Embed Size (px)
DESCRIPTION
sel
Citation preview

KELAINAN SELKelainan Retrogresif, Progresif,
Degenerasi, Nekrosis, Apoptosis, dan Onkogenesis
Bagian Patologi Anatomi
Fakultas Kedokteran Universitas Islam Sumatera Utara
2010 /2011Dr. H. Soekimin, SpPADr. T. Ibnu Alferraly, SpPA

CELL INJURY, DEATH, AND ADAPTATION
DefinitonsPathology is a dicipline bridging clinical practiceand basic sience
To render diagnosis and guide therapy- Identity changes in gross- Morphology ( microscopy ) appearance of cell tissues
The scientific focus of pathology is :Etiology : on the cause of diseasePathogenesis : mechanisme of its development and the pathways by which morphologic changes occur

Normal homeostasis if cell adjusting structure and function to accommodate changing demands andextracellular stresses
Stresses or pathologic stimuli the cell can :- undergo adaptation
eg.: atrophy, hypertrophy, hyperplasia andmetaplasia
- irreversible injury and ultimately dies

Two principal pattern of cell death :
1.NECROSIS, commonly coagulative necrosis
- cellular swelling- protein denaturation- organellar breakdown2. APOPTOSIS, - regulated event
- programmed death

CAUSES OF CELL INJURY1. Hypoxia :-Anemia-Ischemia-Intoxication CO2
-Aerobic oxidative respiration
2. Physical Agent :- mechanical trauma
- extreme temprature- radiation- electric shock- athmosphere pressure
3. Chemical and drugs : - sufficiently concentrated glucose, salt, O2

- air pollutants- insecticides- asbestosis- ethanol
4. Microbilogy Agents- tape worms- rickettsia- virus- bacteria- fungi
5. Immunologic Reaction- anaphylactic reaction- autoimmune diseases

6. Genetic Defects- congenital Malformation- sicle cell anemia
7. Nutritional Inbalance- protein calori insufficiency- vitamins defficiency- diabetes
8. Aging

Mechanism of Cell Injury• Cellular response to injurious stimuli
depens on the : - injury type - duration - severity
• Current Status : ~ nutrional
~ hormonal ~ adaptibility of the cell
• Intercellular systems
> cell membrane integrity> aerobic respiration> protein synthesis> integrity genetic apparatus

• Oxygen and oxygen derived free radicals: ischemic and hypoxic injury
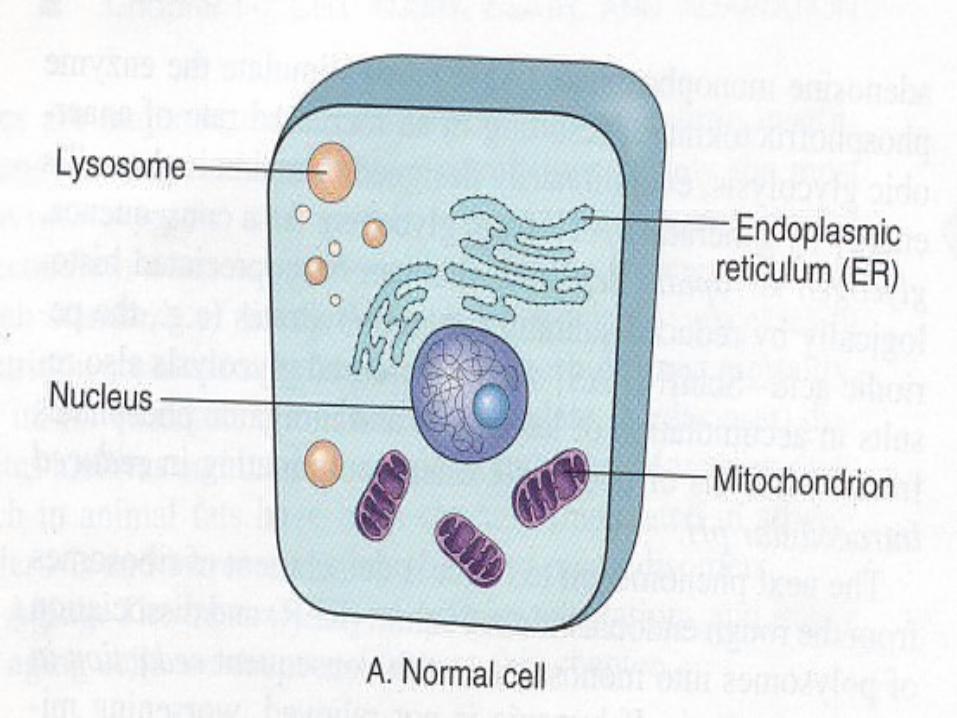
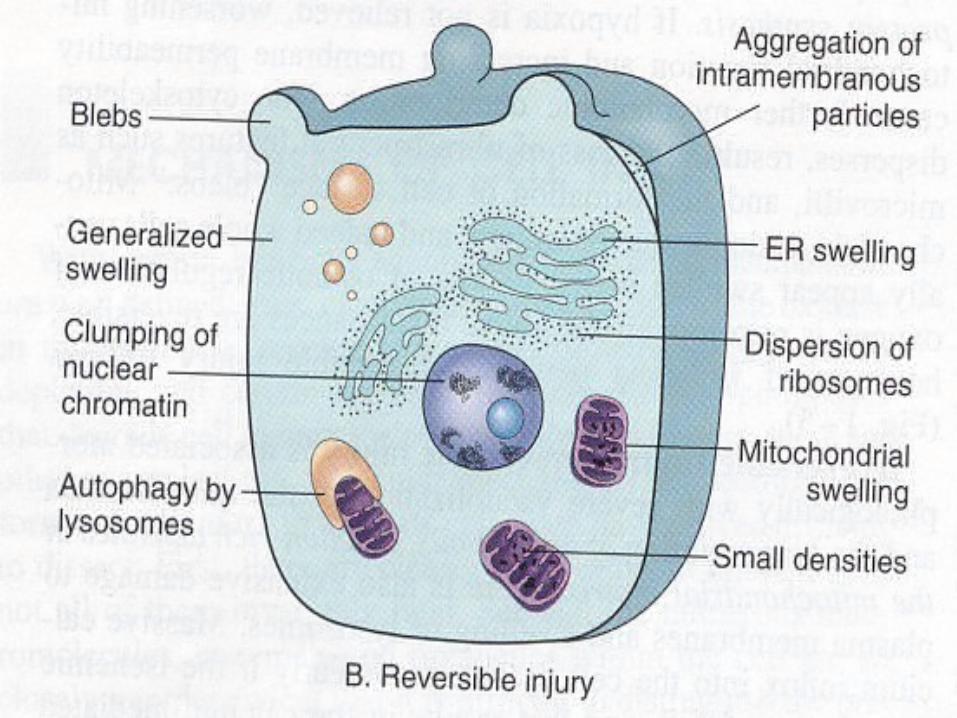
Reversible InjuryRediced oxidative phosphorylation in mitochondria.Activity Natrium Pump is reducedProducing cellular swelling. Loss of microvilli
Glycogen depleted Reduction in protein synthesis Formation of cell surface blebs

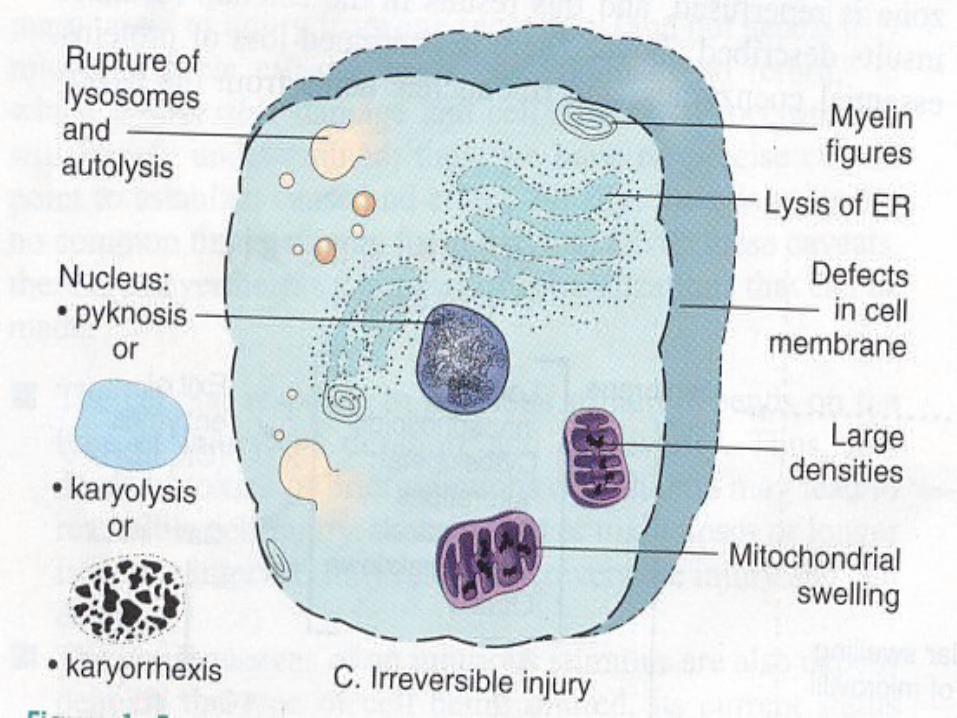
Irreversible Injury Severe vacuolization of the mitochondria Demage of the mitochondrial matrix
Demage of plasma membrane
Swelling of lysosomes
Accumulation of amorphous calcium
Rich dentities in mitochondrial matrix

Forms and Morphology of Cell Injury
1. Reversible acute cell injury
2. Necrosis ( Cell death after irreversible injury )
3. Cell death by suicide = Apoptosis
4. Subcellular alteration as a respond to chronic orpersistent injury stimuli
5. Intracellular accumulations of a number of sub-stances : lipid, carbohidrat, protein, as a result of dearangment
in cell metabolism or excessive storage.

Sublethal Damage
1. Recoverable – necrosis is not
2. Ultrastructural damage to mitochondria
3. Swelling of cellular organelles ( hydrophic deg.)
4. Fatty change is impairment of metabolism

NECROSIS
Refers to a sequence of morphologic changes that follow cell death in living tissue
1. Intense eosinophilia of the dead cell is due to loss of RNA and coagulation of protein.
2. Nuclei undergo phase of pyknosis, karyorhexis and karyolysis leaving a shrunken cell devoid of nucleus
3. Protein may be liberated from the dead cell

The morphologic appearance of necrosis is the result of two essentially processes :
1. Ensymic digestion of the cell2. Denaturation of protein
Autolysis is a dead cell themselves by hydrolitic enzymes
Heterolysis from the lysosomes of invading inflammatory cells

Morphologic Evidence of Necrosis
A Early Change1 – 3 hour before changes of necrosis are
recognizable on electron microscopy6 – 8 hour on light microscopy organelle
degenerationB. Nuclear Change Pyknosis : The chromatine clumps into coare strands
The nucleus becomes a shrunken dense, basophilic mass

Karyorrhexis : The pyknotic nucleus break up
into numerous small basophilic
massKaryolysis : The nucleus lysis as a result
of the action of lysosome deoxy
– ribonucleases
C. Cytoplasmic ChangeAbout 6 hour after cell necrosisCytoplasma becomes homogenous and
deeply` acidophilic. Enzymatic digestion
D. Biochemical ChangesActively transports calcium ions out of the
cell

Tipes of NecrosisDepends on :
1. Cells compotitions2. Speed of necrosis3. Type of injuries
> Coagulative Necrosis
Implies preservation of basic structural outline of the coagulated cell or tissue for a span of days. The structural protein and the enzymatic protein thus blocking cellular proteolysis Coagulation necrosis is cahareteristic of hypoxic death of cells in all tissue except the brain eq. Myocardial Infarction ( occlusion of arterial supply )

Liquefactive or Colliquativa Necrosis
Dead tissue that appears semi liquid as a result of dissolution of tissue by the action of hydrolytic enzymes eq.: - cerebral infarction
- necrosis caused by bacterial inf.
Caseous Necrosis
Dead cell form an amorphous proteinaceaus mass, no original architecture can be seen histologically ( soft and white resembling cream cheese ) Most often in fact of tuberculous infection with central necrosis

> Gumatous Necrosisdescribes dead tissue when it is firm and
rubberylike caseous necrosis in the spirochetal infectionsyphilis.
> Hemorrhagic Necrosisdescribes dead tissue that are suffused withextravasated red cell, when cell death is due toblockage
> Fat Necrosis does not really necrosis. It describes focal areas of fat destruction
tipically occuring following pancreatic injury.
Or after trauma to fat for example in the breast

Describes foci of hard yellow material seen indead adipose tissue
> Fibrinoid Necrosiswhen fibrin is deposited in damage necroticvessel walls in hypertension and vasculitis
> Gangreneextensive tissue necrosis ; is complicated to avariable degree by secondary bacterial
infection

A P O P T O S I Sis responsible for the programmed cell death inseveral important physiology processes
including : The programmed destruction of cells during embryo- genesis as in implantation, organogenesis, and developmental involution Hormon dependent physiologic involution such as the endometrium, lactating, as in the prostate after castration

Cell deletion in proliferating population such as intestinal crypt epithelium or cell dead in tumor
Deletion of autoreactive T cell in the thymus, cell death of cytokine starved lymphocytes

CELL DEATH APOPTOSIS vs. NECROSIS
What is DEATH? (What is LIFE?)
DEATH is IRREVERSIBLE

So the question is….
…NOT what is life or death, but what is REVERSIBLE or IRREVERSIBLE injury

REVERSIBLE CHANGESREDUCED oxidative phosphorylation
ATP depletion
Cellular “SWELLING”

IRREVERSIBLE CHANGESMITOCHONDRIAL IRREVERSIBILITY
IRREVERSIBLE MEMBRANE DEFECTS
LYSOSOMAL DIGESTION

REVERSIBLE = INJURY
IRREVERSIBLE = DEATH
SOME INJURIES CAN LEAD TO DEATH IF PROLONGED
and/or SEVERE enough

INJURY CAUSES (REVERSIBLE)
THE
USUAL
SUSPECTS
But…WHO are
the THREE
WORST?

INJURY CAUSES (REVERSIBLE)
Hypoxia, (decreased O2)
PHYSICAL Agents
CHEMICAL Agents
INFECTIOUS Agents
Immunologic
Genetic
Nutritional

INJURY MECHANISMS (REVERSIBLE)
DECREASED ATP
MITOCHONDRIAL DAMAGE
INCREASED INTRACELLULAR CALCIUM
INCREASED FREE RADICALS
INCREASED CELL MEMBRANE PERMEABILITY

What is Death?What is Life?
DEATH is IRREVERSIBLE MITOCHONDRIAL
DYSFUNCTION PROFOUND MEMBRANE DISTURBANCES
LIFE is……..???

REVERSIBLE IRREVERSIBLEDEATHEMLIGHT MICROSCOPYGROSS APPEARANCES
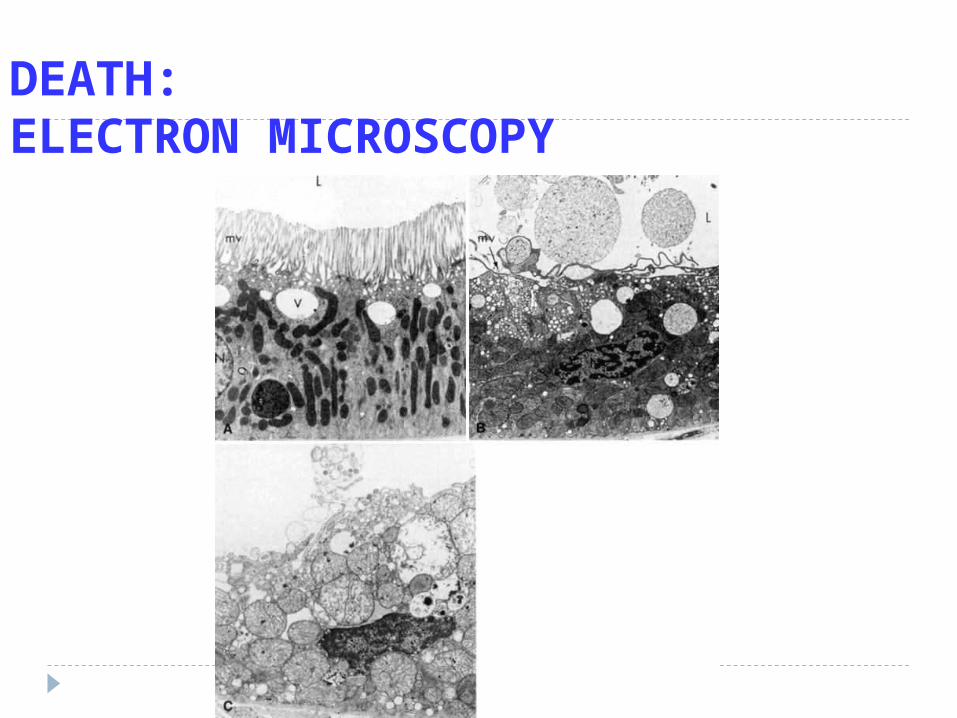
DEATH:ELECTRON MICROSCOPY
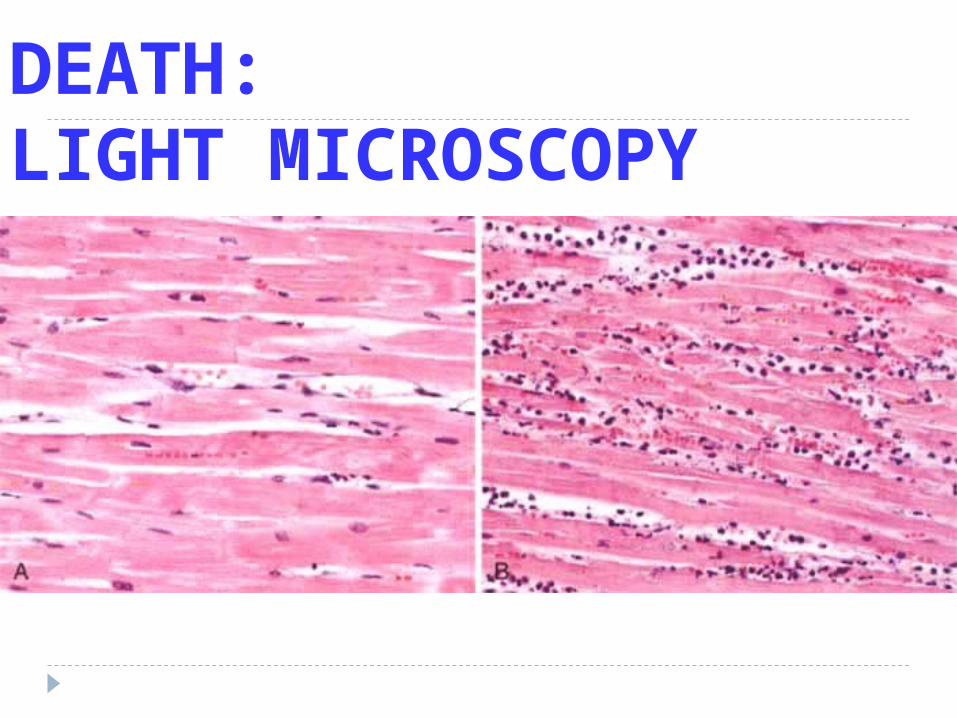
DEATH:LIGHT MICROSCOPY

NECROSIS BROTHERS: Liquefactive (Brain)
Gangrenous (Extremities, Bowel, non-specific) WET DRY
Fibrinoid (Rheumatoid, non-specific)
Caseous (cheese) (Tuberculosis)
Fat (Breast, any fat)
Ischemic (non-specific)
Avascular (aseptic), radiation, organ specific, papillary
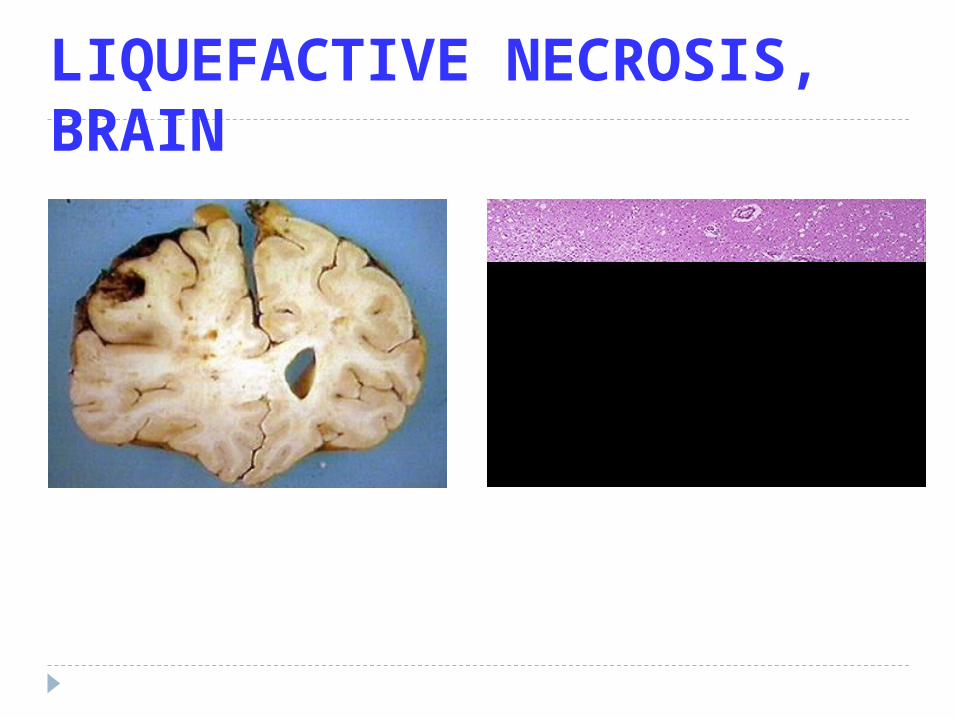
LIQUEFACTIVE NECROSIS, BRAIN
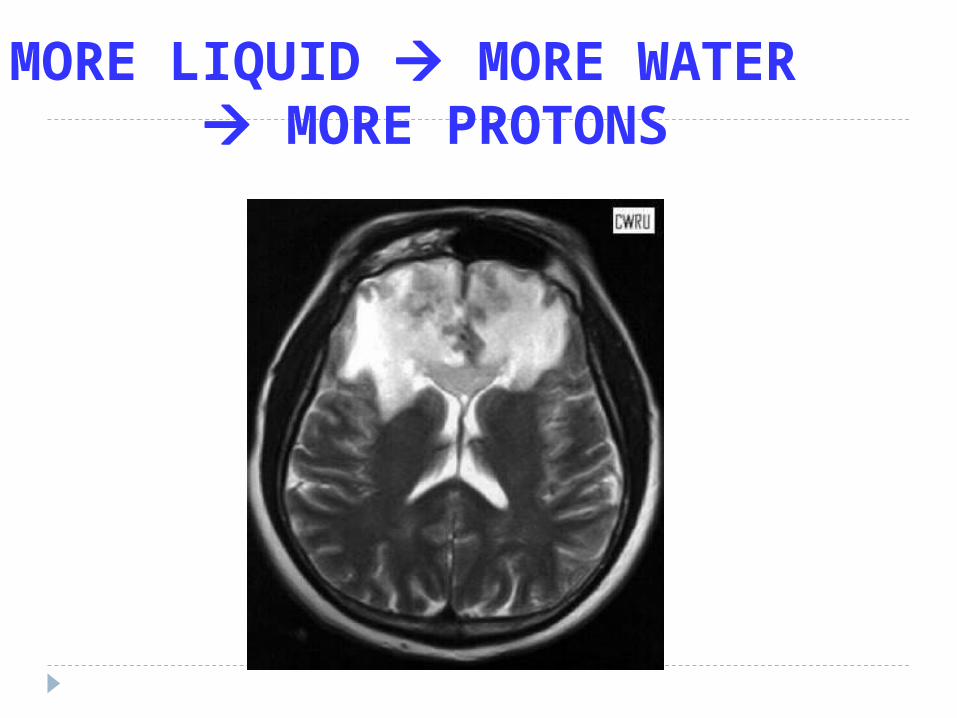
MORE LIQUID MORE WATER MORE PROTONS
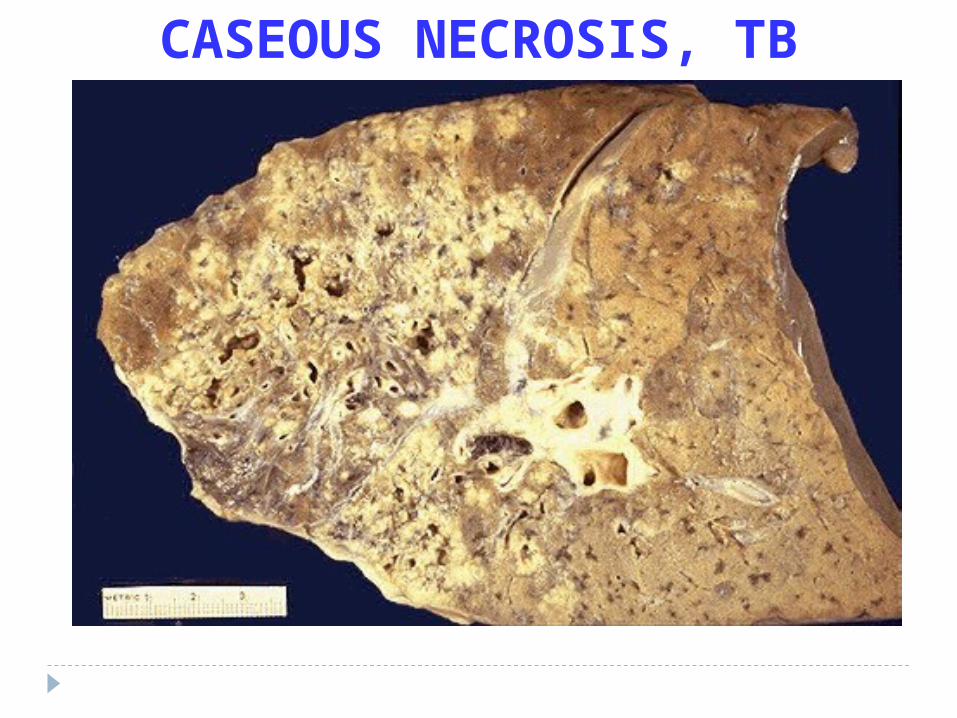
CASEOUS NECROSIS, TB
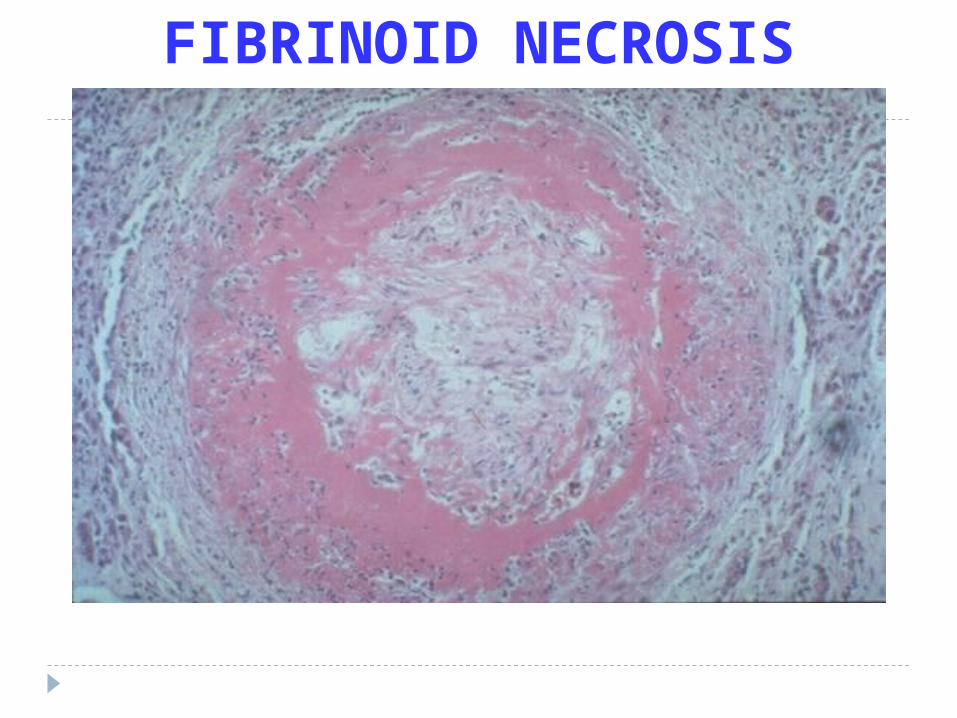
FIBRINOID NECROSIS
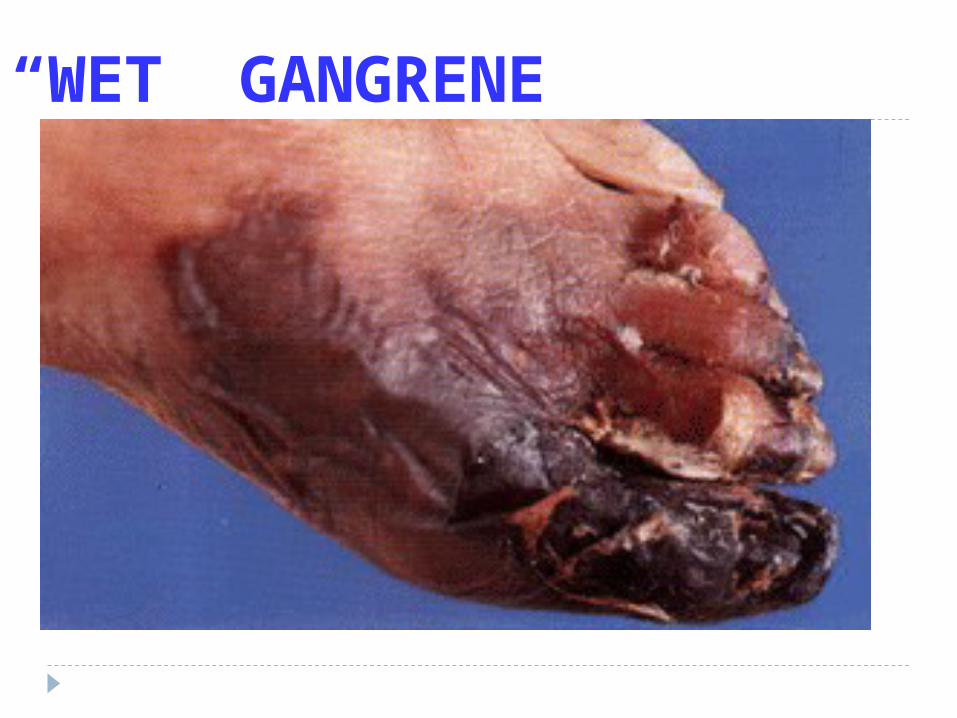
“WET” GANGRENE
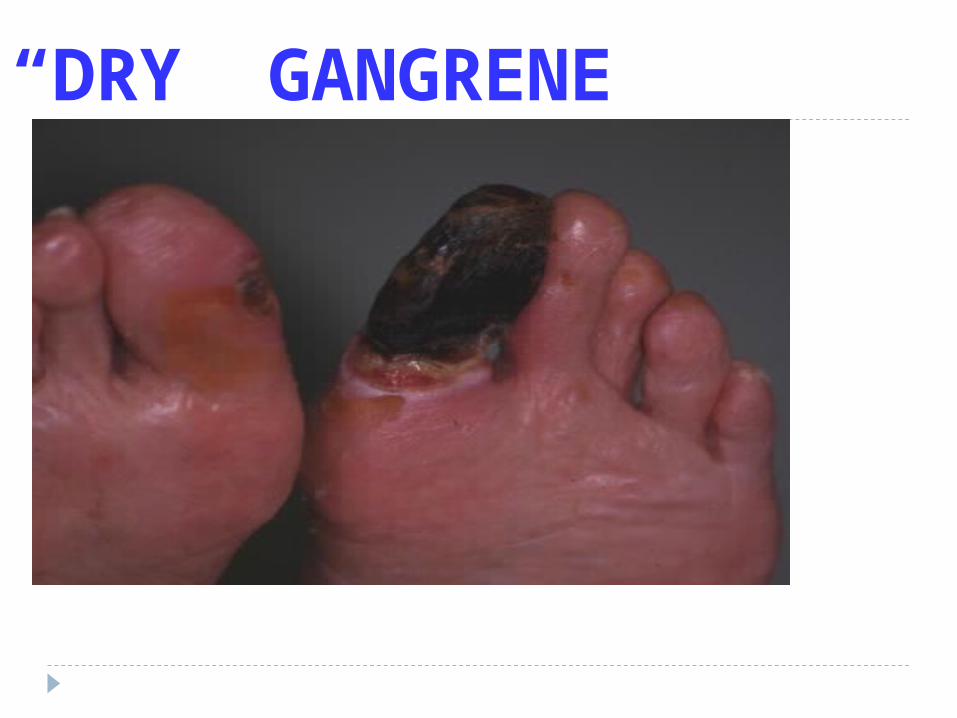
“DRY” GANGRENE

EXAMPLES of Cell INJURY/NECROSIS
Ischemic (Hypoxic)
Ischemia/Reperfusion
Chemical

ISCHEMIC INJURY
REVERSIBLE IRREVERSIBLE
DEATH (INFARCT)

ISCHEMIA/RE-PERFUSION INJURY
NEW Damage “Theory”

CHEMICAL INJURY“Toxic” Chemicals, e.g CCl4 Drugs, e.g tylenol
Dose Relationship
Free radicals, organelle, DNA damage

APOPTOSISNORMAL (preprogrammed)
PATHOLOGIC (associated with Necrosis)

“NORMAL” APOPTOSIS Embryogenesis
Hormonal “Involution”
Cell population control, e.g., “crypts”
Post Inflammatory “Clean-up”
Elimination of “HARMFUL” cells
Cytotoxic T-Cells cleaning up

“PATHOLOGIC” APOPTOSIS
“Toxic” effect on cells, e.g., chemicals, pathogens
Duct obstruction
Tumor cells
Apoptosis/Necrosis spectrum

APOPTOSIS MORPHOLOGYDE-crease in cell size, i.e., shrinkage
IN-crease in chromatin concentration, i.e., hyperchromasia, pyknosis karyorhexis karyolysis
IN-crease in membrane “blebs”
Phagocytosis
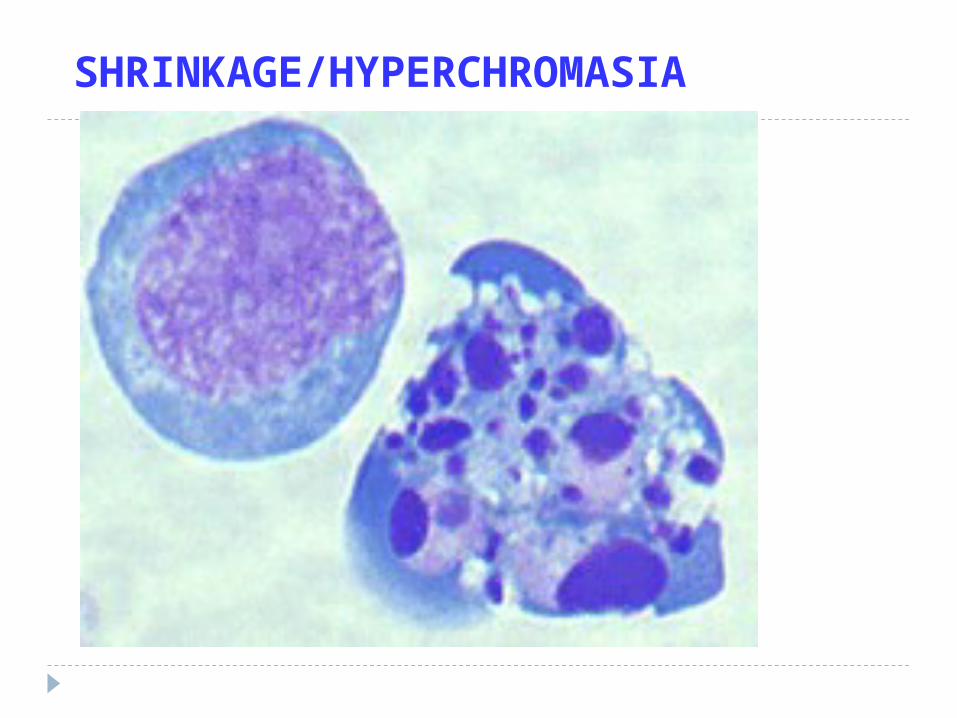
SHRINKAGE/HYPERCHROMASIA
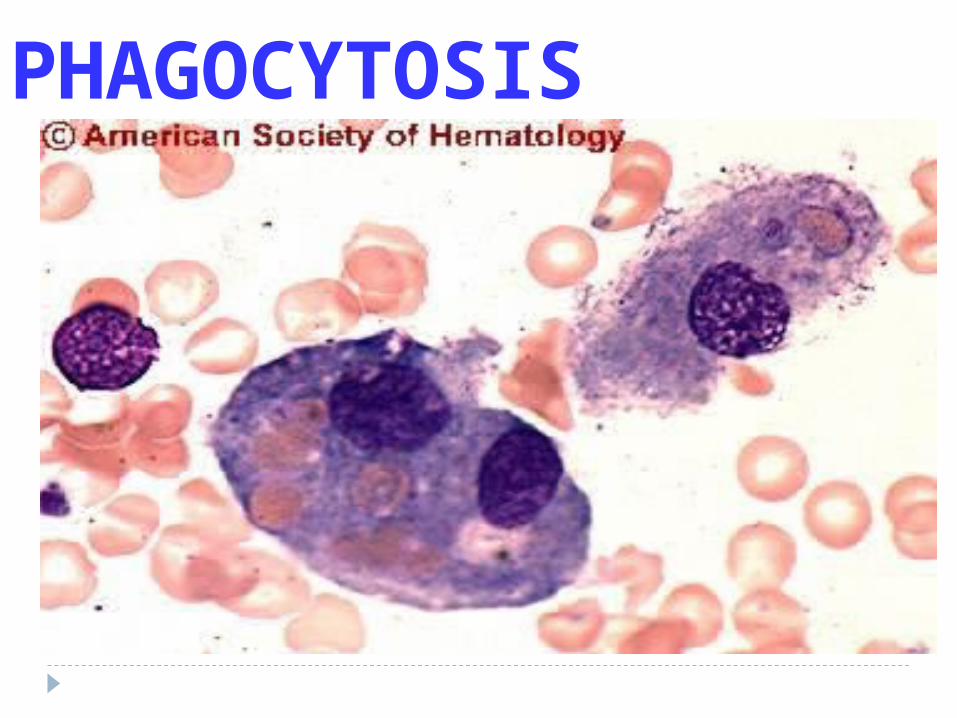
PHAGOCYTOSIS

APOPTOSIS BIOCHEMISTRYProtein Digestion (Caspases)
DNA breakdown
Phagocytic Recognition

SUB-Cellular Responses to Injury(APOPTOSIS/NECROSIS)
Lysosomal Auto-DigestionSmooth Endoplasmic Reticulum (SER) activation
Mitochondrial “SWELLING”Cytoskeleton Breakdown
Thin Filaments (actin, myosin) Microtubules Intermediate Filaments (keratin, desmin,
vimentin, neurofilaments, glial filaments)

INTRAcellular ACCUMULATIONS
Lipids Neutral Fat Cholesterol
“Hyaline” = any “proteinaceous” pink “glassy” substance
GlycogenPigments (EX-ogenous, END-ogenous)
Calcium

LIPID LAW
ALL Lipids are YELLOW grossly and WASHED out (CLEAR) microscopically
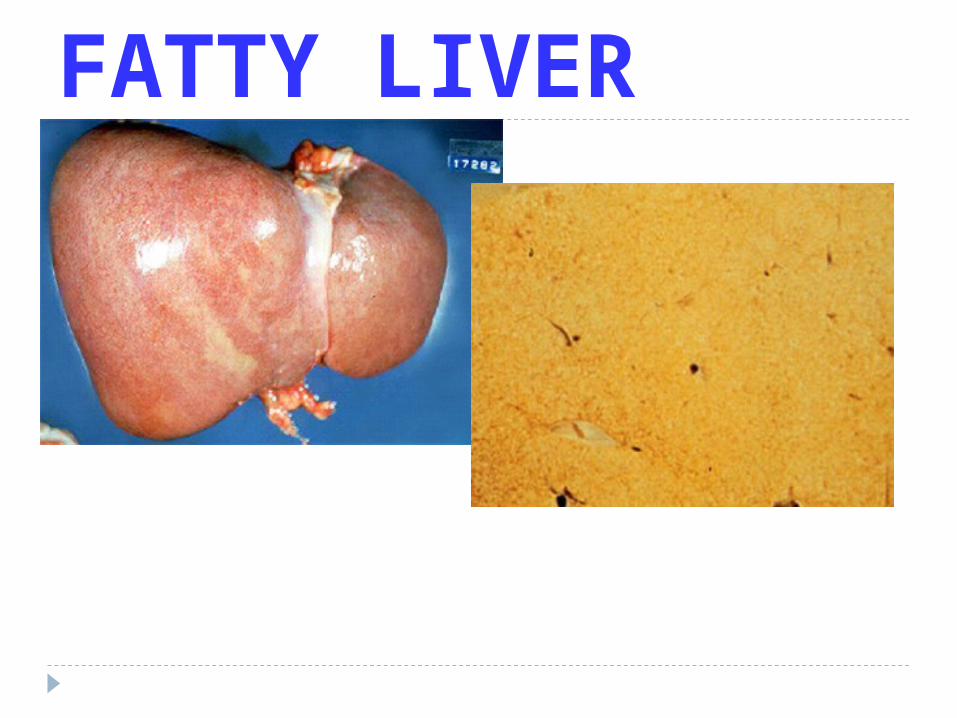
FATTY LIVER
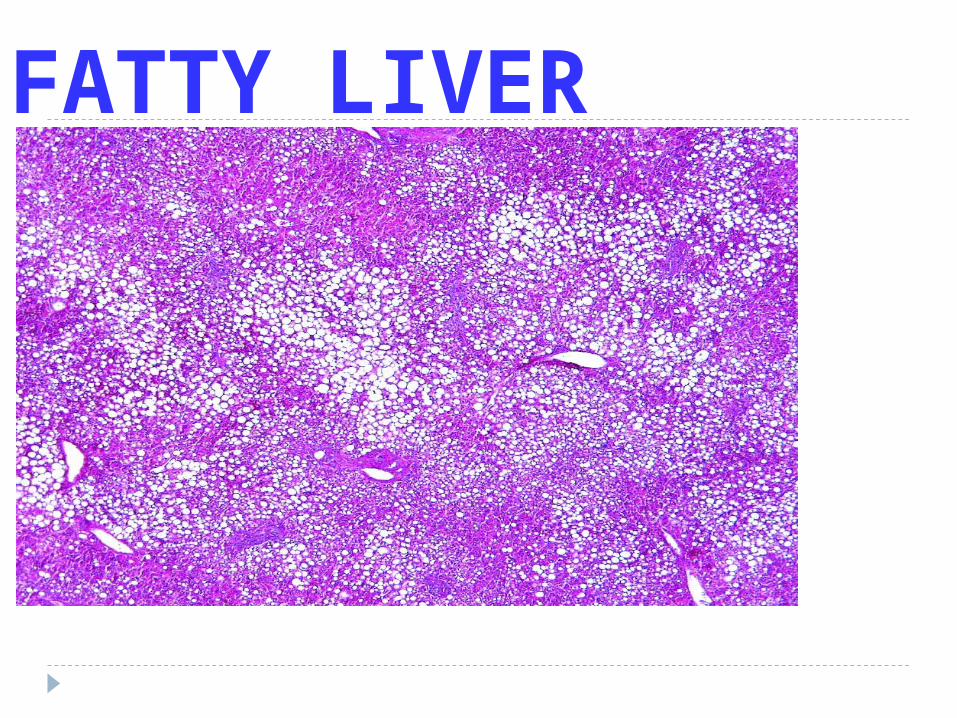
FATTY LIVER
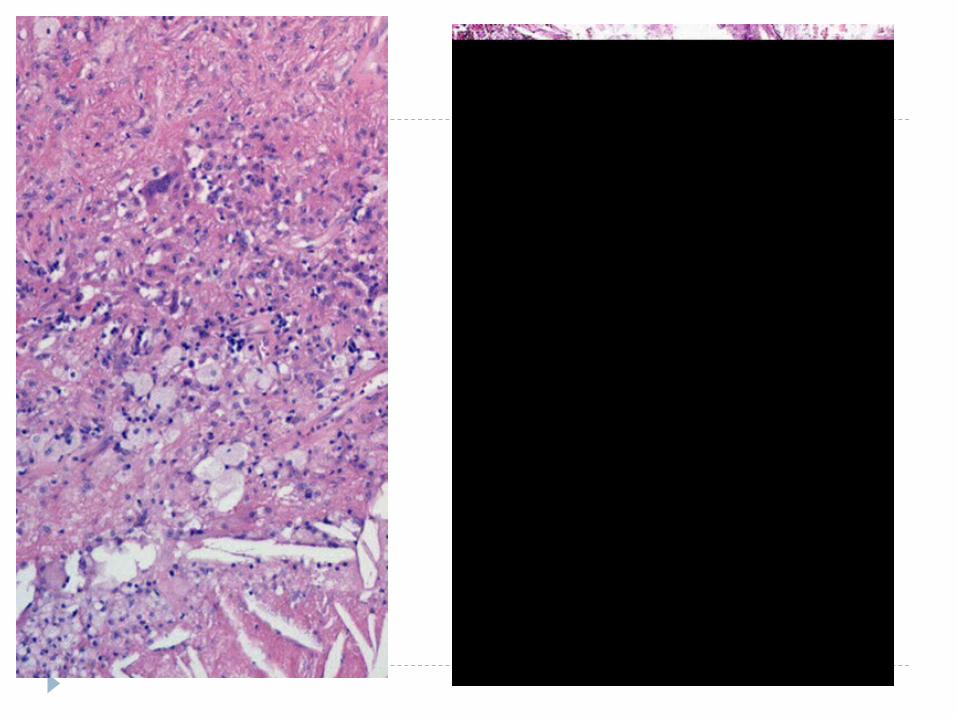

PIGMENTSEX-ogenous--- (tattoo, Anthracosis)
END-ogenous--- they all look the same, (e.g., hemosiderin, melanin, lipofucsin, bile), in that hey are all golden yellowish brown on “routine” Hematoxylin & Eosin (H&E) stains
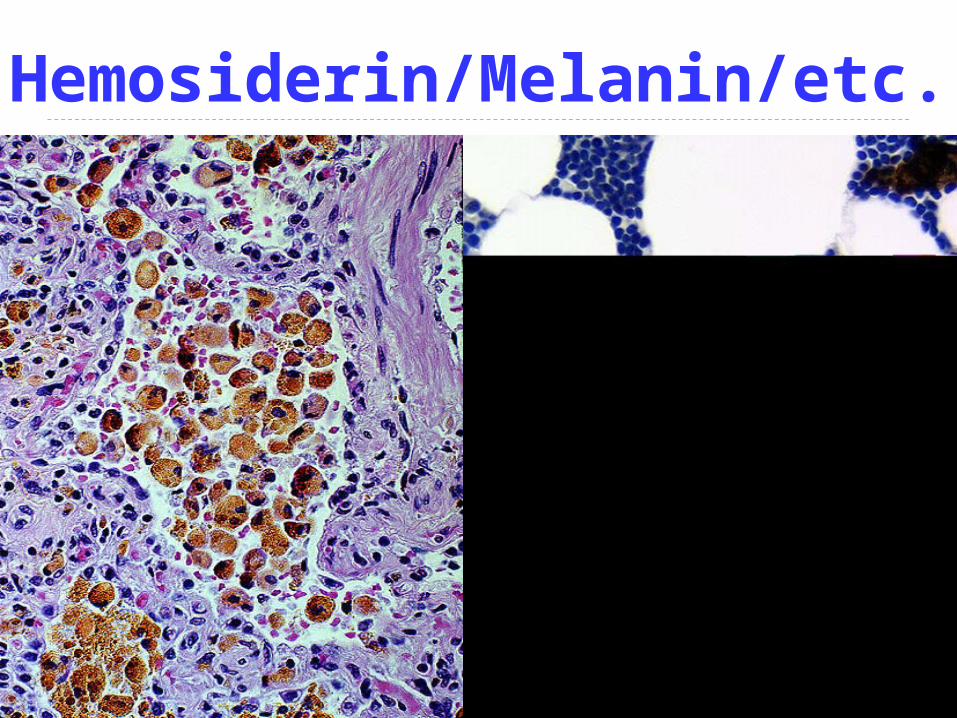
Hemosiderin/Melanin/etc.

CLINICAL EFFECTS OF NECROSIS Abnormal function
Kidney : renal failureCortex in brain : muscle paralysisHeart : heart failureLung : hemoptysis
Bacterial infection : gangrene
Realease of contens of necrotic cellsLiver : elevation SGOTHeart : creatine kinase
Systemic effectsFeverInflamatoir Reaction
Local effectsHemorrhageUlceration
CELLULAR ADAPTATIONS OF GROWTH ANDDIFFENTIATION
Environment adaptation of the cell1. Physiologic Adaptation
- Hormones- Endogenous chemical mediators
2. Pathologic Adaptation- Induction of new protein synthesis by target
cell
Cell Injury :• Death of cells ( permanent organ injury )• Sublethal injury ( adaptation )

Perubahan sel dan jaringan :
1. Agenesis 2. Aplasia3. Hypoplasia4. Atrophy5. Hypertrophy6. Hyperplasia7. Metaplasia8. Dysplasia9. Anaplasia10.Granuloma

The –plasia brothers HYPER- HYPO- (A-) NORMO- META-
DYS- ANA- Frank ANA-

Agenesis Complet absen of an organ Exc : - renal agenesis - ovarial agenesis - tubal agenesis, etc

Aplasia
Anlarge is present but never develops
Exc : lung aplasia with tissue containing rudimentary duct and connective tissue
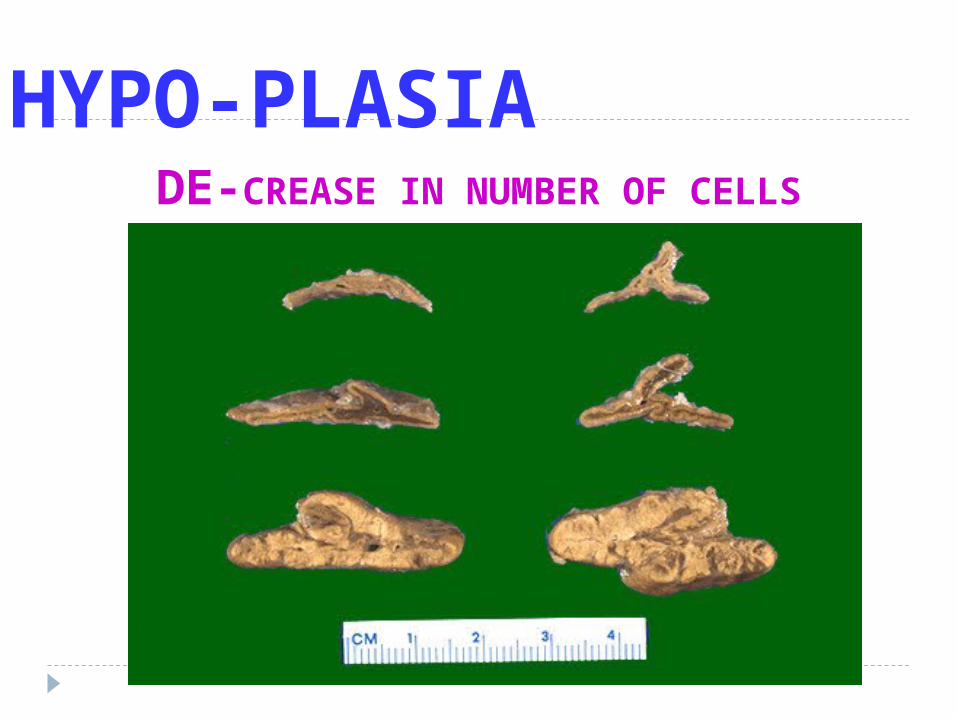
HYPO-PLASIADE-CREASE IN NUMBER OF CELLS

Hypoplasia Anlarge develoved incompletly but the
tissue histhologicaly normal
Exc : microcephaly

The –trophy brothers HYPER- HYPO- (A-)
DYS-

A-TROPHY DE-CREASE IN SIZE OF CELLS? YES
SHRINKAGE IN CELL SIZE DUE TO LOSS OF CELL
SUBSTANCE

ATROPHY
DECREASED WORKLOAD DENERVATION DECREASED BLOOD FLOW DECREASED NUTRITION AGING (involution) PRESSURE

A t r o p h y
is the decrease in the size and a function of a cell but are not dead
Causes of atrophy :

1. Reduced functional demanImmobilitation in fractureProlonged bed rest
2. Inadequate supply of oxygenIschemia
3. Insufficient nutrientsStarvationInadequate nutritionsChronic disease
4. Interrruption of trophic signalsThe functions of many cells depend on signaltransmitted by chemical mediators

-Endocrin system-Neuromucculator transmission eq. Thyroid
Adrenal cortexOvariumTestis
5. Persistant cell injuryCaused by chrinic inflamationeq. - chronic gastritis
- prolonged pressure
6. AgingParticularly in non replacating cells such brain,heart.> Senile Atrophy

The mechanism of atrophy :
• decreased synthesis
• increased catabolism
• influenced by a number of hormones eq. Insulin, tyroid stimulating hormon, glucocorticoids
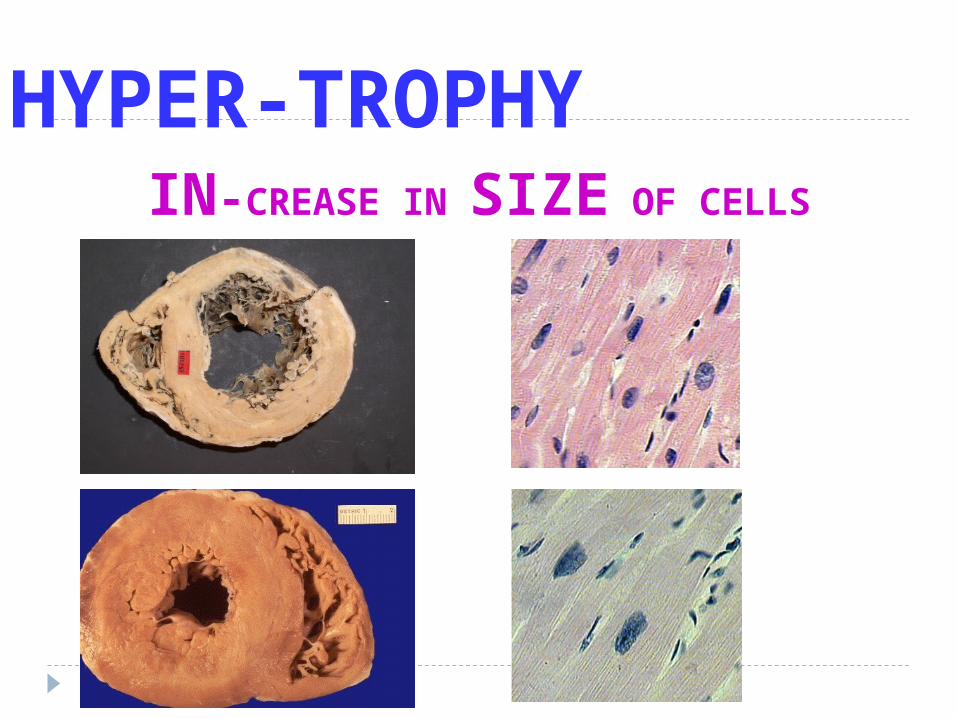
HYPER-TROPHYIN-CREASE IN SIZE OF CELLS

H y p e r t r o p h y
Is an increase in the size of cell accompanied by
Augmented functional capacity
Hypertrophy is a response to trophic signals and
Commonly a normal procesess

I. Physiological ( Hormonal ) hypertrophy- in puberty an icreased production of sex hor –
mon - hypertrophy breast tissue - abnormal hormon production in cancer
II. Increased functional demands- exercise- pathological conditions
eq. Myocardial cell- kidney hypertrophy on surgical removed
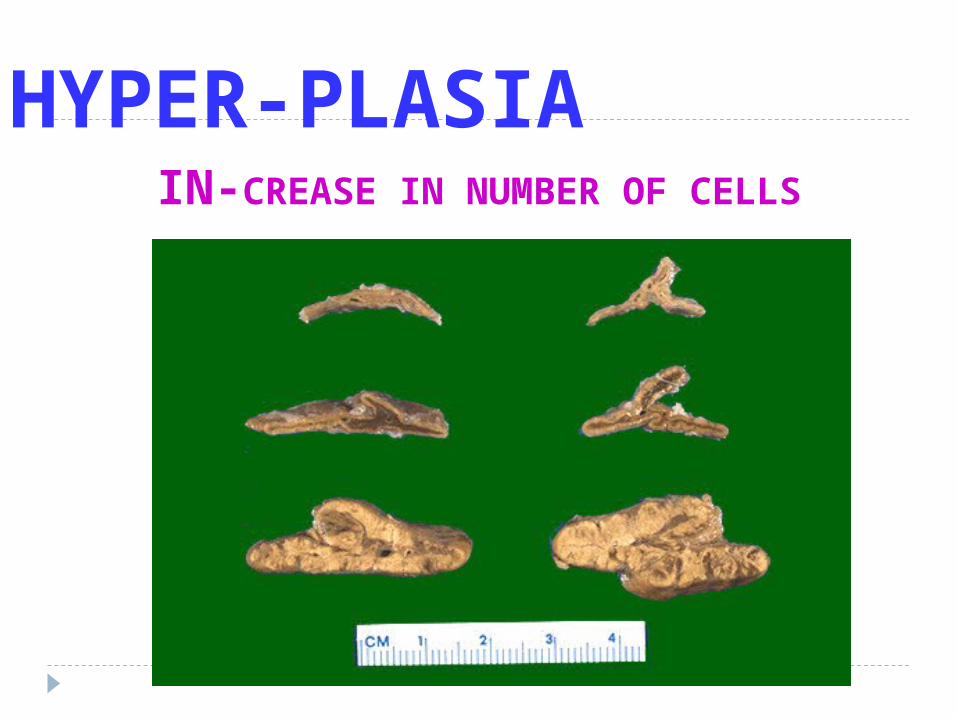
HYPER-PLASIAIN-CREASE IN NUMBER OF CELLS

H Y P E R P L A S I Ais an increase in the number of cells in an organor tissue
Hyperplasia can be :
I . Physiologic hyperplasia- hormonal hyperplasia- compensatory hyperplasia
II. Pathologic hyperplasiaexcessive hormonal or growth factor stimulationeq. Endometrial hyperplasia

1. Hormonal stimulation- estrogen increased endometrium ( hyperplasia)- gynecomastia
2. Increased functional demand- secondary polycytemia- lymphocyte hyperplasia
3. Persistent Cell Injury- chronic inflammation in the skin and the epi –
thelium of viscera- hyperplasia of the bladder epithelium

METAPLASIA A SUBSTITUTION of one NORMAL CELL or TISSUE type, for ANOTHER COLUMNAR SQUAMOUS (Cervix) SQUAMOUS COLUMNAR (Glandular)
(Stomach) FIBROUS BONE
WHY?

M E T A P L A S I Ais a reversible change in which one adult celltype is replace by another adult cell type( Metaplasia is the convertion of one differentia – ted cell type of another )
It is almost invariably a response to persistent injuryand can be thought of as an adaptive mechanism.Most common is the replacment of a glandular epithelium by a squamous cell.
- Squamous metaplasia of the bronchial epithelium to tobacco - lower oesophagus by reflux acidic gastric- endocervical metaplasia
Metaplasia is usually reversible if the stimulus is removed

D Y S P L A S I Acellular dysplasia refers to an alteration in thesize, shape and organization of the cellular com –ponent of a tissue
The cells an epithelium exhibit uniformity of size, shapeAnd nucleus.
Dysplasia mean is disturbed by1. Variation in the size and shape of cells2. Enlargment, irregularity and hyperchromatism of the nuclei.3. Disorderly arrangement of the cells within the epi – thelium

The most common in the cervix and bronchus
It is established that dysplasia is a preneoplastic lession in the sense that it is a necessary stage in the multistep cellular evolution to cancer.
Dysplasia included in the morphological classification Of the stage if intraepithelial neoplasia

Anaplasia normal cell primitive cell Exc : malignan cell (carcinoma, sarcoma, adenocarcinoma, lymphoma, etc)

Granuloma
specialized type of chronic inflamation in tissue reaction
Cause : - infection : tbc, fungal, siphylis, etc - non-infection : sarcoidosis, Crohn’s disease, etc

C E L L U L A R A G I N GAlterations in structure and fuction that may leadto cell death, or at least diminished capacity ofthe cell to respond an injury
Reduced cell in : - pleomorphic vacuolated mitochondria - repair of chromosomal damage
Morphologic alteration in :• pleomorphic vacuolated mitochondria• decreased endoplasmic reticulum• disorted Golgi Apparatus• accumutaion of lipofuscin pigment

Cellular senescence is multifactorial :
1. The cumulative effects of extrinsic influences: free radical damage
2. Intrinsic molecular program of cellular agingcell have a finite life span
tia@pafkusu