Embed Size (px)
Citation preview

J. Am. Chem. SOC. 1981,103, 219-222 219
The yield of higher hydrocarbons is very low. This reflects the fact that in TPDE the primary reaction products are swept from the reactor with very short contact times ( N 1.4 s).13 Although the C balance (based on the collection of the above gases) is usually quite good (>90%), in a few cases it is not, most notably for Rh6(C0)16.15 This could be due to the formation of heavier products (including oxygenates) which are not detected.I6 However, due to the low pressure and short contact times, a more likely explanation may be the formation of some type of Cads (adsorbed carbon) which is unreactive toward H2.l7
A notable feature of Table I is the large amounts of CH4 which are formed, in six cases the conversion of CO to CH4 exceeding 50%. The yield of CH4 does not correlate with the activity of the respective metals for catalytic methanationk8 For example, Ir, the least active of the group 8B metals for methanation, yields far more CHI than Ni, one of the most active metals. Examination of Figures 1-4 shows that the thermal stability of a supported complex is the prime factor in determining the quantity of CH4 formed. Thus Ni(CO),, which decomposes a t a very low tem- perature (Figure l ) , has lost most of its CO before the temperature is high enough to give a reasonable rate of methanation (Figure 2).19 In contrast, Ir4(CO)12 does not lose its CO until 125 OC (Figure 3).2k TPDE in flowing H2 (Figure 4) then yields a curve for CH4 evolution which is remarkably similar to that for CO evolution in H e (Figure 3). Similar correlations are found for the other catalysts.
The two complexes giving the most CH4 (per complex) are Ir4(CO)12 and O S ~ ( C O ) ~ ~ . These are the same two cluster com- plexes which were reported to be active for homogeneous catalytic methanatiomz2 However, in those experiments the total yield of CH4 was only about 4 CH4/complex, whereas several times this amount is now seen to be formed in a purely stoichiometric reaction.z3 Hence, it is possible that the claimed catalytic reaction was in fact stoichiometric (or a heterogeneous r e a c t i ~ n ~ ~ ~ ~ ~ ) and the ability of some cluster complexes (but not mononuclear complexes) to homogeneously yield CH4 may simply reflect their enhanced thermal stability.
TPDE in flowing Hz increases the CH4 yield by 25-fold over TPDE in flowing He.8 In both He8 and Hz (Table I) it is found that the quantity of CHI formed is essentially independent of the nuclearity of a complex. This is contrary to some claims that multinuclear sites are necessary to effect the reduction of COZ6 but consistent with more recent work suggesting that mononuclear complexes can be active for methanation.8~~~ Recent data for the TPDE of Mo(C0)6/Al2O3 in flowing H e and Hz indicate that sintering of mononuclear precursors to polynuclear sites is probably
(1 3) Hydrocarbon synthesis has been reported for several carbonyl cluster complexes supported on wet A1203 and heated in sealed ampules at approx- imately 300 OC for - l@ s.I4 (H2 is generated by the water gas shift reaction.)
(14) Smith, A. K.; Theolier, A,; Basset, J. M.; Ugo, R.; Commereuc, D.; Chauvin, Y. J . Am. Chem. SOC. 1978, 100, 2590.
(15) Rh,(CO),, also gave a particularly poor C balance for reaction in sealed ampule^.'^
(16) Ichikawa, M. Bull. Chem. SOC. Jpn. 1978,51, 2273. (17) TPDE in flowing H, of Mn2(CO),o yielded some CH4 at temperatures
higher than for CO evolution in a sweep gas of He, suggesting some hydro- genation of Cads. However, due to the generally good C balances and the fact that TPDE in Hz did not significantly improve the C balances compared to TPDE in He, it appears that usually only small quantities of Cads can be formed, and it must be fairly unreactive toward H,.
(18) Vannice, M. A. J. Carol. 1975, 37, 449. (19) These results are in a reement with less detailed data previously
published for Ni(CO)4/A1203!o (20) Bjorkland, R. B.; Burwell, R. L., Jr. J . Colloid Interface Sci. 1979,
70, 383. (21) For this experiment the Ir4(C0)12 was dispersed at 125 OC during
which time 0.2 CO/complex was evolved. (22) Thomas, M. G.; Beier, B. F.; Muetterties, E. L. J . Am. Chem. Soc.
1976, 98, 1296. (23) The homogeneous reactions were run at 140 OC in toluene solution
and gave a turnover frequency of 1 X lo-' s-'. At 140 "C the extrapolated turnover frequencies for the stoichiometric hydrogenations of Ir4(C0)12 and O S ~ ( C O ) , ~ are also -1 x 104 s-I.
(24) Bradley, J. S. J . Am. Chem. SOC. 1980, 101, 7419. (25) Doyle, M. J.; Kouwenhoven, A. P.; Cornelis, A.; Oort, B. V. J. Or-
ganomet. Chem. 1979, 174, C55. (26) Muetterties, E. L. Science 1977, 196, 839.
0002-7863/81/1503-219$01 .OO/O 0
not a prerequisite for CH4 formation.8 (Several experiments in flowing Hz at loadings between 0.0036 and 0.76% Mo show that the yield of Cz/Mo is independent of loading, again suggesting that multinuclear sites are not involved.) Hydrogenation is not preceded by the disproportionation of CO since the yield of COz is extremely low. Further, in most cases dissociation of C O followed by hydrogenation of Cads is unlikely, since the CH4 is often formed in a temperature region in which CO is evolved if the TPDE is carried out in flowing He. Hence, it is most likely in these systems that the direct hydrogenation of coordinated CO to CH4 is occurring at discrete subcarbonyl sites.
In addition to the substantial yields of CHI during TPDE in flowing Hz, it is seen (Table I) that the reaction often occurs a t temperatures well below that required for catalytic methanation (200-350 OC).18 An interesting corollary to this result is that a good catalyst for methanation should result if a metal can react with CO to re-form a carbonyl (or subcarbonyl) complex, since a catalytic cycle is now formed. Two of the more active catalysts, Ni and Fe, are in fact the metals which most readily form car- bonyls from CO, whereas many of the catalytically less active metals (Ir, Mn, and Cr) do not undergo this r e a c t i ~ n . ' ~ * ~ ~ However, the most active metal for methanation, Ru, only very slowly forms a carbonyl by exposure to CO under severe condi- tions, although it was suggested that this may be due to adsorption of R U ( C O ) ~ which inhibits further reaction.27 Hydrogenation of carbonyl-like intermediates has been considered as a mechanism for methanation and Fischer-Tropsch synthesis,z8 but currently favored is the dissociation of C O followed by hydrogenation of CadseZ9 Thus, although the facile hydrogenation of coordinated CO is now demonstrated, it is nuclear if this process is important during catalytic methanation. Catalytic methanation over sup- ported carbonyl complexes is currently being studied.
Acknowledgment. Support of this research by the Department of Energy is gratefully acknowledged.
(27) Wender, I.; Pino, P. "Organic Synthesis Via Metal Carbonyls"; In- terscience: New York, 1968; Vol. 1, Chapter 1.
(28) Anderson, R. B. Catalysis 1956, 4, 29. (29) Biloen, P.; Helle, J. N.; Sachtler, W. M. H. J . Caral. 1979, 58, 95.
A Strategy for the Total Synthesis of Jatrophone: Synthesis of Normethyljatrophone Amos B. Smith, III,*t Michael A. Guaciaro, Steven R. Schow, Peter M. Wovkulich, Bruce H. Toder, and Tse Wai Hall
The Department of Chemistry The Laboratory for Research on the Structure of Matter
and The Monell Chemical Senses Center The University of Pennsylvania
Philadelphia, Pennsylvania I91 04 Received October I , 1980
Jatrophone (l), an architecturally interesting macrocyclic diterpene first isolated in 1970 by the late Professor Kupchan from extracts of Jatropha gossypiifolia L. (Euphorbiacae),' merits consideration as a synthetic target in that it displays significant inhibitory activity against a variety of cell lines, including sarcoma 180, Lewis lung carcinoma, P-388 lymphocytic leukemia, and Walker 256 intramolecular carcinsarcoma.2 Indeed, extracts of this plant had long been employed in the treatment of cancerous growths.2
The structure of jatrophone was based on both chemical and X-ray crystallographic studies.' Central to the derived structure
Camille and Henry Dreyfus Teacher-Scholar, 1978-1983; recipient of a National Institutes of Health (National Cancer Institute) Career Devel- opment Award, 1980-1985.
(1) S. M. Kupchan, C. W. Sigel, M. J. Matz, J. A. Saenz Renauld, R. C. Haltiwanger, and R. F. Bryan, J . Am. Chem. Soc., 92,4476 (1970); S. M. Kupchan, C. W. Sigel, M. J. Matz, C. J. Gilmore, and R. F. Bryan, ibid., 98, 2295 (1976).
(2) J. L. Hartwell, Lloydia, 32, 153 (1969).
1980 American Chemical Society
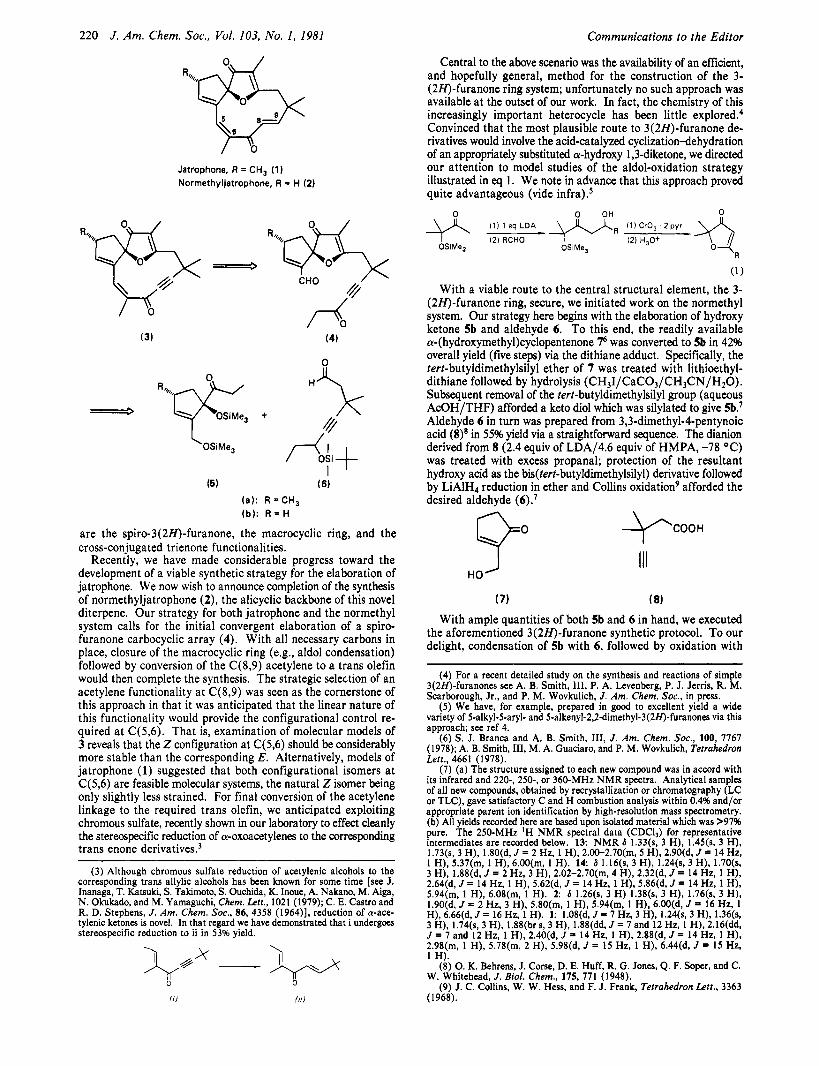
220 J . Am. Chem. SOC., Vol. 103, No. 1, 1981 Communications to the Editor
Central to the above scenario was the availability of an efficient, and hopefully general, method for the construction of the 3- (2H)-furanone ring system; unfortunately no such approach was available at the outset of our work. In fact, the chemistry of this increasingly important heterocycle has been little e ~ p l o r e d . ~ Convinced that the most plausible route to 3(2H)-furanone de- rivatives would involve the acid-catalyzed cyclizationdehydration of an appropriately substituted a-hydroxy 1,3-diketone, we directed our attention to model studies of the aldol-oxidation strategy illustrated in eq 1. We note in advance that this approach proved quite advantageous (vide infra).5
Jatrophone, R = CH, (1) Normethyljatrophone, R = H (2)
(3) (4)
0
T(,hi + I
(5) ( 6) (a): R = C H , (b): R = H
are the spiro-3(2H)-furanone, the macrocyclic ring, and the cross-conjugated trienone functionalities.
Recently, we have made considerable progress toward the development of a viable synthetic strategy for the elaboration of jatrophone. We now wish to announce completion of the synthesis of normethyljatrophone (2), the alicyclic backbone of this novel diterpene. Our strategy for both jatrophone and the normethyl system calls for the initial convergent elaboration of a spiro- furanone carbocyclic array (4). With all necessary carbons in place, closure of the macrocyclic ring (e.g., aldol condensation) followed by conversion of the C(8,9) acetylene to a trans olefin would then complete the synthesis. The strategic selection of an acetylene functionality a t C(8,9) was seen as the cornerstone of this approach in that it was anticipated that the linear nature of this functionality would provide the configurational control re- quired at C(5,6). That is, examination of molecular models of 3 reveals that the Z configuration at C(5,6) should be considerably more stable than the corresponding E. Alternatively, models of jatrophone (1) suggested that both configurational isomers a t C(5,6) are feasible molecular systems, the natural 2 isomer being only slightly less strained. For final conversion of the acetylene linkage to the required trans olefin, we anticipated exploiting chromous sulfate, recently shown in our laboratory to effect cleanly the stereospecific reduction of a-oxoacetylenes to the corresponding trans enone derivative^.^
(3) Although chromous sulfate reduction of acetylenic alcohols to the corresponding trans allylic alcohols has been known for some time [see J. Inanaga, T. Katsuki, S. Takimoto, S. Ouchida, K. Inoue, A. Nakano, M. Aiga, N. Okukado, and M. Yamaguchi, Chem. Lett., 1021 (1979); C. E. Castro and R. D. Stephens, J. Am. Chem. SOC., 86, 4358 (1964)], reduction of a-ace- tylenic ketones is novel. In that regard we have demonstrated that i undergoes stereospecific reduction to ii in 53% yield.
l i l / i i /
( 1 ) 1 eq LDA
( 2 ) R C H O - (2 ) H,Ot OSiMe, OSiMe,
(1 1 With a viable route to the central structural element, the 3-
(2H)-furanone ring, secure, we initiated work on the normethyl system. Our strategy here begins with the elaboration of hydroxy ketone 5b and aldehyde 6. To this end, the readily available a-(hydroxymethy1)cyclopentenone 76 was converted to 5b in 42% overall yield (five steps) via the dithiane adduct. Specifically, the tert-butyldimethylsilyl ether of 7 was treated with lithioethyl- dithiane followed by hydrolysis (CH31/CaC03/CH3CN/H20). Subsequent removal of the tert-butyldimethylsilyl group (aqueous AcOH/THF) afforded a keto diol which was silylated to give 5b.' Aldehyde 6 in turn was prepared from 3,3-dimethyl-4-pentynoic acid (8)* in 55% yield via a straightforward sequence. The dianion derived from 8 (2.4 equiv of LDA/4.6 equiv of HMPA, -78 "C) was treated with excess propanal; protection of the resultant hydroxy acid as the bis(tert-butyldimethylsilyl) derivative followed by LiAlH4 reduction in ether and Collins oxidation9 afforded the desired aldehyde (6).'
oo HO J
k C O O H I Ill
( 7) (8)
With ample quantities of both 5b and 6 in hand, we executed the aforementioned 3(2H)-furanone synthetic protocol. To our delight, condensation of 5b with 6, followed by oxidation with
(4) For a recent detailed study on the synthesis and reactions of simple 3(2H)-furanones see A. B. Smith, 111, P. A. Levenberg, P. J. Jerris, R. M. Scarborough, Jr., and P. M. Wovkulich, J . Am. Chem. SOC., in press.
(5) We have, for example, prepared in good to excellent yield a wide variety of 5-alkyl-5-aryl- and 5-alkenyl-2,2-dimethyl-3(2H)-furanones via this approach; see ief 4.
(6) S. J. Branca and A. B. Smith. 111. J . Am. Chem. Soc.. 100. 7767 (1978); A. B. Smith, 111, M. A. Guaciaro, and P. M. Wovkulich, Tetrahedron Lefr., 4661 (1978).
(7) (a) The structure assigned to each new compound was in accord with its infrared and 220-, 250-, or 360-MHz NMR spectra. Analytical samples of all new compounds, obtained by recrystallization or chromatography (LC or TLC), gave satisfactory C and H combustion analysis within 0.4% and/or appropriate parent ion identification by high-resolution mass spectrometry. (b) All yields recorded here are based upon isolated material which was >97% pure. The 250-MHz 'H NMR spectral data (CDCI,) for representative intermediates are recorded below. 1 3 NMR 6 1.33(s, 3 H), 1.45(s, 3 H), 1.73(s, 3 H), 1.80(d, J = 2 Hz, 1 H), 2.00-2.70(m, 5 H), 2.90(d, J = 14 Hz, 1 H), 5.37(m, 1 H), 6.00(m, 1 H). 14: 6 1.16(s, 3 H), 1.24(s, 3 H), 1.70(s, 3 H), 1.88(d, J = 2 Hz, 3 H), 2.02-2.70(m, 4 H), 2.32(d, J = 14 Hz, 1 H), 2.64(d, J = 14 Hz, 1 H), 5.62(d, J = 14 Hz, 1 H), 5.86(d, J = 14 Hz, 1 H), 5.94(m, 1 H), 6.08(m, 1 H). 2: 6 1.26(s, 3 H) 1.38(s, 3 H), 1.76(s, 3 H), 1.90(d, J = 2 Hz, 3 H), 5.80(m, 1 H), 5.94(m, 1 H), 6.00(d, J = 16 Hz, 1 H), 6.66(d, J = 16 Hz, 1 H). 1: 1.08(d, J = 7 Hz, 3 H), 1.24(s, 3 H), 1.36(s, 3 H), 1.74(s, 3 H), 1.88(br s, 3 H), 1.88(dd, J = 7 and 12 Hz, 1 H), 2.16(dd, J = 7 and 12 Hz, 1 H), 2.40(d, J = 14 Hz, 1 H), 2.88(d, J = 14 Hz, 1 H), 2.98(m, 1 H), 5.78(m, 2 H), 5.98(d, J = 15 Hz, 1 H), 6.44(d, J = 15 Hz, 1 H).
( 8 ) 0. K. Behrens, J. Come, D. E. Huff, R. G. Jones, Q. F. Soper, and C. W. Whitehead, J. Biol. Chem., 175, 771 (1948).
(9) J. C. Collins, W. W. Hess, and F. J. Frank, Tetrahedron Left., 3363 (1968).
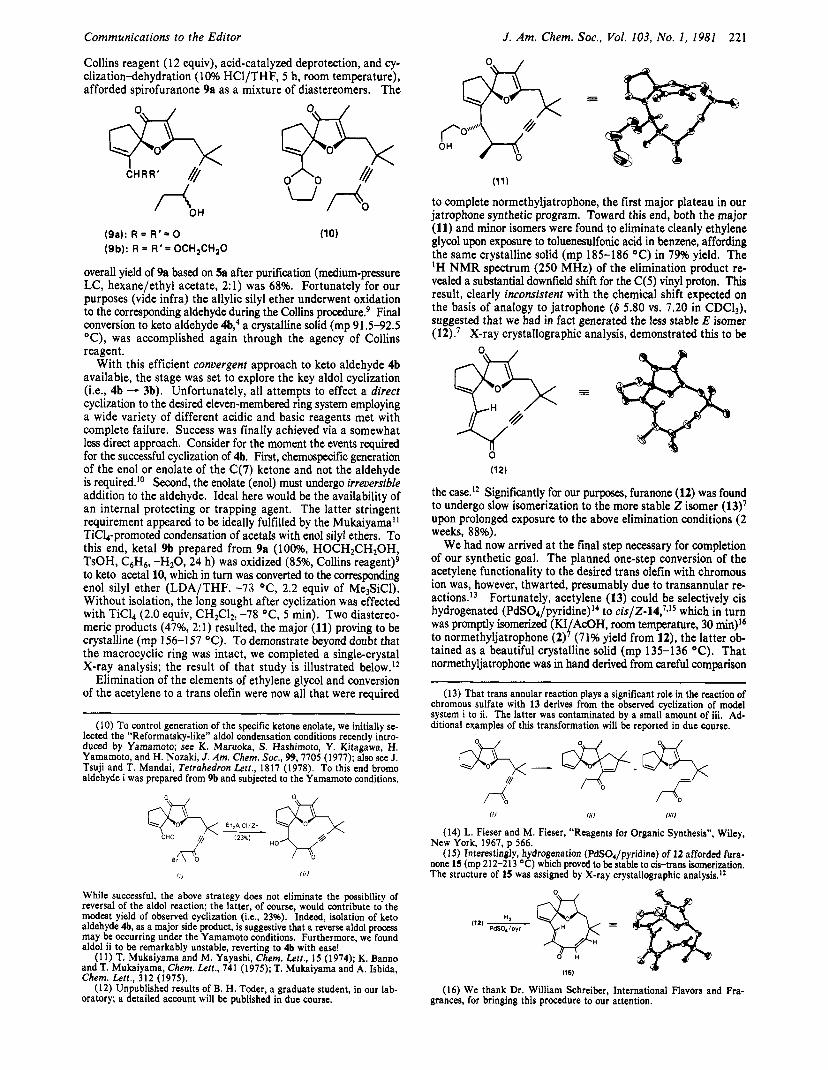
Communications to the Editor
Collins reagent (1 2 equiv), acid-catalyzed deprotection, and cy- clization-lehydration (10% HCl/THF, 5 h, room temperature), afforded spirofuranone 9a as a mixture of diastereomers. The
J. Am. Chem. Sot., Vol. 103, No. 1 , 1981 221
(9a): R = R ' = 0 (9b): R = R ' = OCH,CH,O
i i /+O
overall yield of 9a based on 5a after purification (medium-pressure LC, hexane/ethyl acetate, 2:l) was 68%. Fortunately for our purposes (vide infra) the allylic silyl ether underwent oxidation to the corresponding aldehyde during the Collins procedure? Final conversion to keto aldehyde 4b: a crystalline solid (mp 91.5-92.5 "C), was accomplished again through the agency of Collins reagent.
With this efficient convergent approach to keto aldehyde 4b available, the stage was set to explore the key aldol cyclization (Le., 4b - 3b). Unfortunately, all attempts to effect a direct cyclization to the desired eleven-membered ring system employing a wide variety of different acidic and basic reagents met with complete failure. Success was finally achieved via a somewhat less direct approach. Consider for the moment the events required for the successful cyclization of 4b. First, chemospecific generation of the enol or enolate of the C(7) ketone and not the aldehyde is required.'O Second, the enolate (enol) must undergo irreversible addition to the aldehyde. Ideal here would be the availability of an internal protecting or trapping agent. The latter stringent requirement appeared to be ideally fulfilled by the Mukaiyama' TiC14-promoted condensation of acetals with enol silyl ethers. To this end, ketal 9b prepared from 9a (loo%, HOCH2CH20H, TsOH, C6H6, -H20, 24 h) was oxidized (85%, Collins reagent)9 to keto acetal 10, which in turn was converted to the corresponding enol silyl ether (LDA/THP, -73 O C , 2.2 equiv of Me3SiC1). Without isolation, the long sought after cyclization was effected with TiC14 (2.0 equiv, CHZC12, -78 OC, 5 min). Two diastereo- meric products (47%, 2:l) resulted, the major (11) proving to be crystalline (mp 156-157 "C). To demonstrate beyond doubt that the macrocyclic ring was intact, we completed a single-crystal X-ray analysis; the result of that study is illustrated below.12
Elimination of the elements of ethylene glycol and conversion of the acetylene to a trans olefin were now all that were required
(10) To control generation of the specific ketone enolate, we initially se- lected the "Reformatsky-like" aldol condensation conditions recently intro- duced by Yamamoto; see K. Maruoka, S. Hashimoto, Y. Kitagawa, H. Yamamoto, and H. Nozaki, J . Am. Chem. Soc., 99,7705 (1977); also see J. Tsuji and T. Mandai, Tetrahedron Lett., 1817 (1978). To this end bromo aldehyde i was prepared from 9b and subjected to the Yamamoto conditions.
Et2A1CI/Zn
123%) HO e ti / (lil
While successful, the above strategy does not eliminate the possibility of reversal of the aldol reaction; the latter, of course, would contribute to the modest yield of observed cyclization (Le., 23%). Indeed, isolation of keto aldehyde 4, as a major side product, is suggestive that a reverse aldol process may be occurring under the Yamamoto conditions. Furthermore, we found aldol ii to be remarkably unstable, reverting to 4b with ease!
(1 1) T. Mukaiyama and M. Yayashi, Chem. Lett., 15 (1974); K. Banno and T. Mukaiyama, Chem. Lett., 741 (1975); T. Mukaiyama and A. Ishida, Chem. Lett., 312 (1975).
(12) Unpublished results of B. H. Toder, a graduate student, in our lab- oratory; a detailed account will be published in due course.
(111
to complete normethyljatrophone, the first major plateau in our jatrophone synthetic program. Toward this end, both the major (11) and minor isomers were found to eliminate cleanly ethylene glycol upon exposure to toluenesulfonic acid in benzene, affording the same crystalline solid (mp 185-186 "C) in 79% yield. The 'H N M R spectrum (250 MHz) of the elimination product re- vealed a substantial downfield shift for the C(5) vinyl proton. This result, clearly inconsistent with the chemical shift expected on the basis of analogy to jatrophone (6 5.80 vs. 7.20 in CDC13), suggested that we had in fact generated the less stable E isomer (l2).' X-ray crystallographic analysis, demonstrated this to be
(121
the case.I2 Significantly for our purposes, furanone (12) was found to undergo slow isomerization to the more stable Z isomer (13)' upon prolonged exposure to the above elimination conditions (2 weeks, 88%).
We had now arrived at the final step necessary for completion of our synthetic goal. The planned one-step conversion of the acetylene functionality to the desired trans olefin with chromous ion was, however, thwarted, presumably due to transannular re- ac t ion~. '~ Fortunately, acetylene (13) could be selectively cis hydrogenated (PdS04/pyridine)14 to ci~/Z-14,'*'~ which in turn was promptly isomerized (KI AcOH, room temperature, 30 min)I6
tained as a beautiful crystalline solid (mp 135-136 "C). That normethyljatrophone was in hand derived from careful comparison
(13) That trans annular reaction plays a significant role in the reaction of chromous sulfate with 13 derives from the observed cyclization of model system i to ii. The latter was contaminated by a small amount of iii. Ad- ditional examples of this transformation will be reported in due course.
to normethyljatrophone (2) J/ (71% yield from 12), the latter ob-
(il IiiI IiiiI
(14) L. Fieser and M. Fieser, "Reagents for Organic Synthesis", Wiley, New York, 1967, p 566.
(1 5) Interestingly, hydrogenation (PdSO,/pyridine) of 12 afforded fura- none 15 (mp 212-213 "C) which proved to be stable to cis-trans isomerization. The structure of 15 was assigned by X-ray crystallographic analysis.I2
(16) We thank Dr. William Schreiber, International Flavors and Fra- grances, for bringing this procedure to our attention.
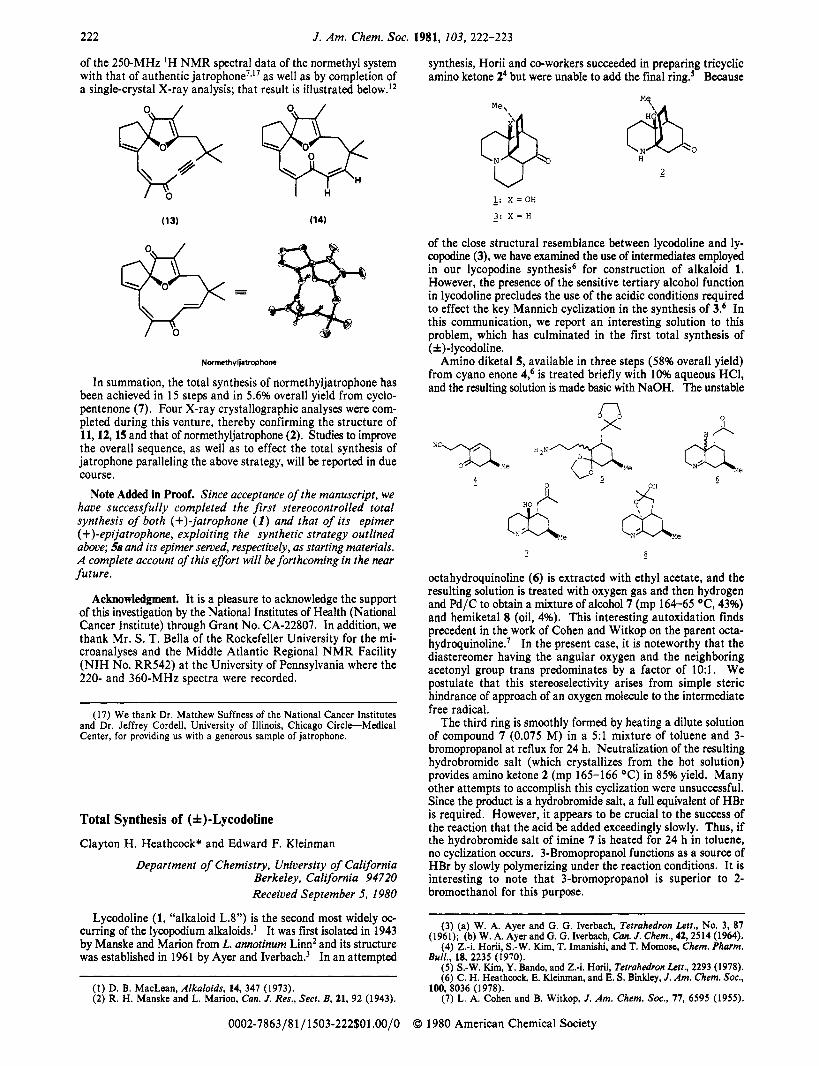
222 J. Am. Chem. SOC. 1981, 103, 222-223
of the 250-MHz ‘H NMR spectral data of the normethyl system with that of authentic jatrophone’,’’ as well as by completion of a single-crystal X-ray analysis; that result is illustrated below.12
Normethyljatrophonc
In summation, the total synthesis of normethyljatrophone has been achieved in 15 steps and in 5.6% overall yield from cyclo- pentenone (7). Four X-ray crystallographic analyses were com- pleted during this venture, thereby confirming the structure of 11, 12, 15 and that of normethyljatrophone (2). Studies to improve the overall sequence, as well as to effect the total synthesis of jatrophone paralleling the above strategy, will be reported in due course.
Note Added in Proof. Since acceptance of the manuscript, we have successfully completed the first stereocontrolled total synthesis of both (+)-jatrophone ( 1 ) and that of its epimer (+)-epijatrophone, exploiting the synthetic strategy outlined above; 5a and its epimer served, respectively, as starting materials. A complete account of this effort will be forthcoming in the near future.
Acknowledgment. It is a pleasure to acknowledge the support of this investigation by the National Institutes of Health (National Cancer Institute) through Grant No. CA-22807. In addition, we thank Mr. S. T. Bella of the Rockefeller University for the mi- croanalyses and the Middle Atlantic Regional N M R Facility (NIH No. RR542) at the University of Pennsylvania where the 220- and 360-MHz spectra were recorded.
(17) We thank Dr. Matthew Suffness of the National Cancer Institutes and Dr. Jeffrey Cordell, University of Illinois, Chicago Circle-Medical Center, for providing us with a generous sample of jatrophone.
Total Synthesis of (f)-Lycodoline
Clayton H. Heathcock* and Edward F. Kleinman
Department of Chemistry, University of California Berkeley, California 94720 Received September 5, 1980
Lycodoline (1, “alkaloid L.8”) is the second most widely oc- curring of the lycopodium alkaloids.’ It was first isolated in 1943 by Manske and Marion from L. annotinum Linn2 and its structure was established in 1961 by Ayer and Iverbach.’ In an attempted
(1) D. B. MacLean, Alkaloids, 14, 347 (1973). (2) R. H. Manske and L. Marion, Can. J . Res., Sect. B, 21, 92 (1943).
synthesis, Horii and co-workers succeeded in preparing tricyclic amino ketone Z4 but were unable to add the final ring.5 Because
0% 1: X = O H
3: X = H -
&*o H
2
of the close structural resemblance between lycodoline and ly- copodine (3), we have examined the use of intermediates employed in our lycopodine synthesis6 for construction of alkaloid 1. However, the presence of the sensitive tertiary alcohol function in lycodoline precludes the use of the acidic conditions required to effect the key Mannich cyclization in the synthesis of 3.6 In this communication, we report an interesting solution to this problem, which has culminated in the first total synthesis of (f)-lycodoline.
Amino diketal5, available in three steps (58% overall yield) from cyano enone 4,6 is treated briefly with 10% aqueous HCl, and the resulting solution is made basic with NaOH. The unstable
n
7 8
octahydroquinoline (6) is extracted with ethyl acetate, and the resulting solution is treated with oxygen gas and then hydrogen and Pd/C to obtain a mixture of alcohol 7 (mp 164-65 OC, 43%) and hemiketal 8 (oil, 4%). This interesting autoxidation finds precedent in the work of Cohen and Witkop on the parent octa- hydroquinoline.’ In the present case, it is noteworthy that the diastereomer having the angular oxygen and the neighboring acetonyl group trans predominates by a factor of 10:l. We postulate that this stereoselectivity arises from simple steric hindrance of approach of an oxygen molecule to the intermediate free radical.
The third ring is smoothly formed by heating a dilute solution of compound 7 (0.075 M) in a 5:l mixture of toluene and 3- bromopropanol a t reflux for 24 h. Neutralization of the resulting hydrobromide salt (which crystallizes from the hot solution) provides amino ketone 2 (mp 165-166 “C) in 85% yield. Many other attempts to accomplish this cyclization were unsuccessful. Since the product is a hydrobromide salt, a full equivalent of HBr is required. However, it appears to be crucial to the success of the reaction that the acid be added exceedingly slowly. Thus, if the hydrobromide salt of imine 7 is heated for 24 h in toluene, no cyclization occurs. 3-Bromopropanol functions as a source of HBr by slowly polymerizing under the reaction conditions. It is interesting to note that 3-bromopropanol is superior to 2- bromoethanol for this purpose.
(3) (a) W. A. Ayer and G. G. Iverbach, Tetrahedron Lett., No. 3, 87 (1961); (b) W. A. Ayer and G. G. Iverbach, Can. J. Chem., 42,2514 (1964). (4) Z.4. Horii, S.-W. Kim, T. Imanishi, and T. Momose, Chem. Pharm.
Bull., 18, 2235 (1970). (5) S.-W. Kim, Y. Bando, and 2.4. Horii, Tetrahedron Lett., 2293 (1978). (6) C. H. Heathcock, E. Kleinman, and E. S. Binkley, J . Am. Chem. Soc.,
lod, 8036 (1978). (7) L. A. Cohen and B. Witkop, J . Am. Chem. SOC., 77, 6595 (1955).
0002-7863/81/1503-222$01.00/0 0 1980 American Chemical Society

3442 J. Org. Chem. 1982,47, 3442-3447
6 0.9 (6 H, t, CHzCH3), 1.2 (19 H, br s, RzCHz, R3CH), 3.7 (3 H, m, RzCHOH, RCHzOH); IR 3400 cm-'.
Anal. Calcd for C14HMOz: C, 72.98; H, 13.12; mol wt, 230. Found: C, 72.81; H, 12.92; mol wt, 230 (MS).
A solution of 500 mg of the mixture of 3a and 3b, 2.5 mL of acetic anhydride, and 0.75 mL of pyridine was kept for 24 h at room temperature under anhydrous conditions. Workup gave 512 mg of the monoacetyl derivative: bp (Kugelrohr) 162-168 "C (0.2 torr); nz4D 1.4430" [lit.21 for 2-pentyl-3-acetoxynonyl heptanoate, bp 160-164 "C (1.0 torr); n'8'5D 1.448401.
Anal. Calcd for CBHUO4: C, 71.83; H, 11.52. Found C, 72.05; H, 11.68.
Oxidation of 3a and 3b. A mixture of 3a and 3b (390 mg) was dissolved in acetone (250 d) and Jones reagentm was added dropwise, with stirring, until a yellow color persisted. The chromium salta were filtered, most of the acetone was removed at reduced pressure, water was added, and the resulting solution was extracted with ether. Drying over sodium sulfate and removal of solvent gave 368 mg of material. Thin-layer chromatographic analysis indicated the presence of two compounds, which were separated by chromatography on silica gel into 73 mg (18%) of a carboxylic acid and 285 mg (76%) of neutral 2-pentyl-3-oxononyl heptanoate (8): bp (Kugelrohr) 138-142 "C (0.1 torr); nuD 1.4440"; 'H NMR 6 0.9 (9 H, t, CHzCH3), 1.3 (24 H, m, CH,), 2.1-2.8 (5 H, m, CHzC=O, CHC=O), 4.1 (2 H, d, CHzOCO); IR 1735,1720 cm-' (ester and ketone C=O).
Anal. Calcd for C21&&: C, 74.07; H, 11.84; mol wt, 340. Found: C, 73.76; H, 11.96; mol wt, 340 (MS).
The acid (9a) was treated with diazomethane and characterized as ita methyl ester, methyl 2-pentyl-3-(heptanoyloxy)nonanoate (9b): bp (Kugelrohr) 143-147 OC (0.2 torr); nuD 1.4410'; 'H NMR 6 0.9 (9 H, t, CHzCH3), 5.0 (1 H, m, RCHOCO); IR 1735 cm-' (ester C=O).
Anal. Calcd for CZzH4204: C, 71.31; H, 11.42; mol wt, 370. Found: C, 71.18; H, 11.42; mol wt, 370 (MS). 2-Pentyl-3-(bromoacetoxy)nonyl heptanoate (4a) and its
isomer (4b) could not be obtained analytically pure. GC of the sample at 245 "C showed both isomers, in a ratio of 3:1, principally 4a, as established by the 'H NMR spectrum. The sample had
(28) Bowers, A,; Halsall, T. G.; Jones, E. R. H.; Lemin, A. J. J. Chem. SOC. 1953, 2548-2560.
the following: bp (Kugelrohr) 187-193 "C (0.15 torr); nuD 1.4558"; IR 1735 cm-'; 'H NMR 6 0.9 (9 H, t, CH,CH3), 1.3 (27 H, br s, RzCHz, R3CH), 2.2 (2 H, t, CH,C=O), 3.8 (2 H, S, COCHzBr), 4.1 (2 H, d, RCHzOC=O), 5.0 (1 H, m, RzCHOC=O). Saponi- fication2' gave heptanoic acid and 2-pentyl-1,3-nonanediol (7); bromoacetic acid was not recovered in the workup.
Anal. Calcd for Cz3H43Br04: mol wt, 462. Found: mol wt, 462 (MS).
2-Pentyl-3-acetoxynonyl heptanoate (5a) and its isomer (5b), a mixture, had the following: bp (Kugelrohr) 162-166 "C (0.1 torr); nz4D 1.4427" [lit.z1 bp 160-164 OC (0.1 torr); n18,5D 1.4484"]; IR 1740 cm-'; 'H NMR 6 0.9 (9 H, t, CHzCH3), 1.3 (27 H, br s, R&Hz,
m, RCHzOC=O), 4.9 (1 H, m, RzCHOC=O). GC of the sample at 195 "C showed both possible isomers in a ratio of 6:l; the 'H NMR spectrum indicated the principal isomer to be 5a.
Anal. Calcd for C23H404: C, 71.83; H, 11.52; mol wt, 384. Found: C, 71.79; H, 11.53; mol wt, 384 (MS).
Saponificationz1 gave heptanoic acid and 2-pentyl-1,3-nonane- diol (7); acetic acid was not recovered in the workup.
2-Pentyl-3-[ (3- hydroxynonanoy1)oxy Inonyl heptanoate (6a), presumably mixed with its positional isomer 6b, had the following: bp (Kugelrohr) 208-214 "C (0.006 torr); nz4D 1.4532"; IR 3450,1735 cm-'; 'H NMR 6 0.9 (12 H, t, CHzCH3), 1.3 (37 H, br s, RzCHz, R3CH), 2.3-2.4 (2 H, m, CHzC=O), 3.9 (1 H, m, R&HOH), 4.0 (2 H, d, RCH,OC=O), 5.0 (1 H, m, RzCHOC=O).
Anal. Calcd for C30H5805: C, 72.24; H, 11.72; mol wt, 498. Found: C, 71.72; H, 11.55; mol wt, 498 (MS).
Saponificationz1 gave 2-pentyl-1,3-nonanediol (7) and two carboxylic acids, which were separated by chromatography into heptanoic acid and 3-hydroxynonanoic acid, mp 58-59 "C (lit.z6 57-59 "C).
Acknowledgment. This work was supported in part by a grant from the National Institute of Allergy and In- fectious Diseases (AI 04769).
Registry No. 1, 26257-80-7; 2,3021-89-4; 2 DNP, 10385-38-3; 3a, 49562-88-1; 3b, 55109-59-6; 4a, 82352-12-3; 4b, 82352-13-4; 5a, 82352-14-5; 5b, 82374-04-7; 6a, 82352-15-6; 6b, 82352-16-7; 7, 55109-63-2; 8, 82352-17-8; 9a, 82352-18-9; 9b, 82352-19-0; heptanal, 111-71-7; ethyl bromoacetate, 105-36-2; heptanoic acid, 111-14-8; 3-hydroxynonanoic acid, 40165-87-5.
RSCH), 2.0 (3 H, 9, OCOCH3), 2.2 (2 H, t, CHzC=O), 4.0 (2 H,
Jatrophone Analogues: Synthesis of cis- and trans -Normet hyljatropholactones
Amos B. Smith, III,*l and Michael S. Malamas Department of Chemistry, The Laboratory for Research on the Structure of Matter and The Monell Chemical
Received February 10, 1982
Senses Center, The University of Pennsylvania, Philadelphia, Pennsylvania 19104
This, a full account, discloses an efficient, convergent synthesis of two novel analogues of the macrocyclic antitumor diterpene jatrophone (1). We term these analogues cis- and trans-normethyljatropholactone (2 and 3, respectively). Our approach in each case begins with the bis(trimethylsily1oxy) ketone 7 and the requisite acetylenic or trans ester-aldehyde, 8 or 12a. Application of our previously developed 3(2H)-furanone synthetic protocol consisting of aldol condensation of the lithium enolate derived from 7 with the respective estepaldehydes 8 or 12a, followed by oxidation (Collins reagent) and acid-catalyzed cyclization-dehydration, affords spirofuranone 6c and 14c, respectively, in 52% and 45% overall yields. Sodium borohydride reduction, ester hydrolysis, and closure of the macrolide by employing the conditions of Mukaiyama (i.e., 1-methyl-2-chloropyridinium iodide- /Et3N/CH3CN) in the case of spirofuranone 14a leads directly to trans-normethyljatropholactone (3), while completion of cis-normethyljatropholactone (2) requires first semihydrogenation; the latter was accomplished by employing PdSOl in pyridine as the catalyst. The overall yields of 2 and 3, based on 7, were 23% and 21 %, respectively.
In connection with a synthetic program which recently culminated in the sucessful stereocontrolled total synthesis
0022-3263/82/ 1947-3442$01.25/0
of (f)-jatrophone i ts epimer (1b),2 and (*)-nor- methyljatrophone ( l ~ ) , ~ we have prepared two architec-
0 1982 American Chemical Society

cis- and trans-Normethyljatropholactones J. Org. Chem., Vol. 47, No. 18, 1982 3443
1 2
(a1 Jatrophone, R = wMe (b) Epijatrophone, R = p-Me (cl Normethyljatrophone, R = H
cis-Normethyljatropholactone
3 4
“xormethyl jatropholactone ( a ) R =n-Pr ( b ) R = protein
turally related analogues which we term cis- and trans- normethyljatropholactone (2 and 3, respectively). We record here a full account of the synthesis of these ana- logues. Our interest in such systems stemmed from the Kupchan observation4 that jatrophone, when treated in neutral or buffered solution with a variety of thiols, in- cluding cysteine hydrochloride and the free thiols of pro- teins such as bovine serum alubumin and DNA-dependent RNA polymerase, led to the formation of unstable adducts possessing the general structure 4. This reactivity was suggested by Kupchan to be responsible for the pro- nounced antileukemic activity (P-388 lymphocytic leuke- mia) and cytotoxicity (KB cell cultures) displayed by ja- trophone.28 It was anticipated that the availability of such synthetic analogues, in conjunction with jatrophone, epi- jatrophone, and normethyljatrophone, would allow further exploration of their relative antitumor properties as well as definition of the site(s) of reactivity with model biologic nucleophiles.
Results and Discussion From the retrosynthetic perspective, acetylenic macro-
lide 5 appeared to be an ideal advanced intermediate from which both 2 and 3 could in turn be elaborated via the stereocontrolled reduction of the acetylenic linkage. Central to this strategy was the prospect of exploiting our recently developed 3(W-furanone synthetic protocol5 for construction of spirofuranone 6a; subsequent macrocyclic lactonization would then afford 5.
(1) Camille and Henry Dreyfus Teacher Scholar, 1978-1983; National Institutes of Health (National Career Institute) Career Development Awardee.
(2) (a) Structure: Kupchan, S. M.; Sigel, C. W.; Matz, M. J.; Saenz Renauld, R. C.; Haltiwanger, R. C.; Bryan, R. F. J. Am. Chem. SOC. 1970, 92,4476. Kupchan, S. M.; Sigel, C. W.; Matz, M. J.; Gilmore, C. J.; Bryan, R. F. Zbid. 1976,98,2295 (b) Synthesis: Smith, A. B., 111; Schow, S. R.; Guaciaro, M. A.; Wovkulich, P. M.; Toder, B. H.; Malamas, M.; Hall, T. W., unpublished results.
(3) Smith, A. B., III; Guaciaro, M. A.; Schow, S. R.; Wovkulich, P. M.; Toder, B. H.; Hall, T. W. J. Am. Chem. SOC. 1981,103, 219.
(4) Kupchan, M. S.; Giacobbe, T. J.; Krull, I. S. Tetrahedron Lett. 1970, 2859. Lillehaug, J. R.: Kleuue. K.: Sigel. C. W.: Kuwhan, S. M. . - Biochem. Biophys. A& 1973,3if 92. ’ - ’
M., Jr.; Wovkulich, P. M. J. Am. Chem. SOC. 1981, 103, 1501. (5) Smith, A. B., III; Levenberg, P. A.; Jerris, P. J., Scarborough, R.
5 ’0
/ R’OOC
6
( a ) R = CH,OH; R ’ = H (b) R = CH,OH; R ’ = Et ( c ) R = CHO; R’ = Et
/ Me,SiO EtOOC
7 8
This scenario calls initially for construction of the silyl protected keto alcohol 7 and acetylenic aldehyde 8, the former prepared previously from a-(hydroxymethy1)- cyclopentenone (9).2bv6 The requisite aldehyde (8) was
/ HO
’r.. II I I R
9 10
(a ) R = H; R‘= COOH (b) R = H; R’=CH,OH
(d) R = COOEt; R ‘= CH,OH (c) R = COOH; R’ = CH2OH
prepared from readily available acetylenic acid loa.’ Reduction with LiA1H4 to the corresponding alcohol lob, followed by generation of the dianion with 2.2 equiv of n-BuLi in THF at -40 OC for 2 h and then addition of solid
(6) Initially, 2-(hydroxymethyl)-2-cyclopentenone (9) was prepared via the keto vinyl anion equivalent methodology developed in our and Swenton’s laboratory. See: Branca, S. J.; Smith, A. B., I11 J. Am. Chem. SOC. 1978,100, 7767. Smith, A. B., 111; Guaciaro, M. A.; Wovkulich, P. M. Tetrahedron Lett. 1978,4661. Also see: Manning, M. J.; Raynolds, P. W.; Swenton, J. S. J. Am. Chem. SOC. 1976,98,5008. Raynolds, P. W.; Manning, M. J.; Swenton, J. S. J. Org. Chem. 1980,45,4467. An altemate more economical preparation of (9) is outlined below: unpublished results of Mr. M. Malamas of our laboratory.
0 0 11 I l I LDA/THF.-7S°C II SePh O,ICH,CI,, -780 c
*OH
I21 PhSeBr
131 LDA/THF, -780 C -
141 HCHO
0
9
(7) Behrens, 0. K., Corse, J.; Huff, D. E.; Jones, R. G., Soper, Q. F.; Whitehead, C. W. J. Bid. Chem. 1948, 175, 771.

3444 J. Org. Chem., Vol. 47, No. 18, 1982 Smith and Malamas
Table I. High-Field (250 MHz) 'H NMR Data for cis- and trans-Normethyljatropholactone (2 and 3 ) and ( & E ) - and (E,Z)-Normethyljatrophone (IC and 11, Respectively)'
chemical shift Hb 2 3 IC 11
1-Ha 1-Hh
2.37 (ddd, 13.4, 8.2, 6.0) 2.13 (ddd. 13.4. 7.4. 3.0)
2-HI 2.62 (m) 2-HL 2.62 ( m j 3 -H 6.14 (br s) 5-HaC 4.98 (dq, 13.9, 1.1) 5-HbC 4.65 (dq, 13.9, 2.3) 7 -H 5.72 (d, 13.6) 8-H 5.84 (d, 13.6) 1 %Ha 2.25 (d, 13.6) 1 O-Hb 2.78 (d, 13.6) 15-CH,d 1.42 (s) 16-CH,d 1.30 (s) 17-CH3 1.64 (s)
2.29 (ddd, 14.1, 7.1, 6.7) 2.04 (ddd, 14.1, 7.1. 3.3) 2.58 (m) 2.58 (m) 6.55 (br s) 4.72 (dq, 13.8, 1.3) 4.27 (d, 13.8) 5.91 (d, 16.1) 6.53 (d, 16.1) 2.31 (d, 12.9) 2.78 (d , 12.9) 1.34 (s)
1.75 (s) 1.21 (s)
2.15 (dd, 13.1, 8.1) 1.85 (dd. 13.1. 6.4) 2.4 (m) 2.2 (m) 5.8 (m) 5.67. (m)
5.86 (d, 16.4) 6.35 (d, 16.4) 2.29 (d, 14.9) 2.77 ( d , 14.9) 1.24 ( s ) 1.13 (s) 1.62 (s)
2.35 (m) 2.10 (m) 2.40 (m) 2.40 (m) 6.08 (m) 6.94 (m)
5.62 (d, 14.0) 5.68 ( d , 14.0) 2.32 (d, 13.2) 2.64 (d, 13.2) 1.16 (s) 1.24 (s) 1.70 (s)
Chemical shifts are iven in parts per million downfield from (CH,),Si (a ) , with multiplicities and coupling constants (in
Assignments hertz) in parentheses. % Numbering for IC and 11 has been altered so that corresponding carbons in 2 , 3, IC, and 11 have the same number. for 15-CH3 and 16-CH, may be reversed in 2, 3, and 11.
Assignments for 5-H, could not be made with confidence in regard to stereochemistry.
Table 11. High-Field (62.9 MHz) NMR Data for cis- and trans-Normethyljatrolactone (2 and 3 ) and
Normethyljatrophone ( 1 ~ ) ~ chemical shift
carbon b f d 2 3 I C
34.2 30.6
122.2 135.6
61.1 165.6 135.8 146.6 38.8 40.4
184.3 112.1 204.8
98.5 34.5 26.4
6.24
35.4 30.6
117.9 136.4 62.5
168.8 144.8 152.4
39.6 42.8
184.1 112.4 205.1
98.7 29.0 24.0
6.55
33.5 30.4
123.1 137.8 140.6 201.1 128.2 158.7 36.3 40.9
182.8 112.0 203.7
98.8 30.0 26.5
5.66
Chemical shifts are reported in parts per million down- field from (CH,),Si (6 ). altered so that corresponding carbons in 2, 3, and IC have the same number. in 2 and 3 may be reversed. paren theses.
COP, afforded after an acidic workup acid 1Oc in 80% yield. This acid was then esterified with EtOH and oxidized with pyridinium chlorochromatea to afford the desired aldehyde 8 in 94% yield.
With ample quantities of both 7 and 8 in hand, we ex- ecuted the 3(2H)-furanone synthetic protocol. To this end, the lithium enolate of 7, generated via addition of 1.3 equiv of LDA in THF at -78 "C, was condensed with aldehyde 8 in THF. Without purification the derived aldol was subjected first to Collins oxidation (ca. 13 e q ~ i v ) ~ and then to acid-catalyzed cyclization-dehydration to afford spiro- furanone 6c in 51% overall yield.
With an efficient, convergent approach to 6c available, the stage was next set for macrocyclic ring closure. Re- duction of 6c with 1.1 equiv of NaBH, in MeOH at -23 "C for 10 min followed by hydrolysis with KzC03 in
Numbering for IC has been
Assignments for carbons 15 and 16 Multiplicity is given in
(8) Corey, E. J.; Suggs, J. W. Tetrahedron Lett. 1975, 2647. (9) Collins, J. E.; Hess, W. W.; Frank, F. 3. Tetrahedron Lett. 1968,
3363.
methanol-water (2:l) gave acid 6a in 77.4% yield for the two steps. Lactonization employing Mukaiyama's proce- dure'O (i.e., 1-methyl-2-chloropyridinium iodide/Et3N/ CH3CN, high dilution) then afforded the pivotal inter- mediate, macrolide 5, in 58% yield.
At this point all that remained to complete the synthesis of cis- and trans-normethyljatropholactone was the ste- reoselective reduction of the C(7,8) acetylenic linkage. As anticipated, semihydrogenation employing PdS04 in pyridine" afforded cis-normethyljatropholactone (2) as a crystalline solid, mp 101-103 OC (ether-hexane). That in fact 2 was in hand derives from careful comparison of the high-field 'H and I3C NMR spectra of 2 with those of la-c, as well as with that of 11, the latter prepared in connection with our normethyljatrophone synthesis (see Tables I and I I ) .~
' 11 12
( a ) R = CHO; R ' = E t (b) R = CH,OH; R ' = H (c) R = CH,OH; R ' = E t
Turning next to the conversion of macrolide 5 to trans-normethyljatropholactone (3), we initially antici- pated exploiting the well-known propensity of chromous sulfate to effect the stereoselective trans reduction of a- acetylenic acids and esters.12 This one-step plan, however, was thwarted presumably due to transannular reaction^.^ Undaunted by lack of initial success, we considered next isomerization of cis-normethyljatropholactone (2) to the desired trans isomer. Such a strategy, of course, was not
(10) Mukaiyama, T.; Usui, M.; Saigo, K. Chem. Lett. 1976, 49. (11) Cram, D. J.; Allinger, N. L. J. Am. Chem. SOC. 1956, 78, 2518.
Also see private communication to L. Fieser: Fieser, L.; Fieser, M. 'Reagents for Organic Synthesis"; Wiley: New York, 1967; p 566.
(12) Castro, C. E.; Stephens, R. D. J. Am. Chem. SOC. 1964,86,4358. Inanaga, J.; Kataski, T.; Takino, S.; Ouchida, S.; Inoui, K.; Nakano, A.; Okudako, N.; Yamaguchi, M. Chem. Lett. 1979,102. For preparation of this reagent see: Castro, C. E. J . Am. Chem. SOC. 1961, 83, 3262.

cis- and trans-Normethyljatropholactones
novel; i t was this approach that we had exploited to great advantage in our jatrophone s y n t h e s i ~ . ~ ~ ~ ~ However, to our dismay all attempts t o effect t h e required isomerization by employing numerous acids, light (with and without Iz), and/or transition-metal catalysts under a wide variety of time, temperature, and solvent regimes proved fruitless. Our only alternative at this point, aside from abandoning
the goal of tram-normethyljatropholacbne, was to prepare trans aldehyde ester 12a and then to subject this inter- mediate to the 3(2H)-furanone synthetic protocol. Toward this end, reduction of the previously prepared hydroxy acid 1Oc with aqueous chromous sulfate for 24 h at room tem- perature proceeded as anticipated to yield 12b in 78% as the sole product. Characteristic of t h e desired trans ole- finic geometry, the 250-MHz NMR spectrum displayed two doublets at 6 5.95 and 6.50 having a coupling constant of 16.2 Hz. Fischer e~ te r i f i ca t ion l~ then gave alcohol 12c in 92% yield, contaminated with 8 % of tetrahydrofuran 13, the latter arising via a facile 5-exo-trigonal cyclization1* of ester 12c.15 Subsequent Collins oxidation of 12c then led to aldehyde 12a.
J . Org. Chem., Vol. 47, No. 18, 1982 3445
presented in due course.
Experimental Section Materials and Methods. Melting points were taken on a
Thomas-Hoover capillary melting apparatus and are corrected. Boiling points are uncorrected. All solvents were distilled prior to use. Solutions were dried over MgSO,. Unless specified otherwise, IR and 'H NMR spectra were obtained for CC14 so- lutions, the former on a Perkin-Elmer Model 337 spectropho- tometer and the latter on either a Varian A-60A (60 MHz), a Varian T60A (60 MHz), or a Bruker WP 250 (250 MHz) spec- trometer. 13C NMR spectra were obtained in CDCl, on either a JEOL PS-100 (25 MHz) or a Bruker WP-250 FT (62.9 MHz) spectrometer; signal multiplicities were determined via off- res- onance decoupling. The internal standard for 'H and 13C NMR experiments was Me4Si. For those compounds containing the TBDMS protecting group, an external reference of either Me& or CHC13 (6 7.24) for CDC13 solutions was used. High-resolution mass spectra were obtained at the University of Pennsylvania Mass Spectrometry Service Center on a Hitachi Perkin-Elmer RMH-2 high-resolution double-focusing electron-impact spec- trometer or a VG micromass 70/70H high-resolution double-fo- cusing electron impact-chemical ionization spectrometer, the latter using isobutane as the reagent gas and each interfaced with a Kratos DS-504 data system. Preparative thin-layer chroma- tography (TLC) was performed on 500- or 1000-fim precoated silica gel plates with fluorescent indicator supplied by Analtech, Inc. Visualization was accomplished with UV light. Flash column chromatography and medium-pressure liquid chromatography were performed with silica gel 60 (0.04-0.063 mm) supplied by E. M. Merck.
Preparation of 3,3-Dimethyl-4-pentyn-l-ol (lob). To a mixture of 100 mL ether and 3.056 g (80.52 mmol) of lithium aluminum hydride under a nitrogen atmosphere at 0 "C was added 5.06 g (40.16 mmol) of 3,3-dimethyl-4-pentynoic acid in 30 mL of ether over a 30-min period. The mixture was stirred for 2 h after the addition. Solid sodium sulfate decahydrate was added to quench the reaction, and the resultant solid was filtered and washed thoroughly with ether. Concnetrated in vacuo afforded 4.28 g of a colorless oil, which was distilled (Kugelrohr, -3 mm, 49 "C) to give 4.21 g (93.5%) of a colorless oil: IR (CHC13) 3635, 3430 (br), 2980,2880 (s), 2120 (w), 1260 (br), 1065,1095 (w), 630 (w), cm-'; NMR (250 MHz, CDC13) 6 1.26 (s, 6 H), 1.72 (t, J = 7 Hz, 2 H), 2.16 (6, 1 H), 2.30 (br s, 1 H), 3.85 (t, J = 7 Hz, 2 H); mass spectrum, m / e 112.0823 (M'; calcd for C7Hlz0 112.0888).
Preparation of Ethyl 4,4-Dimethyl-6-hydroxy-2- hexynoate (loa). To a mixture of 250 mL of THF and 12.47 g of acetylenic alcohol 10b (111.3 mmol) under a nitrogen atmosphere at -42 "C (acetonitrile/COz) was added dropwise 131 mL (256.15 mmol) of n-BuLi. The mixture was stirred 2 h at -42 "C after completion of the n-butyllithium addition. Solid carbon dioxide was then added to the solution, and the thick solution was stirred for 0.5 h followed by acidification with 6 N hydrochloric acid. The aqueous layer was extracted four times with ether and the solution dried. Concentration in vacuo gave 15.7 g of a colorless oil that crystallized upon standing; the latter was employed in the next step without purification.
A mixture of 100 mL of absolute ethanol, 6.8 g (43.58 mmol) of hydroxy acid lOc, and 1 mL of concentrated sulfuric acid was stirred at room temperature for 64 h. Dilution in water and extraction with ether afforded 7.95 g of colorless oil. Purification via flash column chromatography (5:l hexane/ethyl acetate) afforded 7.86 g (98%) of colorless oil: IR (CHCl,) 3630-3450 (br), 2980, 2930 (s), 2210 (s), 1690 (s), 1260 (br), 905 (w) cm-'; NMR
(t, J = 7.25 Hz, 2 H), 2.79 (br s, 1 H), 3.83 (t, J = 7.25 Hz, 2 H), 4.23 (q, J = 7.0 Hz, 2 H); mass spectrum, m / e 184.1100 (M+; calcd for C10H1603 184.1100).
Anal. Calcd for C10H1603: C, 65.20; H, 8.70. Found: C, 65.04; H, 8.79.
Preparation of Ethyl 4,4-Dimethyl-6-oxo-%-hexynoate (8). To a mixture of 160 mL dry methylene chloride and 26.35 g (122.28 mmol) pyridinium chlorochromate under a nitrogen atmosphere at room temperature was added 9.02 g (49.02 mmol) of ester 10d in 50 mL of methylene chloride. The mixture was stirred for 3.5 h at room temperature, diluted in 800 mL ether, and filtered
(250 MHz, CDCl3) S 1.30, (9, 6 H), 1.30 (t, J = 7.0 Hz, 3 H), 1.78
13
R'OOC
14
(a) R = CH,OH; R ' = H (b) R = CH,OH; R ' = E t ( c ) R = CHO; R ' = Et
Executioq of the 3(2H)-furanone synthetic protocol [(a) aldol condensation, (b) Collins oxidation, (c) cyclization- dehydration] as employed above led t o spirofuranone 14c in 45% overall yield. Reduction of t he aldehyde func- tionality with sodium borohydride in methanol (14c - 14b) followed by saponification and macrolactonization of t h e resultant hydroxy acid (14a) again a lb Mukaiyama afforded tram-jatropholactone (3) as a crystalline solid [mp 168-170 "C (ether-hexane)], t he overall yield from spiro- furanone 14c being 42%. Indicative of the trans geometry the olefinic resonances display as two doublets (6 5.91 and 6.53) with t h e characteristic trans coupling of 16.1 Hz.
In summation, an economic convergent synthesis of two analogues of jatrophone has been achieved in 20% and 19% yields (2 and 3, respectively) based on 7. Biological screening data as well as NMR studies t o correlate t he solution conformation vis-&vis the bioactivity will be
(13) Fischer, E. J. Physiol. Chem. 1901, 33, 151. (14) Baldwin, j. E. J. Chem. SOC., Chem. Commun. 1976,734. Bald-
win, J. E.; Cutting, J.; Dupont, W.; Kruse, L.; Silberman, L.; Thomas, R. C. Ibid. 1976,736. Baldwin, J. E. Zbid. 1976,738. Baldwin, J. E.; Thomas, R. C.; Kruse, K. I.; Siverman, L. J. Org. Chem. 1977, 42, 3846.
(15) Interestingly, the propensity with which the analogous hydroxy enone i displayed for this 5-exc-trigonal cyclization (i - ii) precluded its utilization as an intermediate in our jatrophone approach unpublished results of Dr. S. R. Schow of this laboratory.
i ii

3446 J. Org. Chem., Vol. 47, No. 18, 1982
through Florisil(lO0-200 mesh). Removal of the solvent in vacuo and distillation (Kugelrohr, -1 mm, 110-120 "C) gave 8.47 g (93.9%) of a colorless oil: IR (CHC13, 2980, 2930 (s), 2210 (m), 1700 (s), 1260 (br), 1040 (br), 850 (w) cm-l; NMR (250 MHz,
2.0 Hz, 2 H), 4.24 (4, J = 7.25 Hz, 2 H), 9.86 (t, J = 2.0 Hz, 1 H); mass spectrum, m / e 182.0715 (M+; calcd for C10H1403 182.0709).
Preparation of trans -Ethyl 4,4-Dimethyl-6-hydroxy-2- hexenoate (12c). To a mixture of 46 mL of degassed water (nitrogen purge for 2 h) and 4.2 g (26.92 mmol) of hydroxy acid 1Oc was added under a nitrogen atmosphere at room temperature 229 mL of 0.4 M CrS04 over a 30-min period. The reaction solution first became green and then 10 min later turned dark. The mixture was stirred for 12 h at room temperature. The dark reaction solution was then basified with aqueous KOH and vacuum filtered from chromic hydroxide. The filtrate was next acidified (concentrated H2SO4) to pH 1 and extracted with ethyl acetate. The organic extracts were dried over sodium sulfate, filtered, and conentrated in vacuo to afford 3.31 g (78.6%) of 12b as a colorless oil which was used without purification.
A mixture of 100 mL of absolute ethanol, 7.4 g (46.83 mmol) of 12b, and 0.5 mL of concentrated sulfuric acid was stirred at room temperature for 50 h. Dilution in water and extraction with ether afforded 8.53 g (98%) of a colorless oil. Purification via flash column chromatography (101 hexane/ethyl acetate) afforded 7.89 g (90.6%) of 12c as a colorless oil: IR (CC14) 3630,3400 (br), 2980,2930 (s), 1700,1650 (s), 1300,1200 (br), 1035 (br), 995 (w), 870 (w) cm-'; NMR (250 MHz, CDClJ 6 1.1 (s, 6 H), 1.30 (t, J = 7.0 Hz, 3 H), 1.71 (t, J = 7.75 Hz, 2 H), 2.30 (9, 1 H), 3.62 (t, J = 7.75 Hz, 2 H) 4.21 (9, J = 7.0 Hz, 2 H), 5.76 (d, J = 16.2 Hz, 1 H) 6.95 (d, J = 16.2 Hz, 1 H); mass spectrum, m / e 186.1311 (M+; calcd for C10H18O3 186.1290).
Anal. Calcd for Cl0Hl8O3: C, 64.51; H, 9.67. Found C, 64.42; H, 9.69.
Preparation of Ethyl trans-4,4-Dimethyl-6-0~0-2-hex- enoate (12a). To a mixture of 120 mL of methylene chloride and 19.03 g (88.28 mmol) of pyridinium chlorochromate under a ni- trogen atmosphere a t room temperature was added 6.6 g (34.94 mmol) of ester 12c in 40 mL of methylene chloride. The mixture was stirred for 3.5 h at room temperature, diluted in 700 mL of ether, and filtered through Florisil(100-200 mesh). Removal of solvent in vacuo and distillation (Kugelrohr, -1 mm, 115-130 OC) gave 6.07 g (93%) of 12a as a colorless oil: IR (CC14) 2980, 2930 (s), 1700 (s), 1250 (br), 1030 (br) 850 (w) cm-', NMR (250
J = 2.1 Hz, 2 H), 4.24 (4, J = 7.0 Hz, 2 H), 5.81 (d, J = 16.2 Hz, 1 H), 6.90 (d, J = 16.2 Hz, 1 H), 9.75 (t, J = 2.1 Hz, 1 H); mass spectrum, m / e 184.1093 (M+ calcd for C10H16O3 184.1100).
Preparation of Spirofuranone (6c). To a solution of 0.62 mL (4.38 mmol) of diisopropylamine in 3 mL of THF containing a few crystals of 2,2'-dipyridylamine as an indicator at 0 "C under a nitrogen atmosphere was added 2.40 mL (3.41 mmol, 1.42 M solution) of n-butyllithium. The resultant deep red solution was stirred 30 min at 0 "C and then cooled to -78 "C. After 5 min bis(sily1oxy) ketone 7 (765 mg, 2.44 mmol) in THF (3 mL) was added over a period of 5 min. The red mixture was stirred 2 h, and then 896 mg (4.92 mmol) of aldehyde 8 in mL of THF was added over a 50-9 period. The color of the reaction mixture changed to orange-brown; stirring was continued for 2 min, and then saturated ammonium chloride (2 mL) was added. The mixture was diluted with 3 mL of water and extracted thoroughly with ether. The combined extracts were washed with water and brine. Removal of solvent in vacuo gave 1.34 g of yellow oil which was employed in the next step without purification.
To a solution of 7.49 mL (92.61 mmol) of pyridine in 100 mL of methylene chloride was added 4.41 g (44.1 mmol) of chromium trioxide (CrOJ. After the mixture was stirred 20 min at room temperature under a nitrogen atmosphere, the above hydroxyl ketone (1.34 g) in 5 mL of CH2C12 was added and the stirring continued for 3 h. The organic layer was decanted from the black residue and the latter washed with ether. Conventional workup and removal of solvent in vacuo gave 1.09 g of a light brown oil, which was taken up in 50 mL THF with 25 mL of 10% HCl and stirred at room temperature under a nitrogen atmosphere for 48 h. The mixture was then saturated with solid NaCl and diluted with ether. The organic layer was washed and dried. Removal
CDC13) 6 1.31 (t, J , = 7.25 Hz, 3 H), 1.40 (9, 6 H), 2.54 (d, J =
MHz, CDCl3) 6 1.25 (9, 6 H), 1.32, (t, J = 7.0 Hz, 3 H), 2.48 (d,
Smith and Malamas
of the solvent in vacuo gave 657 mg of a viscous yellow oil which was purified via medium-pressure liquid chromatography [hex- ane/ethyl acetate (2:1)] to afford 409.1 mg (50.8% from 7) of 6c as a pale yellow oil: IR (CC14) 2995, 2930 (s), 2210 (s), 1700 (s), 1630 (s), 1250 (br), 1035 (m), 920 (s) cm-'; NMR (250 MHz, CDC13) 6 1.30 (t, J = 7.3 Hz, 3 H), 1.38 (s, 3 H), 1.39 (s, 3 H), 1.79 (s, 3 H), 2.27 (m, 1 H), 2.44 (m, 1 H), 2.70 (s, 2 H), 2.80 (m, 2 H), 4.20 (q, J = 7.0 Hz, 2 H), 7.33 (m, 1 H), 9.64 (s, 1 H); mass spectrum; m / e 330.1403 (M'; calcd for C19H2205 330.1467).
Preparation of Spirofuranone 6b. To a mixture of 4 mL of methanol and 244 mg (0.74 mmol) of spirofuranone 6c under a nitrogen atmosphere at -23 "C (CC14/C02) was added 27.97 mg (0.74 mmol) of sodium borohydride. The mixture was stirred for 15 min at -23 "C before the pH was adjusted to 7 with dilute aqueous HC1. The mixture was extracted with ether, and the organic extract was dried and concentrated in vacuo to afford 219.6 mg of a viscous colorless oil which was purified via medium- pressure liquid chromatography [hexane/ethyl acetate (2:1)] to afford 203 mg (83.2%) of 6b as a colorless oil: IR 3500,3400 (br), 2980, 2930 (s), 2220 (m), 1700 (s), 1630 (s), 1260, 1100 (br), 1030 (m), 750 (br) cm-'; NMR (250 MHz, CDC13) 6 1.29 (t, J = 7.0 Hz, 3 H), 1.40 (9, 3 H), 1.43 (s, 3 H), 1.74 (s, 3 H), 2.21 (m, 1 H), 2.40 (m, 1 H), 2.57 (m, 2 H), 2.71 (s, 2 H), 4.03 (m, 2 H), 4.20 (4, J = 7.0 Hz, 2 H), 6.21 (t, J = 1.8 Hz, 1 H); mass spectrum, m / e 332.1620 (M+; calcd for C19H24O5 332.1624).
Preparation of Spirofuranone 6a. To a solution of 5 mL of MeOH, 2 mL of H20 and 170 mg (0.512 "01) of spirofuranone 6b at room temperature was added a concentrated aqueous K2C03 solution, adjusting the pH of the reaction mixture to 10.5-11.0. The mixture was then stirred 15 h at room temperature under nitrogen and diluted with 5 mL of H20, and the pH was adjusted to 2. The mixture was extracted extensively with ether, and the organic fraction was dried and concentrated in vacuo to afford 144.7 mg (93.0%) of acid 6a: IR 3450,3300,2500 (br), 2980,2930 (s), 2220 (m), 1700, 1630 (s), 1450 (s), 1200 (s), 700 (br) cm-'; NMR
(m, 1 H), 2.40 (m, 2 H), 2.61 (m, 2 H), 2.81 (d, J = 13 Hz, 1 H), 4.05 (m, 2 H), 5.75 (br s, 1 H), 6.20 (t, J = 1.8 Hz, 1 H), COOH not observed.
Preparation of the Macrolide 5. To a solution consisting of 767 mg (3.0 mmol) of 1-methyl-2-chloropyridinium iodide in 73 mL of dry acetonitrile held at reflux was continuously and uniformly added a solution of 230 mg (0.75 mmol) of 6a and 0.84 mL (6 mmol) of triethylamine in 65 mL of dry acetonitrile over a period of 9 h. After one additional hour at reflux, evaporation of the solvent under reduced pressure afforded a residue which was separated via silica gel column chromatography to afford 125.6 mg (57.8%) of 5 as a viscous colorless oil that crystallized upon standing: mp 125-127 "C; IR (CHCl,) 2980, 2930 (s), 220 (m), 1700, 1630 (s), 1500,1400 (m), 1180,1200 (br) cm-'; NMR (250 MHz, CDC1,) 6 1.45 (s, 3 H), 1.49 (s, 3 H) 1.78 (s, 3 H), 2.20 (m, 1 H), 2.41 (m, 1 H), 2.60 (d, J = 13.2 Hz, 1 H), 2.65 (m, 2 H) 2.81 (d, J = 13.2 Hz, 1 H) 4.64 (s, 2 H), 6.47 (t, J = 1.9 Hz, 1 H); mass spectrum, m / e 286.1201 (M+; calcd for C17H1804 286.1205).
Preparation of cis-Normethyljatropholactone (2). A suspension of 10 mg of 5% palladium on barium sulfate in 2 mL of pyridine was stirred under hydrogen at atmospheric pressure for 30 min, whereupon 153 mg of macrolide 5 in 200 mL of pyridine was added and the stirring under hydrogen continued for 15 min. The reaction mixture was then filtered through Celite with a CH2C12 wash. Removal of the solvent in vacuo yielded 2 as a viscous yellow oil that crystallized upon standing. Recrys- tallization from hexane/ethyl ether (1O:l) gave 144.7 mg (94.6%) of a white crystalline solid: mp 101-103 OC; IR (CHC13) 2980, 2930 (s), 1700,1620 (s), 1220 (br), 1090 (m), 715 (br) cm-'; NMR
(m, 1 H), 2.28 (d, J = 13.6 Hz, 1 H), 2.35 (m, 1 H), 2.60 (m, 2 H), 2.75 (d, J = 13.6 H3, 1 H), 4.71 (m, 1 H), 5.05 (m, 1 H), 5.72 (d, J = 13.3 Hz, 1 H), 5.85 (d, J = 13.3 Hz, 1 H), 6.14 (t, J = 1.5 Hz, 1 H); mass spectrum, m / e 288.1351 (M+; calcd for C17H2004 288.1361).
Anal. Calcd for C17H&: C, 70.80; H, 6.95. Found C, 70.60; H, 7.03.
Preparation of Spirofuranone 14c. To a solution of 0.45 mL (3.27 "01) of diisopropylamine in 2.5 mL of THF containing a few crystals of 2,2'-dipyridylamine as indicator at 0 "C under
(250 MHz, CDC13) 6 1.37 (9, 3 H), 1.41 (s, 3 H), 1.70 (s, 3 H), 2.05
(250 MHz, CDC13) 6 1.30 (9, 3 H), 1.42 (s, 3 H), 1.64 (s, 3 H), 2.20

J. Org. C h e m . 1982,47, 3447-3450 3447
a nitrogen atmosphere was added 1.88 mL (2.54 mmol, 1.35 M solution) of n-butyllithium. The resultant deep red solution was stirred 30 min at 0 "C and cooled to -78 "C. After 5 min bis- (silyloxy) ketone 7 (507.2 mg, 1.62 mmol) in 3 mL of THF was added over a period of 5 min. The red mixture was stirred 2 h, and then 675.5 mg (3.67 mmol) of aldehyde 12a in 1 mL of THF was added over a 50-s period. The color of the reaction mixture changed to orange-brown; stirring was continued for 2 min, and then saturated ammonium chloride (2 mL) was added. The mixture was diluted with 3 mL of water and extracted thoroughly with ether. The combined organic extracts were washed with water and dried. Removal of the solvent in vacuo gave 767 mg of yellow oil which was employed in the next step without pu- rification.
To a solution of 5.6 mL (68.6 mmol) of pyridine in 65 mL of methylene chloride was added 3.25 g (32.5 mmol) of chromium trioxide (&Os). After the mixture was stirred 20 min at room temperature under a nitrogen atmosphere, the above hydroxy ketone (767 mg) in 5 mL of CHzClz was then added and the stirring continued for 3 h. The organic layer was decanted from the dark residue, and the latter was washed with ether. A con- ventional workup and removal of solvent in vacuo gave 585 mg of a light brown oil, which was taken up in 40 mL of THF with 20 mL of 5% aqueous HCl and stirred at room temperature under a nitrogen atmosphere for 5 days. The mixture was then saturated with solid NaCl and diluted with ether. The organic layer was washed and dried, and the solvent was removed in vacuo to give 355 mg of a viscous yellow oil, which was purified via medium- pressure liquid chromatography [hexane/ethyl acetate (2:1)] to afford 241.3 mg (45% from 7) of 14c as a pale yellow oil: IR (CHC13) 2980, 2930 (s), 1700, 1630 (s), 1200, 1300 (br), 1040 (s), 920, 840 (m) cm-l; NMR (250 MHz, CDC13) 6 1.12 (s, 6 H), 1.20 (t, J = 7.25 Hz, 3 H), 1.66 (s, 3 H), 2.10 (m, 1 H), 2.35 (m, 1 H), 2.51 (d, J = 10.25 Hz, 2 H), 2.81 (m, 2 H), 4.20 (4, J = 7.25 Hz, 2 H), 5.75 (d, J = 16.2 Hz, 1 H), 6.94 (d, J = 16.2 Hz, 1 H), 7.20 (t, J = 1.5 Hz, 1 H), 9.61 (s, 1 H); mass spectrum, m / e 332.1615 (M+; calcd for ClgH2405 332.1693).
Preparation of Spirofuranone 14b. To a mixture of 4-mL of methanol and 290 mg (0.873 mmol) of spirofuranone 14c under a nitrogen atmosphere at -23 "C (CC14/C02) was added 33.78 mg (0.893 mmol) of sodium borohydride. The mixture was stirred for 15 min at 23 "C before the pH was adjusted to 7 with dilute aqueous HCl. The mixture was extracted extensively with ether. The organic material was then dried and concentrated in vacuo to afford 275.3 mg of a viscous colorless oil which was purified via medium-pressure liquid chromatography [hexane/ethyl acetate (21)] to give 231.3 mg (79.5%) of 14b as a colorless oil: IR (CHC13) 3450,3400 (br), 2980,2930 (s), 1700,1625 (s), 1480 (m), 1200 (br), 1040 (s) cm-'; NMR (250 MHz, CDClJ 6 1.15 (s, 6 H), 1.22 (t, J = 7.8 Hz, 3 HI, 1.72 (s, 3 H), 2.10 (m, 1 H), 2.35 (m, 1 H), 2.60 (br m, 4 H), 3.96 (br d, J = 2 Hz, 2 H), 4.22 (9, J = 7.8 Hz, 2 H),
5.75 (d, J = 16.3 Hz, 1 H), 6.20 (br s, 1 H), 7.03 (d, J = 16.3 Hz, 1 H); mass spectrum, m / e 334.1746 (M'; calcd for CI9Hz6O5
Preparation of Spirofuranone 14a. To a solution of 8 mL of MeOH, 2 mL of HzO, and 250 mg (0.748 "01) of spirofuranone 14b at room temperature was added a concentrated aqueous K2C03 solution, adjusting the pH of the reaction mixture to 10.5-11.0. The mixture was stirred 12 h at room temperature under nitrogen and then diluted with 5 mL of HzO, and the pH was adjusted to 7. The mixture was extracted extensively with ether, and the organic fraction was dried and concentrated in vacuo to afford 231.3 mg (91.5%) of 14a: IR (CHC1,) 3450-2500 (br) 2980-2930 (s) 1700 (s), 1450 (br), 1080 (m), 850 (m) cm-';
H), 2.1 (m, 1 H), 2.35 (m, 2 H), 2.55 (m, 2 H), 2.70 (m, 1 H), 3.59 (br s, 1 H), 3.95 (m, 2 H), 5.18 (br I, 1 H), 5.75 (d, J = 16.2 Hz, 1 H), 6.10 (e, 1 H), 6.50 (d, J = 16.2 Hz, 1 H).
Preparation of trans-Normethyljatropholactone (3). To a solution consisting of 654 mg (2.55 mmol) of 1-methyl-2- chloropyridinium iodide in 65 mL of acetonitrile held at reflux was continuously and uniformly added a solution of 195 mg (0.64 mmol) of spirofuranone 14a and 0.72 mL (5.12 mmol) of tri- ethylamine in 55 mL of dry acetonitrile over a period of 9 h. After one additional hour at reflux evaporation of the solvent under reduced pressure followed via silica gel column chromatography afforded 138 mg of a viscous colorless oil that crystallized upon standing. Recrystallization [hexane/ethyl ether (lOl)] gave 104.0 mg (56.8%) of 3 as a white crystalline solid: mp 168-170 "C; IR 2980,2930 (s), 1700,1630 (s), 1460 (m), 1210 (m), 720 (br) cm-l;
3 H), 1.15 (m, 1 H) 2.28 (m, 1 H), 2.33 (d, J = 13 Hz, 1 H), 2.60 (m, 2 H), 2.80 (d, J = 13 Hz, 1 H), 4.31 (d, J = 14 Hz, 1 H), 4.75 (m, 1 H), 5.95 (d, J = 16.1 Hz, 1 H), 6.50 (d, J = 16.1 Hz, 1 H) 6.51 (t, J = 1.5 Hz, 1 H); mass spectrum, m / e 288.1359 (M'; calcd for C17H20O4 288.1362).
Acknowledgment. It is a pleasure t o acknowledge the support of this investigation by the National Institutes of Health (National Cancer Institute) through Grant Ca- 22807. In addition, we thank Mr. S. T. Bella of t he Rockefeller University for the microanalyses and Drs. G. Furst and T. Terwilliger of t he University of Pennsylvania Spectroscopic Service Centers for aid in recording and interpertation of the high-field NMR and mass spectra, respectively.
Registry No. 2, 82351-40-4; 3, 82398-40-1; 5, 82351-41-5; 6a,
45-9; loa, 67099-40-5; lob, 67099-41-6; lOc, 82351-46-0; 10d, 82351- 47-1; 12a, 82351-48-2; 12b, 82351-49-3; 1212, 82351-50-6; 14a, 82351-
334.1781).
NMR (250 MHz, CDClJ 6 1.15 (9, 3 H), 1.20 (9, 3 H) 1.65 (5, 3
NMR (250 MHz, CDCLJ 6 1.21 (5, 3 H), 1.34 (5, 3 H), 1.75 (5,
82351-42-6; 6b, 82351-43-7; 6 ~ , 82351-44-8; 7, 76445-18-6; 8, 82351-
51-7; 14b, 82351-52-8; 1 4 ~ , 82351-53-9.
Cobalt-Mediated [2 + 2 + 21 Cycloadditions En Route to Natural Products: A Novel Total Synthesis of Steroids via the One-Step Construction of the
B,C,D Framework from an A-Ring Precursor
Ethan D. Sternberg and K. Pe ter C. Vollhardt* Department of Chemistry, University of California, and the Materials and Molecular Research Division,
Received February 9, 1982
Lawrence Berkeley Laboratory, Berkeley, California 94720
The first application of the cobalt-mediated intramolecular cyclization of a&-diynenes to annulated cy- clohexadienes in natural product synthesis is described by demonstrating its feasibility in a versatile and efficient steroid synthesis, including a new total synthesis of the Torgov intermediate, 3-methoxyestra-l,3,5(10),8,14- pentaen-17-one, via a new steroid, 3-methoxyestra-1,3,5(10),8(14),9-pentaen-17-one ethylene ketal. Several model reactions en route to B-homo-7-oxa steroids allow the delineation of some stereochemical details of the tran- sition-metal-catalyzed [2 + 2 + 21 cycloaddition reaction.
We have recently developed methodology based on co- balt-mediated [2 + 2 + 21 cycloadditions of unsaturated
0022-3263/82/1947-3447$01,25/0
substrates which yields annulated and complexed five-l and six-membered2 rings. We believe t h a t this strategy
0 1982 American Chemical Society





























