Embed Size (px)
Citation preview

Small Molecule Therapeutics
Targeting IkB Kinase b/NF-kB Signaling inHuman Prostate Cancer by a Novel IkB Kinaseb Inhibitor CmpdAYanting Zhang1, Rena G. Lapidus1, Peiyan Liu1, Eun Yong Choi1, Samusi Adediran1,Arif Hussain1,2, Xinghuan Wang3, Xuefeng Liu4, and Han C. Dan1,5
Abstract
NF-kB plays an important role in many types of cancer, includ-ing prostate cancer, but the role of the upstream kinase of NF-kB,IKKb, in prostate cancer has neither been fully documented norare there any effective IKKb inhibitors used in clinical settings.Here, we have shown that IKKb activity is mediated by multiplekinases including IKKa in human prostate cancer cell lines thatexpress activated IKKb. IHC analysis (IHC) of human prostatecancer tissue microarrays (TMA) demonstrates that phosphory-lation of IKKa/b within its activation loop gradually increasesin low to higher stage tumors as compared with normal tissue.The expression of cell proliferation and survival markers (Ki-67,Survivin) and epithelial-to-mesenchymal transition (EMT) mar-kers (Slug, Snail), as well as cancer stem cell (CSC)-relatedtranscription factors (Nanog, Sox2, Oct-4), also increase in par-
allel among the respective TMA samples analyzed. IKKb, but notNF-kB, is found to regulate Nanog, which, in turn, modulates thelevels ofOct4, Sox2, Snail, and Slug, indicating an essential role ofIKKb in regulating CSCs and EMT. The novel IKKb inhibitorCmpdA inhibits constitutively activated IKKb/NF-kB signaling,leading to induction of apoptosis and inhibition of proliferation,migration, and stemness in these cells. CmpdA also significantlyinhibits tumor growth in xenografts without causing apparentin vivo toxicity. Furthermore, CmpdA and docetaxel act synergis-tically to inhibit proliferation of prostate cancer cells. These resultsindicate that IKKb plays a pivotal role in prostate cancer, andtargeting IKKb, including in combination with docetaxel, may bea potentially useful strategy for treating advanced prostate cancer.Mol Cancer Ther; 1–11. �2016 AACR.
IntroductionProstate cancer ranks as the second most common cause of
cancer-related deaths among men in the western world (1). Thestandard treatment for metastatic prostate cancer is androgen-deprivation therapy (ADT), but while initially effective in induc-ing a clinical response, invariably fails within 2 years in mostpatients, resulting in castration-resistant prostate cancer (CRPC).Several treatment options are currently available for CRPCpatients, including second-line hormonal therapy, a dendriticcell-based vaccine (sipuleucel-T), bone-targeted agents (radi-um-223), and cytotoxins such as docetaxel and cabazitaxel. How-ever, these treatments ultimately provide limited benefit due tothe inability to provide a long-lasting response (2–5). Thus, to
improve treatment outcomes, identification of relevant signalingpathways associated with the development of CRPC and resis-tance to cytotoxic agents remains a major challenge.
The PI3K signaling pathway plays a critical role in manyphysiologic cellular processes as well as in cancer. Activated PI3Kphosphorylates PtdIns(4,5)P2 to produce phosphoinositidePtdIns(3,4,5)P3 that, in turn, recruits PDK1 and Akt to the plasmamembrane where Akt is phosphorylated and activated by PDK1.The tumor suppressor gene PTEN is a phosphatase that depho-sphorylates PtdIns(3,4,5)P3, thus decreasing Akt activity (6).Inactivation of PTEN mutations is common in cancer, resultingin constitutive activation of Akt. One of the major downstreamsubstrates of Akt is TSC2, which is phosphorylated to activatethe mTOR, another important tumorigenesis-promoting kinase(7–10). PTEN mutations have been identified in over 50% oftumor samples from patients with CRPC. The cumulative evi-dence indicates that loss of PTEN function and subsequentactivation of Akt and mTOR are critical events in the progressionof human prostate cancer (11, 12).
The NF-kB family is composed of five members: RelA (p65),RelB, c-Rel, p50/p105 (NF-kB1), and p52/p100 (NF-kB2). NF-kBsignaling is activated by a variety of inflammatory mediators andgrowth factors through canonical and noncanonical pathways(13, 14). In most signaling cascades, NF-kB is activated by thecanonical pathway that utilizes the IKK complex that containsIKKa, IKKb, and IKKg . IKKa and IKKbdrive the catalytic activity ofIKK, with IKKb playing a dominant role in inflammatory-medi-ated pathways. Normally, p50 and p65 NF-kB heterodimers aresequestered in the cytoplasm in an inactive state by IkBa. Upon
1Marlene and Stewart GreenebaumCancer Center, University of Mary-land School of Medicine, Baltimore, Maryland. 2Baltimore VA MedicalCenter, Baltimore, Maryland. 3Department of Urology, Zhongnan Hos-pital of Wuhan University,Wuhan, Hubei, P.R. China. 4Department ofPathology, Georgetown University Medical Center, Washington, DC.5Department of Pathology,UniversityofMarylandSchool ofMedicine,Baltimore, Maryland.
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
Corresponding Author: Han C. Dan, Marlene and Stewart Greenebaum CancerCenter, University of Maryland School of Medicine, Baltimore, MD 21201. Phone:410-328-0372; Fax: 341-576-873; E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-15-0999
�2016 American Association for Cancer Research.
MolecularCancerTherapeutics
www.aacrjournals.org OF1
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999

stimulation, the IkB kinase (IKK) complex is activated, leading toIkBa phosphorylation, which results in ubiquitination and pro-teasomal degradation of IkBa and subsequent p50 and p65heterodimer translocation to the nucleus. Dysregulation of NF-kB activity is associated with numerous cancers (13, 15), and caninduce gene expression to regulate different cellular processes,including cell proliferation, survival, invasion, metastasis, andresistance to chemotherapy (13–16).
A number of studies have shown that NF-kB signaling plays apivotal role in prostate cancer (17–19). Clinically, prostate cancerpatients with elevated NF-kB have a worse prognosis. The extentof nuclear NF-kB p65 staining has been shown to correlate withtumor grade in prostate cancer (20). In addition, it has beenreported thatNF-kB signaling is upregulated inCRPCpatients andcorrelates with disease progression (21–24). We have previouslyshown that both mTORC1 and NF-kB activity are upregulateddownstream of Akt in PTEN null prostate cancer cells, andmTORC1 interacts with the IKK complex to promote inductionof NF-kB (25, 26). In the present study, we hypothesize that IKKb,the upstream regulator of NF-kB, is an essential driver in prostatecancer.We carried out both in vitro and in vivo studies to determinethe role of IKKb/NF-kB in regulating prostate cancer tumorigen-esis. In addition, we evaluated the targeting of IKKb by the novelIKKb inhibitor CmpdA in prostate cancer cells expressing activat-ed IKKb. Our results suggest that IKKbmay be an important targetin prostate cancer, particularly in the context of functional PTENdeficiency.
Materials and MethodsChemical reagents
The Akt inhibitor Perifosine (KRX-0401) was purchased fromSelleck Chemical. Docetaxel was purchased from Cell SignalingTechnology. The IKKb inhibitor, CmpdA, was kindly provided byDr. Albert Baldwin, University of North Carolina (Chapel Hill,NC). Antibodies against IKKa, IKKb, and Akt were obtained fromUpstate Biotechnology. Antibodies for IHC, including Ki-67,cleaved caspase-3, p-IKKa/b-S177/S181, Slug, and Snail werefrom Abcam. Horseradish peroxidase-labeled anti-mouse andanti-rabbit secondary antibodies were from Santa Cruz Biotech-nology. Antibodies for Oct4 (CST-2750), Sox2 (CST-3728), p65(CST-8242), p-p65 (CST-3033), Survivin (CST-2808), p-Akt-S473 (CST-4058), GAPDH (CST-5174), as well as any additionalantibodies were from Cell Signaling Technology.
Cell lines and cell cultureProstate cancer cell lines, PC3 andDu145,were purchased from
ATCC in 2013, and no authentication was performed in thelaboratory. All cells were maintained in DMEM supplementedwith 10% FBS, 2 mmol glutamine, and 100 U/mL penicillin andstreptomycin. All the cell lines were meticulously passaged andtested for mycoplasma contamination.
Cell lysis and Western blottingCells were grown in 100-mm dishes, rinsed twice with cold
PBS, and then lysed on ice for 20 minutes in 1 mL of lysis buffer[40 mmol/L HEPES (pH 7.5), 120 mmol/L NaCl, 1 mmolEDTA, 10 mmol pyrophosphate, 10 mmol glycerophosphate,50 mmol NaF, 0.5 mmol orthovanadate, and EDTA-free pro-tease inhibitors (Roche Applied Science)] containing 1% TritonX-100 or 0.3% CHAPS. After centrifugation at 13,000 � g for 15
minutes, samples containing 20 to 50 mg protein/lane wereresolved by SDS-PAGE, transferred to Pure Nitrocellulose Mem-brane (Bio-Rad), blocked in 5% nonfat milk, and then blottedwith the indicated antibodies.
RNAisiRNA SMARTpool Raptor, IKKa, IKKb, NF-kB p65, andNanog
were from Dharmacon. Each represents four pooled SMART-selected siRNA duplexes that target the indicated genes. PC3 cellswere transfected with SMARTpool IKKb, p65, or nonspecificcontrol siRNAs using DharmaFECT 1 reagent (Dharmacon)according to the manufacturer's instructions. In brief, a finalconcentration of 20 nmol of siRNA was used to transfect cellsfor 48 to 72 hours.
Cell proliferation assayCell proliferation was measured by MTS assay using the Cell-
Titer 96 Aqueous ONE Solution kit (Promega). Briefly, cells wereseeded into 96-well plates at a density of 5 � 104 cells/mL for 24hours, then the culture media were replaced with fresh mediacontaining the indicated concentrations of CmpdA or vehiclecontrol (DMSO). After incubation for an additional 48 hours,MTS reagent (20 mL) was added to each well and incubated at37�C for 1 to 4 hours. Absorbance at 490 nmwasmeasured usinga microplate reader (Bio-Rad). Three independent experimentswere performed, each in triplicate.
Caspase-3/7 activity assayCells were plated in triplicate at 4� 103 cells/per well in white-
walled 96-well plates (Becton Dickinson). Cells were transfectedwith siRNA as described above and treated with the IKK inhibitorand/or docetaxel as indicated. Caspase-3/7 activity was measuredat 48 hours post-transfection using the Caspase-Glo 3/7 assay(Promega) according to the manufacturer's instructions. TheCaspase-Glo 3/7 assay uses a caspase-3/7 tetrapeptide DEVDsubstrate that produces a luminescent signal on cleavage. Relativelight units were measured on an Lmax Microplate Luminometer(Molecular Devices).
Colony formation assaysOne thousand cells were seeded into 6-well plates and incu-
bated for 5 or 7 days after the appropriate treatment. The colonieswere stained with crystal violet (0.5% w/v) and images takenusing a scanner (Epson).
Analysis of cell cycle and apoptosis via flow cytometryCell-cycle analysis was performed as described previously (27).
In brief, cells were fixedwith 70% cold ethanol at 20 �C for at least1.5 hours and stained with PBS containing 50 mg/mL propidiumiodide (PI) and 30 mg/ml RNase A, and the samples analyzed forDNA content and apoptotic cells (sub-G0/G1 fraction) via a flowcytometer (FACS Canto; Becton Dickinson).
Cell migration assayScratch wound-healing assay was performed to determine cell
migration using confluent cultures (80%–90% confluence). Cellswere photographed under a microscope at 0, 24, and 48 hours.
Real-time cell migration assaySeventy-five thousand cells were plated in serum-free media in
the upper chamber of a CIM plate after serum starvation for 18
Zhang et al.
Mol Cancer Ther; 2016 Molecular Cancer TherapeuticsOF2
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999

hours and CmpdA (2 or 5 mmol/L) or vehicle control were addedsimultaneously. The lower chamber was filled with either serum-free media (negative control) or full media (DMED with 10%FCS). Cells (with or without treatment) were allowed to equili-brate at room temperature for 1 hour and then incubated at 37�Cfor 48 hours in an Xcelligence DP module.
Tumor xenograft studyThe Translational Laboratory Shared Services (TLSS) at the
University of Maryland Greenebaum Cancer Center (Baltimore,MD) performed the in vivo studies using 8-week-old male nu/numice (Harlan Sprague Dawley). A total of 5 � 106 PC3 cells weremixed with 50 mL Matrigel and injected into the flanks of nudemice. Nine days after cell injection, mice were sorted into threebalancedgroups.CmpdA(10or15mg/kg)or vehicle control (10%dimethyl sulfoxide in saline) was given intraperitoneally (IP) twicea week for 3 weeks. The growth of the tumors was measured withelectronic calipers twice perweek for 3weeks. Tumor volumeswerecalculated using the equation Vol ¼ (L � W2)/2 where L is thelongerdimensionandWis the shorter dimension. The tumorswerethen harvested for subsequent analysis.
Tissue microarrayProstate cancer TMA arrays T191 and PR243B were obtained
from US BioMax, Inc.
IHC analysisAll IHC analyses were conducted by the Pathology Core Facility
at the Baltimore VA Medical Center.
Statistical analysisData are presented as mean � SD. Statistical analysis was
performed using GraphPad Prism version 4.0 (GraphPad Soft-ware Inc.). One-way ANOVA followed by Dunnett's multiplecomparisons test was used to analyze the statistical significanceof 3 or more groups. P values <0.05 were considered to bestatistically significant.
ResultsIKKb activity is regulated by Akt, mTORC1, and IKKa inprostate cancer PC3 cells
Our previous studies showed that both mTORC1 and NF-kBactivity were upregulated in AR-negative PTEN null PC3 pros-tate cancer cells (25, 26). In these cells, the IKK complexassociates with the mTORC1 complex downstream of Akt.Moreover, IKKa enhances mTORC1 activity, while mTORC1promotes IKKa and IKKb activity to upregulate NF-kB functionin a positive feedback loop (25, 26). These findings suggestedthat IKKb could be a downstream target of Akt, mTORC1, andIKKa in PC3 cells (Supplementary Fig. S1A). To test this model,we first employed a pan Akt inhibitor, perifosine, that simul-taneously blocks the activity of all three isoforms of Akt (28,29). PC3 cells were treated with different concentrations ofperifosine for 2 hours and phosphorylation of Akt, IKK (bothIKKa and IKKb), S6K (mTOR target), and IkBa and p65 (IKKsubstrates) was determined by Western blot analysis. As shownin Fig. S1B, perifosine dramatically reduced Akt phosphoryla-tion, as well as that of S6K, IKKa/b, IkBa, and p65, confirmingthat both mTORC1 and IKK/NF-kB signaling pathways aredownstream targets of Akt. We previously demonstrated that
depletion of mTORC1 (by mTOR or Raptor knockdown)decreased the activity of both IKKa and IKKb (26), suggestingthat mTORC1 regulates IKKa and IKKb downstream of Akt(Supplementary Fig. S1A). Decreased phosphorylation of S6K,p65, and IkBa occurred after knockdown of IKKa (Supplemen-tary Fig. S1D). Importantly, phosphorylation of IKKb was alsodramatically reduced in a dose-dependent manner upon IKKaknockdown (Supplementary Fig. S1C). Taken together, theseresults suggest that IKKa can affect IKKb through mTORC1-dependent and independent mechanisms (Supplementary Fig.S1A and S1C). In addition, knockdown of IKKb impairs thephosphorylation of both IKKa and p65, yet has no significanteffects on S6K phosphorylation. Thus, IKKb appears to activateNF-kB downstream of IKKa and mTORC1, and is, itself, acti-vated by Akt, mTORC1, and IKKa in PC3 cells (SupplementaryFig. S1A and S1D).
Depletion of IKKb/NF-kB signaling inhibits cell proliferationand migration and leads to apoptosis in PC3 cells
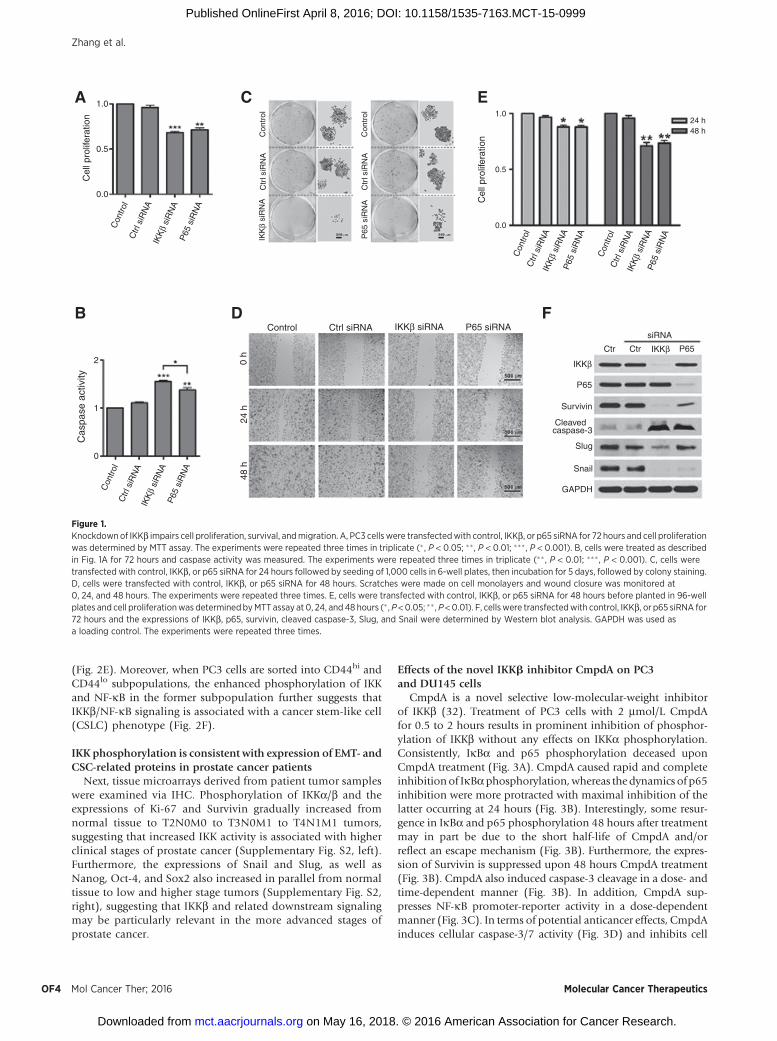
To determine whether the IKKb/NF-kB pathway plays a rolein tumorigenesis, we evaluated the effects of IKKb and NF-kBknockdown on cell proliferation, survival, and migration. Theresults showed that knockdown of either IKKb or p65 leads toinhibition of cell proliferation, with a greater reduction in cellproliferation observed after IKKb knockdown, as comparedwith p65 (Fig. 1A). Reduction in IKKb or p65 expression causeda dramatic increase in apoptosis as well as a significant decreasein PC3 cell colony formation, with greater effects on theseparameters noted with IKKb knockdown (Fig 1B and 1C). PC3cell migration was also strongly suppressed after either IKKb orp65 knockdown (Fig. 1D) in line with the decrease of cellproliferation (Fig. 1E). Consistent with the role of IKKb/p65signaling in cell survival, Western blot analysis revealed thatknockdown of either IKKb or p65 resulted in decreased expres-sion of Survivin and enhanced cleavage of caspase-3 (Fig. 1E).Interestingly, epithelial-to-mesenchymal transition (EMT) mar-kers were differentially affected by IKKb and p65. IKKb knock-down dramatically reduced the expression of both Slug andSnail, whereas knockdown of p65 decreased only Snail expres-sion (Fig. 1E). Taken together, the above studies demonstratethat IKKb and p65 are important in regulating key biologicproperties of PC3 cells, with IKKb, in particular, playing a moreprominent role in these processes.
Knockdown of IKKb/NF-kB inhibits stemness in PC3 cellsTo determine whether the IKKb/NF-kB pathway plays a role in
modulating stemness in prostate cancer cells, the expressionpatterns of the transcription factors Nanog, Oct-4, and Sox2 wereexamined after knockdown of IKKb or p65NF-kB. Knockdown ofIKKb decreased Nanog, Oct-4, and Sox2 levels, whereas knock-down of NF-kB caused only minimal reduction of Sox2 and hadno effects on Oct4 and Nanog expression (Fig. 2A). Previousreports showed that Nanog is upstream of Oct-4 and Sox2(30, 31), as well as Snail and Slug. Knockdown of Nanogdecreased the levels of Oct-4, Sox2, Snail, and Slug in PC3 cellsbut hadnoeffect on IKKb expression (Fig. 2B). Similar resultswereobserved by immunofluorescence (Fig. 2C). Thus, IKKb, in par-ticular, appears to be relevant in the regulation of several mod-ulators of EMT, primarily in a NF-kB–independent manner(Fig. 2D). Furthermore, knockdown of IKKb or NF-kB can sig-nificantly impair the ability of PC3 cells to form tumor spheres
IkB Kinase b in Human Prostate Cancer
www.aacrjournals.org Mol Cancer Ther; 2016 OF3
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999
(Fig. 2E). Moreover, when PC3 cells are sorted into CD44hi andCD44lo subpopulations, the enhanced phosphorylation of IKKand NF-kB in the former subpopulation further suggests thatIKKb/NF-kB signaling is associated with a cancer stem-like cell(CSLC) phenotype (Fig. 2F).
IKK phosphorylation is consistent with expression of EMT- andCSC-related proteins in prostate cancer patients
Next, tissue microarrays derived from patient tumor sampleswere examined via IHC. Phosphorylation of IKKa/b and theexpressions of Ki-67 and Survivin gradually increased fromnormal tissue to T2N0M0 to T3N0M1 to T4N1M1 tumors,suggesting that increased IKK activity is associated with higherclinical stages of prostate cancer (Supplementary Fig. S2, left).Furthermore, the expressions of Snail and Slug, as well asNanog, Oct-4, and Sox2 also increased in parallel from normaltissue to low and higher stage tumors (Supplementary Fig. S2,right), suggesting that IKKb and related downstream signalingmay be particularly relevant in the more advanced stages ofprostate cancer.
Effects of the novel IKKb inhibitor CmpdA on PC3and DU145 cells
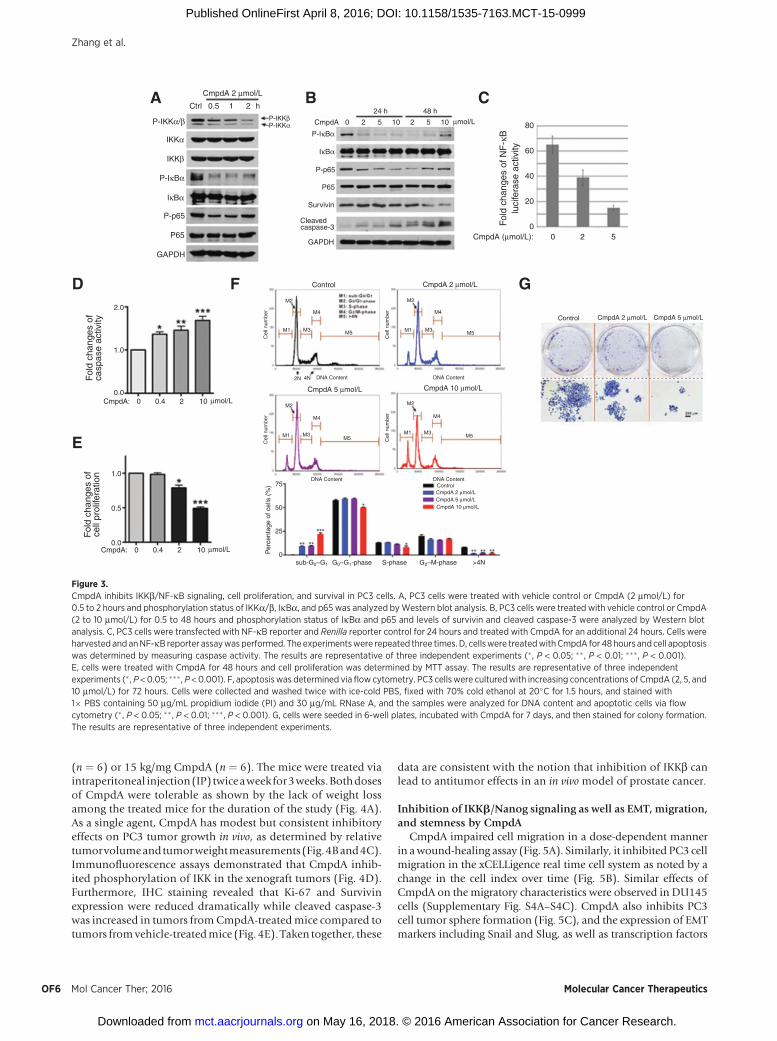
CmpdA is a novel selective low-molecular-weight inhibitorof IKKb (32). Treatment of PC3 cells with 2 mmol/L CmpdAfor 0.5 to 2 hours results in prominent inhibition of phosphor-ylation of IKKb without any effects on IKKa phosphorylation.Consistently, IkBa and p65 phosphorylation deceased uponCmpdA treatment (Fig. 3A). CmpdA caused rapid and completeinhibition of IkBaphosphorylation,whereas thedynamics of p65inhibition were more protracted with maximal inhibition of thelatter occurring at 24 hours (Fig. 3B). Interestingly, some resur-gence in IkBa and p65 phosphorylation 48 hours after treatmentmay in part be due to the short half-life of CmpdA and/orreflect an escape mechanism (Fig. 3B). Furthermore, the expres-sion of Survivin is suppressed upon 48 hours CmpdA treatment(Fig. 3B). CmpdA also induced caspase-3 cleavage in a dose- andtime-dependent manner (Fig. 3B). In addition, CmpdA sup-presses NF-kB promoter-reporter activity in a dose-dependentmanner (Fig. 3C). In terms of potential anticancer effects, CmpdAinduces cellular caspase-3/7 activity (Fig. 3D) and inhibits cell
1.0
0.5
0.0
Con
trol
Con
trol
Con
trol
Ctrl
siR
NA
Ctrl
siR
NA
Ctrl
siR
NAP65
siR
NA
P65
siR
NA
P65
siR
NA
IKK
β si
RN
A
Con
trol
Ctrl
siR
NA
P65
siR
NA
IKK
β si
RN
A
IKK
β si
RN
A
IKK
β si
RN
A
Cel
l pro
lifer
atio
n
Con
trol
Con
trol
Ctr
l siR
NA
Ctr
l siR
NA
Cel
l pro
lifer
atio
n
IKK
β si
RN
A
P65
siR
NA
1.0
0.5
0.0
24 h48 h
2
1
0
Cas
pase
act
ivity
Control Ctrl siRNA P65 siRNAIKKβ siRNA
48 h
24 h
0 h
siRNA
Ctr IKKβ
IKKβ
Ctr P65
P65
Survivin
Cleavedcaspase-3
Slug
Snail
GAPDH
A
B D F
C E
Figure 1.Knockdownof IKKb impairs cell proliferation, survival, andmigration. A, PC3 cells were transfectedwith control, IKKb, or p65 siRNA for 72 hours and cell proliferationwas determined by MTT assay. The experiments were repeated three times in triplicate (� , P < 0.05; ��, P < 0.01; ��� , P < 0.001). B, cells were treated as describedin Fig. 1A for 72 hours and caspase activity was measured. The experiments were repeated three times in triplicate (�� , P < 0.01; ��� , P < 0.001). C, cells weretransfected with control, IKKb, or p65 siRNA for 24 hours followed by seeding of 1,000 cells in 6-well plates, then incubation for 5 days, followed by colony staining.D, cells were transfected with control, IKKb, or p65 siRNA for 48 hours. Scratches were made on cell monolayers and wound closure was monitored at0, 24, and 48 hours. The experiments were repeated three times. E, cells were transfected with control, IKKb, or p65 siRNA for 48 hours before planted in 96-wellplates and cell proliferationwas determinedbyMTT assay at 0, 24, and 48 hours (� ,P <0.05; �� , P<0.01). F, cellswere transfectedwith control, IKKb, or p65 siRNA for72 hours and the expressions of IKKb, p65, survivin, cleaved caspase-3, Slug, and Snail were determined by Western blot analysis. GAPDH was used asa loading control. The experiments were repeated three times.
Zhang et al.
Mol Cancer Ther; 2016 Molecular Cancer TherapeuticsOF4
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999

proliferation (Fig. 3E). A dose-dependent increase in the sub-G0/G1 peak on flow cytometry corroborates the proapoptotic effectsof CmpdA on PC3 cells (Fig. 3F). In addition, at higher concen-trations, CmpdA significantly prevents PC3 cells fromentering theS phase, and also gradually decreases the percentage of cells withDNA content > 4N (Fig. 3F), suggesting that it inhibits G1/Stransition and mitosis in PC3 cells. Consistent with its antipro-liferative and proapoptotic properties, CpmdA inhibits colonyformation of PC3 cells (Fig. 3G).
We extended the above studies to another AR-negative prostatecancer cell line,DU145, that alsopossesses highbasal levels ofNF-kB activity but has wild-type PTEN (33). DU145 cells have highlevels of phosphorylated p65 and IkBa, suggesting that IKK isconstitutively active in these cells (Supplementary Fig. S3A). Weevaluated the antitumor activity of CmpdA in DU145 cells.
Phosphorylation of both NF-kB and IkBa was inhibited byCmpdA in a dose-and time- dependent manner (SupplementaryFig. S3A and S3B). Similar to PC3 cells, CmpdA dramaticallyinhibited DU145 cell proliferation, increased cellular caspase-3/7activity, decreased colony formation, induced cell apoptosis,suppressed expression of Survivin, and increased expression ofcleaved caspase-3 (Supplementary Fig. S3C–S3H).
CmpdA suppresses tumorigenesis in vivoArecentstudydemonstratedthatCmpdA(10mg/kg) inhibited
K-Ras induced lung tumors in amousemodel (34).We evaluatedthe tolerability and antitumor activity of CmpdA in vivo usingmouse xenografts. When tumors reached approximately200 mm3 after PC3 cell inoculations, mice were randomized toone of three treatment groups: vehicle control (n¼ 7), 10 kg/mg
SiRNASiRNA
Ctr CtrCtr Ctr
P65
Control
12 d
ay12
day
6 da
y6
day
Ctrl siRNA P65 siRNA
Nanog siRNA
IKKβ siRNA
IKKβ
IKKβIKKβ
IKKβ
Nanog
Nanog
Nanog Merge Merge/DNA PH
Oct4
Sox2
Slug
Snail
GAPDH
Con
trol
Con
trol
Nan
ogsi
RN
Asi
RN
Asi
RN
AIK
Kβ
P65
Nanog
Oct4
Sox2
GAPDH
600
400
200
0
12 Day
Con
trol
Ctr
l siR
NA
P65
siR
NA
Nan
og s
iRN
A
IKK
β si
RN
A
Sph
ere
num
ber
(>50
μm
ol/L
)
FSC-A
FS
C-W
CD44
Cou
nt
Lo Hi
CD44
P-IKKα/β
IKKα
P-p65
P65
IKKβ
GAPDH
P-IKKβP-IKKα
CD44 Lo
CD44 Hi
A
E D
F
B C
Figure 2.Knockdown of IKKb inhibits stemness via Nanog, Oct-4, and Sox2 in PC3 cells. A, cells were transfected with control, IKKb, or p65 siRNA for 72 hours and theexpressions of IKKb, p65, Nanog, Oct4, and Sox2 were determined by Western blot analysis. GAPDH was used for a loading control. B, cells were transfectedwith control or Nanog siRNA for 72 hours and the expressions of IKKb, Nanog, Oct4, Sox2, Snail, and Slug were determined by Western blot analysis.GAPDHwas used for a loading control. C, cells were transfectedwith control, IKKb, or Nanog siRNA for 72 hours and the expressions of IKKb and Nanog determinedby immunofluorescence (IF). D, proposed model of IKKb regulation of CSC and EMT through NF-kB–dependent and -independent mechanisms. E, cells weretransfected with control, IKKb, p65, or Nanog siRNA, and tumor sphere formation was monitored and counted (�� , P < 0.01; ��� , P < 0.001). F, cells were sortedvia flow cytometry using CD44 antibody, cultured and then analyzed by Western blot analysis with the indicated antibodies.
IkB Kinase b in Human Prostate Cancer
www.aacrjournals.org Mol Cancer Ther; 2016 OF5
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999
(n ¼ 6) or 15 kg/mg CmpdA (n ¼ 6). The mice were treated viaintraperitoneal injection(IP) twiceaweek for3weeks.Bothdosesof CmpdA were tolerable as shown by the lack of weight lossamong the treated mice for the duration of the study (Fig. 4A).As a single agent, CmpdA has modest but consistent inhibitoryeffects on PC3 tumor growth in vivo, as determined by relativetumorvolumeandtumorweightmeasurements (Fig.4Band4C).Immunofluorescence assays demonstrated that CmpdA inhib-ited phosphorylation of IKK in the xenograft tumors (Fig. 4D).Furthermore, IHC staining revealed that Ki-67 and Survivinexpression were reduced dramatically while cleaved caspase-3was increased in tumors fromCmpdA-treatedmice compared totumors fromvehicle-treatedmice (Fig. 4E). Taken together, these
data are consistent with the notion that inhibition of IKKb canlead to antitumor effects in an in vivo model of prostate cancer.
Inhibition of IKKb/Nanog signaling as well as EMT, migration,and stemness by CmpdA
CmpdA impaired cell migration in a dose-dependent mannerin awound-healing assay (Fig. 5A). Similarly, it inhibited PC3 cellmigration in the xCELLigence real time cell system as noted by achange in the cell index over time (Fig. 5B). Similar effects ofCmpdA on the migratory characteristics were observed in DU145cells (Supplementary Fig. S4A–S4C). CmpdA also inhibits PC3cell tumor sphere formation (Fig. 5C), and the expression of EMTmarkers including Snail and Slug, as well as transcription factors
CmpdA 2 μmol/L
CmpdA
Ctrl 0.5 1 2 h
P-IKKβP-IKKα/β
P-IκBα
P-IκBα
IκBα
IκBα
P-p65
P-p65
P65
P65
GAPDH
IKKβ
IKKα
P-IKKα
Survivin
Cleavedcaspase-3
GAPDH
80
60
40
20
00 2 5CmpdA (μmol/L):
24 h 48 h
μmol/L0 2 5 10 2 5 10
Fol
d ch
ange
s of
NF
-κB
luci
fera
se a
ctiv
ity
A B C
CmpdA:
CmpdA:
μmol/L
2.0
1.0
0.00 0.4 2 10
μmol/L0 0.4 2 10
Fol
d ch
ange
s of
casp
ase
activ
ityF
old
chan
ges
ofce
ll pr
olife
ratio
n 1.0
0.5
0.0
Control
Control
CmpdA 2 μmol/L
CmpdA 2 μmol/L
CmpdA 5 μmol/L
CmpdA 5 μmol/L
CmpdA 10 μmol/L
CmpdA 2 μmol/LCmpdA 5 μmol/LCmpdA 10 μmol/L
75
50
25
0Per
cent
age
of c
ells
(%
)C
ell n
umbe
rC
ell n
umbe
r
Cel
l num
ber
Cel
l num
ber
sub-G0−G1 G0−G1-phase G2−M-phaseS-phase >4N
ControlDNA Content DNA Content
DNA ContentDNA Content2N 4N
M1
M1 M1
M1
M2
M2 M2
M2
M3
M3 M3
M4
M5
M3
M4
M4
M4
M5
M5
M5
D F G
E
Figure 3.CmpdA inhibits IKKb/NF-kB signaling, cell proliferation, and survival in PC3 cells. A, PC3 cells were treated with vehicle control or CmpdA (2 mmol/L) for0.5 to 2 hours and phosphorylation status of IKKa/b, IkBa, and p65 was analyzed byWestern blot analysis. B, PC3 cells were treated with vehicle control or CmpdA(2 to 10 mmol/L) for 0.5 to 48 hours and phosphorylation status of IkBa and p65 and levels of survivin and cleaved caspase-3 were analyzed by Western blotanalysis. C, PC3 cells were transfected with NF-kB reporter and Renilla reporter control for 24 hours and treated with CmpdA for an additional 24 hours. Cells wereharvested and anNF-kB reporter assaywas performed. The experimentswere repeated three times. D, cellswere treatedwith CmpdA for 48hours and cell apoptosiswas determined by measuring caspase activity. The results are representative of three independent experiments (� , P < 0.05; �� , P < 0.01; ��� , P < 0.001).E, cells were treated with CmpdA for 48 hours and cell proliferation was determined by MTT assay. The results are representative of three independentexperiments (� , P <0.05; ���, P <0.001). F, apoptosiswas determined via flow cytometry. PC3 cells were culturedwith increasing concentrations of CmpdA (2, 5, and10 mmol/L) for 72 hours. Cells were collected and washed twice with ice-cold PBS, fixed with 70% cold ethanol at 20�C for 1.5 hours, and stained with1� PBS containing 50 mg/mL propidium iodide (PI) and 30 mg/mL RNase A, and the samples were analyzed for DNA content and apoptotic cells via flowcytometry (�, P < 0.05; �� , P < 0.01; ��� , P < 0.001). G, cells were seeded in 6-well plates, incubated with CmpdA for 7 days, and then stained for colony formation.The results are representative of three independent experiments.
Zhang et al.
Mol Cancer Ther; 2016 Molecular Cancer TherapeuticsOF6
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999

Nanog, Oct-4, and Sox2 (Fig. 5D). The expressions of other cancerstem cell (CSC)-related genes were also detected. CmpdAdecreased expression of Gli-1 while having no effect on b-cateninor Notch. Meanwhile, the expression of Snail, Slug, Nanog, Sox2,and CD44 were observed in control groups of xenograft tumorsespecially on the margin of the tumors and were significantlydecreased in CmpdA treated tumors (Fig. 5E).
CmpdA synergizes with docetaxel to induce apoptosis in PC3and DU145 cells
The above series of studies demonstrate that CmpdA as singleagent has significant antitumor activity in prostate cancer cells thathave constitutive IKKb activation. Docetaxel is an FDA-approvedchemotherapy drug used to treat recurrent or metastatic castra-tion-resistant prostate cancer; however, its efficacy is very limitedin patients due to acquired resistance (4, 5). It is important toimprove the efficacy of docetaxel by combining it with othersingle- or multiple-pathway inhibitors. We wanted to determinewhether enhanced antitumor inhibition could occur by dualtargeting of IKKb and the microtubular network through a com-
bination of CmpdA and Docetaxel. Using the combination index(CI)method of Chou and Talalay (35, 36), we tested the effects ofCmpdA and docetaxel combinations on PC3 and DU145 cells.The CI values were determined using the CalcuSyn v2.0 softwarethat provides quantitative definition for additive (CI ¼ 1), syn-ergistic (CI < 1), and antagonistic (CI > 1) effects of drug combi-nations (35, 36). The CI values were less than 1 with the CmpdA/docetaxel combinationwith respect to both PC3 andDU145 cells(Fig. 6A and E), indicating synergistic antiproliferative effectswhen the two agents were combined. CmpdA in combinationwith docetaxel also induced caspase activation and caspase-3cleavage in a dose-dependent manner in both PC3 (Fig. 6B–D)and DU145 cells (Fig. 6F–H). Thus, the combination of CmpdAand docetaxel has strong synergistic antiproliferative activity andinduces apoptosis in prostate cancer cells.
DiscussionActivation of Akt can lead to recruitment of downstream path-
ways that are important in modulating proliferative, anabolic,
33
28
23
18
0 5 10 15 20 25Days after treatment Days after treatment
Tum
or v
olum
e (m
m3 )
Tum
or w
eigh
t (g)
Bod
y w
eigh
t (g)
1,000
800
600
400
200
0
ControlCmpd A 10 mg/kg
Cmpd A 10 mg/kgCmpd A 15 mg/kg
Cmpd A 15 mg/kg
Cm
pd A
15 m
g/kg
Mer
ge
Cle
aved
cas
pase
-3
DN
A
Sur
vivi
nK
i67
P-I
KK
α/β
Cm
pd A
10 m
g/kg
Con
trol
0.8
0.6
0.4
0.2
0.0
ControlCmpd A 10 mg/kgCmpd A 15 mg/kg
Control ControlCmpdA CmpdA treatmentTreatment
ControlA
C D E
B
1 6 9 13 16 20 23
Figure 4.CmpdA suppresses xenograft tumor formation and induces apoptosis in tumors. A, mice inoculated with PC3 cells were treated with different doses of CmpdA.Mouse body weights were monitored twice per week for three weeks (left) and final body weights were determined at the time of sacrifice (right). B,tumor growth curves based on tumor volumesmeasured twiceweekly (� ,P <0.05; �� , P <0.01; ��� ,P <0.001). C, tumorswere photographed andweights recorded atthe time of sacrifice (� , P < 0.05; �� , P < 0.01). D, xenograft tumors harvested from vehicle- or CmpdA-treated mice were stained with p-IKKa/b. E, xenografttumors harvested from vehicle- or CmpdA-treated mice were stained with Ki-67, survivin, or cleaved caspase-3 antibodies, and representative images are shown.
IkB Kinase b in Human Prostate Cancer
www.aacrjournals.org Mol Cancer Ther; 2016 OF7
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999

and antiapoptotic programs in cancer cells (6). Considerableefforts are being made to target Akt for the treatment of cancer.However, to date, strategies to target Akt as an anticancer therapyhave met limited success due to toxicity and recruitment ofcompensatory mechanisms (29, 37, 38). One of the criticaldownstream targets of Akt is mTORC1, which plays an importantrole in controlling Akt-induced oncogenic events (6). However,mTOR inhibitors, such as rapamycin, and their derivatives showmodest benefits in prostate cancer (39, 40). Hence, identificationof other targetable oncogenic pathways that are turned on by lossof PTEN and activation of Akt and mTOR in prostate cancer isimportant to for their potential therapeutic effects in this disease.
NF-kB and its upstream kinase IKKb are upregulated inprostate cancer, especially in a metastatic setting (17, 21, 23,24). We previously reported that IKK/NF-kB signaling is acti-vated by Akt and mTOR in PC3 prostate cancer cells, whichsuggests that IKK/NF-kB is an important downstream target of
the Akt/mTORC1 signaling cascade (25, 26). In the presentstudy, we studied the more detailed regulatory mechanismswhereby IKK/NF-kB is activated downstream of Akt in prostatecancer cells. We now demonstrate that IKKb, the direct kinase ofNF-kB, is activated not only by Akt and mTORC1, but also byIKKa. It should be noted that IKKa regulation of IKKb mayinvolve mTORC1-dependent and/or independent mechanisms;the latter may be particularly relevant in prostate cancer cellssuch as DU145 that have wild-type PTEN and hence, unlikePC3 cells, do not have constitutive activation of Akt/mTOR(Fig. 1A). Regardless of the upstream pathways that may lead toIKKb activation, our studies demonstrate that knockdown ofIKKb inhibits proliferation and migration, and induces apo-ptosis in prostate cancer cells. Mechanistically, knockdown ofIKKb or NF-kB decreases the expression of Survivin, an impor-tant antiapoptotic protein and NF-kB target gene, and inducescaspase-3 cleavage.
ControlCmpdA2 μmol/L
CmpdA5 μmol/L
ControlCmpdA5 μmol/L
CmpdA 12 day
CmpdA
CmpdA10 μmol/LCmpdA
10 μmol/L
48 h
12 d
ay6
day
Cel
l ind
ex0
h
Time (in hours)
Control
Slu
g
Slu
g
Sna
il
Sna
il
CD
44
CD
44
Nan
og
Nan
og
Sox
2
Sox
2
CmpdA treatment
600
400
200
00 5 10 μmol/L
0 2 5 10 2 5 10 μmol/L
Sph
ere
num
ber
(>50
μm
ol/L
)
24 h 48 h
β-Catenin
Nortch
Gli-1
Nanog
Oct4
Sox2
Slug
Snail
GAPDH
DMSO
SFM
CmpdA 2 μmol/LCmpdA 5 μmol/L
A
B
C
E
D
Figure 5.Suppression of cell migration and stemness by CmpdA. A, cells were treated with vehicle control or CmpdA, and scratches were made on cell monolayers. Woundclosure was monitored at 0 and 48 hours. The experiments were repeated three times. B, cells were treated with vehicle control or CmpdA and migration wasmonitored using the xCELLigence System real-time cell analyzer instrument. C, cells were treated with CmpdA and tumor sphere formation was monitored;representative images are shown (� , P < 0.05; �� , P < 0.01). D, cells were treated with different doses of CmpdA (0–48 hours), lysed, and analyzed by Western blotanalysis. The results are representative of three independent experiments. E, xenograft tumors harvested from vehicle- or CmpdA-treated mice werestained with Slug, Snail, Oct-4, Nanog, and Sox2 antibodies, and representative images are shown.
Zhang et al.
Mol Cancer Ther; 2016 Molecular Cancer TherapeuticsOF8
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999

Increasing evidence implicates cancer stem cells (CSC) incancer initiation and growth, and resistance to anticancer therapy.Wnt, b-catenin, and Notch, as well as certain embryonic stem cellself-renewal genes such as NANOG, Oct-4, and Sox2, amongothers, have been shown to play important roles in regulationof some of the biologic properties of CSC. Elevated Nanog isassociated with poorer outcomes in several cancers (11, 30, 31,41–42). Depletion of Nanog results in decreased expressions ofSox2, Oct-4, Snail, and Slug, and impairs the CSC and EMTphenotypes (38). For the first time, our data show that IKKbregulates Nanog, as well as Sox2, Oct-4, Snail, and Slug, most
likely via NF-kB–independent mechanisms. Thus, IKKb may beimportant in regulating CSC and EMT in some forms of prostatecancer.
It should also be noted that compared with knockdown ofp65, knockdown of IKKb causes increased induction of apoptosis(Fig. 1B), decreased protein levels of Slug (Fig. 1E), Nanog, Oct-4,and Nanog (Fig. 2A), and more impairments of tumor sphereformation (Fig. 2E). These data indicate that IKKbmodulates themalignance of prostate cancer most likely through NF-kB–depen-dent and -independent mechanisms, thus demonstrating itspotential as a critical therapeutic target in prostate cancer.
1.00
0.75
0.50
0.25
0.00
Cel
l pro
lifer
atio
n
Cas
pase
act
ivity
Cas
pase
act
ivity
PC3
CA 0 μmol/L CA 0.4 μmol/L CA 2 μmol/L CA 5 μmol/L
DX 0 1 5 0 1 5 0 1 5 0 1 5 nmol/L
CA 0 μmol/L CA 0.4 μmol/L CA 2 μmol/L CA 5 μmol/L
DX 0 1 5 0 1 5 0 1 5 0 1 5 nmol/L
Control
CA 0.4 μmol/L
CA 2 μmol/L
CA 5 μmol/L
CA 2 + DX 1
CA 5 + DX 1 CA 5 + DX 5
CA 5 μmol/L CA 5 + DX 1 CA 5 + DX 5
CA 2 + DX 5
CA 2 μmol/L CA 2 + DX 1 CA 2 + DX 5
CA 0.4 + DX 1 CA 0.4 + DX 5
CA 0.4 μmol/L CA 0.4 + DX 1 CA 0.4 + DX 5
DX 1 nmol/L DX 5 nmol/L
Control DX 1 nmol/L DX 5 nmol/LDU145
1.00
0.75
0.50
0.25
0.00
Cel
l pro
lifer
atio
n
Combination: 1 2 3 4 5 6
Combination: 1 2 3 4 5 6
3
2
1
0DX 0 0 0 0 1 1 1 1
DX 0 0 0 1 1 1 nmol/LCA 0 2 5 0 2 5 μmol/L
CA 0 0.4 2 5 0 0.4 2 5
DX 0 0 0 0 1 1 1 1
DX 0 0 0 1 1 1 nmol/L
CA 0 0.4 2 5 0 0.4 2 5
CA 0 2 5 0 2 5 μmol/L
P-p65
P65
GAPDH
Cleavedcaspase-3
P-p65
P65
GAPDH
Cleavedcaspase-3
3
2
1
0
Cm
pdA
CI ±
1.9
6 s.
d.
Cm
pdA
CI ±
1.9
6 s.
d.
Docetaxcel Fractional effect
Docetaxcel Fractional effect
Normalized isobologram Mixture-Algebraic estimate
Normalized isobologram Mixture-Algebraic estimate
1.0
0.8
0.6
0.4
0.2
0
1.2
0.9
0.6
0.3
0
1.0
0.8
0.6
0.4
0.2
0
0 0.2 0.4 0.6 0.8 1.0 0 0.2 0.4 0.6 0.8 1.0
0 0.2 0.4 0.6 0.8 1.0 0 0.2 0.4 0.6 0.8 1.0
1.0
0.8
0.6
0.4
0.2
0
A
E F G
H
B C
D
Figure 6.Studies with docetaxel and CmpdA combinations. A and E, PC3 (A) and DU145 (E) cells were treated with vehicle control, CmpdA (CA), docetaxel (DX), or acombination of the two, and cell proliferation was assessed by MTT assay. Also shown are the calculated combination index (CI) values (� , P < 0.05; �� , P < 0.01;��� , P < 0.001). B and F, PC3 (B) and DU145 (F) cells were treated as in A and E for 48 hours and cell morphology was assessed. C and G, PC3 (C) andDU145 (G) cellswere treated as in A and E for 48 hours and caspase activity wasmeasured (� , P <0.05; �� ,P <0.01). D andH, PC3 (D) andDU145 (H) cellswere treatedas in A and E for 48 hours and phosphorylation status of p65 as well as cleavage of caspase-3 were determined by Western blot analysis. The experimentswere repeated three times.
IkB Kinase b in Human Prostate Cancer
www.aacrjournals.org Mol Cancer Ther; 2016 OF9
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999

Several IKKb inhibitors have been explored as potential ther-apeutics in recent years. However, to date, no IKKb inhibitor hasbeen successfully developed for clinical use,mainly due to toxicity(43–48). Thus, there remains a significant need to identify novelIKKb inhibitors that can be safely exploited clinically. The novelIKKb-specific inhibitor CmpdA is a competitive ATP inhibitor thatdownregulates IKKb activity and inhibits IkBa phosphorylation.CmpdA has been shown to suppress cell proliferation and sen-sitize several cancer cell lines to chemotherapy in vitro (49). Arecent study showed that CmpdA effectively inhibited K-rasinduced lung cancer in vitro and in vivo (34). To our knowledge,this agent has not yet been evaluated in prostate cancer. In thepresent study, we evaluated the anticancer activity of CmpdA invitro and in vivo in two prostate cancer cell lines that have activatedIKKb. CmpdA led to significant inhibitionof cell proliferation, cellmigration, and EMT transition, andwas associatedwith enhancedapoptosis in prostate cancer cells. Furthermore, CmpdA signifi-cantly inhibited the expression of transcription factors Nanog,Sox-2, and Oct-4 in both prostate cancer cells in cell culture andxenograft tumors, suggesting its potential in suppression of pros-tate cancer stemness. Importantly, CmpdA, as single agent, causedinhibition of tumor xenografts without significant loss of mousebody weight, suggesting that it may be associated with reducedtoxicity in vivo. Given that targeting a single activated pathway isunlikely to provide meaningful long-term tumor control in acomplex disease state such as prostate cancer, we also evaluatedthe effects of combining docetaxel chemotherapy with CmpdA.That significant synergism exists between docetaxel and CmpdAin inhibiting prostate cancer cells suggests that docetaxel, incombination with IKKb targeting, may be a viable strategy toexplore further in future studies.
In summary, we demonstrate that activated IKKbmay representa useful target, thus providing a rationale for exploring thetherapeutic effects of novel IKKb inhibitors with favorable toxicity
profiles, particularly in the subgroups of advanced prostate cancerpatients in whom such pathways may be activated.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: Y. Zhang, H.C. DanDevelopment of methodology: Y. Zhang, X. Wang, H.C. DanAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): Y. Zhang, R.G. Lapidus, P. Liu, E.Y. Choi, S. Adediran,X. Wang, H.C. DanAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): Y. Zhang, P. Liu, E.Y. Choi, A. Hussain, X. Liu,H.C. DanWriting, review, and/or revision of the manuscript: Y. Zhang, R.G. Lapidus,A. Hussain, X. Liu, H.C. DanAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): E.Y. Choi, X. Liu, H.C. DanStudy supervision: X. Liu, H.C. Dan
AcknowledgmentsThe authors thank Dr. Albert Baldwin in University of North Carolina at
Chapel Hill for providing the IKKb inhibitor CmpdA.
Grant SupportThis work was supported, in part, by NIH Grant R00CA149178 and startup
funds from Marlene and Stewart Greenebaum Cancer Center, University ofMaryland School of Medicine (to H.C. Dan) and a Merit Review Award,Department of Veterans Affairs (to A. Hussain).
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received December 21, 2015; revised March 14, 2016; accepted March 28,2016; published OnlineFirst April 8, 2016.
References1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin
2014;64:9–29.2. Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in
prostate cancer. Curr Opin Pharmacol 2008;8:440–448.3. Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and
targeting the androgen receptor therapeutically. J Clin Oncol 2011;29:3651–58.
4. Khemlina G, Ikeda S, Kurzrock R. Molecular landscape of prostate cancer:Implications for current clinical trials. Cancer Treat Rev 2015;41:761–66.
5. Crawford ED, Higano CS, Shore ND, Hussain M, Petrylak DP. Treatingpatients with metastatic castration resistant prostate cancer: a comprehen-sive review of available therapies. J Urol 2015;S0022-5347:04423–27
6. Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in humancancer: rationale and promise. Cancer Cell 2003;4:257–62.
7. Manning BD, Cantley LC. United at last: the tuberous sclerosis complexgene products connect the phosphoinositide 3-kinase/Akt pathway tomammalian target of rapamycin (mTOR) signalling. Biochem Soc Trans2003;31:573–78.
8. Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on geneexpression at a glance. J Cell Sci 2013;126:1713–19.
9. Inoki K, Guan KL. Tuberous sclerosis complex, implication from a raregenetic disease to common cancer treatment. Hum Mol Genet 2009;18:R94–100.
10. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibitedby Akt and suppresses mTOR signalling. Nat Cell Biol 2002;4:648–57.
11. Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: newprospects for old challenges. Genes Dev 2010;24:1967–2000.
12. Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer.Oncogene 2005;24:7465–74.
13. HaydenMS, Ghosh S. Signaling to NF-kappaB. Genes Dev 2004;18:2195–224.
14. Bonizzi G, KarinM. The twoNF-kappaB activation pathways and their rolein innate and adaptive immunity. Trends Immunol 2004;25:280–88.
15. KimHJ,HawkeN, BaldwinAS.NF-kappaB and IKK as therapeutic targets incancer. Cell Death Differ 2006;13:738–47.
16. Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. Distinctroles of the Ikappa B kinase alpha and beta subunits in liberatingnuclear factor kappa B (NF-kappa B) from Ikappa B and in phos-phorylating the p65 subunit of NF-kappa B. J Biol Chem 2002;277:3863–69.
17. Nadiminty N, Gao AC. Mechanisms of persistent activation of the andro-gen receptor inCRPC: recent advances and future perspectives.World JUrol2012;30:287–95.
18. Jin R, Yi Y, Yull FE, Blackwell TS, Clark PE, Koyama T, et al. NF-kappaBgene signature predicts prostate cancer progression. Cancer Res2014;74:2763–72.
19. Jin RJ, Lho Y, Connelly L, Wang Y, Yu X, Saint Jean L, et al. The nuclearfactor-kappaB pathway controls the progression of prostate cancer toandrogen-independent growth. Cancer Res 2008;68:6762–69.
20. Suh J, Payvandi F, Edelstein LC, Amenta PS, Zong WX, Gelinas C, et al.Mechanisms of constitutive NF-kappaB activation in human prostatecancer cells. Prostate 2002;52:183–200.
21. McCall P, Bennett L, Ahmad I, Mackenzie LM, Forbes IW, Leung HY, et al.NFkappaB signalling is upregulated in a subset of castrate-resistant prostate
Zhang et al.
Mol Cancer Ther; 2016 Molecular Cancer TherapeuticsOF10
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999

cancer patients and correlates with disease progression. Br J Cancer2012;107:1554–63.
22. Nguyen DP, Li J, Yadav SS, Tewari AK. Recent insights into NF-kappaBsignalling pathways and the link between inflammation and prostatecancer. BJU Int 2014;114:168–76.
23. Shukla S, MacLennan GT, Fu P, Patel J, Marengo SR, Resnick MI, et al.Nuclear factor-kappaB/p65 (Rel A) is constitutively activated in humanprostate adenocarcinoma and correlates with disease progression. Neo-plasia 2004;6:390–400.
24. Jain G, Cronauer MV, Schrader M, Moller P, Marienfeld RB. NF-kappaBsignaling in prostate cancer: a promising therapeutic target? World J Urol2012;30:303–10.
25. Dan HC, Adli M, Baldwin AS. Regulation of mammalian target of rapa-mycin activity in PTEN-inactive prostate cancer cells by I kappa B kinasealpha. Cancer Res 2007;67:6263–69.
26. Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptorin association with IKK. Genes Dev 2008;22:1490–500.
27. Zhang YT, Ouyang DY, Xu LH, Ji YH, Zha QB, Cai JY, et al. Cucurbitacin Binduces rapid depletion of the G-actin pool through reactive oxygenspecies-dependent actin aggregation in melanoma cells. Acta BiochimBiophys Sin 2011;43:556–67.
28. Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Peri-fosine, a novel alkylphospholipid, inhibits protein kinase B activation.MolCancer Ther 2003;2:1093–103.
29. Nelson EC, Evans CP,Mack PC,Devere-White RW, Lara PN Jr. Inhibition ofAkt pathways in the treatment of prostate cancer. Prostate Cancer ProstaticDis 2007;10:331–39.
30. Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role ofnanog in tumorigenesis and cancer stem cells. Int J Cancer 2014;135:2741–48.
31. Noh KH, Kim BW, Song KH, ChoH, Lee YH, Kim JH, et al. Nanog signalingin cancer promotes stem-like phenotype and immune evasion. J Clin Invest2012;122:4077–93.
32. Ziegelbauer K, Gantner F, Lukacs NW, Berlin A, Fuchikami K,Niki T, et al. Aselective novel low-molecular-weight inhibitor of IkappaB kinase-beta(IKK-beta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br J Pharmacol 2005;145:178–92.
33. Gasparian AV, Yao YJ, Kowalczyk D, Lyakh LA, Karseladze A, Slaga TJ, et al.The role of IKK in constitutive activation of NF-kappaB transcription factorin prostate carcinoma cells. J Cell Sci 2002;115:141–51.
34. BasseresDS, Ebbs A, Cogswell PC, Baldwin AS. IKK is a therapeutic target inKRAS-Induced lung cancer with disrupted p53 activity. Genes Cancer2014;5:41–55.
35. Chou TC.Drug combination studies and their synergy quantification usingthe Chou-Talalay method. Cancer Res 2010;70:440–46.
36. Ashton JC. Drug combination studies and their synergy quantificationusing the Chou-Talalay method–letter. Cancer Res 2015;75:2400.
37. Freudlsperger C, Burnett JR, Friedman JA, Kannabiran VR, Chen Z, VanWaes C. EGFR-PI3K-AKT-mTOR signaling in head and neck squamous cellcarcinomas: attractive targets for molecular-oriented therapy. Expert OpinTher Targets 2011;15:63–74.
38. Argiris A, Cohen E, Karrison T, Esparaz B,Mauer A, Ansari R, et al. A phase IItrial of perifosine, an oral alkylphospholipid, in recurrent or metastatichead and neck cancer. Cancer Biol Ther 2006;5:766–70.
39. Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, et al.mTOR inhibition reverses Akt-dependent prostate intraepithelial neopla-sia through regulation of apoptotic and HIF-1-dependent pathways. NatMed 2004;10:594–601.
40. Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway incastration-resistant prostate cancer. Endocr Relat Cancer 2013;20:R83–99.
41. Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, et al. NANOGpromotes cancer stem cell characteristics and prostate cancer resistance toandrogen deprivation. Oncogene 2011;30:3833–45.
42. Chen X, Rycaj K, Liu X, Tang DG. New insights into prostate cancer stemcells. Cell Cycle 2013;12:579–86.
43. Liu F, Xia Y, Parker AS, Verma IM. IKK biology. Immunol Rev 2012;246:239–53.
44. GambleC,McIntoshK, Scott R,HoKH,Plevin R, Paul A. Inhibitory kappaBKinases as targets for pharmacological regulation. Br J Pharmacol 2012;165:802–19.
45. Suzuki J, OgawaM,Muto S, Itai A, IsobeM,Hirata Y, et al. Novel IkB kinaseinhibitors for treatment of nuclear factor-kB-related diseases. Expert OpinInvestig Drugs 2011;20:395–405.
46. Gasparian AV, Yao YJ, Lu J, Yemelyanov AY, Lyakh LA, Slaga TJ, et al.Selenium compounds inhibit I kappa B kinase (IKK) and nuclear factor-kappa B (NF-kappa B) in prostate cancer cells. Mol Cancer Ther 2002;1:1079–87.
47. Yemelyanov A, Gasparian A, Lindholm P, Dang L, Pierce JW, Kisseljov F,et al. Effects of IKK inhibitor PS1145 onNF-kappaB function, proliferation,apoptosis and invasion activity in prostate carcinoma cells. Oncogene2006;25:387–98.
48. Gasparian AV, Guryanova OA, Chebotaev DV, Shishkin AA, YemelyanovAY, Budunova IV. Targeting transcription factor NFkappaB: comparativeanalysis of proteasome and IKK inhibitors. Cell Cycle 2009;8:1559–66.
49. Bednarski BK, Baldwin AS Jr, Kim HJ. Addressing reported pro-apoptoticfunctions of NF-kappaB: targeted inhibition of canonical NF-kappaBenhances the apoptotic effects of doxorubicin. PloS ONE 2009;4:e6992.
www.aacrjournals.org Mol Cancer Ther; 2016 OF11
IkB Kinase b in Human Prostate Cancer
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999

Published OnlineFirst April 8, 2016.Mol Cancer Ther Yanting Zhang, Rena G. Lapidus, Peiyan Liu, et al.
Inhibitor CmpdAβB Kinase κCancer by a Novel IB Signaling in Human Prostateκ/NF-βB Kinase κTargeting I
Updated version
10.1158/1535-7163.MCT-15-0999doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2016/04/08/1535-7163.MCT-15-0999.DC1
Access the most recent supplemental material at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://mct.aacrjournals.org/content/early/2016/06/08/1535-7163.MCT-15-0999To request permission to re-use all or part of this article, use this link
on May 16, 2018. © 2016 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 8, 2016; DOI: 10.1158/1535-7163.MCT-15-0999


![ikB & 15] 16] 17 O;kdkj.k & ikB & 12] 13] 15 ikB & 14 vkj 15chinmayabokaro.org/ModelQuestions/class 2 all.pdfchinmaya vidyalaya / b s city (cbse new generation school) annual examination,](https://img.pdfslide.us/doc/110x75/5ac0420f7f8b9a4e7c8bab56/ikb-15-16-17-okdkjk-ikb-12-13-15-ikb-14-vkj-2-allpdfchinmaya-vidyalaya-.jpg)