancestry and high incidence of glucose-6-phosphate dehydrogenase (G6PD)
deficiency among Sephardic Jews.chetti tribe in wayanad has sickle cell anemia
running through families
• Age factors: Anemia and jaundice in an Rh-positive infant born to a mother who
is Rh negative or a group A or group B infant born to a group O mother (setting for
a hemolytic anemia)
• History of anemia, jaundice, or gallstones in family
• Persistent or recurrent anemia associated with reticulocytosis
• Anemia unresponsive to hematinics
• Intermittent bouts or persistent indirect hyperbilirubinemia/jaundice
• Splenomegaly.
• Hemoglobinuria
• Presence of multiple gallstones
• Chronic leg ulcers
• Development of anemia or hemoglobinuria after exposure to certain drugs
• Cyanosis without cardiorespiratory distress
• Polycythemia (2,3 Diphosphoglycerate mutase deficiency)

• Dark urine due to dipyrroluria (unstable hemoglobins, thalassemia and ineffective
erythropoiesis).
What are the Laboratory Findings?
Laboratory findings of hemolytic anemia consist of:
• Evidence of accelerated hemoglobin catabolism due to reduced red cell survival
• Evidence of increased erythropoiesis.
Accelerated Hemoglobin Catabolism
Accelerated hemoglobin catabolism varies with the type of hemolysis as follows:
• Extravascular hemoglobin catabolism ()
• Intravascular hemoglobin catabolism ().
The two may not be easily distinguished if the cause for hemolysis is not obvious,
hence the long lists of markers of testing indicated below. The presence of
hemoglobinuria and hemosidenuria and the absence of haptoglobin are the major
markers of intravascular hemolysis in practice.
What are the Markers of Extravascular Hemolysis?
1. Increased unconjugated bilirubin.
2. Increased lactic acid dehydrogenase in serum.
3. Decreased plasma haptoglobin (normal level, 128625 mg/dl).
4. Increased fecal and urinary urobilinogen.
5. Increased rate of carbon monoxide production.
This is a personAL TEACHING FILE WHICH IS NOT INTENDED TO BE
PRINTED SOLD,OR PHOTOCOPIED.
What are the Markers of Intravascular Hemolysis?
1.Increased unconjugated bilirubin.
2. Increased lactic acid dehydrogenase in serum.
3. Hemoglobinuria).
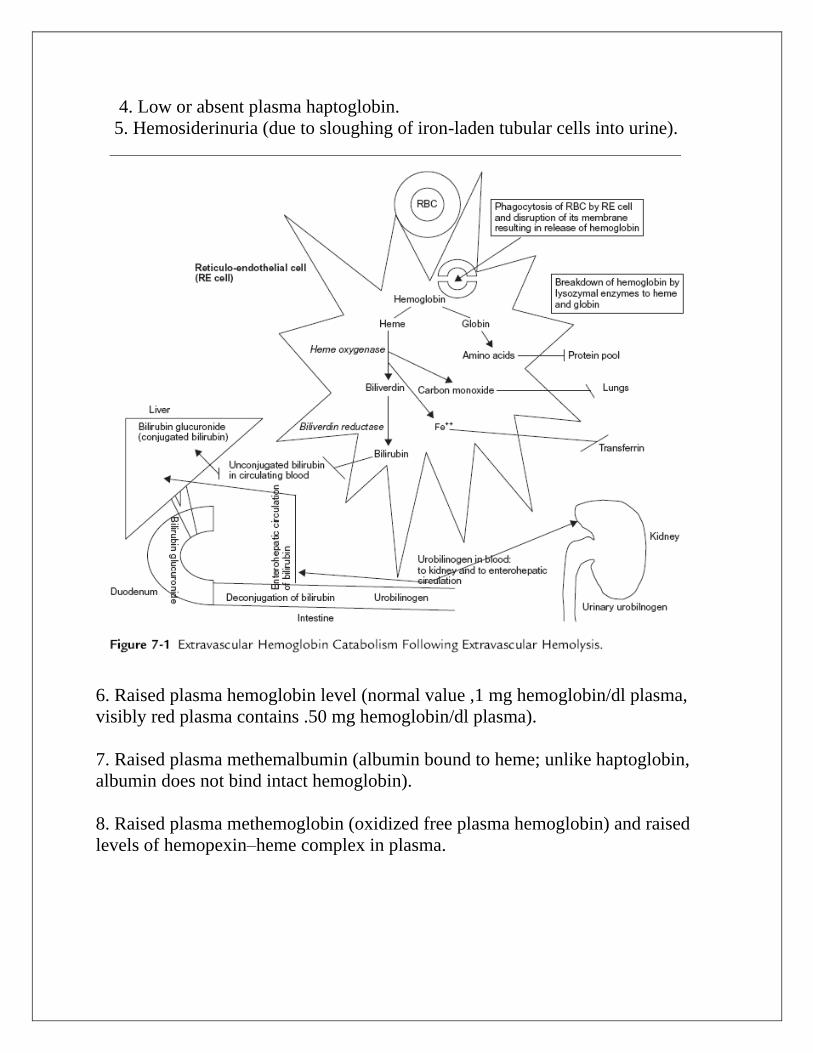
4. Low or absent plasma haptoglobin.
5. Hemosiderinuria (due to sloughing of iron-laden tubular cells into urine).
6. Raised plasma hemoglobin level (normal value ,1 mg hemoglobin/dl plasma,
visibly red plasma contains .50 mg hemoglobin/dl plasma).
7. Raised plasma methemalbumin (albumin bound to heme; unlike haptoglobin,
albumin does not bind intact hemoglobin).
8. Raised plasma methemoglobin (oxidized free plasma hemoglobin) and raised
levels of hemopexin–heme complex in plasma.
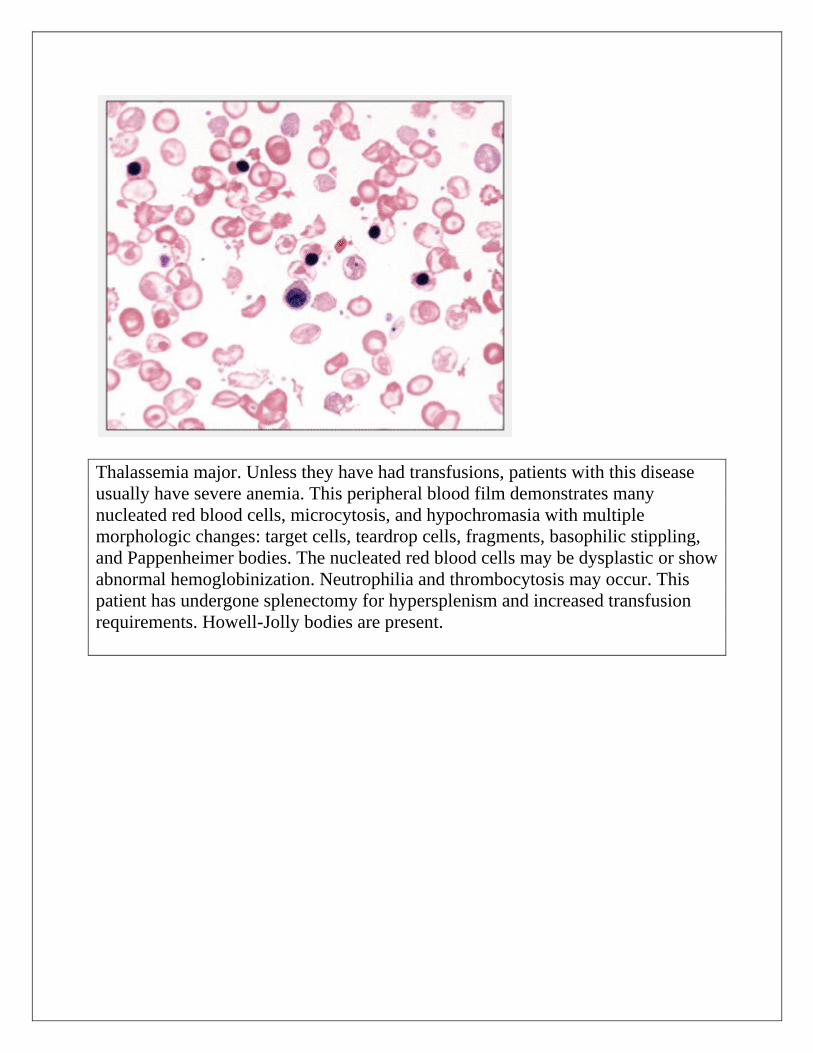
Thalassemia major. Unless they have had transfusions, patients with this disease
usually have severe anemia. This peripheral blood film demonstrates many
nucleated red blood cells, microcytosis, and hypochromasia with multiple
morphologic changes: target cells, teardrop cells, fragments, basophilic stippling,
and Pappenheimer bodies. The nucleated red blood cells may be dysplastic or show
abnormal hemoglobinization. Neutrophilia and thrombocytosis may occur. This
patient has undergone splenectomy for hypersplenism and increased transfusion
requirements. Howell-Jolly bodies are present.
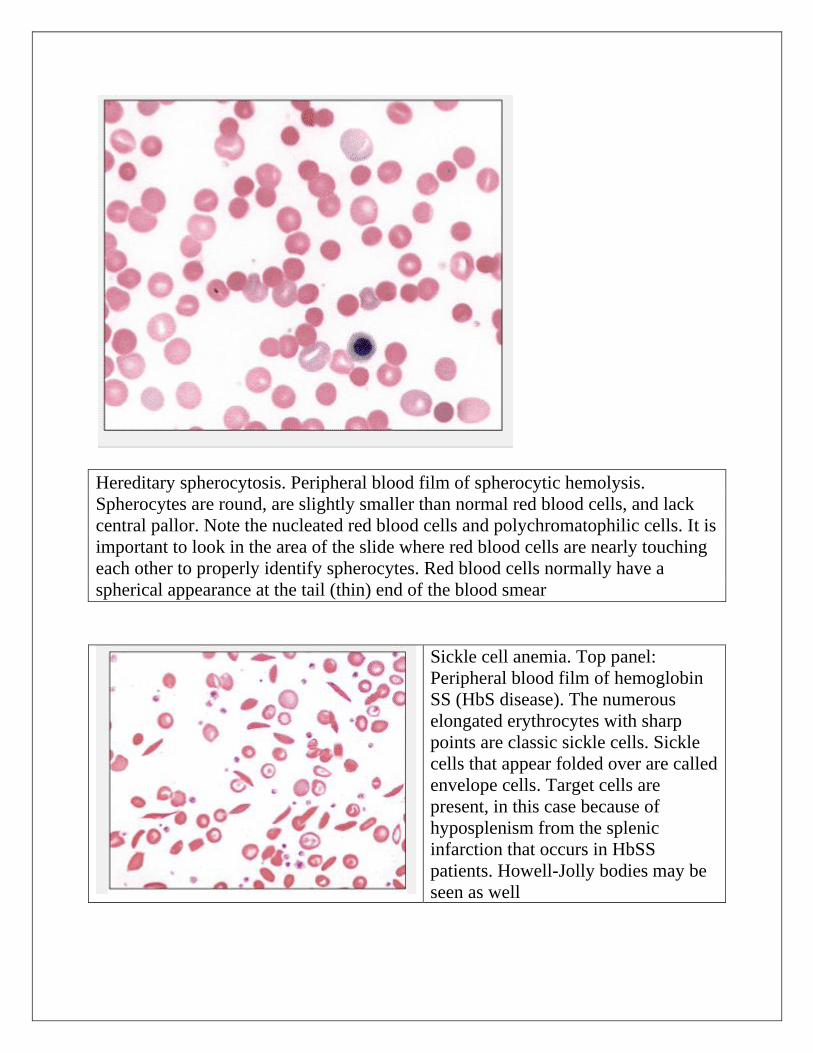
Hereditary spherocytosis. Peripheral blood film of spherocytic hemolysis.
Spherocytes are round, are slightly smaller than normal red blood cells, and lack
central pallor. Note the nucleated red blood cells and polychromatophilic cells. It is
important to look in the area of the slide where red blood cells are nearly touching
each other to properly identify spherocytes. Red blood cells normally have a
spherical appearance at the tail (thin) end of the blood smear
Sickle cell anemia. Top panel:
Peripheral blood film of hemoglobin
SS (HbS disease). The numerous
elongated erythrocytes with sharp
points are classic sickle cells. Sickle
cells that appear folded over are called
envelope cells. Target cells are
present, in this case because of
hyposplenism from the splenic
infarction that occurs in HbSS
patients. Howell-Jolly bodies may be
seen as well

What is the evidence of Increased Erythropoiesis?
Erythropoiesis increases in response to a reduction in hemoglobin and is
manifested by:
• Reticulocytosis: Frequently up to 10–20%; rarely, as high as 80%
• Increased mean corpuscular volume (MCV) due to the presence of reticulocytosis
and increased red cell distribution width (RDW) as the hemoglobin level falls
Supravital stain of reticulocytes with brilliant cresyl blue. The blue-stained
reticular inclusions in the red blood cells represent ribosomes that are precipitated
when exposed to brilliant cresyl blue

Increased normoblasts in peripheral blood
• Specific morphologic abnormalities: Sickle cells, target cells, basophilic
stippling,irregularly contracted cells or fragments (schistocytes), eliptocytes,
acanthocytes and spherocytes
• Erythroid hyperplasia of the bone marrow: Erythroid:myeloid ratio in the marrow
increasing from 1:5 to 1:1
• Expansion of marrow space in chronic hemolysis resulting in:
• Prominence of frontal bones
• Broad cheekbones
• Widened intratrabecular spaces, hair-on-end appearance of skull radiographs
• Biconcave vertebrae with fish-mouth intervertebral spaces.
• Decreased red cell survival demonstrated by 51Cr red cell labeling
• Red cell creatine levels increased.
What are Thalassemia Syndromes ?
Thalassemia refers to genetic disorders in globin chain production. In
individuals with beta thalassemia, there is either a complete absence of β
globin production (β-thalassemia major) or a partial reduction in β globin
production (β thalassemia minor). In alpha thalassemia, there is an absence
of or partial reduction in α globin production. The primary pathology in
thalassemia stems from the quantity of globin production, whereas the
primary pathology in sickle cell disease is related to the quality of globin
produced.
What is the Pathophysiology ?
Two related features contribute to the sequelae of β-thalassemia: inadequate
β-globin gene production leading to decreased levels of normal hemoglobin
(Hb A) and unbalanced α- and β-globin chain production.. In the bone
marrow, thalassemia mutations disrupt the maturation of erythrocytes,
resulting in ineffective erythropoiesis; the marrow is hyperactive, but there
are relatively few reticulocytes and severe anemia exists.
In β-thalassemia, there is an excess of α-globin chains relative to β- and γ-
globin chains, and α-globin tetramers (α4) are formed. These inclusions
interact with the red cell membrane and shorten red cell survival, leading to
anemia and increased erythroid production. The γ-globin chains are
produced in increased amounts, leading to an elevated Hb F (α2γ2). The δ-
globin chains are also produced in increased amounts, leading to an elevated
Hb A2 (α2δ2) in β-thalassemia.
THE THALASSEMIAS
THALASSE
MIA
GLOBIN
GENOTY
PE
FEATURES EXPRESSION
HEMOGLO
BIN
ANALYSIS
α-THALASSEMIA
1 gene
deletion -,α/α,α Normal Normal
Newborn:
Bart's 1-2%
2 gene
deletion trait
-,α/-,α -, -
/α,α
Microcytosis, mild
hypochromasia
Normal, mild
anemia
Newborn:
Bart's: 5-10%
3 gene -,-/-,α Microcytosis, Mild anemia, Newborn:
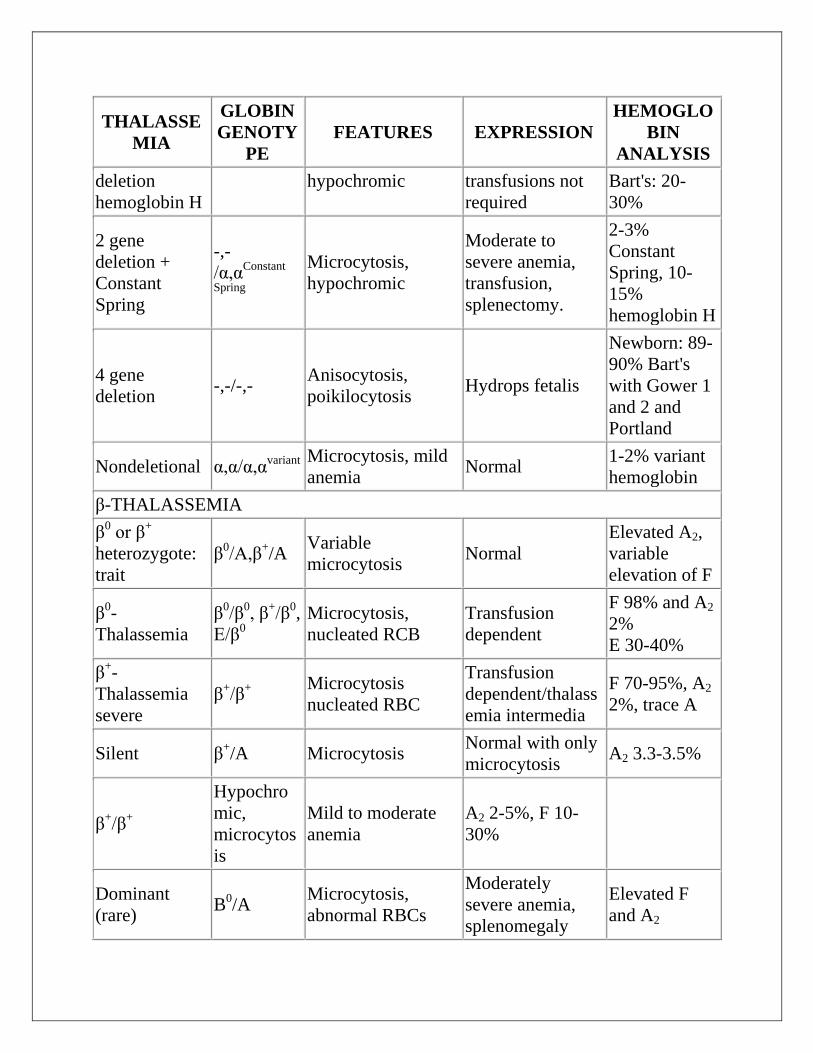
THALASSE
MIA
GLOBIN
GENOTY
PE
FEATURES EXPRESSION
HEMOGLO
BIN
ANALYSIS
deletion
hemoglobin H
hypochromic transfusions not
required
Bart's: 20-
30%
2 gene
deletion +
Constant
Spring
-,-
/α,αConstant
Spring
Microcytosis,
hypochromic
Moderate to
severe anemia,
transfusion,
splenectomy.
2-3%
Constant
Spring, 10-
15%
hemoglobin H
4 gene
deletion -,-/-,-
Anisocytosis,
poikilocytosis Hydrops fetalis
Newborn: 89-
90% Bart's
with Gower 1
and 2 and
Portland
Nondeletional α,α/α,αvariant
Microcytosis, mild
anemia Normal
1-2% variant
hemoglobin
β-THALASSEMIA
β0 or β
+
heterozygote:
trait
β0/A,β
+/A
Variable
microcytosis Normal
Elevated A2,
variable
elevation of F
β0-
Thalassemia
β0/β
0, β
+/β
0,
E/β0
Microcytosis,
nucleated RCB
Transfusion
dependent
F 98% and A2
2%
E 30-40%
β+-
Thalassemia
severe
β+/β
+
Microcytosis
nucleated RBC
Transfusion
dependent/thalass
emia intermedia
F 70-95%, A2
2%, trace A
Silent β+/A Microcytosis
Normal with only
microcytosis A2 3.3-3.5%
β+/β
+
Hypochro
mic,
microcytos
is
Mild to moderate
anemia
A2 2-5%, F 10-
30%
Dominant
(rare) B
0/A
Microcytosis,
abnormal RBCs
Moderately
severe anemia,
splenomegaly
Elevated F
and A2
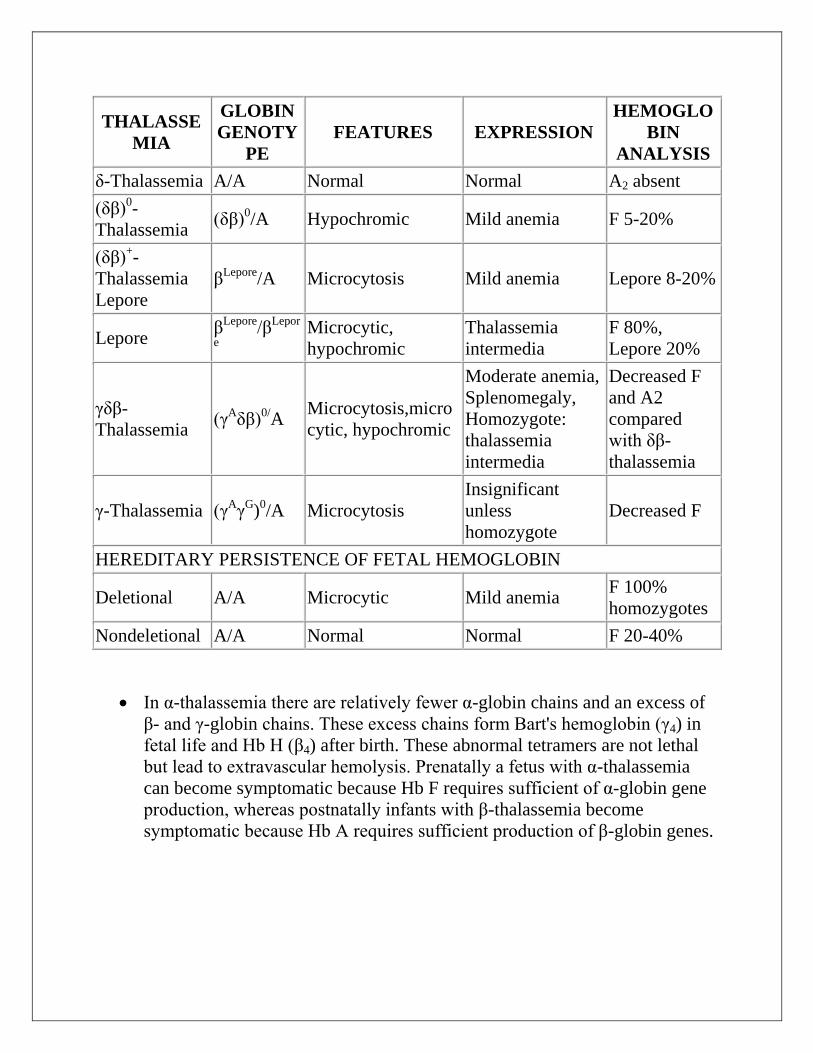
THALASSE
MIA
GLOBIN
GENOTY
PE
FEATURES EXPRESSION
HEMOGLO
BIN
ANALYSIS
δ-Thalassemia A/A Normal Normal A2 absent
(δβ)0-
Thalassemia (δβ)
0/A Hypochromic Mild anemia F 5-20%
(δβ)+-
Thalassemia
Lepore
βLepore
/A Microcytosis Mild anemia Lepore 8-20%
Lepore β
Lepore/β
Lepor
e
Microcytic,
hypochromic
Thalassemia
intermedia
F 80%,
Lepore 20%
γδβ-
Thalassemia (γ
Aδβ)
0/A
Microcytosis,micro
cytic, hypochromic
Moderate anemia,
Splenomegaly,
Homozygote:
thalassemia
intermedia
Decreased F
and A2
compared
with δβ-
thalassemia
γ-Thalassemia (γAγ
G)
0/A Microcytosis
Insignificant
unless
homozygote
Decreased F
HEREDITARY PERSISTENCE OF FETAL HEMOGLOBIN
Deletional A/A Microcytic Mild anemia F 100%
homozygotes
Nondeletional A/A Normal Normal F 20-40%
In α-thalassemia there are relatively fewer α-globin chains and an excess of
β- and γ-globin chains. These excess chains form Bart's hemoglobin (γ4) in
fetal life and Hb H (β4) after birth. These abnormal tetramers are not lethal
but lead to extravascular hemolysis. Prenatally a fetus with α-thalassemia
can become symptomatic because Hb F requires sufficient of α-globin gene
production, whereas postnatally infants with β-thalassemia become
symptomatic because Hb A requires sufficient production of β-globin genes.

Homozygous β-Thalassemia (Thalassemia Major, Cooley Anemia)
Clinical Manifestations
If not treated, children with β-thalassemia usually become symptomatic from
progressive hemolytic anemia, with profound weakness and cardiac
decompensation during the 2nd 6 mo of life. Depending on the mutation and
degree of fetal hemoglobin production, transfusions in β-thalassemia major
are necessary beginning in the 2nd mo to 2nd yr of life, but rarely later. The
decision to transfuse depends on the child's ability to compensate for the
degree of anemia.
Most infants and children have cardiac decompensation at hemoglobins of
4 g/dL or less. Generally, fatigue, poor appetite, and lethargy are late
findings of severe anemia in an infant or child and were more common
before transfusions were standard therapy.
The classic presentation of children with severe disease includes
thalassemic facies (maxilla hyperplasia, flat nasal bridge, frontal bossing),
pathologic bone fractures, marked hepatosplenomegaly, and cachexia and
is now primarily seen in developing countries. The spleen can become so
enlarged that it causes mechanical discomfort and secondary
hypersplenism. The features of ineffective erythropoiesis include expanded
medullary spaces (with massive expansion of the marrow of the face and
skull producing the characteristic thalassemic facies), extramedullary
hematopoiesis, and higher metabolic needs. The hepatosplenomegaly can
interfere with nutritional support. Pallor, hemosiderosis, and jaundice can
combine to produce a greenish brown complexion.
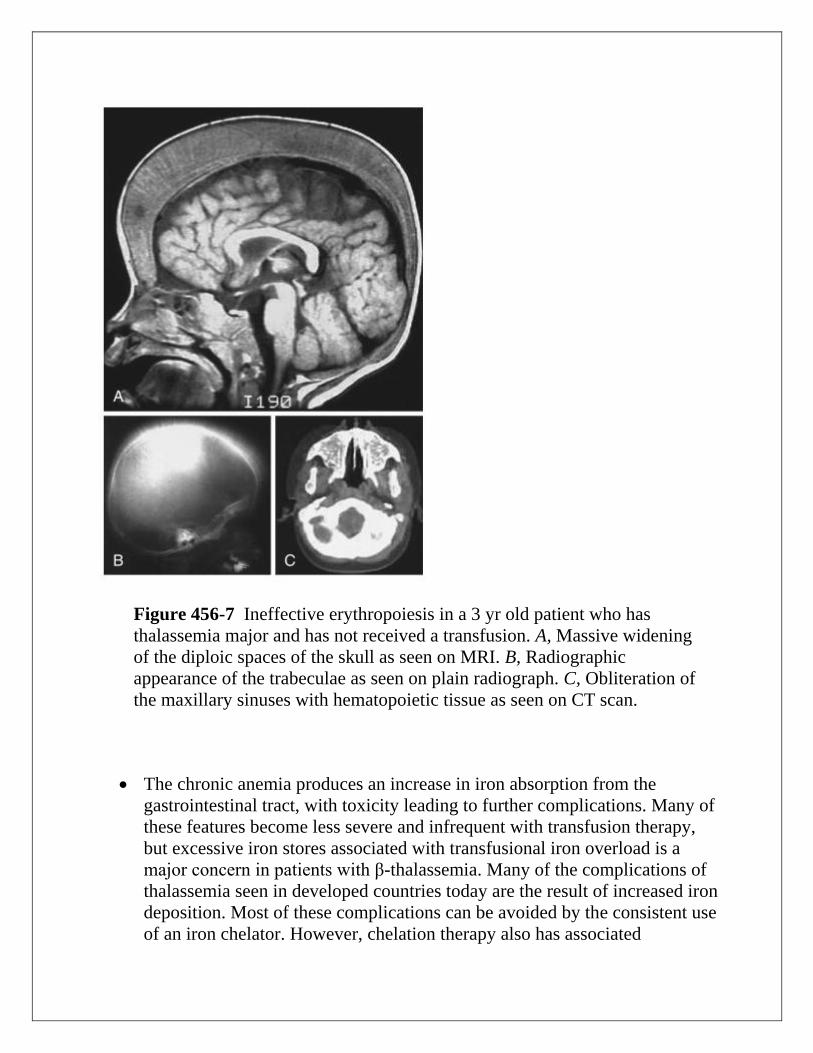
Figure 456-7 Ineffective erythropoiesis in a 3 yr old patient who has
thalassemia major and has not received a transfusion. A, Massive widening
of the diploic spaces of the skull as seen on MRI. B, Radiographic
appearance of the trabeculae as seen on plain radiograph. C, Obliteration of
the maxillary sinuses with hematopoietic tissue as seen on CT scan.
The chronic anemia produces an increase in iron absorption from the
gastrointestinal tract, with toxicity leading to further complications. Many of
these features become less severe and infrequent with transfusion therapy,
but excessive iron stores associated with transfusional iron overload is a
major concern in patients with β-thalassemia. Many of the complications of
thalassemia seen in developed countries today are the result of increased iron
deposition. Most of these complications can be avoided by the consistent use
of an iron chelator. However, chelation therapy also has associated

complications, including hearing loss, peripheral neuropathy, and poor
growth.
Endocrine and cardiac pathology are often associated with excessive iron
stores in patients with β-thalassemia major who are chronically transfused.
Endocrine dysfunction can include hypothyroidism, hypogonadotrophic
gonadism, growth hormone deficiency, hypoparathyroidism, and diabetes
mellitus. Congestive heart failure and cardiac arrhythmias are potentially
lethal complications of excessive iron stores in children with thalassemia.
What are the Laboratory Findings ?
The infant is born only with Hb F or, in some cases, Hb F and Hb E
(heterozygosity for β-thalassemia zero). Eventually, there is severe anemia,
reticulocytopenia, numerous nucleated erythrocytes, and microcytosis with
almost no normal-appearing erythrocytes on the peripheral smear (). The
hemoglobin level falls progressively to <5 g/dL unless transfusions are
given. The reticulocyte count is commonly <8% and is inappropriately low
when compared to the degree of anemia due to ineffective erythropoiesis.
The unconjugated serum bilirubin level is usually elevated, but other
chemistries may be normal at an early stage. Even if the child does not
receive transfusions, eventually there is iron accumulation with elevated
serum ferritin and transferrin saturation. Bone marrow hyperplasia can be
seen on radiographs ().
What is the Treatment ?
Before initiating chronic transfusions, the diagnosis of β-thalassemia major
should be confirmed and the parents counseled concerning this life-long
therapy. Beginning transfusion and chelation therapy are difficult challenges
for parents to face early in their child's life. Before beginning transfusion
therapy, a red-cell phenotype is obtained; blood products that are
leukoreduced and phenotypically matched for the Rh and Kell antigens are
required for transfusion. If a bone marrow transplant is a possibility, the
blood for transfusion should be negative for cytomegalovirus unless the
child has had a previous cytomegalovirus infection. Transfusion therapy
promotes general health and well-being and avoids the consequences of
ineffective erythropoiesis.

A transfusion program generally requires monthly transfusions, with the
pretransfusion hemoglobin level between 9.5 and 10.5 g/dL. In patients
with cardiac disease, higher pretransfusion hemoglobin levels may be
beneficial. Some blood centers have donor programs, pairing donors and
recipients, which decreases the exposure to multiple red cell antigens.
Excessive iron stores from transfusion cause many of the complications of
β-thalassemia major. Accurate assessment of excessive iron stores is
essential to optimal therapy. The serum ferritin is useful in assessing iron
balance trends but does not accurately predict quantitative iron stores.
Undertreatment or overtreatment of presumed excessive iron stores can
occur in managing a patient based on serum ferritin alone.
Quantitative iron by liver biopsy is the standard method for accurately
determining iron store for patients. T2* MRI software is now being used to
estimate iron stores in the liver and heart among patients with β-thalassemia
major. One reason for the preference of T2* MRI over liver biopsy is that
liver iron stores might not accurately reflect cumulative changes in cardiac
iron. Patients can have cardiac iron overload at the time of a safe liver iron
measurement. Many thalassemia centers now monitor cardiac iron with T2*
MRI imaging.
Excessive iron stores can be prevented by the use of deferoxamine
(Desferal) or deferasirox (Exjade). Deferoxamine chelates iron and some
other divalent cations, allowing their excretion in the urine and the stool.
Deferoxamine is given subcutaneously over 10-12 hr, 5-6 days a week. The
side effects include ototoxicity with high-frequency hearing loss, retinal
changes, and bone dysplasia with truncal shortening. The number of hours
that deferoxamine is used daily is more important than the daily dosage.
High dose, short-term infusions increase toxicity with little efficacy.
Plasma non–transferrin bound iron (NTBI) is most likely responsible for
serious iron injury. When deferoxamine is infusing, it binds NTBI. When
deferoxamine is stopped, there are rebound increases in NTBI levels and risk
for injury. In patients with excessive iron stores in the heart resulting in
symptomatic congestive heart failure, 24-hr deferoxamine has been shown to
reverse cardiomyopathy.
The oral iron chelator deferasirox (Exjade) is commercially available For
many patients and families, deferasirox has replaced deferoxamine because
the latter must be given subcutaneously for 10 hr a night, typically 5 of 7
nights a week. Although the optimal dose of deferasirox is well defined,
some patients have a less-than-expected response to the maximum approved
doses (30 mg/kg/day). The optimal dose beyond 30 mg/kg/day is not known,
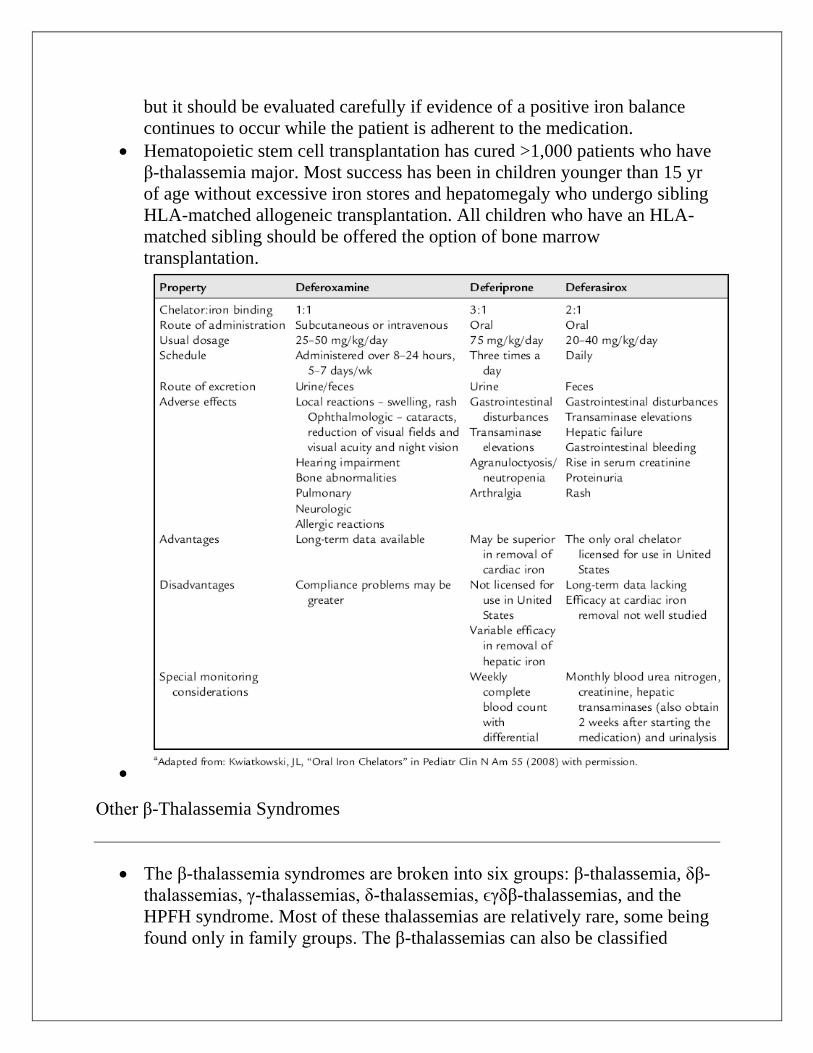
but it should be evaluated carefully if evidence of a positive iron balance
continues to occur while the patient is adherent to the medication.
Hematopoietic stem cell transplantation has cured >1,000 patients who have
β-thalassemia major. Most success has been in children younger than 15 yr
of age without excessive iron stores and hepatomegaly who undergo sibling
HLA-matched allogeneic transplantation. All children who have an HLA-
matched sibling should be offered the option of bone marrow
transplantation.
Other β-Thalassemia Syndromes
The β-thalassemia syndromes are broken into six groups: β-thalassemia, δβ-
thalassemias, γ-thalassemias, δ-thalassemias, ϵγδβ-thalassemias, and the
HPFH syndrome. Most of these thalassemias are relatively rare, some being
found only in family groups. The β-thalassemias can also be classified

clinically as thalassemia trait, minima, minor, intermedia, and major,
reflecting the degree of anemia. The genetic classification does not
necessarily define the phenotype, and the degree of anemia does not always
predict the genetic classification.
Thalassemia intermedia can be any combination of β-thalassemia mutations
(β0/β
+, β
0/β
variant, E/β
0), which will lead to a phenotype of microcytic anemia
with hemoglobin of about 7 g/dL. There is controversy about whether these
children should receive transfusions. They will certainly develop a degree of
medullary hyperplasia, nutritional hemosiderosis perhaps requiring
chelation, splenomegaly, and other complications of β-thalassemia
associated with excessive iron stores. Extramedullary hematopoiesis can
occur in the vertebral canal, compressing the spinal cord and causing
neurologic symptoms; the latter is a medical emergency requiring immediate
local radiation therapy to halt erythropoiesis. Transfusion alleviates the
thalassemic manifestations; the decision to transfuse must be balanced
against the future need for chelation therapy.
Splenectomy may be indicated for patients with thalassemia intermedia who
have a falling steady-state hemoglobin and for transfused patients with rising
transfusion requirements. However, splenectomy can have serious
consequences, including infection, pulmonary hypertension, and thrombosis.
All patients should be fully immunized against encapsulated bacteria before
splenectomy and subsequently should be on long-term penicillin prophylaxis
with appropriate instructions regarding fever management.
The thalassemias classified as minima and minor are usually heterozygotes
(β0/β, β
+/β
+), having a phenotype more severe than trait but not as severe as
intermedia. These children should be investigated for their genotype and
monitored for iron accumulation. The β-thalassemias are influenced by the
presence of α-thalassemia: α-thalassemia trait leading to less severe anemia
and duplicated α genes (ααα/αα) leading to a more severe thalassemia.
Often, patients who are in these groups require transfusions in adolescence
or adulthood; some may be candidates for chemotherapy such as
hydroxyurea.
Thalassemia trait is often misdiagnosed as iron deficiency in children
because the 2 produce similar hematologic abnormalities on CBC, and iron
deficiency is much more prevalent. A short course of iron and re-evaluation
is all that is required to identify children who will need further evaluation.
Children who have β-thalassemia trait have a persistently normal red cell
distribution width and low mean corpuscular volume (MCV). On
hemoglobin analysis, they have an elevated Hb F and diagnostically elevated

Hb A2. There are “silent” forms of β-thalassemia trait, and if the family
history is suggestive, further studies may be indicated.
α-Thalassemia
The same evolutionary pressures that produced β-thalassemia and sickle cell
disease produced α-thalassemia. Infants are identified in the newborn period
by the increased production of Bart's hemoglobin (γ4) during fetal life and its
presence at birth. The α-thalassemias occur most commonly in Southeast
Asia. Deletion mutations are common in α-thalassemia. In addition to
deletional mutations, there are nondeletional α-globin gene mutations, the
most common being Constant Spring (αCS
α); these mutations cause a more
severe anemia and clinical course than the deletional mutations. There are
four α-globin genes and four deletional α-thalassemia phenotypes.
The deletion of one α-globin gene (silent trait) is not identifiable
hematologically. Specifically, no alterations are noted in the mean
corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH).
Persons with this deletion are usually diagnosed after the birth of a child
with a 2-gene deletion or Hb H (β4). During the newborn period, <3% Hb
Bart's is observed. The deletion of one α-globin gene is common in African-
Americans.
The deletion of 2 α-globin genes results in α-thalassemia trait. The α-globin
genes can be lost in a trans-(−α/−α) or cis- (α,α/-SEA
) configuration. The
trans or cis mutations can combine with other mutations and lead to Hb H or
α-thalassemia major. In persons from Africa or of African descent the most
common α-globin gene deletion is in the trans configuration, whereas in
persons from Asia or the Mediterranean region the cis deletion is most
common.
The α-thalassemia traits manifest as a microcytic anemia that can be
mistaken for iron-deficiency anemia). The hemoglobin analysis is normal,
except during the newborn period, when Hb Bart's is commonly <8% but
>3%. Children with a deletion of 2 α-globin genes are commonly thought to
have iron deficiency, given the presence of both low MCV and MCH. The
simplest approach to distinguish between iron deficiency and α-thalassemia
trait is with a good dietary history. Children with iron-deficiency anemia
often have a diet that is low in iron. Alternatively, a brief course of iron
supplementation along with monitoring of erythrocyte parameters might
confirm the diagnosis of iron deficiency, or α-globin gene deletion analysis
may be necessary.

The deletion of three α-globin genes leads to the diagnosis of Hb H disease.
In California, where a large population of Asians resides, ~1 : 15,000
newborns have Hb H disease. The simplest manner of diagnosing Hb H
disease is during the newborn period, when excess in γ-tetramers are present
and Hb Bart's is commonly >25%. Obtaining supporting evidence from the
parents is also necessary. Later in childhood, there is an excess in β-globin
chain tetramers that results in Hb H. A definitive diagnosis of Hb H disease
requires DNA analysis with supporting evidence. Brilliant cresyl blue can
stain Hb H, but it is rarely used for diagnosis. Patients with Hb H disease
have a marked microcytosis, anemia, mild splenomegaly, and, occasionally,
scleral icterus or cholelithiasis. Transfusion is not commonly used for
therapy because the range of hemoglobin is 7-11 g/dL, with MCV 51-73 fl.
The deletion of all four α-globin genes causes profound anemia during fetal
life, resulting in hydrops fetalis; the ζ-globin gene must be present for fetal
survival. There are no normal hemoglobins present at birth (primarily Hb
Bart's, with Hb Gower 1, Gower 2, and Portland). If the fetus survives,
immediate exchange transfusion is indicated. These infants with α-
thalassemia major are transfusion dependent, and hematopoietic stem cell
transplant is the only cure.
The presence of a nondeletional α-globin mutation with a 2-gene deletion
results in a more severe anemia, increased hepatosplenomegaly, increased
jaundice, and a much more severe clinical course than Hb H disease. Hb H
Constant Spring is the most common form (−α/α,αCS
).
Treatment of the α-thalassemia deletion syndromes consists of folate
supplementation, possible splenectomy (with the attendant risks),
intermittent transfusion during severe anemia for the nondeletional Hb
H diseases, and chronic transfusion therapy or bone marrow transplant for
survivors of hydrops fetalis. These children also should not be exposed to
oxidative medications

Hemoglobin H disease. This blood film demonstrates microcytosis,
hypochromasia, and numerous morphologic abnormalities, including target cells,
microspherocytes, and fragments. Basophilic stippling may occur. Polychromasia
is present.





























