Embed Size (px)
Citation preview

HAL Id: jpa-00233373https://hal.archives-ouvertes.fr/jpa-00233373
Submitted on 1 Jan 1935
HAL is a multi-disciplinary open accessarchive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come fromteaching and research institutions in France orabroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, estdestinée au dépôt et à la diffusion de documentsscientifiques de niveau recherche, publiés ou non,émanant des établissements d’enseignement et derecherche français ou étrangers, des laboratoirespublics ou privés.
La diffusion moléculaire de la lumière dans les liquides :effet Cabannes-Daure
A. Rousset
To cite this version:A. Rousset. La diffusion moléculaire de la lumière dans les liquides : effet Cabannes-Daure. J. Phys.Radium, 1935, 6 (12), pp.507-515. �10.1051/jphysrad:01935006012050700�. �jpa-00233373�

LA DIFFUSION MOLÉCULAIRE DE LA LUMIÈRE DANS LES LIQUIDES : EFFET CABANNES-DAUREPar A. ROUSSET.
Assistant à la Faculté des Sciences de Montpellier.
Sommaire. 2014 L’élargissement d’une raie spectrale par diffusion dans un liquide (effet Cabannes-Daure)est un phénomène bien différent du spectre discontinu de rotation qu’on observe par diffusion dans ungaz L’origine des radiations qui le composent n’a pu être précisé qu’après une étude expérimentale trèscomplète : répartition des intensités dans le fond continu (branches P et R) au voisinage de la raie fonda-mentale, comparaison des intensités des branches P, Q et R, largeur de la branche Q.
L’effet Cabannes-Daure doit être attribué à l’étalement d’une partie de la diffusion anisotrope par lesfluctuations d’orientation des molécules L’arrangement des molécules en graupements cybotaeiques, quilimite les orientations relatives des axes des molécules, change le poids statistique des niveaux de rota-tion, de telle sorte que les maxima d’intensité des branches P et R se trouvent ramenés à la premièreraie de rotation la plus proche de la raie fondamentale, c’est-à-dire pratiquement confondus avec
celle ci.En présence de forts momehts permanents, les fluctuations d’orientation d’un grand nombre de
molécules se réduisent à des oscillations de part et d’autre d’une position d’équilibre; la diffusion aniso-trope d’une molécule oscillante se retrouve en entier dans la branche Q; ainsi s’explique la faible inten-sité de l’effet Cabannes-Daure dans les molécules fortement polaires.
Le léger étalement de la branche Q apparaît seulement par diffusion dans un liquide non polaire; iiest donc lié à la rotation des molécules. Le champ de force intermoléculaire provoque la multiplicité desfréquences correspondant aux transitions j ~ j qui donnent la branche Q de rotation.
1. Introduction.
On sait qu’après diffusion dans un liquide une raiespectrale apparaît entourée d’un fond continu d’inten-sité et d’étendue variables suivant le liquide diffusant.Cest l’ensemble des radiations de ce fond continu qu’ondésigne sous le nom d’ « effet Cabannes-Daure ». L’ori-gine de ces radiations a été attribuée à différents phé -nomènes : rotation des molécules, chocs, fluctuationsdu champ moléculaire.Pour Raman et ses collaborateurs [2] [3] ce spectre
est formé par l’ensemble des raies de rotation non réso-lues ; ils expliquent ainsi la dissymétrie (le spectre estplus intense du côté des grandes longueurs d’onde), lefacteur de dépolarisation élevé et la forte intensité pourcertains liquides très anisotropes (benzène, sulfure decarbone).Cabannes et Rocard [4] ont rapproché ces radiations
de celles que donnerait la théorie de l’élargissementdes raies d’émission par les chocs : l’effet d’un choc surun centre émetteur se traduisant par une variation
brusque de l’amplitude et de la phase du train d’ondesémis, la représentation de ces variations par des sériesde Fourier montre que le phénomène équivaut bien àun élargissement des raies. Ils ont fait aussi intervenir,outre les variations non périodiques créées par leschocs directs des molécules, les fluctuations du champmoléculaire, provoquées par les mouvements des molé-cules voisines; en effet, l’amplitude et la direction dumoment électrique induit dans une molécule par lechamp provenant de la polarisation des molécules voi-
sines doivent varier suivant les positions et les orienta-tions de celles-ci.
Après avoir rappelé succintement les conclusions dela théorie des spectres Raman de rotation et celles dela théorie de la diffusion par le champ moléculaire,j’exposerai les résultats de mon étude expérimentale del’effet Cabannes-Daure. Au moyen d’hypothèses sur lesfluctuations d’orientation des molécules dans les
liquides, j’interpréterai ensuite les écarts entre mes
résultats expérimentaux et la théorie des spectresRaman de rotation. Je terminerai par un exposécritique des publications récentes sur l’origine desradiations de l’effet Cabannes-Daure.
Spectres Raman de rotation. - Si l’on supposeles molécules diffusantes d’un gaz animées de mouve-ments de rotation, une partie seulement de la diffu-sion anisotrope conserve même longueur d’onde que laradiation excitatrice (et que la diffusion cohérente dueaux fluctuations de densité) c’est la branche Q. Laplus grande partie apparaît sous forme de raies derotation, de part et d’autre de la raie fondamentale ; cesont les branches P et R (.r-).Des théories classique et quantique des spectres
Raman de rotation relatives aux molécules toupies
(* Les expressions « branches P et B » sont ici. comme danstout cet exposé, des notations rapides pour désigner toutes lesraies de chacune des ailes du sp ctre de rotation pure; elles nene présument rien sur les transitions du quantum de rotationqui leur ont donné naissance.
#
Article published online by EDP Sciences and available at http://dx.doi.org/10.1051/jphysrad:01935006012050700
508
symétriques (4] [5] [6] (axe de symétrie d’ordre supé-rieur ou égal à 3), je rappelle les conclusions suivantes :
1° Les branches P et I? sont dépolarisées : le facteur
de dépolarisation est égal à - si la lumière incidente1
est naturelle et si l’on étudie la lumière diffusée à 90°du faisceau incident.
2° La branche 7~ (du côté des grandes longueursd’ondes) doit être plus intense que la branche Il.
3° La courbe qui donne la distribution des intensitésentre les différentes fréquences des branches P ou Rdoit présenter un maximum d’autant plus rapprochéde la raie fondamentale que le moment d’inertie de lamolécule est plus élevé.
4° L’intensité globale des branches P et R vaut sen-siblement 3 fois l’intensité de la branche Q (exac-tement 3 fois si la molécule est diatomique ou triato-mique linéaire).
Fig.4.
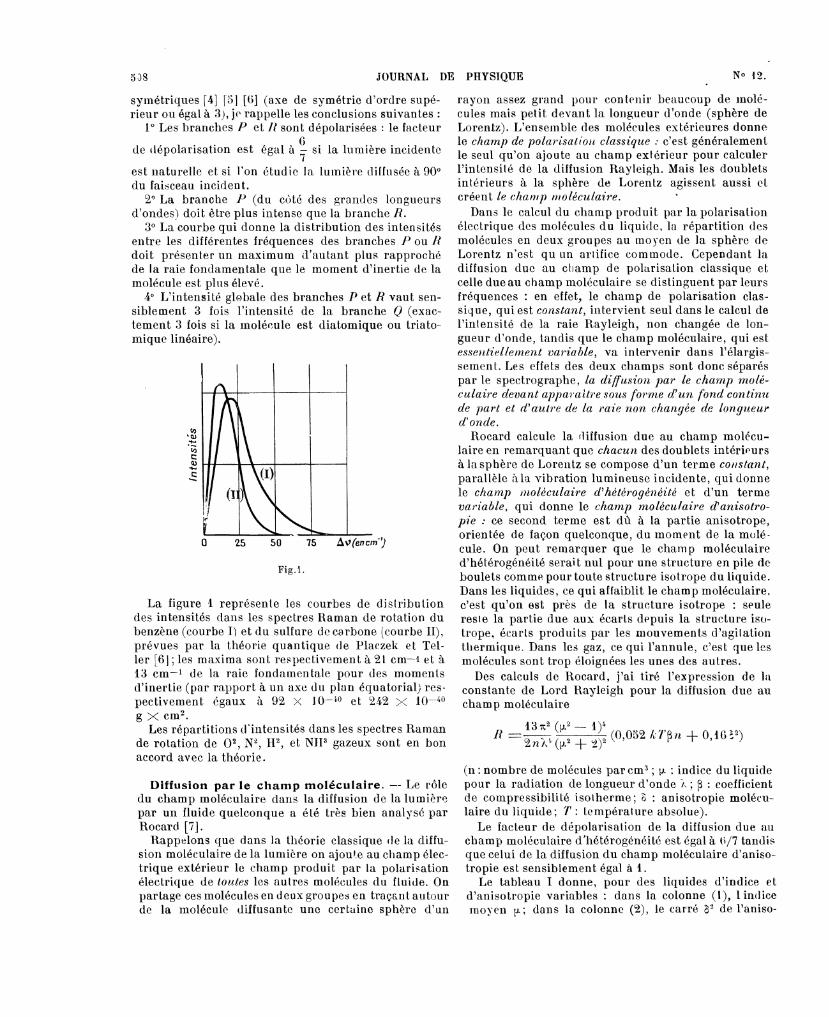
La figure 1 représente les courbes de distributiondes intensités dans les spectres Raman de rotation dubenzène (courbe I) et du sulfure de carbone (courbe II),prévues par la théorie quantique de Placzek et Tel-ler f6] ; les maxima sont respectivement à 21 em-1 et à13 cm-1 de la raie fondamentale pour des momentsd’inertie (par rapport à un axe du plan équatorial) res-pectivement égaux à 92 X 10-fO et 242 X 10-40
g X CM2.Les répartitions d’intensités dans les spectres Raman
de rotation de O2, N2, H2, et NH3 gazeux sont en bonaccord avec la théorie.
Diffusion par le champ moléculaire. -- Le rôledu champ moléculaire dans la diffusion de la lumièrepar un fluide quelconque a été très bien analysé parRocard [7].Rappelons que dans la théorie classique de la diffu-
sion moléculaire de la lumière on ajoute au champ élec-trique extérieur le champ produit par la polarisationélectrique de toutes les autres molécules du fluide. Onpartage ces molécules en deux groupes en traçait autourde la molécule diffusante une certaine sphère d’un
rayon assez grand pour contenir beaucoup de molé-cules mais petit devant la longueur d’onde (sphère deLorentz). L’ensemble des molécules extérieures donnele champ de palar°isatiorc classique : c’est généralementle seul qu’on ajoute au champ extérieur pour calculerl’intensité de la diffusion Rayleigh. Mais les doubletsintérieurs à la sphère de Lorentz agissent aussi etcréent le charup fiioléculaire. °
Dans le calcul du champ produit par la polarisationélectrique des molécules du liquide, la répartition desmolécules en deux groupes au moyen de la sphère deLorentz n’est qu un arlifice commode. Cependant ladiffusion clue au champ de polarisation classique etcelle due au champ moléculaire se distinguent par leursfréquences : en effet, le champ de polarisation clas-siq ue, qui est cor2stant, intervient seul dans le calcul del’intensité de la raie Rayleigh, non changée de lon-gueur d’onde, tandis que le champ moléculaire, qui estesseiitiellement variable, va intervenir dans l’élargis-selnent. Les effets des deux champs sont donc séparéspar le spectrographe, la dilfusioii par le champ rrcolé-culaire devant apparail1’e sous forme fond continuede part et d’autre de la raie non changée de longueurd’onde.Rocard calcule la diffusion due au champ molécu-
laire en remarquant que chacun des doublets intérieursà la sphère de Lorentz se compose d’un terme constant,parallèle àla vibration lumineuse incidente, qui donnele champ nzaléculaire d’hétérogénéité et d’un terme
variable, qui donne le champ moléculaire d’anisotro-pie : .’ ce second terme est dû à la partie anisotrope,orientée de façon quelconque, du moment de la molé-cule. On peut remarquer que le champ moléculaired’hétérogénéité serait nul pour une structure en pile deboulets comme pour toute structure isotrope du liquide.Dans les liquides, ce qui affaiblit le champ moléculaire,c’est qu’on est près de la structure isotrope : seulereste la partie due aux écarts depuis la structure iso-trope, écarts produits par les mouvements d’agitationthermique. Dans les gaz, ce qui l’annule, c’est que lesmolécules sont trop éloignées les unes des autres.
Des calculs de Rocard, j’ai tiré l’expression de laconstante de Lord Rayleigh pour la diffusion due auchamp moléculaire
(n: nombre de molécules par cm3 ; v. : indice du liquidepour la radiation de longueur d’onde î. ; ~3 : coefficientde compressibilité isotherme ; ô : anisotropie molécu-laire du liquide; 1’: température absolue).
Le facteur de dépolarisation de la diffusion due auchamp moléculaire d’hétérogénéité est égal à (i/7 tandisque celui de la diffusion du champ moléculaire d’aniso-tropie est sensiblement égal à 1.Le tableau 1 donne, pour des liquides d’indice et
d’anisotropie variables : dans la colonne (1), 1 indice
mo;en p.; dans la colonne (2), le carré a’ de l’aniso-
509
tropie ; dans les colonnes (3) et (4), les diffusions H etA, en unités arbitraires, dues au champ moléculaired’hétérogénéité et d’anisotropie ; dans la colonne (5),la diffusion due au champ moléculaire total; dans lacolonne (6), le rapport ? de la diffusion par le champmoléculaire total à la diffusion Rayleigh.
TABLEAU 1
Des valeurs de l’ et de 1>, on déduit que l’intensitéde la le molécitlaire, très variabled’un liquide à l’autre, ne que quelques cen-t1"èrnes de lcc diffusion Rayleigh. De plus, elle peutb’étendre sur plusieurs centaines de cm-i [1] ; en con-séquence, on ne peut espérer la mettre en évidenceavec certitude si unP partie de la diffusion anisotropese trouve étalée par la rotation des molécules.
Etude expérimentale de l’efietCabannes-Daure.
1. - La difficulté de l’étude expérimentale de l’effetCabannes-Daure provient du fait que le fond continuqui le constitue entoure une raie beaucoup plus intense
,
et que c’est justement au voisinage immédiat de cetteraie que, d’une part, se trouve la plus grande partiesinon la totalité du fond continu et que, d’autre part,l’étude de la courbe de répartition des intensités est laplus intéressante.Une première condition à réaliser est l’emploi d’un
specl?.ogiapfie à grande J’ai obtenu une
dispersion d’environ 6 1 au mm dans la région 40001en plaçant un téléobjectif (distance focale : 120mm)derrière un train de 2 prismes en flint extra-dense.
Il faut ensuite, autant que possible, réduire l’in-tensité de la raie non changée de longueur d’ondedevant celle du fond continu ( ). Rappelons que la raienon changée de longueur d’onde comprend une partie
(*) Une méthode élégante a été imaginée par Rasetti dans sonétude des spectres de rotation des gaz (Z. 1930, 61,p. 598) : la raie excitatrice est la raie de résonance du mercure,2 536 À; elle est absorbée le long du faisceau diffusé parde la vapeur de mercure. L’absorption de nos liquides nepermet pas d’utiliser cette raie de résonance comme raie exci-tatrice.
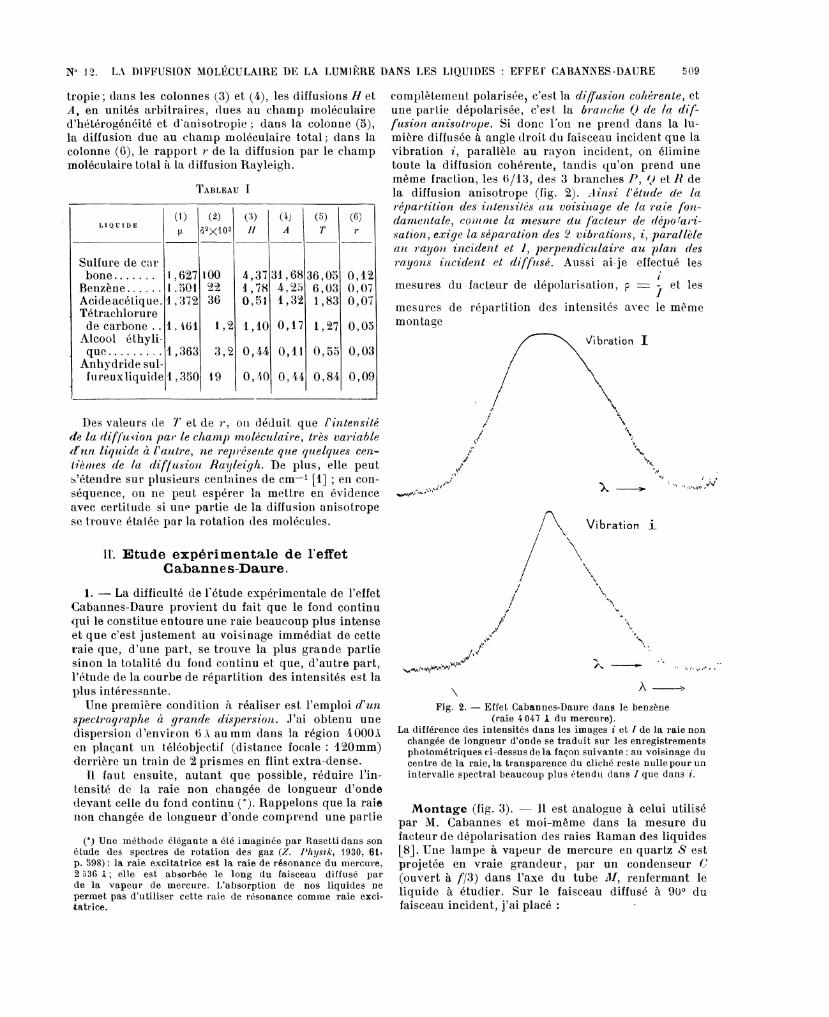
complètement polarisée, c’est la diffusion cohérente, etune partie dépolarisée, c’est la branche Q de la dif-fusion anisotrvpe. Si donc l’on ne prend dans la lu-mière diffusée à angle doit du faisceau incident que lavibration i, parallèle au rayon incident, on éliminetoute la diffusion cohérente, tandis qu’on prend unemême fraction, les 6/>lj, des 3 branches P, 1) et li dela diffusion anisotrope (fig. 2). Ainsi l’élude de la
répartition des intensités au voisinage de la raie fon-damentale, conlfue la niesure du de
sation, exige la séparation des 2 vibrations, i, parallèleait rayon incident et l, »erpeizdiculaiie au plan des
rayons incident et Aussi ai- je effectué lesi
mesures du facteur de dépolarisation, p = Î et les
mesures de répartition des intensités avec le même
montage
Fig. 2. - Effet Cabannes-Daure dans le benzène(raie4047 A du mercure).
La différence des intensités dans les images et 1 de la raie nonchangée de longueur d’onde se traduit sur les enregistrementsphotométriques ci-dessus de la façon suivante : au voisinage ducentre de la raie, la transparence du cliché reste nulle pour unintervalle spectral beaucoup plus étendu dans I que dans i.
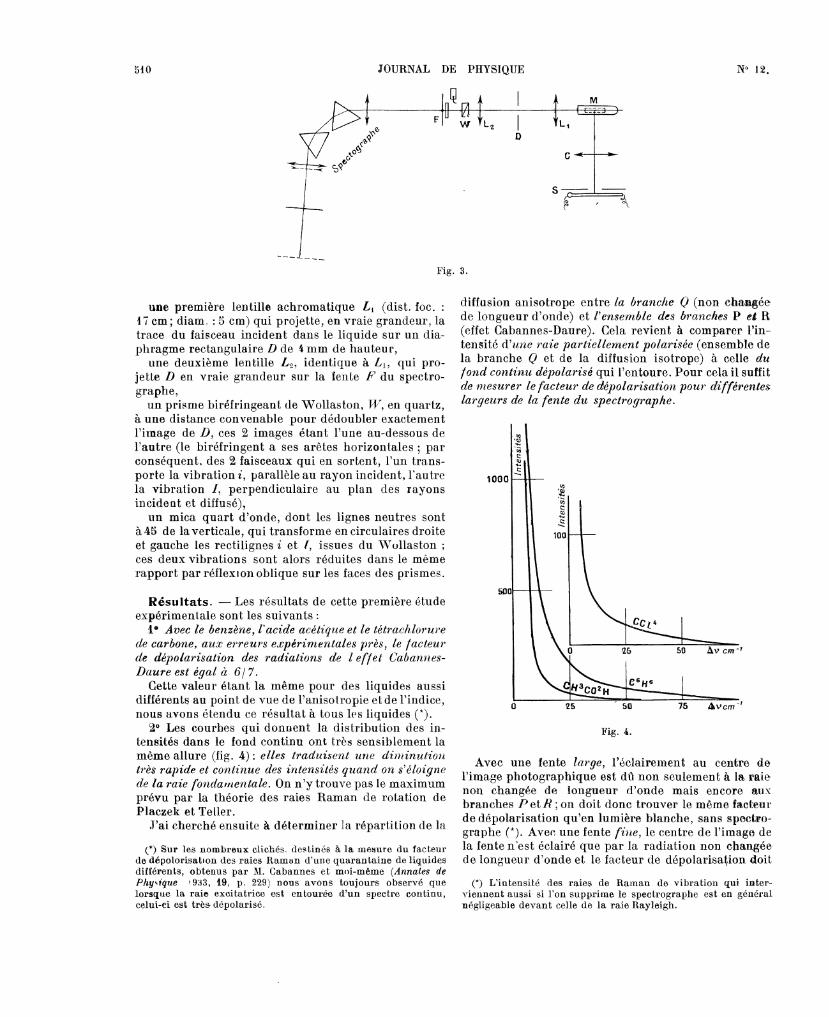
Montage (fig. 3). - Il est analogue à celui utilisépar M. Cabannes et moi-même dans la mesure dufacteur de dépolarisation des raies Raman des liquides[8]. Une lampe à vapeur de mercure en quartz 8 estprojetée en vraie grandeur, par un condenseur C
(ouvert à dans l’axe du tube JI, renfermant le
liquide à étudier. Sur le faisceau diffusé à 9U° dufaisceau incident, j’ai placé : .
510
Fig. 3.
une première lentille achromatique Li (dist. foc. :9 i cm ; diam. : 5 cm) qui projette, en vraie grandeur, latrace du faisceau incident dans le liquide sur un dia-phragme rectangulaire D de 4mm de hauteur,une deuxième lentille L2, identique à L1, qui pro-
jette D en vraie grandeur sur la lente F du spectro-graphe,un prisme biréfringeant de Wollaston, UT, en quartz,
à une dista,nce convenable pour dédoubler exactement
l’image de D, ces 2 images étant l’une au-dessous del’autre (le biréfringent a ses arêtes horizontales ; parconséquent, des 2 faisceaux qui en sortent, l’un trans-porte la vibration i, parallèle au rayon incident, l’autrela vibration I, perpendiculaire au plan des rayonsincident et diffusé),un mica quart d’onde, dont les lignes neutres sont
à 45 de la verticale, qui transforme en circulaires droiteet gauche les rectilignes i et l, issues du Wollaston ;ces deux vibrations sont alors réduites dans le même
rapport par réflexion oblique sur les faces des prismes.
Résultats. - Les résultats de cette première étudeexpérimentale sont les suivants :i Avec le benzène, l’acide acétique et le tétrachloru>.e
de carbone, aux erreurs expérirnentales près, le f acteurde dépolarisation des radiations de l e f f et Cabanlles-Daure est égal à 6j 7.
Cette valeur étant la même pour des liquides aussidifférents au point de vue de l’anisotropie et de l’indice,nous avons étendu ce résultat à tous les liquides (*).
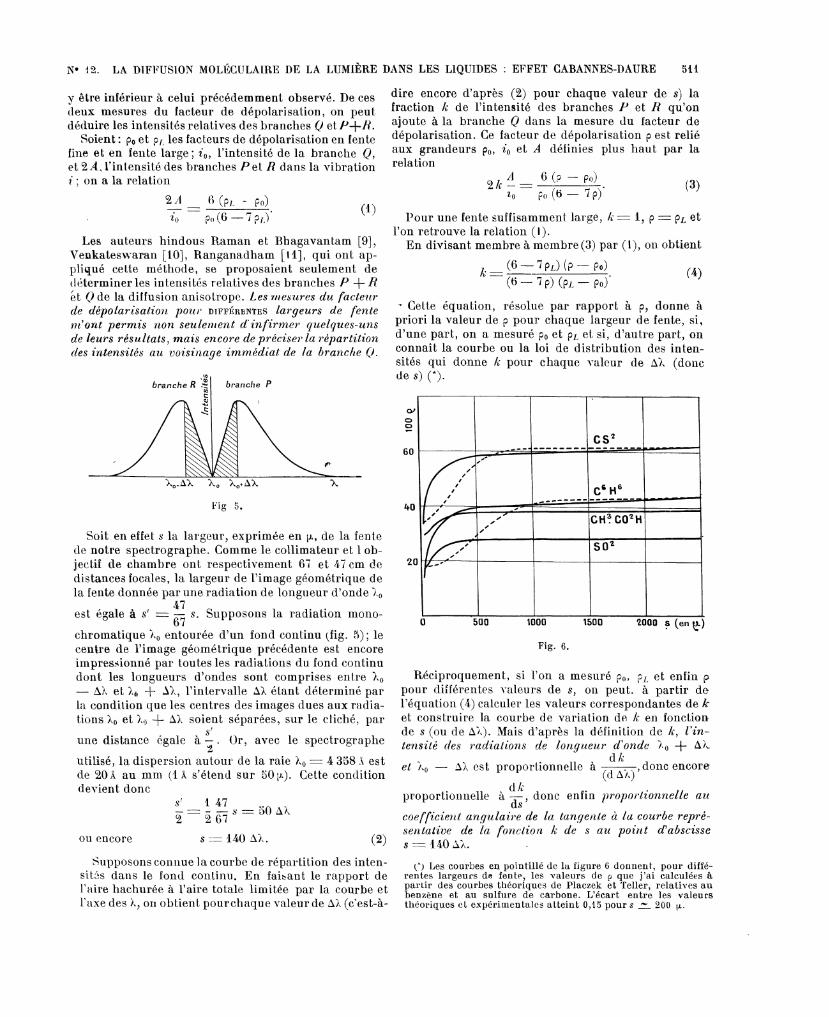
2° Les courbes qui donnent la distribution des in-tensités dans le fond continu ont très sensiblement lamême allure (fig. 4) : elles traduisent une dinzinutiontrès rapide et continue des intensités quand on s’éloignede la r°aie fondaiflentale. On n’y trouve pas le maximumprévu par la théorie des raies Raman de rotation dePlaczek et Teller.
J’ai cherché ensuite à déterminer la répartition de la
(*) Sur les nombreux clichés. deqtinés à la mesure du facteurde dépolorisation des raies Raman d’une quarantaine de liquidesdifférents, obtenus par 31. Cabannes et moi-même (Annales dePhy,ique 1933, 19, p. 229) nous avons toujours observé quelorsque la raie excitatrice est entourée d’un spectre continu,celui-ci est très. dépolarisé.
diffusion anisotrope entre la Q (non changéede longueur d’onde) et l’ensemble des et R
(effet Cabannes-Daure). Cela revient à comparer l’in-tensité d’une raie partiellement polarisée (ensemble dela branche Q et de la diffusion isotrope) à celle dutond contin2c dépolarisé qui l’entoure. Pour cela il suffitde nzesurer le facteur de dé}Jolarisation pour différenteslargeurs de la fente du
Fig. 4.
Avec une fente large, l’éclairement au centre de
l’image photographique est dû non seulement à la raienon changée de longueur d’onde mais encore auxbranches P etR ; on doit donc trouver le même facteurde dépolarisation qu’en lumière blanche, sans spectro-graphe (*). Avec une fente fine, le centre de l’image dela fente n’est éclairé que par la radiation non changée-de longueur d’onde et le facteur de dépolarisation doit
(*) L’intensité des raies de Raman de vibration qui inter-viennent aussi si l’on supprime le spectrographe est en généralnégligeable devant celle de la raie Rayleigh.
511
y être inférieur à celui précédemment observé. De cesdeux mesures du facteur de dépolarisation, on peutdéduire les intensités relatives des branches Q et P+R.
Soient : po et p[, les facteurs de dépolarisation en fentefine et en fente large; io, l’intensité de la branche Q,
l’intensité des branches P et R dans la vibration
i ; on a la relation
Les auteurs hindous Raman et Bhagavantam [9J,Veukateswaran [ 10], Ranganadham [Ii], qui ont ap-pliqué cette méthode, se proposaient seulement dedéterminer les intensités relatives des branches P -~- Rét Q de la diffusion anisotrope. Les rnesures du facteurde dépolarisation DIFFÉRENTES largeurs de fente
permis non seulement d*infirine2- quelques-unsde leurs résultats, mais encore de préciser la répartitionsdes intensités au voisinage immédiat de la branche ~l.
5.
Soit en effet s la largeur, exprimée en ~, de la fentede notre spectrographe. Comme le collimateur et 1 ob-jectif de chambre ont respectivement 67 et 47cm dedistances focales, la largeur de l’image géométrique dela fente donnée par une radiation de longueur d’onde 10
47est égale à s’ _ 47 s. Supposons la radiation mono-67
chromatique Ao entourée d’un fond continu (fig. 5); lecentre de l’image géométrique précédente est encoreimpressionné par toutes les radiations du fond continudont les longueurs d’ondes sont comprises entre Xo- A~, et )~ + Sî., l’intervalle AX étant déterminé parla condition que les centres des images dues aux radia-tions ),o et ),,, -E- soient séparées, sur le cliché, par
rune distance égale à , . Or, avec le spectrographe
y
utilisé, la dispersion autour de la raie ),o == 4 358 A estde 20 À au mm (1 Á s’étend sur 50 ¡;.). Cette conditiondevient donc
ou encore
Supposons connue la courbe de répartition des inten-sités dans le fond continu. En faisant le rapport del’aire hachurée à l’aire totale limitée par la courbe etF axe des 1,, on obtient pour chaque valeur de AX (c’est-à-
dire encore d’après (2) pour chaque valeur de s) lafraction k de l’intensité des branches ~’ et R qu’onajoute à la branche Q dans la mesure du facteur dedépolarisation. Ce facteur de dépolarisation p est reliéaux grandeurs po, io et a définies plus haut par larelation
Pour une fente suffisamment large, k ~ 1, P = PL etl’on retrouve la relation ( I ).En divisant membre à membre (3) par (1), on obtient
- Cette équation, résolue par rapport à p, donne à
priori la valeur de p pour chaque largeur de fente, si,d’une part, on a mesuré po et ~~, et si, d’autre part, onconnaît la courbe ou la loi de distribution des inten-sités qui donne k pour chaque valeur de (doncde s) (*).
Fig, 6.
Réciproquement, si l’on a mesuré po, ~L et enfin ppour différentes valeurs de s, on peut. à partir del’équation (4) calculer les valeurs correspondantes de ket construire la courbe de variation de k en fonctionde s (ou de à5,). Mais d’après la définition de k, l’in-tensité des radiations de longueur- d’onde )’0 -~- Ill....
et î.o - A), est proportionnelle à -(d’ , ,donc encorep 6./-.)
proportionnelle donc enfin proportionnelle auds l
coefficient de la tangente à la courbe repré-sentative de la (onction k de s au point d’abscisses = 140 à~’. _
Les courbes en pointillé de la figure 6 donnent, pour diffé-rentes largeurs de fente, les valeurs de p que j’ai calculées àpartir des courbes théoriques de Placzek et Teller, relatives aubenzène et au sulfure de carbone. L’écart entre les valeursthéoriques et expérimentales atteint 0,15 pour s - 200 y.
512
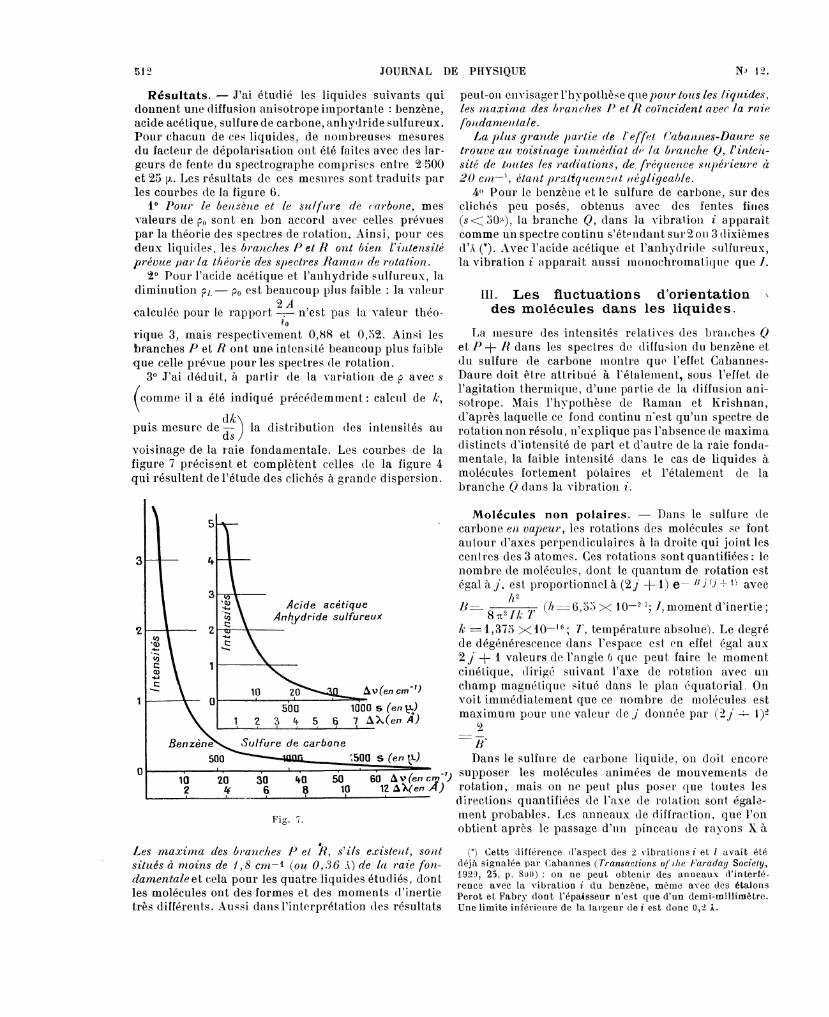
Résultats. - J’ai étudié les liquides suivants quidonnent une diffusion anisotrope importante : benzène,acide acétique, sulfure de carbone, anhydride sulfureux.Pour chacun de ces liquides, de nombreuses mesuresdu facteur de dépolarisation ont été faites avec des lar-geurs de fente du spectrographe comprises entre 2 500et 25 fi.. Les résultats de ces mesures sont traduits parles courbes de la figure 6.
10 le benzène et le sulfure de mes
valeurs de po sont en bon accord avec celles prévuespar la théorie des spectres de rotation. Ainsi, pour cesdeux liquides, les branches P et R ont bien l’intensité
prévue par la théorie des spectres Ralnan de rotation.2° Pour l’acide acétique et l’anhydride sulfureux, la
diminution PL - p0 est beaucoup plus faible : la valeur9- À
calculée pour le rapport - n’est pas la valeur théo-10
rique 3, mais respectivement 0,88 et 0,52. Ainsi lesbranches P et R ont une intensité beaucoup plus faibleque celle prévue pour les spectres de rotation.
3° J’ai déduit, à partir de la variation de p avec s
comme il a été indiqué précédemment: calcul de Iz,
puis mesure de dk) la distribution des intensités au1 ds /
voisinage de la raie fondamentale. Les courbes de la
figure 7 précisent et complètent celles de la figure 4qui résultent de l’étude des clichés à grande dispersion.
Fig. 7.
r
Les maxima des branches P et R, s’ils e,r’isteJlt, sontsitués à moins de 1, 8 cni-1 (ou 0, "î 6 A) de lcc î-aie fon-clamentaleet cela pour les quatre liquides étudiés, dontles molécules ont des formes et des moments d’inertietrès différents. Aussi dansl’interprétation des résultats
peut-on envisager l’hypothèse les les rnaxinla des ¡n’anches Il et R coïncident avec la raie
La plus ,grande partie de l’effet C’abannes-Daure setrouve au voisinage intn1édiat de la branche Q, l’inteil-sité de toutes les radiations, de, (réquence sllpél’ieul’e ii20
41 Pour le benzène et le sulfure de carbone, sur desclichés peu posés, obtenus avec des fentes fines
(s C JO:1.), la branche Q, clans la vibration i apparaîtcomme un spectre con tinu s’étendant surdon 3 dixièmesd’A ("). Avec l’acide acétique et l’anhydride sulfureux,la vibration i apparaît aussi monochromatique que J.
III. Les fluctuations d’orientationdes molécules dans les liquides.
La mesure des intensités relatives des branches Qet P -E- R dans les spectres de tliiiusion du benzène etdu sulfure de carbone montre que l’effet Cabannes-Daure doit être attribué à l’étalement, sous l’effet de
l’agitation thermiques, d’une partie de la diffusion ani-sotrope. Mais l’hypothèse de Raman et Iirishnan,d’après laquelle ce fond continu n’est qu’un spectre derotation non résolu, n’explique pas l’absence de maximadistinets d’intensité de part et d’autre de la raie fonda-mentale, la faible intensité dans le cas de liquides àmolécules fortement t polaires et l’étalement de labranche Q dans la vibration i.
Molécules non polaires. - Dans le sulfure cle
carbone en les rotations des molécules se fontautour d’axes perpendiculaires à la droite qui joint lescentres des 3 atomes. Ces rotations sont quantifiées: lenombre de molécules, dont le quantum de rotation estégal à j, est proportionnel à (2j + 1) avec
IL? , -27.. ,.
1 -. ?t2jiv j1 (h 6,oj X i 0 1, moment d’inertie; ro 7t i /k =1,375) X 10-16 7’, température absolue). Le degréde dégénérescence dans l’espace est en effet égal aux2y + 1 valeurs de l’angle 6 que peut faire le moment
cinétique, dirigé suivant l’axe de rotetion avec un
champ magnétique situé dans le plan équatorial. Onvoit immédiatement que ce nombre de molécules est
maximum pour une valeur de j donnée par (2/ l)-2’
Dans le sulfure de carbone liquide, on doit encore
j supposer les molécules animées de mouvements de
rotation, mais on ne peut plus poser que toutes lesdirections quantifiées de l’axe de rotation sont égala-ment probables. Les anneaux de diffraction, que l’onobtient après le passage d’un pinceau de rayons X à
(") Cette différence d’aspect des 2 vibrations i et I avait étédéjà signalée par Cabannes (Transactions ol’ilie Faraday Society,1929, 25, p. 8uU) : on ne peut obtenir des anneaux d’interfé-rence avec la vibration i du benzène, même avec des étalonsPerot et Fabry dont l’épaisseur n’est que d’un demi-millimètre.Une limite inférieure de la largeur due i est donc 0,’_’ À.

513
travers quelques dixièmes de mm d’un liquide, ontamené Stewart et ses collaborateurs [12] à supposerpour les liquides un état dit « cybotactique » : : il existe-rait un grand nombre de groupements temporaires àl’intérieur desquels 1 orientation des molécules n’est
plus arbitraire, En particulier, du fait qu’on n’obtienten général qu’un anneau dont le diamètre donne pouréquidistance des plans réticulaires le plus pelit dia-mètre des molécules, j’ai conclu que les molécules dia-tomiques ou triatomiques linéaires (comme CS2), assi-milables quant à la forme à des ellipsoïdes allongés,ont tendance à placer leur grand axe clans des plansparallèles : ainsi les seules rotations qui ne détruisentpas les arrangements cybotact iques sont celles autourd’un axe perpendiculaire au plan d’orientation.
Si les axes des ellipsoïdes restaient parallèles à unmême plan P (ou encore si les axes de rotation de molé-cules restaient parallèles entre eux) le nombre demolécules de quantum de rotation j serait propor-tionnel à e- l~ i (j + 1) et ainsi le maxirnum d’intensitése trouverait reporté à la première raie du spectre/Raman de rotation, c’est-à-dire, avec les valeurs éle-vées des moments d’inertie des molécules des liquidesusuels, au voisinage immédiat de la raie fondamen-tale.En réalité, des rotations autour d’axes non perpen-
diculaires au plan P, rotations qui maintiennent l’axede la molécule dans un plan P’ incliné sur le plan P
angle 0, sont encore possibles, mais on doit tes
supposer d’autant moins probables que l’angle 0 est
plus grand : il revient au même d’attribuer à la molecule qui tourne dans le plan P’ une énergie potentielle
qui doit augmenter très rapidement avec 0. Si
1 étierqie ( _ ) d’zcjie molécule qui 1 élte’gie potenÙ>:lle 9) d’une molécule qui tow’nedans un au plan P (c.est-à-direen som)ne qui détruit les arrangements cybotactiques)est beaucoup plus élevée que l’énergie cor’respondaut àUlt degré de liberté (*), le maximll nl d intensité se trou-vera encore rel)oî-té au voisina,qe iuunédiat de la
fondamentale [1, p. 52].L’action des forces intermoléculaires ne se limite pas
à une modification dans la distribution des fréquencesde rotation entre les différentes molécules. Par ana-
logie avec l’effet Stark observé sur les raies émises parles atomes, on peut prévoir que le terme énergiquecorrespondant à une valeur j du quantum de rotationprend des valeurs légèrement différentes suivantsl’orientation relative du moment cinétique et de la nor-male au plan d’orientation. La multiplicité des termesénergétiques entraîne la multiplicité des fréquencescorrespondantes à une même transition j - j . Ainsinon seulement chacune des raies des branches f’ et R
(~j -~ j ± ~) sera remplacée par un spectre continu,
(*) Le fait que les arrangements cyhotactiques ne sont pasdétruits par une élvéation de température, si le volume spéci-fique est maintenu constant I’liyc. Rev., 1934. 46,p. 698 ; BENZ et STEWART. Phys. liev., 1934, 46, p. ’j03), iiiilite enfaveur de cette hypothèse.
mais encore on observera un étalement des fréquencesrelatives à la branche Q (? 2013~ j) C’est seulcnlent dansla vibration i pourra d(Jceler’ sur les clicfiés en
fente fine la lai-geuî- de la bt’unche Q. Pour le benzèneet le suifure de carbone cette largeur est de l’ordre de
ou 1,:S cm-’. Les maxima. supposées placés à la
première raie de rotation, c’est-à dire à cm-1 de laraie fondamentale pour un moment d’inertie de 130 gX cm’ (benzène) disp,-iraissen par suite de l’étaiementde cette raie de rotation et de la branche Q et sontpratiquement reportés sur la raie fondamentale.En résumé, daos les liquides non à 11/olécules
aaisotropes (benzène, sulfure de carbone) les rotationsdes molécules sont encore possibles dans les groupe-ments temporaires caractéristiques de l’état cybotac-ticlue, et par conséquent l’ilileiisilé cle Cabannes-J)aure sera celle prévue par’ la théorie des derotation de Placzeli et ’te 11er , mais la présence du
champ de force intermoléculaire se traduit :1° par un changement dans la répartition des inten--
sités entre les différentes fréquences qui reporte lesmaxima des hranches 1-’ et Il sur la raie fondamentale : -,
2" par un étalement de la branche () (qui s’observedans la yibration i) et de chacune des raies des bran-ches P et Il qui rend impossible la résolution en raiesdistinctes de l’effet Cabannes-Daure.
Molécules polaires. - Par l’analyse aux rayons X.les liquides à molécules polaires ne se distinguent pas,en .principe, des liquides à molécules non polaires.Dans les groupelnents imaginés par Stewart et carac-téristiques de l’état cybotactiquc, les axes cles molé-cules polaires leiident encore à se placer dans des
plans parallèles.Mais les changements d orientation des axes dans
ces plans modifient l’orientation et la position des nio-ments permanents liés aux molécules dans le champcréé par les dipôles des molécules voisines. Les dipôle,et par suite les axes des molécules, tendent à s’orienterdans des positions telles que l’énergie potentielle du
. moment, dans le champ des dipôles voisins, soit mi-nimum. Une rotation de la molécule provoque donc
l’apparition d’un couple de rappel sous l’action duquella molécule va osciller de part et d’autre d’une positiond’équilibre. A cause du déplacement des moléculesvoisines, ce couple change cle valeur, peut s’annuler etainsi une molécule peut effectuer successivement desoscillations et des rotations. Suivant l’importance desdipôles, leur place dans la molécule, la forme aniso-trope de celle-ci, le nombre des rnolécules qui, à unmoment donné, tournent ou oscillent variera d’un
liquide à l’autre. Comme les rotations entre deux chocs,les amplitudes des oscillations seront limitées à unefraction de degré ~~1, p. ~8~, mais les deux hypothèses,rotation ou oscillation, conduisent à des répartitionstrès différentes des intensités entre la raie non changée
°
de longueur d’onde et les radiations que ces fluctua-tions d’orientation font apparaitre dans le spectre de lalumière diffusée.

514
J’ai calculé [1, p. 57] l’intensité de la raie fonda-mentale, de fréquence lV et des raies de fréquencesN ± n qui doivent apparaître dans le spectre de lalumière diffusée par une molécule toupie symétriquequi effectue des oscillations de faihle amplitude de fré-quence n, autour d’un axe du plan équatorial : -.
1° la raie non changée de longueur d’onde a mêmeintensité que dans la diffusion par une molécule im-
2° les raies de fréquences N ± n sont dépolarisées,leur intensité est égale à celle des branches P et R derotation, lJlultipliée par te carré de tarnplitude desoscillations. En adoptant, pour l’amplitude des anglesinférieurs a 1°, cette diffusion supplémentaire ne repré-sente que quelques millièmes de l’intensité du spectreRaman de rotation.
point de vue de la diffusion nlolécule toupie symétrique oscillante peut être con-
fundue avec une niolécule imnlobile.Dans un liquide à molécules hétéropolaires, une
partie seulement des molécules étant animées de mou-vements de rotation, l’importance cles branches P et ~ldoit être diminuée par rapport à celle de la branche rl,le faett-ur de dépolarisation et l’intensité de l’ensembledes 3 branches 1:1, Q et R et de la diffusion cohérenten’en sont pas changés.
Enfin le léger étalement de la branche Q, qui estlié à la multiplicité des niveaux énergétiques corres-pondant à un même quantum de rotation, disparaît spi
la molécule ne tourne pas ; la diffusion anisotroped’une molécule oscillante est monochromatique Enfente très fine, le faible spectre de diffusion des molé-cules qui tournent (branches P et Il et branche Qlégèrement étalée) n’apparaît pas et la vibration i, avec
liquide polaire, seniblei-a aussi nzonochromat£queque la vibration 1, avec un liquide polaire ou non.En résumé, avec des liquides fortement polaires :Il l’effet Cabannes-Daure est faible ;2o les maxima des branches IJ et R sont confondus
avec la raie fondami-ntale ;3° la vibration i paraît monochromatique.Ces conclusions sont en bon accord avec mes nesures
sur l’effet Cabannes-Daure de l’acide acétique et del’anhydride sulfureux liquide dont t les moments per-manents (1,40 et 1,’ i D) sont relativement élevés. Sil’on compare enfin les spectres de diffusion du benzèneet du nitrobenzène, on est frappé par la disparitionpresque totale de l’effet l’abannes Daure dans ce dernier
liquide qui est cependant beaucoup plus anisotropeque le benzène, mais dont les molécules sont fortementpolaires (3,90 D).
IV. Nouvelles recherches sur l’effetCabannes-Daure.
’
De nouvelles recherches sur l’effet Cabannes-Daureont fait l’objet de publications récentes de la part d’au-teurs hindous (Bhagavantam et ses collaboi aieurs) etrusses (Gross et Vuks).
Il Travaux de Bhagavantam. - Les courbes derépartition des intensités dans l’effet Cabannes-Daure,de nombreux liquides ont été déterminées par Bhaga-vantam et ses collaborateurs [13] ; elles diffèrent de lacourbe de répartition prévue par la théorie des spectresde rotation :
1° par l’absence de maxima distincts ;21 par une plus grande intensité au voisinage de la
raie fondamentale ;3° par un plus grand étalement de l’extrémité des ailes.D’autre part, Bhagavantam et A. V. Rao ont montré : --1° qu’une élévation de température (jusqu’au voi-
sinage du point d’ébullition) ne change pas sensible-ment la courbe de répartition et l’intensité totale del’effet Cabannes-Daure [14J ;
2° que les maxima d’intensité caractéristiques desspectres de rotation des gaz, comme C02 et N20,.viennent se confondre avec la raie fondamentale sil’on augmente suffisamment la pression [15].Bhagavantam a donné, de ces résultats, l’interpréta-
tion suivante :La structure des liquides est quasi cristalline et dif-
fère peu de l’état solide. Par extension de la théorie de
Pauling [16] relative aux rotations des molécules dansles cristaux, on peut partager les liquides, comme lessolides, en deux classes :
~° les liquides comme H2, 02, dans lesquels lesmolécules tournent malgré les arrangements quasi-cristallins ;
2’ 1 les liquides à molécules plus lourdes (benzène,sulfure de carbone) dans lesquels la plupart des molé-cules exécutent des oscillations de faible amplitudeautour de positions d’équilibre.
Ainsi la partie la plus importante de l’effet Cabannes-Daure, la plus proche de la raie fondamentale seraitdue à la diffusion des molécules « oscillantes », les« ailes », plus éloignées, seraient constituées par lefaible spectre de rotation des quelques molécules aussilibres dans leur rotation qu’à l’état gazeux.
2. Travaux oie Gross et Vuks. - Gross et Vuks
ayant remarqué qu’une variation de températureest sans effet sur les « ailes » de la raie Rayleigh ontcherché l’origine de ces radiations dans un effet Ramande vibrations faiblement liées [l7]. ~
La plupart des cristaux (phényl-éther, benzène.naphtalène) donnent, dans les spectres de diffusion,moléculaire, des raies Raman de vibration de faible
fréquence, caractérisliques des vibrations du réseaucristallin (et non des oscillations des atomes dans lamolécule). Ces raies s’élargissent jusqu’à la raie fon-damentale en un spectre continu quand le cristal estfondu : oii doit doric çïipposer que les élémertis duréseau subsistent, quoique déformés, à l’état liquide.
D’autre part le fond continu qui s’étend jusqu’à en-viron 20 cm-1 de la raie fondamentale augmente d’in-tensité quand la température s‘élève ; il est constituésans nul doute par un spectre Raman due rotation. Sonintensité paraît liée à l’anisotropie de la molécule.

515
Ainsi pour Gross et Vuks, comme pour Bhagavantam.l’interprétation de l’effet Cabannes-Daure doit êtrerecherchée dans une structure quasi cristalline des
liquides. Il faut cependant distinguer les vibrations
caractéristiques du réseau cristallin et les oscillationsdes molécules autour d’un axe passant par le centrede gravité, ce mode de fluctuations d’orientation pré-sentant, pour les auteurs hindous, l’avantage de relierl’intensité de l’effet Cabannes-Daure à l’anisotropie dela molécule. Mais j’ai montré que l’intensité diffuséepar ces molécules oscillantes, sous forme de fond con-tinu, varie comme le carré de l’amplitude des oscilla-tions : elle est itPgligeable dans le cas des oscillationsde faible llrnplitude intaginées par
L’hypothèse de Gross et Vuks n’est, au contraire,nullement en contracliction avec mes résultats expéri-mentaux. D’après ces auteurs, c’est au voisinage im-inédit de la branche Q qu’il faut rechercher les bran-
ches P et R du spectre de rotation. Si l’on remarqueque le spectre de rotation des molécules non polairesconstitue les 3/4 de la diffusion anisotrope tandis quel’intensité des raies Raman de vibration caractéris-
tiques du réseau cristallin ne vaut que quelques cen-tièmes de l’intensité de la diffusion Rayleigh, on
s’explique la forme de la courbe de répartition de lafigure 7 que j’ai déduite des mesures du facteur dedépolarisation avec des fentes de largeur variable :l’effet Cabannes-Daure est pratiquement limité à20 c»i-1 de part et d’autre de la raie fondamentale-Les extrémités des o ailes », dont l’intensité est beau-
coup plus faible, résultent de la superposition des raies.Raman des vibrations caractéristiques du réseau, etdes raies de fréquences élevées du spectre de rotation.
Je remercie 81. le professeur J. Cabannes qui a bienvoulu s’intéresser à ce travail et m’aider de ses coii,--eils
Manuscrit reçu le 15 octobre 1935.
BIBLIOGRAPHIE
[1] A. ROUSSET. Thèses, Paris 1935.[2] C. V. RAMAN et K. S. KRISHNAN. Proc. Roy. Soc., 1929, A 122,
p. 23.
[3] VENKATESWARAN. Nature, 1934, 128, p. 870.[4] J. CABANNES et Y. ROCARD Journal de Physique, 1929, 10,
p. 32.
[5] C. MANNEBACK. Z. Physik, 1930, 62, p. 224.[6] G. PLACZEK et E TELLER Z. Physik, 1933, 81, p. 209.[7] Y. ROCARD. Annales de Physique, 1928, 10, p. 116.
[ [8] J. CABANNES et A. ROUSSET Annales de Physique, 1933, 19, p. 229.[9] C. V. RAMAN et S. BHAGAVANTAM. Indian Journal of Physics,
1931, 6, p. 353.
[10] S. VENKATESWARAN. Philosophical Magazine, 1932, 14, p. 258.[11] RANGANAOHAM. Indian Journal of Physics, 1932, 7, p. 353.[12] G. W. STEWART. Rev. of Modern Physics, 1930, 2, p. 116.[13] S. BHAGAVANTAM et A. V. RAO. Indian Journal of Physics,
1933, 8, p. 437.[14] S. BHAGAVANTAM et A. V. RAO. Proc. Ind. Acad. Sc., 1935, 1,
p. 419.[15] A. V. RAO. Proc. Ind. Acad. Sc., 1934, 1, p. 274.[16] PAULING Physical Review, 1930, 36, p. 430.
[17] E. GROSS et M. VUKS. Nature, 1935, 135, p. 100 et 431.





























