Embed Size (px)
Citation preview

ORF36 Protein Kinase of Kaposi’s Sarcoma Herpesvirus (KSHV) Activates the c-Jun N-
terminal Kinase (JNK) Signaling Pathway
M. Sabry Hamza1, Richard A. Reyes1, Yoshihiro Izumiya2, Ronald Wisdom3, Hsing-Jien
Kung2 and Paul A. Luciw1,4
1Center for Comparative Medicine, 2Department of Biological Chemistry and Cancer Center,
3Division of Hematology and Oncology, and 4Department of Pathology, University of California,
Davis, CA 95616
Running title: KSHV ORF36 binds and activates JNK
Correspondence footnote:
Paul A. Luciw
Center for Comparative Medicine, University of California
Davis, CA 95616
Telephone: 530-752-3430
1
JBC Papers in Press. Published on July 5, 2004 as Manuscript M400964200
Copyright 2004 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

E-mail: [email protected]
Fax: 530-752-7914
2
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

ABSTRACT
Alpha-, beta- and gamma-herpesviruses encode putative viral protein kinases. The HSV
UL13, VZV ORF 47, and EBV BGLF4 genes all show protein kinase domains in their protein
sequences. Mutational analysis of these herpesviruses demonstrated that the viral kinase is
important for optimal virus growth. Previous studies showed that ORF36 of Kaposi’s sarcoma
herpesvirus (KSHV) has protein kinase activity and is autophosphorylated on serine. The gene
for ORF36 is expressed during lytic growth of the virus and has been classified a late gene.
Inspection of the ORF36 sequence indicated potential motifs that could be involved in activation
of cellular transcription factors. To analyze the function of ORF36, the cDNA for this viral gene
was tagged with the FLAG epitope and inserted into an expression vector for mammalian cells.
Transfection experiments, in 293T and SLK cells demonstrated that expression of ORF36
resulted in phosphorylation of the c-Jun N-terminal kinase. Autophosphorylation of ORF36 is
important for JNK activation because a mutation in the predicted catalytic domain of ORF36
blocked its ability to phosphorylate JNK. Western blot analysis, using phospho-specific
antibodies, revealed that mitogen activated kinases, MKK4 and MKK7, were phosphorylated by
ORF36, but not by the kinase-negative mutant. Binding experiments in transfected cells also
demonstrated that both the wild type and kinase-negative mutant of ORF36 form a complex with
JNK, MKK4 and MKK7. In addition, using a tetracycline inducible Rta BCBL-1 cell line
(TREx BCBL1-Rta), JNK was phosphorylated during lytic replication, and inhibition of JNK
3
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

activation blocked late viral gene expression but not early viral gene expression. In summary,
these studies demonstrate that KSHV ORF36 activates the JNK pathway; thus this cell signaling
pathway may function in the KSHV life cycle by regulating viral and/or cellular transcription.
4
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

INTRODUCTION
Based on molecular and biological properties, the herpesviridae family has been
subdivided into the alpha-herpesvirus (herpes simplex virus, varicella-zoster virus), beta-
herpesvirus (human cytomegalovirus), and gamma-herpesvirus (Epstein-Barr virus, EBV;
Kaposi’s sarcoma associated herpesvirus, KSHV) subfamilies (1). These double-stranded DNA
viruses can remain latent and persist as an episome, expressing a limited number of viral genes,
and thereby establish a lifelong infection in the natural host. Immunosupression or stress can lead
to reactivation of latent virus to produce pathogenic manifestations; examples include, Burkitt’s
lymphoma, nasopharyngeal carcinoma and Hodgkin’s disease associated with EBV (reviewed in
(2)), and Kaposi’s sarcoma, primary effusion lymphomas and multicentric Castleman’s disease
associated with KSHV (reviewed in (3,4)). Reactivation of these latent herpesviruses leads to
sequentially regulated expression of viral genes that play an essential role in the replication and
assembly of infectious virions (2-4) .
During the lytic cycle of infection, herpesviruses express genes which are predicted to
encode protein kinases (reviewed in (5)). These protein kinases are conserved among the
herpesviridae family, and analysis of the amino acid sequence reveals motifs that are shared by
mammalian serine/threonine protein kinases. Located within the catalytic region of these viral
proteins are eleven conserved subdomains that are common to cellular protein kinases. Recent
studies revealed that mutation of a conserved lysine residue within subdomain II abolishes
phosphorylation; this finding shows that this region is necessary for the catalytic activity of these
5
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

protein kinases (6,7). The protein kinases of herpesviruses appear to mimic the function of
cellular protein kinases, including cdc2 and cellular translational factor, EF-1delta, by
phosphorylating the same amino acid residues on their cognate protein targets (5,8).
Furthermore, the viral protein kinases are associated with the virion and may promote
dissociation and entry of the virus particle by phosphorylating the viral tegument proteins (9-
12).
The importance of herpesvirus protein kinases for viral replication and disease has been
investigated. Kinase-null mutants generated in alpha-, beta- and gamma- herpesviruses
demonstrate decreased replication in tissue culture (13-15). Decreased virulence of HSV and
VZV kinase-null mutants has been shown in the mouse model (16-18). However, the protein
kinase of VZV appears to be dispensable for the establishment of latency in rodent models
(14,18).
Studies on cells productively infected with alpha-, beta-, and gamma-herpesviruses
have demonstrated that various members of the stress kinase pathway are activated. HSV
activates JNK, p38, and Ap1 (19-21); CMV activates p38 and JNK(22); and VZV activates
AP1, Jun, Fos, and ATF2 (23). For EBV, the latent membrane protein, LMP2A, regulates c-Jun
through an extracellular signal-regulated kinase (24), and the immediate-early proteins of EBV,
BZLF1 and BRLF1, activate ATF2, p38 and JNK (25). Recent studies have demonstrated the
K15 membrane protein of KSHV activates the mitogen-activated protein kinase and NF-kappaB
pathways (26). However, the role of the viral protein kinase in activating the stress kinase
6
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

pathway for these herpesviruses remains to be determined.
In KSHV, ORF36 encodes a serine protein kinase, which is localized in the nucleus of
infected cells. In vitro kinase assays indicated that this viral protein was autophosphorylated, and
the lysine residue in the kinase subdomain II was essential for protein kinase activity (27).
Conservation of the viral protein kinases amongst the herpesviridae family implies that these
enzymes are indispensable for herpesvirus survival. Based on transcriptional kinetics and
sensitivity to DNA replication inhibitors, ORF36 was classified as a late gene (27,28).
Reactivation of KSHV in BCBL-1 cells (B-lymphocytes latently infected with KSHV) induced
transcription of the viral protein kinase (27). Understanding the cellular events that are regulated
by the KSHV protein kinase may provide a basis for defining molecular mechanisms that control
viral gene expression. Our studies demonstrate the ability of KSHV ORF36 to activate the JNK
pathway and the cellular transcription factor, c-Jun, and inhibtion of JNK activation resulted in
the inhibition of late KSHV viral gene expression.
7
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

EXPERIMENTAL PROCEDURES
Antibodies- Phospho-specific antibodies for SEK1/MKK4 (Thr 261), MKK7 (Ser 271/Thr 275),
SAPK/JNK (Thr 183/Tyr 185), ATF-2 (Thr 71), c-Jun (Ser 63), CREB (Ser 133), Elk-1 (Ser
383), and p38 MAP Kinase (Thr 180/Tyr 182) were obtained from Cell Signaling Technology
(Beverly, MA). Rabbit anti-phosphothreonine and phosphoserine were obtained from Zymed
Laboratories (South San Francisco, CA). MEK-4 (C-20), MEK-7 (H-160), JNK1 (FL), C-Jun
(N), c-Myc, and tubulin (H-300) were obtained from Santa Cruz Biotechnology (Santa Cruz,
CA). Anti-FLAG M2 was obtained from Stratagene (La Jolla, CA). Antibodies to KSHV K8.1
A/B were obtained from Advanced Biotechnologies Incorporated (Columbia, Maryland).
Cloning and Plasmids- The pND vector for expression in mammalian cells (generous gift from
Dr. G. Rhodes, University of California, Davis, CA) was used in the cloning of KSHV ORF 36.
cDNA was synthesized using RNA extracted from BCBL-1 (B-lymphocytes latently infected
with KSHV) cells after 48 hrs of TPA treatment. KSHV ORF36 was amplified by PCR using
appropriate oligonucleotide primers,
5’AAAAGATCTGCCACCATGGATTACAAGGATGACGACGATAAGCGCTGGAAGA
GAATGGAGAGGAG-3’ and 5’-AAAGCGGCCGCTCAGAAAACAAGTCCGCGGGT-3’,
and cloned into pND. The resulting construct was designated pND-nFLAG-KSHV-ORF36.
The forward primer generated a FLAG sequence at the N-terminus. QuickChange XL Site-
8
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Directed Mutagenesis Kit (Stratagene, La Jolla, CA) was used in the construction of pND-
nFLAG-KSHV-ORF36(K108Q). Briefly, pND-nFLAG-KSHV-ORF36 was amplified by
PCR using oligonucleotide primers,
5’-TCCGAGGATCTGTGTGTTGCAGCAGTTTGATAGCCGCCGG-3’ and
5’-CCGGCGGCTATCAAACTTGCTGCACACACAGATCCTCGGA-3’; to generate a
kinase-negative mutant of KSHV ORF36 (lysine at position 108 mutated to glutamine).
Cells and transfections- 293 T and SLK cells (human endothelial cell origin, generous gift from
Drs. Sophie Leventon-Kriss, Tel-Aviv, Israel and Jay A. Levy, UCSF,CA) were cultured in
Dulbecco’s modified essential medium (DMEM) supplemented with 10% fetal calf serum, and
cultures were maintained at 5% CO2 at 370C. Transient expression of KSHV-ORF36 and
KSHV-ORF36 (K108Q) was performed by transfecting pND-nFLAG-KSHV-ORF36 or
pND-nFLAG-KSHV-ORF36 (K108Q) using Fugene-6 transfection reagent according to the
manufactures protocol (Boehringer Mannheim). Cells were used 24hrs after transfection.
Anisomycin treated cells served as positive controls for JNK activation. TREx BCBL1-Rta cell
line (generous gift from Dr. Jae U Jung) were cultured in RPMI supplemented with 20% fetal
calf serum, 250ug/ml blasticidin S HCL (Invitrogen) and 200ug/ml hygromycin B (invitrogen).
KSHV lytic replication was induced in this cell line with1ug/ml of doxycycline (Dox; BD
Clonetech, Palo Alto, CA). Cells were harvested 24hrs post induction.
9
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

JNK inhibition assays- TREx BCBL1-Rta cells were treated with 50nM of JNK inhibitor II
(SP600125) or 20uM of JNK inhibitor negative control (EMD Biosciences, San Diego, CA) in
the presence of 1ug of Dox/ml for 24 hours. Cells were harvested for western blot.
Preparation of cell extracts- For detection of phosphorylated cellular proteins or viral proteins,
cells were treated with SDS-sample buffer (62.5 mM Tris-HCl, 2% SDS, 10% glycerol, 50mM
DTT, 0.01%w/v bromophenol blue), sonicated for 15 seconds, heated for 5 minutes at 950C, and
cooled on ice. For immunoprecipation, cells were treated with cell lysis buffer (20mM Tris-
HCL, 150mM NaCl, 1mM EDTA, 1mM EGTA,1% Triton X-100, 2.5 mM sodium
pyrophosphate, 1mM glycerolphospshate, 1mM Na3VO4, 1ug/ml leupeptin) for 30 minutes at
40C and then centrifuged to remove cell debris.
Western blot analysis- For detection of phospho proteins, cells extracts were electrophoresed on
a 12% SDS-PAGE gel and transferred onto PVDF membrane. Blocking buffer (1X TBS, 0.1%
Tween 20, and 5%w/v nonfat dry milk) was added to the membrane for 30 minutes at room
temperature. Phospho-specific antibodies were diluted in primary antibody dilution buffer
(1XTBS, 0.1% Tween 20, 5% w/v BSA) and membranes were incubated overnight at 40C.
Membranes were washed 3 times with wash buffer (1X TBS, 0.1% Tween 20) and incubated
with rabbit IgG-HRP in blocking buffer for 30 minutes at room temperature. Phospho-proteins
10
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

were detected using SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL).
For detection of total JNK, MKK4 and MKK7, K8.1, K-bZIP, c-Myc (Rta), respective primary
antibodies were diluted in blocking buffer for 30 minutes at room temperature, and then detected
with rabbit or mouse IgG-HRP and SuperSignal Substrate.
11
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

RESULTS
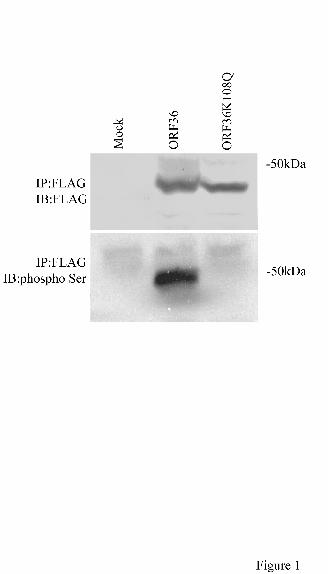
ORF36 viral protein kinase is phosphorylated on serine
The conserved lysine residue in the kinase subdomain II was shown to be essential for
kinase activity and autophosphorylation of ORF36 (27,29). To demonstrate that ORF36 was
phosphorylated on serine and that the lysine at position 108 was important for its
phosphorylation, we transfected 293T cells with plasmids expressing KSHV ORF36 or the
mutant K108Q. Western blot analysis with antibodies that recognized phosphoserine,
phosphothreonine or phosphotyrosine, was used to analyze immunoprecipated FLAG-tagged
ORF36 and the mutant K108Q proteins in transfected cells. A band that had the same molecular
weight as FLAG-tagged ORF36 was detected with phosphoserine antibody (Fig.1), but not with
phosphothreonine or phosphotyrosine antibody (data not shown). The K108Q mutant protein of
ORF36 was not detected with any of these three phospho-specific antibodies (Fig. 1 and data not
shown). This finding demonstrates that the lysine at position 108 is important for the
autophosphorylation of ORF36.
ORF36 activates c-Jun N-terminal kinase
To evaluate the ability of KSHV ORF36 to phosphorylate the MAP kinase pathway, SLK
cells were transfected with plasmids expressing ORF36 or the kinase negative mutant, K108Q.
Twenty four hours post-transfection, cells were lysed to prepare extracts for western blot
analysis. Cells transfected with the empty pND vector served as negative controls, and cells
12
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

treated with anisomycin were positive controls for cell activation. As shown in Fig. 2, ORF 36
activated the p54 isoform of JNK, which was phosphorylated on threonine 183 and tyrosine 185;
the phospho-specific antibody for JNK recognizes JNK only when phosphorylated on threonine
183 and tyrosine 185. The p46 isoform of JNK was not phosphorylated by ORF36. The K108Q
mutant was unable to phosphorylate either the p46 or p54 isoform of JNK. To determine if
ORF36 exclusively activated the SAPK/JNK pathway, the p38 pathway and its downstream
targets, ATF2, CREB and ELK-1, p44/42, MKK3/MKK6 were analyzed. Western blot analysis
demonstrated that these target proteins were not phosphorylated by either the wild type ORF36
or kinase-negative mutant K108Q (Fig. 2).
ORF36 activates JNK via MKK4 and MKK7
MKK4 and MKK7 function as non-redundant activators of JNK in vivo. Synergistic
activation of MKK4 and MKK7 is required for optimal JNK activation. Maximal enzyme
activity of JNK is achieved through phosphorylation of threonine 183 and tyrosine 185, with
MKK4 having a preference for phosphorylating the tyrosine residue, and MKK7 for the
threonine residue (30-33). To determine if the viral kinase utilizes MKK4 and MKK7 in the
activation of JNK, lysates from transfected cells expressing ORF36 or the mutant K108Q were
analyzed by western blot with appropriate antibodies. Fig. 3. demonstrates that both MKK4 and
MKK7 were activated by ORF36, but not by the kinase-negative mutant K108Q. These
observations reveal that the phosphorylation of JNK by ORF36 was dependent on the activation
13
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

of both MKK4 and MKK7, and autophosphorylation of ORF36 at serine was important for this
activity.
Phosphorylation of c-Jun by ORF36 via JNK
Activated JNK regulates transcription by phosphorylating c-Jun, ATF-2 and other
transcription factors. JNK phosphorylates the serine residues 63 and 73 of the transcription factor
c-Jun, which in turn binds to TPA response elements and thereby increases c-Jun expression
(34-37). To demonstrate whether KSHV ORF36 activation of JNK results in c-Jun
phosphorylation, total cell lysates were analyzed by western blot analysis for phosphorylation of
c-Jun. Fig. 4 demonstrates that activation of JNK by KSHV ORF36 results in c-Jun
phosphorylation; the kinase-negative mutant K108Q does not lead to phosphorylation of c-Jun.
ORF36 interacts with MKK4, MKK7 and JNK
Immunoprecipation studies demonstrated that scaffolding proteins, such as JIP-1/2 (JNK
interacting protein 1/2), JSAP-1 (JNK/stress-activated protein kinase associated protein-1) and
beta-arrestin-2, interact physically with their downstream targets. JIP-1 and 2 form complexes
with JNK, MLK (mixed lineage kinase) and MKK7 (38,39), whereas JSAP-1 forms complexes
with MEKK1, MKK4 and JNK(40). These enzyme complexes produce “MAP-kinase modules”,
which facilitate activation and enhance specificity of their respective target proteins (39). To
assess the in vivo association of ORF36 with proteins in the JNK pathway, 293T cells were
14
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

transfected with ORF36 or the mutant K108Q. Immunoprecipation of total cell lysates with the
FLAG, MKK4, MKK7 or JNK antibody demonstrated physical interaction of ORF36 with these
cellular enzymes (Fig. 5). The kinase-negative mutant K108Q was also able to associate with
MKK4, 7 and JNK. Thus, autophosphorylation of ORF36 was not important for association with
MAP-kinase modules, and proceeded in a phosphorylation-independent manner.
Activation of JNK during KSHV reactivation- The KSHV viral protein, Rta, encoded by ORF50
is a transcriptional activator and acts as a molecular switch for KSHV reactivation. Expression of
Rta efficiently induces lytic replication in latently infected KSHV cell lines. The TREx BCBL1-
Rta cell line is a derivative of the BCBL-1 cell line (B-cell lymphoma cell line latently infected
with KSHV), where Rta is under the control of a tetracycline inducible promoter. This cell line
was demonstrated to fully induce lytic replication and produce infectious viral progeny in the
presence of tetracycline. In addition, TREx BCBL1-Rta cell line was demonstrated to induce
KSHV gene expression in a more powerful and efficient manner than TPA stimulation of
BCBL-1 cells (41). Stimulation of TREx BCBL-1-Rta cells with doxycycline for 24 hours
stimulated the expression of Rta, and expression levels did not change with the addition of JNK
inhibitor II (JNKi) or the JNKi negative inhibitor (Figure 6). Anisomycin or untreated cells did
not express Rta. The same cell lysates were used to determine the activation of JNK and c-Jun.
Induction of lytic replication activated JNK and c-Jun, and this activation was abrogated in the
presence of JNKi II. Inhibition of JNK also prevented expression of K8.1 (a KSHV glycoprotein
15
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

that serves as a marker for viral late gene expression) but not K-bZIP (a early lytic gene
identified as a basic-leucine zipper protein) (Figure 6).
16
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

DISCUSSION
KSHV, a gamma-2 herpesvirus, encodes a serine protein kinase (27). Our results support
previously published data, using phosphoamino acid analysis, that this kinase is phosphorylated
on serine (Fig.1) (27). Autophosphorylation was abrogated by mutating the lysine residue in the
catalytic subdomain II of ORF36 (Fig. 1). In this study, we established a mechanism by which
the viral protein kinase activates the mitogen activated stress kinase pathway. Using transient
transfection experiments, JNK was activated by KSHV ORF36, but not by the kinase-negative
mutant, K108Q (Fig 2). This finding indicates that autophosphorylation of ORF36 was important
for JNK activation. The p44/42, MKK3/6 and p38 pathways were not activated by ORF36,
showing specificity for JNK activation (Fig.2). Both MKK4 and MKK7 act synergistically to
activate JNK (30-32). Figure 3 demonstrates that ORF36 activates MKK4 and MKK7. These
findings imply that JNK activation by ORF36 is via phosphorylation of MKK4 and MKK7.
Recent data show that scaffolding proteins, such as JIP-1/2 and JSAP-1, associate with
MEKK1, MLK, MKK4, MKK7 and JNK, forming enzyme complexes and facilitating the
activation of the mitogen/stress activated protein kinase pathway (38-40). To demonstrate
whether ORF36 associates with these complexes, immunoprecipation experiments were
performed with antibodies specific for MKK4, MKK7 or JNK. These studies showed that KSHV
ORF36 was associated with MKK4, MKK7 and JNK, and that this association was independent
of the phosphorylation state of ORF36. The significance of ORF36 activation of JNK is
attributed to the phosphorylation and activation of the transcription factor, c-Jun. In addition, we
17
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

have demonstrated that inhibiting the activation of JNK during KSHV reactivation severely
represses expression of the late lytic viral gene, K8.1, but had no effect on the expression level of
the early lytic gene, K-bZIP (Figure 6). This demonstrates that initiation of lytic infection and
the transcription of early lytic genes may not require the activation of the JNK pathway.
Conversely, expression of K8.1, a structural glycoprotein component of KSHV particles, is
dramatically reduced in expression levels in the presence of SP600125.
The physiological role of ORF36 for KSHV replication is not defined, but possible clues
to its function have emerged from studies performed on homologous protein kinases found in
other herpesviruses. Previous work demonstrated that alpha-, beta- and gamma-herpesviruses
encode viral protein kinases, which are represented by HSV-1 ULl3, VZV ORF47, HCMV
UL97, HHV-6 U69, EBV EGLF4, KSHV ORF36 and RRV ORF36 (5,42,43). Similar to
cellular serine/threonine protein kinases, these viral kinases contain eleven subdomains; a
conserved lysine residue in subdomain II was important for kinase activity (44,45). Conservation
of these viral protein kinases amongst the herpesviridae family, and their homology to cellular
serine kinases, indicate indispensability for herpesvirus survival. A number of studies using
kinase-null viral mutants demonstrated the importance of the kinase for regulating viral gene
expression, replication, or tissue tropism. HSV UL13 kinase mediates post-translational
processing and influences expression of several viral genes (46). HCMV UL97 kinase-null
mutants show replication deficiency in tissue culture, and the UL97 protein kinase is important
for phosphorylation of multiple cellular targets (13,47,48). VZV ORF47 null mutants show
18
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

replication deficiency and the inability to phosphorylate several viral proteins (15,49,50). In
addition to phosphorylating viral targets, cellular targets including CKII beta, EF-1delta, p60,
and RNA pol II, serve as substrates for herpesvirus protein kinases (5,42,51-53). To demonstrate
conserved function between herpesvirus protein kinases, chimeric viruses expressing substituted
protein kinases from other herpesviruses were partially able to compensate for lost function (54).
In addition, these viral protein kinases were found in purified virus particles, suggesting a role in
the assembly of virions or in virion entry into cells (10,12).
Activation of the mitogen activated stress kinase pathway is important for enhancing
herpesvirus replication. Infection with HSV-1 resulted in the activation of JNK, c-Jun, p38,
AP-1 and enhancement of viral replication (19,20), and the phosphorylation of the transcription
factor, Sp1 (21). During VZV infection, activation of c-Jun, Fos, ATF-2 and AP-1 was
important for regulating viral genes (23), whereas activation of stress-activated MAP kinases
upregulated transgenes in CMV infection(22). These observations on other herpesviruses have
implications for our findings on the KSHV ORF36 kinase. Accordingly, ORF36 might enhance
viral replication by activating c-Jun N-terminal kinase, and hence modulating cellular
transcription by activation of transcription regulated by c-Jun. KSHV and EBV also encode
proteins that are capable of activating the mitogen-activated stress kinase pathway (24-26).
Functional studies to demonstrate the significance of these findings will provide clues to the
relevance of the mitogen-activated stress kinase pathway in herpesvirus survival and
transcription. Such studies could be based on the analysis of kinase-null mutants of the virus,
19
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

dominant negative JNK pathway mutants, and specific inhibitors for the JNK pathway.
Another possible role for ORF36 phosphorylation of JNK may be the activation of
transcription factors encoded by KSHV. Merek’s disease virus, also a gamma-herpesvirus,
encodes MEQ, which mimics c-Jun. The MEQ protein shares extensive homology with the
Jun/Fos family of transcription factors, within the basic region-leucine zipper (bZIP) domain. In
addition, MEQ dimerizes with itself and other cellular transcription factors, and can functionally
substitute for c-Jun (55). KSHV also encodes a gene with a basic region-leucine zipper domain;
this protein has been designated K-bZIP and shows homology with BZLF1, an EBV protein
essential for viral reactivation and replication (56,57). These observations suggest that ORF36
plays a role in the activation of KSHV transcription by activating viral homologs of cellular c-
Jun. Consequently, KSHV ORF36 (a viral late protein) might target an immediate-early viral
protein, such as, K-bZIP. This proposed model is supported by the finding that herpesvirus
protein kinases are found in purified virions (10,12). In fact, using in vitro kinase assays, ORF36
was shown to phosphorylate K-bZIP (these authors, unpublished results). Future studies to
define the role of ORF36 in KSHV replication and transcription will provide clues into the
mechanism by which herpesviruses modulate host cell signaling pathways and maintain their
survival within the infected host.
20
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

FOOTNOTES
We thank Dr. Albert Van Geelen for helpful discussions. The research in this paper was
supported by grants from the NCI (CA91574) to H.J.K., the California Universitywide AIDS
Research Program (R00-D-034) to H.J.K., and NCRR (RR00169) to P.A.L.
21
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

ABBREVIATIONS
The abbreviations used in this paper are: HSV, herpes simplex virus; VZV, Varicella-Zoster
virus; EBV, Epstein-Barr virus; KSHV, Kaposi’s sarcoma herpesvirus; HCMV, human
cytomegalovirus; RRV, rhesus rhadinovirus; JNK, c-Jun N terminal kinase; SAPK, stress
activated protein kinase, MAPK, mitogen activated protein kinase; MEKK4, SAPK/Erk kinase 1;
MKK7, MAP kinase kinase 7.
22
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

FIGURE LEGENDS
FIG. 1. Autophosphorylation and the conserved lysine residue in the catalytic subdomain of
KSHV ORF36 protein kinase. 293T cells transfected with pND (empty vector), or with plasmids
expressing wild type ORF36 or mutant K108Q, were lysed, and cell extracts were
immunoprecipated with anti-FLAG antibody and protein A sepharose beads. Immunoprecipated
proteins were detected by western blot analysis using anti-FLAG or anti-phosphoserine
antibody(see Materials and Methods). The control is lysate from pND transfected cells (lane 1).
Wild type ORF36 protein kinase was detected using anti-phospho-serine antibody (lane 2), but
this same antibody did not detect the K108Q mutant (lane 3). Equal amounts of ORF36 and
K108Q proteins were detected using anti-FLAG antibody. Phospho-serine proteins were
detected using chemiluminescence and FLAG tagged proteins were detected using colorimetry.
FIG. 2. Autophosphorylation of KSHV ORF36 and JNK activation. Total protein extracts from
SLK cells transfected with wild type ORF36 or mutant K108Q were analyzed by western blot
using antibodies that recognized the activated forms of proteins within the p44/42, MKK3/6,
p38, JNK and ERK pathway (see Materials and Methods). For controls, lysates were prepared
from mock transfected cells (empty pND vector, lane 1) or cells treated with anisomycin (JNK
activator, lane 2). JNK was specifically phosphorylated by wild type ORF36 (lane 3).
Phosphorylation of JNK was not detected in cells transfected with mutant K108Q (lane 4). Equal
loading of lysates was confirmed with antibodies that recognized total cellular JNK. Expression
23
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

of ORF36 and K108Q was detected by anti-FLAG antibody. KSHV ORF36 and K108Q did not
phosphorylate other members of the MAP kinase pathway as demonstrated by western blot
analysis using phospho-specific antibodies for ATF-2, p38, MKK3/6 and p44/42.
FIG. 3. Synergistic activation of MKK4 and MKK7 by KSHV ORF36. Lysates of transfected
SLK cells were analyzed by western blot using antibodies that recognized the phosphorylated
forms of MKK4 and MKK7 (see Materials and Methods). ORF36 activated both MKK4 and
MKK7 (lane 3), whereas the mutant K108Q was unable to activate either MKK4 or MKK7 (lane
4). These results demonstrate that synergistic activation of MKK4 and MKK7 was important for
the activation of JNK, and this activation was dependent on the phosphorylation state of ORF36
(lane 4). Equal loading of lysates was confirmed using antibodies that recognized total cellular
MKK4 and MKK7 protein.
FIG. 4. Phosphorylation of c-Jun transcription factor by KSHV ORF36. Activation of JNK
results in the phosphorylation and activation of its substrate, c-Jun. To demonstrate whether
JNK was catalytically active, phosphorylation of c-Jun was demonstrated in cells transfected
with ORF36 (lane 3), but not in cells transfected with the mutant K108Q (lane 4) by western blot
analysis using anti-phospho-c-Jun antibody. Lane 1 (negative control) and lane2 (positive
control-anisomycin) served as controls for c-Jun phosphorylation. Equal loading of protein
lysates was demonstrated by western blot analysis using anti-c-Jun antibody.
24
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

FIG. 5. Phosphorylation state and association of KSHV ORF36 with MKK4, MKK7 and JNK.
(A) Transfected 293T cells were lysed and immunoprecipated with FLAG antibody. Western
blot analysis of the immunoprecipated FLAG-tagged ORF36 protein revealed association with
MKK4, MKK7 and JNK (lanes 2 and 3). (B). Transfected 293T cells were immunoprecipated
with either MKK4, MKK7 or JNK antibodies. Western blot analysis using FLAG antibody
detected FLAG-tagged ORF36 (lanes 2,3,5,6,8 and 9). The phosphorylation state of KSHV
ORF36 was not essential for this association (lanes 3,6, and 9).
FIG. 6. Phosphorylation of JNK and c-Jun during KSHV reactivation and their significance in
lytic viral gene expression. KSHV reactivation and viral lytic gene expression was induced in the
TREx-BCBL-1 Rta cell line using doxycycline (1ug/ml) for 24 hours (lanes 3, 4 and 5). In
addition to doxycycline treatment, cells were treated with 50nM of JNK inhibitor II (JNKi II,
SP600125) (lane 4) or 20uM JNK inhibitor II negative control (lane5). Cells were not induced
(lane 1) or treated with anisomycin (lane 2). Doxycycline-induced expression of Rta was
detected in lanes 3, 4 and 5. Expression of Rta and lytic replication phosphorylated JNK and c-
Jun (lane 3) in these cells. Expression of Rta, in the presence or absence of JNKi II resulted in
the expression of K-bZIP (lanes 3, 4 and 5). Inhibition of K8.1 expression in the presence of Rta
and K-bZIP expression was observed in the presence of JNKi II (lane 4). Equal loading of cell
lysates was confirmed using anti-tubulin antibody.
25
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

REFERENCES
1. Roizman, B., and Pellet, P. E. (2001) in Fields Virology (Knipe, D. M., and Howley, P. M., eds) Vol. 2, pp. 2381-2397, 2 vols., Lippincott Williams and Wilkins
2. Kieff, E., and Rickinson, A. (2001) in Fields Virology (Knipe, d., and Howley, p., eds) Vol. 2 fourth edition, pp. 2576-2673, 2 vols., Lippincott Williams and Wilkins
3. Bubman, D., and Cesarman, E. (2003) Hematol Oncol Clin North Am 17, 717-7454. Cotter, M. A., 2nd, and Robertson, E. S. (2002) Front Biosci 7, d358-3755. Kawaguchi, Y., and Kato, K. (2003) Rev Med Virol 13, 331-3406. Johnson, L. N., Lowe, E. D., Noble, M. E., and Owen, D. J. (1998) FEBS Lett 430, 1-117. Smith, R. F., and Smith, T. F. (1989) J Virol 63, 450-4558. Kawaguchi, Y., Matsumura, T., Roizman, B., and Hirai, K. (1999) J Virol 73, 4456-44609. Morrison, E. E., Wang, Y. F., and Meredith, D. M. (1998) J Virol 72, 7108-711410. Overton, H. A., McMillan, D. J., Klavinskis, L. S., Hope, L., Ritchie, A. J., and Wong-
kai-in, P. (1992) Virology 190, 184-19211. Wolf, D. G., Honigman, A., Lazarovits, J., Tavor, E., and Panet, A. (1998) Arch Virol
143, 1223-123212. van Zeijl, M., Fairhurst, J., Baum, E. Z., Sun, L., and Jones, T. R. (1997) Virology 231,
72-8013. Prichard, M. N., Gao, N., Jairath, S., Mulamba, G., Krosky, P., Coen, D. M., Parker, B.
O., and Pari, G. S. (1999) J Virol 73, 5663-567014. Sato, H., Pesnicak, L., and Cohen, J. I. (2003) J Virol 77, 11180-1118515. Besser, J., Sommer, M. H., Zerboni, L., Bagowski, C. P., Ito, H., Moffat, J., Ku, C. C.,
and Arvin, A. M. (2003) J Virol 77, 5964-597416. Coulter, L. J., Moss, H. W., Lang, J., and McGeoch, D. J. (1993) J Gen Virol 74 ( Pt 3),
387-39517. Heineman, T. C., and Cohen, J. I. (1995) J Virol 69, 7367-737018. Moffat, J. F., Zerboni, L., Sommer, M. H., Heineman, T. C., Cohen, J. I., Kaneshima, H.,
and Arvin, A. M. (1998) Proc Natl Acad Sci U S A 95, 11969-1197419. McLean, T. I., and Bachenheimer, S. L. (1999) J Virol 73, 8415-842620. Zachos, G., Clements, B., and Conner, J. (1999) J Biol Chem 274, 5097-510321. Kim, D. B., and DeLuca, N. A. (2002) J Virol 76, 6473-647922. Bruening, W., Giasson, B., Mushynski, W., and Durham, H. D. (1998) Nucleic Acids Res
26, 486-48923. Rahaus, M., and Wolff, M. H. (2003) Virus Res 92, 9-2124. Chen, S. Y., Lu, J., Shih, Y. C., and Tsai, C. H. (2002) J Virol 76, 9556-956125. Adamson, A. L., Darr, D., Holley-Guthrie, E., Johnson, R. A., Mauser, A., Swenson, J.,
and Kenney, S. (2000) J Virol 74, 1224-1233
26
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

26. Brinkmann, M. M., Glenn, M., Rainbow, L., Kieser, A., Henke-Gendo, C., and Schulz, T. F. (2003) J Virol 77, 9346-9358
27. Park, J., Lee, D., Seo, T., Chung, J., and Choe, J. (2000) J Gen Virol 81, 1067-107128. Sun, R., Lin, S. F., Staskus, K., Gradoville, L., Grogan, E., Haase, A., and Miller, G.
(1999) J Virol 73, 2232-224229. Smith, G. A., Young, P. L., and Mattick, J. S. (1991) Arch Virol 119, 199-21030. Lisnock, J., Griffin, P., Calaycay, J., Frantz, B., Parsons, J., O’Keefe, S. J., and LoGrasso,
P. (2000) Biochemistry 39, 3141-314831. Lawler, S., Fleming, Y., Goedert, M., and Cohen, P. (1998) Curr Biol 8, 1387-139032. Fleming, Y., Armstrong, C. G., Morrice, N., Paterson, A., Goedert, M., and Cohen, P.
(2000) Biochem J 352 Pt 1, 145-15433. Kishimoto, H., Nakagawa, K., Watanabe, T., Kitagawa, D., Momose, H., Seo, J.,
Nishitai, G., Shimizu, N., Ohata, S., Tanemura, S., Asaka, S., Goto, T., Fukushi, H., Yoshida, H., Suzuki, A., Sasaki, T., Wada, T., Penninger, J. M., Nishina, H., and Katada, T. (2003) J Biol Chem 278, 16595-16601
34. Hibi, M., Lin, A., Smeal, T., Minden, A., and Karin, M. (1993) Genes Dev 7, 2135-214835. Galcheva-Gargova, Z., Derijard, B., Wu, I. H., and Davis, R. J. (1994) Science 265,
806-80836. Diehl, A. M., Yin, M., Fleckenstein, J., Yang, S. Q., Lin, H. Z., Brenner, D. A.,
Westwick, J., Bagby, G., and Nelson, S. (1994) Am J Physiol 267, G552-56137. Westwick, J. K., Weitzel, C., Minden, A., Karin, M., and Brenner, D. A. (1994) J Biol
Chem 269, 26396-2640138. Ikeda, A., Hasegawa, K., Masaki, M., Moriguchi, T., Nishida, E., Kozutsumi, Y., Oka, S.,
and Kawasaki, T. (2001) J Biochem (Tokyo) 130, 773-78139. Whitmarsh, A. J., Cavanagh, J., Tournier, C., Yasuda, J., and Davis, R. J. (1998) Science
281, 1671-167440. Ito, M., Yoshioka, K., Akechi, M., Yamashita, S., Takamatsu, N., Sugiyama, K., Hibi,
M., Nakabeppu, Y., Shiba, T., and Yamamoto, K. I. (1999) Mol Cell Biol 19, 7539-754841. Nakamura, H., Lu, M., Gwack, Y., Souvlis, J., Zeichner, S. L., and Jung, J. U. (2003) J
Virol 77, 4205-422042. Kawaguchi, Y., Kato, K., Tanaka, M., Kanamori, M., Nishiyama, Y., and Yamanashi, Y.
(2003) J Virol 77, 2359-236843. Kato, K., Kawaguchi, Y., Tanaka, M., Igarashi, M., Yokoyama, A., Matsuda, G.,
Kanamori, M., Nakajima, K., Nishimura, Y., Shimojima, M., Phung, H. T., Takahashi, E., and Hirai, K. (2001) J Gen Virol 82, 1457-1463
44. Chee, M. S., Lawrence, G. L., and Barrell, B. G. (1989) J Gen Virol 70 ( Pt 5), 1151-1160
45. Hanks, S. K., Quinn, A. M., and Hunter, T. (1988) Science 241, 42-5246. Purves, F. C., Ogle, W. O., and Roizman, B. (1993) Proc Natl Acad Sci U S A 90, 6701-
6705
27
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

47. Talarico, C. L., Burnette, T. C., Miller, W. H., Smith, S. L., Davis, M. G., Stanat, S. C., Ng, T. I., He, Z., Coen, D. M., Roizman, B., and Biron, K. K. (1999) Antimicrob Agents Chemother 43, 1941-1946
48. Littler, E., Stuart, A. D., and Chee, M. S. (1992) Nature 358, 160-16249. Ng, T. I., and Grose, C. (1992) Virology 191, 9-1850. Stevenson, D., Colman, K. L., and Davison, A. J. (1994) J Gen Virol 75 ( Pt 2), 317-32651. Kawaguchi, Y., Van Sant, C., and Roizman, B. (1998) J Virol 72, 1731-173652. Bruni, R., Fineschi, B., Ogle, W. O., and Roizman, B. (1999) J Virol 73, 3810-381753. Biggar, R. J., Engels, E. A., Whitby, D., Kedes, D. H., and Goedert, J. J. (2003) J Infect
Dis 187, 12-1854. Ng, T. I., Talarico, C., Burnette, T. C., Biron, K., and Roizman, B. (1996) Virology 225,
347-35855. Liu, J. L., and Kung, H. J. (2000) Virus Genes 21, 51-6456. Lin, S. F., Robinson, D. R., Miller, G., and Kung, H. J. (1999) J Virol 73, 1909-191757. Izumiya, Y., Lin, S. F., Ellison, T., Chen, L. Y., Izumiya, C., Luciw, P., and Kung, H. J.
(2003) J Virol 77, 1441-1451
28
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Kung and Paul A. LuciwMuhammad S. Hamza, Richard A. Reyes, Yoshihiro Izumiya, Ronald Wisdom, Hsing-Jien
N-terminal kinase (JNK) signaling pathwayORF36 protein kinase of kaposi's sarcoma herpesvirus (KSHV) activates the c-Jun
published online July 5, 2004J. Biol. Chem.
10.1074/jbc.M400964200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on August 18, 2019
http://ww
w.jbc.org/
Dow
nloaded from



![[PPT]FAMILIA: HERPESVIRIDAE - CAO-Blog 4toAño ... · Web viewFAMILIA: HERPESVIRIDAE ESTRUCTURA: ADN CAPSIDE ENVOLTURA 162 CAPSOMER ESPIGAS DE GLICOPROTEINAS 150 – 300 NM LINEAL](https://img.pdfslide.us/doc/110x75/5ae5426c7f8b9ae1578c114a/pptfamilia-herpesviridae-cao-blog-4toao-viewfamilia-herpesviridae-estructura.jpg)