Embed Size (px)
Citation preview

Kinetics of CO Oxidation over Pt-Modified CuO Nanocatalysts
Luu C. Loc1,+, Nguyen Tri2, Hoang T. Cuong1 and Ha C. Anh2
1Institute of Chemical Technology, Vietnam Academy of Science and Technology,01 Mac Dinh Chi Str., Ho Chi Minh City, 70100 Vietnam2Ho Chi Minh City University of Technology, Vietnam National University - Ho Chi Minh City,268 Ly Thuong Kiet Str., Ho Chi Minh City, 70100 Vietnam
Three Pt-CuO nanocatalysts PtCu/Al, PtCu/CeAl and PtCu/Ce have been successfully prepared. The characterization of the catalysts wasexamined by X-ray powder diffraction (XRD), transmission electron microscopy (TEM), X-ray energy dispersive analysis (EDS), temperature-programmed reduction (TPR), nitrogen physisorption measurements, and IR-CO adsorption. The kinetics of CO oxidation using these catalystswas studied in a gradientless flow-circulating system at 398498K. The obtained kinetic equation confirmed that the reaction proceeds inmedium surface coverage with the participation of CO molecules and oxygen atoms. [doi:10.2320/matertrans.MA201545]
(Received January 30, 2015; Accepted May 29, 2015; Published August 25, 2015)
Keywords: carbon monoxide oxidation, kinetics, platinum-modified copper oxide nanocatalysts
1. Introduction
The advantage of low-temperature oxidation is to reducefuel consumption for conversion of large volume of pollutedair. Metal oxides and multioxide owning high activity andthermal stability are considered as alternative catalyst for theexisting expensive noble metals. In fact, promising resultswere obtained by adding a small amount of noble metalsto metal oxide catalysts. Particularly, the highest activityin oxidation of CuO/CeO2 catalysts modified with Pt isrationalized by the strong link between the Pt with CuO/CeO2.1) The synergic effects between metal oxides and noblemetals results in the increase of reducibility, which mayenhance the oxygen transfer from the metallic oxides to thenoble metals.2)
Research on kinetics of oxidation of single CO on noblemetal and oxide catalysts have intensively studied.311)
Among various forms of suggested kinetic equations forthe oxidation of CO, power-law kinetic expressions wererepeatedly proposed. Indeed, first-order of oxygen and zero-order of CO concentrations for CO oxidation on bulk copperoxide were reported by Garner et al.3) In contract, over asilica-supported copper oxide catalyst, first-order of CO andzero-order of oxygen concentrations were observed and Eley-Rideal mechanism was proposed.4) In addition, a power-lawrate equation was found to satisfactorily fit the experimentaldata of carbon monoxide oxidation with CO at partialpressure ranging from 0.0015 to 0.0125 atm over CuOsupported on nanosized CeO2.5) Kinetics of CO oxidation inthe CO-PROX process (H2-rich gases) has been investigatedin a fixed-bed reactor by Caputo et al.6) and a power-law rateequation was found. On the basis of Langmuir-Hinshelwoodmechanism, the following expression for the reaction rate ofCO oxidation on CuO/£-Al2O3 was proposed by Vanniceet al.:7)
r ¼ kKCOPCO
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiKO2
PO2
p
ð1þKCOPCO þ ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiKO2
PO2
p Þ2 ð1Þ
where PCO, PO2are the partial pressures of CO and O2; KCO
and KO2-adsorption coefficients for CO and O species,
respectively. However, order of O2 pressure was found to benear zero and the equation became power-law kineticexpressions:7)
r ¼ kðKCOPCOÞ0:7 ð2ÞIn our previous publications,8,9) oxide catalysts 10mass%CuO/£-Al2O3 (Cu/Al), 10mass% CuO/20mass% CeO2 +£-Al2O3 (Cu/CeAl) have been reported to be the mostactive and stable catalysts in the complete oxidation of CO.Furthermore, high active catalysts at low temperature reactionwas obtained when Pt of 0.1mass% was introduced to CuOcatalysts.10) The kinetics of deep oxidation of CO8) andp-xylene and its mixtures with CO9) over Cu/Al and Cu/CeAl have been investigated at the temperature range of 473543K. The following rate equations were achieved for deepoxidation of sole CO:8)
rCO ¼ kCOPCOP0:5O2
ð1þ k1P0:5O2
þ k3PCO2Þ ð3Þ
and p-xylene:9)
rxyl ¼kxylPxylP
0:5O2
ðP0:5O2
þ k3PCO2þ k4Pxyl þ k5PH2OÞ
ð4Þ
It has been revealed that a complicated mutual effectassociated with the formation of new intermediates takesplace in the simultaneous oxidation of CO and p-xylene tochange the reaction kinetics. The following rate equationswere obtained for deep oxidation of CO and p-xylene in theirmixture on the Cu/CeAl catalyst:9)
r�i ¼k�i PiP
0:5O2
ð1þ k�1P0:5O2
þ k�2PCO þ k�3PCO2þ k�4Pxyl þ k�5PH2OÞ
� k��i PCOPxyl
ð1þ k�2PCOÞð1þ k�6PCOPxylÞð5Þ
where ri* reaction rate of CO or p-xylene oxidation intheir mixture; ki*, ki** constants; Pi partial pressure of icomponents; i-CO or p-xylene.
The aim of this study is to establish the kinetics of COoxidation using Pt-modified CuO catalysts.
+Corresponding author, E-mail: [email protected]
Materials Transactions, Vol. 56, No. 9 (2015) pp. 1403 to 1407Special Issue on Nanostructured Functional Materials and Their Applications©2015 The Japan Institute of Metals and Materials
2. Experimental Procedure
Two catalysts: 0.1mass% Pt + 10mass% CuO/£-Al2O3
(PtCu/Al) and 0.1mass% Pt + 10mass% CuO/(20mass%CeO2 + 69.9mass% £-Al2O3) (PtCu/CeAl) have been pre-pared by sequential impregnations as described in ourprevious works.10,12) Catalyst 0.1mass% Pt + 7.5mass%CuO/CeO2 (PtCu/Ce) was prepared by the urea nitratescombustion method described by H. Matralis et al.11)
with molar ratios of urea/nitrate = 4.17. Ce(NO3)3·6H2O,Cu(NO3)2·3H2O, H2PtCl6·6H2O complex, urea (CO(NH2)2)and £-Al2O3 were purchased from Merck. All of theprecursors were used without further purification. The IRspectra were recorded from apparatus Nicolet Spectrometer460 in the range of 4000400 cm¹1 with a resolution of4 cm¹1. The catalyst samples were pretreated in a pureoxygen flow of velocity 5 L/h for 1 h at 873K for metaloxide catalyst and at 573K for Pt containing sample. Thekinetics of CO oxidation was studied in a gradientless flow-circulating system at 398498K. Ranges of initial partialpressures of CO, O2 and CO2 were 2.520, 35140 and 025(hPa), respectively. The follow gas have been used: O2
(99.999%); N2 (99.999%); Air (21mol% O2 + 79mol% N2);mixture CO (6mol%) + N2 (94mol%); and mixture CO2
(6mol%) + N2 (94mol%).
3. Results and Discussions
3.1 Physico-chemical characteristics and activity of theobtained catalysts
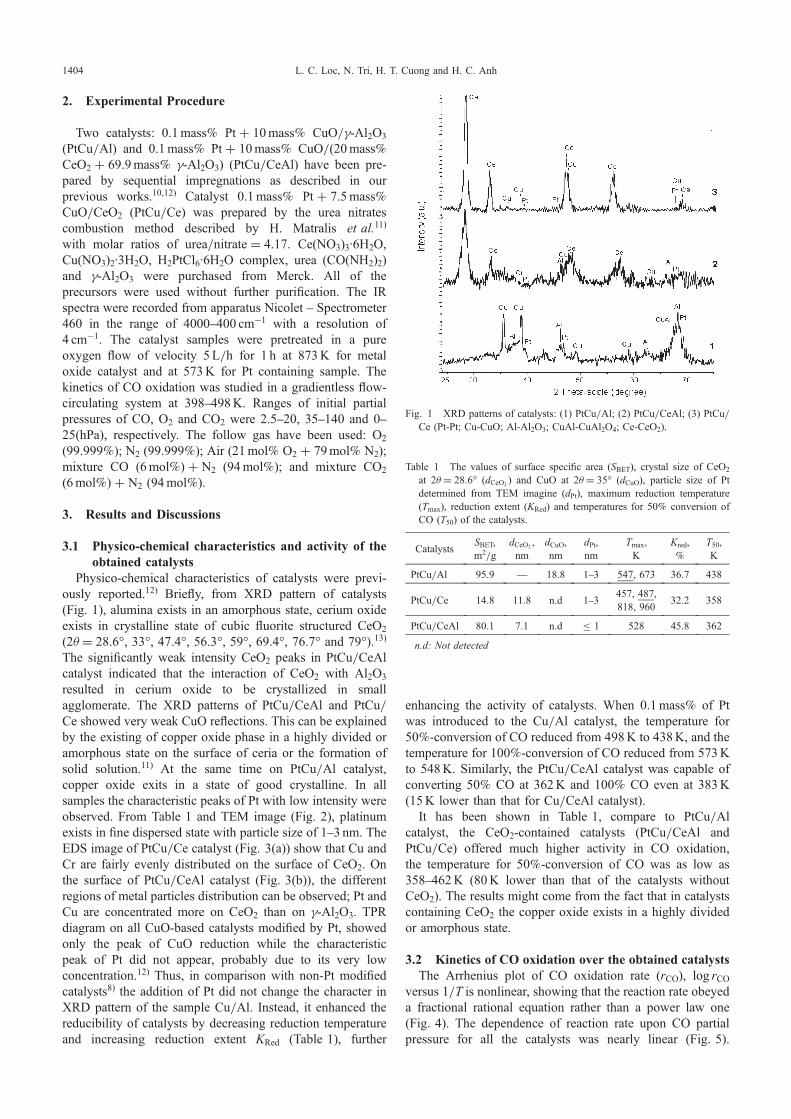
Physico-chemical characteristics of catalysts were previ-ously reported.12) Briefly, from XRD pattern of catalysts(Fig. 1), alumina exists in an amorphous state, cerium oxideexists in crystalline state of cubic fluorite structured CeO2
(2ª = 28.6°, 33°, 47.4°, 56.3°, 59°, 69.4°, 76.7° and 79°).13)
The significantly weak intensity CeO2 peaks in PtCu/CeAlcatalyst indicated that the interaction of CeO2 with Al2O3
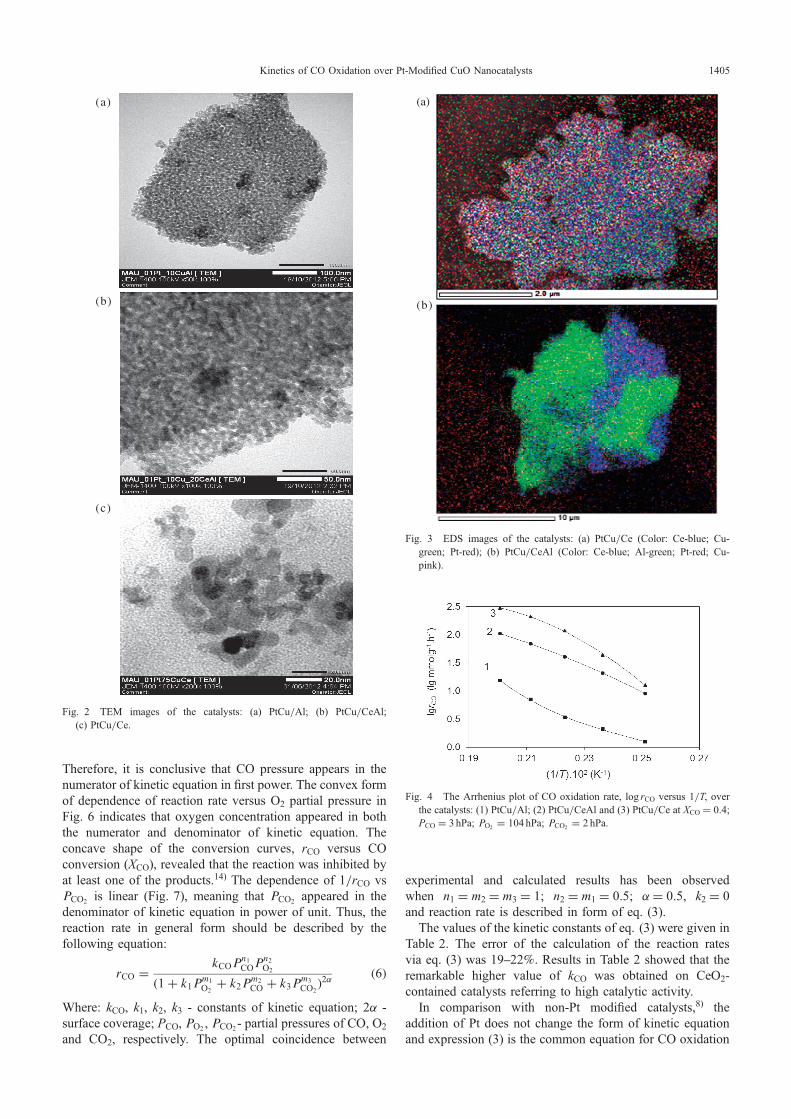
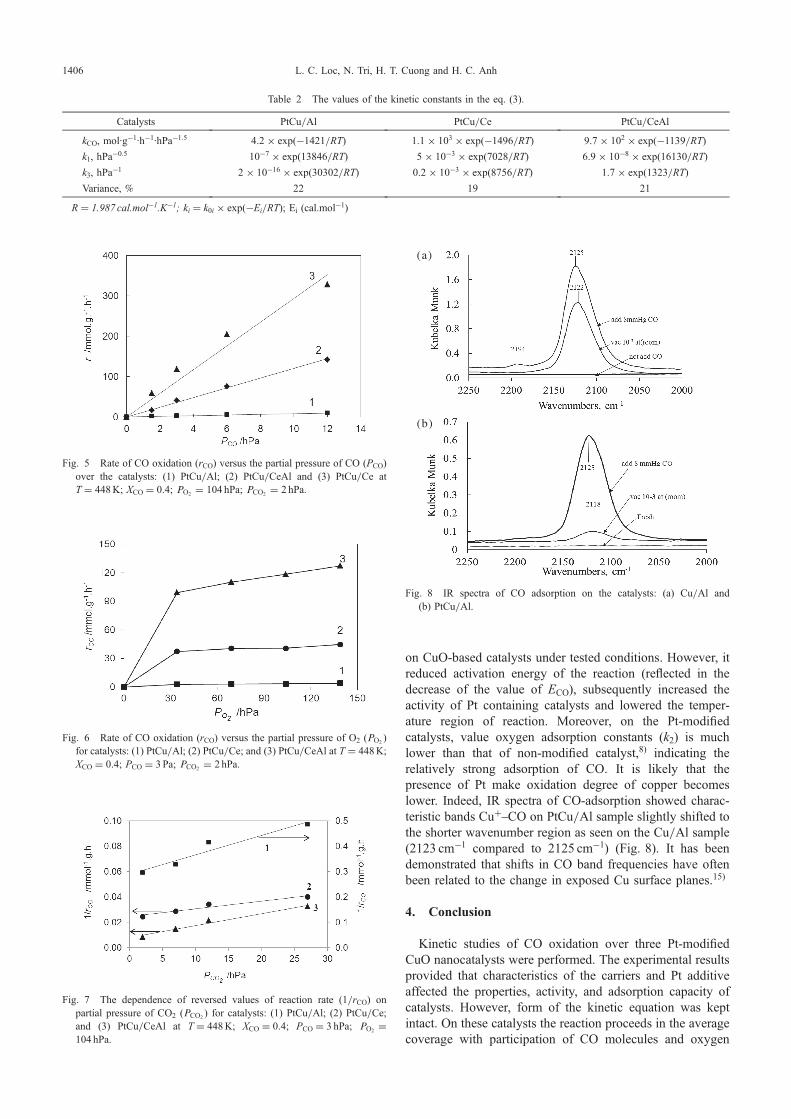
resulted in cerium oxide to be crystallized in smallagglomerate. The XRD patterns of PtCu/CeAl and PtCu/Ce showed very weak CuO reflections. This can be explainedby the existing of copper oxide phase in a highly divided oramorphous state on the surface of ceria or the formation ofsolid solution.11) At the same time on PtCu/Al catalyst,copper oxide exits in a state of good crystalline. In allsamples the characteristic peaks of Pt with low intensity wereobserved. From Table 1 and TEM image (Fig. 2), platinumexists in fine dispersed state with particle size of 13 nm. TheEDS image of PtCu/Ce catalyst (Fig. 3(a)) show that Cu andCr are fairly evenly distributed on the surface of CeO2. Onthe surface of PtCu/CeAl catalyst (Fig. 3(b)), the differentregions of metal particles distribution can be observed; Pt andCu are concentrated more on CeO2 than on £-Al2O3. TPRdiagram on all CuO-based catalysts modified by Pt, showedonly the peak of CuO reduction while the characteristicpeak of Pt did not appear, probably due to its very lowconcentration.12) Thus, in comparison with non-Pt modifiedcatalysts8) the addition of Pt did not change the character inXRD pattern of the sample Cu/Al. Instead, it enhanced thereducibility of catalysts by decreasing reduction temperatureand increasing reduction extent KRed (Table 1), further
enhancing the activity of catalysts. When 0.1mass% of Ptwas introduced to the Cu/Al catalyst, the temperature for50%-conversion of CO reduced from 498K to 438K, and thetemperature for 100%-conversion of CO reduced from 573Kto 548K. Similarly, the PtCu/CeAl catalyst was capable ofconverting 50% CO at 362K and 100% CO even at 383K(15K lower than that for Cu/CeAl catalyst).
It has been shown in Table 1, compare to PtCu/Alcatalyst, the CeO2-contained catalysts (PtCu/CeAl andPtCu/Ce) offered much higher activity in CO oxidation,the temperature for 50%-conversion of CO was as low as358462K (80K lower than that of the catalysts withoutCeO2). The results might come from the fact that in catalystscontaining CeO2 the copper oxide exists in a highly dividedor amorphous state.
3.2 Kinetics of CO oxidation over the obtained catalystsThe Arrhenius plot of CO oxidation rate (rCO), log rCO
versus 1/T is nonlinear, showing that the reaction rate obeyeda fractional rational equation rather than a power law one(Fig. 4). The dependence of reaction rate upon CO partialpressure for all the catalysts was nearly linear (Fig. 5).
Fig. 1 XRD patterns of catalysts: (1) PtCu/Al; (2) PtCu/CeAl; (3) PtCu/Ce (Pt-Pt; Cu-CuO; Al-Al2O3; CuAl-CuAl2O4; Ce-CeO2).
Table 1 The values of surface specific area (SBET), crystal size of CeO2
at 2ª = 28.6° (dCeO2) and CuO at 2ª = 35° (dCuO), particle size of Pt
determined from TEM imagine (dPt), maximum reduction temperature(Tmax), reduction extent (KRed) and temperatures for 50% conversion ofCO (T50) of the catalysts.
CatalystsSBET,m2/g
dCeO2,
nmdCuO,nm
dPt,nm
Tmax,K
Kred,%
T50,K
PtCu/Al 95.9 ® 18.8 13 547, 673 36.7 438
PtCu/Ce 14.8 11.8 n.d 13457, 487,818, 960
32.2 358
PtCu/CeAl 80.1 7.1 n.d ¯ 1 528 45.8 362
n.d: Not detected
L. C. Loc, N. Tri, H. T. Cuong and H. C. Anh1404
Therefore, it is conclusive that CO pressure appears in thenumerator of kinetic equation in first power. The convex formof dependence of reaction rate versus O2 partial pressure inFig. 6 indicates that oxygen concentration appeared in boththe numerator and denominator of kinetic equation. Theconcave shape of the conversion curves, rCO versus COconversion (XCO), revealed that the reaction was inhibited byat least one of the products.14) The dependence of 1/rCO vsPCO2
is linear (Fig. 7), meaning that PCO2appeared in the
denominator of kinetic equation in power of unit. Thus, thereaction rate in general form should be described by thefollowing equation:
rCO ¼ kCOPn1COP
n2O2
ð1þ k1Pm1
O2þ k2P
m2
CO þ k3Pm3
CO2Þ2¡ ð6Þ
Where: kCO, k1, k2, k3 - constants of kinetic equation; 2¡ -surface coverage; PCO, PO2
, PCO2- partial pressures of CO, O2
and CO2, respectively. The optimal coincidence between
experimental and calculated results has been observedwhen n1 = m2 = m3 = 1; n2 = m1 = 0.5; ¡ = 0.5, k2 = 0and reaction rate is described in form of eq. (3).
The values of the kinetic constants of eq. (3) were given inTable 2. The error of the calculation of the reaction ratesvia eq. (3) was 1922%. Results in Table 2 showed that theremarkable higher value of kCO was obtained on CeO2-contained catalysts referring to high catalytic activity.
In comparison with non-Pt modified catalysts,8) theaddition of Pt does not change the form of kinetic equationand expression (3) is the common equation for CO oxidation
(a)
(b)
(c)
Fig. 2 TEM images of the catalysts: (a) PtCu/Al; (b) PtCu/CeAl;(c) PtCu/Ce.
Fig. 4 The Arrhenius plot of CO oxidation rate, log rCO versus 1/T, overthe catalysts: (1) PtCu/Al; (2) PtCu/CeAl and (3) PtCu/Ce at XCO = 0.4;PCO = 3 hPa; PO2
¼ 104 hPa; PCO2¼ 2 hPa.
(a)
(b)
Fig. 3 EDS images of the catalysts: (a) PtCu/Ce (Color: Ce-blue; Cu-green; Pt-red); (b) PtCu/CeAl (Color: Ce-blue; Al-green; Pt-red; Cu-pink).
Kinetics of CO Oxidation over Pt-Modified CuO Nanocatalysts 1405
on CuO-based catalysts under tested conditions. However, itreduced activation energy of the reaction (reflected in thedecrease of the value of ECO), subsequently increased theactivity of Pt containing catalysts and lowered the temper-ature region of reaction. Moreover, on the Pt-modifiedcatalysts, value oxygen adsorption constants (k2) is muchlower than that of non-modified catalyst,8) indicating therelatively strong adsorption of CO. It is likely that thepresence of Pt make oxidation degree of copper becomeslower. Indeed, IR spectra of CO-adsorption showed charac-teristic bands Cu+CO on PtCu/Al sample slightly shifted tothe shorter wavenumber region as seen on the Cu/Al sample(2123 cm¹1 compared to 2125 cm¹1) (Fig. 8). It has beendemonstrated that shifts in CO band frequencies have oftenbeen related to the change in exposed Cu surface planes.15)
4. Conclusion
Kinetic studies of CO oxidation over three Pt-modifiedCuO nanocatalysts were performed. The experimental resultsprovided that characteristics of the carriers and Pt additiveaffected the properties, activity, and adsorption capacity ofcatalysts. However, form of the kinetic equation was keptintact. On these catalysts the reaction proceeds in the averagecoverage with participation of CO molecules and oxygen
Fig. 5 Rate of CO oxidation (rCO) versus the partial pressure of CO (PCO)over the catalysts: (1) PtCu/Al; (2) PtCu/CeAl and (3) PtCu/Ce atT = 448K; XCO = 0.4; PO2
¼ 104 hPa; PCO2¼ 2 hPa.
Fig. 6 Rate of CO oxidation (rCO) versus the partial pressure of O2 (PO2)
for catalysts: (1) PtCu/Al; (2) PtCu/Ce; and (3) PtCu/CeAl at T = 448K;XCO = 0.4; PCO = 3Pa; PCO2
¼ 2 hPa.
Fig. 7 The dependence of reversed values of reaction rate (1/rCO) onpartial pressure of CO2 (PCO2
) for catalysts: (1) PtCu/Al; (2) PtCu/Ce;and (3) PtCu/CeAl at T = 448K; XCO = 0.4; PCO = 3 hPa; PO2
¼104 hPa.
Table 2 The values of the kinetic constants in the eq. (3).
Catalysts PtCu/Al PtCu/Ce PtCu/CeAl
kCO, mol·g¹1·h¹1·hPa¹1.5 4.2 © exp(¹1421/RT) 1.1 © 103 © exp(¹1496/RT) 9.7 © 102 © exp(¹1139/RT)
k1, hPa¹0.5 10¹7 © exp(13846/RT) 5 © 10¹3 © exp(7028/RT) 6.9 © 10¹8 © exp(16130/RT)
k3, hPa¹1 2 © 10¹16 © exp(30302/RT) 0.2 © 10¹3 © exp(8756/RT) 1.7 © exp(1323/RT)
Variance, % 22 19 21
R = 1.987 cal.mol¹1.K¹1; ki = k0i © exp(¹Ei/RT); Ei (cal.mol¹1)
(a)
(b)
Fig. 8 IR spectra of CO adsorption on the catalysts: (a) Cu/Al and(b) PtCu/Al.
L. C. Loc, N. Tri, H. T. Cuong and H. C. Anh1406

atoms. Furthermore, CeO2 depressed the formation ofmassive CuO leading to the increase in catalyst reductionand reaction rate. Addition of 0.1mass% Pt decreased theactivation energy of the reaction and increased COadsorption, leading increased the activity of CuO-basedcatalysts.
Acknowledgments
This work was supported by the Vietnam NationalFoundation for Science and Technology Development(NAFOSTED) under grand No. 104.03-2012.60.
REFERENCES
1) C. R. Jung, A. Kundu, S. W. Nam and H.-I. Lee: Appl. Catal. A 331(2007) 112120.
2) M. Ferrandon: Ph.D. Dissertation, Royal Institute of Technology,Stockholm, (2001).
3) W. E. Garner, F. S. Stone and P. F. Tiley: Proc. R. Soc. A 221 (1952)472489.
4) R. A. Prokopowicz, P. L. Silveston, R. R. Hudgins and D. E. Irish:React. Kinet. Catal. Lett. 37 (1988) 6370.
5) J. L. Ayastuy, A. Gurbani, M. P. González-Marcos and M. A. Gutiérrez-Ortiz: Ind. Eng. Chem. Res. 48 (2009) 56335641.
6) T. Caputo, L. Lisi, R. Pirone and G. Russo: Ind. Eng. Chem. Res. 46(2007) 67936800.
7) K. I. Choi and M. A. Vannice: J. Catal. 131 (1991) 2235.8) L. C. Loc, H. T. Cuong, N. Tri and H. S. Thoang: J. Exper. Nanosci. 6
(2011) 631640.9) L. C. Loc, N. Tri, H. T. Cuong, H. S. Thoang, Y. A. Agafonov, N. A.
Gaidai, N. V. Nekrasov and A. L. Lapidus: Kinet. Catal. 55 (2014) 611619.
10) L. C. Loc, D. T. T. Mai, N. Tri, H. T. Cuong, B. T. Huong and H. S.Thoang: J. Chem. 48 (2010) 8489 (in Vietnamese).
11) G. Avgouropoulos, T. Ioannides and H. Matralis: App. Catal. B: Env.56 (2005) 8793.
12) L. C. Loc, N. Tri, H. T. Cuong, H. M. Nam and H. C. Anh: Adv. Nat.Sci.: Nanosci. Nanotechnol. 6 (2015) doi:10.1088/2043-6262/6/1/015011.
13) H. I. Chen and H. Y. Chang: Solid State Commun. 133 (2005) 593598.
14) S. G. Bashkirova and S. L. Kiperman: Kinet. i Katal. 11 (1970) 631637 (in Russian).
15) N. Y. Topsoe and H. Topsoe: Topics Catal. 8 (1999) 267270.
Kinetics of CO Oxidation over Pt-Modified CuO Nanocatalysts 1407

















![Carbon Monoxide and Formic Acid Oxidation at Rh@Pt … · 2019. 12. 19. · Rh@Pt NP is only available for a 1 ML shell [15], and formic acid oxidation has only been reported for](https://img.pdfslide.us/doc/110x75/6027ce9e56f0065e972ce381/carbon-monoxide-and-formic-acid-oxidation-at-rhpt-2019-12-19-rhpt-np-is-only.jpg)











