Embed Size (px)
Citation preview

Eur. J. Biochem. 22 (1971) 166-172
Isolation and Properties of tRNA Nucleotidyl Transferase from Yeast
Hans STERNBACH, Friedrich VON DER HAAR, Eckhard SCHLIMME, Erika GAERTNER, and Friedrich CRAMER
Max-Planck-Institut fur Experimentelle Medizin, Abteilung Chemie, Gottingen
(Received June 4, 1971)
1. tRNA nucleotidyl transferase has been purified 5000-fold from commercial bakers’ yeast. The isolation procedure includes ammonium sulfate fractionation and chromatography on DEAE- cellulose, CM-cellulose, Biogel and phosphocellulose.
2. The enzyme is homogeneous on analytical centrifugation and by sodium dodecylsulfate gel electrophoresis.
3. The molecular weight of the native and denatured enzyme M:=O is 71000 as determined by ultracentrifuge measurements and 70000 as determined by sodium dodecylsulfate gel electrophoresis.
4. On micro isoelectric focusing the enzyme shows an isoelectric point a t about pH 7.5. 5. The enzyme catalyses the incorporation of ATP and CTP into the 3‘-end of tRNA, and,
in the presence of pyrophosphate, the pyrophosphorolyses of adenylic and cytidylic acid residues from the 3’-end of tRNA.
tRNA nucleotidyl transferase has been purified from different sources [l - 121. Hitherto the enzyme from yeast was only partially purified and charac- terized by Morris et al. [13] but it is desirable for many reasons to have homogeneous enzyme. In vitro the enzyme catalyses the reaction
tRNA . . . + 2 CTP + I ATP + tRNApCpCpA + 3 PPi.
I n the presence of pyrophosphate this reaction is completely reversible. The physiological role of tRNA nucleotidyl transferase is as yet uncertain. Its most plausible function seems to be its capability to com- plete the processing of the precursor tRNA by adding the 3’-end.
The tRNA nucleotidyl transferase recognizes not only the different tRNAs in homologous systems, but is also active with all tRNAs in heterologous systems. I n comparison with the aminoacyl-tRNA synthetases the specificity of this enzyme is less mark- ed and its structure probably less complicated. There- fore this enzyme is most useful for studies of specific interactions with natural and modified tRNAs. Another important point is that the enzyme is able to incorporate modified nucleotides a t the 3‘-end of tRNA [14,15], thereby allowing one to introduce certain labels or substituted nucleotides, which may be useful for structural studies on the tRNA. Since unnatural nucleotides are usually incorporated at slower rates it is essential to have an enzyme free
Enzymes. tRNA nucleotidyl transferase or ATP, CTP : tRNA nucleotidyl transferase or -C-C-A pyrophosphorylase (EC 2.7.7.-); DNA-dependent RNA polymerase (EC n n n c , 4 . t . t . U ) .
from nucleolytic contaminations in order to avoid side reactions.
In this paper we described an isolation procedure and some properties of tRNA nucleotidyl transferase from commercial bakers’ yeast.
EXPERIMENTAL PROCEDURES
MATERIALS
The source of the enzyme was commercial bakers’ yeast from C. Langemeyer (BRD-4532 Mettingen). Analytically pure ammonium sulfate was purchased from J. T. Baker Chemicals (N. V. Deventer, Hol- land). Whatman microgranular DEAE-cellulose DE- 52 (1 .O mequiv./g dry), Whatman microgranular carboxymethyl-cellulose CM-52 (1.0 mequiv./g dry), and Whatman cellulose-phosphate Pll(7.4 mequiv./g dry) were purchased from L. Hormuth-Vetter (BRD- 6800 Heidelberg-Wiesloch) and Biogel P150 (100 to 200 mesh) from Biorad Lab. (BRD-8000 Miinchen 66). Yeast bulk tRNA (analytical grade) was a product from Boehringer Mannheim GmbH (BRD-6800 Mann- heim, Germany). Dithiothreitol (Cleland’s reagent) was purchased from E. Barth (BRD-6804 Ilvesheim) ; acrylamide, N,N’-methylene-bis-acrylamide, N,N, N’,N’-tetramethylethylenediamine and coomassie brillant blue R-250 from Serva Entwicklungslabor (BRD-6900 Heidelberg) ; LKB-Ampholine 400/, pH 3- 10 from Colora Messtechnik (BRD-7073 Lorch). Labelled and unlabelled nucleotides were products from Schwarz Bioresearch Inc. (Orangeburg, U.S.A.), pancreatic DNAase I (2500 units/mg) from Worthing- ton Biochemical Corp. (Freehold, U.S.A.) and bovine

Vo1.22, No.2.1971 H. STERNBACII, F. VON DER HAAR, E. SCHLIMME, E. GAERTNER, and F. CRAMER 167
serum albumin RHD 04 from Behring-Werke (BRD- 3550 MarburglLahn). The glass beads were purchased from B. Braun (BRD-3508 Melsungen). All other chemicals were of the highest purity available from commercial sources. Doubly distilled water was used for all solutions. The DNA-dependent RNA poly- merase was prepared according to the procedure of Burgess [ 161.
Buffers Buffer A contained 0.2 M Tris pH 8.5, 0.02M
MgCI,, 0.1 M NH,Cl, 0.001 M EDTA. Buffer B con- tained 0.05 M Tris pH 7.6, 0.02 M MgCl,, 0.03 M NH,CI, 0.001 M EDTA. Buffer C contained 0.03 M potassium phosphate pH 7.3, 0.001 M EDTA, 0.1 mM dithiothreitol. Buffer D contained 0.05 M sodium phosphate pH 7.2, 0.001 M EDTA, 0.001 M dithiothreitol.
METHODS
Enzyme Assay The method used is modified from that of Miller
et al. [lo] and Carre et al. [ll], and is based on the incorporation of labelled AMP or CMP from ATP or CTP into the 3'-terminal position of tRNA. The reaction mixture, unless indicated otherwise, con- tained in a final volume of 50 pl: 6 pmoles of Tris- HCI pH 9.0, 2 pmoles of KCI, 0.4 pmole of MgSO,, 0.032 pmole of CTP, 0.1 mg of bulk tRNA from yeast, 0.05 pmole [3H]ATP (100000 counts/min per 0.01 pmole), and enzyme solution (1-30 pg protein). The reaction was carried out a t 32 "C for different times, after which normally 10 p1 of the mixture were spotted on a Whatman 3MM paper disc. The discs were washed twice during 20 min with 5*/, trichloro- acetic acid and finally with ethanol and ether. After drying the discs were placed in 7ml toluene-based scintillator solution and counted in a Packard Tricarb liquid scintillation counter (Model 3375). One unit of the enzyme is defined as the amount of enzyme able to incorporate 1 nmole of C3H]ATP into tRNA in one minute a t 32 "C under the conditions mentioned above. Protein was determined by the procedure of Lowry et al. [17] using serum albumin as a standard. The protein concentration of the purified enzyme was measured by the absorbance at 280 nm and 260 nm according to Warburg et al. [18].
Column Chromatography Whatman DEAE- and CM-cellulose were washed
extensively with buffer C before they were poured into the columns. The phosphocellulose was first washed with 0.3 N NaOH for 20 to 30 min and rinsed until the pH was about 8.0. Then the cake was resus- pended in 0.3 N HCI for 20 to 30 min and rinsed again until the pH was about 5.0. The resin was
titrated with 3.0N KOH to pH 7.2 before it was poured into the column. The column was equilibrated a t 2 "C with buffer D until the pH of the eluate was identical to that of the buffer D. The Biogel Pi50 was suspended in buffer D being additionally 0.1 M in KCI and the slurry was poured into the column.
Isolation Procedure Preparation of the Crude Extract. All steps were
carried out a t 2 "C. 1000 g yeast were suspended in 600ml buffer A. The suspension was homogenized continuously in the presence of glass beads (0.3 mm diameter) and under a nitrogen atmosphere in the Vibrogen-Zellmuhle (E. Buhler, BRD-7400 Tubingen) a t a flow rate of 750ml/h. The homogenate was centrifuged at 19000 rev.lmin for 2 h at 2 "C using a Spinco R19 rotor. The supernatant normally had a pH of about 6.0 and was brought to pH 7.6 with concentrated ammonia. The pellets were discarded. For optimal DEAE-chromatography it is necessary to digest the main part of the nucleic acids. Therefore 5 ml of a freshly prepared 2 mg/ml solution of DNAase I in buffer B were added with gentle mixing for 1 h. During this time the temperature should rise to about 10 "C.
Ammonium ii'ulfate Fractionation and Dialysis. 225 g of solid ammonium sulfate per 1000 ml of the crude extract were added slowly with stirring to give a saturation of approximately 38O/,. The pH was prevented from dropping below 7.6 by addition of ammonia. The solution was stirred for 1 h and the precipitate removed by centrifuging a t 13 000 rev./ min for 40 min in a Sorvall GSA rotor. To the super- natant 115 g of solid ammonium sulfate per 1000 ml was added to give a 55O/, saturated solution. The precipitate contained nearly all the enzyme and was stirred and centrifuged as above. Subsequently it was dissolved in the minimum necessary volume of buffer C. The solution was placed in dialysis tubing and exhaustively dialysed (about 6 h) against buffer C until no sulfate was detectable.
DEAE-Cellulose Chromatography. 1000 g of equi- librated Whatman DE-52 cellulose were poured into a column (6 x55 cm). 180 to 220 ml dialysate were applied to this column and eluted with buffer C at a flow rate of 80 ml/h. 18 ml fractions were collected. The fractions containing the majority of the enzyme activity were pooled for the next step.
CM-Cellulose Chromatography. A 4.5 x 40 cm co- lumn filled with about 500g of well equilibrated Whatman CM-52 cellulose was used. The pooled fractions from the DEAE-cellulose column were applied and the cellulose was rinsed with buffer C until the eluate had virtually no absorption at 280 nm. The retained enzyme was subsequently eluted (7 ml fractions) with buffer C being additionally 0.15 M in KCI. The enzyme-containing fractions were

168 tRNA Nucleotidyl Transferase from Yeast Eur. J. Biocham.
pooled and the protein precipitated by 65 satura- tion with ammonium sulfate. The precipitate was pelleted hard (15000 rev./min, 20 min, Sorvall 5534 rotor) and dissolved in a few ml of buffer D.
Gel Filtration. The solution of the ammonium sulfate precipitate was applied to a 3 x 6 0 cm column filled with Biogel PI50 (100-200 mesh). Chromatography was carried out with buffer D being additionally 0.1 M in NaCI. Fractions of 5 ml each were collected.
Phosphocellulose Chromatography. The combined fractions from the previous step were immediately applied to a phosphocellulose column (2 x 16 om). The column was washed with buffer D until the eluate had no absorption at 280nm. Then a linear NaCl gradient was applied, composed of 120ml buffer D being additionally 0.1 M in NaCl and 120 ml buffer D being additionally 0.5M in NaCl. The fractions containing enzyme activity, each of 3 ml, were combined and diluted with three volumes buffer D. The diluted fractions were applied again to a phosphocellulose column (1 x 7 cm) in order to con- centrate the enzyme. The enzyme was eluted with buffer D being additionally 0.5 M in NaC1. For storage an equal volume of doubly distilled glycerol was added to this solution. In this glycerol solution the enzyme can be stored a t -20 "C for months with no measurable loss of activity.
Sodium Dodecylsulfate Electrophoresis This was performed in 5 O / , gels in the absence of
potassium ions according to Shapiro et al. 11191. For the electrophoresis the protein solutions (minimal concentration : 0.5 mglml) contained 1 O l i o sodium dodecylsulfate, 1 mercaptoethanol and 2001, gly- cerol. These solutions were immediately heated to I00 "C for 1 min [20] and applied directly to the gel under the normal conditions [Zl]. After electro- phoresis the gels were placed over night in a 20°/, sulfosalicylic acid for fixing the protein bands and then stained with coomassie brillant blue R-250. Destaining was carried out immediately with 7 O / , acetic acid solution.
Sedimentation Equilibrium Centrifugation The sedimentation experiments were carried out
according to the method of Yphantis [22] in a Spinco Model E ultracentrifuge equipped with a monochromator, photoelectric scanner, multiplexer and a RTIC-temperature control unit. Double sector cells with 12 mm optical path and sapphire windows were used.
Micro Isoelectric Focusing This was carried out in polyacrybmide gel columns
as described by Hayes et al. [23]. The polymeriza- tion mixture contained (final concentrations) : 7.5
acrylamide, 0.2O/, N,N'-methylene-bisacrylamide, 0.058 N , N,N', N'-tetramethylethyleneditmine, 2.0°/, carrier ampholyte (pH 3-10), 30 pg/ml pro- tein and 0.0015°/, riboflavine. After deaeration under reduced pressure for 2 min the polymerization mix- ture was pipetted into glass tubes ( 5 ~ 1 2 0 m m ) coated with dimethyldichlorosilane. The gel solution was polymerized within 20min by exposure to a! daylight-fluorescence lamp. The gels were allowed to equilibrate a t 4 "C for 10 min, and the isoelectric focusing was performed in the bath assembly used for disc electrophoresis: l o l o sulfuric acid was used at the anode (top) and 2.g0/, ethanolamine a t the ca- thode. Voltage was applied and increased to a maxi- mum of 120 volts so that the current did not exceed 1.5 mA per gel. After 12 h focusing was complete, and the gels were immediately removed. Focusing was performed with a t least three gels a t a time, one of which was used for staining, one to determine the pH gradient, and one to detect the enzymatic activity.
The gels were fixed in a 5O/, trichloroacetic acid/ 5 O / , sulfosalicyclic acid aqueous solution for 2 h. They were then thoroughly washed in water for 2 h and stained in a solution 0f0.066~/, coomassie brillant blue R-250 in 0.2 M Tris-HC1 pH 7.7 for several hours. Destaining was accomplished by washing the gels in a 701, acetic acid solution.
The pH-gradient was determined as described by Catsimpoolas [24]. After electrofocusing an un- stained gel was cut into slices of about 1.2 mm thick- ness [25]. Consecutive two slice fractions were sus- pended in 1 .O ml of water for 4 h. Afterwards the pH of the solution was measured a t 25 "C with a microelectrode exactly calibrated for the required pH range.
Another unstained gel was slices as described above and every two slices were suspended in the buffered standard assay mixture. After incubation at 32 "C for 30 min samples of 10 pl were withdrawn, and the enzymatic activity was determined as de- secribed.
RESULTS Purification of the tRNA Nucleotidyl Transferase A typical purification of the enzyme from 1000 g
bakers' yeast is summarized in the Table. Crude extract obtained by the equipment de-
scribed above in the presence of glass beads gave the best yield of enzymatic activity. The employment of buffer A is necessary, because phosphate buffer of similar ionic strength leads to worse yield. The specific activity of the crude extract is very reproduc- ible. The value determined may be falsified by nu- cleases, other enzymes and ATP.
Ammonium sulfate fractionation and extensive dialysis of the precipitate are necessary in order to
Vol.22, No.2,1971 H. STERNBACH, F. VON DER HAAR, E. SCKLIMME, E. GAERTNER, and F. CRAMER 169
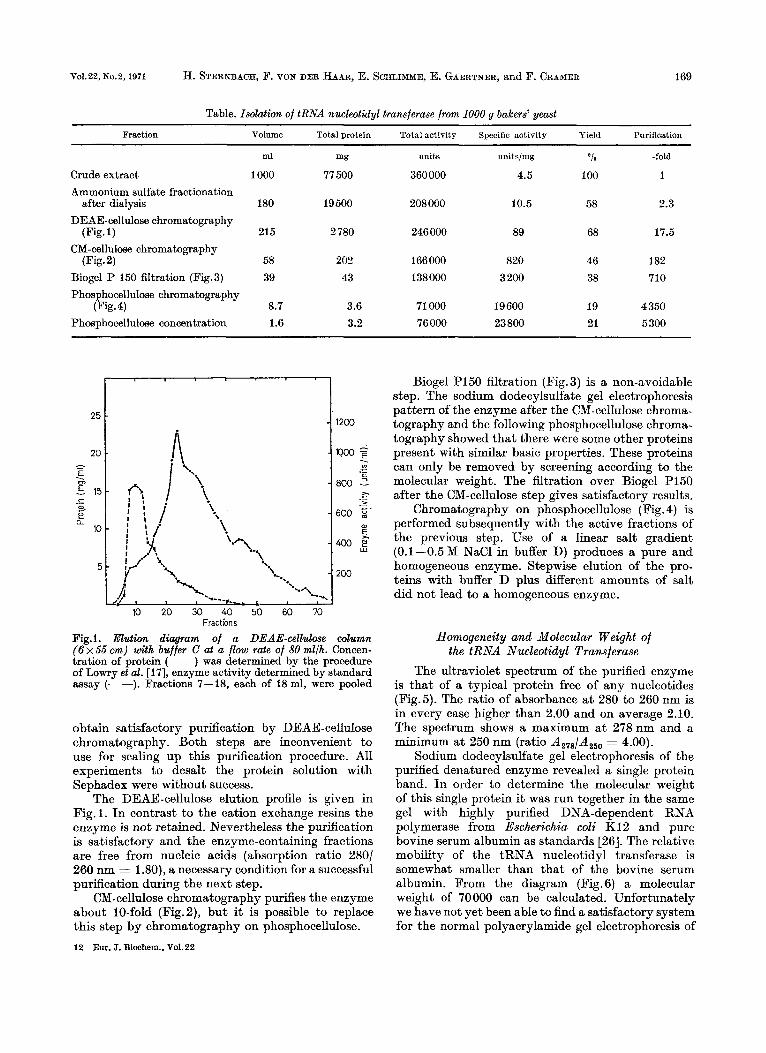
Table. Isolation of tRNA nucleotidyl transferase from 1000 g bakers’ yeast
Fraction Volume Total protein Total activity Specific activity Yield Purification
ml
1000
mg
77 500
units
360000
unitslmg
4.5
OIO -fold
100 1 Crude extract Ammonium sulfate fractionation
after dialysis 180 19500 208 000 10.5 58 2.3
DEAE-cellulose chromatography (Fig. 1) 215 2 780 246 000 89 68 17.5
CM-cellulose chromatography (Fig. 2) 58
39
203 43
166000
138000
820
3 200
46 182
38 710 Biogel P 150 filtration (Fig.3) Phosphocellulose chromatography
(Fig.4) 8.7
1.6 3.6
3.2
71 000
76000
19600
23 800 19 4 350
21 5300 Phosphocellulose concentration
Biogel PI50 filtration (Fig. 3) is a non-avoidable step. The sodium dodecylsulfate gel electrophoresis pattern of the enzyme after the CM-cellulose chroma- tography and the following phosphocellulose chroma- tography showed that there were some other proteins present with similar basic properties. These proteins can only be removed by screening according to the molecular weight. The filtration over Biogel P150 after the CM-cellulose step gives satisfactory results.
Chromatography on phosphocellulose (Fig. 4) is performed subsequently with the active fractions of the previous step. Use of a linear salt gradient (0.1-0.5M NaCl in buffer D) produces a pure and homogeneous enzyme. Stepwise elution of the pro- teins with buffer D plus different amounts of salt did not lead to a homogeneous enzyme.
c= E . m
E 15 c ._ s e Q
10
5
Fractbns Fig.1. Elution diuqram of a DEAE-cellulose column (6 x55 em) with buffer C at a flow rate of 80 mllh. Concen- tration of protein (-) was determined by the procedure of Lowry et al. [17], enzyme activity determined by standard assay (---). Fractions 7-18, each of 18 ml, were pooled
Homogeneity and Molecular Weight of the tRNA Nucleotidyl Transferase
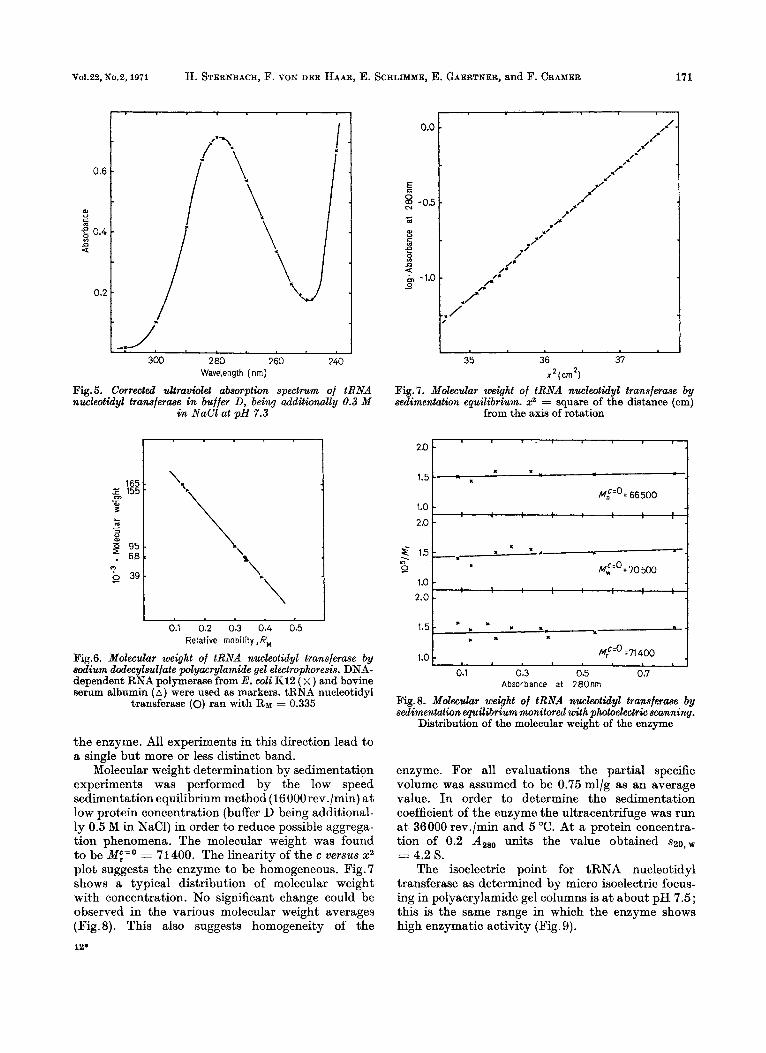
The ultraviolet spectrum of the purified enzyme is that of a typical protein free of any nucleotides (Fig.5). The ratio of absorbance a t 280 to 260 nm is in every case higher than 2.00 and on average 2.10. The spectrum shows a maximum a t 278nm and a minimum a t 250 nm (ratio A,,8/A250 = 4.00).
Sodium dodecylsulfate gel electrophoresis of the purified denatured enzyme revealed a single protein band. In order to determine the molecular weight of this single protein it was run together in the same gel with highly purified DNA-dependent RNA polymerase from Escherichia coli K12 and pure bovine serum albumin as standards [26]. The relative mobility of the tRNA nucleotidyl transferase is somewhat smaller than that of the bovine serum albumin. From the diagram (Fig. 6) a molecular weight of 70 000 can be calculated. Unfortunately we have not yet been able to find a satisfactory system for the normal polyacrylamide gel electrophoresis of
obtain satisfactory purification by DEAE-celluIose chromatography. Both steps are inconvenient to use for scaling up this purification procedure. All experiments to desalt the protein solution with Sephadex were without success.
The DEAE-cellulose elution profile is given in Fig. 1. In contrast to the cation exchange resins the enzyme is not retained. Nevertheless the purification is satisfactory and the enzyme-containing fractions are free from nucleic acids (absorption ratio 280/ 260 nm = 1 .SO), a necessary condition for a successful purification during the next step.
CM-cellulose chromatography purifies the enzyme about 10-fold (Fig.2), but it is possible to replace this step by chromatography on phosphocellulose. 12 Em. J. Biochem., Yo1.22

170 tRNA Nucleotidyl Transferase from Yeast Eur. J. Biochem.
2 500
2000 2 . Ln
c 3 - __
1500 - 5 .- - u m
1000 0,
E, c w
500
10 20 30 40 50 60 X3 80 90 Fractions
Fig.2. Elution diagram of a CM-cellulose column (4.5 x 40 cm) with buffer C or buffer C being additionally 0.15 M in KCl (arrow) at a flow rate of 40 mllh. Concentration of protein (-) was determined by the procedure of Lowry et al. [17],
enzyme activity determined by standard assay (----). Fractions 66-73, each of 7 ml, were collected
Fractions
Fig.3. Eiogel P150 filtration pattern (column 3 ~ 6 0 c m ) , with buffer D being additionally 0.1 M in NaCl at a flow rate of 30 mllh. Concentration of protein (-) was determined by the procedure of Lowry et al. [17], enzyme activity determined
by standard assay (----). Fractions 9-15, each of 5 ml, were pooled
I ’ ’ I 1.1 1
10000
6000 .$ ._ - u m
m g. 2000
Fractions Fig.4. Elution diagram of a phosphocellulose column ( 2 ~ 1 6 c m ) with a linear 0.1-0.5 M NaCl gradient in buffer D at a flow rate of 20 mllh. Concentration of protein (-) was calculated from ultraviolet monitoring, enzyme activity determined by
standard assay (----). Fractions 47-49, each of 3 ml, were pooled and concentrated
V01.22, No.2.1971 H. STERNBACH, F. VON DER HAAR, E. SCELIMME, E. GAERTNER, and F. CRAMER 171
OX
al " c m
0.4 :: n a
0.;
280 260 240
-* */, 300
Wavelength (nrn)
Fig.5. Corrected ultraviolet absorption spectrum of tRNA nucleotidyl tranaferaae in buffer D, being additionally 0.3 M
in NaCl at pH 7.3
Relative mobility ,RM
Fig.6. Molecular weight of tRNA nucleotidyl transferme by sodium dodecylsuljate polyacrylamide gel electrophoresis. DNA- dependent RNA polymerase from E. wli Ki2 ( x ) and bovine serum albumin ( A ) were used as markers. tRNA nucleotidyl
transferase (0) ran with RM = 0.335
the enzyme. All experiments in this direction lead to a single but more or less distinct band.
Molecular weight determination by sedimentation experiments was performed by the low speed sedimentation equilibrium method (16000rev./min) at low protein concentration (buffer D being additional- ly 0.5 M in NaC1) in order to reduce possible aggrega- tion phenomena. The molecular weight was found to be ME,=' = 71400. The linearity of the c versus xR plot suggests the enzyme to be homogeneous. Fig. 7 shows a typical distribution of molecular weight with concentration. No significant change could be observed in the various molecular weight averages (Fig.8). This also suggests homogeneity of the 12'
I 35 36 37
2 (cm
Pig. 7. Molecular weight of tRNA nucleotidyl transferme by sedimentation equilibrium. x2 = square of the distance (cm)
from the axis of rotation
I Mt'0=66500 I
1 .o 2.0
I 1.5
M:" ~ 7 1 4 0 0 1.0
0.1 0.3 0.5 0.7 Absorbance at 280nrn
Fig.8. Holecular weight of tRNA nuclwtidyl tramferaae by sedimentation equilibrium monitored with photoelectric scanning.
Distribution of the molecular weight of the enzyme
enzyme. For all evaluations the partial specific volume was assumed to be 0.75 ml/g as an average value. In order to determine the sedimentation coefficient of the enzyme the ultracentrifuge was run a t 36000 rev./min and 5 "C. At a protein concentra- tion of 0.2 A,,, units the value obtained ~ 2 0 , ~
= 4.2 S. The isoelectric point for tRNA nucleotidyl
transferase as determined by micro isoelectric focus- ing in polyacrylamide gel columns is a t about pH 7.5; this is the same range in which the enzyme shows high enzymatic activity (Fig. 9).
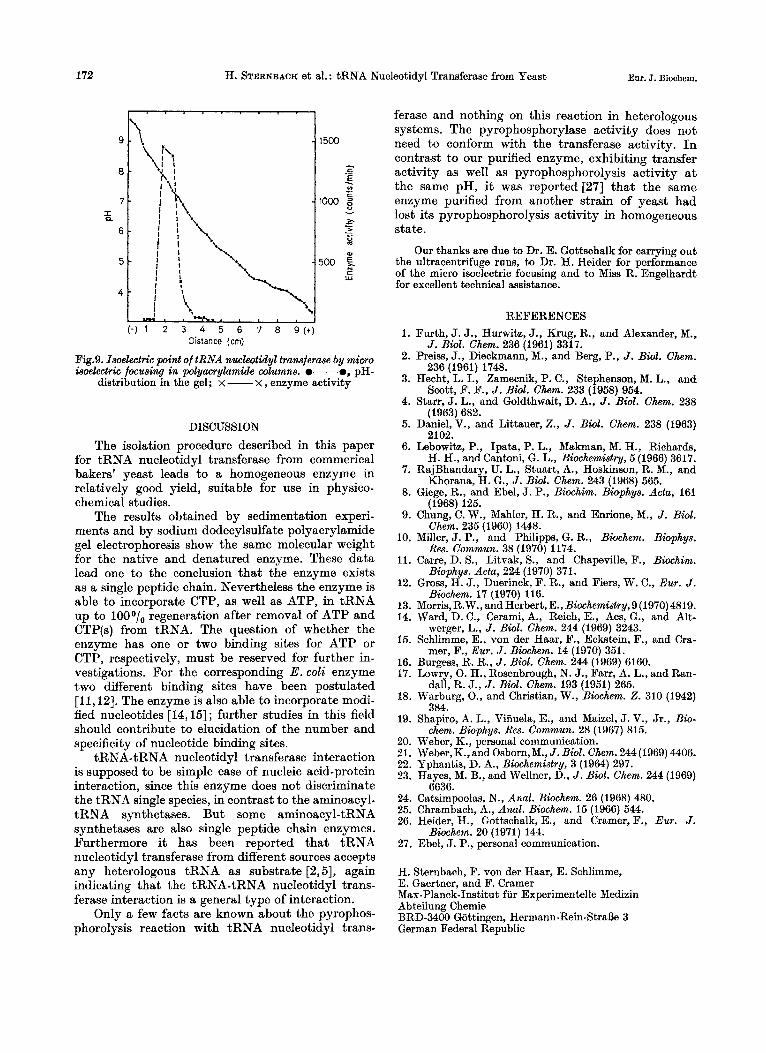
172 Eur. J. Biochem. H. STERNBACH et al. : tRNA Nucleotidyl Transferase from Yeast
I . . . . . . . I . 1
Distance (cm)
Fig.9. Isoelectric point of tRNA nucleotidyl transferase by micro isoelectric focusing in polyacrylamide wlumns. e-e, pH-
distribution in the gel; x - x , enzyme activity
DISCUSSION The isolation procedure described in this paper
for tRNA nucleotidyl transferase from commerical bakers' yeast leads to a homogeneous enzyme in relatively good yield, suitable for use in physico- chemical studies.
The results obtained by sedimentation experi- ments and by sodium dodecylsulfate polyacrylamide gel electrophoresis show the same molecular weight for the native and denatured enzyme. These data lead one to the conclusion that the enzyme exists as a single peptide chain. Nevertheless the enzyme is able to incorporate CTP, as well as ATP, in tRNA up to 1000/o regeneration after removal of ATP and CTP(s) from tRNA. The question of whether the enzyme has one or two binding sites for ATP or CTP, respectively, must be reserved for further in- vestigations. For the corresponding E . coli enzyme two different binding sites have been postulated [11,12]. The enzyme is also able to incorporate modi- fied nucleotides [14,15] ; further studies in this field should contribute to elucidation of the number and specificity of nucleotide binding sites.
tRNA-tRNA nucleotidyl transferase interaction is supposed to be simple case of nucleic acid-protein interaction, since this enzyme does not discriminate the tRNA single species, in contrast to the aminoacyl- tRNA synthetases. But some aminoacyl-tRNA synthetases are also single peptide chain enzymes. Furthermore it has been reported that tRNA nucleotidyl transferase from different sources accepts any heterologous tRNA as substrate [2,5], again indicating that the tRNA-tRNA nucleotidyl trans- ferase interaction is a general type of interaction.
Only a few facts are known about the pyrophos- phorolysis reaction with tRNA nucleotidyl trans-
ferase and nothing on this reaction in heterologous systems. The pyrophosphorylase activity does not need to conform with the transferase activity. I n contrast to our purified enzyme, exhibiting transfer activity as well as pyrophosphorolysis activity a t the same pH, it was reported [27] that the same enzyme purified from another strain of yeast had lost its pyrophosphorolysis activity in homogeneous state.
Our thanks are due to Dr. E. Gottschalk for carrying out the ultracentrifuge runs, to Dr. H. Heider for performance of the micro isoelectric focusing and to Miss R. Engelhardt for excellent technical assistance.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13. 14.
15.
16. 17.
18.
19.
20. 21. 22. 23.
24. 25. 26.
27.
REFERENCES Furth, J. J., Hurwitz, J., Krug, R., and Alexander, M.,
J . Bwl. Chem. 236 (1961) 3317. Preiss, J., Dieckmann, M., and Berg, P., J . Bwl. Chem.
236 (1961) 1748. Hecht, L. I., Zamecnik, P. C., Stephenson, M. L., and
Scott, F. F., J . Biol. Chem. 233 (1958) 954. Starr, J. L., and Goldthwait, D. A., J . Biol. Chem. 238
(1963) 682. Daniel, V., and Littauer, Z., J . Biol. Chem. 238 (1963)
2102. Lebowitz, P., Ipata, P. L., Makman, M. H., Richards,
H. H., and Cantoni, G. L., Biochemistry, 5 (1966) 3617. RajBhandary, U. L., Stuart, A., Hoskinson, R. M., and
Khorana, H. G., J . Biol. Chem. 243 (1968) 565. Giege, R., and Ebel, J. P., Biochim. Biophys. Acta, 161
(1968) 125. Chung, C. W., Mahler, H. R., and Enrione, M., J . Biol.
Chem. 235 (1960) 1448. Miller, J. P., and Philipps, G. R., Biochem. Bwphys.
Res. Commun. 38 (1970) 1174. Carre, D. S., Litvak, S., and Chapeville, F., Biochim.
Biophys. Acta, 224 (1970) 371. Gross, H. J., Duerinck, F. R., and Fiers, W. C., Eur. J .
Biochem. 17 (1970) 116. Morris,R.W., and Herbert,E., Biochemistry,9(1970)4819. Ward, D. C., Cerami, A., Reich, E., Acs, G., and Alt-
werger, L., J. Bwl. Chem. 244 (1969) 3243. Schlimme, E., von der Haar, F., Eckstein, F., and Cra-
mer, F., Eur. J . Biochem. 14 (1970) 351. Burgess. R. R.. J . Biol. Chem. 244 (1969) 6160. L O G , 0. H., Rosenbrough, N. J., Farr, A. L., and Ran-
dall, R. J., J . Biol. Chem. 193 (1951) 265. Warburg, 0.; and Christian, W., Biochem. 2. 310 (1942)
Shapiro, A. L., Viiiuela, E., and Maizel, J. V., Jr., Bio-
Weber, K., personal communication. Weber, K., and Osborn,M., J . Bid . Chem. 244 (1969) 4406. Yphantis, D. A., Biochemistry, 3 (1964) 297. Hayes, M. B., and Wellner, D., J . Biol. Chem. 244 (1969)
Catsimpoolas, N., Anal. Biochem. 26 (1968) 480. Chrambach, A., Anal. Biochem. 15 (1966) 544. Heider, H., Gottschalk, E., and Cramer, F., Eur. J .
Ebel, J. P., personal communication.
384.
chem. Biophys. Res. Commun. 28 (1967) 815.
6636.
Biochem. 20 (1971) 144.
H. Sternbach, F. von der Haar, E. Schlimme, E. Gaertner, and F. Cramer Max-Planck-Institut fur Exmrimentelle Medizin Abteilung Chemie BRD-3400 Gottingen, Hermann-Rein-StraOe 3 German Federal Republic