Embed Size (px)
Citation preview

Vol. 110 (2006) ACTA PHYSICA POLONICA A No. 6
Large-Scale Nanosecond Simulations
of the Dynamics of the Xe + H2O System
K. Kholmurodov∗
Joint Institute for Nuclear Research, Laboratory of Radiation Biologyand “Dubna” University, Moscow Reg., Dubna 141980, Russia
Z. Seifert, K. Raczkiewicz and J. Nitka
Faculty of Physics, Adam Mickiewicz UniversityUmultowska 85, 61-614 Poznan, Poland
(Received February 23, 2006)
Large-scale molecular dynamics simulations of the high-pressure trans-
formations of the xenon/water system were performed involving special pur-
pose molecular dynamics machines. We investigated several systems of dif-
ferent sizes and geometry at the suitable simulational conditions (density,
temperature, etc.), which are similar to the experiments conducted on the
xenon hydrates. A binary mixture (ice water + Xe) undergoes at high pres-
sure a long evolution and the Xe-guest atoms, enclosed inside the water
molecules, were observed and analyzed. Even for the thin slabs, starting
with the capture of the guest atoms by the water molecules, the water clus-
ters around the xenon atoms are formed. The results show that such a
hydrate-like formation preserves its structural stability over a long period of
the simulation time of order of nanosecond. The molecular dynamics sim-
ulations were performed on a basis of the MDGRAPE-2 modifications of
the DL POLY general purposes package, with the efficient treatment of the
Ewald real and reciprocal-space components of the Coulombic and Van der
Waals forces. The MDGRAPE-2 accelerates the calculations of the Coulomb
and Van der Waals forces, without applying a spherical cut of a fixed dis-
tance.
PACS numbers: 31.15.Qg, 61.20.–p, 61.50.Ah, 81.10.Aj, 91.60.Gf
1. Introduction
Understanding mechanisms of the high pressure–temperature reactions ofgases with water is one of fundamental topics in hydrate phenomena [1–5]. Themost challenging question, regarding hydrates, is the formation of the hydrates[3, 4, 6]. The hydrates kinetics, as a basically time-dependent phenomena, isgenerally characterized through the nucleation, growth, and dissociation processes.It is worth noting that the time-dependent hydrate formation phenomena are
∗corresponding author; e-mail: [email protected]
(759)

760 K. Kholmurodov et al.
substantially more challenging than time-dependent phenomena of structure andthermodynamics [4, 7–14].
The initial process of the hydrates formation in the system guestmolecule/water is a topic of the intense research and it is a task of great practicalinterest. The formation of gas hydrate possesses a stochastic nature, the hydratecrystallization process demands a very long timescale to observe. One of the mostpowerful tools in the theoretical studies on the hydrate phenomena is moleculardynamics (MD) method [2, 4, 10–16]. The aim of our present work is to investi-gate, through the large-scale nanosecond MD simulations, the initial stage of thegas(xenon)/water interaction process. We will consider the high pressure condi-tions, closely to the experimental situations, which are conducted on the xenonhydrates.
It is worth noting that MD method has been used widely in the hydrate stud-ies from the different aspects. MD simulations have been performed in [10−12]on the hydrate lattice stability, where the calculation results have been used toestimate the macroscopic and spectroscopic properties. The approaches employedin these studies include the Raman spectra along with the estimation of the elec-tronic and vibrational properties of the guest atoms in a clathrate hydrate, etc.The structural stability aspects and the effects of the guest molecules on the hostlattice structure were studied in [13, 14], through the molecular dynamics, andin [15, 16], using the lattice dynamics method. In Ref. [15] the motion of thehost and the guest within cages, depending on the guest size ratios (larger thanmethane within the 512 cavity), was investigated. Some other findings, regardingthe application of the MD in hydrates, cover the investigation of the affect of wa-ter molecules outside the cavity, the hydrate lattice stretch in correlation with theguest size, the vibrations of guests within the different cavities, the guest–guestinteractions, the hydrogen bonding in the hydrate and in the hydrate crystal in-terface, and others [4, 17–20].
Our goal is to present clustering processes of the water around the guest(xenon) molecules and on the time-dependent behavior of the hydrate-like forma-tion in the gas/water system. In the xenon hydrate experiments (see, for exam-ple [5]), structure I xenon clathrate was observed to be stable up to 1.8 GPa. Inthe case of Xe hydrates, the cavities are filled with 46H2O molecules and eightguest molecules. At the room temperatures, growth of crystals from the initialXe + H2O mixture was observed by increasing pressure to 0.7–0.8 GPa. In thepresent MD study large-scale transformations of the xenon and water system wereinvestigated for different sizes and geometry. To compare the system behavior atdifferent pressures, we simulated the xenon/water dynamics at the low and highgas densities. The evolution of the initial configuration of the xenon + water mix-ture was estimated at the temperatures 240 K and 280 K. The several events ofhydrate-like formation are observed at both temperatures. Once the guest xenonatom has been captured by the water molecules, a hydrate-like formation preserves

Large-Scale Nanosecond Simulations . . . 761
a structural stability over a timescale of order of nanosecond. In the calculationswe used a special purpose MD machine MDGRAPE-2, with the efficient treatmentof the Ewald real- and reciprocal-space components of the Coulombic and Van derWaals forces [21].
The paper is organized as follows. First, we present model and the simulationmethod, then calculation results and finally, we summarize the key points of thispaper and discuss the results.
2. Simulation method and systems2.1. Systems
The MD simulations have been performed on five model systems of xenonand water. In Fig. 1 the snapshots of the five simulated systems (numbered I, II,
Fig. 1. Five configurations of the water + Xe mixtures are shown. As described in
the text, the top figures are systems I (left) t = 75 ps and II (right) t = 315 ps; the
middle figures are systems III (left) t = 29 ps and IV (right) t = 21 ps and the bottom
is system V t = 10 ps. The water molecules are shown as dots, the xenon as large gray
circles.

762 K. Kholmurodov et al.
III, IV, V) are presented. The first two pictures (systems I and II, top) are theslabs of the Xe + water mixtures, contained in the volumes of V = L3 = (42 A)3
and V = L3 = (82 A)3, respectively (L = Lx = Ly = Lz). For these twoconfigurations we have the same number of particles: the TIP4P water molecules= 2240, the Xe atoms = 320 (9280 total atomic sites). The xenon for the system Iis a high density gas, for the system II — a low density gas. The initial water/gasvolume proportions in these configurations are: water = 5/6(V ), Xe gas = 1/6(V ).For the next two pictures (system III and IV, middle) we have the same volumeV = L3 = (82 A)3, but different numbers of particles: the water molecules = 2560,the Xe atoms = 1920 (12160 total atomic sites, left figure); the water molecules= 4480, the Xe atoms 1280 (19200 total atomic sites, right). The last simulatedsystem (system V) was contained in the volume of V = L3 = (43 A)3, with 2240TIP4P water molecules and 320 Xe atoms which gives 9280 total atomic sites.
2.2. Simulation methodWe have used the Lenard–Jones potential and TIP4P water model to describe
Xe–Xe and Xe–water interactions. The equations of motions were integrated usingVerlet algorithm and the temperature was controlled by a Nose–Hoover thermo-stat. The trajectories were generated in the (N, V, T ) ensemble, with the integra-tion time step of 1 fs, and the periodic boundary conditions were applied in alldirections. The Ewald method was used for calculation of the electrostatic forces.In all performed MD simulations, the water + Xe system was gradually equili-brated at a given temperature in a step of 10 K and for the several hundred timesteps.
3. Simulation results
In Fig. 2, the results of the MD simulations, performed at 280 K on thesystem I, are presented. The calculations have been done for about one million timesteps. During the system’s evolution several Xe atoms are seen to be surrounded bythe water molecules. The events of water clustering around the xenon atoms havebeen monitored in real time. The mobility of the Xe atoms, which are capturedin the water phase, are essentially low in comparison with the atoms in the gasphase. Once the Xe was enclosed by the water molecules, a hydrate-like structurehas been formed, which preserves a stability over a long period of simulation time(the timescale in Fig. 2 is of an order of nanosecond).
We investigated in detail the above Xe + water clustering formation by thecrystal CERIUS2 Visualiser. In Fig. 3 the structure of the Xe + water cluster,surrounded by a hydrogen-bond network, is shown. This structure, as analysisshows, closely resembles a hydrate-like formation (Fig. 3). However, the systemstemperature seems too be too high to form a crystal, with a good structural stabil-ity. For system I we have performed the same simulation also at the temperatureof 240 K. Nevertheless, no events of the water clustering around the xenon atomshave been observed.

Large-Scale Nanosecond Simulations . . . 763
Fig. 2. The six sequential snapshots of the MD simulation are shown for the system I
as described in the text. The temperature is 280 K. The water molecules are shown as
dots, the xenon as large gray circles. The initial configuration t = 0, t = 1.5 ns is the
final state, the other snapshots correspond to 0.3 ns, 0.6 ns, 0.9 ns, 1.2 ns.
Fig. 3. The Xe + water clustering structure is presented. The Xe atoms (large circles)
with the water molecules (red oxygen, white hydrogen), surrounded by hydrogen bonds
(yellow dotted line), are shown.

764 K. Kholmurodov et al.
About 1.5 nanosecond calculations showed no results, like those presentedin Figs. 2 and 3. Probably, at lower temperatures for this system a much longersimulation is necessary for the hydrate to form.
In Fig. 4 the results of MD simulations are presented for the system II at240 K. The calculations have been performed for Nstep > 106 time steps. Startingwith the well-separated states, the pockets of Xe enclosed by water inside the slabare formed.
Fig. 4. Six sequential snapshots of the MD simulations are shown for the system II,
as described in the text. The temperature is 240 K. The final configuration is for time
moment of 1.7 nanosecond. The water molecules are shown as dots, the xenon as large
gray circles. The initial configuration t = 0, t = 1.5 ns is the final state, the other
snapshots correspond to 0.3 ns, 0.6 ns, 0.9 ns, 1.2 ns.
The number of the Xe atoms, clustering in the water phase, changes withtime. For some Xe atoms from the water–gas interface there is a possibility toreturn back to the gas phase. However, those Xe atoms, which are surroundedby the water molecules at the center of the slab, are “long-lived” objects. Forexample, the Xe, which is marked in black in Fig. 4, has remained in the waterphase during about 1 million time steps of the system’s dynamics. (The finalconfiguration in Fig. 4 corresponds to a 1.5 nanosecond state.) The Xe atoms,surrounded by the water molecules, possess a very low mobility, in comparisonwith the atoms in gas phase.
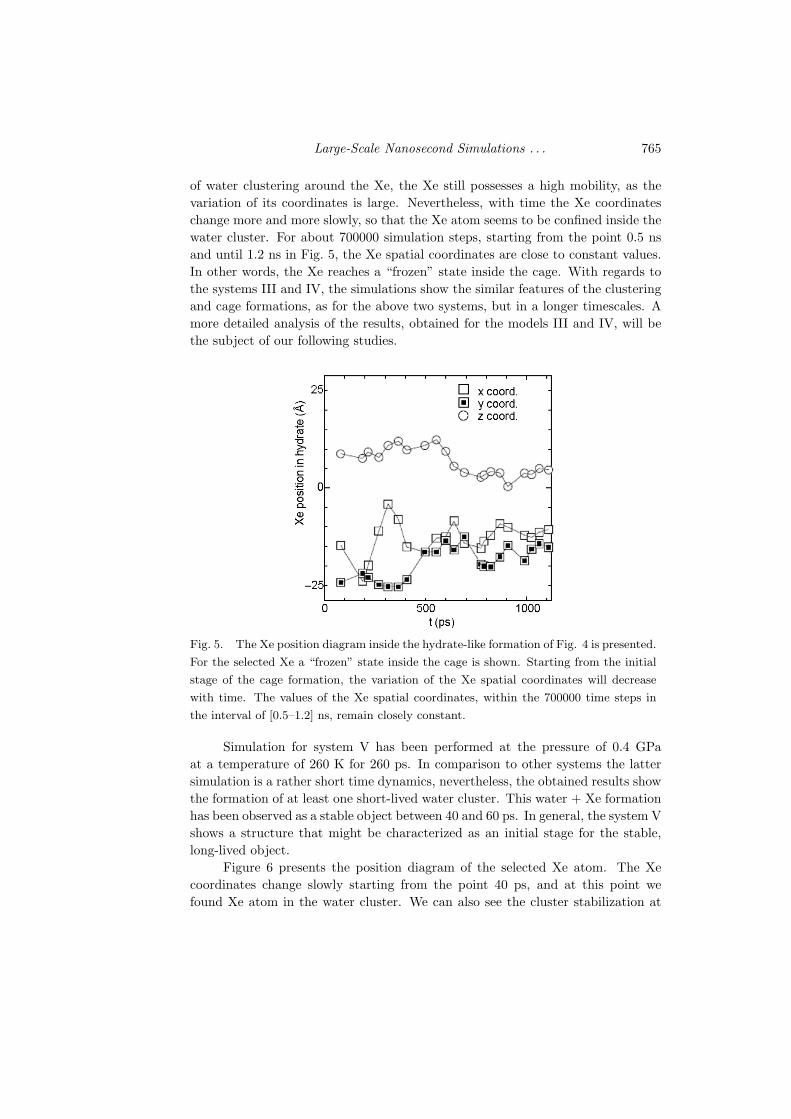
Figure 5 demonstrates the position diagram of the selected Xe (was shown inFig. 4 in black) inside the water cage. As it is seen from Fig. 5, at the initial stage
Large-Scale Nanosecond Simulations . . . 765
of water clustering around the Xe, the Xe still possesses a high mobility, as thevariation of its coordinates is large. Nevertheless, with time the Xe coordinateschange more and more slowly, so that the Xe atom seems to be confined inside thewater cluster. For about 700000 simulation steps, starting from the point 0.5 nsand until 1.2 ns in Fig. 5, the Xe spatial coordinates are close to constant values.In other words, the Xe reaches a “frozen” state inside the cage. With regards tothe systems III and IV, the simulations show the similar features of the clusteringand cage formations, as for the above two systems, but in a longer timescales. Amore detailed analysis of the results, obtained for the models III and IV, will bethe subject of our following studies.
Fig. 5. The Xe position diagram inside the hydrate-like formation of Fig. 4 is presented.
For the selected Xe a “frozen” state inside the cage is shown. Starting from the initial
stage of the cage formation, the variation of the Xe spatial coordinates will decrease
with time. The values of the Xe spatial coordinates, within the 700000 time steps in
the interval of [0.5–1.2] ns, remain closely constant.
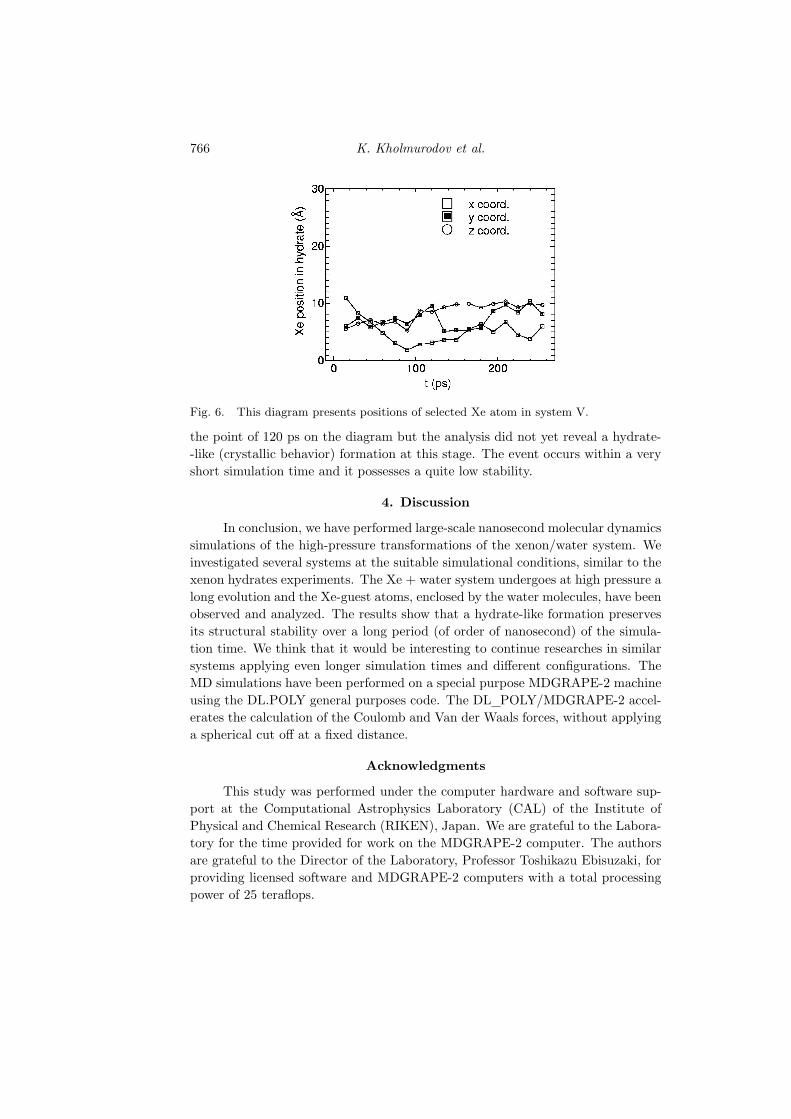
Simulation for system V has been performed at the pressure of 0.4 GPaat a temperature of 260 K for 260 ps. In comparison to other systems the lattersimulation is a rather short time dynamics, nevertheless, the obtained results showthe formation of at least one short-lived water cluster. This water + Xe formationhas been observed as a stable object between 40 and 60 ps. In general, the system Vshows a structure that might be characterized as an initial stage for the stable,long-lived object.
Figure 6 presents the position diagram of the selected Xe atom. The Xecoordinates change slowly starting from the point 40 ps, and at this point wefound Xe atom in the water cluster. We can also see the cluster stabilization at
766 K. Kholmurodov et al.
Fig. 6. This diagram presents positions of selected Xe atom in system V.
the point of 120 ps on the diagram but the analysis did not yet reveal a hydrate--like (crystallic behavior) formation at this stage. The event occurs within a veryshort simulation time and it possesses a quite low stability.
4. Discussion
In conclusion, we have performed large-scale nanosecond molecular dynamicssimulations of the high-pressure transformations of the xenon/water system. Weinvestigated several systems at the suitable simulational conditions, similar to thexenon hydrates experiments. The Xe + water system undergoes at high pressure along evolution and the Xe-guest atoms, enclosed by the water molecules, have beenobserved and analyzed. The results show that a hydrate-like formation preservesits structural stability over a long period (of order of nanosecond) of the simula-tion time. We think that it would be interesting to continue researches in similarsystems applying even longer simulation times and different configurations. TheMD simulations have been performed on a special purpose MDGRAPE-2 machineusing the DL.POLY general purposes code. The DL POLY/MDGRAPE-2 accel-erates the calculation of the Coulomb and Van der Waals forces, without applyinga spherical cut off at a fixed distance.
Acknowledgments
This study was performed under the computer hardware and software sup-port at the Computational Astrophysics Laboratory (CAL) of the Institute ofPhysical and Chemical Research (RIKEN), Japan. We are grateful to the Labora-tory for the time provided for work on the MDGRAPE-2 computer. The authorsare grateful to the Director of the Laboratory, Professor Toshikazu Ebisuzaki, forproviding licensed software and MDGRAPE-2 computers with a total processingpower of 25 teraflops.

Large-Scale Nanosecond Simulations . . . 767
References
[1] D.W. Davidson, in: Water: A Comprehensive Treatise, Ed. F. Pranks, Plenum,
New York 1973, p. 115.
[2] Y.P. Handa, J.S. Tse, D.D. Klug, E. Whalley, J. Chem. Phys. 94, 623 (1991).
[3] Y.A. Dyadin, E.G. Larionov, T.V. Mikina, L.I. Starostina, Mendeleev Commun.
7, 74 (1997).
[4] E.D. Sloan, Jr., Clathrate Hydrates of Natural Gases, Marcel Dekker, Inc., New
York 1998.
[5] C. Sanloup, H.-K. Mao, R.J. Hemley, Proc. Natl. Acad. Sci. USA 99, 25 (2002).
[6] J.P. Long, E.D. Sloan, Mol. Simul. 11, 145 (1993).
[7] J.P. Long, E.D. Sloan, Int. J. Thermophys. 17, 1 (1996).
[8] L.A. Baez, P.A. Clancy, Ann. New York Acad. Sci. 715, 177 (1994).
[9] J.S. Parent, M.Sc. Thesis, University of Calgary, 1993.
[10] J.S. Tse, M.L. Klein, J. Phys. Chem. 91, 5789 (1987).
[11] J.S. Tse, M.A. White, J. Phys. Chem. 92, 5006 (1988).
[12] J.S. Tse, Ann. New York Acad. Sci. 715, 187 (1994).
[13] P.M. Rodger, J. Phys. Chem. 94, 6080 (1990).
[14] P.M. Rodger, Mol. Simul. 5, 315 (1990).
[15] K. Koga, H. Tanaka, J. Chem. Phys. 104, 263 (1996).
[16] K. Koga, H. Tanaka, K. Nakanishi, Mol. Simul. 12, 241 (1994).
[17] K.A. Sparks, J.W. Tester, J. Phys. Chem. 96, 11022 (1992).
[18] M.J. Hwang, Ph.D. Thesis, University of Pittsburgh, Pittsburgh (PA) 1989.
[19] M.J. Hwang, G.D. Holder, S.R. Zele, Fluid Phase Equil. 93, 437 (1993).
[20] T. Carver, M. Drew, M. Rodger, in: Proc. 2nd Int Conf. on Natur. Gas Hydrat.,
Toulouse 1996, Ed. J.P. Monfort.
[21] K. Kholmurodov, W. Smith, K. Yasuoka, T. Ebisuzaki, Comput. Phys. Commun.
125, 167 (2000); in: Proc. 6th World Multiconf. SCI’2002, Orlando (USA) 2002.





























