Embed Size (px)
Citation preview

Proc. Natl. Acad. Sci. USAVol. 76, No. 7, pp. 3083-3086, July 1979Biochemistry
L-Glucosylceramide: Synthesis, properties, and resistance tocatabolism by glucocerebrosidase in vitro
(glucocerebroside/Gaucher disease/animal model/chemical sphingolipidosis)
ANDREW E. GAL, PETER G. PENTCHEV, JANICE M. MASSEY, AND ROSCOE 0. BRADY
Developmental and Metabolic Neurology Branch, National Institutes of Neurological and Communicative Disorders and Stroke, National Institutes of Health,Bethesda, Maryland 20205
Contributed by Roscoe 0. Brady, March 16, 1979
ABSTRACT Procedures for the synthesis and radioactivelabeling of L-glucosylceramide are described. This compoundis a stereoisomeric analogue of D-glucosylceramide which oc-curs in nature and accumulates in pathological quantity in theorgans and tissues of patients with Gaucher disease. The prop-erties of L-glucosylceramide that have been examined so farhave been found to be indistinguishable from those of the nat-urally occurring glycolipid. However, L-glucosylceramide iscompletely refractory to enzymatic hydrolysis by purified pla-cental glucocerebrosidase and enzyme(s) present in whole tissueextracts. It is anticipated that L-glucosylceramide will be auniquely useful substance for exploring pathogenetic processesin animal analogues of Gaucher disease.
The pathogenesis of most lysosomal storage disorders is poorlyunderstood at this time although clear definitions of the primaryenzymatic lesions often exist (1). Because of this situation, in-formation relating to the pathologic manifestations of thesediseases has depended primarily on descriptions of clinicalcourses in patients afflicted with these disorders and on thefindings at postmortem examinations. The pathogenetic basisof the anatomical and physiological alterations of these disordershas been largely confined to speculation. Gaucher disease is aclassic example of this dilemma. This disorder is caused by adeficiency of glucocerebroside-3-glucosidase activity in thetissues of affected individuals (2). It has recently been shownin a patient with non-neuropathic form of the disorder (typeI) that this deficiency arises from a structural mutation in theenzyme that results in markedly decreased hydrolysis of glu-cosylceramide without apparent effect on the affinity of theenzyme for the substrate (3). The molecular bases that distin-guish the two neuropathic forms of Gaucher diseases (types IIand III) from type I Gaucher disease are not known. In addition,the pathophysiologic consequences of the accumulation ofglucosylceramide are poorly understood. For example, it hasbeen observed that there is a wide variation in the quantity ofglucosylceramide in the liver of patients with Gaucher diseaseand that there often is little or no correlation of this accumu-lation with (i) the level of residual glucocerebrosidase activityor (ii) the severity of the clinical manifestations (4).The availability of an experimental model of Gaucher disease
seems to be a paramount requirement for answering many ofthese puzzling questions. Although a strain of mutant BALB/cmice with hepatic and splenic accumulation of glucocerebrosidehas recently been reported, the clinical manifestations of thedisease in these mice are not typical of Gaucher disease (5).Investigators have attempted to inhibit glucocerebrosidase
activity by the administration of conduritol-3-epoxide to inducea syndrome resembling Gaucher disease in mice (6). However,the quantity of glucosylceramide that accumulated in liver andspleen was only 2- to 3-fold greater than that in normal miceand is therefore far below that found in patients with Gaucherdisease (4). In addition, the typical Gaucher bodies or tubularstructures characteristic of the disorder were not observed inspleen, liver, or bone marrow of mice treated with this reagent(7).
Because of the lack of a suitable Gaucher disease model atthe enzyme level, investigators have attempted with littlesuccess, to produce Gaucher disease-like lesions by the ad-ministration (intraperitoneally or orally) of high levels ofgalactosylceramide to rats and rabbits (8). When radioactiveglucosylceramide was administered intravenously to rats, it wasrapidly cleared from the plasma and taken up by the reticulo-endothelial system where it was quickly catabolized, and noaccumulation could be demonstrated (unpublished data). Thepronounced susceptibility of glucosylceramide to normal cat-abolic processes pointed to the desirability of attempting toprepare a substrate analogue with the chemical and physicalproperties of the natural glycolipid but with the added featureof its being resistant to enzymatic hydrolysis. It was consideredlikely that glucosylceramide containing L-glucose instead ofthe naturally occurring D-glucose might be useful in this re-gard.We describe here the syntheses of 1-O-3-D-glucosyl-N-
palmitoyl-DL-sphingosine and 1-O-3-L-glucosyl-N-palmi-toyl-DL-sphingosine and compare the chemical and physicalproperties of the two stereoisomers. In addition, the catabolicinertness of L-glucosylceramide in vitro is demonstrated.
MATERIALS AND METHODSMaterialsSilica gel 60 (E. Merck) plates were used for thin-layer chro-matography. The migration of glycolipids was visualized bycharring with ammonium bisulfate (9). The silica gel used forcolumn chromatography was Bio-Sil HA (minus 35 mesh;Bio-Rad) that was activated prior to use by heating at 1100Cfor 12 hr. Hydrogen bromide in acetic acid was obtained fromKoch-Light Labs (Colnbrook, England). Radioactive mea-surements were carried out in a Searle mark III liquid scintil-lation system using Aquasol (New England Nuclear). 3-0-Benzoyl-N-palmitoyl-DL-sphingosine, mp 77-78, and 1-0-3-D-glucopyranosyl-N-[1-14Clstearoyl-DL-sphingosine (0.315mCi/mmol; 1 Ci = 3.7 X 1010 becquerels) were kindly providedby D. Shapiro. L-[1-14C]Glucose was purchased from NewEngland Nuclear. Unlabeled L-glucose was obtained fromSigma. Radioscans were made with a series 6000 Varian
3083
The publication costs of this article were defrayed in part by pagecharge payment. This article must therefore be hereby marked "ad-vertisement" in accordance with 18 U. S. C. §1734 solely to indicatethis fact.
Proc. Natl. Acad. Sci. USA 76 (1979)
(Berthold) radioscanner. Melting points were taken on aThomas-Hoover melting point apparatus and are reportedcorrected.Synthesis of glucocerebrosides
1,2,3,4,6-Penta-O-acetyl-,-L-glucopyranoside. A mixtureof 5.4 g (30 mmol) of L-glucose, 30 ml of acetic anhydride, and2.5 g of previously melted sodium acetate was heated at 950Cfor 2 hr. The mixture was decomposed with 12.0 g of ice water,stirred for 3 hr at 00C, and kept for 1 day at 250C. The pre-cipitate was filtered and washed with 600 ml of water, driedon the filter, and washed with 60 ml of benzene/hexane, 1:1(vol/vol). The product was recrystallized from ethanol (600 ml).The yield was 5.8 g (50%) having a melting point of 130-1310C.On thin-layer chromatography in cyclohexane/benzene/iso-propylether/pyridine, 115:20:20:25 (vol/vol), the pentaacetatehad RF 0.33. Pentaacetyl-D-glucopyranoside had the same RFvalue in this system:
2,3,4,6-Tetra-O-acetyl-a-L-glucopyranosyl Bromide. Asolution of 3.9 g (10 mmol) of L-pentaacetylglucose in 20 ml of45% (wt/vol) hydrogen bromide in acetic acid was kept at 4VCfor 18 hr and subsequently at 250C for 4 hr. The mixture wasthen decomposed by the addition of 16 ml of ice water. Theproduct was extracted with 300 ml of ether, and this solutionwas washed at 00C with three consecutive 400-ml portions ofa 0.5 M potassium bicarbonate. The product was dried overanhydrous calcium chloride, the solvent was evaporated, andthe residue was recrystallized from 20 ml of isopropyl ether.The yield was 1.95 g (47%) with a melting point of 88-890C.Thin-layer chromatography in the above described system gavean RF 0.47 for the bromide. The melting point and RF for theD-isomer were the same.
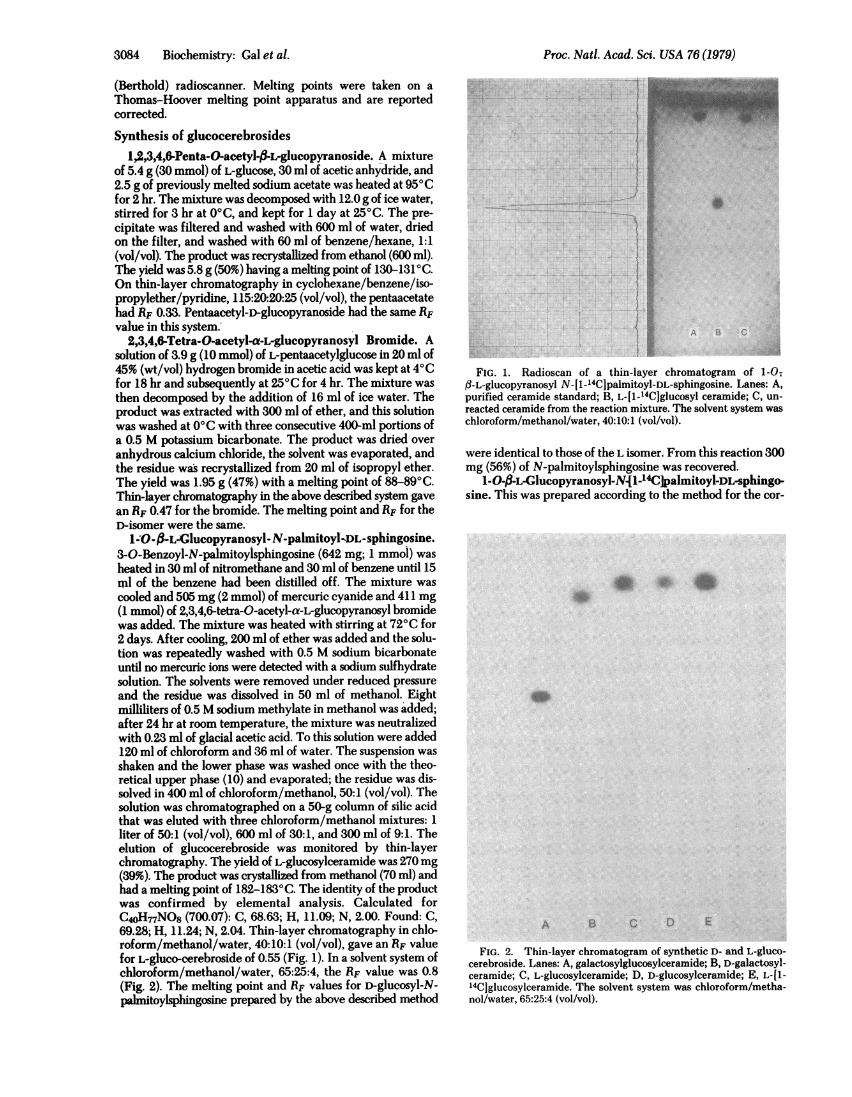
1-O-,-.L-GIucopyranosyI-.N-palmitoyl-DL-sphingosine.3-O-Benzoyl-N-palmitoylsphingosine (642 mg; 1 mmol) washeated in 30 ml of nitromethane and 30 ml of benzene until 15ml of the benzene had been distilled off. The mixture wascooled and 505 mg (2 mmol) of mercuric cyanide and 411 mg(1 mmol) of 2,3,4,6-tetra-0-acetyl-a-L-glucopyranosyl bromidewas added. The mixture was heated with stirring at 720C for2 days. After cooling, 200 ml of ether was added and the solu-tion was repeatedly washed with 0.5 M sodium bicarbonateuntil no mercuric ions were detected with a sodium sulfhydratesolution. The solvents were removed under reduced pressureand the residue was dissolved in 50 ml of methanol. Eightmilliliters of 0.5 M sodium methylate in methanol was added;after 24 hr at room temperature, the mixture was neutralizedwith 0.23 ml of glacial acetic acid. To this solution were added120 ml of chloroform and 36 ml of water. The suspension wasshaken and the lower phase was washed once with the theo-retical upper phase (10) and evaporated; the residue was dis-solved in 400 ml of chloroform/methanol, 50:1 (vol/vol). Thesolution was chromatographed on a 50-g column of silic acidthat was eluted with three chloroform/methanol mixtures: 1liter of 50:1 (vol/vol), 600 ml of 30:1, and 300 ml of 9:1. Theelution of glucocerebroside was monitored by thin-layerchromatography. The yield of L-glucosylceramide was 270mg(39%). The product was crystallized from methanol (70 ml) andhad a melting point of 182-1830C. The identity of the productwas confirmed by elemental analysis. Calculated forC4oH-nNO8 (700.07): C, 68.63; H, 11.09; N, 2.00. Found: C,69.28; H, 11.24; N, 2.04. Thin-layer chromatography in chlo-roform/methanol/water, 40:10:1 (vol/vol), gave an RF valuefor L-gluco-cerebroside of 0.55 (Fig. 1). In a solvent system ofchloroform/methanol/water, 65:25:4, the RF value was 0.8(Fig. 2). The melting point and RF values for D-glucosyl-N-Palinitoylsphingosine prepared by the above described method
A B c
FIG. 1. Radioscan of a thin-layer chromatogram of 1-0;f3-L-glucopyranosyl N-[1-14C]palmitoyl-DL-sphingosine. Lanes: A,purified ceramide standard; B, L-[1-'4CJglucosyl ceramide; C, un-reacted ceramide from the reaction mixture. The solvent system waschloroform/methanol/water, 40:10:1 (vol/vol).
were identical to those of the L isomer. From this reaction 300mg (56%) of N-palmitoylsphingosine was recovered.
1-O-3-L-Glucopyranosyl-N{1-14C palmitoyl-DL-sphingo-sine. This was prepared according to the method for the cor-
A B C D E
FIG. 2. Thin-layer chromatogram of synthetic D- and L-gluco-cerebroside. Lanes: A, galactosylglucosylceramide; B, D-galactosyl-ceramide; C, L-glucosylceramide; D, D-glucosylceramide; E, L-Il-14Cjglucosylceramide. The solvent system was chloroform/metha-nol/water, 65:25:4 (vol/vol).
3084 Biochemistry: Gal et al.

Proc. Natl. Acad. Sci. USA 76 (1979) 3085
responding nonradioactive cerebroside described abov. Thefirst step of the synthesis used 540.5 mg (3 mmol) of L-[1-14C]glucose (1.17 mCi/mmol). The yield of tetraacetyl-L-[1-14C]glucopyranosyl bromide was 222 mg (0.55 mmol). Thecompound appeared to be homogeneous by thin-layer chro-matography, with a single corresponding peak of radioactivity(Fig. 1).
1-O-f-D-Glucopyranosyl-N-palmitoyl-DL-sphingosine. Thestarting material was purified tetraacetyl-D-glucopyranosylbromide (11), 62 mg (0.15 mmol); all other reagents used were1/10th the quantities used for the synthesis of the L isomer. Theyield was 18 mg; the product melted at 182-183°C.Assay of glucocerebrosidase activityThe enzyme source consisted of either highly purified humanplacental glucocerebrosidase (12) or whole mouse tissue extractsprepared as 20% (wt/vol) homogenates in water. Enzymaticactivity was measured in 50 mM potassium phosphate, pH 5.90.12% Cutscum/0.05% crude sodium taurocholate (Difco) ina final volume of 0.2 ml. The reaction was initiated by the ad-dition of 54 nmol of D-[1-'4C]glucosylceramide as a 5-,ul samplein a solution of sodium taurocholate. After incubation for 1 hrat 370C, the reaction was stopped by the addition of 1.0 ml ofcooled bovine serum albumin (10 mg/ml) followed by 0.1 mltrichloroacetic acid (1 g/ml, aqueous solution). The mixturewas swirled and then centrifuged at 3000 X g for 10 min. Theenzymatically liberated [1-14C]glucose in the acid-soluble su-
pernatant was measured by liquid scintillation spectroscopy.
RESULTSSynthesis of Glucocerebroside Analogues. 1,2,3,4,6-
Penta-O-acetyl-fl-L-glucopyranose and 2,3,4,6-tetra-0-ace-tyl-a-L-glucopyranosyl bromide were prepared in good yieldsby a modification of the method of Potter et al. (13). Themelting points of these compounds were the same as those ofthe D-isomers. The syntheses of the D and L glucosylceramideswere based on the method of Shapiro (14). The melting pointsand RF values for the stereoisomeric cerebrosides were iden-tical. L-[1-'4C]Glucosylceramide was prepared on a smallerscale in good yield.
Effect of Purified Glucocerebrosidase on the D- and L-Glucosylceramides. No release of labeled hexose could bedetected when L-[1-'4C]glucosylceramide was incubated withthe enzyme under conditions routinely used for the hydrolysisof D-glucosylceramide. When the amount of enzyme and timeof incubation were adjusted to achieve nearly complete hy-drolysis of the D isomer, there was still no detectable hydrolysisof L-glucosylceramide (Fig. 3). Furthermore, the addition ofthe L isomer did not influence the hydrolysis of D-glucosyl-ceramide when the glycolipids were incubated together inequimolar concentration, indicating that the analogue con-
taining L-glucose does not interact with the active site on theenzyme.
Incubation of Tissue Extracts with D- and L-Glucosylcer-amides. When the glycolipids were incubated with wholemouse tissue extracts under conditions that allow expression ofmost lysosomal hydrolases, there was substantial ,B-glucosidaseactivity observed with the D isomer but no detectable hydrolysisof the L isomer (Table 1). Because the normal mode of cleavageof glucosidase did not appear to be functioning with L-glu-cosylceramide, the possibility that the hydrolysis of the amidelinkage resulted in the formation of glucosylsphingosine (psy-chosine) and free fatty acid was investigated. Examination ofmixtures of tissue extracts incubated with L-[1-'4C]glucosyl-ceramide by thin-layer chromatography revealed only a singleradioactive area that corresponded with the migration of glu-
I-.I..
A B C D
FIG. 3. Incubation (24 hr) of D- and L-glucocerebroside with10,000 units of purified human placental glucocerebrosidase. Thereaction was stopped by the addition of chloroform/methanol, 2:1(vol/vol); after partitioning, the contents of the lower phase wereanalyzed by thin-layer chromatography. Lanes A and B represent D-and L-glucosylceramide, respectively, in the absence of glucocere-brosidase. Lanes C and D represent D- and L-glucosylceramide in thepresence of glucocerebrosidase. The dark spot in the middle of theplate corresponds to the RF value for glucosylceramide standard. Thedark spot at the top of lane C corresponds to ceramide. The otherlipids seen on the plate originate from the partially purified sodiumtaurocholate used in the assay.
cosylceramide standard. There was no indication of the for-mation of a labeled reaction product in the region of psychosine(Fig. 4).
DISCUSSIONThe stereochemistry of the D- and L-glucosylceramides issomewhat complex. Due to the additional asymmetric natureof sphingosine, the two cerebrosides are not pure enantio-morphs. The synthetic sphingosine with which the respectivecerebrosides were prepared was, itself, a racemic mixture ofD- and L-sphingosine. The conjugation to this racemic mixtureof an additional optically active molecule (D- or L-glucose)results in an equimolar mixture of diastereoisomers. Conse-quently, the stereochemical relationship between the synthetic
Table 1. Incubation of mouse tissue extracts withD- or L-glucosylceramide
Glucosylceramide hydrolyzed,nmol/g (wet)/hr
Tissue D-Glucose isomer L-Glucose isomer
Liver 4800 0Spleen 5300 0Kidney 2200 0Intestine 1500 0Thymus 4000 0Brain 3000 0Lung 2300 0Plasma 5* 0
Frozen mouse tissues were thawed and homogenized by hand in 4vol of distilled water with a tightly fitting all-glass homogenizer. A25-pu aliquot of the whole homogenate was incubated with D- or L-[1-14C]glucosylceramide.* Shown as nmol/ml per hr.
p .,II$pw
Biochemistry: Gal et al.

Proc. Natl. Acad. Sci. USA 76 (1979)
..!in.....
.W.iusp
I
A B
FIG. 4. Incubation of L-[1-C14Jglucosylceramide with a crudemouse liver homogenate. Lane A represents a mixture of the followingstandard glycolipids in order of diminishing RF values: glucosylcer-amide, galactosylceramide, galactosylglucosylceramide, galactosyl-galactosylglucosylceramide, and N-acetylgalactosaminylgalactosyl-galactosylglucosylceramide (globoside). Lane B represents a chloro-form/methanol extract of a 24-hr incubation of mouse whole liverhomogenate (20%, wt/vol) with L-[1-14C]glucosylceramide. Thechromatogram was made on a silica plate in a solvent system ofchloroform/methanol/water, 40:10:1 (vol/vol), scanned for radioac-tivity, and subsequently visualized by charring. Psychosine(glucosyl)has been shown (15) to chromatograph with a RF value close to thatof globoside.
D- and L-glucosylceramide preparations consists of 50% en-
antiomorphism and 50% diastereoisomerism. Although a lackof sufficient material limited a rigorous analysis of the potentialchemical and physical differences due to diastereoisomerism,the D- and L-glucosylceramide preparations appeared to beidentical on the basis of their chemical properties describedabove. An additional example of their similarity was the iden-tical migration of D- and L-glucosylceramides in thin-layerchromatographic systems (Fig. 2). The resolution obtained bythis system can be judged from the decidedly slower migrationof galactosylceramide than its 4-epimeric analogue glucosyl-ceramide.
Because the refractoriness of L-glucosylceramide to enzy-matic hydrolysis at both the glucosidic and amide linkages hasbeen demonstrated in vitro, use of this glycolipid may beconsidered for a number of physiological and pathological in-vestigations. The anticipated catabolic inertness of the L-hexosylglycolipid in vsvo may enable one to mimic the storage of D-glucosylceramide that occurs in Gaucher disease with a highdegree of fidelity. An investigation of the chronology of the
appearance of cellular responses such as lysosomal hypertrophyinduced by the accumulation of L-glucosylceramide should beparticularly instructive. The influence of the form in which theglycolipid is administered (free, associated with lipoproteins,or incorporated into erythrocytes or leukocytes) may pro-foundly affect the uptake of the glycolipid. Furthermore, it willbe possible to carry out examinations of the potential intercel-lular exchange of L-glucosylceramide between cells of the re-ticuloendothelial system with parenchymal cells (e.g., Kupffercells and hepatocytes) and the possibility of the excretion of thislipid via the bile. It will also be of considerable interest to de-termine if L-glucosylceramide can enter anabolic pathways.The formation and disposition of such a "hybrid" syntheticnatural product could provide considerable insight concerningthe source, transport, and fate of complex sphingolipids.
In sum, the numerous investigative possibilities concerningthe metabolism of L-glucosylceramide might be thought of asrepresenting a reversal of natural processes. Metabolic disordersoccur because mutant enzymes are no longer capable of in-teracting properly with their normal substrates. One can nowattempt to alter physiological processes by presenting a "mu-tant" substrate to normal enzymes.
1. Brady, R. 0. (1976) Science 193,733-739.2. Brady, R. O., Kanfer, J. N. & Shapiro, D. (1965) Biochem. Bio-
phys. Res. Commun. 18,221-225.3. Pentchev, P. G., Brady, R. O., Blair, H. E., Britton, D. E. & Sorrell,
S. H. (1978) Proc. Nati. Acad. Sci. USA 75,3970-3973.4. Pentchev, P. G., Barranger, J. A., Gal, A. E., Furbish, F. S. &
Brady, R. 0. (1979) in Glycoproteins and Glycolipids in DiseaseProcesses, ACS Symposium Series, No. 80, ed. Walborg, E. F.,Jr. (Am. Chem. Soc., Washington, DC), p. 156.
5. Pentchev, P. G., Boothe, A. D., Gal, A. E., Omodeo-Sale, F.&Brady; R. 0. (1979) in Enzyme Therapy in Genetic Diseases, 2ndInternational Symposium, Hilton Head, South Carolina, March3-7 (The National Foundation, New York), p. 106 (abstr.).
6. Stephens, M. C., Bernatsky, A., Burachinsky, V., Legler, L. &Kanfer, J. (1978) J. Neurochem. 30, 1023-1027.
7. Adachi, M. & Volk, B. W. (1977) Arch. Pathol. Lab. Med. 101,255-259.
8. Burton, R. M. & Sodd, M. A. (1969) Lipids 4,496-500.9. Gal, A. E. (1968) Anal. Biochem. 24,452-461.
10. Folch, J., Lees, M. & Stanley, G. H. S. (1957) J. Biol. Chem. 226,497-509.
11. Gal, A. E., Pentchev, P. G. & Fash, F. J. (1976) Proc. Soc. Exp.Biol. Med. 153,363-366.
12. Furbish, F. S., Blair, H. E., Shiloach, J., Pentchev, P. G. & Brady,R. 0. (1977) Proc. Nati. Acad. Sci. USA 74,3560-3563.
13. Potter, A. L., Sowden, J. C., Hassid, W. Z. & Doudoroff, M. (1948)J. Am. Chem. Soc. 70, 1751-1752.
14. Shapiro, D. (1969) in Chemistry of Sphingolipids (Hermann,Paris), p. 100.
15. Neskovic, N. M., Nussbaum, J. L. & Mandel, P. (1970) J. Chro-matogr. 49,255-261.
3086 Biochemistry: Gal et al.



















![Ch 5: ARIMA model · 1.1 Non-Stationary Data [ToC] Dow Jones Index From Aug. 28 to Dec. 18, 1972 l l l l l ll l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l](https://img.pdfslide.us/doc/110x75/5ee0213ead6a402d666b5f8b/ch-5-arima-model-11-non-stationary-data-toc-dow-jones-index-from-aug-28-to.jpg)








