Embed Size (px)
Citation preview

1
DIAGNOSIS OF KALA-AZAR FROM BUFFY COAT USING DIFFERENT TECHNIQUES INCLUDING
POLYMERASE CHAIN REACTION (PCR)
DR. RISHDINA MIRZA M.B.B.S
DEPARTMENT OF MICROBIOLOGY MYMENSINGH MEDICAL COLLEGE
MYMENSINGH, BANGLADESH
JULY 2010
id1515859 pdfMachine by Broadgun Software - a great PDF writer! - a great PDF creator! - http://www.pdfmachine.com http://www.broadgun.com

2
TO WHOM IT MAY CONCERN
This is to certify that Dr. Rishdina Mirza, a student of thesis part has completed
the thesis entitled �Diagnosis of Kala-azar from buffy coat using different
techniques including Polymerase chain reaction (PCR)� in the Department of
Microbiology, Mymensingh Medical College under my guidance and supervision
and this is up to my satisfaction. Her protocol was approved by protocol approval
committee of the Department of Microbiology and Ethical review committee of
Mymensingh Medical College.
Dated, Mymensingh The
Professor Dr. Md. Akram Hossain Head of the Department of Microbiology
Mymensingh Medical College Mymensingh, Bangladesh

3
ACKNOWLEDGEMENTS
First of all thanks to Almighty Allah, the merciful and the beneficent, for giving
me the opportunity and providing me the ample energy and patience to complete
the entire thesis work.
I am very grateful and deeply indebted to my honourable teacher and guide
Professor Dr. Md. Akram Hossain, Head of the Department of Microbiology,
Mymensingh Medical College. It is my great pleasure to express my deepest
regards and whole hearted indebtedness to him for his inspiring encouragement,
continuous guidance, active cooperation, constant supervision, valuable
suggestions, constructive criticism and help in carrying out this work successfully.
I convey my deepest regards and gratitude to my respected teacher Professor Dr.
A.K.M. Shamsuzzaman, Head of the Department of Microbiology, Begum
Khaleda Zia Medical College, Dhaka for his cordial cooperation and valuable
suggestions.
I would like to express my gratitude to Dr. Md. Chand Mahmud, Assistant
Professor, of the Department of Microbiology, Mymensingh Medical College for
his valuable suggestions. My cordial respect and complements to Dr. Shyamal
Kumar Paul, lecturer of the Department of Microbiology, for his cooperation and
efforts for this thesis. I am thankful to the honorable members of the Ethical
review committee for giving kind approval of my thesis protocol. I am also
obliged to Professor Dr. Md. Aminul Haque, Principal, Mymensingh Medical
College, for his kind permission to conduct the present study in this institution.
I express my grateful thanks to Dr. Md. Abdur Razzaque Mia, Associate Professor
and Head of the Department of Biochemistry, Mymensingh Medical College, for
his cordial cooperation and valuable suggestion.
I would like to express my gratitude to all other Lecturers, M.Phil Students in the
Department of Microbiology, Mymensingh Medical College.

4
I would like to thanks Abu Hanif Md. Al-Rafi, Laboratory Technologist and also
thanks to all other Laboratory Technologists and supporting staffs of
Microbiology Department, Mymensingh Medical College. I must thanks to
Librarian of Mymensingh Medical College for providing me journals, books and
necessary articles.
I owe my heartfelt gratitude and sincere thanks to my brother Mirza Masrur Huq
my sister Dr. Nadia Mirza and my brother-in-low Dr. Sirazul Kader, for their
affectionate support.
I would like to convey my greatest regards and sincere thanks to my dear husband
Dr. Md. Mazharul Alam for his patience, devoted cooperation and assistance in
collecting specimens and sensible sharing of my pains and pleasures.
I remained incomplete if I do not express wholehearted thanks and gratitude to my
beloved mother Dr. Anwara Khanam, for her blessings and sparing me so much
time in this job from all sorts of social and familial responsibilities.
Last but not the least, I am indebted to those cases and controls form whom I have
collected specimens and pray to Almighty for their quick recovery.
Lastly my profound gratitude and heartfelt regards to my beloved father Prof. Dr.
Mirza Hamidul Huq, ex. Principal, Mymensingh Medical College, Mymensingh
of who always inspired me for higher studies and is my constant source of
inspiration and encouragement in every step of my life.
Thanks again to all.
Mymensingh, July 2010 Dr. Rishdina Mirza

5
CONTENTS
PAGE NO.
LIST OF TABLE vii
LIST OF FIGURE viii
LIST OF ABBREVIATION IX
SUMMARY xiii
CHAPTER 1 INTRODUCTION 1
OBJECTIVES 7
CHAPTER 2 REVIEW OF LITERATURE 8
CHAPTER 3 MATERIALS AND METHODS 52
CHPATER 4 RESUTLS 63
CHAPTER 5 DISCUSSON 85
CHAPTER 6 CONCLUSION 93
BIBLIOGRAPHY 94
APPENDICES 127
11aaaa

6
LIST OF TABLES
TABLE NO. TITLE PAGE NO.
Table 1 Age and sex distribution of subjects 64
Table 2 Housing condition among study population 66
Table 3 Comparison between low and middle socioeconomic status in Kala-azar patients 68
Table 4 Duration of fever in Kala-azar cases 70
Table 5 Hepato-splenomegly in Kala-azar cases 72
Table 6 Haematological findings of (41) confirmed cases of kala-azar 74
Table 7 Prevalence of kala-azar cases among study population 76
Table 8 Sensitivity and specificity of LD body microscopy from buffy coat in confirmed KA cases 78
Table 9 Sensitivity and specificity of buffy coat PCR in confirmed KA cases 80
Table 10 Sensitivity and specificity of buffy coat microscopy in ICT positive KA cases 82

7
LIST OF FIGURES
FIGURE NO. TITLE PAGE NO.
Figure 1 Distribution of kala-azar cases in hospitals 84
Figure 2 Buffy-coat 139
Figure 3 Buffy-coat 139
Figure 4 Buffy-coat smear 140
Figure 5 Leishmania donovani in buffy coat smear at 100X (oil immersion) lens under microscope 141
Figure 6 Leishmania donovani in buffy coat smear at 100X (oil immersion) lens under microscope 141
Figure 7 Leishmania donovani in buffy coat smear at 100X (oil immersion) lens under microscope 142
Figure 8 Leishmania donovani in buffy coat smear at 100X (oil immersion) lens under microscope 142
Figure 9 PCR amplification product of Leishmania donovani from buffy coat samples, Lane MW = 100 bp DNA ladder, Lane 1-15 patients buffy coat sample. Products were analyzed by electrophoresis in 1% agarose gel containing 0.5 g of ethidium bromide/ml in TBE buffer and photographed under UV illumination. 143
Figure 10 PCR amplification product of Leishmania donovani from buffy coat samples, Lane MW = 100 bp DNA ladder, Lane 1-8 patients buffy coat sample; Lane NC = Negative control. Products were analyzed by electrophoresis in 1% agarose gel containing 0.5 g of ethidium bromide/ml in TBE buffer and photographed under UV illumination. 144
Figure 11 rK39 immunochromatographic dipstick test for kala-azar 145
Figure 12 Bone marrow smears showing amastigote form of Leishmania donovani 146

8
LIST OF ABBREVIATIONS Abbreviation Expanded version
ACP = Acid phosphatase
AIDS = Acquired immune deficiency syndrome
AMP = Adenosine monophosphate
APC = Antigen presenting cell
AQP = Aquaglyceroprotein
AT = Aldehyde test
ATP = Adenosine triphosphate
bp = Base pair
CCIEP = Counter-current immunoelectrophoresis
CD = Cluster of differentiation
CFT = Complement fixation test
CL = Cutaneous Leishmaniasis
cm = Centimeter
CRP = C-reactive protein
CSF = Colony stimulating factor/Cerebrospinal fluid
DAT = Direct agglutination 0C = Degree centigrade
DDT = Dichloro Diphenyl Trichloroethane
DGHS = Director General for Health Services
DNA = Deoxy ribonucleic acid
dNTP = Deoxynucleotide triphosphate
DTH = Delayed type of hypersensitivity
EDTA = Ethylenediamine tetra-acetic acid
ELISA = Enzyme linked immunosorbant assay
et al = at alia (all others)
FCS = Foetal calf serum
GBP = Gene B protein
GCS = Gamma-glutamylcysteine synthetase
G-CSF = Granylocyte colony stimulating factor
GKO = Gene knockout

9
LIST OF ABBREVIATIONS (CONTD.) Abbreviation Expanded version
GM-CSF = Granulocyte-Macrophage colony stimulating factor
gp = Glycoprotein
Hsp = Heat shock protein
HCl = Hydrochloric acid
ICDDRB = International Center for Diarrhoeal Diseases and Research, Bangladesh
ICT = Immunochromatography test
IFAT = Indirect fluorescent antibody test
IgG = Immunoglobulin G
IL = Interleukin
INF = Interferon
IU = International unit
KA = Kala-azar
KCl = Potassium chloride
kDa = Kilo-dalton
kDNA = Kinetoplast DNA
Kg = Kilogram
L = Leishmania
LAAMB = Lipid associated amphotericin B
LACK = Leishmania homologue of receptor for Activared C Kinase
LAT = Latex agglutination test
< = Less then
LD bodies = Leishmania donovani bodies
LdMT = L. donovani miltefosine transporter
LdRos = L. donovani Ros protein
LMDR = Leishmania multi-drug resistant
Ln-PCR = Leishmania nested Polymerase chain reaction
LPG = Lipophosphoglycin
LST = Leishmanin skin test

10
LIST OF ABBREVIATIONS (CONTD.) Abbreviation Expanded version
MCL = Mucocutaneous Leishmaniasis
mM = Millimole
mg = Milligram
µgm = Microgram
ml = Millilitre
µl = Microlitre
µm = Micrometer
MIC = Minimum inhibitory concentration
M-CSF = Macrophage colony stimulating factor
MHC = Major Histocompetibility complex
MIF = Macrophage inhibitory factor
MgCl2 = Magnesium chloride
> = More than
medRNA = Mini exon derived RNA
MDR = Multi-drug resistant
n = Number
NNN = Novy McNeal Nicolle
NO = Nitric oxide
NK = Natural killer
ODC = Ornithine-decarboxylase
PCR = Polymerase chain reaction
PKDL = Post kala-azar dermal leishmaniasis
PCR-SHEL = PCR solution hybridisation enzyme linked assay
PCR-SSCP = PCR single stranded conformation polymorphisms
PAGE = Polyacrylamide gel electrophoresis
PBF = Peripheral Blood Film
% = Percent
RNA = Ribonucleic acid

11
LIST OF ABBREVIATIONS (CONTD.)
Abbreviation Expanded version
r = Recombinant
SAG = Sodium antimony gluconate
Sb = Sodium stibogluconate
SD = Sodium deviation
SSU-rRNA = Small subunit ribosomal RNA
SDS = Sodium dodecyl sulphate
TNF = Tumour necrosis factor
TDR = Tropical disease research
Th = Helper T
TCR = T-cell receptor
TGF = Transforming growth factor
UV = Ultra violet
VL = Visceral Leishmaniasis
WHO = World Health Organization
Zn = Zinc

12
SUMMARY
Diagnosis of kala-azar is problematic because of a variety of reasons. Definitive
diagnosis of Kala-azar requires tissue specimens, which are conventionally
obtained by organ needle aspiration for microscopic demonstration of amastigote
forms in stained smears. Organ aspiration and accurate examination of smears
also require technical skills that are not uniformly available in rural areas. PCR
testing of aspirate material improves parasitologic yield, but these methods are
seldom undertaken outside the research laboratories. Another interesting field for
definitive diagnosis of Kala-azar might be the detection of LD body from buffy
coat smear of peripheral blood. This cross-sectional study was conducted in the
Department of Microbiology, Mymensingh Medical College during the period
from July 2009 to June 2010. This study included 106 kala-azar cases were from
rural hospital (66.98%) and urban hospital (33.02%). Among them, 41 were
confirmed Kala-azar cases on the basis of LD body positive bone marrow in
microscopy. Another 65 Kala-azar cases were included on the basis of ICT
positive results for anti-Leishmanial antibody (rk39). The control group (n=44)
comprised of age and sex matched 44 healthy persons (Non kala-azar fever
patients) in the endemic area. Majority of the cases (58%) were in the age group
of 2-12 years. Male to female ratio of cases was 1.46:1. Most (68.87%) of the
Kala-azar cases were having earthen houses. All cases of Kala-azar were having
low-grade irregular fever with a mean duration of 3.77±1.32 months. Mean
enlargement of liver and spleen was found as 5.19±2.03 cm and 6.98±2.58 cm
respectively. Anaemia, leucopoenia, Thrombocytopenia were found in 34(83%),
18(44%), 33(80.48%) of confirmed cases of kala-azar respectively. Sensitivity
and specificity for buffy coat microscopy for LD body of confirmed Kala-azar
cases were found as (92.7%) and (95.45%) respectively. Sensitivity and
specificity of buffy coat PCR in confirmed kala-azar cases were 95.12% and

13
100% respectively. Out of 65 ICT positive cases 92.30% yielded LD body
positive in buffy coat smear microscopy and 95.38% yielded PCR positive from
buffy coat. LD body detection in buffy coat smear became highly sensitive and
specific. Analyzing the findings of the present study, use of modified blood buffy
coat smear for confirmatory diagnosis of Kala-azar in endemic areas is
recommended. This study, also evaluated a prospective field for molecular
diagnosis of Kala-azar by an easy and accessible approach.

14
CHAPTER 1
INTRODUCTION
The term leishmaniasis refers to various clinical syndromes caused by obligate
intracellular protozoa of the genus Leishmania. Sir William Leishman (British
Army Pathologist) reported about the parasite in 1903 and in the same year the
discovery was verified by Captain Charles Donavan (Professor of Physiology at
Madras University in India). Henceforth the parasite was named as the Leishman-
Donovan body or Leishmania donovani (Gibson, 1983).
Visceral leishmaniasis (VL) or Kala-azar is a re-emerging serious public health
problem in the Indian sub-continent targeting the poor (Joshi et al., 2008). Nearly
350 million people are at risk in 88 countries around the world. Currently an
estimated 12 million people are infected and around 2 million infections occur
each year (Rai et al., 2008). Of all the cases, 90% occur in India, Bangladesh,
Nepal, Sudan and Brazil (Ahasan et al., 2008). The Kala-azar cases estimation
was made for Bangladesh, India and Nepal and it shows that annual total number
of Kala-azar cases occurred from Bangladesh, India and Nepal are 136,500,
270,900 and 12,600 respectively (Joshi et al, 2008; Rijal et al, 2006).
The parasitic disease Kala-azar was first described in 1824, in Jessore district.
Bengal in what is now Bangladesh (Bern and Chowdhury, 2006). Previous
epidemiological studies show that 34 out of 64 districts of Bangladesh reported
kala-azar cases but 90% of them are from 10 districts. The cumulative reported
incidence of kala-azar by district from 1994 � 2004 shows that the worst hit area
in Bangladesh is Mymensingh followed by Pabna, Tangail, Jamalpur, Sirajganj,
Gazipur, Natore, Naogaon, Manikganj, Rajshahi and Naawabgaj (Rahman et al.,
2008) .

15
In Mymensingh district, only 5 of 12 thanas reported kala-azar cases in recent
years. Using the population of the respective thana the denominator, the incidence
of kala-azar in Fulbaria thana ranged from 30 to 33/10,000/year since 2000, while
that in Trishal, the next most affected thana, ranged from 21 to 26/10,000/year.
Over the same period of time, the incidence in the other 3 endemic thanas,
Bhaluka, Muktagacha, and Goforgaon, ranged from 5 to 15 cases/10,000/year. For
the 5 endemic thanas of Mymensingh district, the mean annual reported incidence
since 2000 was 17/10,000/year, compared to 8.3/10,000 when the total district
population was used (Alam et al., 2009).
Kala-azar carries a high mortality rate ranging from 80% to 100% and even with
treatment, case fatality rates in excess of 10% are common (Salam et al., 2009).
Leishmaniasis development depends on several risk factors such as malnutrition,
immunosuppression, age, immunological status and genetic factors (Assimina et
al., 2008).
The risk of Kala-azar was highest for people in the 3- to 14-year and 15- to 45-
year age groups (Bern et al., 2005). Kala-azar mainly affects infants and young
children and male sex predominance (Uzair et al., 2004).
Kala-azar is typically caused by the Leishmania donovani complex, which
includes three species: L. donovani, Leishmania infantum, and Leishmania
chagasi. On the Indian subcontinent, the disease is almost exclusively caused by
L. donovani. The initial report of Leishmania tropica causing Kala-azar in India
was refuted by us and others. L. infantum is responsible for Kala-azar in children
in the Mediterranean basin (Begum et al., 2002).

16
It is transmitted to man by phlebotomus (sandfly) in the Old World and
Lutzomiyia in the New World Sergentomyia is another vector found in
Baluchistan (Brito et al., 2000; EI-Hasan et al., 1995).
Occasional nonvector transmissions also have been reported through blood
transfusions, sexual intercourse, organ transplants, excrements of dogs, and
sporadically outside endemic areas. Congenital Kala-azar was described first in
1926 by Low and Cooke (Christoph et al., 1999). The incubation period is 3-6
months, though 10-24 years is also re-ported (Arias et al., 1996).
The disease is characterized by fever, hepatosplenomegaly, anaemia, leucopoenia
and hypergammaglobulinemia. Serious complications are cancrum oris,
dysentery, pneumonia, anaemia, agranulocytosis, jaundice, severe haemorrhage
and anasarca. Pulmonary tuberculosis may occur with Kala-azar (Cruz et al.,
2006).
The diagnosis of Kala-azar is complex because its clinical features are shared by a
host of other commonly occurring diseases, such as malaria, typhoid, and
tuberculosis; many of these diseases can be present along with Kala-azar (in cases
of coinfection); sequestration of the parasite in the spleen, bone marrow, or lymph
nodes further complicates this issue.
Laboratory diagnosis of leishmaniasis can be made by the following:
(i) demonstration of parasite in tissues of relevance by light microscopic
examination of the stained specimen, in vitro culture, or animal inoculation; (ii)
detection of parasite DNA in tissue samples; or (iii) immunodiagnosis by
detection of parasite antigen in tissue, blood, or urine samples, by detection of
nonspecific or specific antileishmanial antibodies (immunoglobulin), or by assay
for Leishmania-specific cell-mediated immunity (Sundar and Rai 2002).

17
Definitive diagnosis of Kala-azar is made by demonstration of Leishmania
amastigotes in aspirates from lymph node, bone marrow or spleen. These
procedures have a sensitivity 58%, 60-85% and 96%, respectively (Zijlstra et al.,
1992). Direct demonstration of parasites in tissue smears, a technique that is
invasive and requires considerable expertise, by a �field test� (Boelaert et al.,
2007). Through splenic aspirate have high sensitivity, is associated with risk of
fatal hemorrhage in inexperienced hands (Kager et al., 1983). Marrow aspiration
either from illiac crest or from sternum is very much painful (Boelaert et al.,
1999; Chowdhury et al., 1993). In paediatric patients, detection rate of LD body
from bone marrow is higher because of higher parasitization (DGHS, 2009).
The Aspirates can also be used to culture leishmanian protozoas in NNN media
(1% defibrinated Rabbit blood + salt azar + Penicillin) and it takes about 1-3
weeks to grow. Other medias include, Grace�s culture media (Growth occurs
within 72 hours) and Sneader�s culture media. Culture is costly, time consuming
and requires sophisticated laboratory settings, so not a useful diagnostic tool
(Sundar and Rai, 2002).
The development of PCR has provided a powerful approach to the application of
molecular biology techniques to the diagnosis of leishmaniasis. Primers designed
to amplify conserved sequences found in minicircles of KDNA of leishmanias of
different species were tested in various tissues of relevance. In recent years PCR
based diagnostic methods with a wide range of sensitivities and specificities have
been described. The sensitivity of PCR with whole blood from Kala-azar patients
was 96%. PCR assay with buffy coat preparation to detect Leishmania was 10
times more sensitive than that with whole blood preparation and particularly good
results were obtained when proteinase K based methods were used. Proteinase K

18
based PCR was able to detect 10 parasites/ml (Sundar and Rai 2002; Motazedian
et al., 2002; Schonioan et al., 2003).
The serodiagnosis of leishmaniasis (based on antibody detection) appears to be an
alternative to parasitological diagnosis. A number of serological techniques have
been developed for diagnosis of Kala-azar including ELISA, dot ELISA and
direct agglutination test. The sensitivity and specificity of such diagnostic
methods depends on the type, source and purity of antigen employed, as some of
Leishmania antigens have common cross-reactive epitopes shared with other
microorganisms (Sarkari et al., 2005). rk39 ICT strip test is easy to perform at
field. It shows high sensitivity and specificity (Salam et al., 2009; Bern et al.,
2000).
Antigen detection is more specific than antibody-based immunodiagnostic tests. A
new latex agglutination test (KATEX) for detecting leishmanial antigen in urine
of patients with Kala-azar has showed sensitivities between 68 and 100% and a
specificity of 100% in preliminary trials. The antigen is detected quite early
during the infection and the results of animal experiments suggest that the amount
of detectable antigen tends to decline rapidly following chemotherapy. The test
performed better than any of the serological tests when compared to microscopy.
Large field trials are under way to evaluate its utility for the diagnosis and
prognosis of Kala-azar (Attar et al., 2001; Fakhar et al., 2006).
Another interesting field for definitive diagnosis of Kala-azar might be the
detection of LD body from buffy coat smear of peripheral blood. Because, when
blood is settled in presence of an anticoagulant, the WBCs are deposited and
concentrated in buffy coat at the interface of RBCs and plasma. If buffy coat is
considered for making a smear, it is supposed to show high content of WBCs
including monocytes. The monocytes parasitized by LD bodies, if present, will be

19
higher in the smear. Thus the rate of LD body positivity is expected to be higher
(Shamsuzzaman et al., 2007).
Considering the background described above definitive diagnosis of kala-azar is
technically difficult job. LD body from blood buffy coat is an easy and
noninvasive approach for definitive diagnosis of Kala-azar. The present study
explored the reliability and effectiveness of LD body detection in blood buffy coat
smear and also to identify Leishmania donovani kinetoplast DNA from blood
buffy coat of Kala-azar patients by Nested PCR (Ln-PCR) which is non-invasive,
reliable, sensitive and specific. Therefore the present study was designed with the
following objectives.

20
OBJECTIVES
General objective: To evaluate buffy coat- a noninvasive method for definitive
diagnosis of kala-azar.
Specific Objectives
i. To diagnose kala-azar by detection of LD bodies from bone marrow
aspiration in clinically suspected cases.
ii. To evaluate buffy coat smear for detection of LD bodies in clinically
suspected cases.
iii. To identify Leishmania donovani kinetoplast DNA from blood buffy
coat of Kala-azar patients by Nested PCR (Ln-PCR)
iv. To detect anti rK39 antibody by ICT in Kala-azar patients
v. To determine the sensitivity and specificity of buffy coat smear for
diagnosis of kala-azar
vi. To find out haematological changes in kala-azar patients

21
CHAPTER 2
REVIEW OF LITERATURE
2.1 Historical background
The organism of kala-azar was first described in 1903 by Sir William Leishman
who examined the spleen of a British soldier stationed at Dumdum near Calcutta,
India. Later the same year, Charles Donovan verified this finding and the
organism in often called Leishman-Donovan (LD) body (Chulay, 1991). The
genus Leishmania was created by Ross in 1903 to include Leishmania donovani.
In India, the disease is known a kala-azar, meaning �black sickness or fever� as
the disease turns the color (pigmentation) of the skin black, the word �kala� means
�black and �azar� means �deadly�, thereby signifying a fatal illness (Chatterjee,
1982). In 1904, Roger observed the conversion of amastigotes of promastigotes in
culture (Chulay, 1991). A similar parasite was observed in a disease of children in
the Mediterranean countries by Nicolle in 1908, proposed the name of infantile
kala-azar for this disease and designated the parasite as L. infantum (Chatterjee,
1982). Further investigation revealed that it was a strain of L. donovani. The
parasite of South American Kala-azar originally named as L. chagasi in 1937, was
also found to be identical of L. donovani. Promastigotes were found in sandflies
by Alder and Theodor in 1925 (Chulay, 1991). In 1940, it was demonstrated the
Plebotomus argentipes was the vector of Indian kala-azar (Birley, 1993) and in
1942, in India kala-azar was transmitted experimentally to human volunteer by
sandfly bite (Swaminathan et al., 1942).
Irregular epidemic waves have swept through Assam, Bengal and Bihar since the
1800s with a frequency of 15-20 years. In 1890-1900 an epidemic swept Assam
which depopulated whole villages and reduced populations over large areas. In
1917 another epidemic started in Assam and Bengal and reached its height about

22
1925 and mysteriously subsided, until by 1931, it was almost gone. In 1937 a new
outbreak began in Bihar (Birley, 1993). Before the Second World War, kala-azar
was endemic in Assam, Bengal (a part of which is now Bangladesh), Bihar and
some other parts of Indian subcontinent. Due to insecticide spraying as a part of
malaria eradication campaign in 1958-1964, kala-azar almost disappeared from
chasten states of Indian and Bangladesh but with the cessation of insecticide
spraying there has been a resurgence of Kala-azar in these regions (Thakur et al.,
1981; Rahman and Islam, 1983).
2.2 World�s situation of Leishmaniasis
Kala-azar is endemic in the tropical and sub-tropical regions of Africa, Asia, the
Mediterranean, Southern Europe, South and Central America. The distribution of
Kala-azar in these areas however is not uniform; it is patchy and often associated
with areas of drought, famine and densely populated villages with little or no
sanitation (WHO, 1991; Rab and Evans, 1995).
In Pakistan 239 cases of Kala-azar due L. infantum were reported between 1985
and 1995. Off those, 52% were children below the age of 2 years and 86% were
below 5 years. This figure represented an increased of ten-fold in infantile Kala-
azar cases over the 10 year period from 0.2 to 2 per 100 000 population and male
cases out numbered female cases by three times (Rab and Evans, 1995). Kala-azar
has been known to exist in the Himalayas in Pakistan for over three decades.
However recently sporadic cases are beginning to appear in the North West
Frontier Province, Punjab and Azad Jammu and Kashmir. All of these areas are
mountainous and contain large farming communities (Rab and Evans, 1995).
In neighbouring India Kala-azar is endemic in the states of Bihar, Uttar Pradesh
and West Bengal. One of the largest epidemics occurred in 1978 in North Bihar

23
were over half a million people fell victim to Kala-azar. In the first eight months
of 1982, 7500 cases were reported in India and in one year alone between 1987
and 1988, 22 000 cases of Kala-azar were registered (WHO, 1991). In Bangladesh
cases of Kala-azar greatly declined between 1953-1970, probably as a result of
mass chemotherapy with pentavalent antimonials and wide spread spraying with
DDT to control malaria. Following the end of the malaria control programme in
1970, sandfly vector populations increased and so did the cases of Kala-azar and
currently appear at a rate in excess of 15000 per year (Al-Masum et al., 1995). In
Brazil, Kala-azar is distributed widely in the south, east and the central regions of
the country. Kala-azar commonly affects poor and malnourished children below
the age of 15 years (Arias et al., 1996). The disease is highly endemic in the states
of Bahia and Ceara, which together account for 70% of the total cases of Kala-
azar in Brazil, upto 1989, 15000 cases of Kala-azar was recorded in multiple
states (WHO, 1991). Recently the foci of Kala-azar has shifted from rural villages
to large cities probably as a result of migration of settlers from villages into these
cities creating dense population and living in sub-standard house with improper
sanitation and keeping farm animals in their gardens. The two cities of Teresin
and Sao Luis together accounted for 40-50% of the total number of Kala-azar
cases. During 1993 and 1994, approximately 3000 cases per year were recorded
(Arias et al., 1996). In Central America, the disease has been increasing in Costa
Rica, Honduras and Nicaragua. This is most probably due to an increase in the
human population and their movements in and out of these areas (Carreira, 1995).
First case of Kala-azar in Sudan was reported in 1938.
Since then the disease has become widespread and endemic in south and eastern
parts of the White Nile and Upper Nile states (Hashim et al., 1995). Other
sporadic areas include the provinces of Kasala, Jonglei and Kapoeta in the south,
E1 Fasher an E1 Nahud in the west and also north of Khartoum (WHO, 1991). In

24
most countries, males are almost twice more likely to be Kala-azar than females,
with young children being at the highest risk. In the village of Um-Salala in
eastern Sudan, the average age of Kala-azar patients was found to be 6.6 years
with a male to female ratio of 1.8:1. Annual incidence was recorded as 38.4 per
1000 population between 1991 and 1992 (Zijlstra et al., 1994). The first case of
Kala-azar in Ethiopia was documented in 1942 in the southern parts of the
country. Since then the disease has spread to become endemic in the Segen, Woito
and Gelana river valleys. The highest incidence was recorded in Aba Roba area
(WHO, 1991). It was found that 58% of the people affected were children below
15 years of age with the lowest risk groups being males above the age of 39 years
and females above 24 years (Ali and Ashford, 1994). In Somalia, Kala-azar is also
endemic with higher prevalence among children below the age of 15 years. Males
are three times more susceptible than females (Shiddo et al., 1995).
2.2.1 Epidemiology
Kala-azar is a re-emerging serious public health problem in the Indian sub-
continent targeting the poor (Joshi et al., 2008). Nearly 350 million people are at
risk in 88 countries around the world. Currently an estimated 12 million people
are infected and around 2 million infections occur each year (Rai et al., 2008). Of
all the cases, 90% occur in India, Bangladesh, Nepal, Sudan and Brazil (Ahasan et
al., 2008).
The parasitic disease Kala-azar was first described in 1824, in Jessore district.
Bengal in what is now Bangladesh (Bern and Chowdhury, 2006). Previous
epidemiological studies show that 34 out of 64 districts of Bangladesh reported
kala-azar cases but 90% of them are from 10 districts. The cumulative reported
incidence of kala-azar by district from 1994 � 2004 shows that the worst hit area
in Bangladesh is Mymensingh followed by Pabna, Tangail, Jamalpur, Sirajganj,

25
Gazipur, Natore, Naogaon, Manikganj, Rajshahi and Naawabgaj (Ahasan et al.,
2008) .
In Mymensingh district, only 5 of 12 thanas reported kala-azar cases in recent
years. Using the population of the respective thana the denominator, the incidence
of kala-azar in Fulbaria thana ranged from 30 to 33/10,000/year since 2000, while
that in Trishal, the next most affected thana, ranged from 21 to 26/10,000/year.
Over the same period of time, the incidence in the other 3 endemic thanas,
Bhaluka, Muktagacha, and Goforgaon, ranged from 5 to 15 cases/10,000/year. For
the 5 endemic thanas of Mymensingh district, the mean annual reported incidence
since 2000 was 17/10,000/year, compared to 8.3/10,000 when the total district
population was used (Bern and Chowdhury, 2006).
The resurgence of kala-azar was associated with lack of spraying which allowed
the build-up of sand fly population. It also revealed that about 90% of cases of
kala-azar came from low-income group who were living in the houses plastered
with mud and cow-dung. It was also noted that the disease were low or rare in
malaria endemic areas where regular DDT spraying was going on (Masum et al.,
1991).
2.2.2 Mode of Transmission
Transmission of kala-azar is caused by the bite of infected female sandfly of the
genera Phlebotomus and Lutzomyia. Some of the species feed on man and also a
variety of warm and cold-blooded animals, which is an important factor in
spreading the disease. A sandfly become infected 14-18 days after the ingestion of
the infected blood meal and remains infected throughout its lifetime and are
capable of infecting several persons (Cheesbrough, 1999).

26
Transmission of Kala-azar may take place by contamination of bite wound or
contact when the insect is crushed during the time of biting (Park and Park, 2000).
Accidental inoculation of parasites during laboratory work may result in
leishmaniasis. Transmission by blood transfusion has also been recorded.
Congenital infection from an infected mother may occur rarely. Transmission of
infection to fetus in utero may occur due to placental defect (Bahr and Bell, 1987).
Transmission during coitus was also recorded (Chatterjee, 1980).
A case of fatal leishmaniasis was also noted in renal transplant patient due to
kidney transplantation (Vanorshovan et al., 1979).
2.2.3 Vectors
Sandfly species and subspecies of Phlebotomus in the Old World (Asia, Africa
and Europe) and Lutzomyia in the New World (Central and South America) are
the only proven vectors of Leishmania (WHO, 1990).
In the Central and South America Lutzomyia longipalpis is the main vector for
transmission of Kala-azar. In the Mediterranean region, Phlebotomus perniciousus
and Phlebotomus ariasi are important vectors while in China Phebotomus
chinensis and Phlebotomus alexandri are the proven vectors. In East Africa
including Sudan Phlebotomus martini and Phlebotomus orientalis are considered
as vectors for the diseases. In India, for the anthropologic form of Kala-azar the
proven vector is Phlebotomus argentipes (WHO, 1990; Swaminathan et al.,
1942). Besides these many other species have been implicated as vectors of
leishmaniasis.
In Bangladesh, during an outbreak of kala-azar at Kalihati Thana of Tangail
districts sandfly species of Phlebotomus argentipis, Segentomyia babu babu,
Sergentomyia barrudi and Sergentomyia shortill were identified (Masum et al.,

27
1990a). The sandflies collected from Thakurgaon district of Bangladesh during an
outbreak of Kala-azar were identified as Phlebotomus argentipes, Phlebotomus
papatasi, Sergentomyia babu babu and Sergentomyia barrudi (Masum et al.,
1990b).
An entomological study on vector of kala-azar in Shahjadpur thana of Sirajgonj
district showed that the species were Phlebotomus argentipes, Phlebotomus
malabaricus and Phlebotomus minutus. Among these Phlebotomus argentipes
was 70.6% (Ahmed and Ahmed, 1983).
In Bangladesh on effort so far has been made to implicate sandflies as vector(s) of
leishmaniasis, however, Phlebotomus argentipes is a recognized vector of kala-
azar in India and in Bangladesh this species occurs in areas where cases of kala-
azar have been found (Elias et al., 1989).
2.2.4 Reservoirs
Broadly speaking, there are two types of Kala-azar, namely zoonotic and
anthroponotic Kala-azar (WHO, 1990). In the anthroponotic form, human, act as
reservoir and in the zoonotic form animals are the reservoir.
Human- In the anthroponotic form, which is prevalent in India, Bangladesh and
some countries of East Africa, humans are directly involved as reservoir (WHO,
1990). All efforts to find a zoonotic reservoir in Indian Kala-azar has so far been a
failure (Srivastava and Chakravarty, 1984). Since PKDL may persist for up to 20
years such patients may act as a chronic reservoir of infection (Chulay, 1991) and
an outbreak of Kala-azar has been traced to case of PKDL (Addy and Nandy,
1992).

28
Dogs- In the zoonotic form, dogs are the principal reservoir in Europe and Africa
around the Mediterranean regions. Dogs are also important reservoirs in China
and South and Central America.
Wild canines- In southern France and central Italy foxes with inapparent
infection are the reservoir. Foxes are also reservoir in Brazil and jackals are
probably an important source of sporadic mainly rural cases that occur in the
Middle East and Central Asia.
Rodents and others- Rattus rattus, the common peridomestic rat in many
countries, has been found to be infected with various Leishmania species in both
Old World and New World. While its role in the maintenance of parasite
populations has not been fully established, it is strongly suspected as a secondary
reservoir host of L. infantum in Italy (WHO, 1990). L. donavani has been isolated
from Arvicanthis nilotica and other rodents in Sudan and rodents are probably
important in maintaining enzoonotic foci in interepidemic period.
2.3 Taxonomy of Leishmania (WHO, 1990)
Order : Kinetoplastida
Family : Trypansomatidae
Genus : Crithidia, Leptomanas, Herpetomonas, Blastocrithidia,
Leishmania, Sauroleishmania, Trypanosoma, Phytomonas
and Endotrypamm
Subgenus : Leishmania, Viannia
Complex : L. donovani, L. tropical. L. major, L. aethiopica, L. mexicana,
L. braziliensis, L. guyanensis.
Species : L. archibaldi, L. killicki, L. major, L. aethiopica, L.
amazonensis, L. braziliensis, L. guyanensis, L. panamensis, L.
panamensis, L. changasi, L. tropica, L. garnhami, L.
Peruviana, L. donovani, L. mexicana, L. infantum, L. pifanoi,
L. arabica, L. venezuelensis, L. gerbilli, L. enriettii.

29
(The classification of genera and subgenera is based on extrinsic characters and
that of the complexes mainly on intrinsic characters-isoenzymes).
2.4 Aetiology
Causative Agents
Seven complexes consisting of 17 Leishmania species i.e. Leishmania donovani
complex, Leishmania tropica complex, Leishmania major complex, Leishmania
aethiopica complex, Leishmania mexicana complex, Leishmania braziliensis
complex and Lcishmania guyanensis complex have been identified as causative
agents of leishmaniasis all over the world (WHO, 1990).
Kala-azar is caused by parasite species of the Leishmania donovani complex
which include Leishmania donovani donovani, Leishmania donovani infantum
and Leishmania donovani chagasi (in this thesis the three species has been
referred to as L. donovani, L. infantun and L. chagasi respectively). Some
undefined or unspecified species belonging to the L. donovani complex has been
isolated from Kala-azar patients in Kenya, Ethiopia and Somalia (Pearson and
Sousa, 1990).
Beside these species mentioned above, other species generally associated with
cutaneous leishmaniasis have been isolated from Kala-azar patients in different
regions of the world (Sacks et al., 1995) isolated L. tropica from patient with
classical Indian Kala-azar. L. major has been isolated from a patient of Kala-azar
in Israel (WHO, 1990). Kala-azar caused by L. amazoniensis has been reported
from Bahia state in Brazil (Pearson and Sousa, 1996). Viscerotropic syndrome
caused by L. tropica affected a small number of American troops during
Operation Desert Storm (Magill et al., 1993).

30
Old World, anthroponotic Kala-azar is caused by L. donovani. In India,
Bangladesh, Nepal parts of China and East Africa Kala-azar is believed to be
caused by L. donovani but other species has been identified as mentioned above.
Zoonotic Kala-azar in Mediterranean region, China, Middle East and parts of Sub-
Saharan Africa is caused by L. infantum with dog being the principal reservoir.
New World zoonotic Kala-azar is caused by L. chagasi and L. infantum, here as
well, dogs are the main reservoir.
2.5 Morphological Forms
Leishmania are digenetic (existing two forms) protozoa which exists as:
i. Amastigote form and
ii. Promastigote form.
Staining characteristics- Romanowsky dyes stains chromatin of the nucleus and
nucleic acid containing kinetoplast a brilliant red or violet, whereas the cytoplasm
is stained pale blue (Neva and Sacks, 1990).
With Leishman�s stain the cytoplasm appears blue, the nucleus pink or violet and
the kinetoplast, bright red (Chatterjee, 1982).
i. Amastigote form- Amastigote are aflagellar, obligate intracellular form
which reside within mononuclear phagocytes of their vertebrate hosts
including man. Amastigotes appear as round or oval bodies ranging
from 2-3 m in major diameter. The size of amastigote from different
species is known to vary. The cytoplasm of the amastigote often stain
the same as the host cell cytoplasm and only the nucleus and kinetoplast
distinguished. The nucleus of the parasite occupies a central position or
along side of the cell membrane. The kinetoplast lies adjacent to the
nucleus either tangentially or right-angle to it. The kinetoplast stains
more densely than the nucleus and it is variable in shape being round,

31
rod-shaped or curved in profile (Chaterjee, 1982; Neva and Sacks,
1990).
ii. Promastigote form- Promastigotes are flagellated extracellular forms
which are found in the gut of sandflies and in vitro culture. The
flagellar promastigote from measure 10-20 m in length not including
the length of the flagellum which may equal the body length. The pale
blue staining cytoplasm contains a centrally placed nucleus. The kin
kinetoplast lies about 2 m from the anterior end and the flagellum
emerges anteriorly. The overall shape is that of a spindle with the
posterior end gradually tapering to a point (Neva and Sacks, 1990).
2.6 Life-Cycle
In sandfly host- The amastigotes are ingested with the first blood meal of the
female sandfly in which they become transformed almost immediately into
promastigotes which multiply in the midgut and then migrate forwards to the
anterior part of the thoracic midgut or �cardia�. From here they move forward to
contaminate the mouth parts to be regurgitated into the wound caused by the bite
at the second blood meal. In all cases the infection is transmitted by bite.
Development in the sandfly from amastigotes to infective promastigote stage
(metacyclic promastigotes) varies from 5-10 days (Manson-Bahr and Bell, 1991).
In mammalian host- After inoculation by the sandfly, either into a capillary or
the dermal tissue, the promastigotes encounter macrophages which actively search
them out and phagocytose them by receptor-medicated endocytosis. The
promastigote changes into amastigotes in a phagolysosome where it multiplies by
binnary fission. Multiplication goes on continuously till the cell becomes packed
with parasites. The host cell in thereby enlarged and eventually ruptures or the
amastigotes leave the macrophages by penetrating the cell membrane. The
amastigotes thus released into the circulation are again either taken up by, or

32
invade fresh macrophages and the cycle is repeated. In this way the entire
reticuloendothelial system becomes progressively infected. In the blood stream,
some of the free amastigote are phagocytosed by the neutrophilic granulocytes
and monocytes (macrophages). A blood-sucking sandfly draws these free
amastigote forms as well as those within the monocytes during its blood-meal
(Chatterjee, 1982; Manson-Bahr and Bell, 1991).
2.7 Determinants of virulence
2.7.1 Surface Lipophosphoglycan (LPG)
Lipophosphoglycan (LPG) are heterogenous molecules and constitute the most
abundant surface component; each call has more then 106 copies, LPG is present
in both leishmanial stages and is released as �excretory factors�. The molecular
structure of LPG consists of three portions; terminal repeats of phosphorylated
saccharides, a phosphorylated heptasaccharide core and lyso-phosphotodylinositol
(Turco et al., 1987). The terminal saccharides repeating units are also structurally
unique and vary with Leishmania species. The difference in this portion of the
LPG distinguishes antigenically different Leishmania species. Antigenic
heterogeneity is further contributed to, probably even more significantly, by
another group of related surface inositol glycolipids. The tissue tropism of
different Leishmania species may be related to the variations in their surface
glycolipids (Handaman et al., 1987).
LPG and related glycolipids play important biological roles in Leishmania
macrophage interactions. The receptor for LPG appear to be complement receptor
(CR) 3 and p150, 95 (CR4) (Chang et al., 1990). In addition, LPG enhances the
survival of promastigotes in macrophages (Handman et al., 1986). Experimental
evidence suggests that the mechanism of this protection may be based on the
action of LPG to scavenge oxygen free radicals (Chan et al., 1989) and or to

33
inhibit relevant enzymes, e.g. liposomal glycosidase and protein kinase C. Other
proposed biological action of LPG includes inhibition of lymphoproliferative
response and activation of T suppressor cells (Chang et al., 1990). Leishmanial
virulence has been related to quantities and qualitative changes of promastigote
LPG.
The Leishmania LPG and related glycolipids are highly immunogenic owing to
their unique structural features (Tolson et al., 1989). They have long been
exploited as excretory factors for serotyping of Leishmania species (Turco, 1988).
2.7.2 Glycoprotein (gp) 63
Surface glycoprotein known as gp63 was initially recognized as a major surface
antigen of promastigotes by using monoclonal antibodies and surface radio
iodination of living cells (Chang et al., 1990). It constitutes about1% of the total
cellular proteins or 500,000 copies per cell (Bordier, 1987; Bouvier, et al., 1985).
The gp63 protein is seen to migrate on SDS polyacrylamide gels between 60-65
kD depending on the species of Leishmania (Wilson et al., 1989) and which is the
major antigenic protein of most promastigotes (Colomer-Gould et al., 1985). Gp
63 has been found in all major pathogenic Leishmania species (Bourver et al.,
1987), and in both stages (Colomer-Gould et al., 1985) endowed with proteolytic
activity (Etges et al., 1986). Gp63 has been shown to cleave C3 into C3b and
other C3 products (Chaudhuri and Chang, 1988).
By virtue of its proteolytic activity, universal presence, abundance and surface
localization, gp63 is thought to be a Leishmania virulence factor. The abundance
of gp63 is often correlated with infectivity (Kweider et al., 1989; Wilson et al.,
1989) and host-parasite interactions (Russell et al., 1986; Wilson et al., 1988).
Because of gp63�s proteolytic activity, it may function in the CR-mediated

34
endocytosis of promastigote (Da Silva et al., 1989) and protection of Leishmania
from intralysosomal microbicidal factor (Chang et al., 1990). Leishmania gp63 is
immunogenic; anti-gP63 antibodies have been reported in sera from patients with
leishmaniasis (Heath et al., 1987; Reed et al., 1987; Murray, et al., 1989). Gp63 is
potentially useful antigens for immunodiagnosis (Reed et al., 1987) and
immunoprophylaxis (Yang et al., 1990).
2.7.3 Acid phosphatases
The cell surface acid phosphatases (ACP) was initially discovered on the
promastigote stage using ultrastructure cytochemistry. Two forms of ACP
namely, membrane-bound ACP and secretary ACP, are present. The two forms
are antigenically distinct and each probably exists as multiple isoenzyme present
in both stages of most Leishmania species. One form of the membrane-bound
ACP and the secretory ACP have been purified from L. donovani and found to be
homodimers of 120 kD and 134 kD (Chang et al, 1990). All these different forms
of ACP are nonspecific monoesterase capable of hydrolyzing a variety of
phosphorylated substrates.
The membrane-bound ACP purified from promastigotes reduced the respiratory
burst of neutrophils (Remaley et al., 1985) and is itself resistant to oxidative
metabolites (Saha et al., 1985). In addition, it has been shown to dephosphorylate
certain phospholipids and phosphoroteins. Thus, the ectoenzyme is thought to
protect Leishmania species by interfering with the regulatory mechanism of the
macrophages that produces microbial free radicals (Glew et al., 1988). Its
production in large quantity elicits humoral immune response of the host and may
conceivably to the pathobiology in leishmaniasis (Chang et al., 1990).

35
2.7.4 Nucleotidases
5� nucleotidase and 3� nucleotidase/nuclease have been reported to exist on the
surface of some trypanosomatid protozoa including Leishmania species. The 5�
nucleotidase has an electrophoretic mobility of about 70 kD on SDS-PAGE and is
active in degrading both ribo-and deoxyribonucleotides. The 3� nucleotidase
/nuclease of 43 kD is more abundant. The latter enzyme is most active with 3�
AMP and prefers RNA over DNA as substrate. The alkaline pH optima of both
enzymes suggest that they probably serve such functions for promastigotes in the
sandfly gut better than for amastigotes in the phagolysosomes of the macrophages
(Chang et al., 1990).
2.7.5 Transporters
The plasma membrane of Leishmania has been shown biochemically and
genetically to possess the transport systems for folate, glucose, nucleosides,
proline and ribose. They also possess a cation or proton transporting ATPase,
which is apparently crucial for the homeostasis of Leishmania species and the
transport of nutrients necessary for their adaptation to the changing environment
in their life-cycle (Chang et al., 1990).
2.7.6 Cystenic proteinase and megasomes
The cysteine proteinase, a conserved and wide-spread enzyme, has been found in
Leishmania species generally associated with cutaneous form of leishmaniasis of
South American origin. It is present in an unusual organelle, or megasome a
modified lysosome, which is noticeable in the promastigotes grown to stationary
phase and becomes fully developed as they differentiable into amastigotes. This
enzyme is proposed to sever a degenerative role, possibly for the nutritional
benefits of the amastigotes and for releasing ammonia or other amines to

36
modulate that host lysosomal activity for the parasites� intracellular survival and
is functionally important to amastigotes (Chang et al., 1990).
2.7.7 Heat-shock proteins
Exposure of promastigotes to elevated temperatures results in an over-expression
of the classic heat-shock genes other genes. Multiple protein bands emerge shortly
after heat-shock and their number varies with different species. Only one of these
proteins has been positively identified immunologically as equivalent to Hsp70
protein. Most intruging is the increase in the virulence seen with briefly heat-
shocked promastigotes. What molecular changes in these promastigotes account
for virulence remains uncertain (Chang et al., 1990).
2.7 Pathogenesis
Despite the wide range of variation of the geographical distribution, clinical
manifestations and species involved, leishmanial infection shares a common
feature, namely the parasitizaiton of the phagocytic cells of major organs of the
reticuloendothelial (RE) system such as spleen, liver, blood, bone morrow and
skin (WHO, 1990).
With the bites of infected sandfly, the metacyclic (infective) forms of parasite are
inoculated into the microwound of the skin. Here the promastigotes are exposed to
IgG and IgM which opsonize them and are killed by activation the membrane
attach complex of complement through the classical pathway (Pearson and
Steigbige, 1980). Those escaping the lethal effect of serum bin to macrophages.
Lipophosphoglycan 9LPG) and gp63 has been considered parasite ligands
(Handman and Goding, 1985; Rizvi et al., 1988; Russell and Wright, 1988), with
complement receptor CR 1 and CR 3 and mannose-fucose receptor as the
corresponding macrophage receptors (Blackwell et al., 1985; Mosser and Edelson,
1985; Da Silva et al., 1989). After receptor-mediated endocytosis the parasite

37
resides within the phagolysosomes, thus are sheltered from the body�s immune
system. Side by side, they overcome the microbicidal conditions of the
macrophage phagolysosomes. Amastigotes and metacyclic promastigotes appear
to evade oxygen-dependent destruction by triggering a minimal respiratory burst
during infection due to the use of C3 receptors for internalization (Da Silva et al.,
1989). LPG scavenges oxygen free radicals (Chan et al., 1989) and inhibits
relevant enzymes e.g. lysosomal glycosidase and protein kinase C which may
enhance the survival of promastigotes in macrophages (Chang et al., 1990). Gp63,
a parasite ectoenzyme, has been shown to protect lipid-protein substrates from
intralysosomal degradation within macrophages. Protection against low pH that
exists within the phagolysosmes, is accomplished by the action of a membrane
proton translocating ATPase which is located on the cytoplasmic side of the
parasite surface membrane and acts by coupling ATP hydrolysis to proton-
pumping activity (Chang et al., 1990).
The parasites multiply by binary fission within the phagolysosomes. The infected
macrophages secrete colony-stimulating factors which stimulate precursor cells of
macrophages thereby providing new target cells for the parasites and form a
granuloma with epitheliod cells and giant�s cells in the dermatotropic species and
hyperplasia of the reticuloendothelial cells in the viscerotropic species (WHO,
1990). In Kala-azar, the major host response is cellular and the amastigotes in
macrophages are killed due to increased production of oxygen and nitric oxide
metabolites in the macrophages (Liew et al., 1990) stimulated by lymphokines
particularly interferon- (IFN-), from activated T-helper 1 cells generated during
the immune response. The released amastigotes are destroyed extracellularly with
the appearance of delayed type hypersensitivity. So, in majority cases, the
infections are mild and self-limiting (WHO, 1990). A small fraction of individuals
who develop specific suppression of cell-mediated immunity permits the

38
dissemination and uncontrolled multiplication of parasites leading to disease and
complications.
2.9 Immunology of leishmaniasis
2.9.1 Natural Immunity
There is considerable variation in the immune response to infection in Kala-azar.
Some animals are completely resistant to infection and disease in man is very
variable. Resistance to infection with Leishmania donovani subspecies infection
in mice has been shown to be under genetic control. Infection with all forms of
leishmaniasis which is allowed to run its natural course immunizes against
reinfection with the homologous strain. Once recovered from infection either
naturally or by chemotherapy, established immunity is life long and their are no
reliable records of second attack of Kala-azar (Manson-Bahr and Bell, 1991).
2.9.2 Acquired immunity
In Kala-azar there is a wide spectrum of disease varying from self-healing and
subclinical infection to overt kala-azar syndrome. The majority of infection is
self-healing and subclinical. In a study in Italy it was found that clinical illness
developed in only 3% of those infected (Pampiglione et al. 1975), while others
suggested that upto 85% of infected individuals may spontaneously control
infection (Evans et al. 1992; Holaday et al. 1993).
Infection with subspecies of Leishmania donovani complex is associated with a
profound impaired cellular response but marked humoral response. Kala-azar
patients fail to respond to L. donovani antigens in term of delayed type
hypersensitivity (Rezai et al., 1978; Ho et al., 1983; Halder et al., 1983)
lymphocyte proliferation in response to both mitogens (Ghose et al., 1979) and
parasite antigens (Halder et al., 1983) and IL-2 and IFN- production in vitro and

39
in vivo (Carvalho et al., 1985). However, these immune responses are restored
after successful chemotherapy (Manson-Bahr and Bell, 1991).
2.9.3 Genetic regulation of Leishmaniasis
Several different host genes control the infection of distinct species of
Leishmania. In the murine model, two stages of genetic regulation have been
identified-primary innate susceptibility or resistance and subsequent acquired
immunity. This division is particularly clear in Kala-azar where innate
susceptibility or resistance is controlled by expression of a single autosomal
dominant gene locus designated Lsh/Ity/Bcg (Bradley, 1977; Plant et al., 1982).
Natural resistance-associated macrophage protein gene 1 (Nramp1) which had
been isolated as a candidate gene has been established as an allele of Lsh/Ity/Bcg
(Formica et al., 1994). Acquired immunity is controlled by at least three
additional genes Rld-1 (lined to major his compatibility loci, H-2), H-11 linked
gene and Ir-2 (Leiw and O�Donnell, 1993).
As yet little is known about the effect of genetic variation on leishmaniasis in
humans and genetic determinants of human leishmaniasis have yet to be
elucidated. However both visceral and cutaneous leishmaniasis can exhibit a wide
clinical spectrum in human. In mucocutaneous leishmaniasis caused by L.
braziliensis, sign of genetic variation have been recorded (Walton and Valverde,
1979).
2.9.4 Humoral immune response
A marked hypergammaglobulinemia and absence of detectable cell-mediated
immunity are the principal immunological features of Kala-azar. There is
significant increase in IgG and IgM levels but not of IgA levels (Aikat et al.,
1979; Galvao-Castro et al., 1984). The rise in IgM level is less than that of IgG.

40
Elevated level of IgE was also detected in sera of Kala-azar patients (Peleman et
al., 1989).
Both Leishmania-specific and non-specific antibodies are produced during
infection with Leishmania species. Although both IgM and IgG antileishmanial
antibodies were detected in sera from Kala-azar patients, IgG titre higher than
those of IgM. No correlation was found between the titre of IgG or IgM
antileishmanial antibodies and the total serum levels of IgG or IgM (Galvao-
Castro et al., 1984). By immunoblot analysis Leishmania specific antibodies in
the IgG and IgM classes were observed but there was no reactivity in the IgA and
IgE classes (Rolland-Burger et al., 1991).
Non-specific antibodies reacting to haptens, foreign proteins and autoantigens
have been detected in sera of Kala-azar patients. Autoantibodies to IgG, smooth
muscles and single-stranded DNA was observed in many instances (Aikat et al.,
1979; Galva-Castro et al., 1984).
The marked increase of immunoglobulin levels in patients with Kala-azar and
demonstration of variety of antibodies against proteins unrelated to parasite and
against autoantigens is suggestive of polyclonal B cell activation (Weintraub et
al., 1982; Campos Neto and Bunn-Moreno, 1982). The mechanisms that may
induce polyclonal B cell activation are not completely understood but it has been
demonstrated that parasite-derived mitogens from promastigotes induce
polyclonal B cell activation in animal�s activation (Weintraub et al., 1982;
Campos Neto and Bunn-Moreno, 1982). Activated T cells were demonstrated
in Kala-azar patients which secrete cytokines involved in B cell growth and
differentiation and thus may contribute to the polyclonal B cell activation
(Raziuddin et al., 1992).

41
Antileishmanial antibodies have been shown in vitro to lyses promastigotes in
presence of complements (Pearson and Steigbigel, 1980; Mosser and Edelson,
1984) to promote phagocytosis (Herman, 1980). However there is little evidence
for a corresponding role in-vivo for antibody in determining the outcome of
leishmanial infection. Available evidence argues strongly against a protective role
in controlling leishmaniasis.
The increased level of Leishmania-specific and non-specific antibodies has been
exploited in the various serological diagnosis of Kala-azar.
Hypergammaglobulinemia has formed the basis of aldehydes or formol gel test
used for identifying patients of Kala-azar. Cross-reactive antibodies gives rise to
positive results in complement fixation test using mycobacterial antigens.
Leishmania-specific antibodies are detected by ELISA, IFAT and DAT using
either whole or whole cell lysate as antigens and are used for specific diagnosis of
leishmaniasis. In polypeptides are detected for the purpose of diagnosis of
leishmaniasis.
2.9.5 Cell-mediated immune response
Leishmaniasis has emerged as a model system for study of T-cell mediated
immunity. Several cell types may have a role in cell-mediated immune response
in leishmaniasis but the principal role is played by CD4+ T helper cells.
Natural killer (NK) cells
Little work has been done on the possible role of NK cells in leishmaniasis. It has
been shown that mice profoundly deficient in NK cell activity are modestly
capable of eliminating L. donovani infection than the normal controls. Therefore
NK cells may be involved in the response to Kala-azar. It has been shown that
early presence of IFN- favors CD4+ Th1 cell differentiation from precursor from
precursor cells (Bray, 1985) and IFN- may be derived from NK cells (Scott,

42
1993), thereby may help in developing protective immunity in Leishmania
infection.
T cells
Activated T cells were demonstrated in the peripheral blood of patients with
Kala-azar (Raziuddin et al., 1992) secreting elevated levels of cytokines involved
in B cell growth and differentiation which may contribute to the polyclonal B cell
activation associated with Kala-azar.
CD8+ T cells
Cytotoxic T lymphocytes have traditionally been associated with resistance to
viral infections but three is now increasing evidence that these cells my also have
an important role in immunity to intracellular microbes (Kaufmann, 1988). This
may be due to direct cytotoxicity or through cytokine production. CD8+ T cells
produce IFN- and TNF- (Fong and Mosmann, 1990). Both these lymphokines
are known to be important in activating macrophages to kill Leishmania.
Therefore, CD4+T cells and CD8+T cells may interact to kill Leishmania-infected
macrophages both by production of lymphokines and by direct lysis. Therefore it
is likely that a greater role of CD8+T cells in the control of leishmaniasis will be
apparent.
CD4+T cells
Although a role for CD8+T cells in leishmaniasis appears to be likely the
important cell in mediating protective immunity in leishmaniasis is undoubtedly
the CD4+T cells. The subsets of CD4+T cell are directly related to both protective
immunity and disease exacerbation. Resistance of Leishmania infection and early
cure is associated with a T helper (Th)-1 cell response while disease progression
is related to Th2 type response.

43
For a clear understanding of the involvement of the subsets of CD4+T helper cells
in cell-mediated immune response in leishmaniasis a short overview of CD4+T
helper cells is given below.
CD4+T Helper Cells in Leishmaniasis
Evidences has accumulated that the differential immune response observed in
resistant and susceptible mice and healed and non-healed mice was due to
activation of different CD4+T cell subsets. Although there is a difference between
mice and humans in terms of the immune response to infection with different
Leishmania species, several important principles have emerged. Resolution of
leishmanial infection and protection against reinfection in susceptible humans and
mice is governed by the expansion of Leishmania-specific helper T cells of the
CD4+ Th1 cell type that produce IFN-. When present these cells activate
macrophages to kill intracellular amastigotes (Murray et al., 1983; Sundar et al.,
1994). IL-2 appears to play an important role in promoting the development of
protective Th1 responses (Murray and Hariprasad, 1995). TNF- also appears to
exert its leishmanicidal activity by activating macrophages, rather than acting
directly on the parasite (Liew et al., 1990). Membrane-associated TNF- on
Leishmania specific CD4+T cells may be important in sending activation signals
to infected macrophages, resulting in parasite killing (Sypek and Wyler, 1991).
During progressive systemic infections in mice, there is expansion of CD4+T cells
of the Th2 type that secrete IL-4, but not INF- or IL-2 inc response to
leishmanial antigens. IL-4 suppresses the development of murine Th1 response
and activation of macrophages by INF- (Liew et al., 1989). Similar reciprocal
activity of IL-4 and INF- was obtained with human monocytes infected with L.
donovani (Lehn et al., 1989). In human with Kala-azar, IL-10 rater than IL-4 may
be responsible for the suppression of potentially protective Th1 responses

44
(Holaday et al., 1993) but others suggested that IL-4 may play a more prominent
role than IL-10 in Indian kala-azar (Sundar et al., 1997).
2.9.6 Clinical features
Kala-azar has got various other names. These include Dum-dum fever, Sikari
disease, Burdwan fever and Shahib�s disease. However, the most commonly used
term is Kala-azar, which in Hindi means black sickness or black fever. The terms
originally referred to Indian. Kala-azar due to its characteristic symptoms,
blackening or darkening of the skin of the hands, feet face and the abdomen
(Lainson and Shaw, 1987). Typical Kala-azar usually occurs after an incubation
period of 2-6 months depending on the patient�s age, immune status and the
species of Leishmania. In endemic cases of Kala-azar, the disease is chronic and
onset is gradual. Although people of all ages are susceptible in the old world,
children below the age of 15 are more commonly affected with L. infantum being
largely responsible (Rab and Evans, 1995). The symptoms of Kala-azar vary
between individuals and according to geographical foci. Common symptoms
included high undulating fever often with 2-3 peaks in 24 hours and drenching
sweats which can easily be misdiagnosed as malaria. Chills, rigors, weight loss,
fatigue, cough, burning feet, insomnia, abdominal pain, joint pain, epistaxis and
diarrhoea might be associated. Most commonly observed clinical signs include
massive splenomegaly, hepatomegaly and anaemia. The duration of the disease
can be 1-20 weeks (Hashim et al., 1995). Splenic enlargement along with
hepatomegaly causes an abdominal protuberance in these patients.
While the disease progresses, the spleen extends well below the costal margin; it
is usually firm or hard in consistency but soft spleen may be encountered in acute
disease (Bryceson, 2000; Sundar et al., 2001). Late in the course, epistaxis and
gingival bleeding caused by severe thrombocytopenia may occur; oedema and

45
ascites may also develop. Jaundice with mildly elevated enzyme levels is rarely
seen and is considered to be a bad prognostic sign. Commonly encountered
laboratory findings include normocytic-normochromic anaemia, neutropenia,
thrombocytopenia, hypergammaglobulinemia as a result of polyclonal B cell
activation and hypoalbuminemia. Serum levels of hepatic transaminases may be
elevated (Totan et al., 2002; Haider et al., 2001; Peacock, 2001; Maltezou et al.,
2000; Minodier and Garnier, 2000).
2.10 Complications of Kala-azar
Kala-azar is commonly complicated by secondary infections, such as pneumonia,
bronchial infections, tuberculosis, malaria, diarrhoea or dysentery, viral
infections, bacterial skin infections, otitis media and Cancarum oris.
Thrombocytopenia may cause epistaxis or bleeding from other sites and this may
precede death. Leishmania enteritis may be a cause of diarrhoea and
malabsorption and pulmonary involvement may mimic pneumonia, Mortality is
related to immunosuppression causing secondary infections and hemorrhage and
in untreated cases mortality ranges from 75-95% (WHO, 1996; Peter, 2000).
2.11 Sequalae of Kala-azar and post Kala-azar dermal leishmaniasis (PKDL)
PKDL has been reported from India (Prasad, 1999) occur in approximately 10%
of cases after the treatment of Kala-azar. Lesions can develop at late as 1-2 years
after treatment for the original disease and manifest on the face, trunk or
extremities and may persist for as long as 20 years. In Africa, it has reported that
dermal lesions occur in only 2% of cases and tend to appear during or shortly after
the treatment and persist only for a few months (Zijlstra et al., 2000; Prasad,
1999). PKDL is characterized by a patchy and raised maculopapular rash and
changes in skin colour. Late manifestations are papules, papules or nodules. It
may be confused with lepromatous leprosy, fungal infections, diffuse cutaneous

46
leishmaniasis or other skin disorders. Pentavalent antimony at a dose of 20mg/kg/
day for 4 months or longer is used to treat Indian PKDL. In Ethiopia, Kenya and
Sudan, PKDL is treated for 2-3 months. Once lesions improve clinically treatment
may be stopped, as PKDL very rarely relapse (WHO, 1996).
2.12 Differential diagnosis of Leishmaniasis
The differential diagnosis of Kala-azar includes malaria, tropical splenomegaly
syndrome, schistosomiasis, cirrhosis with portal hypertension, African
trypanosomiasis, milliary tuberculosis, brucellosis, typhoid fever, bacterial
endocarditis, histoplasmosis, malnutrition, lymphoma and leukemia. Similarly
numerous primary and secondary skin disease and conditions are frequently
misdiagnosed as early lesions of cutaneous leishmaniasis. Some of the common
conditions that that should be differentiated from cutaneous leishmaniasis are
tropical ulcers due to other causes, impetigo, infected insect bits, leprosy, lupus
vulgaris, yaws, blastomycosis and skin cancer (Herwaldt, 1999). Mucocutaneous
leishmaniasis is sequelae of new world cutaneous leishmaniasis and results from
direct extension or hematogenous or lymphatic metastasis to the nasal or oral
mucosa. Paracoccidioidomycosis, polymorphic reticulosis, Wegener�s
granulomatosis, lymphoma, histoplasmosis, yaws, tuberculosis, nasopharyngeal
carcinoma and other destructive lesions are frequently misdiagnosed as early
lesions of mucocutaneous leishmaniasis (Herwaldt, 1999; El-Hassan and Zijlstra,
2001). Hence other diagnostic methods are required to confirm the clinical
suspicion (Herwaldt, 1999).
2.13 Laboratory diagnosis
Laboratory diagnosis of leishmaniasis can be made by the following:
(i) demonstration of parasite in tissues of relevance by light microscopic
examination of the stained specimen, in vitro culture, or animal inoculation; (ii)

47
detection of parasite DNA in tissue samples; or (iii) immunodiagnosis by
detection of parasite antigen in tissue, blood, or urine samples, by detection of
nonspecific or specific antileishmanial antibodies (immunoglobulin), or by assay
for Leishmania-specific cell-mediated immunity (Sundar and Rai, 2002).
2.13.1 Microscopic examinations
The commonly used method for diagnosing Kala-azar has been the demonstration
of parasites in splenic or bone marrow aspirate. The presence of the parasite in
lymph nodes, liver biopsy, or aspirate specimens or the buffy coat of peripheral
blood can also be demonstrated. Amastigotes appear as round or oval bodies
measuring 2 to 3 µm in length and are found intracellularly in monocytes and
macrophages. In preparations stained with Leishman�s stain, the cytoplasm
appears pale blue, with a relatively large nucleus that stains red. In the same plane
as the nucleus, but at a right angle to it, is a deep red or violet rod-like body called
a kinetoplast.
After identification, parasite density can be scored microscopically by means of a
logarithmic scale ranging from 0 (no parasite per 1,000 oil immersion fields) to +6
(>100 parasites per field) (Chulay and Bryceson, 1983). The sensitivity of the
bone marrow smear is about 70% or lower (Boelaert et al., 1999 and Chowdhury
et al., 1993). Splenic aspirate, though associated with rise of fatal hemorrhage in
inexperienced hands, is one of the most valuable methods for diagnosis of kala-
azar, with a sensitivity exceeding 90% (Zijlstra et al., 1992 and Kager et al.,
1983). It required no special equipment, from the patient�s standpoint is generally
preferable than the more painful bone marrow aspirate, and has proven to be safe
and relatively easy to perform in experimented hands. For patients suspected of
have Kala-azar, splenic aspirate can be performed even when spleen is not
palpable, after demarcating the area of splenic dullness by percussion. The only

48
risk of splenic puncture is bleeding from a soft and enlarged spleen. Occasionally
amastigotes have also been demonstrated in liver biopsy (50-80% sensitivity) and
lymph node aspirate (56% sensitivity) (Zijlstra et al., 1992).
Blood buffy coat:
Another interesting field for definitive diagnosis of Kala-azar might be the
detection of LD body from buffy coat smear of peripheral blood. Because, when
blood is settled in presence of an anticoagulant, the WBCs are deposited and
concentrated in buffy coat at the interface of RBCs and plasma (Shamszzaman et
al., 2007). Under all aseptic precautions (cleaning the area with povidone iodine
followed by adequate rubbing by alcohol pad containing 60% isopropyl) 3 ml of
venous blood will be collected from median ante-cubital or appropriate veins by
gentle suction. The collected blood sample will immediately be introduced into a
3.2% Tri-sodium citrate coated vacutainer. In a sterile round bottom disposable
test tube (10 ml capacity), 2 ml of Histopaque will be pipette carefully. Then 2 ml
of citrated blood from the vacationer will be poured slowly by the side of the test
tube so that the blood completely overlay the separation fluid with out any
mixing. The test tube will now be centrifuged for 15 minutes. 3000 rpm referable
in a swinging centrifuge machine. After centrifugation, a thick buffy coat will be
seen in the interface of plasma and Histopaque. RBCs will be settled at the bottom
part of the test tube. Now the plasma will be gently sucked by a micropipette of
1ml capacity and will be preserved in an eppendrof tube. Then by another of 100-
200 µl capacity, the buffy coat material will be sucked out very gently. Part of the
material will be used in another eppendrof containing 0.5 ml of 95% alcohol for
analysis by PCR (DGHS, 2009).
If buffy coat is considered for making a smear, it is supposed to show high content
of WBCs including monocytes. The monocytes parasitized by LD bodies, if

49
present, will be higher in the smear. Thus the rate of LD body positivity is
expected to be higher (Shamszzaman et al., 2007).
2.13.2 Culture of Leishmanial parasites
Culture of parasite and improve the sensitivity of parasite isolation but
Leishmania culture is rarely needed in routine clinical practice. However, cultures
are required for (i) obtaining a sufficient number of organisms to use an antigen
for immunologic diagnosis and speciation (ii) obtaining parasites to be used in
inoculating susceptible experimental animals (iii) in vitro screening of drugs (iv)
accurate diagnosis of the infection with the organism. Leishmania strains can be
maintained as promastigotes in artificial culture medium. The culture media used
may be monophonic (Schneider�s insect medium, M199 or Grace�s medium) or
diphasic (NNN and Tobies medium). Most preferable diphasic medium is that
which contain modified diphasic rabbit blood agar overlaid with RPMI 1640 from
Gibco BRI, Grand Island, N.Y for primary isolation and M199 medium
containing 20% fetal calf serum to amplify parasite numbers (Sundar et al., 2001).
Hockmeyer�s medium, which is Schneider�s commercially prepared culture
medium supplemented with 30% heat-inactivated fetal calf serum with 100 IU of
penicillin and 100 µg of streptomycin, is simple to use and satisfactory for
diagnosis of Kala-azar but it is expensive. Culture tubes are inoculated with 1 to 2
drops of bone marrow or splenic aspirate and incubated at a temperature between
22 0C and 280C. The tubes are examined weekly for the presence of promastigotes
by phase contrast microscopy or by wet mount of culture fluid for 4 weeks before
being discarded as negative. If promastigotes are present, they are maintained by
weekly passage to fresh medium. Blood can also be used to isolate the parasite but
the method is slow and takes longer. Aseptically collected blood (1 to 2 ml) is
diluted with 10 ml of citrated saline and the cellular deposit obtained after
centrifugation is inoculated in culture media. Contamination of the culture media

50
by bacteria of yeast species or other fungi usually complicates the culture but can
be avoided by use of good sterile techniques and be the addition of penicillin (200
IU/ml), streptomycin (200µg/ml) and 5-flucytosine (500µg/ml) to the medium
(Schur and Jacobson, 2001). In vitro culture of the amastigotes is done for
chemotherapeutic studies and to study the interrelationship of the amastigotes and
macrophages. The amastigotes are grows in tissue or macrophage culture. These
cell lines are produced from (i) human peripheral blood monocytes by density
sedimentation with lymphocyte separation medium from LSM; Organon-Teknika,
Durham, N.C (Grogl et al., 1993) (ii) macrophage cell lines, e.g., P388D and
J774G8 lines from mice; and (iii) dog sarcoma and hamster peritoneal exudates of
cell line, in which case continuous culture can be achieved (Schur and Jacobson,
2001).
Species recognition:
In areas of endemicity, recognition of species of Leishmania is rarely required.
However, identification of an organism to the species level is helpful
epidemiologically and is also important for the treatment and prognosis
determination for global travelers who are not immune to the parasite and tend to
develop unusual manifestations of the disease (Magill et al., 1993). Identification
of species of the L. donovani complex is particularly difficult, because
morphologically the species are almost indistinguishable from each other. For
species level identification, a large amount of promastigotes is obtained by culture
of the organism and the species-specific isoenzyme pattern is analyzed by
cellulose acetate electrophoresis (Kerutzer et al., 1993). Typing of washed live
promastigotes by direct agglutination test with species-specific monoclonal
antibodies is another highly sensitive taxonomic tool frequently utilized for this
purpose (Jaffe and Sarfstein, 1987). Species-level identification can also be done
by analysis of amplified minicircle kinetoplast DNA (kDNA), by choosing

51
primers from conserved regions of different Leishmania species kDNA
minicircles (Sacks et al., 1995; Smyth et al., 1992). Yet another method used for
identification of species of Leishmania is the analysis of the in vitro promastigotes
released antigenic factors, which are different for different leishmanial species
(IIg and Stierhof, 1994).
2.13.3 Immune diagnosis
Specific serological tests
Antigen Detection
The antigen detection is an ideal method of diagnosing an infection. Antigen
levels are expected to broadly correlate with the parasite load as well. Antigen
detection is more specific than antibody-based immunodiagnostic tests (De
Colmenares et al., 1995; Vinayak et al., 1994). This method is also useful in the
diagnosis of disease in cases where there is deficient antibody production (as in
AIDS patients). De Colmenares et al., (1995) from Spain have reported two
polypeptide fractions of 72-75kDa and 123 kDa in the urine of kala-azar patients.
The sensitivities of the 72-75-kDa fractions were 96% and the specificities were
100%. Besides, these antigens were not detectable within 3 weeks of anti-kala-
azar treatment, suggesting that the test has a very good prognostic value.
However, detection of antigen in the patient�s serum is complicated by the
presence of high level of antibodies, circulating immune complexes, serum
amyloid, rheumatoid factor and autoantibodies; all of which may mask
immunologically important antigenic determinants or competitively inhibit the
binding of free antigen (Senaldi et al., 2001). A new latex agglutination test
(KATEX) for detecting leishmanial antigen in urine of patients with Kala-azar has
showed sensitivities between 68 and 100% and a specificity of 100% in
preliminary trials. The antigen is detected quite early during the infection and the
results of animal experiments suggest that the amount of detectable antigen tends

52
to decline rapidly following chemotherapy. The test performed better than any of
the serological tests when compared to microscopy. Large field trials are under
way to evaluate its utility for the diagnosis and prognosis of Kala-azar (Attar et
al., 2001). Whether the test the applications for the detection of asymptomatic
cases of Kala-azar and monitoring therapy is yet to be confirmed. There are no
antigen detection systems currently available for CL and MCL.
Enzyme Linked Immunosorbent Assay:
The Enzyme Linked Immunosorbent Assay (ELISA) is a valuable tool in the
serodiagnosis of leishmaniasis. The test is useful for laboratory analysis as well as
for field applications. However, the sensitivity and specificity of ELISA is greatly
influenced by the antigen used. More recently, several recombinant antigens like
rGBP form L. donovani, rORFF from L. infantum, rgp63, rK26 and rK39 from L.
chagasi have been developed and tested (Montoya et al., 1997; Martin et al.,
1998; Jensen et al., 1999; Rajasekariah et al., 2001; Raj et al., 1999; Bhatia et al.,
1999). Of these, the rK39 antigen is found to be highly sensitive and predictive of
onset of disease manifestation of Kala-azar patients. In contrast, it doe not show
detectable antibodies in CL or MCL. The antibody titres to this antigen directly
correlate with active disease and have potential in monitoring the chemotherapy
and in predicting the clinical relapse. In addition rK39 ELISA has a high
predictive value for detecting Kala-azar in immunocompromised person, like
those with AIDS. A kit (InBios, USA) using this antigen and based on lateral flow
is now commercially available in the form of antigen-impregnated nitrocellulose
paper strips adapted for the use under field conditions. The rK39 strip test has
been found highly sensitive has a reliable indicator of KA in Indian subcontinent
(Burns et al., 1993; Singh et al., 1995; Singh et al., 2002; Houghton et al., 1998).
Due to lack of facilities and expertise to perform skin biopsy, smears and cultures
the proper diagnosis of cutaneous leishmaniasis remains under reported. The use

53
of serological tools in the diagnosis of CL has also been assessed by a number of
studies. ELISA using few recombinant antigens viz., gene B protein (GBP) from
L. major recombinant major surface glycoprotein, gp63, from L. major and 2
recombinant proteins, T26-U2 and T26-U4 from Viannia peruviana have been
tried but have been found less suitable for diagnosing CL (Schalling et al., 2001;
Zijlstra et al., 2001). Laboratory diagnosis of MCL has also taken a back seat and
most of the cases are diagnosed clinically.
Antibody detection
Conventional methods for antibody detection included gel diffusion, complement
fixation test, indirect haemagglutination test, IFA test, and countercurrent
immuoelectrophoresis (Bray et al., 1985, Haldar et al., 1981 and Hockmeyer et
al., 1984).
Indirect fluorescent antibody test (IFAT):
The Indirect fluorescent test is one of the most sensitive tests available. The test is
based on detecting antibodies which are demonstrated in the very early stages of
infection and are undetectable six to nine months after cure. If the antibodies
persist in low titers, it is a good indication of a probable relapse a probable
relapse. Titers above 1:20 are significant and above 1: 128 are diagnostic
(Davidson, 1998). There is a possibility of a cross reaction with trypanosomal
sera. However this can be overcome by using Leishmania amastigotes as the
antigen instead of the promastigotes (Weigle et al., 1987). Although this test is
more sensitive (96%) and specific (98%) than soluble antigen of ELISA, it is
cumbersome and not suitable for field conditions (Sassi et al., 1999).
Direct agglutination test (DAT):
The direct agglutination test is a highly specific and sensitive test. It is
inexpensive and simple to perform making it ideal for both field and laboratory

54
use. The method uses whole, stained promastigotes either as a suspension or in a
freeze-dried form. The freeze-dried form is heat stable and facilitates the use of
DAT in the field. However, the major disadvantage of DAT is the relative long
incubation time of 18 hours and the need for serial dilutions of serum (Gari-
Toussaint et al, 1994; Schallig et al., 2001). DAT has no prognostic value too.
The test may remain positive for several years after cure. Recently, Schoone et al.
(2001) have developed a fast agglutination-screening test (FAST) for the rapid
detection (<3 hours) of anti-leishmania antibodies in serum samples and on blood
collected on filter paper. The FAST utilizes only one serum dilution leading to
qualitative results. It offers the advantages of DAT based on the freeze-dried
antigen regarding reproducibility, specificity and sensitivity.
DAT has been evaluated for the diagnosis of CL using L. major promastigotes and
was shown to be highly (90.5%) specific and sensitive (Schallig et al., 2001).
Immuno-chromatography test (ICT):
A recombinant antigen, rK39 has been shown to be specific for antibodies in
patients with Kala-azar caused by members of the L. donovani complex (Badaro
et al., 1996; Bern et al., 2000; Burns et al., 1993). This antigen, which is
conserved in the kinesin region, is highly sensitive and predictive of the onset of
acute disease. The antigen is derived from L. chagasi, which in the Untied States
is used for veterinary purposes, thought it is not approved for human use. High
antibody titers in immunocompetent patients with Kala-azar have been
demonstrated. This antigen has been reported to be 100% sensitive and 100%
specific in the diagnosis of Kala-azar and PKDL by ELISA (Kumar et al., 2001;
Singh et al., 1995). Another important facet of anti-rK39 antibody is that the titer
correlates directly with the disease activity, indicating its potentiality for use in
predicting response to chemotherapy. It was previously shown that anti-rK39

55
antibody titers were 59-fold higher than those of antibody against CSA at the time
of diagnosis, and with successful therapy, it fell sharply at the end of treatment
and fell further during follow-up monitoring. In patients who experience disease
relapse, the titer rose steeply again (Kumar et al., 2001). The diagnostic and
prognostic utility of rK39 for HIV-infected patients has also been demonstrated
(Houghton et al., 1998). Because of the conditions prevailing in areas of
endemicity, any sophisticated method cannot be employed on a wider scale. There
is a need for a simple rapid and accurate test with good sensitivity and specificity,
which can be used without any specific expertise. A promising ready to use
immunochromatographic strip test based on rK39 antigen has been developed as a
rapid test for use in difficult field conditions. The recombinant antigen is
immobilized on a small rectangular piece of nitrocellulose membrane in a band
form and goat-anti-protein A is attached to the membrane above the antigen band.
After the finger is pricked, half a drop of blood is smeared at the tip of the strip,
and the lower and end of the strip is allowed to soak in 4 to 5 drops of phosphate-
buffered saline, placed on a clean glass slide or tube. If the antibody is present, it
will react with the conjugate (protein A colloidal gold) that is predried on the
assay strip. The mixture moves along the strip by capillary action and reacts with
rK39 antigen on the strip, yielding a pink band. In the strip of patients who are
infected, two pinkish lines, appear in the middle of the nitrocellulose membrane
(the upper pinkish band serves as a procedural control). In the first extensive field
trial in 323 patients, we found the strip test to be 100% sensitive and 98% specific
(Sundar et al., 1998). Several studies from the Indian subcontinent reported the
test to be 100% sensitive (Bern et al., 2000; Sundar and Rai, 2002; Sundar et al.,
1998). However, when evaluated in Sudan, the sensitivity of the test was only
67%. In the Sudan study, all the parasitologically confirmed Kala-azar patients
who tested negative by the rK39 strip test showed IgG against rK39 by micro-

56
ELISA at lower titers (Zijlstra et al., 2001). In a study done in southern Europe,
the rK39 strip test results were positive in only 71.4% of the cases of Kala-azar
(Jelinek et al., 1999). These differences in sensitivity may be due to differences in
the antibody responses observed in different ethnic groups (Singh et al., 1995).
When tested for PKDL, the test had 91% sensitivity (Salotra et al, 2001). High
levels of specificity (97 to 100%) have been reported uniformly for this test;
however, with a later version of the rK39-treated strips, some (12.5%) healthy
endemic control subjects also tested positive ( Sundar and Rai, 2002). While such
reactions might be considered to be false positive, these probably represent
subclinical infections: PCR assay for L. donovani was positive in a few of these
cases (Salotra et al., 2001; Sundar and Rai, 2002). Anti-rK39 IgG may be present
in serum for an extended period after successful treatment for Kala-azar; thus,
patients with suspected relapse of Kala-azar with a past history of infection would
not be candidates for diagnosis by strip testing. Another drawback of this format
is that an individual with a positive rK39 strip test result may suffer from an
illness (malaria, typhoid fever or tuberculosis) with clinical features similar to
those of Kala-azar yet be misdiagnosed as suffering from Kala-azar. Besides these
limitations, the rK39 ICT strip test has proved to be versatile in predicting acute
infection. It is the only available format for diagnosing Kala-azar with acceptable
sensitivity and specificity. It is also inexpensive and simple (Sundar and Rai,
2002).
Complement Fixation Test (CFT):
Complement fixation test was introduced more than 60 years ago in the diagnosis
of Kala-azar (Smith et al., 1984). Since then it has been used as a serodiagnostic
tool in different countries. Although, CFT becomes positive in the early stage of
infection (in 3 weeks) it is a non-specific test. Sensitivity of this test varies with
the source and method of extraction of antigen and also with different batches of

57
antigen (Aikat et al., 1979) and false positive results are encountered. In addition,
the test has shown to be of limited value in routine laboratory diagnosis (Duxbury
and Sadun, 1964) and large-scale epidemiological studies (Hommel et al., 1978).
Due to its complicated procedure that test can only be performed in reference
laboratories (Rahman and Islam, 1979).
Immunoblotting:
Most of the work concerning the use of immunoblotting in the diagnosis of
leishmaniasis has been done on Kala-azar. In CL, anti-leishmanial antibodies
though detectable, are present in low titres, Hence Immunoblotting is not widely
adopted for diagnosing CL. The biggest advantage of immunoblotting, as a
general rule, is that various antigens expressed and antibodies recognized during
the course of infection can be documented. It also has an advantage of permanent
documentation. However, the technique is not user-friendly and limited only to
research laboratories (Herwaldt, 1999; Weigle et al., 1987; Maalej et al., 2003;
Brito et al., 2000).
Non-specific tests
Formol gel test
The formol get test has the advantage of being cheap and simple to perform.
Serum obtained from about 5 ml of blood is mixed with one drop of 30%
formaldehyde. A positive reaction is shown if the mixture solidifies and forms a
white opaque precipitate within 20 minutes. A positive test can not be detected
until 3 months after infection and becomes negative 6 months after cure. The test
is non specific since it is based on detecting raised levels of IgG and IgM which
also result from African trypanosomiasis, malaria and schistosomiasis (WHO,
1991).

58
Skin testing (Cell-mediated immunity)
Leishmanin Skin Test (LST):
Delayed hypersensitivity is an important feature of cutaneous forms of human
leishmaniasis and can be measured by the leishmanin test, also known as the
Montenegro reaction. Leishmanin is a killed suspension of whole (0.5-1×10/ml)
or disrupted (250 g protein/ml) promastigotes in pyrogen-free phenol saline
(Palma and Gutierrez, 1991). No cross-reactions occur with Chagas� disease but
some cross-reactions are found with cases of glandular tuberculosis and
lepromatous leprosy. Leishmanin skin test in usually used as an indicator of the
prevalence of cutaneous and mucocutaneous Leishmaniasis in human and animal
populations and successful cures of Kala-azar (Weigle et al., 1987; Singh, 1999;
Manson-Bahr, 1987). During active KA disease there will be no or negligible cell
mediated immune response. However, the leishmanin antigen is not commercially
available and no field study has been carried out in India.
2.13.4 Molecular method
Microscopy and culture have the limitations of low sensitivity and are time
consuming. The immunological methods fail to distinguish between past and
present infections and are not very reliable in immunocompromised patients.
While the molecular approach is capable of detecting nucleic acids unique of the
parasite, it would address these limitations. A variety of nucleic and detection
methods targeting both DNA and RNA have been developed. Recently molecular
probes, using kinetoplast DNA (kDNA), ribosomal RNA (rRNA), mini exon
derived RNA (med RNA) and genomic repeats have been evaluated and used to a
much higher sensitivity and specificity. The distinct advantages of DNA
hybridization are that large numbers of samples can analyzed quickly without
compromising efficiency and many different types of samples can be processed

59
including blood spots, tissue samples, splenic and bone marrow aspirates (Wilson,
1995). The probes are more specific since they are made to target regions on the
kDNA or rRNA, which are unique to a particular Leishmania species, complex or
isolate (Williams et al., 1995). A probe has been developed to recognize the
genomic DNA repeat Lmer2 that is specific for L. donovani. This probe has been
used to diagnose infection in human Kala-azar patients and in P. martini, the
sandfly vector of L. donovani (Wilson, 1995). The early DNA probes were
labeled with radioactive 32p, which were easy to use and gave high signals with
low background interference. However, these were for laboratory use only and
were not suitable as diagnostic kits, since 32p has a short half-life of about 14 days
and being radioactive they presented safety problems. Recently, a
chemiluminescent hapten digoxigenin labeled kDNA probes have been
developed. These probes are as sensitive as the 32p labeled probes (can detect as
low as 100 parasites) and are stable enough to use in diagnostic kits, which are
more practical in the field. However, high background signals make result
interpretation difficult (Wilson, 1995; Williams et al., 1995). The latest
developments in molecular diagnostic technology have come about as a direct
result of the advent of the polymerase chain reaction (PCR). The PCR is able to
amplify small amounts of DNA or RNA to larger usable quantities (Nuzum et al.,
1995; Wilson, 1995). Although the PCR is able to detect a single copy of target
DNA, repeat sequences are used to improve sensitivity. The PCR assay can detect
parasite DNA or RNA a few weeks ahead of appearance of any clinical signs or
symptoms. Different DNA sequence in the genome of Leishmania like it�s region,
gp63 locus, telomeric sequences, sequence targets in rRNA genes such as 18s
rRNA and SSU-rRNA and both conserved and variable regions in kinetoplast
DNA (kDNA) minicircles are being used by various workers (Santos-Gomes et
al., 2000; El Tai et al., 2001; Pizzuto et al., 2001; Wortman et al., 2001; Ostria

60
and Tezeda, 2002). An improved PCR solution hybridization enzyme linked assay
(PCR-SHELA) was developed and has been used to diagnose infection of L.
donovani in patients in India, Kenya and Brazil with 90% sensitivity and 100%
specificity (Nuzum et al., 1995). The PCR-SSCP technique has been developed
for the detection of sequence variation in rRNA genes within the L. donovani
species. In addition, it can be performed easily and rapidly from clinical samples
without prior need of cultivation of the parasite. PCR assay has also been used for
post-therapeutic follow-up and for detection of relapses among HIV-infected
patients using SSU rRNA gene target. The use of fluorogenic real-time PCR using
SSU-rRNA gene added with complete automation has made quantification of
parasite burden possible. Beside it is a rapid, sensitive and highly specific test
(Attar et al., 2001; Santos Gomes et al., 2000; El Tai et al., 2001; Pizzuto et al.,
2001; Wortman et al., 2001). Recently, primers was developed that can
differentiate the Indian strains causing Kala-azar and PKDL. An Alu-PCR-like
amplification was performed from the cultured L. donovani isolated from Kala-
azar and PKDL patients. The banding pattern of PCR amplicons could clearly
group all the PKDL strains in one group while Kala-azar stains had intro-species
heterogeneity as reported by us earlier (Ostria and Tezeda, 2002).
2.14 Treatment of Kala-azar
The main objectives in the treatment of kala-azar are as follows:
To cure the patient of an intracellular parasitic infection
To prevent relapse and the development of unresponsiveness
To keep hospitalization and treatment cost to minimum
The first-line drugs:
After initial assessment of the patient, start with standard first line drugs
pentavalent antimonials, sodium stibogluconate (SSG). The aim of the first line

61
therapy is to supply an adequate daily dose of SSG for long enough to achieve
parasitological cure without relapse (Chowdhury et al., 1993).
The drugs are administered parentally and are the safest currently available drugs
since they are rapidly excreted by the kidneys and there is virtually on
accumulation in the body. Potential side effects include nausea, vomiting,
diarrhoea, ECG changes and convulsions. Sodium stibogluconate is usually
administered at a dose of 20 mg/Kg body weight daily in a single dose for 28
days. Relapse cases can be treated with same therapy for 40-60 days (Abdullah,
2002).
The second-line drugs:
Second line drugs are used for resistant Kala-azar cases. Drug resistance can be
primary or secondary. Resistance can be caused by delayed diagnosis (prolong
duration of illness), interrupted and low dose treatment, immunological failure,
emergence of resistant strains of parasites and leishmaniasis associated with
AIDS. Leishmaniasis in AIDS, mainly reported from southern Europe, now is
observed in other part of the world, including South America, Asia and North
Africa. Patients with an HIV co-infection can develop unusual manifestations of
leishmaniasis. Parasitologic diagnosis by peripheral blood smear and buffy coat
smear is more rewarding in HIV co-infection because parasites are found more
commonly in the monocytes of these patients. Resistance to stibogluconate should
be treated with alternate agents, such as:
Liposomal amphotericin (0.5-3 mg/kg) on alternate days until a dose of 20
mg/kg. Liposomal amphotericin is a lipid complex with amphotericin that
is more active than amphotericin B. The 3 preparations formulated are
amphotericin B lipid complex, liposomal amphotericin B and amphotericin
B colloidal dispersion. Cure rates of more than 90% have been observed in

62
various studies. The high cost of these drugs is a disadvantage to its use in
areas where Kala-azar is prevalent.
Pentamidine (2-4 mg/kg) on alternate days for 15 doses. Pentamidine is
available in 2 preparations, pentamidine isethionate (Pentam 300) and
pentamidine dimethane sulphonate (Lomidine). Lomidine given in the
same schedule is more effective than pentamidine isethionate. The serious
adverse effects associated with pentamidine include fall in blood pressure
with an intravenous injection, hypoglycaemia, pancreatitis and reversible
renal dysfunction.
Combination of stibogluconate with drugs such as aminosidine and
interferon gamma, also has produced good results in cases showing poor
response to stibogluconate therapy alone. Aminosidine, an
aminoglycosides identical to paromomycin also has been found effective in
trials from India (http://www.emedicine.com).
Oral drugs have obvious advantages over injectables, and several oral
formulations lie allopurinal, fluconazole and autovaquone have been tested.
Stimaquine and miltefosine are two promising oral anti-leishmanial compounds.
But recent from Brazil showed poor efficacy of stimaquine compounded with
nephrotoxicity. Miltefosine, an alkyl phospholipids, developed as an antitumour
agent, is the first oral compound which has been successfully developed in India
for the treatment of Kala-azar. A daily dose of 100-150 mg for four weeks cured
>95% patients. Recommended daily dose is 100 mg for adults weighing more
than 25 kg and 50 mg for those weighing <25 kg for four weeks (Sundar, 2003).
Kala-azar treatment failure (KATF): When kala-azar does not respond to usual
therapy with sodium stibogluconate. It is usually due to inappropriate or sub-

63
optimal dose or irregular therapy. Treatment is usually with second line drugs like
resistant kala-azar cases.
Pentavalent antimonials remain the drugs of choice for treating PKDL. Injection
sodium stibogluconate at a dose of 20 mg/kg of body weight daily for 20 days.
Then 10 days interval. This is one cycle. Total 6 cycles are needed. If a second
course is required, it should be given after an interval to two months. Pentamidine
is infective in PKDL (Abdullah, 2002).
Diet: High protein and high calorie diet during the course of treatment is required
(http://www.emedicine.com).
The emergence of drug resistance in Leishmania parasites is a major obstacle to
their control. The lack of response to pentavalent antimonials in Kala-azar has
been a problem for many years and is increasing world wide, occurring in about
5-70% patients in some endemic areas (Quellette and Papadopoulou, 1993).
Prognosis to kala-azar depends upon the immune status of the host and effective
treatment. In a person who is non-immune to kala-azar, the introduced
promastigote changes to amastigote, increase in number and spread to distant
organs without hindrance and manifests the disease. If such as patient is left
untreated their mortality is very high from 90 to 95 percent in adults and from 75
to 80% in children. Death usually follows within 2 years of the onset mainly due
to some complications such as diarrhoea, amoebic and bacillary dysentery,
pneumonia, pulmonary percentage of cases, kala-azar may be self-limiting and
spontaneous cure is possible (Chowdhury et al., 1993).
2.15 Control measures for Kala-azar
Elimination of sandfly vectors:
In endemic areas such as Bangladesh and India sandfly control is often combined
with malaria control and Brazil with malaria and chagas disease control

64
programmes (WHO, 1997; Al-Masum et al., 1995). This is cost effective method
given the expense of mass spraying with chemicals such as DDT, Malathion,
Fenitrothion Propoxur and Diazinon. In endemic areas this has to be an on-going
process and continued surveillance is required to keep the vector populations low
(Al-Masum et al., 1995). In epidemics large scale spraying is required. Spraying
should particularly include animal shelters, inside of buildings and the immediate
surrounding areas. Self-protection is important, several methods are available to
aviod being bitten by sandflies including repellents such as diethyltoluamide
(DEET) applied to the exposed areas of the body and clothing. Fine mesh screens
(less than 16-mesh) can be applied to doors and windows and bed nets should be
used impregnated with insecticides such as permethrin and deltamethrin.
Mosquito nets are not effective since sandflies can get through the holes.
Elimination of reservoir hosts:
In areas of anthroponotic foci of Kala-azar active case detection and treatment is
imperative, especially asymptomatic seropositive patients. A recent study in Israel
showed that the asymptomatic or sub-clinical cases are 4-30 times greater than
those with the Kala-azar symptoms (Ephros et al., 1994). The IFAT, ELISA,
DAT, the western blot and the Leishmania skin test have all been useful in
detecting seropositive cases of Kala-azar (Rab and Evans, 1995; Shiddo et al.,
1995). In endemic areas of zoonotic Kala-azar, elimination of wild animals and
stray doges can be carried out by shooting and by using poisoned baits
impregnated with strychnine. Screening and treatment of domestic animals
especially dogs is recommended using the various available techniques including
the IFAT, DAT and ELISA (Shiddo et al., 1995).

65
CHAPTER 3
MATERIALS AND METHODS
3.1 Place and period
The study was conducted in the department of Microbiology, Mymensingh
Medical College, during the period from July 2009 to June 2010. Cases were
selected from Mymensingh Medical College Hospital and Trishal Thana Health
Complex.
3.2 Type of the study
The study was designed as a cross sectional study.
3.3 Study population
106 cases and 44 healthy controls were included in this study.
3.4 Subjects
Group I (confirmed Kala-azar cases): Clinically suspected febrile patients,
subsequently confirmed as Kala-azar by detection of LD bodies from bone
marrow aspirates by microscopy (n=41).
Group II (Serologically diagnosed Kala-azar cases): Clinically suspected
febrile patients, subsequently diagnosed as Kala-azar by detection of
antileishmanial antibodies using rK-39 antigen based ICT (n=65).
Group III (Controls): People from endemic area those were free from fever,
physically healthy and comparable to age and sex of the Kala-azar cases were
included as healthy controls (n=44).

66
3.4.1 Inclusion criteria
A total of 106 patients (clinical isolates) irrespective of age and sex were selected
from Mymensingh Medical College Hospital and Trishal Thana Health Complex
on the basis of following criteria:
Patients had history of prolonged low grade irregular fever.
Weight loss
Normal or increased appetite
Pallor
Bleeding
Organomegaly
Blackening of skin
3.4.2 Exclusion criteria of cases
Febrile persons showing negative results either in ICT or LD body were excluded
from the study.
3.5 Controls
People from endemic area those were free from fever, physically healthy and
comparable to age and sex of the Kala-azar cases were included as healthy
controls.
3.6 Data collection and analysis
Relevant history, clinical findings, laboratory records and follow-up of every case
was collected and recorded in a per-designed data sheet. Subsequently, data were
analyzed by computer programmed SPSS version 12.0.
3.7 Collection of specimen
Sample or specimen: Blood, bone marrow.

67
(a) Collection of bone marrow aspirates: The preferred site for bone marrow
aspiration include sternum preferably manubrium of the sternum, iliac crest and
sometimes tibia. The patient was admitted in medicine and pediatric unit in
Mymensingh Medical College Hospital. The purpose of bone marrow collection
was explained clearly to the patient and the attendant. After adequate motivation,
written consent was taken from the legal guardian or from the patient. Keeping the
patient lying on the side, site was palpated and foxed. The area was cleaned by
tincture iodine and rectified spirit. Injection Jasociane (1%) 1.5-2.0 ml was pushed
into the site as local anesthetic. Allowing 5-10 minutes time, appropriate bone
marrow needle was inserted up to the marrow cavity. Then up to 1 ml of marrow
material was aspirated by gentle suction through a 10 ml disposable syringe. After
completing aspiration, the syringe was removed from the needle and the needle
was drawn out. The local wound was sealed by sterile gauge and microspore
adhesive tape. Immediately after aspiration of bone marrow, thin film of the
aspirate was made on at least three microscopic slides. Films were allowed to dry
in air and the back of the slide was rubbed rapidly with a finger to slightly warm
the glass and drive off moisture (Dey et al., 1998). With proper labeling bone
marrow smear was carried to the laboratory for staining and microscopic
examination.
(b) Collection of blood: Under all aseptic precautions (cleaning the area with
povidone iodine followed by adequate rubbing by alcohol pad containing 60%
isopropyl) 5 ml of venous blood was collected from median ante-cubital or
appropriate veins by gentle suction. The collected blood sample was immediately
be introduced into a 3.2% Tri-sodium citrate coated vacutainer. Rest of the blood
was used to prepare peripheral blood film, ICT strip test, estimation of
hemoglobin and ESR. Immediately after collection with proper labeling, blood
was carried to the laboratory for buffy coat preparation.

68
3.8 Laboratory methods
3.8.1 Staining of marrow film: At first marrow film was covered with Leishman
stain (Appendix II) and kept for one minute. Then double quantity of distilled
water was added and kept for 12 minutes. Finally more distilled water was flowed
over the marrow film for washing and dried in the air (Duguid, 1996).
3.8.2 Microscopic examination of marrow film: At least 1000 fields per slide
preferably around the edges of the preparation were examined under oil
immersion lens to detect amastigote from of Leishmania donovani. Leishmania
amastigote appeared as small round or oval organisms about 3µm x 5 µm size
inside or outside macrophages. Each amastigote was containing a red-mauve
nucleus, a smaller more deeply staining red-mauve kinetoplast and pale blue
cytoplasm. The particular diagnostic feature to look for in LD bodies was the
presence of nucleus and kinetoplast. After identification, parasite density can be
scored microscopically by means of a logarithmic scale ranging from 0 (no
parasite per 1,000 oil immersion fields) to +6 (>100 parasites per field). The
grading of parasitemia is done by the criteria suggested by the WHO (10x
eyepiece and 100x oil immersion lens):
6+ >100 parasites/fields
5+ 10-100 parasites fields
4+ 1-10 parasites fields
3+ 1-10 parasites/ 10 fields
2+ 1-10 parasites/ 100 fields
1+ 1-10 parasites 1000 fields
0 0 parasites 1000 fields (Source : WHO TRS 793)
3.9 Making buffy coat (density gradient centrifugation)
In a sterile round bottom disposable test tube (10 ml capacity), 2 ml of Histopaque
(Appendix II) pipetted carefully. Then 2 ml of citrated blood from the vacutainer

69
was poured slowly by the side of the test tube so that the blood completely
overlay the separation fluid without any mixing. The test tube was centrifuged for
15 minutes. 3000 rpm referable in a swinging centrifuge machine.
3.9.1 Taking buffy coat material and preparation of smears
After centrifugation, a thick buffy coat was seen in the interface of plasma and
Histopaque. RBCs were settled at the bottom part of the test tube. Now the plasma
was gently sucked by a micropipette of 1ml capacity and was preserved in an
eppendrof tube. Then by another of 100-200 µl capacity/micropipette, the buffy
coat material was sucked out very gently. Part of the material was used in another
eppendrof containing 0.5 ml of 95% alcohol for analysis by PCR (DGHS, 2009).
3.9.2 Staining of buffy coat smears
Buffy coat films were stained by Leishman stain.
Procedure of Leishman staining
A air dried thin film was made and placed on a staining rack and flooded with
Leishman stain. The slide was left for 2 minutes to fix. Double amount distilled
water was added over the flooded stain from a plastic wash bottle for better
mixing of the solution. The stain was left for 10 minutes. The stain was washed
with running tap water and air dried again the smear to examine under oil
immersion objectives (DGHS, 2009).
3.9.3 Microscopic examination of the buffy coat film
At least 1000 fields per slide were examined under oil immersion lens to detect
amastigote form of Leishmania donovani. Amastigotes appear as round or oval
bodies measuring 2 to 3 ìm in length and are found in inside or outside the

70
monocytes and macrophages. In preparations stained with Leishman stain, the
cytoplasm appears pale blue, with a relatively large nucleus that stains red. In the
same plane as the nucleus, but at a right angle to it, is a deep red or violet rod-like
body called a kinetoplast. If any amastigote in 1000 fields was found, the slide
was reported as positive. Grading of positive smear was done according to
standard chart.
Score 0 = No LD bodies in 1000 fields
Score 1 = 1-10 LD bodies in 1000 fields
Score 2 = 1-10 LD bodies in 100 fields
Score 3 = 1-10 LD bodies in 10 fields
Score 4 = 1-10 LD bodies per fields
Score 5 = 10-100 LD bodies per fields
Score 6 = 100 LD bodies per fields (DGHS 2009)
3.10 Nested PCR for Leishmania Donovani Kinetoplast DNA (Kdna)
Amplification:
Four major steps:
A. DNA extraction from samples,
B. DNA amplification in thermal cycler,
C. Gel electrophoresis,
D. Visualization / documentation under UV light.
3.10.1 DNA extraction from samples:
1. Buffy coat was taken for DNA extraction
2. DNA was extracted by phenol-chloroform method.
3. 200 ìl buffy coat was homogenized with
-200 ìl lysis buffer [50Mm Tris-HCL (pH7.6),1Mm EDTA]
-10 ìl proteinase K in a 1.5ml microcentrifuge tube.

71
4. The homogenate was then incubated at 370C overnight
5. 200ìl of a phenol-chloroform-isoamyl alchohol mixture (25:24:1by volume)
was added.
6. Mixed gently for 5 minutes and micro centrifuged for 10 minutes at 10,000 rpm
at room temperature.
7. The top (aqueous) phase containing the DNA was removed carefully and
transferred to a new tube. The steps were repeated 1-3 times.
8. An equal volume of chloroform-isoamyl alchohol (24:1) was added. Mixed
gently for 2 minutes and spined for 1 minute at 10000 rpm.
9. Then 300 ìl supernatant, 400 ìl ice cold (100%) ethanol and 10 ìl NaCl was
mixed and kept overnight at -200C or in -700C for 1 hour.
10. It was spined for 20 min in a microcentrifuge at maximum speed and remove
the supernatant was removed.
11. 1ml of (70%) ethanol was added at room temperature.
12. The pellet was dried at 370C temp. for about half an hour.
13. The pellet was dissolved in 20-50 ìl TBE buffer pH 8.0 (10mM Tris HCL,
and 1mM EDTA) (Fakhar et al., 2006).
Preparation of PCR mixture:
1. DDW (double Distilled water) -------------- 38.5 µl, 1 = 38.5 µl
Buffer solution
(Contained 10 mM Tris-HCl, pH 8.3,
50 mM KCl, 1.5mM MgCl2) ------------------------- 5µl, 1 = 5µl
dNTP ---------------------------------------------------- 4µl, 1 = 4µl
Primer (1st pair) ----------------------------------------- 1µl, 1= 1µl
= 48.5µl
Taq Polymerase ------------------------------------------------ 0.5µl

72
In each PCR tube:
Reaction mixture ---------------------------------------------- 49 µl
Extracted DNA -------------------------------------------------- 1 µl
Total volume in each PCR tube --------------------------- = 50 µl
3.10.2 DNA amplification in thermal cycle
Amplification
50 µl amplification mixer was dispensed in thermocycler (Model-Master cycler
personal) tubes. 1.0 µl template was added to each of the tubes and mixed with
pipette tips. Amplification was carried out according to the following directions.
PCR tubes were placed in thermal cycler: Programme was planned
Denaturation at 940C ------------------ 30 seconds
Annealing at 450C --------------------- 30seconds
Extension at 720C --------------------- 1 minute
Final extension at 720C --------------- 3 minutes have been performed
In Nested PCR the amplified product of 1st PCR is used as extracted DNA with
the reaction mixture which contains the 2nd pair of Primer for nested PCR.
Reaction mixture for PCR tubes of 50 µl volume:
DDW (Double Distilled Water) ------------- 38.5 µl, 1 = 38.5 µl
Buffer solution
(Contained 1OmM Tris-HCl, pH 8.3,
50 mM KCl, 1.5mM MgCl2) ----------------- 5µl, 1 = 5µl
dNTP -------------------------------------------- 4µl, 1 = 4µl
Primer (2nd pair) -------------------------------- 1µl, 1= 1µl
= 48.5µl
Taq Polymerase ------------------------------------------------ 0.5 µl
= 49 µl
Cycle repeated for 30 times

73
In reaction mixture there was 2nd pair of primer of was added for Nested
PCR
In each PCR tube:
Reaction mixture ----------------------------------------------- 49 µl
1st amplified product (used as Extracted DNA) ------------ 1 µl
Total volume in each PCR tube ----------------------------- 50 µl
In Nested PCR the following programme is done in thermal cycle:
Denaturation at 940C ------------------ 1`minutes
Annealing at 450C --------------------- 1 minute
Extension at 720C --------------------- 1.5 minute
Final extension at 720C --------------- 3 minutes
3.10.3 Agarose gel electrophoresis
The amplification reaction was visualized on a 2% agarose gel; a 100-bp DNA
ladder (Pharmacia, Uppsala, Sweden) was used as a marker. Samples were scored
as positive when a PCR product of 385 bp could be detected.
3.10.3.1 Preparation of agarose gel
Two percent agarose gel was prepared by melting 1.0 gm agarose in 50 ml 1X
TBE buffer using a microwave oven or gas burner. The melted agarose was
allowed to cool to about 500C and was poured into gel electrophoresis unit with
spacers and comb. After solidification of the gel, the comb was removed and
wells were found.
3.10.3.2 Loading and running the sample into gel electrophoresis
Four µL of PCR product was mixed with 2.0 µL of gel loading dye. The mixture
was slowly by loaded into the well using micropipette tips. Mark DNA of known
size (100 bp ladder) was loaded in one well to determine the size of the PCR
products. Electrophoresis was carried out at 100 volts for 45 minutes.
Cycle repeated for 36 times

74
3.11 Visualization/documentation under UV light
Products were analyzed by electrophoresis in 1% agarose gel containing 0.5,
µg/ml ethidium bromide, in TBE buffer and photographed under UV illumination
(Maury et al., 2005).
3.12 Primers used for PCR in Kala-azar
Target Primer Sequence Tm (0C) Fragment length
Ldl primers_L. donovani
kinetoplast
minicircle
sequene
(SEOID No.1)
(SEQID No. 2)
692 bp
5�-AAA TCG GCT CCG AGG CGG GAA AC-3,
5�-GGT ACA CTCT TCAGTAG CAC-3�
640C
56.50C
PCR product 600bp
385bp 5�-TCG GAC GTG TGT GGA TAT GGC-3� 60.40C
PCR product 385bp
Nested PCR (from Indian study)
592bp 5�-CCG ATA ATAT GTAT CTC CCG-3�
54.60 C
3.13 Immuno-Chromatography Test ICT)
Procedure for antibody detection by ICT as per kit manual
3.13.1 Principle of the test: ICT for Kala-azar is a qualitative, membrane based
immunoassay for the detection of antibodies in Kala-azar in human serum. The
membrane is pre-coated with a novel recombinant Kala-azar (rk39) antigen on the
test line region and chickens anti-protein A on the control region. During testing,
serum sample reacts with the dye conjugate (protein A colloidal gold conjugate),
which has been pre-coated in the test device. The mixture then migrates upward
on the membrane chromatographically by capillary action to react with
recombinant Kala-azar antigen on the membrane and generates a red line.
Presence of this red line indicates a positive result, while its absence indicates a
negative result. Regardless of the presence of antibody to Kala-azar antigen, as the
mixture continues to migrate across the membrane to the immobilized chicken
anti-protein A region, a red line at the control line region will always appear. The

75
presence of this red line serves as verification for sufficient sample volume and
proper flow and as a control for the reagents.
3.13.2 Test Procedure: Serum and Chase Buffer were allowed to reach room
temperature prior to testing. The ICT test strip for Kala-azar was removed from
the foil pouch and 20 µl of serum was added to the test strip in the area beneath
the arrow. The test strip was placed into a test tube so that the end of the strip was
facing downward as indicated by the arrows on the strip. Then 150 µl of the
Chase Buffer solution provided with the test kit was added. The result was taken
within 10 minutes. Background was clear before the result of the test was taken.
Results interpreted after 10 minutes could be misleading.
3.13.3 Results
A) Positive result: The test was positive when a coloured control line and
coloured test line appeared in the test area. A positive result indicated that the
ICT detected antibodies to members of L. donovani complex. A faint line was
considered a positive result. The red colour in the test region will vary
depending on the concentration of anti-leishmanial antibodies present.
B) Negative result: The test was negative when only the coloured control line
was appeared. A negative result indicated that the ICT did not detect
antibodies to members of L. donovani complex.
C) Invalid result: When no coloured lines appeared either at the control line or at
the line areas, the test was taken as invalid. The test was also invalid if no
control line was appeared but a test line was seen, retest using a new test strip
and fresh serum was recommended (Inbios Inc., USA).

76
CHAPTER 4
RESULTS
The present study was conducted on a total of 150 subjects. Among them, 106
were cases of Kala-azar and 44 were age and sex matched healthy controls. Out of
106 cases, 41 were confirmed as kala-azar by detection of LD bodies from bone
marrow aspirates by microscopy and 65 were ICT positive clinically diagnosed
cases of Kala-azar.
Age and sex distribution of subjects is shown in the following table 1. Majority of
the cases (58%) and controls (22) were in the age group of 2-12 years. Number of
cases and controls gradually declined with increase in age. Minimum member of
case (02) and control (01) was in the age group of 57 years and above. Total male
were 63 (59.43%) and female 43 (40.57%) in the cases group and corresponding
values for controls were 26 (59.09%) and 18(40.91%) respectively. Male to
female ration of cases was 1.46:1.
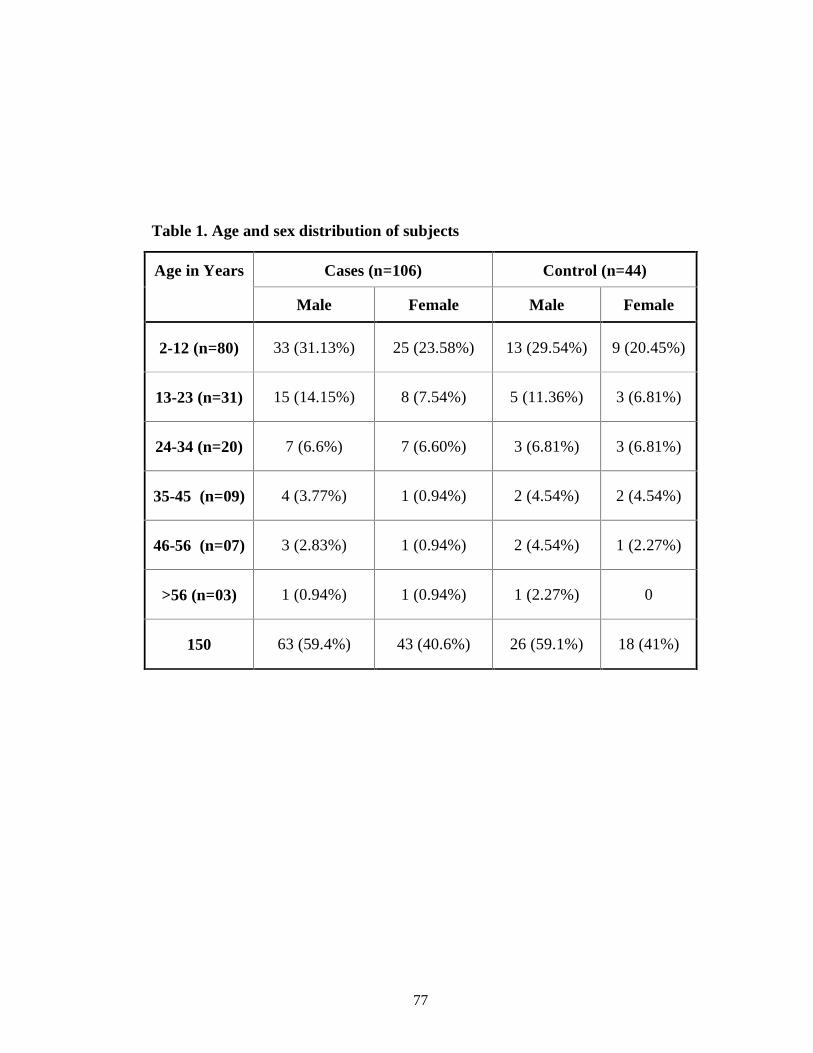
77
Table 1. Age and sex distribution of subjects
Cases (n=106) Control (n=44) Age in Years
Male Female Male Female
2-12 (n=80) 33 (31.13%) 25 (23.58%) 13 (29.54%) 9 (20.45%)
13-23 (n=31) 15 (14.15%) 8 (7.54%) 5 (11.36%) 3 (6.81%)
24-34 (n=20) 7 (6.6%) 7 (6.60%) 3 (6.81%) 3 (6.81%)
35-45 (n=09) 4 (3.77%) 1 (0.94%) 2 (4.54%) 2 (4.54%)
46-56 (n=07) 3 (2.83%) 1 (0.94%) 2 (4.54%) 1 (2.27%)
>56 (n=03) 1 (0.94%) 1 (0.94%) 1 (2.27%) 0
150 63 (59.4%) 43 (40.6%) 26 (59.1%) 18 (41%)

78
Table 2 shows the housing condition of Kala-azar cases. Earthen house and tin
shade were in 68.87% and 31.13% respectively.
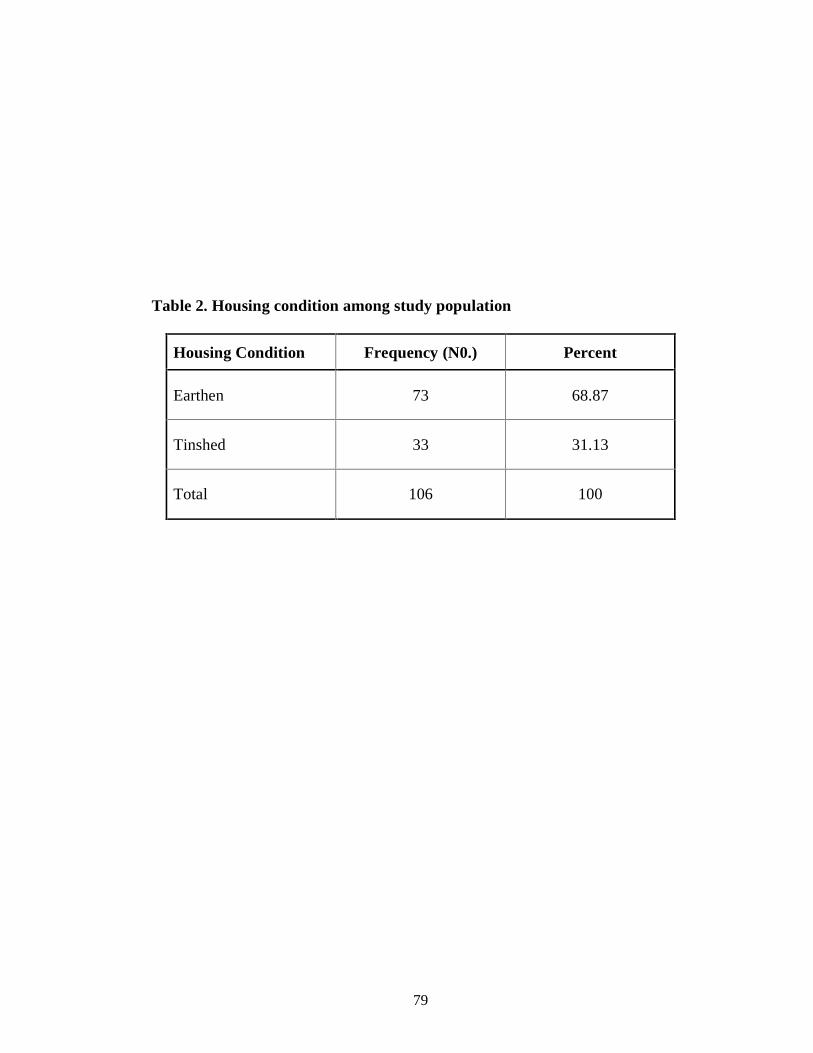
79
Table 2. Housing condition among study population
Housing Condition Frequency (N0.) Percent
Earthen 73 68.87
Tinshed 33 31.13
Total 106 100

80
Table 3 Shows Comparison between low and middle socioeconomic status in
Kala-azar cases. Out of 106 Kala-azar cases 64 (60%) were from low economic
status, 36 (34%) were from middle economic status and 6 (6%) were from upper
economic status.
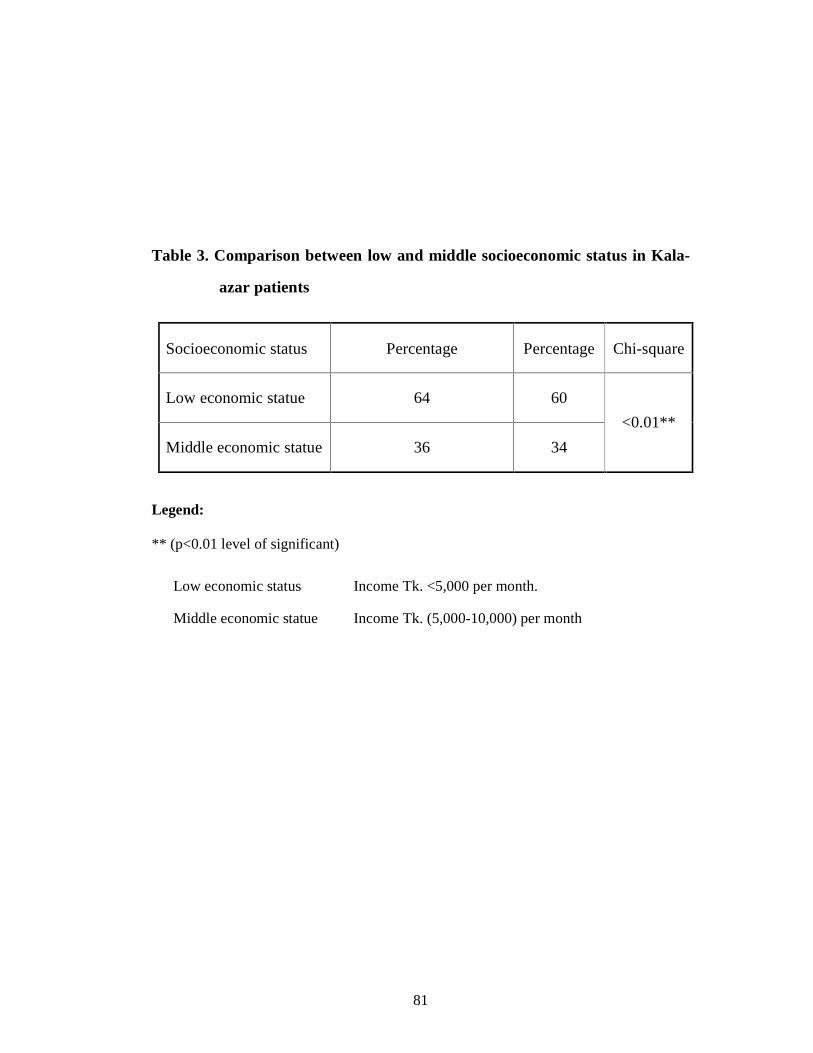
81
Table 3. Comparison between low and middle socioeconomic status in Kala-
azar patients
Socioeconomic status Percentage Percentage Chi-square
Low economic statue 64 60
Middle economic statue 36 34
<0.01**
Legend: ** (p<0.01 level of significant)
Low economic status Income Tk. <5,000 per month.
Middle economic statue Income Tk. (5,000-10,000) per month

82
Table 4 shows Kala-azar cases as per duration of fever. Majority of cases (29.2%)
had fever for 4 months duration followed by duration of 2 months (25.5%) and
duration of 3 and 5 months (18.9%) and (16%). Only 3.8% cases had duration of
fever for >6 months. Mean duration ±SD of fever was found as 3.77±1.32 month.
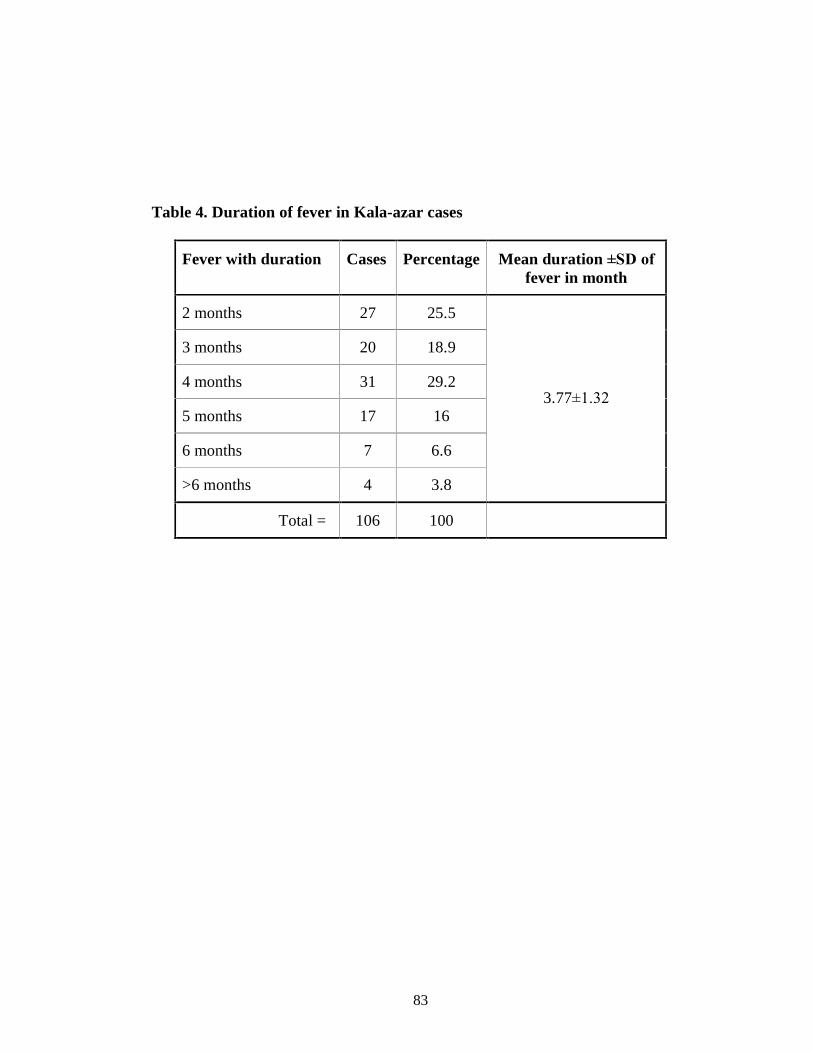
83
Table 4. Duration of fever in Kala-azar cases
Fever with duration Cases Percentage Mean duration ±SD of
fever in month
2 months 27 25.5
3 months 20 18.9
4 months 31 29.2
5 months 17 16
6 months 7 6.6
>6 months 4 3.8
3.77±1.32
Total = 106 100

84
Enlargement (in cm) of liver (hepatomegaly) and spleen (splenomegaly) of Kala-
azar cases are given in Table 5. Mean enlargement ±SD of liver and spleen was
found as 5.19±2.03 and 6.98±2.58 respectively. Regarding hepatomegaly, highest
enlargement as 12.5 cm was found in (2.8%) and lowest as 2.5 cm in (17%).
Majority (67.9%) cases showed enlargement by 5 cm. Regarding splenomegaly,
highest enlargement as 12.5 cm were found in (4.7%) and lowest as 2.5 cm in
(8.5%). In (34.9%), (30.2%) and (21.7%) cases enlargement were found as 5 cm,
7.5- cm and 10 cm respectively.
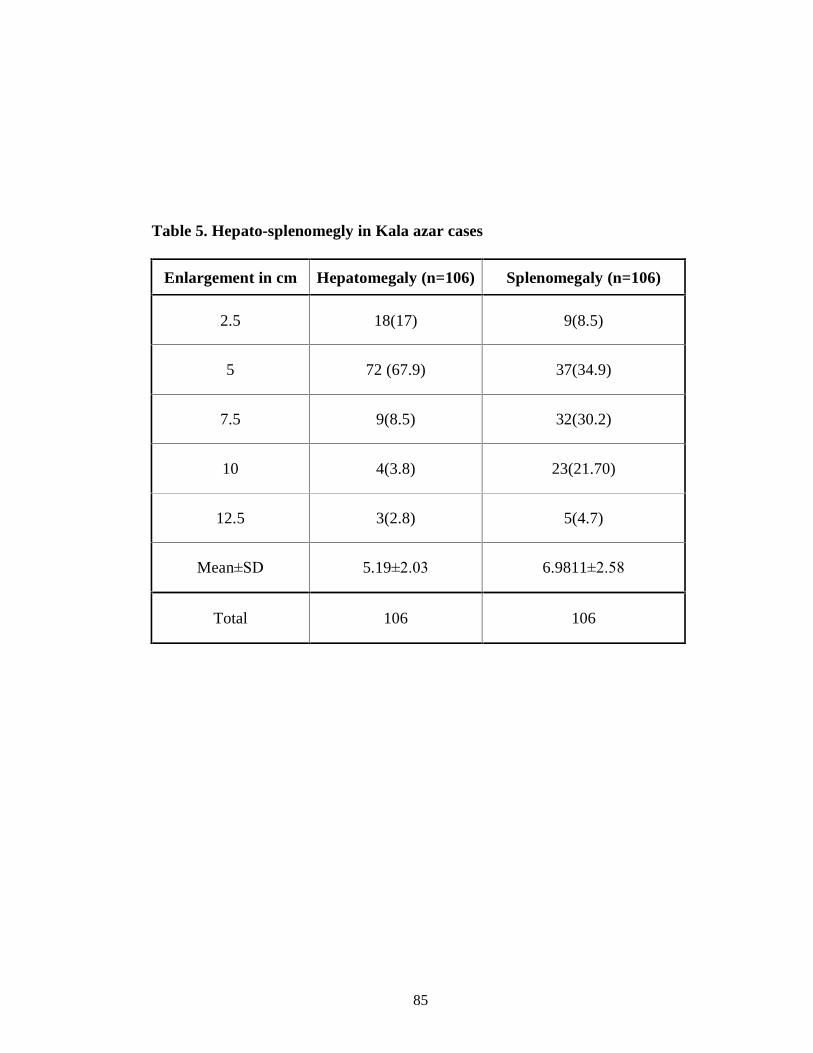
85
Table 5. Hepato-splenomegly in Kala azar cases
Enlargement in cm Hepatomegaly (n=106) Splenomegaly (n=106)
2.5 18(17) 9(8.5)
5 72 (67.9) 37(34.9)
7.5 9(8.5) 32(30.2)
10 4(3.8) 23(21.70)
12.5 3(2.8) 5(4.7)
Mean±SD 5.19±2.03 6.9811±2.58
Total 106 106

86
Haematological findings of (41) confirmed cases of kala-azar cases are given in
Table 6. Anaemia, leucopoenia, Thrombocytopenia were found in 34(83%),
18(44%), 33(80.48%) of confirmed cases of kala-azar cases respectively.
Lymphocyte count was found to be above normal level (>45%) in 87.71 %
patients. Erythrocyte Sedimentation Rate (ESR) was raised in 95% of the patients
in this study.
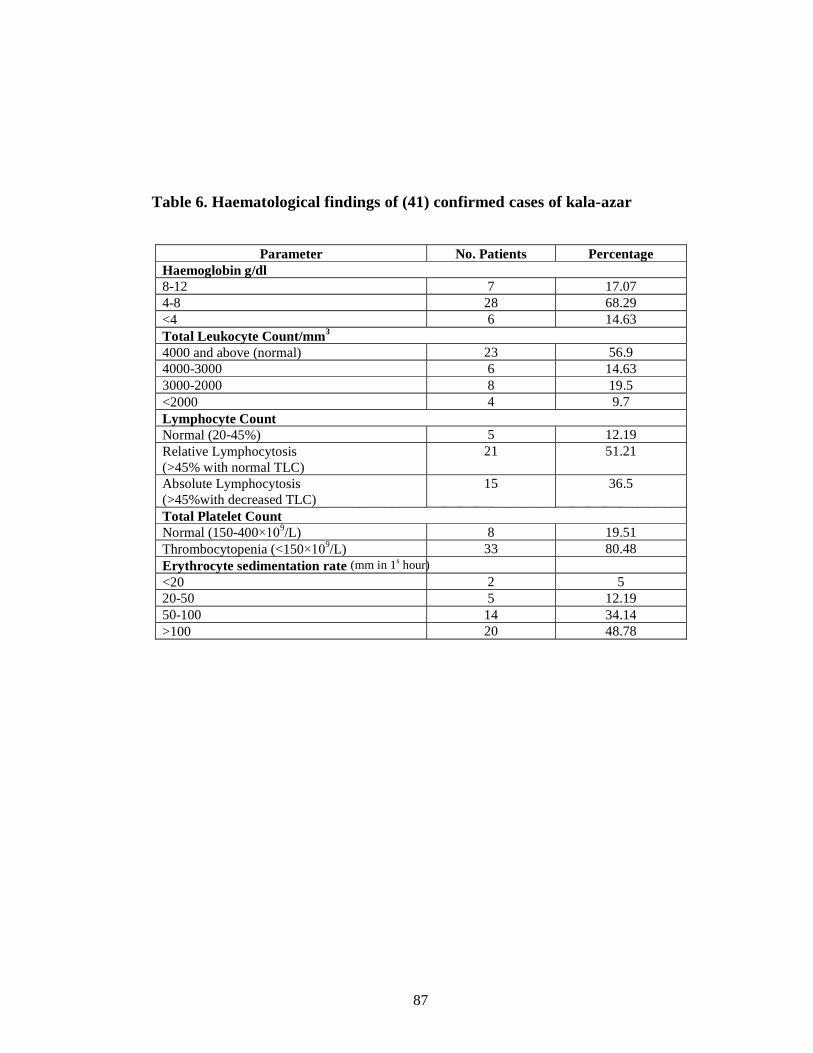
87
Table 6. Haematological findings of (41) confirmed cases of kala-azar
Parameter No. Patients Percentage
Haemoglobin g/dl 8 - 12 7 17.07 4 - 8 28 68.29 <4 6 14.63 Total Leukocyte Count/mm 3 4000 and above (normal) 23 56.9 4000 - 3000 6 14.63 3000 - 2000 8 19.5 <2000 4 9.7 Lymphocyte Count Normal (20 - 45%) 5 12.19 Relative Lymphocytosis (>45% with normal TLC)
21 51.21
Absolute Lymphocytosis (>45%with decreased TLC)
15 36.5
Total Platelet Count Normal (150 - 400×10 9 /L) 8 19.51 Thrombocytopenia (<150×10 9 /L) 33 80.48 Erythrocyte sedimentation rate (mm in 1s hour) <20 2 5 20 - 50 5 12.19 50 - 100 14 34.14 >100 20 48.78

88
Table 7 indicates prevalence of kala-azar cases among study population. Among
41 bone marrow positive confirmed Kala-azar cases, buffy coat microscopy for
LD was positive in 38 (92.7%) confirmed Kala-azar cases and PCR from buffy
coat was positive in 39(95.12%) confirmed Kala-azar cases. Among 65 ICT
positive KA cases, buffy coat microscopy for LD was positive in 60(92.30%) and
PCR from buffy coat was not done. Out of 44 endemic controls, 02(4.54%) were
positive by buffy coat microscopy and 0.00(0.00%) were positive by buffy coat
PCR.
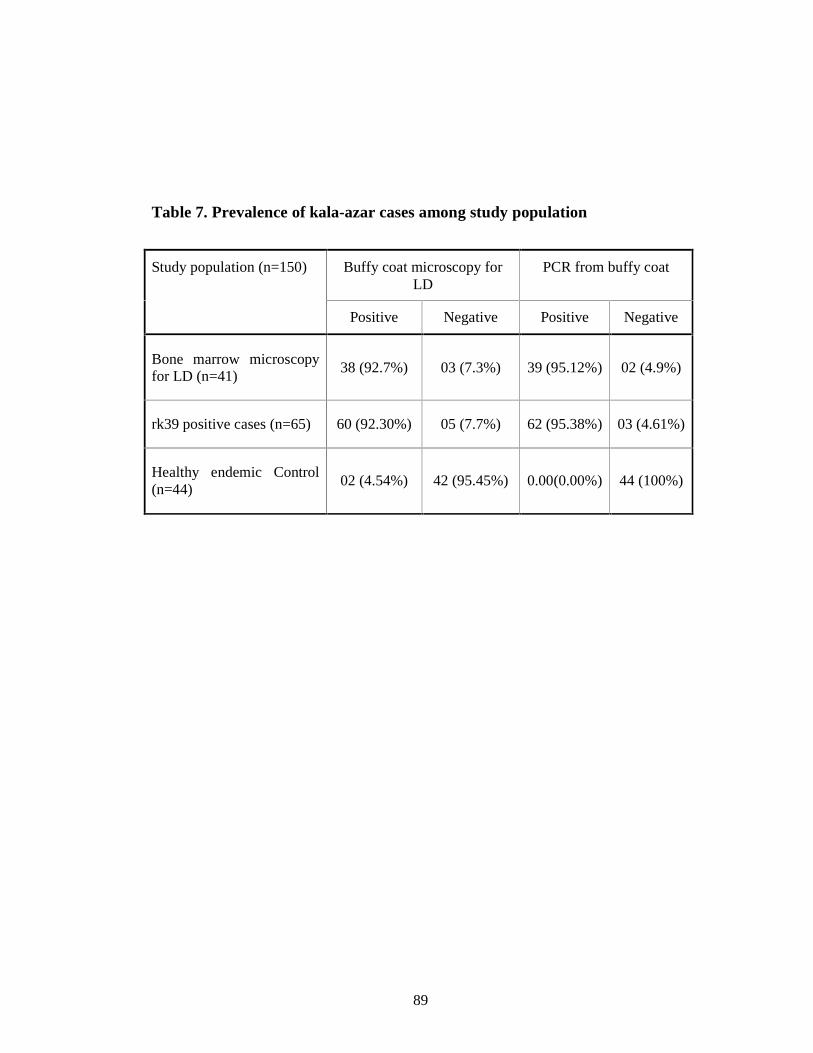
89
Table 7. Prevalence of kala-azar cases among study population
Buffy coat microscopy for LD
PCR from buffy coat Study population (n=150)
Positive Negative Positive Negative
Bone marrow microscopy for LD (n=41)
38 (92.7%) 03 (7.3%) 39 (95.12%) 02 (4.9%)
rk39 positive cases (n=65) 60 (92.30%) 05 (7.7%) 62 (95.38%) 03 (4.61%)
Healthy endemic Control (n=44)
02 (4.54%) 42 (95.45%) 0.00(0.00%) 44 (100%)

90
Table 8 indicates Sensitivity and specificity of LD body in buffy coat of
confirmed Kala-azar cases. Buffy coat microscopy for LD were positive in 38
(92.7%) and were negative in 03(7.3%) of confirmed Kala-azar cases .In controls,
42(95.45%) were negative and 02(4.54%) were positive. Accordingly, Sensitivity
and Specificity were 92.7% and 95.45% respectively.
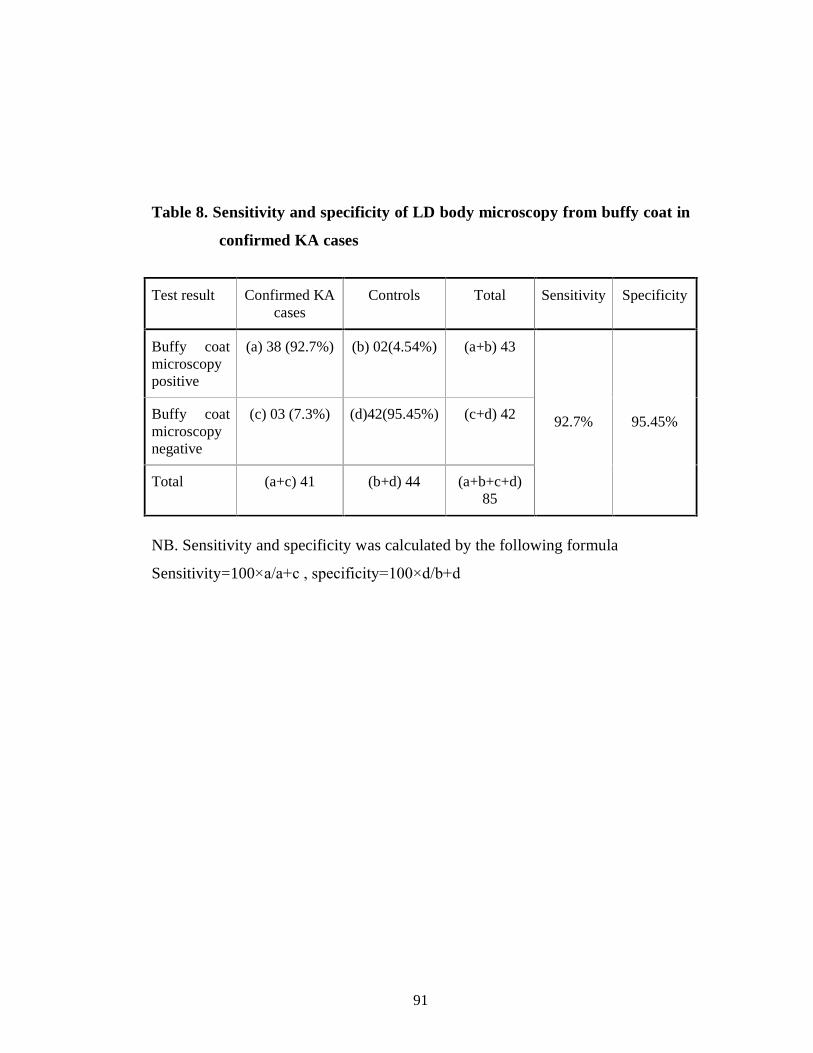
91
Table 8. Sensitivity and specificity of LD body microscopy from buffy coat in
confirmed KA cases
Test result Confirmed KA cases
Controls Total Sensitivity Specificity
Buffy coat microscopy positive
(a) 38 (92.7%) (b) 02(4.54%) (a+b) 43
Buffy coat microscopy negative
(c) 03 (7.3%) (d)42(95.45%) (c+d) 42
Total (a+c) 41 (b+d) 44 (a+b+c+d) 85
92.7% 95.45%
NB. Sensitivity and specificity was calculated by the following formula
Sensitivity=100×a/a+c , specificity=100×d/b+d

92
Table 9 indicates Sensitivity and specificity of buffy coat PCR in confirmed KA
cases. PCR from buffy coat was positive in 39(95.12%) and were negative in 02
(4.9%) of confirmed Kala-azar cases. In controls, 44(100%) were negative and
0.00(0.00%) were positive. Accordingly, Sensitivity and Specificity were 95.12%
and 100% respectively.
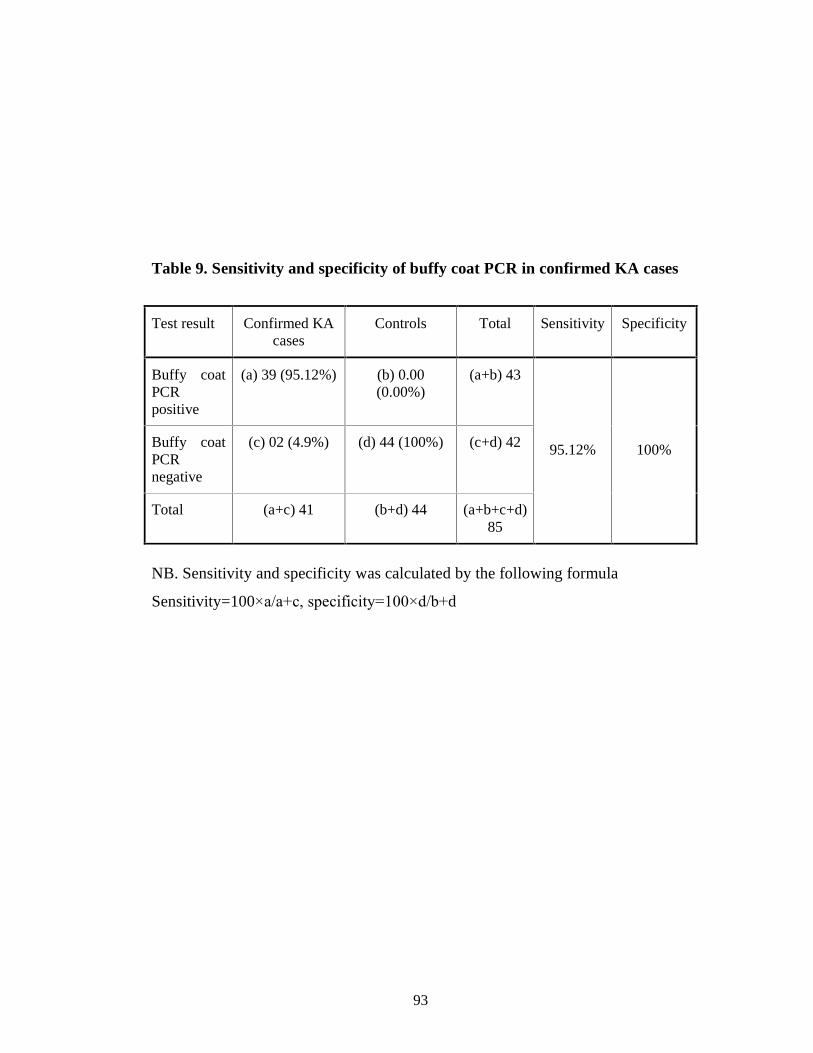
93
Table 9. Sensitivity and specificity of buffy coat PCR in confirmed KA cases
Test result Confirmed KA cases
Controls Total Sensitivity Specificity
Buffy coat PCR positive
(a) 39 (95.12%) (b) 0.00 (0.00%)
(a+b) 43
Buffy coat PCR negative
(c) 02 (4.9%) (d) 44 (100%) (c+d) 42
Total (a+c) 41 (b+d) 44 (a+b+c+d) 85
95.12% 100%
NB. Sensitivity and specificity was calculated by the following formula
Sensitivity=100×a/a+c, specificity=100×d/b+d

94
Table 10 indicates Sensitivity and specificity of LD body in buffy coat of ICT
positive KA cases. Buffy coat microscopy for LD was positive in 60(92.30%) out
65 cases. In controls, 42(95.45%) were negative and 02(4.54%) were positive.
Accordingly, Sensitivity and Specificity were 92.30% (p<0.001) and 95.45%
(p<0.001) respectively.
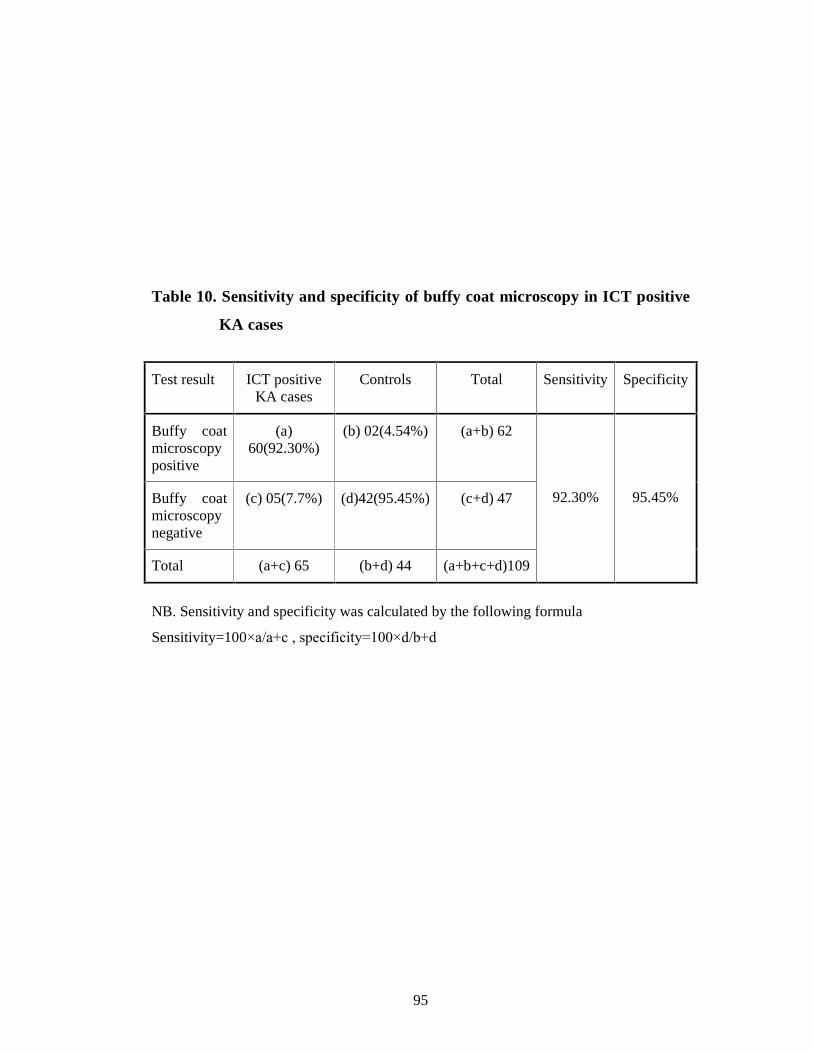
95
Table 10. Sensitivity and specificity of buffy coat microscopy in ICT positive
KA cases
Test result ICT positive KA cases
Controls Total Sensitivity Specificity
Buffy coat microscopy positive
(a) 60(92.30%)
(b) 02(4.54%) (a+b) 62
Buffy coat microscopy negative
(c) 05(7.7%) (d)42(95.45%) (c+d) 47
Total (a+c) 65 (b+d) 44 (a+b+c+d)109
92.30% 95.45%
NB. Sensitivity and specificity was calculated by the following formula
Sensitivity=100×a/a+c , specificity=100×d/b+d

96
Distribution of kala-azar cases in hospitals are shown in figure 1. Out of 106 kala-
azar cases 71 (66.98%) were from rural hospital (Trishal Thana Health Complex)
and 35 (33.02%) were from urban hospital (Mymensingh Medical College
Hospital).
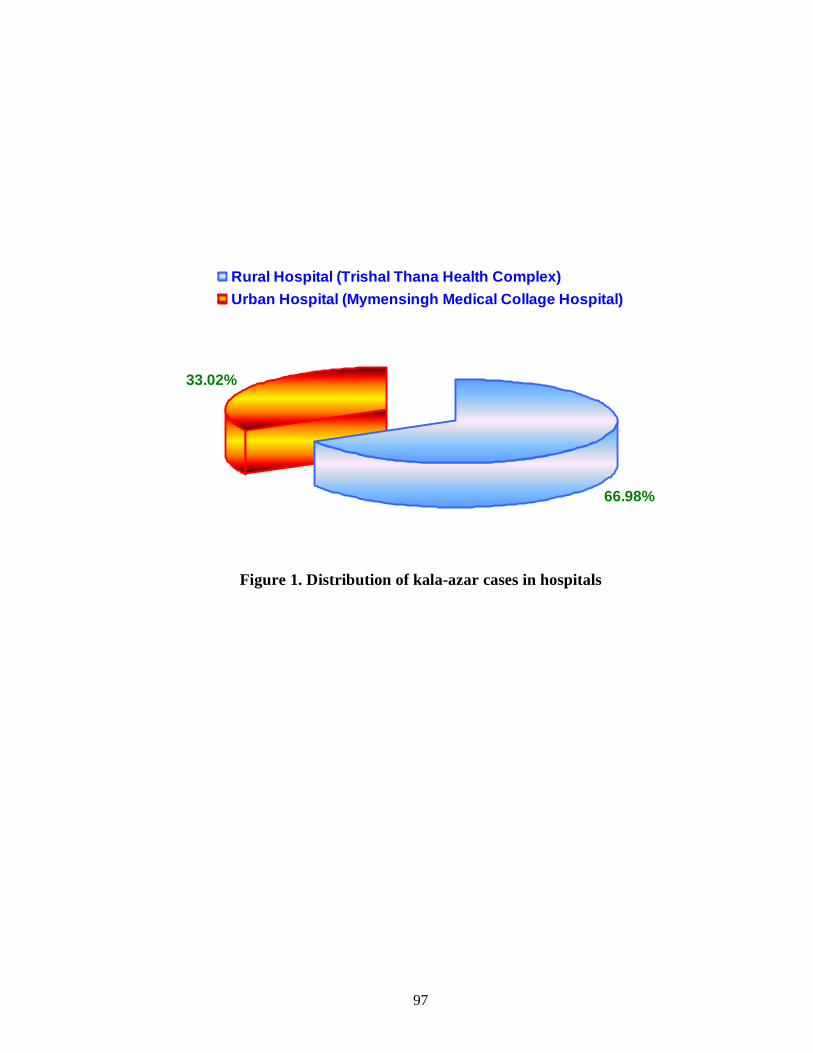
97
66.98%
33.02%
Rural Hospital (Trishal Thana Health Complex)
Urban Hospital (Mymensingh Medical Collage Hospital)
Figure 1. Distribution of kala-azar cases in hospitals

98
CHAPTER 5
DISCUSSION
Kala-azar is a poverty-related disease. Since the poorest segment of the
community mostly suffer from this disease, so due attention is usually not paid on
behalf of the health authority. Moreover, development researches are also difficult
to perform because of remote and backward approach to reach the endemic
locality. Thereby, it has appeared as a neglected disease, though it has been
constituting a major health problem at regional level in the Indian sub-continent.
It is the essential demand of the time to of control Kala-azar. For this purpose,
timely and adequate treatment may contribute significantly to reducing the human
reservoir of this anthroponotic disease. Chemotherapy is critically important in
reducing the burden of disease and antimonials are the first-line drugs for all
clinical forms. Treatment is long, expensive and not devoid of adverse side
effects. In many states of India, treatment failure is already well documented due
to low responsiveness of parasites to sodium antimony gluconate. In Bangladesh,
resurgence of Kala-azar has been noticed in the decade of 1970 that has gradually
increased to an epidemic form in many areas. Currently Kala-azar appears at a
rate in excess of 15000 per year (Al-Masum et al., 1995; DGHS, 1995). A large
number of studies were carried out mostly on epidemiological, clinical, diagnostic
and therapeutic aspects. In diagnostic aspects, definitive diagnosis of Kala-azar is
a technically difficult job. Because, collection of splenic or bone marrow aspirate
is painful and risky, these samples are not collected or examined at community,
upzila health care center. This study has described the technical aspects of an easy
definitive diagnostic method from blood buffy coat smear and usefulness of
Leishmania nested polymerase chain reaction (Ln-PCR) for diagnosis of kala-azar

99
from the same specimen. We selected majority of Kala-azar cases from Thishal
Thana Health Complex.
In the present study, out of 106 cases, 63 (59.4%) were male and 43 (40.6%) were
female giving a male to female ratio of 1.46:1 (Table 1). Other studies from home
also recorded almost similar ratio between male to female e.g. ICDDRB (2002)
and ICDDRB (2003) found male to female ratio of 1.2:1 and 1.04:1 respectively.
Several local studies were also consistent as showing male to female ratio of
1.38:1 (Sarker et al., 2003), 1.8:1 (Musa et al., 1996), 2:1 (Talukder et al., 2003)
and 2.4:1 (Shamsuzzaman et al., 2003). Similar finding was reported from abroad
where ratio between male to female was 1.3:1 (Antonio et al., 2002). In a study,
from Sudan, it was reported that like many countries males are almost twice
(1.8:1) more likely to be affected by Kala-azar than females (Zijlstra et al., 1994).
Males are infected more often then females, most likely because of their increased
exposure to sandflies due to professional activate (Berman, 1997).
Kala-azar affects various age groups depending on the infecting species,
geographic location, disease reservoir and host immuno-competence. In Indian
type of Kala-azar, children between 5 and 15 years of age are affected more
(Gradoni et al., 1995).
In our study, majority of cases (54.7%) were in the 2-12 years age group (Table
1). Almost similar findings were also found in many studies. Ali and Ashford
(1994) from Ethiopia in a study found 142 cases of Kala-azar, where 58%
children below 15 years. In study by Nuttal et al., (2002) observed that majority
patients of Kala-azar were in the age group of 5-15 years. In one local study on 65
patients, over 60 cases of KA were under 20 years of age (Talukder et al., 2003).
On the other hand, higher number (86%) of KA cases were found under 5 years
children in Pakistan during the period between 1985-1995. This figure was

100
relatively higher in comparison to that of the present study (Rab and Evans,
1995). Higher incidence of kala-azar in children might be due to the reason that
children of poor family generally suffer from malnutrition. Consequently their
immunity is hampered increasing the risk of getting kala-azar infection and
developing severe disease (Desjeux, 2004). Moreover adult population in the
endemic locality might develop protective immunity from previous infection that
reduces the chance of reinfection (Gregory et al., 2008).
In our study, living houses of most (68.87%) Kala-azar cases were made of
earthen walls (Table 2). Due to low socio-economic condition people made their
houses with mud. Easily developed tiny cracks and crevices in the earthen house
are the favourable places for sandflies (Desjeux, 2004). So, people residing in
earthen houses must bear more risk for being infected by KA agents.
In our study, most of the Kala-azar patients 64(60%) were from low
socioeconomic status (Table 3). This finding also correlates with others studies
from Bangladesh who reported that there was a strong association between Kala-
azar and poverty (Bern et al., 2005). Similar findings were also reported by
(Thakur, 2003) and Bern et al., (2000), we also observed the effect of
socioeconomic factors on kala-azar patients.
The extent and presentation of the disease depends on several factors, including
the humoral and cell-mediated immune response of the host, the virulence of the
infecting species and the parasite burden. The remarkable presentation of kala-
azar patients is prolonged low-grade irregular fever associated with organomegaly
of spleen and liver. Anaemia and darkening of skin are also found in majority of
cases (Gradoni et al., 1995).

101
In our study, prolonged low-grade irregular fever was found in 100% cases and
majority of cases (29.2%) had fever for 4 months duration (Table 4). Similar
findings were also reported by other authors both in home and in abroad
describing association of fever in 95-100% cases of kala-azar (ICDDRB, 2003;
WHO, 1996; Maltezou et al., 2000; Sarker et al., 2003). Mean duration of fever in
month of kala-azar cases in the present study was found ad 3.77±1.32. Multiple
authors reported duration of fever in kala-azar cases as over 3 months period
keeping very good agreement with that of ours (Talukder et al., 2003; ICDDRB,
2003; WHO, 1996). The explanation for relatively longer period of fever in kala-
azar cases is well explained by the fact that the onset of fever is usually insidious
with irregular rise and fall. The patient usually does not become prostrated or
anorexic. So, the patient remain ambulatory without hampering physical work.
For this reason, patients remain reluctant to seek medical advice or to be
investigated to elucidate the causes of fever.
In our study, hepatomegaly with mean enlargement of 5.19±2.03 cm and
splenomegaly with a mean enlargment of 6.98±2.58 cm were found in 100% cases
(Table 5). ICDDRB (2003) and Sarker et al., (2003) reported splenomegaly in
100% cases but hepatomegaly in 79% cases. WHO (1996) reported 98%
hepatosplenomegaly from India. Hepatomegaly was found in 85% and
splenomegaly in 99% cases by Maltezou et al., (2000). However, rate of
hepatomegaly obtained in the present study was not showing exact accordance
with other studies. The reason behind this outcome, the delay in part of the
patients came to seek medical advice. Our majority patients attended while they
were in a febrile stage of more than 2 months. The 2 months period is sufficient
enough for development of palpable organomegaly (Oliaro and Bryceson, 1993).
That was why we got splenohepatomegaly in 100% cases.

102
Each of confirmed kala-azar cases (41) showed a high ESR, reduced haemoglobin
concentrations, reduced TLC, increased percentages of lymphocytes in DLC and
reduced TPC (Table 6). Majority of them (68.29%) were having moderate
anaemia. Similar findings have been reported by Rai et al., 2008; Hassan et al.,
1995; Saleem et al., 1986.
In this study, 43.83% patients showed leucopoenia and 52.86% were neutropenic.
Infections were common and severe in these neutropenic patients. A study from
Armed Forces Institute of Pathology in 1986, showed leucopoenia in 50% and
neutropoenia in 100% cases, (Saleem et al., 1986). Similar findings have been
reported by Manzoor et al., 2008.
Lymphocyte count was found to be above normal level (>45%) in 87.71 %
patients. Similar finding was also reported by (Manzoor et al., 2008).
Thrombocytopenia was noted in 80.48% patients in out study. This
thrombocytopenia was the main cause of bleeding in our patients. These findings
about platelet count resemble the findings in the study done by Saleem (1986) in
which 91% cases showed platelet count of <100,000/cmm. Erythrocyte
Sedimentation Rate (ESR) was raised in 95% of the patients in our study. In a
study from Institute of Medical Sciences of Pakistan 1995, ESR was found raised
in 88% of cases.
In the study, out of 150 kala-azar cases, 41 were bone marrow positive cases, 65
were rk39 positive cases and 44 were healthy endemic control (Table 7). Among
these 41 bone marrow positive cases buffy coat microscopy for LD body were
positive in 38 (92.7%) cases and buffy coat PCR were positive in 39 (95.12%)
cases.

103
Out of 65 ICT positive cases buffy coat microscopy were positive in 60 (92.30%)
and PCR from baffy coat were positive in 62 (95.38%).
In this study, in 41 confirmed kala-azar cases, sensitivity and specificity of the
buffy coat microscopy for LD body was found as 92.7% and 95.45% respectively
(Table 8). Almost similar findings were reported by another study done in
Mymensingh region, where buffy coat smear positive result is 92.94% in
confirmed kala-azar cases (Shamsuzzaman, et al., 2007). However, there was no
other local study report to see LD bodies in blood buffy coat smear to compare
with the finding of present study. Williams et al., (1995) made comments that
finding of Leishmania parasite in blood is sometimes possible in paticnt with kala-
azar from Kenya and India.
The present study yielded LD body detection at a relatively higher rate (>92%).
This is well explained by the fact that density gradient centrifugation technique
was adopted in this study using lymphoprep to prepare thick buffy coat in
comparison to that found in wintrobes tubes. As a result more macrophage as well
as more LD bodies were concentrated in buffy coat (DGSH, 2009).
Out of 41 confirmed cases 03 (7.3%) were negative in buffy coat smear may be
due to the lower parasitic load in the circulating blood than in the bone marrow
(Lachoud et al., 2002). However, other reasons, such as temporary may also exist
absence of parasites from the blood of kala-azar cases as reported by Le Fichoux
et al., (1999).
In our study 44 healthy endemic controls, 2 (4.54%) were positive by buffy coat
microscopy. It might be due to asymptomatic carriers (Sundar and Rai, 2002).
The sensitivity and specificity of PCR were 95.12% and 100% respectively as per
shown in Table 9. Different studies found sensitivity and specificity values as

104
follows 97% sensitivity and 100% specificity by Salam et al., (2009), 99% and
100% by Maury et al., (2005). The best sensitivity was obtained in the study
because PCR assay was done in the buffy coat.
PCR assay with buffy coat preparations to detect Leishmania was 10 times more
sensitive then that with whole blood preparation (Sundor and Rai, 2002).
Leishmania as are obligate intracellular parasites (in the vertebrate host) and
therefore are supposedly more concentrated in the buffy coat. On the other hand
Buffy coat prepared with PK-based methods gave the best results (Lachoud et al.,
2000).
Out of 41 confirmed cases 02 (4.9%) were negative by PCR in this study.
Negative PCR results have been reported in a few patients with positive
parasitological proof (Maury, 2005; Cortes et al., 2004).
Low parasitc load could be the cause of less sensitivity of the PCR method (Salam
et al., 2009). In our study, PCR was found negative in only two patients with
grade 1 parasite burden but none of the patients with more then grade 1 parasite
burden was negative by PCR. Therefore, it is reasonable to speculate that negative
PCR might be due to low parasite burden in kala-azar patients. However, other
reasons, such as temporary absence of parasites from the blood of kala-azar cases
as reported by Le Fichouk et al., (1999), cannot be excluded. It is also possible
that inhibitor elements that could be present in the Kala-azar patients sample
might result in negative PCR results.
In this study among ICT positive cases sensitivity of buffy coat microscopy were
92.30% and 95.45% respectively (Table 10). Similar values obtained from buffy
coat smear for LD body among ICT positive cases were reported by
(Shamsuzzaman et al., 2007) in the same institute. Buffy coat smear for LD body

105
were examined among all ICT positive found and some 53 (92.98%) were found
positive (Shamsuzzaman, et al., 2007). Out of 65 ICT positive cases 05 (7.7%)
were negative by buffy coat microscopy. The fact is that an individual with a
positive rk39 strip test result may suffer from an illness (malaria, typhoid fever or
tuberculosis) with clinical features similar to these of kala-azar yet be
misdiagnosed as suffering from kala-azar (Sundor and Ray, 2002).
On the other hand, manufacturer stated that antibody to rk39 antigen could found
in cases of malaria and rheumatoid arthritis (InBios, USA).
Analyzing the findings of the study, it can be concluded that Kala-azar affected
more males than females and majority of them were children under 12 years of
age. In this study explored the reliability and effectiveness of LD body detection
in blood buffy coat smear for definitive diagnosis of Kala-azar and compared the
positivity of the PCR carried out in DNA extracted from buffy coat. The
advantages of performance of PCR on blood buffy coat are non-invasive, reliable,
sensitive and specific.

106
CHAPTER 6
CONCLUSION
Analyzing the findings of the present study, it can be concluded that Kala-azar
remain to be as an endemic disease in this locality. Detection of LD body from
buffy coat by microscopy is more easy, non-invasive and highly sensitive and
specific method. PCR method from same specimen for diagnosis of Kala-azar was
also found highly sensitive and specific.
Thus, efforts should be made to establish LD body detection from buffy coat
smear method at least in the tertiary care hospitals, if possible, at the district-level
hospitals, especially in the endemic areas of Bangladesh and PCR method at least
in the tertiary care hospitals.

107
BIBLIOGRAPHY
Aikat, BK, Shegal, S, Mahjan, RC, Pathnia, AGS, Bhattacharya, PK &
Sahya, S 1979, �The role of counter-immunoelectectrophoresis as a
dignostic tool in kala-azar� Indian Journal of Medical Research, vol. 70,
pp. 592-597.
Ahmed, TU & Ahmed, RU 1983, �An entomological investigation of kala-azar
focus in Bangladesh�, Journal of Preventive and Social Medicine, vol. 2,
pp. 31-36.
Addy M & Nandy A 1992, �Ten years of kala-azar in West-Bengal, part I. Did
post-kala-azar dermal leishmaniasis initiate the outbreak in 24-Parganas�.
Bull. vol. 70, pp. 341-346.
Ali, A & Ashford, RW 1994, �Visceral leishmaniasis in Ethiopia. III. The
magnitude and annual incidence of infection, as measured by serology in
an endemic area�, Ann Trop Med Parasitol, vol. 88, pp. 43-47.
Al-Masum, MA, Evans, DA, Minter, DM & El-Harith, A 1995, �Visceral
leishmaniasis in Bangladesh: the value of DAT as a diagnostic too�, Trans
R Sco Trop Med Hyg, vol. 89, pp. 185-186.
Arias, JR, Monteiro, PS & Zicker, F 1996, �The reemergence of visceral
leishmaniasis in Brazil�, Emerg Infect Dis., vol. 2, pp. 145-146.
Attar, ZJ, Chance, ML, el-Safi, S, Carney, J, Azazy, A, El-Hadi, M, Dourado,
C & Hommel M 2001, �Latex agglutination test for the detection of
urinary antigens in visceral leishmaniasis�, Acta Trop, vol. 78, pp. 11-16.

108
Abdullah, ABM 2002, �Short Cases in Clinical Medicine�, 2nd edition, pp. 219-
221.
Antonio, CP, Cristina, MAJ, Gabriel, WO, Magda, MS and Sampio, C 2002,
�Visceral leishmaniasis: clinical and laboratorial aspects.� J. Pediatr. Vol.
78, pp. 120-127.
Ahasan, HN, Ayaz, K & Bari, SM 2008, �Current diagnosis and treatment of
kala-azar Bangladesh perspective�, Journal Medicine, vol. 9, pp. 45-49.
Assimina, Z, Charilaos, K & Fotoula, B 2008, �Leishmaniasis: An overlooked
public Health concern�, Journal Health Science. vol-2.
Alam M, Shamsuzzaman A K, K Kuhls1 and Scho¨nian, 2009. �PCR diagnosis
of visceral leishmaniasis in an endemic region,Mymensingh district,
Bangladesh�. Tropical Medicine and International Health, vol. 14 no. 5, pp.
499�503
Bradley, DJ 1977, �Regulation of Leishmania populations within the host. II.
Genetic control of acute susceptibility of mice of Leishmania donovani
infection�, Clinical and Experimental Immunology, vol. 30, pp. 130-140.
Bouvier, J, Etges, RJ & Bordier, C 1985, �Identification and purification of
membrane and soluble form of the major surface protein of Leishmania
promastigotes�, Journal of Biological Chemistry, vol. 260, pp.15504-
15509.

109
Bray, RS 1985, �Immnuodiagnosis of leishmaniasis�, In S. Cohen and E.H. Sadun
(ed.), Immunology of parasitic infections, Blackwell Scientific
Publications, Oxford, Untied Kingdom.
Blackwell, JM, Ezekowitz, RAB, Roberis, MB, Channon, JY, Sim, RB &
Gordon, S 1985, �Macrophage complement and lectin-like receptors bind
Leishmania in absence of serum� J Exp Med, vol. 162, pp. 324-331.
Bordier, C 1987, �The promastigote surface protease of Leishmania. Parasitology
Today, vol. 3, pp. 1417-1418.
Bouvier J, Etges RJ & Bordier C 1987, �Identification of the promastigote
suface protease in seven species of Leishmania�. Molecular and
Biochemical Parasitology, vol. 24, pp. 73-79.
Bahr, M & Bell, DR 1987, �Visceral leishmaniasis�, In: Manson�s Tropical
disease, 19th ed. pp. 87-113.
Birley, MH 1993, �An historical review of malaria, kala-azar and filariasis in
Bangladesh in relation to the Flood Action Plan�, Annals of Tropical
Medicine and Parasitology, vol. 87, no. 4, pp. 319-334.
Burns, JM, Shreffler, WG, Benson, DR, Ghalib, HW, Badaro, R & Reed, SG
1993, �Molecular characterization of a kinesing-related antigen of
Leishmania chagasi that detects specific antibody in both African and
American visceral leishmaniasis�, Proc Natl Acad Sci USA, vol. 90, pp.
775-790.

110
Badaro, R, Benson, D, Eulalio, MC, Freire, M, Cunha, S, Netto, EM, Pedral-
Sampaio, D, Madureira, C, Burns, JM, Houghton, RL, David, JR &
Reed SG 1996, �rK39: a cloned antigen for Leishmania chagasi that
predicts active visceral leishmaniasis� J. Inftect. Dis, vol. 173, pp. 758-761.
Berman, JD 1997, �Human Leishmaniasis: Clinical, Diagnostic and
Chemotherapeutic Development in the last 10 years.� Clinical Infectious
Diseases. vol. 24, pp. 684-703.
Bhatia, A, Daifalla, NS, Jen, S, Badaro, R, Reed, SG & Skeiky, YA 1999,
�Cloning, characterization and serological evaluation of K9 and K26: two
related hydrophilic antigens of Leishmania chagasi�, Mol. Biochem
Parasitol, vol. 102, pp. 249-261.
Boelaert, M, El-Safi, S, Mousa, H, Githure, J, Mbati, P, Gurubacharya, V,
Shrestha, J, Jacquet, D, De Muynck, A, Le Ray D, & Vander Stuyft, P
1999, �Multicenter evaluation of repeatability and reproducibility of the
direct agglutination test for visceral leishmaniasis�, Trop. Med. Int. Health,
vol. 4, pp. 31-37.
Brito, ME, Mendonca, MG, Gomes, YM, Jardim, ML & Abath, FG 2000,
�Identification of potentially diagnostic Leishmania braziliensis antigens in
human cutaneous leishmaniasis by immunoblot analysis�, Clin Diagn Lab
Immunol, vol. 7, pp. 318-321.
Bryceson, A 2000, �Visceral leishmaniasis in India�, Lancet, vol. 356, pp. 1933.
Bern, C, Jha, SN, Joshi, AB, Thakur, GD & Bista, MB 2000, �Use of the
recombinant k39 dipstick test and the direct agglutination test in a setting

111
endemic for visceral leishmaniasis in Nepal�, Am. J. Trop. Med. Hyg, vol.
63, pp. 153-157.
Begum, N.; Nasum, M.A.; Mamoon, A.B.; Begum A.,2002, �Visceral
leishmaniasis among the suspected febrile partients�. Bangladesh Journal
of Medicine. (jan), vol.13, pp. 21-25.
Bern C, Hightower AW & Chowdhury R 2005, �Risk factors for kala-azar in
Bangladesh�. Emerging Infectious Diseases, vol. 11, pp. 655�662.
Bern, C & Chowdhury, R 2006, �The epidemiology of visceral leishmaniasis in
Bangladesh prospects for improved control�, Indian Journal Med. Res.,
vol. 123, pp. 275-288.
Boelaert, M ,Sujit Bhattacharya, François Chappuis, Sayda H. El Safi, Asrat
Hailu, Dinesh Mondal, Suman Rijal, Shyam Sundar, Monique
Wasunna & Rosanna W. Peeling, 2007, �Evaluation of rapid diagnostic
tests: visceral leishmaniasis�. Nature Reviews Microbiology, S30-
S39 ,doi:10.1038/nrmicro1766
Chatterjee 1980, �Dermal leishmanoid�. In: Parasitology. 12th (ed.) Calcutta:
Chatterjee Medical Publishers, pp. 64-5.
Campos-Neto, A & Bunn-Moreno, MM 1982, �Polyclonal B cell activation in
hamster infected with parasites of the genus Leishmania�, Infection and
Immunity, vol. 38, p. 871.

112
Chatterjee, KD 1982, �Parasitology (Protozoology and Heminthology) in
Relation to Clinical Medicine�, 12th edition. Chatterjee Medical Publishers,
Calcutta, India, pp. 53-69 and 208-223.
Chulay JD & Brycesson ADM 1983, �Quantifications ofamastigotes of
leishmania donovani in smear of spleniceaspirates of patients with visceral
leishmaniasis�. Am. J. Trop.Med. Hyg., vol. 3, p. 2
Carvalho, EM, Badaro, R, Reed, S, Jones, TC & Johnson, WD 1985,
�Absence of gamma-interferon and interleukin-2 production during active
visceral leishmaniasis�, J. of Clin Invest, vol. 76, pp. 2066-2069.
Colomer-Gould, V, Quitao, LG, Keithly, J & Nogueira, N 1985, �A common
major surface antigen an amatigate and promastigates of Leishmania
species�, Journal of Experimental Medicine, vol. 162, pp. 902-916.
Chan, J, Fujiwara, T, Brennan, P, McNeil, M & Turco, SJ 1989, �Microbial
glycolipids: Possible virulence factors that scavenge oxygen radicals�
Proceedings of National Academy of Science, USA. vol. 86, pp. 2453-
2457.
Chaudhuri, G & Chang, KP 1988, �Acid protease activity of a major surfece
membrane glycoprotein (gp63) from Leishmania mexicana promastigotes�,
Molecular and Biochemical Parasitology, vol. 27, pp. 43-52.
Chang, KP, Chaudhuri, G & Fong, D 1990, �Molecular determinates of
Leishmania Virulence�, Annual Review of Microbiology, vol. 44, pp. 499-
529.

113
Chulay, JD 1991, �Leishmaniasis�. Editor G. Thomas Strickland in �Hunter�s
Tropical Medicine� 7th edition. W.B. Saunders Company, Philadelphia,
U.S.A pp. 638-642.
Chowdhury, MS, Ahmed, MTU & Hussain, AMZ 1993, �Visceral
leishmaniasis its control�, 1st ed. IEDCR, Dhaka. pp. 4-28.
Carreira, PF, Maingon, R, Ward, RD, Noyes, H, Ponce, C, Belli, A, Arana, B,
Zeledon, R & Sousa, OE 1995, �Molecular techniques in the
characterization of Leishmania isolates from Central America�, Ann Trop
Med and Parasitology, vol. 89, pp. 31-36.
Cheesbrough, M 1999, �District Laboratory Practice in Tropical Countries,
Combridge University Press�, Low Price edition.
Christoph, K, Meinecke, MD, Justus S, Linda, O & Bernhard FMD 1999,
�Congenital transmission of visceral leishmaniasis (Kala-azar) from an
asymptomatic mother to her child�. Pediatrics, vol. 104, pp 65.
Cortes S, Rolão N, Ramada J & Campino L 2004, �PCR as a rapid and
sensitive tool in the diagnosis of human and canine leishmaniasis
using Leishmania donovani s.l.-specific kinetoplastid primers�. Trans. R.
Soc. Trop. Med. Hyg., vol. 98, pp. 12-17.
Cruz I, Nieto, Moreno J, Canavate C, Desjeun, P & Alver J 2006,
�Leishmania/HIV co-infection in the second decade�, Indian J Med,
vol.123, pp.357-88.

114
Duxbury, RE & Sadun, EH 1964, �Flourescent antibody test for scrodiagnosis of
visceral leishmaniasis�, American Journal of Tropical Medicine and
Hygiene, vol. 13, pp. 523-529.
Da Silva, RP, Hall, BF, Joiner, KA & Sacks, DL 1989, �CR1, the C3b receptor,
mediates binding of invective Leishmania major metacyclic promastigotes
to human macrophages�, Journal of Immunology, vol. 143, pp. 617-622.
De Colmenares, M, Portus, M, Riera, C, Gallego, M, Aisa, MJ, Torras, S &
Munoz, C 1995, �Detection of 72-75 kD and 123 kD fractions of
leishmania antigen in urine of patients with visceral leishmaniasis�, Am. J.
Trop. Med. Hyg., vol. 52, pp. 427-428.
DGHS 1995, �Kala-azar: A training module for medical officers�. Dhaka: DGHS;
pp. 1-30.
Duguid JP 1996, �Staining methods-Leishman�s stain, Mackie and McCartney-
Practical Medical Microbiology. 14th (edn.), Macgrow-Hill Companies Inc,
New York NY, USA: pp 808.
Dey NC, Dey TK, Dey M & Sinha D 1998, �Bone Marrow Biopsy; Practical
Microbiology: Protozoology and Parasitology, Protozoal Parasites�.
14,151.
Davidson, RN 1998, �Practical guide for the treatment of leishmaniasis�, Drugs,
vol. 56, pp. 1009-1018.
Desjeux P 2004, �Leishmaniasis: current situation and new perspectives�. Comp
Immunol. Microbiol. Infect. Dis., vol. 27, pp. 305-318.

115
DGHS, 2009, �Training module on �LD body detection from blood buffy coat�
Mohakhali, Dhaka 1212, Bangladesh.
Etges, R, Bouvier, J & Boedier, C 1986, �The major surface protien of
Leishmania promastigotes is a protease�, Journal of Biological Chemistry,
vol. 261, pp. 9098-9101.
Elias, M, Rahman, AJMM & Khan, NI 1989, �Visceral leishmaniasis and its
control in Bangladesh�, Bulletin of the World Health Organization, vol. 67,
pp. 43-49.
Evans, TG, Teixeira, MJ, McAulliffe, IT, Vasconcelos, AW, Sousa, AQ &
Lima, JW 1992, �Epidemiology of visceral leishmaniasis in northeast
Brazil�, Journal of Infect Dis., vol. 166, pp. 1124-1132.
Ephros M, Paz A & Jaffe CL 1994, �Asymptomatic visceral leishmaniasis in
Israel�. Trans. R. Soc. Trop. Med. Hyg., vol. 88(6), pp. 651-652.
El-Hassan AM, Zijlstra EE, Ismael A & Ghalib HW 1995, �Recent
observations on the epidemiology of kala-azar in the Eastern and Central
States of the Sudan�. Tropical and Geographical Medicine,vol 47,pp.
151±156.
El-Hassan AM & Zijlstra EE 2001, �Leishmaniasis in Sadan. Mucosal
leishmaniasis�. V. Trans. R. Sco. Trop. Med. Hyg., vol. 95, pp 105-108.
El Tai, NO, El Fari, M, Mauricio, I, Miles, MA, Oskan, L & El Safi, SH 2001,
�Leishmania donovani: intraspecific polymorphisms of Sudances isolates

116
revealed by PCR-based analyses and DNA sequencing�, Exp. Parasitol.,
vol. 97, pp. 35-44.
Fong, TA & Mosmann, TR 1990, �Alloreactive marine CD+ T cell clones secrete
the Th1 pattern of cytokine�, Journal of Immunology, vol. 144, pp. 1744-
1752.
Formica, S, Roach, TIA & Blackwell, JM 1994, �Interaction with extracellualr
matrix proteins influences Lsh/Ity/Bcg (candidate Nramp) gene regulation
of macrophage priming/activation for tumor necrosis factor-á and nitrite
release�, Immunology, vol. 82, pp. 42-50.
Fakhar, K, Asgari, Q, Motazediam, MH, Mohebali, M, Hatam, GhR &
Alborzi, AV 2006, �First report of Kala-azar from qeshm island in Persian
gulf�. Iranian J. Parasitol. Vol. No. 1, pp. 53-56.
Ghose, AC, Halder, JP, Pal, SC, Mishra, BP & Mishra, KK 1979,
�Phytohaemagglutinin-induced lymphocyte transformation test in Indian
kala-azar�, Transactions of the Royal Society of Tropical Medicine and
Hygiene, vol. 73, pp. 725-726.
Gibson EM 1983, �The identification of Kala-azar and the discovery of
Leishmania donovani�. Medical History, vol. 27, pp. 203-213.
Galvao-Castro, B, Saferreira, JA, Marzochi, KF, Marzochi, MC, Countinho,
SG & Lambert, PH 1984, �Polyclonal B-cell activation, circulating
immune complexes and autoimmunity in human visceral leishmaniasis
Clinical and Expermental Immunology�, vol. 56, pp. 58-66.

117
Glew, RH, Saha, AK, Das, S & Remaley, AT 1988, �Biochemistry of
Leishmania species�, Microbiological Reviews, vol. 52, pp. 412-432.
Grogl, M, Daugirda, JL, Hoover, DL, Magill, AJ & Berman, JD 1993,
�Survivability and infectivity of visccrotoropic Leishmania tropica from
operation desert storm participants in human blood product maintained
under blood bank conditions�, Am. J. Trop. Med. Hyg., vol. 49, pp. 308-
315.
Gari-Toussaint, M, Leliever, A, Marty, P & Le-Fichoux, Y 1994,
�Contribution of serological tests of the diagnosis of visceral leishmaniasis
in patients infected with the human immunodeficiency virus�, Trans. R.
Sco. Trop. Med. Hyg., vol. 88, pp. 301-302.
Gradoni, L, Bryceson, A & Desjeux, P 1995, �Treatment of Mediterranean
visceral leishmaniasis.� Bull World Health Organ. Vol. 73, pp. 191-197.
Gregory DJ, Sladek R, Olivier M & Matlashewski G 2008, �Comparison of the
effects of Leishmania major or Leishmania donovani infection on
macrophage gene expression�. Infect Immun, vol. 76, pp. 1186-1192.
Hommel, M, Peters, W, Ranque, J, Quilici, M & Lanotte, G 1978, �The micro
ELISA technique in the serodiagnosis of visceral leishmaniasis�, Annals of
Tropical Medicine and Parasitology, vol. 72(3), pp. 142-452.
Herman, R 1980, �Cytphilic and opsonic antibodies in visceral leishmaniasis in
mice�, Infection and Immunity, vol. 28, pp. 585-593.

118
Haldar, JP, Ghose, S, Saha, KC & Ghose, AC 1981, �Serological profiles in
Indian post kala-azar dermal leishmaniasis�, Transaction of the Royal
Society of Tropical Medicine and Hygiene, vol. 75, pp. 514-517.
Halder, JP, Ghose, S, Saha, KC & Ghose, AC 1983 �Cell-mediated immune
response in Indian kala-azar dermal leishmaniasis�, Infections and
Immunity, vol. 42, pp. 702-707.
Ho, M, Koech, DK, Iha, DM & Bryceson, ADM 1983, �Immunosuppression in
Kenyan visceral leishmaniasis�, Clinical and Experimental Immunology,
vol. 51, pp. 207-214.
Hockmeyer, WT, Wellde, BT, Sabwa, CL, Smith, DH, Rees, PH & Kager, PA
1984, �A complement of fixation test for visceral leishmaniasis using
homologous parasite antigen I�, Ann. Trop. Med. Prasasitol, vol. 78, pp.
489-493.
Handman, E & Goding, JW 1985, �The Leishmania receptor for macrophage is
a lipid-containing glycoconjugate�, EMBO J., vol. 4, pp. 329-336.
Handman E, Schnur LF, Spithill TW & Mitchell GF 1986, J. Immunol., vol.
137, pp. 3608-3613.
Handman, E, McCnoville, MJ & Goding, JW 1987, �Carbohydrate antigens as
possible parasite vaccines A case for the Leishmania glycolipid�,
Immunology Today, vol. 8, pp. 181-185.

119
Heath, S, Chance, ML, Hommel, M & Cramption, JM 1987, �Cloning of gene
enoding the immunodominent surface antigen of hidnovani Molecular
Biochemical Parasitology�, pp. 23, pp. 211-222.
Holaday, BJ, Pompeii, M, Evans, T, Braga, D, Texeira, M & Sousa, A 1993,
�Correlates of Leishmania-specific immunity in clinical spectrum of
infection with Leishmania chagasi�, J of Infect Dis., vol. 167, pp. 411-417.
Hashim, FA, Ali, MS, Satti, M, El-Hassan, AM, Ghalib, HW, El-Safi, S & El-
Hag, IA 1995, �An outbreak of acute kala azar in a nomadic tribe in
western Sudan: features of the disease in a previously non-immune
population�, Trans. R. Soc. Trop. Med. Hyg., vol. 88, pp. 431-432.
Houghton, RL, Petrescu, M, Benson, DR, Skeiky, YA, Scalone, A & Badaro,
R 1998, �Cloned antigen (recombinant K39) of Leishmania chagasi
diagnostic for visceral leishmaniasis in human immunodeficiency virus
type 1 patients and a prognostic indicator for monitoring patients
undergoing drug therapy�, J. Infect. Dis., vol. 177, pp. 1339-1344.
Herwaldt, BL 1999, �Leishmaniasis�, Lancet, vol. 354, pp. 1191-1199.
Haidar, NA, Diab, ABL & El-Sheikh, AM 2001, �Visceral leishmaniasis in
children in the Yemen�, Saudi Med. J., vol. 22, pp. 516-519.
IIg, T, Stierhof, YD, Wiese, M, McConville, MJ & Overath, P 1994,
�Characterization of phosphoglycan containing secretary products of
leishmania�, Parasitology, vol. 108, pp. 563-571.

120
ICDDR, B 2002, �Clinical characteristics of Visceral Leishmaniasis in an
Endemic Community in Bangladesh.� ICDDRB Publication. vol. 3, 6-9.
ICDDR, B 2003, �Centre for health and population research�. Health and Science
Bulletin. vol. 1, pp. 1-6.
Indios Inc., USA Inbios International, Inc. 5621st Ave. South, Suite 600 Seattle,
WA 98104 USA 206-344-5821 P/N 900003.8
Jaffe, CL & Sarfstein, R 1987, �Species-specific antibodies to Leishmania
tropica (minor) recognize somatic antigens and exometabolites�, J.
Immunol., vol. 139, pp. 1310-1319.
Jehinek, T, Eichenlub, S & Loscher, T 1999, �Sensitivity and specificity of a
rapid immuochromatographic test for diagnosis of visceral leishmaniasis�,
Eur. J. Clin. Microbiol. Infect. Dis., vol. 18, pp. 669-670.
Jensen, AT, Gasim, S, Moller, T, Ismail, A, Gaafar, A, Kemp, M & Hussar,
AM 1999, �Serodiagnosis of Leishmania donovani infections: assessment
of enzyme-linked immunosorbent assays using recombinant L. donovani
gene B protein (GBP) and a peptide sequence of L. donovani (GBP)�,
Trans. R. Soc. Trop. Med. Hyg., vol. 93, pp. 157-160.
Joshi, A, Narain, JP, Prasittisuk, C, Bhatia, R, Hashim, G, Jorge, A, Banjara,
M & Kroeger A 2008, �Can visceral leishmaniasis be eliminated from
Asia�, Journal Vector Borne Dis., vol. 45, pp. 105-111.
Kager, PA & Rees, PH 1983, �Splenic aspiration. Review of literature�, Trop
Geogr. Med., vol. 35, pp. 111-124.

121
Kaufmann, SHE 1988, �T lymphocytes in intracellular microbial infections�,
Immunology Today, vol. 9, pp. 168-174.
Kweider, M, Lemesre, JL, Santoro, F, Kusnier, ZF, Sadigursky, M &
Capron, A 1989, �Development metacyclic Leishmania is associated with
the increasing of GP65, the major surface antigen Parasite Immunology�,
vol. 11, pp. 197-209.
Kreutzer, RD, Grogl, M, Neva, FA, Fryauff, DJ, Magill, AJ & Aleman-
Munoz, MM 1993, �Identification and genetic comparison of leishmanial
parasites causing viscerotropic and cutaneous disease in soldiers returning
from Operation Desert Storm�, Am. J. Trop. Med. Hyg., vol. 49, pp. 357-
363.
Kumar, R, Pai, K, Pathak, K & Sundar, S 2001, �Enzyme-linked
immunosorbent assay for recombinant K39 antigen in diagnosis and
prognosis of Indian visceral leishmaniasis�, Clin. Diagn. Lab. Immuno.,
vol. 8, vol. 1220-1224.
Lainson, R & Shaw, JJ 1987, �Evolution, Classification and Geographical
distribution. In: Peters W and Killick-Kedric R (eds)�, The Leishmaniases
in Biology and Medicine. vol. 1 London Academic Press. pp 1-120.
Lehn, M, Weiser, WY, Engelhorn, S, Gillis, S & Remold, HG 1989, �IL-4
inhibits H2O2 production and antileishmanial capacity of human cultured
monocytes mediated by IFN-gamma�, The Journal of Immunology, vol.
143, pp. 3020-3024.

122
Leiw, FY, Millott, S, Lelchuk, R, Chan, WL & Ziltener, H 1989, �Macrophage
activaiton by interfecron-gamma from host-protective T-cells is inhibited
by interleukin (IL)-3 and IL-4 produced by disease promoting T-cells in
leishmaniasis�, Europenan Journal of Immmunology, vol. 19, pp. 1227-
1232.
Liew, FY, Millott, S, Parkinson, C, Palmer, RMJ & Moncada, S 1990,
�Macrophage killing of Leishmania parasites in vivo is mediated by nitric
oxide from L-arginine�, J. of Immunolo., vol. 144, pp. 4794-4797.
Leiw, FY & O�Donnell, CA 1993, �Immunology of Leishmaniasis�, Advances in
Parasitology, vol. 32, pp. 161-259.
Le Fichoux Y, Quaranta JF, Aufeuvre JP, Lelièvre A, Marty P & Suffia I
1999, �Occurrence of Leishmania infantum parasitemia in asymptomatic
blood donors living in an area of endemicity in southern France�. J. Clin.
Microbiol., vol. 37, pp. 1953-1957.
Lachaud, L, Dereure, J, Chabbert, E, Reynes, J, Mauboussin, JM, Oziol, E,
Dedet JP & Bastien P 2000, �Optimized PCR Using Patient Blood
Samples For Diagnosis and Follow-Up of Visceral Leishmaniasis, with
Special Reference to AIDS Patients�. J. Clin. Microbiol. vol. 38, pp. 236-
240
Lachoud L 2002, �Value of two PCR methods for the diagnosis of canine visceral
leishmaniasis and the detection of asymptomatic carriers�. Parasitology,
London, vol. 125, pp. 197-207.

123
Murray, HW, Rubin, BY & Rothermel, CD 1983, �Killing of intracellualr
Leishmaina donovani by lymphokine-stimulated human mononuclear
phagocytes. Evidence that interferon-gamma is the activaing cytokine�,
Journal of Clinical Investigation, vol. 72, pp. 1506-1510.
Mosser DM & Edelson PJ. 1984, �Activation of the alternative complement
pathway by Leishmania promastigotes: parasite lysis and attachment to
macrophages�. J. Immunol., vol. 132(3), pp. 1501�1505
Mosser DM & Edelson PJ 1985, �The mouse macrophage receptor for C3bi
(CR3) is a major mechanism in the phagocytosis of Leishmania
promastigotes�. J. Immunol. vol. 135, pp. 2785-2789.
Manson-Bahr, PEC 1987, �Old World leishmaniasis, In: FEG Cox (ed.), The
Welcome Trust illustrated history of tropical diseases�, The Welcome Trust
London United Kingdom. pp 206-217.
Murray HW, Oca MJ, Granger AM & Schreiber RD 1989, �Requirement for
T cells and effect of lymphokines in successful chemotherapy for an
intracellular infection. Experimental visceral leishmaniasis�. J. Clin.
Invest., vol. 83(4), pp. 1253�1257.
Masum, C, Lamouroux, D, Dunan, S & Quilici, M 1990a, �An epidemiological
investigation of a kala-azar outbreak at Kalhati upazilla of Tangial District,
Bangladesh�, Journal of Preventive and Social Medicine, vol. 4-9, pp. 13-
15.
Masum, MA, Chowdhury, MS, Ahmed, RU & Mia, MAH 1990b, �Outbreak
Diseases�, Editor-in-chief P.R. Murray, Editors, E.J. Baron, M.A. Pfaller,

124
F.C. Tenover and R.H. Yolken, In �Manual of Clincal Microbiology� 6th
edtion. American Society for Microbiology, Washinton D.C.; USA. pp
110-122.
Masum, MA & Chowdhury, MS 1991, Journal of Preventive and Social
Medicine, vol. 4(9), pp. 48-53.
Manson Bahr, PEC & Bell, DR 1991, �Manson�s Tropical Diseases.� 19th eds.
Bailliere Tindall, Philadelphia. U.S.A. pp. 87-113.
Magill, AJ, Grogl, M, Gasser, RA, Wellington, S & Oster, CN 1993,
�Viscerotropic leishmaniasis caused by Leishmania tropica in soldiers
returning from Operation Desert Storm�, N. Engl. J. Med., vol. 328, pp.
1383-1387.
Murray, HW & Hariprasad, J 1995, �Interleukin 12 is effective treatment for an
established systemic intracellular infection: experimental visceral
leishmaniasis�, Journal of Experimental Medicine, vol. 181, pp. 387-391.
Musa, AK, Alam, MN, Tabassum, S &Rahman, M 1996, �Study on cell
mediated immunity in patients of visceral leishmaniasis.� p. 73.
Montoya, Y, Leon, C, Talledo, M, Nolasco, O, Padilla, C & Munoz-Najar, U
1997, �Recombinant antigens for specific and sensitive serodiagnosis of
Latin American tegumentary leishmaniasis�, Trans R Soc Trop Med Hyg.,
vol. 91, pp. 674-676.
Martin, SK, Thuita-Harun, L, Adoyo-Adoyo, M & Wasunna, KM 1998, �A
diagnostic ELISA for visceral leishmaniasis, based on antigen from media

125
conditioned by Leishmania donovani promastigotes�, Ann. Trop. Med.
Parasitiol., vol. 92, pp. 571-577.
Maltezou, HC, Siafas, C, Mavrikou, M, Spyridis, P, Stavrinadis, C &
Karpathios, TH 2000, �Visceral leishmaniasis during childhood in
southern Greece�, Clin. Infect. Dis., vol. 31, pp. 1139-1143.
Minodier, P & Garnier, JM 2000, �La leishmaniose viscerale infantile en
Provence�, Arch pediatr, vol. 7, pp. 572-577.
Motazedian H, Karamian M, Noyes HA & Ardehali S 2002, �DNA extraction
and amplification of Leishmania from archived,Giemsa-stained slides, for
the diagnosis of cutaneous Leishmaniasisby PCR.� Annals of Tropical
Medicine and Parasitology, vol. 96, pp. 31�34.
Maalej, IA, Chenik, M, Louzir, H, Ben, SA, Bahloul, C & Amri, F 2003,
�Comparative evaluation of ELISA based on ten recombinant or purified
Leishmania antigens for the serodiagnosis of mediterrean visceral
lesihmaniasis�, Am. J. Trop. Med. Hyg., vol. 68, pp. 312-320.
Maury, R, Singh, RK, Kumar, B, Salotra, P, Rai, M & Sundar, S 2005,
�Evaluation of PCR of diagnosis of Indian Kala-azar and Assessment of
cure.� J. Clin. Microbiol. vol. 43(7), pp. 3038-3041.
Manzoor, ER, Zardad, M, Javed S & Azhar MQ 2008, �Haematological
findings in relation to clinical findings of visceral leishmaniasis in hazara
division�. J. Ayub Med. Coll. Abbottabad, vol. 3, pp 20-24.

126
Neva, F & Sacks, D 1990, �Leishmaniasis�. Editors K.S. Warren and A.A.F.
Mahmoud. In �Tropical and Geographical Medicine� 2nd edition. McGraw-
Hill Inc. USA. pp. 296-308.
Nuzum, E, White, F & Thakur, C 1995, �Diagnosis of symptomatic visceral
leishmaniasis by use of the polymerase chain reaction and patient bolld�, J.
Infec. Dis., vol. 171, pp. 751-754.
Nuttall SD 2002, �A naturally occurring NAR variable domain against the
Gingipain K protease from Porphyromonas gingivalis.� FEBS Lett. vol.
516, pp. 80-86
Oliaro PL & Bryceson ADM 1993, �Practical progress and new drugs for
changing patterns of leishmaniasis�. Parasitology, vol. 9, p. 323.
Ostria, M & Tezeda, SG 2002, �Molecular probes and polymerase chain reaction
for detection and typing of Leishmania species in Mexico. Trans R Soc
Trop Med Hyg., vol. 96, pp. 101-104.
Pampiglione, S, Manson-Bahr, PEC, La Placa, M & Borgatt, MA 1975,
�Studies in Meditteranean Leishmaniasis 3�, The leishmanin skin test.
Transaction of Royal Society of Tropical Medicine and Hygiene, vol. 69,
pp. 60-68.
Pearson, RD & Steigbigel, RT 1980, �Mcchanism of effect of human sera upon
Leishmania donovani�, J. of Immunolo., vol. 125, pp. 2195-2201.

127
Plant, JE, Blackwell, JM, O�Brien, AD, Bradley, DJ & Glynn, AA 1982, �Are
the Lash and Ity disease resistance genes at one locus on muse
chromosome 1? Nature, vol. 297, pp. 510-511.
Peleman, R, Wu, J, Fargeas, C & Deiespesse, G 1989, �Recombinant
interleukin 4 suppresses the production of interferon gamma by human
mononuclear cells�, Journal of Experimental Medicine, vol. 170, pp. 1751-
1756.
Pearson RD & Souza AQ 1990, �Leishmania species: visceral (kala-azar),
cutaneous and mucosal leishmaniasis�. In: Mandell GL, Douglas RG,
Bennett JB, eds. Principles and practice of infectious diseases. 3rd ed. New
York: Churchill Livingstone, pp. 2066-2077.
Palma, G & Gutierrez, Y 1991, �Laboratory diagnosis of Leishmania�, Clin Lab
Med., vol. 11, pp. 909-922.
Prasad, LS 1999, Kala-azra. Indian J. Pediatr., vol. 66, pp. 539-546.
Peter, CM 2000, �Leishmaniasis. In: Behrman RE, Kliegman RM, Jenson HB,
editors, Nelson Textbook of Pediatrics. 16th edn. Philadelphia WB
Saunders 2000.
Park, JE & Park, K 2000, �Park�s Textbook of Preventive and Social Medicine,
16th ed. M/S Bannasidas Bhanot Publishers, Jabalpur, India, pp. 231-233.
Pizzuto, M, Piazza, M, Senese, D, Scalamogna, C, Calattini, S & Corsico, L
2001, �Role of PCR in diagnosis and prognosis of visceral leishmaniasis in

128
patients co-infected with human immunodeficiency virus type 1�, J. Clin.
Microbiol., vol. 39, pp. 351-361.
Peacock, CS, Collins, A, Shaw, M, Silveira, F, Costa, J & Coste, CH 2001,
�Genetic epidemiology of visceral leishmaniasis in northeastern Brazil�,
Nenet Epidemiol., vol. 20, pp. 383-396.
Quellette, M & Papadopoulou, B 1993, �Mechanisms of drug resistance in
leishmania�, Parasitology Today, vol. 9(5), pp. 150-153.
Rezai, HR, Ardekali, SM, Amirhakimi, G & Kharazmi, A 1978,
�Immunological features of kala-azar�, American Jounral of Tropical
Medicine and Hygiene, vol. 27, pp. 1079-1083. .
Rahman, KM & Islam, N 1979, �Recent advances in kala-azar�, Bangladesh
Medical Journal, vol. 8(1), pp. 13-19.
Rahman, KM & Islam, N 1983, �Resurgence of visceral leishmaniasis in
Bangladesh�, Bull World Health Organ, vol. 61(1): pp. 113-116.
Remaley, AT, Glew, RH, Kuhns, DB, Basford, RE & Waggoner, AS 1985,
�Leishmania donovani: Surface membrane acid phosphatase blocks
neutrophil oxidative metabolite productions�, Experimental Parasitology,
vol. 60, pp. 331-341.
Russell DG & Wilhelm H 1986, �The involvement of a major surface
glycoprotein (gp63) of Leishmania promastigotes in attachment to
macrophages�. J. Immunol., vol. 136, pp. 2613-2620.

129
Reed, SG, Badaro, R & Lloyd, RMC 1987, �Identification of specific and corss
reactive antigens of Leishmania donovani chagasi by human infection
sera�, Journal of Immunology, vol. 138, pp. 1596-1601.
Rizvi, FS, Quassi, MA, Marty, B, Santoro, F & Capron, A 1988, �The major
surface protein of Leishmania promastigotes is a fibronectin-like
molecule�, European Journal of Immunology, vol. 18, pp. 473-478.
Russell, DG & Wright, SD 1988, �Complement receptor type 3 (CR3) binds to
an Arg-Gly-Asp-containing region in the major surface glycoprotein, gp63,
of Leishmania promastigotes�, J. of Experimental Medicine, vol. 168, pp.
279-292.
Rolland-Burger, L, Rolland, X, Greive, W & Monjour, L 1991, �Immunoblot
analsysis of the humorla response to Leishmania donovani infantum
polypeptides in human visceral leishmaniasis�, Journal of Clinical
Microbiology, vol. 29(7), pp. 1429-1435.
Raziuddin, S, Telmasani, AW, El-Awad, MEH, Al-Amari, O & Al-Janadi, M
1992, �Gamma- T cell and the immune response in visceral
leishmaniasis�, European Journal of Immunology, vol. 22, pp. 1143-1148.
Rab, MA & Evans, DA 1995 �Leishmania infantum in the Himalayas�, Trans R
Soc Trop Med Hyg., vol. 89, pp. 27-32.
Raj, VS, Ghosh, A, Dole, VS, Madhubala, R, Myler, PJ & Stuart, KD 1999,
�Serodiagnosis of leishmaniasis with recombinant ORFF antigen�, Am. J.
Trop. Med. Hyg., vol. 61, pp. 482-487.

130
Rajasekariah, GH, Ryan, JR, Hillier, SR, Yi, LP, Stiteler, JM & Cui, L 2001,
�Optimisation of an ELISA for the serodiagnosis of visceral leishmaniasis
using in vitro derived promastigote antigens�, J. Immunol Methods, vol.
252, pp. 105-119.
Rijal S, Koirala S, Vander Stuyft P, Boeleart M 2006, �The economicburden of
visceral leishmaniasis for households in Nepal�. Trans R Soc Trop Med
Hyg., vol. 100(9): pp 838�841
Rai, ME, Muhammad, Z, Sarwar, J & Qureshi, AM 2008, �Haematological
findings in relation to clinical findings of visceral leishmaniasis hazara
division�. Journal Ayub Coll Abbottabad, vol. 3, pp 20-22.
Rahman, M & Ahmed, B 2008, �Kala-azar Elimination program in Bangladesh.
In: Internal conference of combat neglected.� Tropical Diseases, vol. 41,
pp. 212-215.
Swaminathan, CS, Shortt, HE & Anderson, LA 1942, �Transmission of Indian
kala-azar to man by the bites of Phlebotomus argentipes�, Indian Journal of
Medical Research, vol. 30, pp. 473-477.
Saleem M, Anwar CM & Malik IA 1986, �Visceral Leishmaniasis in children. A
new focus in Azad Kashmir�. J. Pak. Med. Assoc. vol. 36, pp. 230�234.
Srivastava, S & Chaknnvarty, AK 1984, �Investigation of possible zoonotic
reservoir of Indian kala-azar�, Annals of Tropical Medicine and
Parasitology, vol. 78(5), pp. 501-504.

131
Smith, DH, Wellde, BT, Sabwa, CL, Reardon, MJ & Hockmeyer, WT 1984,
�A complement fixation test for visceral leishmaniasis using homologous
parasitic antigen�, II. Results in an endemic area in Kenya. Annals of
Tropical Medicine and Parasitology, vol. 78(5), pp. 495-500.
Saha, AK, Das, S, Glew, RH & Gottieb, M 1985, �Resistance of leishmanial
phosphatases to inactivation by oxygen metabolites�, Journal of Clinical
Microbiology, vol. 22, pp. 329-332.
Sypek JP, Matzilevich MM & Wyler DJ 1991, �TH2 lymphocyte clone can
activate macrophage antileishmanial defense by a lymphokine-independent
mechanism in vitro and can augment parasite attrition in vivo�. Cell.
Immunol., vol. 132, p. 178.
Smyth, AJ, Gosh, A, Hassan, MO, Bass, D, De Bruijn, MH, Adhya, S, Mallik,
KK & Barker, DC 1992, �Rapid and sensitive detection of Leishmania
kinteoplast DNA from spleen and blood samples of kala-azar patients�,
Parasitology, vol. 105, pp. 183-192.
Scott P 1993, �Selective differentiation of CD4+ T helper cell subsets�. Current
Opinion in Immunology, vol. 5, pp. 391-397.
Sunder, S, Rosenkaimer, F & Murray, HW 1994, �Successful treatment of
refractory visceral leishmaniasis in India using antimony plus interferon-�,
Journal of Infectious Diseases, vol. 170, pp. 992-996.
Sacks, DL, Kenney, RT, Kreutzer, RD, Jaffe, CL, Gupta, AK and Sharma,
MC 1995, �Indian kala-azar caused by Leishmania tropical�, Lancet, vol.
345(8955), pp. 959-961.

132
Singh, S, Gilman-Sachs, A, Chang, KP & Reed, SG 1995, �Diagnostic and
prognostic value of K39 recombinant antigen in Indian leishmaniasis�, J.
Parasitol., vol. 81, pp. 1000-1003.
Shiddo, SA, Aden-Mohamed, A, Akuffo, HO, Mohamud, KA &
Thorstensson, R, 1995, �Visceral leishmaniasis in Somalia: prevalence of
markers of infection and disease manifestations in a village in an endemic
area�, Trans R Soc Trop Med Hyg., vol. 89, pp. 361-365.
Sunder, S, Reed, SG, Sharma, S, Mehrotra, A & Murray, HW 1997.
�Circulating T helper 1 (Th1) cell- Th2 cell associated cytokines in Indian
patients with visceral leishmaniasis�, American Journal of Tropical
Medicine and Hygiene, vol. 56(5), pp. 522-525.
Sundar, S, Reed, SG, Singh, VP, Muma, PCK & Murray, HW 1998, �Rapid
accurate field diagnosis of visceral leishmanisis�, Lancet, vol. 351, pp. 563-
565.
Singh, S 1999, �Diagnostic and Prognostic markers of anti-kala-azar therapy and
vaccination�, Proceeding of V Round Table Conference Series. No. 5
Gupta S and Sood OP (Eds). New Delhi: Ranbaxy Science Foundation. pp
95-114.
Sassi, A, Louzir, H, Ben, SA, Mokni, M, Ben, OA & Dellagi, K 1999,
�Leishmanin skin test lymphoproliferative responses and cytokine
production after symptomatic or asymptomatic Leishmania major infection
in Tunisisa�, Clin. Exp. Immunol., vol. 116, pp. 127-132.

133
Santos-Gomes, G, Gomes-Pereira, S, Campino, L, Araujo, MD & Abranches,
P 2000, �Performance of immunoblotging in diagnosis of visceral
Leishmaniasis in human immunodeficiency virus Leishmania sp-co
infected patients�, J. Clin. Microbiol., vol. 38, pp. 175-178.
Schoone GJ, Hailu A, Kroon CC, Nieuwenhuys JL, Schallig HD & Oskam L
2001, �A fast agglutination screening test (FAST) for the detection of anti-
Leishmania antibodies�. Trans. R. Soc. Trop. Med. Hyg., vol. 95, pp. 400-
401.
Schur, LF & Jacobson, RL 2001, �Parasitological techniques�, In: Peters W and
Killick Kendrick R (ed.). Leishmaniasis in biology and medicine, vol 1
Academic Press New York NY. pp. 500-541.
Salotra, P, Sreenivas, G & Pogue, GP 2001, �Development of a speceis-specific
PCR assay for detection of Leishmania donovani in clinical samples from
patients with kala-azar and post kala-azar dermal leishmaniasis�, J. Clin.
Microbiol., vol. 39, pp. 849-854.
Schallig, HD, Schoone, GJ, Kroon, CC, Hailu, A, Chappuis, F & Veeken, H
2001, �Development and application of �simple� diagnostic tools for
visceral leishmaniasis�, Med Microbiol Immunol (Berl)., vol. 190, pp. 69-
71.
Senaldi, G, Xiao-su, H, Hoessli, DC & Bordier, C 2001, �Serological diagnosis
of visceral leishmaniasis by a dot-enzyme immunoassay for the detection
of a Leishmani donovani related circulating antigen�, J. Immunol. Methods,
vol. 193, pp. 9-15.

134
Sundar, S, Pai, K, Kumar, R, Tripathi, KP, Gam, AA, Roy, M & Kenny, RT
2001, �Resistance to treatment in kala-azar: speciation of isolates from
Northeast India�, Am J Trop Med Hyg., vol. 65, pp. 193-196.
Sundar, S & Rai, M 2002, �Laboratory Diagnosis of Visceral Leishmaniasis�,
Clin Diagn Lab Immunol., vol. 9(5), pp. 951�958.
Singh, S, Kumari, V & Singh, N 2002, �Predicting kala-azar disease
manifestations in asymptomatic patients with latent Leishmania donovani
infection by detection of antibody against recombinant K39 antigen�, Clin.
Diagn. Lab. Immunol., vol. 9, pp. 568-572.
Sundar, S 2003, �Indian Kala-azar-Better Tools Needed for Diagnosis and
Treatment�, Journal of Postgraduate Medicine, vol. 49, pp. 29-30.
Schonian G, Nasereddin A, Dinse N et al. 2003. �PCR diagnosisand
characterization of Leishmania in local and imported clinicalsamples�.
Diagnostic Microbiology and Infectious Disease, vol. 47, pp 349�358.
Sarker, CB, Momen, A, Jamal, MF, Siddiqui, NI, Siddiqui, FM, Chowdhury,
KS, Rahman, S & Talukder, SI 2003, �Immunochromatographic (rk39)
strip test in the diagnosis of visceral leishmaniasis in Bangladesh.
Mymensingh Medical J. vol. 12, pp. 93-97.
Shamsuzzaman, AK, Hossain, MA, Musa, AK, Hasan, MU, & Dhar, DK
2003, �A preliminary report of culture of Leishmania donovani in
Mymensingh Medical College and evaluation of new
immunochromatography test (ICT).� Mymesningh Med. J. vol. 12, pp. 51-
54.

135
Sarkari, B, Chance, M & Hommel, M 2005, �A capture ELISA for the diagnosis
of visceral leishmaniasis using a monoclonal antibody against a
lesihmanial urinary antigen�, Iranian Biomedical Journal, vol. 9(3), pp.
117-122.
Shamszzaman, AKM, Mahmud, MC, Akther, S, Musa, AKM & Hossain, MA
2007, �LD bodies from blood Buffy coat: An easy approach for definitive
diagnosis of visceral leishmaniasis�, Bangladesh J. Med. Microbiol., vol.
01(02), pp. 43-47.
Salam, AM, Mondul, D, Kabir, M & Haque, R 2009, �rK39 enzyme-linked
immunosrbent assay for the diagnosis of Kala-azar in an endemic zone of
Bangladesh�, Pak. J. Med. Sci., vol. 25, No. 4, pp. 635-640.
Salam, MA, Mondal, D, Kabir, M, Ekarm, AR & Haque, R 2009, �PCR for
diagnosis and assessment of cure in kala-azar patients in Bangladesh�, Acta
Trop., vol. 113(1), pp. 5-55.
Thakur, CP, Kumar, M & Pathak, PK 1981, �Kala-azar hits, again�, Journal of
Tropical Medicine and Hygiene, vol. 84, pp. 271-276.
Turco, SJ, Hull, SR, Orlandi, PAJr, Sheperd, SD & Homans, SD 1987,
�Structure of major carbohydrate fragment of the Leishmania donovani
lipophosphoglycan of National Academy for Science, USA. vol. 76, pp.
2143-4354.
Turco, SJ 1988, �The lipophosphoglycan of Leishmania�, Parasitology Today,
vol. 4, pp. 255-257.

136
Tolson, DL, Turco, SJ, Beecroft, RP & Pearson, TW 1989, �The
immunochemical structure and surface arrangement of Leishmania
donovani lipophosphoglycan determined using monoclonal antibodies�,
Molecular and Biochmicla Parasitology, vol. 35, pp. 109-118.
Totan, M, Dagdemir, A, Muslu, A and Albayrak, D 2002, �Visceral childhood
leishmaniasis in Turkey�, Acta Paediatr, vol. 91, pp. 62-64.
Thakur, Chandreshwar Prasad; Narayan, Shyam, Ranjan & Alok 2003,
�Kala-Azar (Visceral Leishmaniasis) and HIV Coinfection in Bihar, India:
Is This Combination Increasing�. AIDS Journal of Acquired Immune
Deficiency Syndromes, vol. 32, pp 572-573
Talukder, SI, Huq, MH, Rahman, S, Haque, MA, Sarker, CB, & Ali, MS
2003, �Epidemiological characteristics of sixty five cases of Kala-azar
attending to a laboratory in Mymensingh.� Mymensingh Med. J. Vol. 12,
pp. 89-92.
Uzair, M, Khan, SJ, Munib, S, Raheem, F & Shah, HS 2004, �Visceral
leishmaniasis (kala-azar): presentation, diagnosis and response to therapy
(an experience of ten cases in adults)�, Gomal Journal of Medical Sciences,
vol. 2, No. 1.
Vanorshovan, BA, Michielsen, P & Vandepitte, J 1979, �Fatal leishmaniasis in
renal transplant patient, Lancet, pp. 740-747.
Vinayak, VK, Mahajan, D, Sobit, RC, Singh, N & Sunder, S 1994, �Anti-66
kDa anti leishmanial antibodies as specific immunodianostic probe for
visceral leishmaniasis�, Indian J Med Res., vol. 99, pp. 109-114.

137
Walton, BC & Valverde, L 1979, �Racial differences in espundia�, Annals of
Tropical Medicine and Parasitology, vol. 73, pp. 23-29.
Weintraub, J, Gotlier, M. and Weintbaum, F.I. (1982). Leishmania tropica:
assocoiaiton of a B-cell mitogen with hypergammaglobulinaemia in mice.
Experimental Parasitology. vol. 53, p 87.
Weigle, KA, de Davalos, M, Heredia, P, Molineros, R, Saravia, NG &
D�Alessandro, A 1987, �Diagnosis of cutaneous and mucocutaneous
leishmaniasis in Colombia: a comparison of seven methods�, Am J Trop
Med Hyg., vol. 36, pp. 489-496.
Wilson, ME & Hardin, KK 1988, �The major concavalin A-binding glycoprtein
of Leishmania donovani chagasi promastigotes is involved in attachment to
human macrophage�, Journal of Immunology, vol. 141, p. 265.
Wilson, ME, Hardin, KK & Donelson, JE 1989, �Expression of the major
surface glycoprotein of Leishmania donovani chagasi in virulent and
attenuated promastigotes�, Journal of Immunology, vol. 143, pp. 678-684.
World Health Organization 1990, �The Leishmaniases� Report of a WHO
Expert Committee, Technical Report Series 701, WHO, Geneva, 1984.
World Health Organization 1991, Control of the leishmaniasis. Technical report
series. vol. 793, pp. 1-9.
Willams, JE 1995, �Leishmania JE and Trypanosoma�, In: Medical parasitology.
A practical approach. Gillespie SH, Hawkey PM (Eds.). London: Oxford
University Press. pp. 51-64.

138
Wilson, SM 1995, �DNA-based methods in the detection of Leishmania parasites:
Fields applications and practicalities�, Ann Trop Med Parasitol., vol. 89,
pp. 95-100.
World Health Organization 1996, Manual on Visceral leishmaniasis control.
World Health Organization, Division of Control of Tropical Diseases,
Geneva.
World Health Organization 1997, 13th Programme Report, Special Programme
for Research and Training in Tropical Diseases, WHO Geneva,
Switzerland. pp. 1-60.
Wortman, G, Sweeney, C, Houng, HS, Aronson, N, Stiteler, J & Jackson, J
2001, �Rapid diagnosis of Leishmaniasis by fluorogenic polymerase chain
reaction�, Am J Trop Med Hyg., vol. 65, pp. 583-587.
Yang, DM, Fairweather, N, Button, LL, McMaster, WR, Kahl, LP & Liew,
FY 1990, �Oral Salmonella typhimurium (AroA) vaccine expressing a
major leishmanial surface protein (gp63) preferentially induces T-helper 1
cells and protective immunity against leishmaniasis�, Journal of
Immunology, vol. 145, pp. 2281-2285.
Zijlstra, EE, Ali, MS, Hassn, AM, Toum, AI, Satti, M, Ghalib, HB & Kager,
PA 1992, �Kala-azar; a comparative study of parasitological methods and
the direct agglutination test in diagnosis�, Trans. R. Soc. Trop. Med. Hyg,
vol. 86, pp. 505-507.
Zijlstra EE, El-Hassan AM, Ismael A & Ghalib HW 1994, �Endemic kala-azar
in eastern Sudan: a longitudinal study on the incidence of clinical and

139
subclinical infection and post-kala-azar dermal leishmaniasis�. Am. J. Trop.
Med. Hyg., vol. 51, pp. 826�836.
Zijlstra EE, Khalil EAG & Kager PA 2000, �Post-kala-azar dermal
leishmaniasis in the Sudan: clinical presentation and differential diagnosis�.
Brit. J. Dermatol., vol. 143, pp. 136-143.
Zijlstra, EE, Nur, Y, Desjeux, P, Khalil, EA, El-Hassan, AM & Groen, J
2001, �Diagnosing visceral leishmaniasis with the recombinant K39 strip
test: experience from the Sudan�, Trop Med Int Health, vol. 6, pp. 108-113.

140
APPENDICES
APPENDIX I
DATA SHEET
SI.No:
Name: Age: Sex:
Father�s name:
Mother�s name:
Address:
Village: PO: Thana: District:
Hospital Particulars: Regn: Ward: Bed: Unit:
Socio-economic Condition: Poor/ Average.
Housing: Tinshade/ Earthen/ Others. Family history: Positive/ Negative.
Clinical information
Irregular fever: weeks, Emaciation: no/ mild / remarkable
Bleeding: yes / no Appetite: Normal / Decreased /Increased. Jaundice..................
Blackening of skin: yes / not Weight: ........ Anaemia: no / mild /remarkable
Spleen: palpable /not palpable, if palpable: ....cm from left costal margin.
Liver: palpable / not palpable, if palpable: .....cm from right costal margin.
Previous treatment for kA: no/ yes. if yes: Miltefosine/ SAG/ Duration and dose..........
Ongoing treatment:

141
Laboratory information
Haemoglobin: ...... gm/dl.
Platelets: ................... ./cu mm.
TLC: / cu mm of blood.
DLC- N% L% E% M% B%
ESR :..................min in the 1st hour.
Prothrombin time: not done/ sec
BT: not done/ min sec
CT: not done/ min sec
ICT strip: positive/ negative
Spleen aspirate: not done/score
BM: not done / score
Buffy coat: not found / score
Comment:
Signature of data collector

142
APPENDIX II
Making buffy coat (density gradient centrifugation)
In a sterile round bottom disposable test tube (10 ml capacity), 2 ml of Histopaque
pipetted carefully.
Substance Name:
Histopaque 1077
Ingredient
Diatrizoic acid sodium salt dehydrate 9.000%
Ficoll 5.7%
Water 85.300%
Then 2 ml of citrated blood from the vacutainer was poured slowly by the side of the test
tube so that the blood completely overlay the separation fluid without any mixing. The
test tube was centrifuged for 15 minutes. 3000 rpm referable in a swinging centrifuge
machine.
Taking buffy coat material and preparation of smears
After centrifugation, a thick buffy coat was seen in the interface of plasma and
Histopaque. RBCs was settled at the bottom part of the test tube. Now the plasma was
gently sucked by a micropipette of 1ml capacity and was preserved in an eppendrof tube.
Then by another of 100-200 µl capacity/micropipette, the buffy coat material was sucked
out very gently. Part of the material was used in another eppendrof containing 0.5 ml of
95% alcohol for analysis by PCR (DGHS, 2009).
Staining of buffy coat smears
Buffy coat film were stained by Leishman stain.

143
Preparation of Leishman stain:
This stain was prepared by 0.2 gm of powdered dye & was transferred to a conical flask
of 200-250 ml capacity.
100 ml of methanol was added and warmed the mixtures to 500C for 15 mins,
occasionally shaking it
The flask was allowed to cool & filtered. It was then ready for use
Procedure of Leishman staining
A air dried thin film was made and placed on a staining rack and flooded with Leishman
stain. The slide was left for 2 minutes to fix. Double amount distilled water was added
over the flooded stain from a plastic wash bottle for better mixing of the solution. The
stain was left for 10 minutes. The stain was washed with running tap water and air dried
again the smear to examine under oil immersion objectives (DGHS, 2009).

144
APPENDIX III
Equipments for PCR
Centrifuge 5415 R with refrigerator
Vortex Mixer (Digi system VM-2000)
Micropipettee (Company-Eppendrof)
Laminar Flow Cabinet (Company stremline)
Microwave oven for gel preparation
Electric heated dryer (Model Jo2714)
Thermal cycler (Model-Master cycler personal)
Gel apparatus (Major science Minis-300)
Major science Minis UVDI
Preparation of PCR mixture:
DDW (double Distilled water) -------------- 38.5 µl, 1 =38.5 µl
Buffer solution
(Contained 10 mM Tris-HCl, pH 8.3,
50 mM KCl, 1.5mM MgCl2) ------------------ 5µl, 1 = 5µl
dNTP ---------------------------------------------- 4µl, 1 = 4µl
Primer (1st pair/2nd pair) -------------------------1µl, 1= 1µl
= 48.5µl
Taq Polymerase ---------------------------------------------0.5µl
In each PCR tube:
Reaction mixture --------------------------------------------49 µl
Extracted DNA ----------------------------------------------1 µl
Total volume in each PCR tube -------------------------= 50 µl

145
APPENDIX IV
To perform Hemoglobin estimation: Requirements:
Equipments Consumables Refrigerator Colorimeter,
Micropipettes
Gloves, Tissue Paper, Labels, Test tubes � 7/10ml Pipettes � 5/10ml Cuvette Micro tips Measuring cylinder 500ml Drabkin�s solution Hemoglobin control Discard bin & Hypochlorite
Principle:
The method is based on the determination of cyanmethemoglobin
In this method, hemoglobin is oxidized by ferricyanide to methemoglobin.
The methemoglobin is finally converted by cyanide to the stable
cyanmethemoglobin
The concentration of cyanmethemoglobin is directly proportional to
hemoglobin concentration in the blood.
The OD is measured in the Colorimeter at wavelength 540 nm
Procedure
The colorimeter was switched on and adjusted at the wavelength 540.
The samples of blood was arranged to be tested in order and the protocol
sheet was filled in details.
7/10 ml test tubes were labeled with sample number; one tube was
allocated for testing control.

146
5 ml Drabkin�s solution was dispensed to the test tubes, using a pipette and
rubber bulb.
The EDTA tubes containing blood was shacked to ensure thorough mixing
of the cells and plasma.
By using a micropipette and tip, 20ìl of control was added to the test tube
marked �Control.�
This step was repeated till all the samples have been added to the tubes
bearing the corresponding number.
Test tubes were Shacked well (the tube was gently tapped on the palm or
the mouth of the tube was covered with Para film and the tube was
inverted) and was kept at room temperature for 3-5 minutes.
The colorimeter was brought to zero with reagent blank.
The OD of the control solution was taken and then serially the OD of the
samples.
The color intensity was stable for 1 hour; however, the reading was taken
at the same time (either 5 minutes or 10 minutes) for all the patients to
ensure uniformity.
The Optical density value was documented on the protocol sheet
immediately.

147
To perform platelet counts Requirements:
Equipments Consumables
Refrigerator,
Counting chamber
and cover slip
Micropipettes
Gloves, Tissue Paper, Labels,
Platelet diluting fluid, Micro tips
Discard bin & Hypochlorite
Principle:
Whole blood is diluted 1 in 200 in platelet diluting fluid
Platelets are counted microscopically using an Improved Neauber ruled
counting chamber (haemocytometer)
The number of platelets is calculated by adjusting for the dilution and the
volume of fluid used.
Procedure:
The blood was collected up to mark 0.5 from anticoagulated blood
Blood was wiped off from outside the pipette
Platelet diluting fluid was sucked up to mark 11 above
It was then mixed well by rotating the pipette for a few seconds between the
fingers
The cover slip was placed on the counting chamber
The pipette was rotated again for few times and the fluid was discarded
from the stem and a little from the bulb
The tip of the pipette was touched at the edge of the cover slip at about 45º
angle and slowly the fluid was introduced
The chamber was filled by negative pressure

148
Care was taken to ensure that there was no air bubble under the cover slip
The chamber was left undisturbed for 20 minutes for the cells to settle
The chamber was placed under microscope and was focused with 10X
objective.
The cells were then counted with 40X objective in five small squares of the
central 25 square grids.
Platelets were seen as small bright retractile fragments.

149
To perform WBC counts
Requirements:
Equipment Consumables Refrigerator, Counting chamber and cover slip Micropipettes
Manual counter Microscope
Gloves, Tissue Paper, Labels, WBC Diluting fluid, Micro tips Discard bin & Hypochlorite,
Principle:
Whole blood is diluted 1 in 20 in an acid reagent which haemolyze the red
cell, leaving the white cells
WBC is counted microscopically using an Improved Neauber ruled
counting chamber (haemocytometer)
The number WBC per liter of blood is calculated
Procedure:
The blood was collected up to mark 0.5 or 1 either from oxalated or EDTA
mixed blood or directly from a finger prick
Blood was wiped off from outside the pipette
WBC fluid was sucked up to mark 11 above
It was then mixed well by rotating the pipette for a few seconds between the
fingers
The cover slip was placed on the counting chamber
The pipette again was rotated for few times and the fluid was discarded
from the stem and a little from the bulb.
The tip of the pipette was touched at the edge of the cover slip at about 450
angle and the fluid was slowly introduced.
The chamber was filled by negative pressure.

150
Care was taken to ensure that there was no air bubble under the cover slip.
The chamber was left undisturbed for 2-5 minutes for the cells to settle.
The chamber was placed under microscope and the chamber was focused
with 10X objective.
The cells were then counted with 40X objective in four large square
corners.
The cells was counted that touched the upper and right hand margins.
The cells were avoided that touched the left and bottom margins.

151
Determination of ESR
Principal: If blood containing anticoagulant is allowed to stand in a tube, place
vertically. The R.B.C settle down gradually to the bottom since their specific
gravity is greater than that of plasma. The rate in which the R.B.C settle is noted.
Apparatus and reagents:
i) Westergren ESR tube
ii) ESR stand
iii) Disposable syringe with needle
iv) Cotton
v) Test tube
vi) 3.8% sodium citrate
vii) 70% ethanol
Procedure: 0.4 ml of 3.8% sodium citrate was taken in a sterile test tube. Then
1.6 ml of venous blood was added. Test tube was shaken gently to mix the blood
with anti-coagulant properly. Then blood was drawn-from test tube into the
westergren ESR tube upto the mark �O�. Westergren tube was placed vertically by
ESR stand and was allowed to settle down of R.B.C. Then reading was taken at
the end of 1st hour.

152
APPENDIX V
Figure 2. Buffy-coat
Figure 3. Buffy-coat

153
APPENDIX VI
Figure 4. Buffy-coat smear
Unstained buffy-coat smear Leishman stained buffy-coat smear

154
APPENDIX VII
Figure 5. Leishmania donovani in buffy coat smear at 100X (oil immersion) lens
under microscope
Figure 6. Leishmania donovani in buffy coat smear at 100X (oil immersion) lens
under microscope

155
APPENDIX VIII
Figure 7. Leishmania donovani in buffy coat smear at 100X (oil immersion) lens
under microscope
Figure 8. Leishmania donovani in buffy coat smear at 100X (oil immersion) lens
under microscope
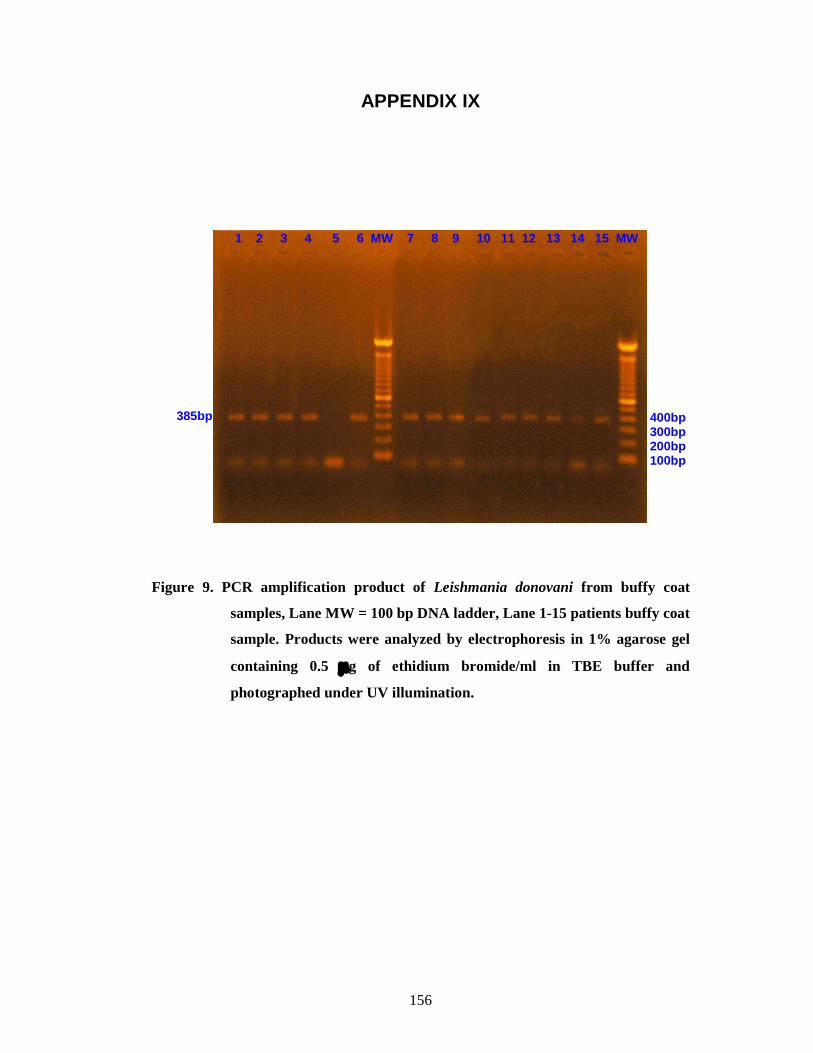
156
APPENDIX IX Figure 9. PCR amplification product of Leishmania donovani from buffy coat
samples, Lane MW = 100 bp DNA ladder, Lane 1-15 patients buffy coat
sample. Products were analyzed by electrophoresis in 1% agarose gel
containing 0.5 g of ethidium bromide/ml in TBE buffer and
photographed under UV illumination.
1 2 3 4 5 6 MW 7 8 9 10 11 12 13 14 15 MW
400bp 300bp 200bp 100bp
385bp

157
Figure 10. PCR amplification product of Leishmania donovani from buffy coat
samples, Lane MW = 100 bp DNA ladder, Lane 1-8 patients buffy coat
sample; Lane NC = Negative control. Products were analyzed by
electrophoresis in 1% agarose gel containing 0.5 g of ethidium
bromide/ml in TBE buffer and photographed under UV illumination.
MW 1 2 3 4 5 6 7 8 NC NC MW
400bp 300bp
200bp
100bp
385bp

158
APPENDIX X
Figure 11. rK39 immunochromatographic dipstick test for kala-azar
The test is considered positive with the appearance of two red lines (one in the control area and another in the test area)
The test is considered negative with the appearance of a single red line in the control area

159
APPENDIX XI
Figure 12. Bone marrow smear showing amastigote form of Leishmania
donovani

160
APPENDIX VI
STATISTICAL FORMULA Formula for mean
-
x = n
x
-
x = mean of observations
x = individual observations
n = number of observation
Formula for Standard Deviation
SD = 1
)x -(x 2-
n
SD = Standard deviation
(n-1) = applicable for sample -
x = mean of observations
x = individual observations
n = number of observation
Formula for Chi-square test (2):
2=
E
E) - (O 2
Here,
O = Observed frequencies
E = Expected frequencies
df = Degrees of freedom = (c-1) (r-1)
Operational definition:
Persons living in a family having a per capita income per month of less than 1500
TK. were considered as low economic status.