Embed Size (px)
Citation preview

THE JOURNAL OF B%oLwfr.a CI<EMISTRY 0 1994 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 269, No. 40, Issue of October 7, pp. 24920-24927. 1994 Prrnted in U.S.A.
Direct Square-Wave Voltammetry of Superoxidized [4Fe-4SI3+ Aconitase and Associated 3Fe/4Fe Cluster Interconversions*
(Received for publication, May 25, 1994, and in revised form, July 11, 1994)
Jiajie Tong and Benjamin A. FeinbergS From the Department of Chemistry, University of Wisconsin, Milwaukee, Wisconsin 53201
We report a direct square-wave voltammetric study of the iron-sulfur enzyme, aconitase, at the pyrolytic graphite edge electrode. .New and established redox driven reactions were observed and the equilibrium re- duction potential for each couple was determined:
+IO0 mV, EFj,,,, = -281 mV, and putatively, E&,4spl2- =I
-1000 mV, all uersus normal hydrogen electrode. Most importantly we have directly observed the superoxi- dized [4Fe-4SIS' form of aconitase (originally proposed by Emptage, M. H., Dreyer, J.-L., Kennedy, M. C., and Beinert, H. (1983) J. Biol. Chen. 258, 11106-11111) and directly followed its conversion to the [3Fe-4SI1+ form; this intermediate is required for the deactivation of aconitase. Without exogenous ferrous iron, [3Fe-4Slo aconitase is apparently super-reduced at very negative potentials to the [3Fe-4S12- form and the concomitant formation of [4Fe-4SI2+ aconitase was followed over time. It is the apparent decomposition of super-reduced [3Fe-4SJ2- aconitase that provides the source of ferrous iron for the interconversion of [3Fe-4Slo aconitase to the [4Fe-4SI2+ form. Voltammetry of free and substrate bound [4Fe-4SI2+ aconitase showed that the latter is less susceptible to oxidation but, surprisingly, has the same
E[sFe-4S)l+f0 = -268 mv, E$Fe-48]2+/1+ = -450 mv7 E6Fe-4S]S+12+ 0
0 E[4Fe.4S]5+/2+*
~ ~- ~ ~ ~
Aconitase (citrate (isocitrate) hydrolase, EC 4.2.1.3) is a key [4Fe-4S] enzyme that catalyzes the stereospecific dehydration- rehydration of citrate to isocitrate via the dehydrated interme- diate cis-aconitate (1). These catalyzed transformations consti- tute the second and third steps of the Krebs cycle as shown in Scheme I.
HO E::; H o - p co; co; co,
Citrate cis-Aconitate Isocitrate SCHEME I
The cubane-like [4Fe-4SIZ+ cluster of active aconitase has three of its irons coordinated to cystinate residues and the fourth iron, also termed Fe, (2), which is coordinated to HO-. Upon addition of substrate (citrate), Fe, coordinates with the hydroxyl oxygen and the @-carboxyl oxygen of citrate, and the HO- is protonated to H,O, which remains coordinated to Fe, (3-5). Much of what we know about aconitase comes from the
GM 48598 (to B. A. F.). The costs of publication of this article were * This work was supported by National Institutes of Health Grant
defrayed in part by the payment of page charges. This article must therefore be hereby marked "aduertisernent" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ 'Ib whom correspondence should be addressed: Dept. of Chemistry, P. 0. Box 413, University of Wisconsin, Milwaukee, WI 53201. Tel.: 414-229-4169; Fax: 414-229-5530.
work of Beinert and Kennedy, and co-workers, with the details of the catalytic mechanism and cluster redox chemistry re- viewed by Beinert and Kennedy ( 9 , Kennedy and Stout (11, Emptage (61, and Lauble et al. (3). The redox states and prop- erties of aconitase are of great interest since they virtually determine the catalytic activity of aconitase (71, its degrada- tion, reconstitution, and interconversion to other forms. When the enzyme is isolated under aerobic conditions, the cluster is isolated in the [3Fe-4S11+ form which is catalytically inactive (8, 9) and, at best, binds substrate weakly (5). Upon reduction in the presence of Fez+, the cluster is interconverted to the 14Fe- 4S12+ form, and the enzyme then has its maximal activity; al- ternatively, in the absence of ferrous ion, about 75% of maximal activity can be obtained (10). There are also experiments that show that the [4Fe-4SI1+ form has about 30% of the maximal aconitase activity (7), but this activity may arise from partial reoxidation. Of great interest for this work is the "superoxi- dized" form of aconitase, i.e. [4Fe-4SI3+ aconitase, proposed by Emptage et al. (7). This reactive intermediate was proposed to be an unstable form of aconitase that interconverted to the inactive [3Fe-4SI1+ form, but to date no direct evidence of its existence has been obtained. These are all redox driven inter- conversions and as such, are ideal candidates for voltammetrid electrochemical study. Making use of visible, electron paramag- netic, and Mossbauer spectroscopies, Emptage et al. (7) determined the identities and redox states of the species below, excepting the proposed superoxidized [4Fe-4S13+ form. A sum- mary of their observations are shown in Scheme 11, with the proposed [4Fe-4S13+ intermediate bracketed.
[3Fe-4Sla [4Fe-4S12+
-e +e I 1 I&
[4Fe-4S11+ SCHEME I1
In this paper we report our direct electrochemical study of aconitase using square-wave voltammetry at the pyrolytic graphite edge electrode. Square-wave voltammetry (SWV)' pro- vides significantly better signal to noise ratio than cyclic vol- tammetry, and the experiment is often much faster (11). The results presented here demonstrate both the plasticity of the
The abbreviations used are: SWV, square-wave voltammetry; E,, initial potential; E,, final potential; PIPES, 1,4-piperazinediethanesul- fonic acid; TAPS, 3-([2-hydroxy-l,l-bis~hydroxymethyl~ethyllam~no~-1- propanesulfonic acid.
24920
Direct Electrochemistry of Aconitase 24921
redox driven reactions of aconitase and the advantages of using square-wave voltammetry to elucidate them. In addition to the stable redox couples described by Kennedy et al. (10) and Emptage et al. (7) shown in Scheme 11, we have also directly observed for the first time two reactive intermediates of aconi- tase: superoxidized and super-reduced aconitase and their in- terconversion to the [3Fe-4SI1+ and [4Fe-4S12+ forms, respec- tively. This voltammetric study also sheds new light on: ( a ) the difficulty of reducing [4Fe-4SI2+ aconitase to the [4Fe-4SI1+ form and ( b ) the nature of substrate stabilization of the aconitase [4Fe-4SI2+ cluster.
EXPERIMENTAL PROCEDURES Buflers-The standard buffer used throughout this work, if not
otherwise specified, was 50 mM Tris, pH 8, that was 100 mM in NaCl. Tris-HC1, Tris Base, and NaCl were all obtained as ultra-pure reagents from J. T. Baker. Double-distilled deionized water that had been passed through a Milli-Q Water Purification System to a conductivity of 18 Mohmdcm was used throughout. For the experiments with the 13Fe-4SI form of aconitase, the lyophilized enzyme was dissolved in the standard buffer, so that the final aconitase concentration was typically about 1 m. The pH dependence studies of [3Fe-4S] aconitase were camed out in three different buffers all obtained from Sigma: PIPES, pK 6.8; HEPES, pK 7.5; and TAPS, pK 8.4.
Aconitase Preparations: [3Fe-4SI1+ and [4Fe-4SF' Forms-Porcine mitochondrial heart aconitase was obtained from Sigma (A5384) in the lyophilized [3Fe-4S11+ form. The enzymatically inactive [3Fe-4S11+ form of aconitase was used as received from Sigma without further purifica- tion. The spirane-like, linear form of aconitase, represented as Cys,FeS,FeS,FeCys,, was prepared in buffer at pH 9 and 6 M urea as described by Kennedy et al. (12). The activated [4Fe-4SI2+ aconitase was obtained by following the activation procedure of Kennedy et al. (10) in a Coy anaerobic chamber. Typically for our experiments, 24 mg of aconitase was dissolved in 200 pl of standard buffer that was also 50 mM in dithiothreitol (dithiothreitol, Fisher, electrophoresis grade) to form a
Fe(NH,),(SO,), (Analytical Reagent Grade, Fisher) was added to the 1.4 mM enzyme solution. Then 30 pl (a 10-fold excess) of 0.1 M
above aconitase solution. After incubation for 4 h at 7 "C, the solution was dialyzed against 200 ml of standard buffer overnight at the same temperature, using Spectrapor (Los Angeles) dialysis tubing with a 3.5 kDa molecular mass cutoff. Before performing any electrochemical ex- periments, this dialysis procedure was repeated a second time for an additional 8 h to remove all traces of reducing agent andlor ferrous iron from the activated aconitase solution.
Actiuity of[4Fe-4SpAconitase-To verify that the reconstitution and preparation of the [4Fe-4SI2+ aconitase was effective, its activity was determined using the method of Henson and Cleland (13), in which visible spectroscopy is used to determine the rate at which citrate is transformed to cis-aconitate, which absorbs at 240 nm. The activity of the reconstituted Sigma [4Fe-4SIZ+ aconitase was 10 times that reported by Sigma. Experiments were also done to examine the activity of any [4Fe-4SlZ+ aconitase produced via electrochemically driven interconver- sion. For these experiments, [3Fe-4SI1+ aconitase was reduced at an applied potential of -1100 mV at the electrode for 5 min, then held for 5 min at -400 mV (to reoxidize any [4Fe-4SI1+ aconitase that may have been formed to the active [4Fe-4SI2+ form), followed by 5 min with no applied potential to allow [4Fe-4S12+ aconitase generated at the elec- trode to diffuse into bulk solution. 10 pl of this very partially electro- lyzed solution were removed under anaerobic conditions and placed into a cuvette with the appropriate concentration of citrate for determina- tion of its activity.
Square-wave Voltammetry-Square-wave voltammetry is a rapid and sensitive electrochemical method which provides especially good signal to noise response. Osteryoung and co-workers (11,14) have been largely responsible for developing square-wave voltammetry and making it accessible for electroanalytical research. In square-wave voltammetry, a potential is applied over time in the form of a square-wave superim- posed on a stair case (see Fig. 1). The applied potential is progressively stepped in fixed increments, hE, (the step potential), and pulsed posi- tively and negatively at each step with potential E,,, the square-wave amplitude. This is all done at a particular frequency f = i', where T is the time for one full cycle. The resultant current is measured at the end (0) of each forward and reverse pulse. Throughout this paper the posi- tive direction is the anodic or oxidizing direction and the negative di- rection is the cathodic or reducing direction. Overall, the voltammetric scan is either in the positive or negative direction. For each applied
-..- I
-0.1 0 I- I '
-0.05 1 5.1 5.2 5.3
TIME (seconds)
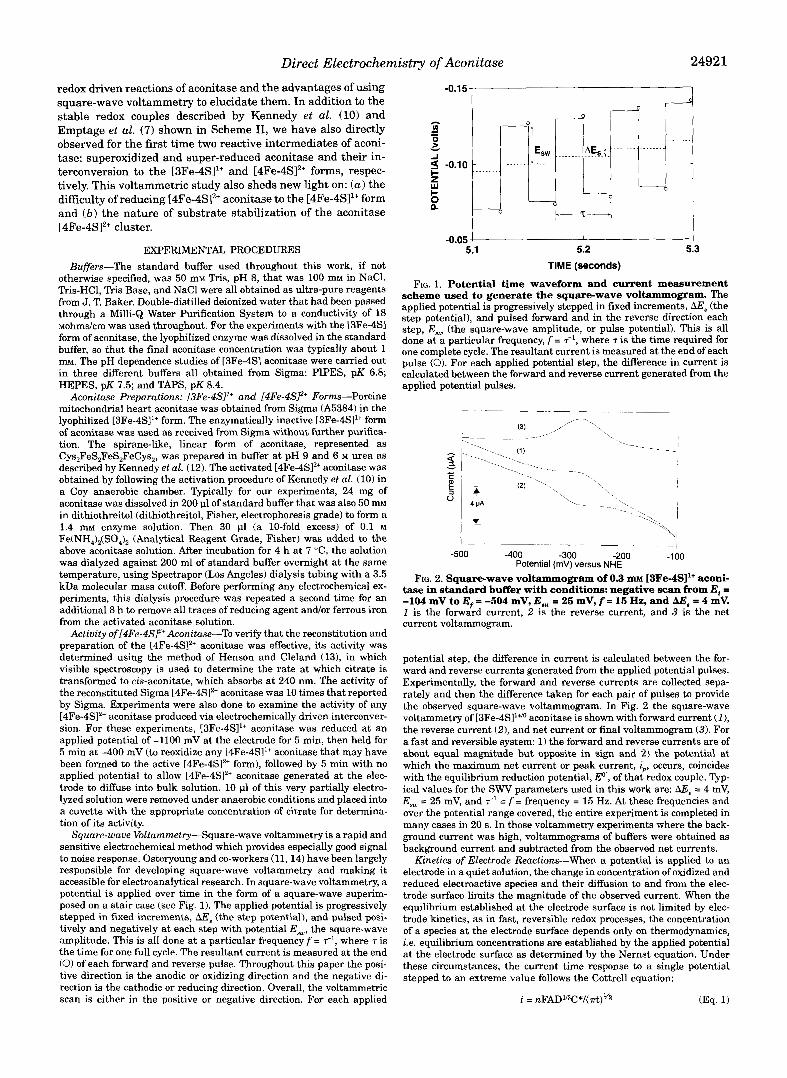
FIG. 1. Potential time waveform and current measurement scheme used to generate the square-wave voltammogram. The
step potential), and pulsed forward and in the reverse direction each applied potential is progressively stepped in fixed increments, AE, (the
step, E, (the square-wave amplitude, or pulse potential). This is all done at a particular frequency, f = T-~, where T is the time required for one complete cycle. The resultant current is measured at the end of each pulse (0). For each applied potential step, the difference in current is calculated between the forward and reverse current generated from the applied potential pulses.
-500 -400 -300 -200 -1 00 L~- - ___~ ~~~ ~ ~ ~ ~ _ _ _ ~ _ ~ ~ ~ . ~ 1
Potential (mV) versus NHE
tase in standard buffer with conditions: negative scan from Ei = FIG. 2. Square-wave voltammogram of 0.3 m~ [3Fe-4SI1+ aconi-
-104 mV to E, = -504 mV, E,, = 25 mV, f = 15 Hz, and AE, = 4 mV. 1 is the forward current, 2 is the reverse current, and 3 is the net current voltammogram.
potential step, the difference in current is calculated between the for- ward and reverse currents generated from the applied potential pulses. Experimentally, the forward and reverse currents are collected sepa- rately and then the difference taken for each pair of pulses to provide the observed square-wave voltammogram. In Fig. 2 the square-wave voltammetry of [3Fe-4SI1+" aconitase is shown with forward current (I), the reverse current (2), and net current or final voltammogram (3). For a fast and reversible system: 1) the forward and reverse currents are of about equal magnitude but opposite in sign and 2) the potential at which the maximum net current or peak current, i,,, occurs, coincides with the equilibrium reduction potential, E", of that redox couple. Typ- ical values for the SWV parameters used in this work are: AE, = 4 mV, E,, = 25 mV, and = f = frequency = 15 Hz. At these frequencies and over the potential range covered, the entire experiment is completed in many cases in 20 s. In those voltammetry experiments where the back- ground current was high, voltammograms of buffers were obtained as background current and subtracted from the observed net currents.
Kinetics of Electrode Reactions-When a potential is applied to an electrode in a quiet solution, the change in concentration of oxidized and reduced electroactive species and their diffusion to and from the elec- trode surface limits the magnitude of the observed current. When the equilibrium established at the electrode surface is not limited by elec- trode kinetics, as in fast, reversible redox processes, the concentration of a species at the electrode surface depends only on thermodynamics, i.e. equilibrium concentrations are established by the applied potential at the electrode surface as determined by the Nernst equation. Under these circumstances, the current time response to a single potential stepped to an extreme value follows the Cottrell equation:
i = ~ F A D W * / ( & ) ~ ~ (Eq. 1)
24922 Direct Electrochemistry of Aconitase
10.0 r
9.0 I
-500 -400 -300 -200 -100 ~ -I
Potential (mv) versus NHE
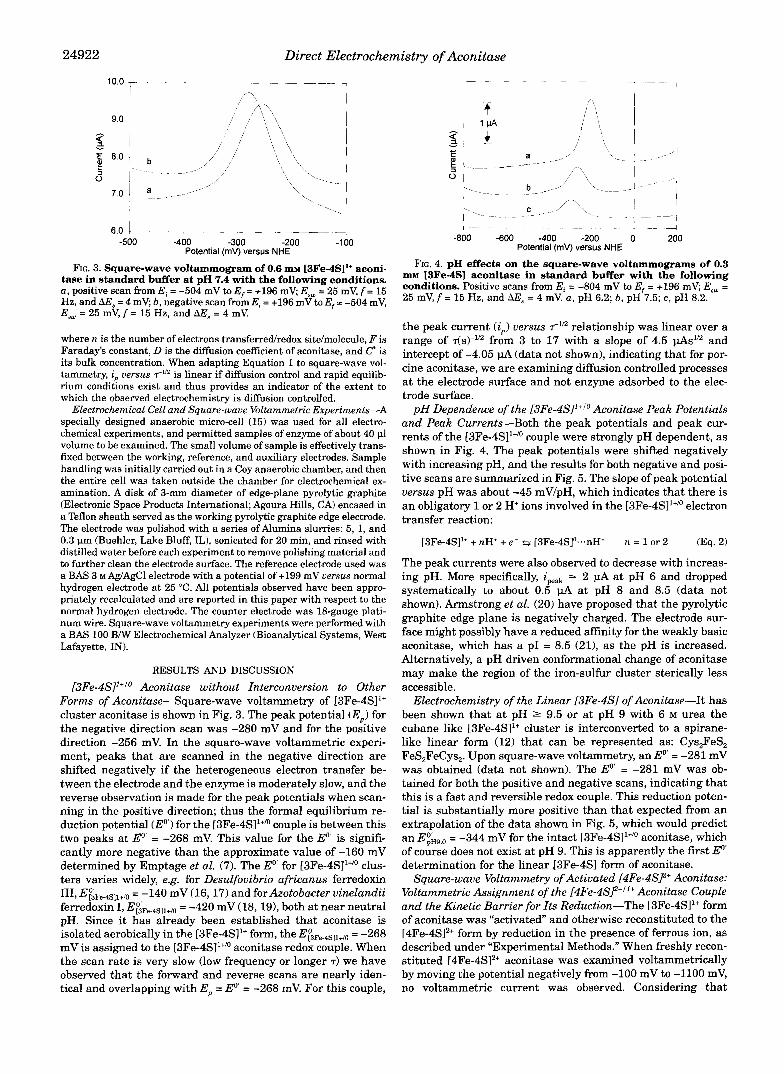
FIG. 3. Square-wave voltammogram of 0.6 m~ [3Fe-4S11+ aconi- tase in standard buffer at pH 7.4 with the following conditions. a , positive scan from Ei = -504 mV to E, = +I96 mV; E,, = 25 mV, f = 15 Hz, and A E s = 4 mV; b, negative scan from E, = +I96 mVto E f = -504 mV, E,, = 25 mV, f = 15 Hz, and AE, = 4 mV.
where n is the number of electrons transferredlredox site/molecule, F is Faraday's constant, D is the diffusion coefficient of aconitase, and c" is its bulk concentration. When adapting Equation 1 to square-wave vol- tammetry, i, uersus T-" is linear if diffusion control and rapid equilib- rium conditions exist and thus provides an indicator of the extent to which the observed electrochemistry is diffusion controlled.
Electrochemical Cell and Square-waue Voltammetric Experiments-A specially designed anaerobic micro-cell (15) was used for all electro- chemical experiments, and permitted samples of enzyme of about 40 1.11 volume to be examined. The small volume of sample is effectively trans- fixed between the working, reference, and auxiliary electrodes. Sample handling was initially carried out in a Coy anaerobic chamber, and then the entire cell was taken outside the chamber for electrochemical ex- amination. A disk of 3-mm diameter of edge-plane pyrolytic graphite (Electronic Space Products International; Agoura Hills, CA) encased in a Teflon sheath served as the working pyrolytic graphite edge electrode. The electrode was polished with a series of Alumina slurries: 5, 1, and 0.3 pm (Buehler, Lake Bluff, IL), sonicated for 20 min, and rinsed with distilled water before each experiment to remove polishing material and to further clean the electrode surface. The reference electrode used was a BAS 3 M Ag/AgCl electrode with a potential of +I99 mV uersus normal hydrogen electrode at 25 "C. All potentials observed have been appro- priately recalculated and are reported in this paper with respect to the normal hydrogen electrode. The counter electrode was 18-gauge plati- num wire. Square-wave voltammetry experiments were performed with a BAS 100 BiW Electrochemical Analyzer (Bioanalytical Systems, West Lafayette, IN).
RESULTS AND DISCUSSION
[3Fe-4S]1+'o Aconitase without Interconversion to Other Forms of Aconitase-Square-wave voltammetry of [3Fe-4S11+ cluster aconitase is shown in Fig. 3. The peak potential (E,) for the negative direction scan was -280 mV and for the positive direction -256 mV. In the square-wave voltammetric experi- ment, peaks that are scanned in the negative direction are shifted negatively if the heterogeneous electron transfer be- tween the electrode and the enzyme is moderately slow, and the reverse observation is made for the peak potentials when scan- ning in the positive direction; thus the formal equilibrium re- duction potential (E") for the [3Fe-4SI1+" couple is between this two peaks at Eo' = -268 mV. This value for the Eo' is signifi- cantly more negative than the approximate value of -160 mV determined by Emptage et al. (7). The Eo' for [3Fe-4SI1+" clus- ters varies widely, e.g. for Desulfovibrio africanus ferredoxin 111, E~~Fe.4Sll+/o = -140 mV (16, 17) and for Azotobacter vinelandii ferredoxin I, E&e.4sll+~o = -420 mV (18, 191, both at near neutral pH. Since it has already been established that aconitase is isolated aerobically in the [3Fe-4SI1+ form, the E&e.4Sll+/0 = -268 mV is assigned to the [3Fe-4SI1+/O aconitase redox couple. When the scan rate is very slow (low frequency or longer T ) we have observed that the forward and reverse scans are nearly iden- tical and overlapping with E, = Eo' = -268 mV. For this couple,
I ...
I --.
-800 -600 -400 -200 0 200 Potential (mv) versus NHE
m~ RFe-4SI aconitase in standard buffer with the following FIG. 4. pH effects on the square-wave voltammograms of 0.3
conditions. Positive scans from E, = -804 mV to E, = +196 mV; E,, = 25 mV, f = 15 Hz, and AE, = 4 mV. a , pH 6.2; b, pH 7.5; c, pH 8.2.
the peak current (i,) versus T - ~ ~ relationship was linear over a range of ds)-'" from 3 to 17 with a slope of 4.5 pAs" and intercept of -4.05 pA (data not shown), indicating that for por- cine aconitase, we are examining diffusion controlled processes at the electrode surface and not enzyme adsorbed to the elec- trode surface.
pH Dependence of the [3Fe-4S]l+Io Aconitase Peak Potentials and Peak Currents-Both the peak potentials and peak cur- rents of the [3Fe-4SI1+/O couple were strongly pH dependent, as shown in Fig. 4. The peak potentials were shifted negatively with increasing pH, and the results for both negative and posi- tive scans are summarized in Fig. 5. The slope of peak potential versus pH was about -45 mVIpH, which indicates that there is an obligatory 1 or 2 H+ ions involved in the [3Fe-4S11+/o electron transfer reaction:
[3Fe-4SI1+ + nH' + e - s [3Fe-4SIo...nH+ n = 1 or 2 (Eq. 2)
The peak currents were also observed to decrease with increas- ing pH. More specifically, ipeak = 2 pA at pH 6 and dropped systematically to about 0.5 pA at pH 8 and 8.5 (data not shown). Armstrong et al. (20) have proposed that the pyrolytic graphite edge plane is negatively charged, The electrode sur- face might possibly have a reduced affinity for the weakly basic aconitase, which has a PI = 8.5 (21), as the pH is increased. Alternatively, a pH driven conformational change of aconitase may make the region of the iron-sulfur cluster sterically less accessible.
Electrochemistry of the Linear r3Fe-4~91 of Aconitase-It has been shown that at pH 2 9.5 or at pH 9 with 6 M urea the cubane like [3Fe-4SI1+ cluster is interconverted to a spirane- like linear form (12) that can be represented as: Cys,FeS, FeS,FeCys,. Upon square-wave voltammetry, an Eo = -281 mV was obtained (data not shown). The Eo' = -281 mV was ob- tained for both the positive and negative scans, indicating that this is a fast and reversible redox couple. This reduction poten- tial is substantially more positive than that expected from an extrapolation of the data shown in Fig. 5, which would predict an E&,o = -344 mV for the intact [3Fe-4S]1+/o aconitase, which of course does not exist at pH 9. This is apparently the first E'" determination for the linear [3Fe-4S] form of aconitase.
Square-wave Voltammetry of Activated [4Fe-4S/2' Aconitase: Voltammetric Assignment of the [4Fe-4S/2tJ1+ Aconitase Couple and the Kinetic Barrier for Its Reduction-The [3Fe-4S11+ form of aconitase was "activated and otherwise reconstituted to the [4Fe-4SI2+ form by reduction in the presence of ferrous ion, as described under "Experimental Methods." When freshly recon- stituted [4Fe-4SI2+ aconitase was examined voltammetrically by moving the potential negatively from -100 mV to -1100 mV, no voltammetric current was observed. Considering that
Direct Electrochemistry of Aconitase 24923
I I
I
,-> a ' I
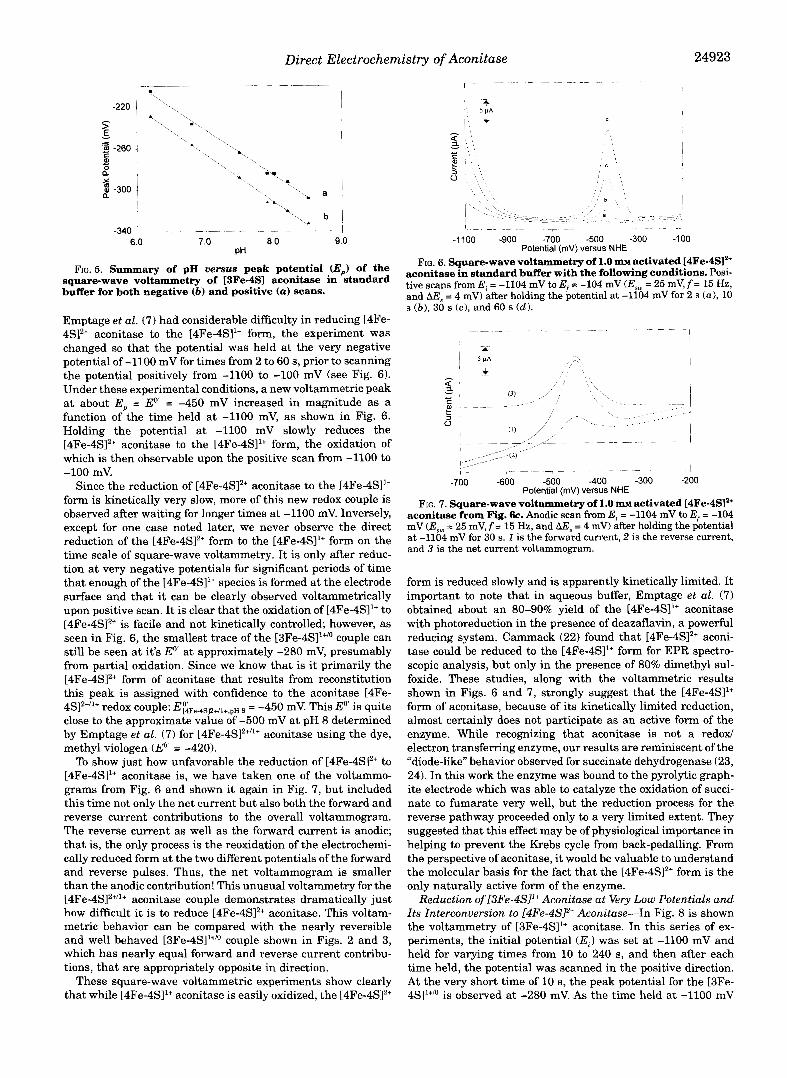
FIG. 5. Summary of pH uersus peak potential (E,) of the
buffer for both negative (b) and positive (a) scans. square-wave voltammetry of I3Fe-4SI aconitase in standard
Emptage et al. (7) had considerable difficulty in reducing [4Fe- 4SI2+ aconitase to the [4Fe-4S11+ form, the experiment was changed so that the potential was held at the very negative potential of -1100 mV for times from 2 to 60 s, prior to scanning the potential positively from -1100 to -100 mV (see Fig. 6). Under these experimental conditions, a new voltammetric peak at about E, = Eo' = -450 mV increased in magnitude as a function of the time held at -1100 mV, as shown in Fig. 6. Holding the potential at -1100 mV slowly reduces the [4Fe-4SI2+ aconitase to the [4Fe-4SI1+ form, the oxidation of which is then observable upon the positive scan from -1100 to -100 mV.
Since the reduction of [4Fe-4S12+ aconitase to the [4Fe-4S11+ form is kinetically very slow, more of this new redox couple is observed after waiting for longer times at -1100 mV. Inversely, except for one case noted later, we never observe the direct reduction of the [4Fe-4S12+ form to the [4Fe-4S11+ form on the time scale of square-wave voltammetry. It is only after reduc- tion at very negative potentials for significant periods of time that enough of the [4Fe-4S11+ species is formed at the electrode surface and that it can be clearly observed voltammetrically upon positive scan. It is clear that the oxidation of [4Fe-4S11+ to [4Fe-4S12+ is facile and not kinetically controlled; however, as seen in Fig. 6, the smallest trace of the [3Fe-4S]1+/o couple can still be seen at it's Eo' at approximately -280 mV, presumably from partial oxidation. Since we know that is it primarily the [4Fe-4S12+ form of aconitase that results from reconstitution this peak is assigned with confidence to the aconitase [4Fe-
close to the approximate value of -500 mV at pH 8 determined by Emptage et al. (7) for [4Fe-4S12+"+ aconitase using the dye, methyl viologen (E'' = -420).
To show just how unfavorable the reduction of [4Fe-4SI2+ to [4Fe-4S11+ aconitase is, we have taken one of the voltammo- grams from Fig. 6 and shown it again in Fig. 7, but included this time not only the net current but also both the forward and reverse current contributions to the overall voltammogram. The reverse current as well as the forward current is anodic; that is, the only process is the reoxidation of the electrochemi- cally reduced form at the two different potentials of the forward and reverse pulses. Thus, the net voltammogram is smaller than the anodic contribution! This unusual voltammetry for the [4Fe-4S]2+'1+ aconitase couple demonstrates dramatically just how difficult it is to reduce [4Fe-4SI2+ aconitase. This voltam- metric behavior can be compared with the nearly reversible and well behaved [3Fe-4S11+/0 couple shown in Figs. 2 and 3, which has nearly equal forward and reverse current contribu- tions, that are appropriately opposite in direction.
These square-wave voltammetric experiments show clearly that while [4Fe-4SI1+ aconitase is easily oxidized, the [4Fe-4SI2+
4S]2+/'+ redox couple: E ~&e.4s12+/l+,pH = -450 mV. This Eo' is quite
d
FIG. 6. Square-wave voltammetry of 1.0 m~ activated [4Fe-4S12+ aconitase in standard buffer with the following conditions. Posi- tive scans from E, = -1104 mV to E, = -104 mV (Esw = 25 mV, f = 15 Hz, and AE, = 4 mV) after holding the potential at -1104 mV for 2 s (a), 10 s ( b ) , 30 s (c), and 60 s (d).
r
w ~~ ~~~ ~~I - ~- ~ - 1 - - - 7 ~ ~- r~ ~ ~
-700 -600 -500 -400 -300 -200 Potential (mV) versus NHE
aconitase from Fig. 6c. Anodic scan from E, = -1104 mV to Ef = -104 FIG. 7. Square-wave voltammetry of 1.0 m~ activated [4Fe-4SI2+
mV (Esm = 25 mV, f = 15 Hz, and AE, = 4 mV) after holding the potential a t -1104 mV for 30 s. 1 is the forward current, 2 is the reverse current, and 3 is the net current voltammogram.
form is reduced slowly and is apparently kinetically limited. It important to note that in aqueous buffer, Emptage et al. (7) obtained about an 80-90% yield of the [4Fe-4S11+ aconitase with photoreduction in the presence of deazaflavin, a powerful reducing system. Cammack (22) found that [4Fe-4S12+ aconi- tase could be reduced to the [4Fe-4SI1+ form for EPR spectro- scopic analysis, but only in the presence of 80% dimethyl sul- foxide. These studies, along with the voltammetric results shown in Figs. 6 and 7, strongly suggest that the [4Fe-4S11+ form of aconitase, because of its kinetically limited reduction, almost certainly does not participate as an active form of the enzyme. While recognizing that aconitase is not a redox/ electron transferring enzyme, our results are reminiscent of the "diode-like'' behavior observed for succinate dehydrogenase (23, 24). In this work the enzyme was bound to the pyrolytic graph- ite electrode which was able to catalyze the oxidation of succi- nate to fumarate very well, but the reduction process for the reverse pathway proceeded only to a very limited extent. They suggested that this effect may be of physiological importance in helping to prevent the Krebs cycle from back-pedalling. From the perspective of aconitase, it would be valuable to understand the molecular basis for the fact that the [4Fe-4S12+ form is the only naturally active form of the enzyme.
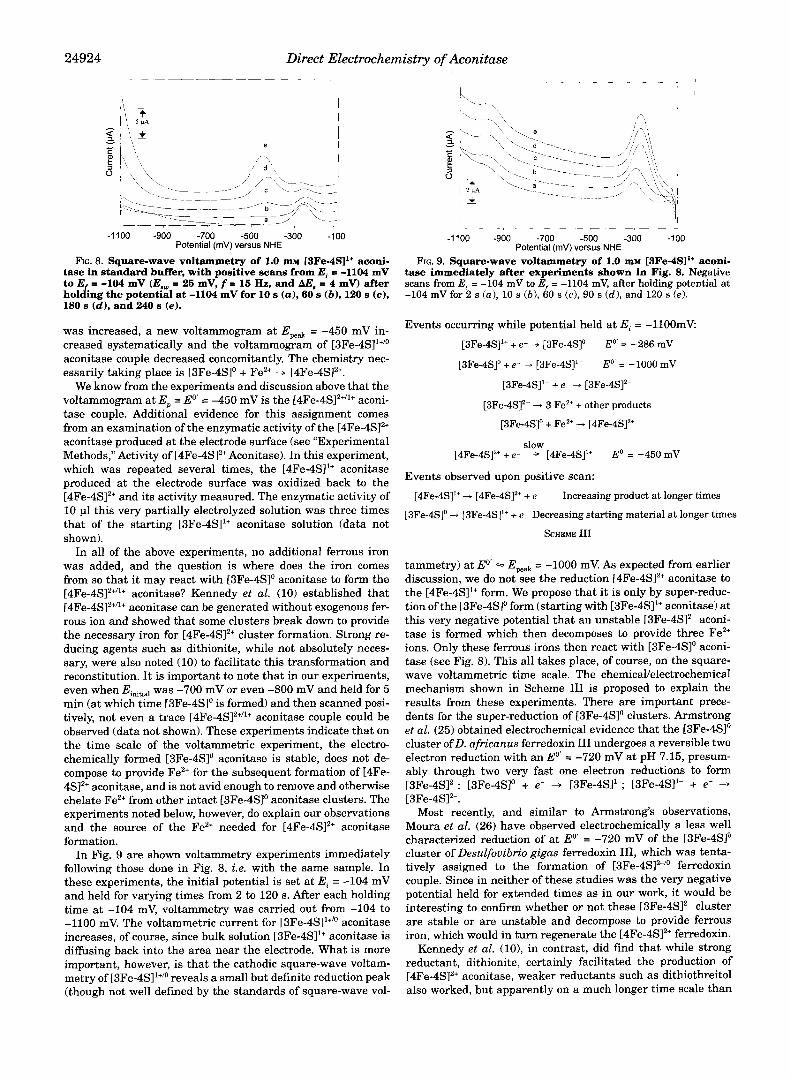
Reduction of [3Fe-4S11+ Aconitase at Very Low Potentials and Its Znterconversion to [4Fe-4SP Aconitase-In Fig. 8 is shown the voltammetry of [3Fe-4S11+ aconitase. In this series of ex- periments, the initial potential (E,) was set at -1100 mV and held for varying times from 10 to 240 s, and then after each time held, the potential was scanned in the positive direction. At the very short time of 10 s, the peak potential for the [3Fe- 4SI'"" is observed at -280 mV. As the time held at -1100 mV
24924 Direct Electrochemistry of Aconitase
It,, i", t ! ",, 5 PA < # ' , * - $, " e c e, i... ~ ?\, I
~ \, ~'\\ \,, -"".""/-y'c"----" 1::: .""_ _ _ ~ ~ -~~ ,'" /r'L~- r ' -==-"~- ----+7\~- -1
5 1 I \ '
"" I
/ \"/
,-~-" __.___l
-1100 -900 -700 -500 -300 -100 Potentlal (mV) versus NHE
FIG. 8. Square-wave voltammetry of 1.0 m~ [3Fe-4S11+ aconi- tase in standard buffer, with positive scans from Ei c -1104 mV to E = -104 mV (Eaw = 25 mV, f = 15 Hz, and AE, = 4 mv) after holding the potential at -1104 mV for 10 s (a), 60 s (b) , 120 s (c), 180 s (d) , and 240 s (e).
was increased, a new voltammogram at Epeak = -450 mV in- creased systematically and the voltammogram of [3Fe-4Sl'+/o aconitase couple decreased concomitantly. The chemistry nec- essarily taking place is [3Fe-4Slo + Fez+ + [4Fe-4S12+.
We know from the experiments and discussion above that the voltammogram at E, = Eo' = -450 mV is the [4Fe-4S]2+/1+ aconi- tase couple. Additional evidence for this assignment comes from an examination of the enzymatic activity of the [4Fe-4S12+ aconitase produced at the electrode surface (see "Experimental Methods," Activity of [4Fe-4SlZ+ Aconitase). In this experiment, which was repeated several times, the [4Fe-4SI1+ aconitase produced at the electrode surface was oxidized back to the [4Fe-4SlZ+ and its activity measured. The enzymatic activity of 10 pl this very partially electrolyzed solution was three times that of the starting [3Fe-4SI1+ aconitase solution (data not shown).
In all of the above experiments, no additional ferrous iron was added, and the question is where does the iron comes from so that it may react with [3Fe-4Slo aconitase to form the [4Fe-4Sl2+''+ aconitase? Kennedy et al. (10) established that [4Fe-4SI2+/'+ aconitase can be generated without exogenous fer- rous ion and showed that some clusters break down to provide the necessary iron for [4Fe-4SJz+ cluster formation. Strong re- ducing agents such as dithionite, while not absolutely neces- sary, were also noted (10) to facilitate this transformation and reconstitution. It is important to note that in our experiments, even when Einitid was -700 mV or even -800 mV and held for 5 min (at which time [3Fe-4SIo is formed) and then scanned posi- tively, not even a trace [4Fe-4S]2+"+ aconitase couple could be observed (data not shown). These experiments indicate that on the time scale of the voltammetric experiment, the electro- chemically formed [3Fe-4SI0 aconitase is stable, does not de- compose to provide Fez+ for the subsequent formation of [4Fe- 4S12+ aconitase, and is not avid enough to remove and otherwise chelate Fez+ from other intact [3Fe-4SIo aconitase clusters. The experiments noted below, however, do explain our observations and the source of the Fez+ needed for [4Fe-4SI2+ aconitase formation.
In Fig. 9 are shown voltammetry experiments immediately following those done in Fig. 8, i.e. with the same sample. In these experiments, the initial potential is set a t Ei = -104 mV and held for varying times from 2 to 120 s. After each holding time a t -104 mV, voltammetry was carried out from -104 to -1100 mV. The voltammetric current for [3Fe-4SI1+" aconitase increases, of course, since bulk solution [3Fe-4S11+ aconitase is diffusing back into the area near the electrode. What is more important, however, is that the cathodic square-wave voltam- metry of [3Fe-4SI1+" reveals a small but definite reduction peak (though not well defined by the standards of square-wave vol-
1 - - - " - - - " ,I
-1100 -900 -700 -500 -300 -100 Potentlal (mV) versus NHE
tase immediately after experiments shown in Fig. 8. Negative FIG. 9. Square-wave voltammetry of 1.0 m~ [3Fe-4SI1+ aconi-
scans from E, = -104 mV to E = -1104 mV, after holding potential at -104 mV for 2 s (a), 10 s ( b ) , 60 s (c), 90 s (d) , and 120 s (e ) .
Events occurring while potential held a t Ei = -1100mV:
[3Fe-4SI1+ + e- + [3Fe-4SIo Eo = -286 mV
[3Fe-4SJ0 + e- - [3Fe-4S]" EO' = -1000 mV
[3Fe-4S]" + e- - [3Fe-4SI2-
[3Fe-4SI2- + 3 Fez+ + other products [3Fe-4SIo + Fez+ - [4Fe-4SI2'
[4Fe-4SI2+ + e- + [4Fe-4SI1+ Eo = -450 mv slow
Events observed upon positive scan: [4Fe-4SI1+ -j [4Fe-4SI2+ + e ~ Increasing product at longer times
[3Fe-4SIo + [3Fe-4SI1+ + e - Decreasing starting material at longer times SCHEME I11
tammetry) at Eo' ICpeak = -1000 mV. As expected from earlier discussion, we do not see the reduction [4Fe-4S12+ aconitase to the [4Fe-4SI1+ form. We propose that it is only by super-reduc- tion of the [3Fe-4SIo form (starting with [3Fe-4SI1+ aconitase) at this very negative potential that an unstable [3Fe-4SI2- aconi- tase is formed which then decomposes to provide three Fez+ ions. Only these ferrous irons then react with [3Fe-4SIo aconi- tase (see Fig. 8). This all takes place, of course, on the square- wave voltammetric time scale. The chemicaUelectrochemica1 mechanism shown in Scheme I11 is proposed to explain the results from these experiments. There are important prece- dents for the super-reduction of [3Fe-4Slo clusters. Armstrong et al. (25) obtained electrochemical evidence that the [3Fe-4Slo cluster ofD. africanus ferredoxin I11 undergoes a reversible two electron reduction with an Eo' = -720 mV a t pH 7.15, presum- ably through two very fast one electron reductions to form [3Fe-4S12-: [3Fe-4SJ0 + e- + [3Fe-4SI1-; [3Fe-4S11- + e- 4 [3Fe-4S12-.
Most recently, and similar to Armstrong's observations, Moura et al. (26) have observed electrochemically a less well characterized reduction of a t Eo' = -720 mV of the [3Fe-4SI0 cluster of Desulfouibrio gigas ferredoxin 111, which was tenta- tively assigned to the formation of [3Fe-4S]2-/0 ferredoxin couple. Since in neither of these studies was the very negative potential held for extended times as in our work, it would be interesting to confirm whether or not these [3Fe-4S12- cluster are stable or are unstable and decompose to provide ferrous iron, which would in turn regenerate the [4Fe-4S12+ ferredoxin.
Kennedy et al. (lo), in contrast, did find that while strong reductant, dithionite, certainly facilitated the production of [4Fe-4SJ2+ aconitase, weaker reductants such as dithiothreitol also worked, but apparently on a much longer time scale than
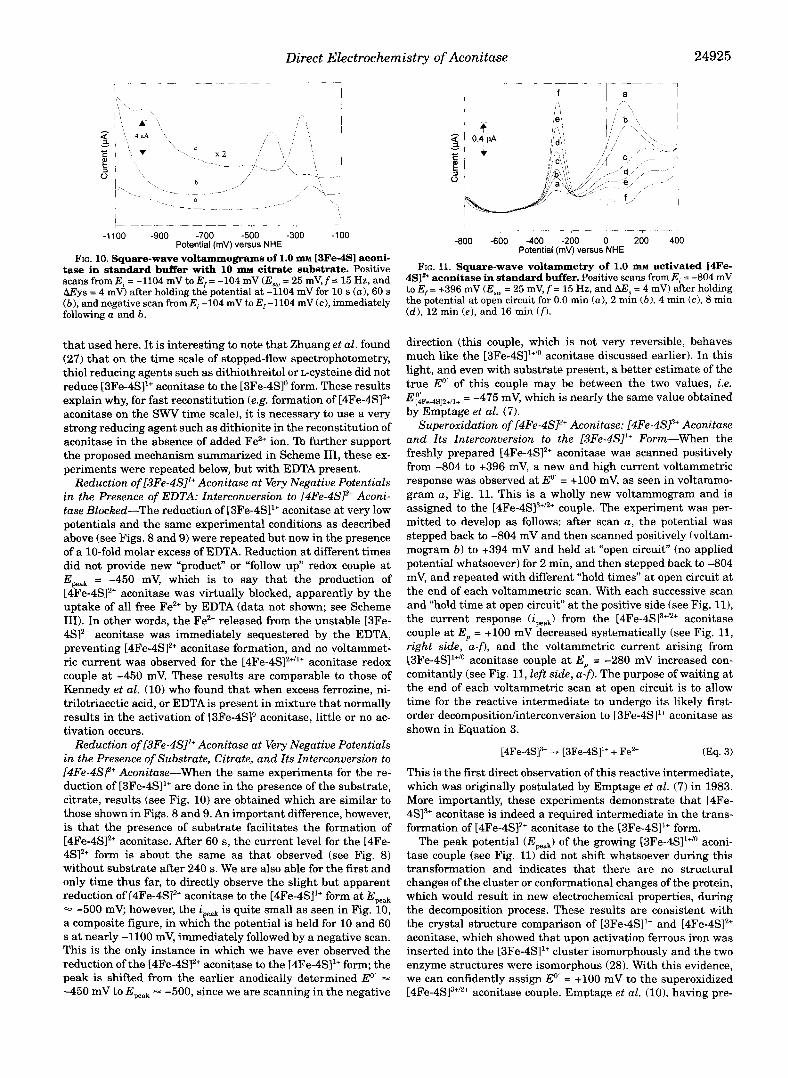
Direct Electrochemistry of Aconitase 24925
tase in standard buffer with 10 m~ citrate substrate. Positive FIG. 10. Square-wave voltammograms of 1.0 m~ 13Fe-4SI aconi-
scans from Ei = -1104 mV to E,. = -104 mV (Esw = 25 mV, f = 15 Hz, and AEys = 4 mV) after holding the potential at -1104 mV for 10 s (a), 60 s ( b ) , and negative scan from E, -104 mV to E,-1104 mV (c), immediately following a and b.
that used here. It is interesting to note that Zhuang et al. found (27) that on the time scale of stopped-flow spectrophotometry, thiol reducing agents such as dithiothreitol or L-cysteine did not reduce [3Fe-4SI1+ aconitase to the [3Fe-4Slo form. These results explain why, for fast reconstitution (e.g. formation of [4Fe-4S12+ aconitase on the SWV time scale), it is necessary to use a very strong reducing agent such as dithionite in the reconstitution of aconitase in the absence of added Fez+ ion. To further support the proposed mechanism summarized in Scheme 111, these ex- periments were repeated below, but with EDTA present.
Reduction of [3Fe-4SI1+ Aconitase at Very Negative Potentials in the Presence of EDTA: Znterconversion to (4Fe-4SP Aconi- tase Blocked-The reduction of [3Fe-4SI1+ aconitase at very low potentials and the same experimental conditions as described above (see Figs. 8 and 9) were repeated but now in the presence of a 10-fold molar excess of EDTA. Reduction at different times did not provide new “product” or “follow up” redox couple at E,,, = -450 mV, which is to say that the production of [4Fe-4S12+ aconitase was virtually blocked, apparently by the uptake of all free Fez+ by EDTA (data not shown; see Scheme 111). In other words, the Fez+ released from the unstable [3Fe- 4S12- aconitase was immediately sequestered by the EDTA, preventing [4Fe-4S12+ aconitase formation, and no voltammet- ric current was observed for the [4Fe-4S12+”+ aconitase redox couple at -450 mV. These results are comparable to those of Kennedy et al. (10) who found that when excess ferrozine, ni- trilotriacetic acid, or EDTA is present in mixture that normally results in the activation of [3Fe-4Slo aconitase, little or no ac- tivation occurs.
Reduction of [3Fe-4SI1+ Aconitase at Very Negative Potentials in the Presence of Substrate, Citrate, and Its Znterconversion to [4Fe-4SF Aconitase-When the same experiments for the re- duction of [3Fe-4SI1+ are done in the presence of the substrate, citrate, results (see Fig. 10) are obtained which are similar to those shown in Figs. 8 and 9. An important difference, however, is that the presence of substrate facilitates the formation of [4Fe-4S12+ aconitase. After 60 s, the current level for the [4Fe- 4S12+ form is about the same as that observed (see Fig. 8) without substrate after 240 s. We are also able for the first and only time thus far, to directly observe the slight but apparent reduction of [4Fe-4S12+ aconitase to the [4Fe-4S11+ form at Epeak - -500 mV; however, the ipeak is quite small as seen in Fig. 10, a composite figure, in which the potential is held for 10 and 60 s at nearly -1100 mV, immediately followed by a negative scan. This is the only instance in which we have ever observed the reduction of the [4Fe-4SlZ+ aconitase to the [4Fe-4SI1+ form; the peak is shifted from the earlier anodically determined Eo’ = -450 mV to Epeak 4 -500, since we are scanning in the negative
~ ~ 7 - ~- 1- ~ ’ ~ ~~~ 7~ ~ ~.
-800 -600 -400 -200 0 200 400 Potential (mv) versus NHE
4SIa+ aconitase in standard buffer. Positive scans from E, = -804 mV FIG. 11. Square-wave voltammetry of 1.0 m~ activated [4Fe-
to E,. = +396 mV (Esw = 25 mV, f = 15 Hz, and LW, = 4 mV) after holding the potential at open circuit for 0.0 min (a ) , 2 min ( b ) , 4 min (c), 8 min (d) , 12 min ( e ) , and 16 min Cf?.
direction (this couple, which is not very reversible, behaves much like the [3Fe-4SI1”’ aconitase discussed earlier). In this light, and even with substrate present, a better estimate of the true Eo’ of this couple may be between the two values, i.e. E~~Fe.4S12+/1+ = -475 mV, which is nearly the same value obtained by Emptage et al. (7).
Superoxidation of [4Fe-4SP Aconitase: [4Fe-4SP Aconitase and Its Znterconversion to the [3Fe-4SI1+ Form-When the freshly prepared [4Fe-4S12+ aconitase was scanned positively from -804 to +396 mV, a new and high current voltammetric response was observed at Eo’ = +lo0 mV, as seen in voltammo- gram a , Fig. 11. This is a wholly new voltammogram and is assigned to the [4Fe-4S]3+’2+ couple. The experiment was per- mitted to develop as follows: after scan a , the potential was stepped back to -804 mV and then scanned positively (voltam- mogram b) to +394 mV and held at “open circuit” (no applied potential whatsoever) for 2 min, and then stepped back to -804 mV, and repeated with different “hold times” at open circuit at the end of each voltammetric scan. With each successive scan and “hold time at open circuit” at the positive side (see Fig. l l ) , the current response ( i W J from the [4Fe-4SI3+”+ aconitase couple at E,, = +lo0 mV decreased systematically (see Fig. 11, right side, a-f), and the voltammetric current arising from [3Fe-4Sl1+’’ aconitase couple at E, = -280 mV increased con- comitantly (see Fig. 11, left side, a-f). The purpose of waiting at the end of each voltammetric scan at open circuit is to allow time for the reactive intermediate to undergo its likely first- order decompositiodinterconversion to [3Fe-4S11+ aconitase as shown in Equation 3.
[4Fe-4SI3+ -, [3Fe-4Sl1’ + Fez+ (Eq. 3)
This is the first direct observation of this reactive intermediate, which was originally postulated by Emptage et al. (7) in 1983. More importantly, these experiments demonstrate that [4Fe- 4S13+ aconitase is indeed a required intermediate in the trans- formation of [4Fe-4S12+ aconitase to the [3Fe-4SI1+ form.
The peak potential (E,,,,) of the growing [3Fe-4Sl‘+/o aconi- tase couple (see Fig. 11) did not shift whatsoever during this transformation and indicates that there are no structural changes of the cluster or conformational changes of the protein, which would result in new electrochemical properties, during the decomposition process. These results are consistent with the crystal structure comparison of [3Fe-4SI1+ and [4Fe-4SI2+ aconitase, which showed that upon activation ferrous iron was inserted into the [3Fe-4S11+ cluster isomorphously and the two enzyme structures were isomorphous (28). With this evidence, we can confidently assign Eo’ = +lo0 mV to the superoxidized [4Fe-4S13+’2+ aconitase couple. Emptage et al. (lo), having pre-

24926 Direct EZectrochemistry of Aconitase
dicted the likely existence of this intermediate and using the relatively strong oxidizing agent, ferricyanide (Eo = +400 mV) as the oxidant, made every effort to create this species in bulk and obtain its EPR spectrum, but without success. Having now determined that E&+4sp+12+ = +lo0 mV, it is likely that the bulk oxidation of the [4Fe-4SI2+ aconitase (to form the [4Fe-4SI3+ aconitase) may be more successful with weaker oxidizing agents whose E'" values are in the vicinity of +I50 mV. Such oxidizing agents are only slightly more positive than the Eo' for the [4Fe-4S]3+'2+ aconitase couple and are thus less aggressive oxidizing agents than ferricyanide. We expect the observed EPR spectrum for this superoxidized, paramagnetic [4Fe-4SI3+ form to be similar to that of high potential iron-sulfur protein.
During the superoxidation of [4Fe-4SI2+, the electrochemis- try of the [4Fe-4SI3+ form of aconitase becomes more complex after its initial formation (see Fig. 11, right side, c-f). Initially, as [4Fe-4S13+ aconitase is formed, a single voltammogram with E'' = +lo0 mV is observed. Upon successive oxidations and more importantly over time, the superoxidized form (which is all this time at or near the electrode surface) apparently inter- converts into two, and probably very similar, reactive interme- diates as evidenced by two new and cIearly observable voltam- mograms with E" = 0 mV and E" +225 mV, respectively. These results indicate that the superoxidized form is proceed- ing through two electrochemically distinguishable intermedi- ates, both of which interconvert to the same product, [3Fe-4SI1+ aconitase. While we do not at present understand this phenom- enon, it is possible that with the additional positive charge density on Fe, its coordination by both water and hydroxide can be voltammetrically differentiated.
Superoxidation of the [4Fe-4@+ Form ofAconitase with Sub- strate Present-With the substrate, citrate, in 10-fold molar excess and bound to the [4Fe-4S12+ aconitase, it was very diffi- cult to oxidize or even detect the [4Fe-4S13+ aconitase at the pyrolytic graphite edge electrode (data not shown), and even when holding the potential at +393 mV for extended periods of time the voltammetric peak current for [3Fe-4S11+ aconitase product was also quite small. These results suggest that with the substrate bound the cluster is much less accessible to oxi- dation. It is likely that binding the negatively charged sub- strate, citrate (which has three negatively charged carboxyl anions at pH 8) at the active site of the enzyme, blocks the favored interactions between the aconitase cluster and the elec- trode surface. This conclusion was supported by experiments that were carried out with the electrochemical promoter neo- mycin (29); this and other promoters are redox inactive but facilitate interaction of the redox species with the electrode surface. Under these conditions (1 mM promoter), an increased voltammetric response for the [4Fe-4SI3+'+ aconitase couple was observed, and the voltammetric experimental results with the substrate-bound aconitase, shown in Fig. 12, were very similar to those without substrate (see Fig. 11). There are, however, four important differences: 1) all reactions were slowed substantially and longer oxidation times at +394 mV were required to produce the [3Fe-4SI1+ aconitase product, 2) the [4Fe-4SI3+ aconitase interconverts to two new reactive in- termediates as before: Eo' = 0 mV and Eo = +225 mV, 3) the peak currents (iJ for the [3Fe-4SI1+'* and [4Fe-4S13+/2+ aconitase couples are reduced by more than 50% compared to the experi- ments without substrates, and 4) the peak potentials (E,) for all the redox couples, including the "split" redox couples, are virtually identical to those observed when no substrate was present.
These experiments indicate that after substrate binds to [4Fe-4SI2+ aconitase, it is more difficult to oxidize the [4Fe-4S12+ aconitase to the superoxidized form, as evidenced by lower
t " - " - - - " " - - ~
-800 -600 400 -200 0 200 400 Potential (mv) versus NHE
FIG. 12. Square-wave voltammetry of 1.0 IUM activated L4Fe- 4S]'+ aconitase in standard buffer that is 10 IIIM in citrate and 1 LUM in neomycin promoter. Anodic scan from E: = -804 mV to E - +396 mV, after holding at +396 mV for 0 min (a ) , 4 min ( b ) , 8 min fci 12 min (dl , and 20 min (e).
observed voltammetric peak currents. However, there is no shift of the peak potential. I t is important to remember that to carry out the oxidation of [4Fe-4SI2+ aconitase with substrate the use of the electrochemical promoter, neomycin, was re- quired. The lower peak currents and the requirement for neo- mycin suggest that the reduced peak currents arises primarily from steric blocking of the active site cluster. In contrast, we noted earlier that the super-reduction of [3Fe-4S11+ aconitase (with subsequent [4Fe-4S12+ aconitase formation) is facilitated by the presence of substrate (see Figs. 8-10).
It is also possible that citrate coordination to Fe, provides thermodynamic stabilization of either [4Fe-4S13+ or [4Fe-4S12+ aconitase, or both. The fact that these peak potentials are not shifted indicates that substrate must stabilize both [4Fe-4S13+ and [4Fe-4S12+ aconitase equally, i.e. Fe,-ligand interaction is of equal strength both for redox states. If one redox state was stabilized more than the other, then the observed voltammetric peak potential for the [4Fe-4SI3+"+ aconitase couple(s) would be shifted to a more positive or negative potential, depending on which was stabilized more; this was not observed. From a somewhat different perspective, since the [4Fe-4S]3+n+ aconi- tase redox couple and its associated interconversion to two new species are all at the same potentials as those observed without substrate, then it follows that the binding of substrate to ac- onitase does not change the manner by which [4Fe-4SI3+ aconi- tase is formed or alter the mechanism of its subsequent inter- conversion to the [3Fe-4SI1+ form. WhiIe it appears that substrate blocks access to the active site cluster, it remains to be demonstrated whether or not the rate of interconversion of [4Fe-4SI3+ aconitase to the [3Fe-4SI1+ in the presence of sub- strate is really slower or not. Kennedy et al. (10) have shown that the substrate-bound active [4Fe-4SP aconitase is much more stable to oxygen and utilized this behavior to isolate 90% active aconitase aerobically.
CONCLUSIONS
In conclusion, and without summarizing all of the results of this study, direct square-wave voltammetry at the pyrolytic graphite edge electrode of pig heart aconitase has proven to be a powerful methodology, and one which, in addition to clarifjr- ing the known redox coupleshnterconversions of aconitase, has revealed clearly for the first time two reactive intermediates. The reactive intermediate, [4Fe-4S13+ aconitase, was directly observed, as was its subsequent interconversion to the [3Fe- 4SI1+ form. This intermediate is required for the oxidative de- activation of [4Fe-4S12+ aconitase to the [3Fe-4S11+ form. The [4Fe-4SI3+'+ aconitase couple has additional complexity by vir- tue of splitting into two new redox couples, i.e. two new reactive

Direct Electrochemistry of Aconitase 24927
intermediates. Evidence for the formation of super-reduced [3Fe-4SI2- aconitase, its decomposition, and the subsequent for- mation of [4Fe-4SI2+ aconitase was obtained by following prod- uct formation as a function of time reduced at very low poten- tials. Experiments are now under way, using the methods of modern electroanalytical chemistry and bioelectrochemistry, to determine the homogenous rate constants for all of the inter- conversions discussed in this paper, in addition to further study of both superoxidized and super-reduced forms of aconitase. Lastly, it is important to recognize that this is an enzyme with a molecular mass of 83 kDa, and thus, a seemingly unlikely candidate for reversible and quasi-reversible voltammetry; whether aconitase is a special case remains to be seen. It seems clear that modern bioelectrochemistry really can serve us in understanding not only simple redox and metalloproteins, such as ferredoxin, but also complex and high molecular weight metalloenzymes such as aconitase.
Acknowledgments-We thank Professors Kristene Surerus, Univer- sity of Wisconsin-Milwaukee, Michael Ryan, Marquette University, and Janet Osteryoung, North Carolina State University, for their helpful comments on this article.
REFERENCES 1. Kennedy, M. C., and Stout, C. D. (1992) in Aduances in Inorganic Chemistry
2. Emptage, M. H., Kent, T. A,, Kennedy, M. C., Beinert, H., and Munck, E. (1983)
3. Lauble, H., Kennedy, M. C., Beinert, H., and Stout, C. D. (1992) Biochemistry
4. Werst, M. M., Kennedy, M. C., Beinert, H., and Hoffman, B. M. (1990) Bio-
6. Emptage, M. H. (1988) in Metal Clusters in Proteins (Que, L., Jr., ed) Vol. 372, 5. Beinert, H., and Kennedy, C. (1989) Eur. J. Biochem. 186 ,515
pp. 343471, ACS Series Symposium 372, American Chemical Society, Wash., D. C.
(Cammack, R., ed) p. 323439, Academic Press, Inc., New York
Proc. Natl. Acad. Sci. U. S. A. 80, 46744678
31,2735-2748
chemistry 29,10526-10532
7. Emptage, M. H., Dreyer, J.-L., Kennedy, M. C., and Beinert, H. (1983) J. Biol. Chem. 258,11106-11111
8. Beinert, H., Emptage, M. H., Dreyer, J. -L., Scott, R. A,, Hahn, J. E., Hodgson,
9. Beinert, H., andThomson,A. J. (1983)Archiu. Biochem. Biophys. 222,333461 K. O., and Thomson, A. J. (1983) Proc. Natl. Acad. Sci. U. S. A. 80,393-396
10. Kennedy, M. C., Emptage, M. H., Dreyer, J.-L., and Beinert, H. (1983) J. Biol.
11. Osteryoung, J. G., and ODea, J. J. (1986) in Electroanalytical Chemistry: a Chem. 268,11098-11105
Series ofAduances (Bard, A. J., ed) Vol. 14, pp. 209-308, Marcel Dekker, Inc., New York
12. Kennedy, M. C . , Kent, T. A,, Emptage, M., Merkle, H., Beinert, H., and Munck,
13. Henson, C. P., and Cleland, W. W. (1967) J. Biol. Chem. 242,3833-3838 14. Osteryoung, J. G., and Osteryoung, R. A. (1985) Anal. Chem. 57, 101-110 15. Smith, E. T., Bennett, D. W., and Feinberg, B. A. (1991)Anal Chim. Acta 251,
E. (1984) J. Biol. Chem. 259; 14463-14471
27-33 16. George, S. J., Armstrong, F. A,, Hatchikian, E. C., and Thomson, A. J. (1989)
Biochem. J . 264, 265-273 17. George, S. J., Armstrong, F. A,, Hatchikian, E. C., and Thomson, A. J. (1989)
Biochem. J. 264,275-284 18. Iismaa, S. E., Vaquez, A. E., Jensen, G. M., Stephens, P. J., Butt, J. N.,
Armstrong, F. A,, and Burgess, B. K. (1991) J. Biol. Chem. 266, 21563- 21571
19. Yoch, D. C., and Carithers, R. P. (1978) J. Bacteriol. 136,8224324 20. Armstrong, F. A,, Bond, A. M., Hill, H. A. O., Oliver, B. N., and Psalti, I. S. M.
21. Kennedy, C., Rauner, R., and Gawron, 0. (1972) Biochem. Biophys. Res. Com-
23. Sucheta, A,, Ackrell, B. A. C., Cochran, B., and Armstrong, F. A. (1992) Nature 22. Cammack, R. (1973) Biochem. Biophys. Res. Cornnun. 54, 548-554
24. Cammack, R. (1992) Nature 356, 288 25. Armstrong, F. A., Butt, J. N., George, S. J., Hatchikian, E. C., and Thomson, A.
J. (1989) FEBS Letts. 269, 15-18 26. Moreno, C., Macedo, A. L., Moura, I., LeGall, J., and Moura, J. J. G. (1994) J.
Inorg. Biochem. 53,219-234 27. Zhuang, H. -Y., Faridoon, K. Y., and Sykes, A. G. (1992) Inorg. Chem. Acta 201,
239-242 28. Robbins, A. H., and Stout, C. D. (1989) Proc. Natl. Acad. Sci. U. S. A. 86,
3639-3643 29. Armstrong, F. A. (1990) in Bioinorganic Chemistry, Structure and Bonding
(Armstrong, F. A,, Banci, L., Bertini, I., Cooper, S. R., Luchinat, C., Mingos,
Verlag, New York D. M. P., Rawle, S. C., and Zhenyang, L., eds) Vol. 72, pp. 137-221, Springer-
(1989) J. Am. Chem. Soc. 110,9165-9189
mun. 47, 740-745
3643,361462










![A Square Wave Voltammetry Study on the …downloads.hindawi.com/journals/jchem/2019/8706061.pdfconcentrate [19, 20], 14.3 and 122.8μmol TE per gram of lemon/grape juice concentrate](https://img.pdfslide.us/doc/110x75/5e5f7033c6c38a52df6a7f3a/a-square-wave-voltammetry-study-on-the-concentrate-19-20-143-and-1228mol.jpg)


















