Embed Size (px)
Citation preview

Kinetics of the oxidation of diphenyl sulfide with hydrogen peroxide catalyzed by sodium metavanadatel
YOSHIRO OGATA~ AND KAZUSHIGE TANAKA Department of Applied Chemistry, Faculty of Engineering, Nagoya University, Chikusa-ku, Nagoya, Japan
Received April 6, 19813
YOSHIRO OGATA and KAZUSHIGE TANAKA. Can. J. Chem. 60,848 (1982). The oxidation of diphenyl sulfide (Ph,S) by hydrogen peroxide in the presence of a catalytic amount of sodium metavanadate
(NaVO,) has been studied kinetically by means of iodometry of hydrogen peroxide. The reaction rate is expressed as: u = k[NaV03b,[Ph,S]2, when the concentrationof catalyst is very low and [PhzS]oIIHzO,]o > 2, where [ L,and [ lo mean stoichiometric and initial concentration, respectively. The effective oxidant may consist of polymeric as well as monomeric peroxyvanadate in view of the effect of concentration of catalyst on the rate. The main oxidizing species at low concentration of catalyst seems to be diperoxyvanadate V0,-. The rate constant k, in u = k,[Ph,SIz tends to decrease with initial concentration of H,O,, which is present in excess of the catalyst. A probable mechanism for the ox~dation is discussed.
YOSHIRO OGATA et KAZUSHIGE TANAKA. Can. J. Chem. 60,848 (1982). Faisant appel I'iodomttrie du peroxyde d'hydrogkne, on a realise une etude cinktique de I'oxydation du sulfure de biphenyle
(Ph2S) par le peroxyde d'hydrogkne en presence d'une quantitt catalytique de metavanadate de sodium (NaVO,). On exprime la vitesse de la reaction par I'kquation: u = k[NaV03]9t [Ph2SI2, lorsque la concentration du catalyseur est tres faible et lorsque [Ph2S]~/[H,O2I0 > 2 ou [ l, et [ 1, signifiient respectivement les concentrations stochiometriques et initiales. L'oxydant effectif peut i t re le peroxyvanadate sous la forme polymCrisCe aussi bien que sous la forme monomere compte tenu de I'effet de la concentration du catalyseur sur la vitesse. Pour une faible concentration du catalyseur, I'oxydant principal semble &tre de diperoxyvanadate V0,-. La constante de vitesse k2 dans I'expression u = k2[Ph,SI2 tend a diminuer avec la concentration initiale de H202 qui est en excks par rapport au catalyseur. On discute du mecanisme probable de I'oxydation.
[Traduit par le journal]
Introduction Vanadic acid is converted to its peroxy acids on
treatment with H20,. Beg and Ahmad (1) have reported on the kinetics and mechanism of epox- idation of maleic and fumaric acids by H202 catalyzed by sodium orthovanadate, in which the main oxidant is monoperoxyvanadic acid H,VO,. On the other hand, Modena and co-workers have reported the oxidation of sulfides with tert-butyl hydroperoxide (2) or H20z (3) in the presence of bis(acety1acetonato) oxovanadium(1V) VO(acac), in alcohol (ROH) as solvent, where the main oxidant is said to be complexed monoperoxy- vanadic acid VO(OR),OOH produced by conver- sion of VO(OR),. These reports, however, paid little attention to the rate of diperoxyvanadic acid.
The treatment of metavanadate with H,02 pro- duces peroxy acids consisting mainly of diperoxy- vanadate V0,- and polymeric peroxyvanadate (4), so that these peroxyvanadates may participate in this oxidation. The present paper describes the rate data and some factors influencing the rate for the NaV0,-catalyzed oxidation of diphenyl sulfide by hydrogen peroxide in 98 wt.% methanol (with 2 wt.% water).
Results and discussion Peroxy vanadate
Generally, metavanadate ion is known to exist mainly as V30,,- in neutral solution (5). The ion V30g3- is converted to monoperoxyvanadate ion V03+ and diperoxyvanadate ion V0,- on treat- ment with hydrogen peroxide as follows (6).
Here, the formation of V0,- is influenced by [H+] (see eq. [3]). But the main species present in the system with < 0.015 M H202 seems to be V0,- based on the fact that the color of the reaction mixture is yellow, that no decomposition of H202 is observed on standing for ca. 8 h at 25OC, and that the rate becomes first-order with respect to sub- strate (Ph2S) by addition of strong acid (H2S04) as stated later.
The identification of VO,+ and V0,- bv means of 'Contribution NO. 289. polarography and uv spec~roscopy failed because 2Author to whom all correspondence should be addressed. of the complex reaction at the electrode (7) and 3Revision received August 26, 1981. partial overlapping of the spectra (8), respectively.
0008-4042182IO70848-05$01.00/0 01982 National Research Council of CanadalConseil national de recherches du Canada
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
SAN
DIE
GO
ST
AT
E U
NIV
ER
SIT
Y o
n 11
/11/
14Fo
r pe
rson
al u
se o
nly.
OGATA AN1 D TANAKA 849
Kinetics The oxidation of diphenyl sulfide gave diphenyl
sulfoxide together with a small amount of diphenyl sulfone (eq. [I]). Most of our kinetic experiments were done under conditions in which the formation of diphenyl sulfone was negligible; i.e., in the presence of a large excess of sulfide over H202. The rate was followed by iodometry of active oxygen, since the rate constants k2 (in u = k2[Ph2- SI2) calculated by the iodometry were practically in agreement with those calculated on the basis of the consumption of Ph2S, determined by means of glc; i.e., 6.52 x M-I s-I (by iodometry) and 6.15 x 10-4 M-1 S-l (by glc).
Though vanadate is a weak oxidant, diphenyl sulfide is oxidized by neither metavanadate nor H202 alone in this solvent (98 wt.% methanol) at 25°C. The spontaneous decomposition of H202 is negligible under these conditions.
The effect of initial concentration of diphenyl sulfide on the rate was examined, the results being
i shown in Table 1. The rate is expressed as u = k2[Ph2S12, if [Ph2SloI[H20210 > 2 and [H20210 -
1 1.15 x M. This result is different from that of I sodium tungstate, where the rate expression u =
k,[Ph2S] is observed. The term [Ph2SI2 in the rate 1 expression suggests that peroxyvanadate and di- phenyl sulfide form a 1:l coordination complex, (V0,-Ph2S)-, which may act as a strong oxidant for Ph2S.
If [Ph2S], is lower than 2 x lop2 M, further oxidation giving a considerable amount of Ph2S02 occurs, by which the rate constant decreases.
Effects of concentration of catalyst and H202 The effect of the initial concentration of sodium
metavanadate [NaVO,], relative to that of H202,
TABLE 1. Rate dataa for NaV0,-catalyzed oxidation of Ph,S with H,O, at 25°C in 98 wt.% MeOH
Initial concn. x lo3 M I 1
[NaV0310 [H2O2I0 [Ph,Slo k, x lo4 M-Is-I
0.5 4.06 30 7.30 0.5 4.10 25 7.60 0.5 5.83 30 6.95 0.5 6.02 25 6.46
1 0.5 11.4 25 5.09 0.5 11.6 30 5.36 0.5 11.8 35 5.19 0.5 11.7 40 5.18 1 .O 11.3 25 6.52 1 .O 11.3 30 7.06 1.0 11.6 35 7.11 1.0 11.6 40 6.92
I "u = k2[Ph2Sl'.
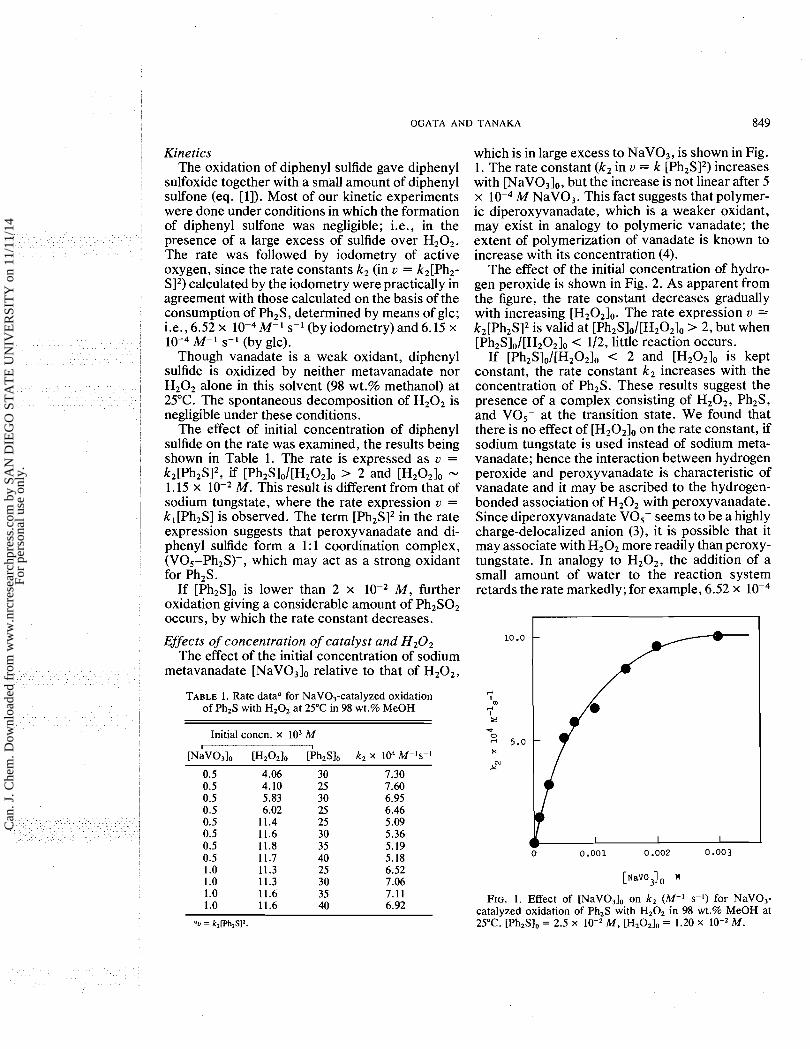
which is in large excess to NaVO,, is shown in Fig. 1. The rate constant (k2 in u = k [Ph2SI2) increases with [NaVO,],, but the increase is not linear after 5 x M NaV0,. This fact suggests that polymer- ic diperoxyvanadate, which is a weaker oxidant, may exist in analogy to polymeric vanadate; the extent of polymerization of vanadate is known to increase with its concentration (4).
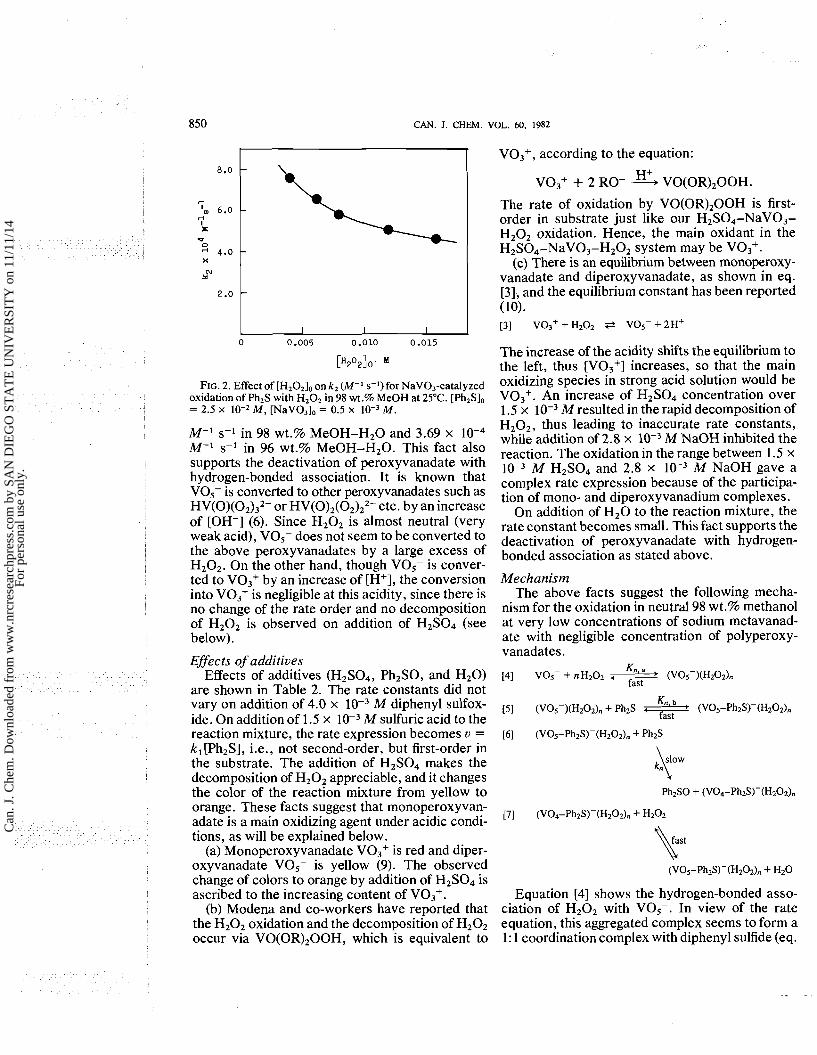
The effect of the initial concentration of hydro- gen peroxide is shown in Fig. 2. As apparent from the figure, the rate constant decreases gradually with increasing [H2O2I0. The rate expression u = k2[Ph2SI2 is valid at [Ph2S]o/[H202]o > 2, but when [Ph2S],/[H202], < 112, little reaction occurs.
If [Ph2S],I[H202], < 2 and [H202], is kept constant, the rate constant k2 increases with the concentration of Ph2S. These results suggest the presence of a complex consisting of H202, Ph2S, and V0,- at the transition state. We found that there is no effect of [H202], on the rate constant, if sodium tungstate is used instead of sodium meta- vanadate; hence the interaction between hydrogen peroxide and peroxyvanadate is characteristic of vanadate and it may be ascribed to the hydrogen- bonded association of H202 with peroxyvanadate. Since diperoxyvanadate V0,- seems to be a highly charge-delocalized anion (3), it is possible that it may associate with H202 more readily than peroxy- tungstate. In analogy to H202, the addition of a small amount of water to the reaction system retards the rate markedly; for example, 6.52 x
C N ~ V O J O
FIG. 1 . Effect of [NaVO,], on k, (M-' s-') for NaV0,- catalyzed oxidation of Ph,S with H202 in 98 wt.% MeOH at 25°C. [Ph,S], = 2.5 x lo-, M, [H202], = 1.20 x M .
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
SAN
DIE
GO
ST
AT
E U
NIV
ER
SIT
Y o
n 11
/11/
14Fo
r pe
rson
al u
se o
nly.
850 CAN. J. CHEM. VOL. 60, 1982
FIG. 2. Effect of [H,O,], on k , (M-I ssl)for NaV03-catalyzed oxidation of Ph2S with HZOZ in 98 wt.% MeOH at 25°C. [Ph2Sl0 = 2.5 x M, [NaVO3lo = 0.5 x M .
M-I s-I in 98 wt.% MeOH-H20 and 3.69 x M-I s-I in 96 wt.% MeOH-H20. This fact also supports the deactivation of peroxyvanadate with hydrogen-bonded association. It is known that V0,- is converted to other peroxyvanadates such as HV(0)(02)32- or HV(0)2(02)22- etc. by an increase of [OH-] (6). Since H202 is almost neutral (very weak acid), V0,- does not seem to be converted to the above peroxyvanadates by a large excess of H202. On the other hand, though VOS- is conver- ted to V03+ by an increase of [H+], the conversion into V03+ is negligible at this acidity, since there is no change of the rate order and no decomposition of H202 is observed on addition of H2S04 (see below).
Effects of additives Effects of additives (H2S04, Ph2S0, and H20)
are shown in Table 2. The rate constants did not vary on addition of 4.0 x M diphenyl sulfox- ide. On addition of 1.5 x M sulfuric acid to the reaction mixture, the rate expression becomes v = k,[Ph2S], i.e., not second-order, but first-order in the substrate. The addition of H2S04 makes the decomposition of H202 appreciable, and it changes the color of the reaction mixture from yellow to orange. These facts suggest that monoperoxyvan- adate is a main oxidizing agent under acidic condi- tions, as will be explained below.
(a) Monoperoxyvanadate V03+ is red and diper- oxyvanadate V0,- is yellow (9). The observed change of colors to orange by addition of H2S04 is ascribed to the increasing content of V03+.
(b) Modena and co-workers have reported that the H202 oxidation and the decomposition of H202 occur via VO(OR),OOH, which is equivalent to
V03+, according to the equation:
V03+ + 2 RO- VO(OR)200H.
The rate of oxidation by VO(OR)200H is first- order in substrate just like our H2S04-NaV03- H202 oxidation. Hence, the main oxidant in the H2S0,-NaV03-H202 system may be V03+.
(c) There is an equilibrium between monoperoxy- vanadate and diperoxyvanadate, as shown in eq. [3], and the equilibrium constant has been reported (10). [3] VOsf + H202 e V05- + 2H+
The increase of the acidity shifts the equilibrium to the left, thus [V03+] increases, so that the main oxidizing species in strong acid solution would be V03+. An increase of H2S04 concentration over 1.5 x M resulted in the rapid decomposition of H202, thus leading to inaccurate rate constants, while addition of 2.8 x M NaOH inhibited the reaction. The oxidation in the range between 1.5 x
M H2S04 and 2.8 x M NaOH gave a complex rate expression because of the participa- tion of mono- and diperoxyvanadium complexes.
On addition of H 2 0 to the reaction mixture, the rate constant becomes small. This fact supports the deactivation of peroxyvanadate with hydrogen- bonded association as stated above.
Mechanism The above facts suggest the following mecha-
nism for the oxidation in neutral 98 wt.% methanol at very low concentrations of sodium metavanad- ate with negligible concentration of polyperoxy- vanadates.
[4] V05- + n Hz02 & , a , fast
(VO~-)(H202)n
Equation [4] shows the hydrogen-bonded asso- ciation of H202 with VOS-. In view of the rate equation, this aggregated complex seems to form a 1: 1 coordination complex with diphenyl sulfide (eq.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
SAN
DIE
GO
ST
AT
E U
NIV
ER
SIT
Y o
n 11
/11/
14Fo
r pe
rson
al u
se o
nly.

OGATA AND TANAKA 85 1
TABLE 2. Effecta of additives for NaV0,-catalyzed oxida- Also, the reactivity of free V05- is much reduced tion of Ph2S with H202 at 25°C in 98 wt.% MeOH by association with H202, i.e.,
x 10, M koKo-b >> k,K,,aK,,b[H2021 + .... ~ d d i t i v e [NaVO,], [~ , t ) , l , k ,b x 10S S-' + knKn,aKn,b[H2021n; . . - . - - - -
H2S04 (1.5) 0.5 11.8 8.80 consequently, H2SO.i (1.5) 0.5 35.5 8.60 ~ $ 0 4 (1.5) 0.5 48.4 8.44
k2= x lo4 M - I S-I 9 kiKi,aKi.b[H2021i = k0K0.b i =O
Ph2S0 (4.0) 1.0 11.4 6.57 H 2 0 (3.0)d 1 .O 11.5 4.84 Hence, H 2 0 (4.0)d 1 .O 11.5 3.69 None 1 .O 11.3 6.52
a[Ph,Sl, = 2.5 x lo-' M. = k,[Ph,S].
' u = k,[Ph2Sl1. "Weight %.
[5]) and this complex seems to react with another molecule of diphenyl sulfide to give diphenyl sul- foxide (eq. [6]). The bulky Ph2S molecule would permit the second molecule of Ph2S to attack on the peroxy oxygen atom but not on the vanadium atom. Since monoperoxyvanadate seems to be unstable in this system, (V04-Ph2S)-(H202), formed in eq. [6] would be rapidly converted to (~0,-P~,s)-- (H202), by excess H202 (eq. [7]).
This mechanism involving a rate-determining attack of complex (V0,-Ph2S)-(H202), on di- phenyl sulfide to form diphenyl sulfoxide leads to a rate expression as follows.
,2
Here, KO,, = 1 and ki represents the ith rate constant, Kiss the ith equilibrium constant of eq. [4], and Ki,b, the ith equilibrium constant of eq. [S]. Since NaVO, reacts rapidly with H202 and since the uv spectrum of peroxyvanadates does not change with increasing concentration of H202 which is present in excess of the catalyst, NaVO, may be completely converted to peroxyvanadates (mainly V0,-). Hence, the stoichiometric initial concentration of NaVO, is approximated as: [NaV0310 [V05-Is,. On the other hand, the association of V05- with H202 at various molar ratios is expressed as:
Introduction of eq. [9] into eq. [8] leads to
[Ill u - kOKO,b{[VOS-Ist K~.~[H~O~I ' [VOS-I}
x [Ph2SI2 = k2[Ph2SI2
This expression is consistent with the experimental results. The apparent rate constant k2 decreases with increasing [H2O,l0., since the concentration or the sum of concentrations of associated peroxy- vanadates, i.e.,
increases with [H2O2I0. On the other hand, in the presence of H2S04, the
following mechanism may be operating as a main pathway as stated above.
[12a] v30g3- + 3 H202 + 6 H2SO4 3 V03+ + 6 H20 + 6 HS04-
(1261 V03+ + Ph2S + V02+ + PhzSO
But the direct oxidation with acidic H202 and spontaneous decomposition of H202 render the reaction kinetics complex. It has been reported (10) that both peroxy species VO,+ and V05- partici- pate in the oxidation of iodide ion in the [Hf] range of 0.1-1.0.
Experimental Materials
Methanol was ~urified bv fractionation ( b ~ 6 6 7 ° C ) and it . - was used as 98 wt.% solvent to make the oxidation system homogeneous. Diphenyl sulfide was purified by vacuum distilla- tion (bp 118-119°C. 4 Tom). Sodium metavanadate was of commercial, guaranteed reagent grade, quality.
Kinetics A mixture of NaVO, and H202 in 98 wt.% MeOH was kept
standing at 25°C for 5 min, then the reaction was started by addition of Ph2S. Aliquots were taken out at appropriate intervals of time, and their peroxide contents were determined iodometrically. The concentrations of Ph2S were calculated based on the H20, concentration after subtraction of metavan- adate concentration. The consumption of Ph2S was measured by glc (a Yanagimoto G 180 gas chromatograph with FID) with a column (2.5 mm x 1 m) packed with PEG 20M.
Acknowledgments The authors wish to thank Mr. M. Inaishi for his
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
SAN
DIE
GO
ST
AT
E U
NIV
ER
SIT
Y o
n 11
/11/
14Fo
r pe
rson
al u
se o
nly.

852 CAN. 1. CHEM. VOL. 60, 1982
assistance in preparation of the manuscript and to Mitsubishi Gas Chem. Co. for their gift of 90% Hz02
1. M. A. BEG and I. AHMAD. J. Org. Chem. 42, 1590 (1977). 2. R. CURICI, F. DI FURIA, R. TESTI, and G. MODENA. J.
Chem. Soc. Perkin Trans. 11, 752 (1974). 3. (a) 0. BORTOLINI, F. DI FURIA, P. SCRIMIN, and G.
MODENA. J. Mol. Catd . 7,59(1980); (6) F. DI FuRIAand G. MODENA. In Fundamental research in homogeneous cata- lysis. Vol. 3. Edited by M. Tsutsui. Plenum Press, New York. 1979. p. 433.
4. C. F. BELL and K. A. K. L o n . Modern approach to inorganic chemistry. 2nd ed. London. 1%6.
5. (a) P. Z. DULLBERG. Z. Phys. Chem. 45, 129 (1903); (6) K. SCHILLER and E. THILO. Z. Anorg. Allg. Chem. 310, 261 (1961); (c) R. A. ROBINSON and D. A. SINCLAIR. J. Chem. SOC. 642 (1934).
6. GMELIN, 48, V[B], 162. 7. I. M. KOLTHOFF and E. P. PARRY. J. Am. Chem. Soc. 73,
5315 (1951). 8. 0. BORTOLINI, F. DI FURIA, G. MODENA, and P. SCRIMIN.
J. Mol. Catal. 9, 323 (1980). 9. M. ORHANOVIC and R. G. WILKINS. J. Am. Chem. Soc. 89.
278 (1967). 10. F. S ~ c c o . Inorg. Chem. 19,2722 (1980).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
SAN
DIE
GO
ST
AT
E U
NIV
ER
SIT
Y o
n 11
/11/
14Fo
r pe
rson
al u
se o
nly.
















![Lab 28 Polybrominated diphenyl ethers [PDF - 732.9 KB]](https://img.pdfslide.us/doc/110x75/62061c2e8c2f7b173004ae98/lab-28-polybrominated-diphenyl-ethers-pdf-7329-kb.jpg)












