Embed Size (px)
Citation preview

Cancer Letters, 26 (1985) 191-200 Elsevier Scientific Publishers Ireland Ltd.
191
LACK OF SPECIFIC INHIBITION OF DNA REPAIR IN WI-38 HUMAN DIPLOID FIBROBLASTS BY SODIUM SACCHARIN
JULIE A. SKARE and TERESA K. WONG
The Procter & Gamble Company, Miami Valley Laboratories, P. 0. Box 39175, Cincinnati, OH 45247 (U.S.A.)
(Received 23 October 1984) (Revised version received 31 December 1984) (Accepted 7 January 1985)
SUMMARY
The ability of sodium saccharin (NaS) to inhibit the repair of DNA damaged by UV irradiation was examined in cultured WI-38 human diploid fibroblasts. Cesium chloride density gradient ultracentrifugation was used to measure DNA repair and DNA replication. NaS (lO-10,000 pg/ml) did not specifically inhibit UV light-induced DNA repair. At doses of NaS (1785 and 10,000 pg/ml) that caused a 62-67s inhibition of semiconserva- tive DNA replication, there was little or no inhibition of DNA repair syn- thesis. In cell cultures not exposed to UV irradiation, NaS failed to induce DNA repair. RNA synthesis and protein synthesis were unaffected by NaS at all doses tested. The inhibition of semiconservative DNA replication at higher doses of NaS may be a manifestation of cytotoxicity. In contrast to results with NaS, WI-38 cells were very sensitive to DNA repair inhibition by the well-studied inhibitor quinacrine_HCl. These results do not support mechanisms of saccharin-induced tumorigenesis involving either direct induction of DNA damage or inhibition of the repair of DNA damage caused by other agents.
INTRODUCTION
Inhibition of DNA repair synthesis was proposed in the early 1970s by Gaudin et al. as a possiblemechanism of tumor promoter or cocarcinogen action [6-81. These investigators reported that materials as diverse as phorbol esters, steroid hormones and DNA-binding drugs all inhibited DNA repair in UV-irradiated human lymphocytes. It was reported subsequently, however, that these chemicals did not have a specific effect on DNA repair in human lymphocytes, HeLa cells, or human fibroblasts; either semicon- servative DNA replication was preferentially inhibited or both DNA replica-
0304-3835/85/$03.30 o 1985 Elsevier Scientific Publishers Ireland Ltd. Published and Printed in Ireland

192
tion and DNA repair were inhibited equally [ 4,181. The synthesis of other macromolecules, such as RNA and protein, was also inhibited [ 181. Further. more, a non-promoting phorbol ester analogue was nearly as effective in inhibiting DNA repair synthesis as was the potent promoter 12-O-tetra- decanoylphorbol-13-acetate [ll] . These findings indicated that inhibition of DNA repair synthesis may not be important or at least not sufficient as a mechanism of action for tumor promoters or cocarcinogens.
Lindberg and Pinnas have reported data indicating that saccharin totally inhibited DNA repair in human lymphocytes without affecting DNA repli- cation [15]. To our knowledge, a full report of this work has not been published nor have the results been reproduced by other investigators. In the studies reported here we sought to determine if a specific inhibition of DNA repair synthesis could be caused by NaS in WI-38 human diploid fibroblasts. Repair synthesis in WI-38 cells has been well characterized previously [ 12-141. In the present studies, DNA repair synthesis and DNA replication were measured by cesium chloride density gradient ultra- centrifugation. This method provides an unambiguous means to differentiate these two processes.
MATERIALS AND METHODS
Cells WI-38 CCL75 (human diploid fetal lung) cells at their 15th passage
(population doubling 21.7) were obtained from the American Type Culture Collection in Rockville, MD. Cells were grown and maintained as previously described [ 51. Cells were screened for mycoplasma and found to be free of contamination.
Chemicals and enzymes 5-Bromodeoxyuridine (BUdR), 5-fluorodeoxyuridine (FUdR), chloro-
quine-HCl and ribonuclease A were purchased from Sigma Chemical Company St. Louis, MO. ~-[4,5-~H] leucine (50 Ci/mmol), [5,6-3H] uridine (43 Ci/mmol) and Nuclear Chicago Solubilizer were purchased from Amersham, Arlington Heights, IL. [methyl-3H] Thymidine (20 Ci/mmol) was purchased from New England Nuclear, Boston, MA. Whatman GF/C filters (2.4 cm) were purchased from Scientific Products, Columbus, OH. NaS (Lot no. 1648, 99.3% pure) was obtained from Sherwin Williams, Cincinnati, OH. Isoamyl alcohol and chloroform were purchased from MCB Manufacturing Chemists, Inc. Gibbstown, NJ. 3,5-Diaminobenzoic acid dihydrochloride was purchased from Aldrich Chemicals, Milwaukee, WI. Cesium chloride and proteinase K were made by E. Merck, Darmstadt, F.R.G. and were purchased from MCB Manufacturing Chemists, Inc., Gibbstown, NJ.
Toxicity assay Cultures were prepared by seeding WI-38 cells onto 100 mm Corning

193
tissue culture plates. After the cells reached confluence, the complete medium (Eagle Basal Medium (BME) + 10% fetal bovine serum (FBS)) was replaced with 10 ml of BME containing 0.5% FBS and the cultures were maintained for 7-10 days before use. This procedure slowed the growth of the cultures. Growth arrested cultures were used in the toxicity assay in order to duplicate the assay conditions used in the DNA repair studies. The toxicity assay was performed by exposing the cultures for 4 h to lo-fold serial dilutions of NaS from 10,000-0.01 pg/ml. Exposure was done either in the presence or absence of an S-9 metabolic activation system consisting of a 9000 X g supernate prepared from liver homogenates of Sprague- Dawley rats induced with Aroclor 1254 [l] . Following exposure to the test material, the cultures were examined using a phase contrast microscope. The presence of cell debris and/or rounded, constricted, or floating cells were scored as signs of cytotoxicity.
DNA repair synthesis and DNA replication Cells were grown to confluence in 100 mm Petri dishes. After the cells
reached confluence, the complete medium (BME + 10% FBS) was replaced with 10 ml of BME containing 0.5% FBS. Cells were maintained in low serum medium for 7-10 days prior to use. Maintenance of cultures in low serum medium reduced the rate of cell proliferation such that the incidence of S-phase cells was approximately 5-10%. For 1 h before UV-irradiation, the cells were exposed to 10 PM BUdR and 2 FM FUdR in conditioned low serum medium. The medium was conditioned by maintaining the cells in it for 7-10 days.
The conditioned medium was removed from the cultures and saved after the preincubation with BUdR and FUdR. The cultures were rinsed with phosphate buffered saline (PBS) and overlayed with 0.2 ml of PBS. Cultures were exposed at room temperature to UV irradiation from a GE no. G30T8 germicidal lamp. Incident radiation was measured using a Blak-Ray UV meter (Ultra-Violet Products, San Gabriel, CA). UV fluence rate was approximately 12 ergs/s per mm 2. In studies measuring DNA replication, the UV-irradiation step was omitted. PBS was replaced with the conditioned medium containing BudR and FUdR, and 20 pCi/ml [3H] thymidine and one one-hundredth volume of a concentrated aqueous solution of test material (NaS or quinacrine-HCl) were added immediately. Cultures were incubated at 37°C for 4 h in a 5% CO* in air atmosphere. The medium was removed and the cells were washed twice with ice-cold Tris-buffered saline. Approxi- mately 0.5 ml of 0.1% sodium dodecyl sulphate (SDS) in 0.01 M Tris-HCl, 5 mM EDTA (pH 7.9) was added to each culture. Cultures were stored at -80°C until processed for density gradient ultracentrifugation. Five or six cultures were pooled for each dose level.
Cultures were processed for neutral cesium chloride density gradient ultracentrifugation as described previously [ 51, except that polyallomer tubes were used instead of nitrocellulose tubes. In one experiment the samples of normal density DNA were recentrifuged under alkaline conditions

194
to confirm that the radiolabel was incorporated into non-replicating DNA. The alkaline gradients were also prepared as previously described [5].
Gradients were fractionated and aliquots of the fractions were analyzed for tritium content by liquid scintillation counting and for DNA content by enhancement of diaminobenzoic acid dihydrochloride fluorescence
WI *
RNA and protein synthesis Cultures were prepared by maintenance in low serum medium as described
for the cytotoxicity and DNA repair assays. Cells were incubated for 4 h in conditioned medium containing either 4 pCi/ml of [ 3H] uridine or 4 pCi/ml of [ 3H] leucine. NaS was added to the cultures immediately before addition of the radiolabel. After incubation the cultures were washed twice with PBS. The cells were scraped from each of the plates into a l-ml volume of PBS and the cell suspensions were sonicated. Radiolabel incorporated into trichloroacetic acid (TCA)-precipitable material was measured in samples which were precipitated by adding 2 ml of an ice-cold solution containing 10% TCA in 20 mM sodium pyrophosphate along with 300 pg of bovine serum albumin as a carrier. Precipitates were collected on Whatman GF/C filters, and the filters were washed 6 times with 2 ml volumes of 10% TCA in 20 mM sodium pyrophosphate, then twice with 2 ml volumes of 95% ethanol. After the filters were airdried, 0.5 ml of Nuclear Chicago Solubilizer was added to solubilize the radioactivity. Ten microliters of glacial acetic acid was added and the samples were analyzed by liquid scintillation count- ing. The incorporation of radiolabel was normalized on the basis of DNA content by analyzing parallel aliquots of the sonicated cell samples in the diaminobenzoic acid dihydrochloride fluorescence enhancement assay. Expression of these data relative to total DNA content is comparable to normalization of RNA or protein synthesis on a per cell basis. Measurements of RNA and protein synthesis were obtained from each of 3 individual culture dishes per dose group. Data were subjected to analysis of variance.
RESULTS
Cy totoxicity None of the doses of NaS tested for cytotoxicity (O.Ol-10,000 pg/ml),
either in the presence of absence of S-9, showed any evidence of a toxic effect when cultures were examined by phase contrast microsocopy. How- ever, microscopic evaluation is a crude means of assessing viability. Effects on DNA synthesis described below provided a more sensitive means for assessing the cytotoxicity of NaS. All subsequent studies were conducted in the absence of S-9.
Effect of NaS on UV light-induced DNA repair In unirradiated cultures incubated in the presence of [ 3H] thymidine and
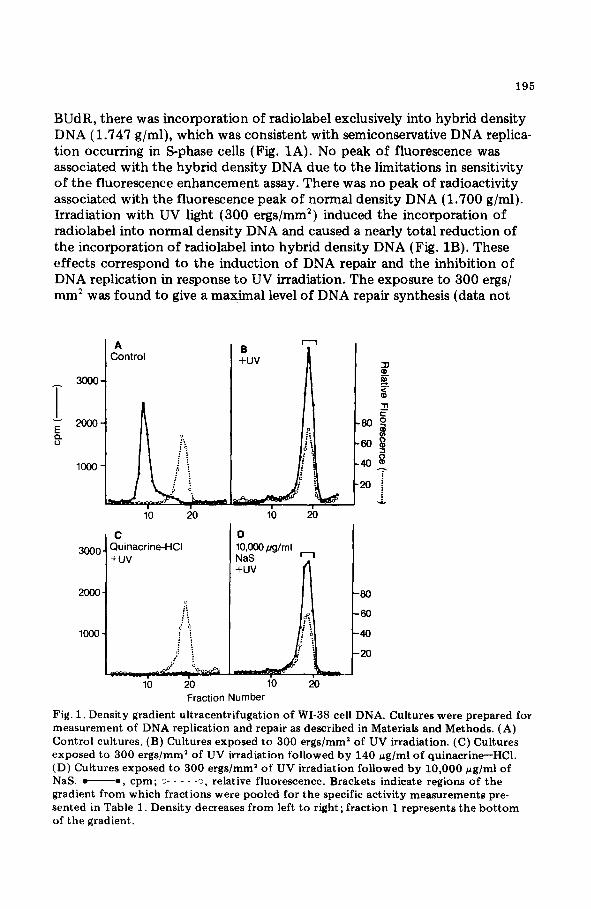
195
BUdR, there was incorporation of radiolabel exclusively into hybrid density DNA (1.747 g/ml), which was consistent with semiconservative DNA replica- tion occurring in S-phase cells (Fig. 1A). No peak of fluorescence was associated with the hybrid density DNA due to the limitations in sensitivity of the fluorescence enhancement assay. There was no peak of radioactivity associated with the fluorescence peak of normal density DNA (1.700 g/ml). Irradiation with UV light (300 ergs/mm2) induced the incorporation of radiolabel into normal density DNA and caused a nearly total reduction of the incorporation of radiolabel into hybrid density DNA (Fig. 1B). These effects correspond to the induction of DNA repair and the inhibition of DNA replication in response to UV irradiation. The exposure to 300 ergs/ mm2 was found to give a maximal level of DNA repair synthesis (data not
I A Control
I C
3ooo Quinacrine-HCI +uv
Fraction Number
Fig. 1. Density gradient ultracentrifugation of WI-38 cell DNA. Cultures were prepared for measurement of DNA replication and repair as described in Materials and Methods. (A) Control cultures. (B) Cultures exposed to 300 ergs/mm2 of UV irradiation. (C) Cultures exposed to 300 ergs/mm2 of UV irradiation followed by 140 rg/ml of quinacrine-HCl. (D) Cultures exposed to 300 ergs/mm’ of UV irradiation followed by 10,000 pg/ml of NaS. -0, cpm; ii- - - - -3, relative fluorescence. Brackets indicate regions of the gradient from which fractions were pooled for the specific activity measurements pre- sented in Table 1. Density decreases from left to right; fraction 1 represents the bottom of the gradient.

196
shown). When cultures were exposed to the well-studied DNA repair inhib- itor, quinacrine-HCl, there was a total inhibition of UV light-induced DNA repair synthesis (Fig. 1 C) .
In contrast, the incorporation of radiolabel into normal density DNA in response to UV irradiation was apparently unaffected by exposure to NaS at concentrations of lo-10,000 pg/ml. An example of a typical gradient profile is shown in Fig. 1D. The amount of DNA repair synthesis was calcu- lated in pooled fractions of normal density DNA in order to quantitatively compare the NaS-treated cultures to control cultures receiving only UV irradiation (Table 1). Doses of NaS ranging from lo-1785 pg/ml had no inhibitory effect on the amount of DNA repair synthesis occurring in response to UV irradiation. A decrease in DNA repair synthesis of approxi- mately 15% was observed in cultures exposed to 10,000 pg/ml of NaS. Although this pattern of response was reproduced in 2 independent experi- ments, the decrease at the high dose was not statistically significant when the data from these 2 experiments were subjected to analysis of variance. all dose levels, rebanding of the normal density DNA peak under alkaline conditions, which enhances the purification of DNA containing repaired regions, confirmed that the incorporation of radiolabel observed in the
TABLE 1
NaS EFFECTS ON MACROMOLECULAR SYNTHESIS
At
NaS dose
(pglmi)
UV-induced DNA replica- RNA synthesis Protein synthesis DNA repair tion (cpm/pg (cpm/crg DIWC (cpm/rg DNAJd (cpmlrg DNAY DNA)b
0 762 + 17 374 1487 + 260 2821 -I 221 10 753f61 297 1767 f 158 3419 f 505 57 766 + 54 334 1707 f 138 3221 f 477
319 740f24 344 1335 f 86 2287 f 330 1785 760 f 28 123 1239+ 90 2189 f 126
10,000 642 f 10 141 1391 f 117 2765 + 218
a[3H]Thymidine incorporation was determined in fractions of non-replicating normal density DNA pooled from the region of the gradient indicated by brackets in Fig. 1. The values represent the mean f range of specific activity measurements from 2 indepen- dent experiments. Data were subjected to analysis of variance.
b [“H]Thymidine incorporation was expressed as cpmlrg DNA (radioactivity in hybrid density DNA/total amount of normal density DNA recovered from the gradient). One determination was made for each gradient in Fig. 2.
‘RNA synthesis was measured by [‘Hluridine incorporation into TCA-precipitable material. Values represent the mean f S.E. for 3 individual culture dishes. Data were subjected to analysis of variance.
dProtein synthesis was measured by [ “Hlleucine incorporation into TCA-precipitable material. Values represent the mean + S.E. for 3 individual culture dishes. Data were subjected to analysis of variance.
197
cultures treated with both UV irradiation and NaS was due to DNA repair synthesis (data not shown).
Effect of NaS on DNA repair and DNA replication Cultures were exposed to NaS without prior UV irradiation to determine
if NaS induced DNA repair or affected DNA replication. In NaS-treated cultures there was no peak of radiolabel coincident with the peak of normal density DNA, indicating that NaS did not induce DNA repair. At the 2 higher doses tested (1785 and 10,000 ccg/ml) there was an apparent decrease in the incorporation of radiolabel into hybrid density DNA. Both the counts per minute (cpm) of radioactivity in the fractions containing hybrid density DNA and the DNA content in the fractions containing normal density DNA were summed. The amount of DNA replication was quantitated as cpm per pg of DNA, assuming that the quantity of DNA present in the normal density peak was a measure of the total DNA applied to the gradient. As shown in Table 1, there was a 62-67s inhibition in the amount of DNA replication at the higher doses of NaS (1785 and 10,000 fig/ml). The reduc- tion in DNA replication at higher doses was reproducible in a second experi- ment in which DNA replication was inhibited by 47% and 57%, respectively, at doses of 1785 and 10,000 pg/ml of NaS (data not shown).
Effect of NaS on RNA synthesis and protein synthesis The incorporation of [ 3H] uridine and [ 3H] leucine into TCA-precipitable
material was measured in control cultures and in cultures exposed to NaS
25
01 b 10 loo
s Ouinacrine Concentration
0 (M/ml)
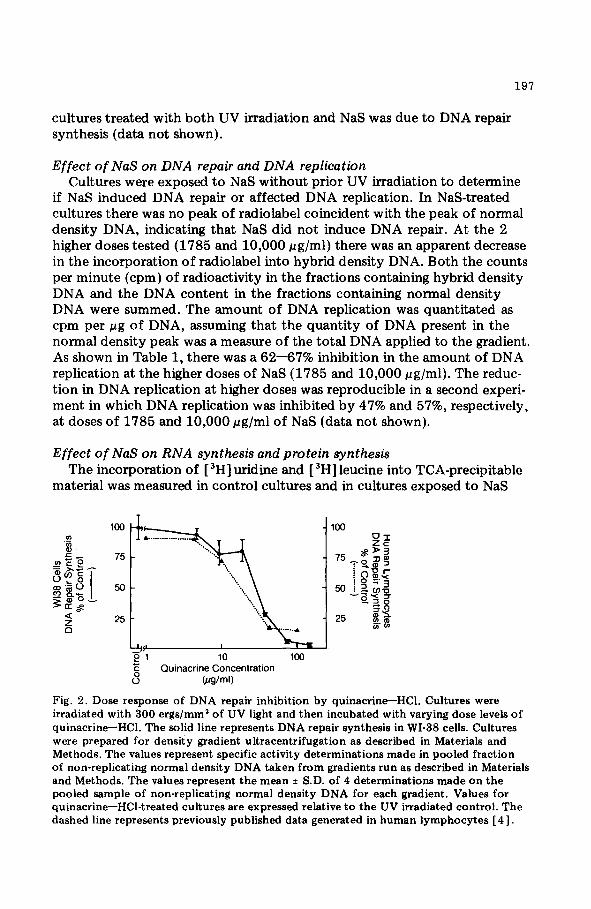
Fig. 2. Dose response of DNA repair inhibition by quinacrineHC1. Cultures were irradiated with 300 ergs/mm* of UV light and then incubated with varying dose levels of quinacrine-HCl. The solid line represents DNA repair synthesis in WI-38 cells. Cultures were prepared for density gradient ultracentrifugation as described in Materials and Methods. The values represent specific activity determinations made in pooled fraction of non-replicating normal density DNA taken from gradients run as described in Materials and Methods. The values represent the mean f S.D. of 4 determinations made on the pooled sample of non-replicating normal density DNA for each gradient. Values for quinacrine-HCl-treated cultures are expressed relative to the UV irradiated control. The dashed line represents previously published data generated in human lymphocytes [ 41.

198
(lo-10,000 pg/ml). Analysis of variance showed that treatment with NaS did not significantly affect either [‘HI uridine or [ 3H] leucine incorporation (Table 1).
Sensitivity of WI-38 cells to quinacrine-HCl The inhibition of DNA repair in UV-irradiated WI-38 cells as a function of
quinacrine-HCl concentration was investigated using the methods employed in Fig. 1. The dose response relationship is plotted in Fig. 2. Doses of 9.4 and 18.8 E.cg/ml both resulted in a 20% inhibition of DNA repair synthesis, while a dose of 37.5 pg/ml caused a 70% inhibition. Doses of 75 and 150 c(g/ ml caused total inhibition. In a study comparing effects on DNA repair and replication, exposure to 30 pg/ml of quinacrine-HCl with or without UV light caused a 78% inhibition of DNA repair, while DNA replication was inhibited by 96% (data not shown).
DISCUSSION
The data presented here indicate that NaS does not specifically inhibit DNA repair synthesis. These results, therefore, do not support the hypo- thesis that saccharin acts as a cocarcinogen or tumor promoter by a specific inhibition of DNA repair. Furthermore, these results are in general agree- ment with the conclusions that have been drawn concerning a variety of other types of chemicals including steroid hormones, DNA binding agents, phorbol esters, vitamin A alcohol, and inhibitors of cellular respiration such as potassium cyanide [4,18]. That is, any effect of these agents on DNA excision repair is non-specific and involves an equivalent or greater effect on other cellular processes (DNA replication, RNA synthesis, protein synthesis). A similar conclusion was drawn in a recent review article concerning DNA repair inhibitors [23. In the case of NaS, the only substantial effect on macromolecular synthesis is the inhibition of DNA replication at the higher dose levels. Even at doses that cause a substantial inhibition of DNA replica- tion, NaS has little or no effect on DNA repair synthesis. RNA synthesis and protein synthesis are also unaffected. The effect on DNA replication may be an indication of a cytotoxic or cytostatic response to the high levels of NaS. These concentrations of NaS (1785 and 10,000 I.cg/ml) would be expected to significantly alter the ionic composition and total osmolarity of the culture medium which may in turn affect cell membrane properties and ion transport. Changes in monovalent cation (Na+, K’) transport have been implicated in the regulation of cell cycle progression including the G, /S phase transition and progression through S phase [ 31.
The results of these studies in cultured human fibroblasts differ from those reported in human peripheral lymphocytes, where a low dose of saccharin (10 pg/ml) caused a total inhibition of DNA repair synthesis without affecting DNA replication stimulated by the mitogen phytohemag- glutinin [15]. The explanation for the disparity between Lindberg and

199
Pinnas’ results and the results described here is unknown. Although it is possible that cell types differ in their sensitivity to a particular inhibitor of DNA repair, we consider that explanation unlikely. Cleaver and Painter found that the effects on DNA replication and repair of 14 chemicals of diverse classes were very similar in human lymphocytes and in HeLa cells [4]. The concentration of the tumor promoter 12-O-tetradecanoylphorbol- 13acetate that caused a 50% inhibition of repair synthesis in WI-38 cells was similar to the half-maximal inhibitory concentration in human lympho- cytes, despite the fact that different methods for measuring repair synthesis were used [7,18]. We have shown that the inhibition of DNA repair by quinacrine-HCl in WI-38 cells follows a dose response curve nearly identical to that reported for this chemical in human lymphocytes [4]. Moreover, as with lymphocytes, DNA replication in WI-38 cells is inhibited to a greater extent at a given concentration of quinacrine-HCl than is DNA repair.
Conflicting reports have appeared in the literature regarding the DNA- damaging capacity of saccharin. In a preliminary report, Ochi and Tonomura claimed that saccharin induced unscheduled DNA synthesis in cultured human fibroblasts, although thedata appearing in the abstract showed no appreciable effect above background [ 171. Using alkaline sucrose gradient analysis, Miyata et al. found no evidence that saccharin had any effect on the integrity of DNA in rat bladder epithelial cells [16]. The studies reported here demonstrate that NaS does not induce DNA repair in WI-38 cells.
To summarize, these results do not support a mechanism involving the direct induction of DNA damage by NaS. These results also cast doubt upon the hypothesis that DNA repair inhibition is involved as a mechanism of action for saccharin-induced tumorigenesis. However, a definitive test of this hypothesis would require studies in the target tissue, the bladder epithelium.
ACKNOWLEDGEMENTS
We thank Drs. W.J. Coppinger, E.D. Thompson, R.A. LeBoeuf and R.L. Anderson for critical review of this manuscript. We are indebted to Ms. L.R. Schroer for assistance in preparation of the manuscript.
REFERENCES
1 Ames, B.N., McCann, J. and Yamasaki, E. (1975) Methods for detecting carcinogens and mutagens with the SalmoneZla/mammalian-microsome mutagenicity test. Mutat. Res., 31, 347-364.
2 Bernheim, N.J. and Falk, H. (1983) Chemical, physical, and genetic factors interfering with DNA repair - a review. J. Am. COB. Toxicol., 2,23-54.
3 Boonstra, J., Mummery, C.L., van Zoelen, E.J.J., van der Saag, P.T. and de Laat, S.W. (1982) Monovalent cation transport during the cell cycle. Anticancer Res., 2, 265- 274.
4 Cleaver, J.E. and Painter, R.B. (1975) Absence of specificity in inhibition of DNA repair replication by DNA-binding agents, cocarcinogens, and steroids in human cells. Cancer Res., 35, 1773-1778.

200
5 Coppinger, W.J., Wong, T.K. and Thompson, E.D. (1983) Unscheduled DNA syn- thesis and DNA repair studies of peroxyacetic and monoperoxydecanoic acids.
Environ. Mut., 5, 177-192. 6 Gaudin, D., Gregg, R.S. and Yielding, K.L. (197 1) DNA repair inhibition: a possible
mechanism of action of co-carcinogens. Biochem. Biophys. Res. Commun., 45, 630-636.
7 Gaudin, D., Gregg, R.S. and Yielding, K.L. (1972) Inhibition of DNA repair by cocarcinogens. Biochem. Biophys. Res. Commun., 48, 945-949.
8 Gaudin, D., Gregg, R.S. and Yielding, K.L. (1972) Inhibition of DNA repair replica- tion by DNA binding durgs which sensitize cells to alkylating agents and X-rays. Proc. Sot. Exp. Biol. Med., 141, 543-547.
9 Gaudin, D., Guthrie, L. and Yielding, K.L. (1974) DNA repair inhibition: a new mechanism of action of steroids with possible implications for tumor therapy. Proc. Sot. Exp. Biol. Med., 146, 401-405.
10 Kapp, L.N., Brown, S.L. and Klevecz, R.R. (1974) Detecting small quantities of DNA on DNA on CsCl gradients. Biochim. Biophys. Acta, 361, 140-143.
11 Langenbach, R. and Kuszynski, C. (1975) Nonspecific inhibition of DNA repair by promoting and nonpromoting phorbol esters. J. Natl. Cancer Inst., 55, 801-802.
12 Lieberman, M.W. and Poirier, M.C. (1973) DeoxyribonucIeoside incorporation during DNA repair of carcinogen-induced damage in human diploid fibroblasts. Cancer Res., 33, 2097-2103.
13 Lieberman, M.W. and Poirier, M.C. (1974) Distribution of deoxyribonucleic acid repair synthesis among repetitive and unique sequences in the human diploid genome. Biochemistry, 13, 3018-3023.
14 Lieberman, M.W. and Poirier, M.C. (1974) Base pairing and template specificity during deoxyribonucleic acid repair synthesis in human and mouse cells. Biochem-
istry, 13, 5384-5388. 15 Lindberg, R.E. and Pinnas, J.L. (1980) Inhibition of DNA repair by tobacco smoke
and saccharin. Fed. Proc., 39, 1022. 16 Miyata, Y., Hagiwara, A., Nakatsuka, T., Murasaki, G., Arai, M. and Ito, N. (1980)
Effects of caffeine and saccharin on DNA in the bladder epithelium of rats treated with N-butyl-N-(3-carboxypropyl)-nitrosamine. Chem.-Biol. Interact., 29, 291-302.
17 Ochi, H. and Tonomura, A. (1978) Presence of unscheduled DNA synthesis in cultured human cells after treatment with sodium saccharin. Mutat. Res., 54, 224.
18 Poirier, M.C., De Cicco, B.T. and Lieberman, M.W. (1975) Nonspecific inhibition of DNA repair synthesis by tumor promotors in human diploid fibroblasts damaged with Nacetoxy-2-acetylaminofluorene. Cancer Res., 35, 1392-1397.





























