Embed Size (px)
Citation preview

University College Cork, Ireland
August 28th - Sept 1st 2016
Book of Abstracts

Proceedings of ISC 2016, Cork, Ireland 2016
SYMPOSIUM ABSTRACT BOOK
Title: 31st International Symposium on Chromatography - Congress Proceedings
Editor: Jeremy D. Glennon
Apryll M. Stalcup
Subject: Chromatography - International
Published by: Publisher - Apryll M. Stalcup and Jeremy D. Glennon
ALL RIGHTS RESERVED. This book contains material protected under International and Federal Copyright Laws and Treaties. Any unauthorized reprint or use of this material is prohibited. No part of this book may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, or by any information storage and retrieval system without express written permission from the author / publisher.
NOTICE: Neither the publisher, editors, publishing staff nor authors assume any liability for any injury or damage whatsoever to any persons, animals or property arising out of or relating to any use of the material contained in this publication. Where trade names appear, no discrimination is intended, and no endorsement either by the authors, editors or publisher is implied. Readers should make their own enquiries to ensure that the information contained in this publication is relevant to their individual circumstances and is current. All material published in these proceedings represents the opinions of the authors and does not necessarily reflect the opinions of the Editors or the institutions with which the authors are affiliated.


3www.ISC2016.ie @isc2016 #isc2016 /isc2016
Pharmaceutical Analysis
COMMITTEES
Local Organising Committee (LOC)Apryll M. Stalcup (DCU)
Eileen O’Callaghan (UCC)
Jeremy D. Glennon (UCC)
International Scientific Committee (Member of the Permanent Scientific Committee*)
Boguslaw Buszewski* (Torun)
Attila Felinger* (Pécs)
Gert Desmet* (Brussels)
Tony Edge* (Runcorn)
Michael Lämmerhofer* (Tübingen)
Uwe Karst (Münster)
Didier Thiebaut* (Paris)
Alain Berthod (Lyon)
Wolfgang Lindner (Vienna)
David McCalley (Bristol)
Brett Paull (Tasmania)
Karen Phinney (Gaithersburg)
Chao Yan (Shanghai)
Irena Vovk (Slovenia)
Supalax Srijaranai (Khon Kaen)
Gerard Rozing (Karlsruhe)
W. John Lough (Sunderland)
Monika Dittmann (Germany)
National Organising Committee, ISSC:Dara Fitzpatrick (UCC)
Mila Pravda (UCC)
Eric Moore (UCC)
Elizabeth Gilchrist (UCC)
Elizabeth Guihen (UL)
Brendan O’Connor (DCU)
Dermot Brabazon (DCU)
Brian Kelleher (DCU)
Andreas Heise (RCSI)
David Collins (DCU)
Damien Connolly (WIT)


5www.ISC2016.ie @isc2016 #isc2016 /isc2016
Pharmaceutical Analysis TABLE OF CONTENTS
pg
Plenary Speakers 6
Keynote Speakers 15
Oral Speakers 36
Tutorials 109
Short Courses 116
Posters 121
Late Breaking Posters 372
TABLE OF CONTENTS

www.ISC2016.ie @isc2016 #isc2016 /isc20166
PLENARY ABSTRACTS
PL01 - Practice and ramifications of ultra fast chiral and achiral separations
Author: Daniel W. Armstrong, The University of Texas at Arlington, Department of Chemistry & Biochemistry, Arlington, TX
Abstract: A variety of brush-type stationary phases (CSPs) were synthesized using superficially porous particles (SPPs) and sub 2 micron particles. Given their high efficiencies and relatively low back pressures, columns containing SPP particles were particularly advantageous for difficult ultrafast separations in the 2 to 40 second range. Further, they can be used in all mobile phase modes and with high flow rates and pressures. When operating under these conditions, both instrumentation and column packing must be modified or optimized so as not to limit separation performance and quality. Further, frictional heating results in axial thermal gradients of up to 16° C and radial temperature gradients up to 8° C which can produce interesting effects in enantiomeric separations. It is shown that the kinetic behavior of various stationary phases can differ from one another as much as they differ from the well-studied C-18 reversed phases. Three additional interesting aspects of this work are: a) the first kinetic evidence of two different chiral recognition mechanisms, b)a demonstration of increased efficiencies at higher flow rates for specific separations, and c) the lowest reduced plate height yet reported for a non C18 phases.
PL02 - More than a Gut Feeling: Harnessing the Microbiome for Better Brain Health
Author: John F. Cryan, University College Cork, Cork, Ireland
Abstract: The brain-gut-microbiota axis is emerging as a research area of increasing interest for those investigating the biological and physiological basis of neurodevelopmental, age-related and neurodegenerative disorders. The routes of communication between the gut and brain include the vagus nerve, the immune system, tryptophan metabolism, via the enteric nervous system or by way of microbial metabolites such as short chain fatty acids. Studies in animal models have shown that the development of an appropriate stress response is dependent on the microbiota. Developmentally, a variety of factors can impact the microbiota in early life including mode of birth delivery, antibiotic exposure, mode of nutritional provision, infection, stress as well as host genetics. At the other extreme of life, individuals who age with considerable ill health tend to show narrowing in microbial diversity. Stress can significantly impact the microbiota-gut-brain axis at all stages across the lifespan Recently, the gut microbiota has been implicated in a variety of conditions including autism, schizophrenia and Parkinson’s disease. Moreover, fundamental brain processes from adult hippocampal neurogenesis to myelination to microglia activation have been show to be regulated by the microbiome. Further studies will focus on understanding the mechanisms underlying such brain effects

7www.ISC2016.ie @isc2016 #isc2016 /isc2016
PLENARY ABSTRACTS
PL03 - Porous polymer-based monolithic columns: A success story
Author: F. Svec*
Abstract: The modern monoliths emerged 25 years ago. While the early polymer-based monoliths were used in columns for the rapid separations of proteins, current literature describes a number of different applications in addition to typical chromatography demonstrating versatility of the monolithic materials. Several new formats have emerged recently that extend the use of monoliths in areas different from column chromatography. For example, polymer-based monolithic columns prepared using novel approaches enable efficient and rapid separation of small molecules. New chemistries have also been developed to afford monolithic columns for the separation in various modes and to control their selectivity. Modification of pore surface with nanoparticles and metal-organic frameworks is a current trend that extends applications of monoliths in the arena of highly selective fishing-out systems. Thin monolithic layers are also gaining more attention since they allow efficient separations of proteins combined with mass spectrometry using very simple means. Monoliths also serve as supports for immobilization of enzymes to form very active enzymatic reactors.Disclosure of Interest: None DeclaredKeywords: Chromatography, History, Monolith
PL04 - Chromatographic-mass spectrometric methods in sports drug testing
Author: Mario Thevisa,b
a Institute of Biochemistry – Center for Preventive Doping Research, German Sport University Cologne, Germany
b European Monitoring Center for Emerging Doping Agents (EuMoCEDA), Cologne/Bonn, Germany
Abstract: Sports drug testing laboratories are facing multifaceted challenges including the misuse of naturally/endogenously occurring substances, non-approved/discontinued drug candidates, urine manipulation, etc. In order to provide best-possible analytical performance, mass spectrometry-based approaches are predominantly utilized to detect prohibited substances and methods of doping. With the constantly increasing analytical requirements concerning the number of target compounds, the complexity and range of physico-chemical properties of analytes (e.g., inorganic ionic transition metals, gases, lipids, alkaloids, peptides, proteins, DNA/RNA-based drugs, etc.) as well as the desire to accelerate analyses and obtain information allowing also for retrospective data mining, high resolution/high accuracy mass spectrometry has become a mainstay in doping controls. In that context, various assays have been reported, enabling either multi-component analyses of low- or high molecular mass measurands or the specific and dedicated (confirmatory) detection of prohibited substances. Selected applications will be presented reporting on examples of recent findings in routine sports drug testing, demonstrating both the inventiveness of cheating individuals that undermine current anti-doping efforts as well as

www.ISC2016.ie @isc2016 #isc2016 /isc20168
PLENARY ABSTRACTS
the relevance of in-depth investigations into unusual findings, where the athletes’ innocence was to be shown albeit prohibited substances were unequivocally identified in their doping control urine samples.
PL05 - Automated data acquisition and interpretation underpin the evaluation of glycosylation in big data, personal patient profiles and mammalian cell culture
Authors: Dr. Roisin O’Flaherty, Dr. Mark Hilliard, Dr. Radka Saldova, Dr Henning Stockmann, Dr. Mohankumar Muniyappa, Sinead Hallinan, Dr. Ian Walsh and Prof. Pauline M RuddNational Institute for Bioprocessing Research and Training, Fosters Avenue, Dublin
Abstract: The future success of personalised medicine depends on the development of tailor made therapies and diagnostics coupled with an understanding of nodes where disease pathways might be effectively interrupted in individual patients. We have developed automated glycan analytical platforms to monitor critical quality attributes in biopharmaceutical drugs and to provide holistic approaches to disease. The N-glycan workflow which involves automated glycan release, labelling, coupled LC/MS and computer assisted data analysis has been embedded into the Waters Corporation UNIFI platform. Further, we have used our high throughput glycan release platform to analyse the glycomes of thousands of individual serum and IgG samples. We have associated specific glycan pools with other –omics data to investigate pathways of disease. References: 1: Haakensen VD, Steinfeld I, Saldova R, Shehni
AA, Kifer I, Naume B, Rudd PM, Børresen-Dale AL, Yakhini Z. Serum N-glycan analysis in breast cancer patients--Relation to tumour biology and clinical outcome. Mol Oncol. 2016 (1):59-72.
2: Stöckmann H, Duke RM, Millán Martín S, Rudd PM. Ultrahigh throughput, ultrafiltration-based N-glycomics platform for ultraperformance liquid

9www.ISC2016.ie @isc2016 #isc2016 /isc2016
PLENARY ABSTRACTS
chromatography (ULTRA(3)). Anal Chem. 2015 87(16):8316-22.
3: Saldova R, Asadi Shehni A, Haakensen VD, Steinfeld I, Hilliard M, Kifer I, Helland A, Yakhini Z, Børresen-Dale AL, Rudd PM. Association of N-glycosylation with breast carcinoma and systemic features using high-resolution quantitative UPLC. J Proteome Res. 2014 13(5):2314-27.
4: Lauc G, Adamczyk B, Rudd PM, Rudan I. et al., Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet. 2013;9(1):
PL06 - SFC and SFC-MS in drugs analysis
Authors: JeanLuc Veuthey, Alexandre Grand-Guillaume-Perrenoud, Lucie Novakova, Vincent Desfontaine, Davy Guillarme1 School of Pharmaceutical Sciences, 2 University of Geneva,3 University of Lausanne, Boulevard d’Yvoy 20, 1211
Geneva 4, Switzerland
Abstract: Supercritical fluid chromatography (SFC) is now fully competitive with current LC approaches thanks to the recent introduction of modern SFC platforms. This statement is especially true when using columns packed with sub-2-µm fully porous or sub-3 µm superficially porous particles, known as ultra-high performance supercritical fluid chromatography (UHPSFC).
The applicability of UHPSFC will be demonstrated through the separation of various mixtures of compounds, including basic, acidic, neutral, hydrophilic and lipophilic molecules present in different matrices. As illustrated with these examples, the main advantage of UHPSFC is related to the direct compatibility of SFC mobile phase with a wide spectrum of stationary phase chemistries offering almost infinite possibilities to play with selectivity.
The successful coupling of SFC with MS has already been described, but its use for the analysis of drugs in biological materials has been scarcely reported, while extended selectivity and sensitivity can be provided by MS and MS/MS detection. The use of UHPSFC-ESI-MS/MS will be demonstrated for the analysis of numerous drugs in different complex matrices (biological fluids and plant extracts). Detection sensitivity,

www.ISC2016.ie @isc2016 #isc2016 /isc201610
PLENARY ABSTRACTS
matrix effect and overall analytical performance achieved with UHPSFC-ESI-MS/MS will be discussed and compared to that of UHPLC-ESI-MS/MS.
PL07 - Are There Remaining Challenges of Chromatographic Enantiomer Separations?
Authors: W. Lindner1, M. Kohout2, K. Hamase3, A.Péter4 and I. Ilisz4
1 Institute of Analytical Chemistry, University of Vienna, Austria;
2 Dept. of Organic Chemistry, University of Chemistry and Technology, Prague, Czech Republic
3 Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka, Japan
4 Department of Inorganic and Analytical Chemistry, University of Szeged, Hungary
Abstract: Over the years we reached the perception that chromatographic enantiomer separations moved from art to routine thanks to an exceptional success story. This is true whereby the academia and the pharmaceutical industry were the drivers working in a fruitful symbiosis with chromatographic material manufactures leading to a large portfolio of so-called “chiral” columns on the market.
However, there are still remaining challenges to be mastered in the field:• Enantiomer separations of highly polar
analytes• (Ultra)-trace analysis of enantiomeric
impurities• Sensitive enantioselective analysis of
target compounds in complex matrices• High speed enantioselective analysis
to improve the number of analyses per time unit (important for biomarker monitoring, omics fields, etc.)
• Shift from the “trial and error” approach of enantiomer separations to knowledge based predictive concepts (implies understanding of enantioselective molecular recognition phenomena which includes dedicated synthesis, the use of spectroscopic tools, modelling, etc.)

11www.ISC2016.ie @isc2016 #isc2016 /isc2016
PLENARY ABSTRACTS
As can be extracted there remains a lot to do for the future and it becomes also evident that Enantioselective Separation Sciences (ESS) are of interdisciplinary character on the one side and when application oriented it may be multidimensionally oriented in the separation space.
In this lecture we will address most of the raised issues and discuss then on the basis of representative examples touching very practical but also mechanistic aspects. Particular focus will be given towards variants of enantioselective amino acid and small peptides analysis concepts also in context with our developments of chiral ion exchanger based “chiral” columns.
PL08 - Modeling and understanding dispersion in liquid chromatography: 60 years past, 60 years aheadAuthor: Gert Desmet*
Abstract: The band broadening processes occurring in liquid chromatography are very complex. Due to the combined effort of many great scientists building upon each other’s work in the past decades, the most important parts of the puzzle have now been solved. To date, this has produced a relatively clear picture of the different individual band broadening contributions, how they are linked together, and how they can be used to qualitatively explain most of the experimental facts and findings. At the conference, an overview of the major theoretical breakthroughs made in the past will be given and will be used in a didactic way to explain a number of contemporary issues (the power of core-shell particles, the sense and non-sense of monolithic columns, the difference in column efficiency between hilic and reversed-phase columns,…)
Despite the many achievements made in the past, there are still a number of important and intriguing questions that remain: is there a direct link between particle size distribution and column efficiency?, is there a possibility to quantitatively relate the values of the A, B, and C-term to the bed structure?, how does radial dispersion exactly contribute to the observed eddy-dispersion?,… Answering all these questions might leave work for maybe another 60 years of research and improvements. Combining a full and detailed understanding of the different band broadening sources, with the new materials engineering possibilities that

www.ISC2016.ie @isc2016 #isc2016 /isc201612
PLENARY ABSTRACTS
will undoubtedly emerge, it is written in the stars that our successors will be using columns providing much higher speed and efficiency than conceivable today. At present, we can speculate on the potential use of nanotechnology, microfabrication, 3D-printing,… but the future is bound to bring even new fabrication opportunities.Keywords: Modelling, Theory
PL09 - Prediction of Retention times in Reversed - Phase, Ion-Exchange and Hydrophilic Interaction Liquid Chromatography Modes Based on Chemical Structures
Authors: P. R. Haddad*, S. H. Park 1, M. Taraji 1, Y. Wen 1, E. Tyteca 1, M. Talebi 1, R. Amos 1, R. A. Shellie
2, R. Szucs 3, J. W. Dolan 4, C. A. Pohl 5
1 University of Tasmania, Hobart 7001, 2 Trajan Scientific and Medical, Melbourne, Australia, 3 Pfizer Worldwide R&D, Sandwich, United Kingdom, 4 LCResources, McMinnville, 5 ThermoFisher Scientific, Sunnyvale, United States
Abstract: This presentation will report a large-scale academic-industry collaborative study on the prediction of analyte retention times based solely on chemical structures. The goal of this study is facilitate rapid selection of the optimal chromatographic mode and separation conditions. This study uses Quantitative Structure-Retention Relationships (QSRR) whereby molecular modelling is utilised to generate molecular descriptors of analytes based on their chemical structures. From the large number of descriptors generated, the most relevant descriptors are then determined and a mathematical relationship for an individual unknown analyte is generated which relates the selected descriptors and measured retention times for a test set of analytes. Finally, this relationship can then be used to predict the retention time for the unknown analyte, again based only on its chemical structure.
The above approach has been applied to the prediction of retention times in reversed-phase liquid chromatography (RPLC) using moderately sized retention

13www.ISC2016.ie @isc2016 #isc2016 /isc2016
PLENARY ABSTRACTS
databases (approximately 100 compounds) either obtained in-house or from the open literature. The same methodology has also been applied to retention time prediction in hydrophilic interaction liquid chromatography (HILIC) using a new experimentally-derived database of retention times under HILIC conditions. Finally, the method has been adapted for ion chromatography (IC) using a dataset of inorganic and low molecular weight organic anions and a separate dataset of organic cations for which the slope and intercepts have been measured for the linear relationship between log(retention factor) and log(eluent concentration).
In this talk, the overall project will be described and the critical questions underlying the success of the project will be discussed. A particular focus will be the role of the structural similarity of the unknown test compound (compared to the structures of the analytes contained in the database) in the QSRR process. Disclosure of Interest: None DeclaredKeywords: Prediction of retention times, QSRR, Structural similarity

www.ISC2016.ie @isc2016 #isc2016 /isc201614
Invited Dinner Speaker
The adventures of a bioanalytical chemist in the never-never land of antidepressants: Finding a way out of the “ketamine paradigm”
Author: Irving W. Wainer, PhD, DHCChief Scientific Officer, Mitchell Woods Pharmaceuticals
(R,S)-Ketamine is a chiral phencyclidine derivative that produces rapid and short-lived anesthesia via inhibition of the N-methyl-D-aspartate (NMDA) receptor. The anesthetic activity of this drug has been associated with the parent compound and to a lesser degree with the N-demethylated metabolite (R,S)-norketamine, while other (R,S)-ketamine metabolites were designated as “inactive”, the “Ketamine Paradigm”. Recent studies demonstrated that a single sub-anesthetic dose of (R,S)-ketamine produces rapid and profound antidepressant response. As part of our studies of this effect in patients with treatment-resistant and bipolar depression, we developed a stereoselective lc/ms/ms assay capable of quantifying all of the major (R,S)-ketamine metabolites. The analysis of plasma samples demonstrated that (R,S)-ketamine was rapidly transformed in to multiple metabolites including (2S,6S;2R,6R)-hydroxynorketamine and that these metabolites were associated with clinical response. The hydroxynorketamine metabolites are weak NMDA inhibitors, raising the question of the viability of the “Ketamine Paradigm” as an explanation of (R,S)-ketamine’s antidepressant effects. The pharmacological effects produced by sub-anesthetic dosing of (R,S)-ketamine were studied using non-targeted and targeted pharmacometabolomics analysis,
which identified a relationship between basal D-serine plasma levels and response in which reduced D-serine plasma levels predict and track a positive antidepressant response. A similar relationship was observed for the positive response to (R,S)-ketamine treatment in patients suffering from Post-Traumatic Stress Disorder (PTSD). Cell-based studies using enantioselective CE-LIF analysis and Western blot were used to elucidate the pharmacological mechanism and to establish the pharmacological effects of the hydroxynorketamine metabolites. The antidepressant activity of (2S,6S)- and (2R,6R)-hydroxynorketamine were studied in multiple mouse models of depression and the data demonstrate that both compounds produce antidepressant effects with the (2R,6R) isomer being the more potent agent. This talk will address how our ability to accurately measure chemical and biological changes led us to a new “Ketamine Paradigm” and to the identification of the ketamine metabolites responsible for the drugs antidepressant activity. The presentation will also discuss the development of a pharmacophore model that is being used to design a new family of antidepressant drugs.

Keynote Abstracts

www.ISC2016.ie @isc2016 #isc2016 /isc201616
KEYNOTE ABSTRACTS
KN01 - Modern liquid chromatographic approaches for the characterization of biopharmaceuticalsAuthors: Davy Guillarme, Jean-Luc Veuthey, Szabolcs FeketeSchool of Pharmaceutical Sciences, University of Geneva, University of Lausanne, Boulevard d’Yvoy 20, 1211 Geneva 4, Switzerland
Abstract: The characterization of therapeutic monoclonal antibodies (mAbs) and antibody-drug conjugates (ADCs), which combines the specificity of a mAb with a potent cytotoxic agent covalently bound via a linker to the antibody, is a tremendous challenge to state-of-the-art separation technologies. Indeed, subtle changes in these large (> 145 kDa) molecules at the amino acid level can have profound effects on efficacy and pharmacokinetic properties, thus it is important to have the ability to rapidly and accurately assess changes in the distribution of different isoforms (e.g., glycosylation, oxidation, deamidation, lysine truncation…) of such biomolecules.
Today, one of the most widely used analytical technique for therapeutic protein characterization is liquid chromatography, probably due to the remarkable developments of the past few years, enabling a new level of chromatographic performance. Recent developments in LC, such as ultra-high-pressure LC (UHPLC), columns packed with wide-pore superficially porous particles and organic monolith columns now allow a dramatic increase in separation efficiency, even with large intact biomolecules.
The aim of this presentation will be to review the possibilities and trends of current state-of-the-art LC column technology applied
for different modes of chromatography for the characterization of therapeutic proteins. Therefore, the recent trends in size exclusion chromatography (SEC), ion exchange chromatography (IEX), hydrophobic interaction chromatography (HIC), reversed phase liquid chromatography (RPLC) and hydrophilic interaction chromatography (HILIC) for analysis, at the protein level, of biopharmaceuticals including therapeutic proteins, mAbs and ADCs will be critically discussed.
KN02 – Polymeric surfactants for the development of macroporous polymer monoliths
R. Dario Arruaa, Aminreza Khodabandehb, Fotouh R. Mansourb,c and Emily F. Hildera a Future Industries Institute, University of South Australia,
Mawson Lakes Campus, Mawson Lakes 5095, Australiab Australian Centre for Research on Separation Science
(ACROSS), School of Physical Sciences, University of Tasmania, Private Bag 75, Hobart 7001, Australia
c Department of Pharmaceutical Analytical Chemistry, Tanta University, Tanta, Egypt
Abstract: Monolithic columns have been proven to be a promising alternative to traditionally used packed columns. High permeability and low resistance to mass transfer are some of the advantages these materials present. Among the different ingredients present in the polymerization mixture, the porogen selection and its composition is known for being one of the main parameters to affect the porous architecture of the resultant monolith. Aiming to explore new materials as porogens, different research groups reported the use of ionic liquids, polymers, and surfactants as pore forming agents for the synthesis of monolithic HPLC columns.

17www.ISC2016.ie @isc2016 #isc2016 /isc2016
KEYNOTE ABSTRACTS
The use of surfactants, in particular, has been shown to have a significant effect on the morphology and pore structure of the materials. This presentation will introduce the use of polymeric surfactants for the synthesis of two types of polymer monoliths: highly cross-linked poly(polyethylene glycol diacrylate) (polyPEGDA) monoliths and polymerised high internal phase emulsions (poly(HIPEs)) foams.
In the first part of the lecture, the effect of the use of different surfactants on the porous properties and chromatographic performance of polyPEGDA monoliths will be presented. Furthermore, due to the ability of polymeric surfactants to dissolve polar and non-polar compounds, the use of the same surfactant-containing porogenic mixture to prepare a variety of different polymeric monoliths will be discussed. This approach shows the potential of using a ‘generic porogen mixture’ for the preparation of polymer monoliths with different chemistries.
In the second part of the presentation the synthesis of well-defined diblock copolymers to be used as stabilisers for the preparation of hydrophilic and hydrophobic polyHIPEs will be discussed. The effect of the different polymerisation conditions (role of initiator, organic modifier and the polymerization rate) on the morphology of the resulting materials will be presented.
KN03 - Chemical and kinetic characterization of stationary phases for fast liquid chromatography Author: Attila Felinger Department of Analytical and Environmental Chemistry, University of Pécs, Ifjúság útja 6, H-7624 Pécs, Hungary email: [email protected]
Abstract: During the recent years, a number of chromatographic columns with various types of packing materials dedicated for UHPLC instrumentation have been introduced. The most frequently used columns in this area are narrow and short (2.1 mm ID and 50 mm length), and are packed with sub-3-µm or sub-2-µm particles. Besides the use of the well-known and widely used fully-porous or core-shell packing materials, another potential is in the development of monolithic silica columns, where improved efficiency can be achieved and moderate column pressures are sufficient owing to the small skeleton size and large (through-pore size)/(skeleton size) ratios.
The kinetic performance of reversed phase columns packed with fully porous and core-shell particles with various particle diameters and the performance of a monolithic silica columns can be rather different. We also compared the kinetic performance of the same type monolithic column mounted by the producer or by the user. In addition to the results obtained with van Deemter plots and kinetic plots, we suggest a column-reversal method to examine the bed heterogeneity at the column ends and explain the reduction of column efficiency for early eluting solutes.
The monolithic column shows systematically better efficiency for early

www.ISC2016.ie @isc2016 #isc2016 /isc201618
KEYNOTE ABSTRACTS
eluting compounds than the packed columns, therefore an additional band broadening effect is suspected for the packed columns. The effect of the presence of the frits and the bed heterogeneity of the columns near the frits can characterized by a column-reversal method. It has been shown that significant differences can exist between the two ends of the packed columns, while the monolithic column shows rather similar performance at either column end.
The flow reversal method is useful to characterize the sample band broadening in the immediate vicinity of the column ends. Although flow reversal has a peak compression effect, and the peaks observed with reversed flow are always narrower and more symmetrical than the peaks obtained without reversing the flow, flow reversal can be a useful tool to show the differences between the intrinsic plate heights of the columns and also for showing the difference between the two respective column ends. The local plate height was found much smaller in every case than the one we get for the unretained thiourea after a simple injection without arrested and reversed flow.
The Tanaka test is a simple characterization measurement for stationary phases. By means of a few simple chromatographic test injections we will get information about surface area, surface coverage, amount and functionality of silanols.
According to the above-defined results the six studied columns have similar properties, even though those stationary phases are fundamentally different – such as monolithic, core-shell or fully porous packings.
KN04 – The Development of Multiplexed Liquid Chromatography–Mass Spectrometry Approaches for –Omics Investigations
Author: Gérard HopfgartnerLife Sciences Mass Spectrometry, Department of Inorganic and Analytical Chemistry, University of Geneva, 20 Bd d’Yvoy, CH1211-Geneva 4, Switzerlandemail: [email protected]
Abstract: Due to its high sensitivity, selectivity and sample throughput liquid chromatography (LC) coupled to low or high resolution tandem mass spectrometry (LC-MS/MS) MS plays more and a major role as a tool to identify and quantify metabolomic and proteomic biomarkers. Biomarker discovery can by performed unsupervised by analysing hundreds or thousands of analytes in a limit set of samples. On the other hand biomarkers discovery can be performed with the target determination of metabolites or proteins selected from previous knowledge or hypothesis driven. Samples throughput and chemical is becoming an additional challenge in the field calling for multiplexed liquid chromatographic and mass spectrometric approaches with improved selectivity. These approaches can include hyphenation of sample preparation, dual LC separations systems, Data Independent Acquisition, MRM-Cubed and Ion mobility.
Recent instrumental improvements in high resolution mass spectrometry (HRMS) have enabled data independent information acquisition (DIA) schemes, such as MSEverything or SWATH. SWATH-MS has emerged as a key technique for peptides quantification taking the advantage of the Selected Reaction Monitoring

19www.ISC2016.ie @isc2016 #isc2016 /isc2016
KEYNOTE ABSTRACTS
KN05 – Bayesian statistics: a new concept for automated analysis of megavariate LCxLC and GCxGC chromatographic dataAuthor: G. Vivó-Truyols 1,*, M. Woldegebriel 1, A. Barcaru 1, M. Lopatka 2
1 van ‘t Hoff Institute for Molecular Sciences, 2 kdvI institute for Mathematics, University of Amsterdam,
Amsterdam, Netherlands
Abstract: Data analysis methods applied to chromatographic data are a routine part of most modern analytical workflows. With the emergence of hyphenation (especially mass spectrometry) and two-dimensional methods (e.g. GCxGC and LCxLC) new challenges for the data analysis are emerging. Analysing these enormous and complex quantities of data becomes a tremendous challenge, especially because of the need to do it automatically.
Traditionally, chromatographic data has been processed using the so-called frequentist approach. With this approach, we get just a final answer about hypothesis we are testing, but we don’t get any information about its probability of being true. Bayesian statistics offers however a very interesting alternative, since it delivers an answer with its associated probability, e.g. “the probability that a certain hypothesis is true is x%”. This way of thinking opens a new world of possibilities, especially in the area of automated massive chromatographic data treatment.
We have applied this way of thinking to a broad range of examples in which the automation of the data treatment was a must. One example concerns peak detection in two-dimensional chromatography (LCxLC and GCxGC), in which the different
(SRM) mode with post-acquisition quantification on almost any analyte (metabolite, peptide) previously identified. Also SWATH/MS offers unique features for metabolomics and drug metabolism in the High Resolution - SRM mode for qualitative and quantitative analyses (QUAL/QUAN). While for exometabolites structural elucidation is possible based on the constraints of the parent drug, endogenous metabolites identification remains challenging. The use of annotated MS/MS spectral libraries is therefore becoming mandatory for exploiting untargeted approaches. Considering the complexity of biological samples (urine, plasma, cells) the separation power of one dimensional liquid chromatography is far not sufficient to achieve a good analyte dynamic range. Sample fractionation is one possible approach but on cost of sample throughput. Comprehensive LCxLC could be another solution but remains technical demanding in particular considering the transfer of the analyte. Ion mobility spectrometry (IMS) has become available on commercial instruments over the last years and can be used as an additional separation dimension. The addition of modifiers in differential mobility spectrometry (DMS) is particularly interesting as it allows to tune the separation resolving power.

www.ISC2016.ie @isc2016 #isc2016 /isc201620
KEYNOTE ABSTRACTS
peak arrangements might be possible, and their probability of being true is analysed. Another example concerns the screening of toxicological compounds, in which the objective was to assess the probabilities of a list of compounds being present in the sample, analysed with LC-MS. Another example concerns the comparison of GCxGC-MS chromatograms, in which small differences between samples are to be found. Finally, a Bayesian method for base-line correction in GC-MS is going to be discussed. In all cases, the algorithm applied involves a paradigm shift in the way the chromatographer is working on data analysis, speeding up the process of automated data treatment.Disclosure of Interest: None DeclaredKeywords: Chemometrics, GCxGC, LCxLC
KN06 – New bonded phases for chromatography of proteins
Mary J. Wirth, Jonathan Yasosky and Alexis HuckabeePurdue University
Abstract: It is urgent to improve proteins separations because proteins play so many roles in human health, especially protein drugs and protein biomarkers for diseases. For chromatography of small molecules, much attention has been given to particle morphology, with the introduction of sub-2 mm particle more than a decade ago and the more recent introduction of superficially porous particles. Our own group has introduced submicrometer particles for the ultimate reduction in mass transport distances. Protein separations are now largely limited by the bonded phase. The bonded phase limits resolution for the diverse types of protein chromatography used for therapeutic monoclonal antibodies and for LCMS used in proteomics. We present improved protein separations from new bonded phases.

21www.ISC2016.ie @isc2016 #isc2016 /isc2016
KEYNOTE ABSTRACTS
KN07 - A New Particle For Chromatography
Authors: Peter Myers, Haifei ZhangDepartment of Chemistry University of Liverpool
Abstract: In recent years there has been a renewed interest in particle technology for both HPLC and UHPLC. Prior to this renewed interest there was very little novel development in HPLC particle technology, since the primary changes related to reducing the particle size from 5micron down to 1.7microns. The purity of the underlying silica has become purer, for example the sodium levels have been reduced from around 1500ppm down to 10ppm, which has resulted in more robust chromatography, but has not intrinsically affected the structure of the particles.
We have also seen new hybrid silicas but again the particle size and particle structure has not resulted in a step change in performance from previous offerings.
The reintroduction of pellicular or core shell particles however was a great step change in particle design and brought a new interest into particles. Recent papers have shown that these core shell particles when used in the separation of small analytes benefit from their very narrow particle size distribution that leads to exceptionally well-packed columns, and also the reduced dead volume caused by the solid core.
This presentation outlines a totally new particle called a Sphere-on-Sphere particle1 manufactured using a one-step synthesis delivering a near monodispersed particles and core shell morphology.1 Patent Pending ThermoFisher Scientific University of Liverpool
The morphology of the particle has been designed to deliver the real advantages of the core-shell particles.
The total diameter of the particle and the core diameter can be controlled together with the effective pore diameters on the surface of the material. All the particles are solid and so the pores are as a result of the interconnectivity of the outer smaller spheres attached to the large single central solid core. The total particle size can be varied from 3 to 10 microns with an effective pore size range up to 1.5 microns.
Examples will be shown of these S-O-S particles used in normal phase and reversed phase separations of small molecules and also proteins.
Further example will be shown of how these particles can be used as scaffold particles or carrier particles in which new, novel stationary phase can be trapped in the outer layer.
Fig 1 The new Sphere-on-Sphere particles
Fig 1 The new Sphere-on-Sphere particles

www.ISC2016.ie @isc2016 #isc2016 /isc201622
KEYNOTE ABSTRACTS
KN08 – Exploiting Ionic Liquids and Polymeric Ionic Liquids in Multidimensional Gas Chromatography and Sample Preparation
Author: Jared L. AndersonDepartment of Chemistry, Iowa State University, Ames, IA 50011 U.S.A.
Abstract: Ionic liquids (ILs) can be designed to exhibit unique properties for their use in a number of applications in analytical and bioanalytical chemistry. This talk will focus on the design and synthesis of ILs, magnetic ionic liquids (MILs) and polymeric ionic liquids (PILs) and the use of these materials in a number of applications within sample preparation and multidimensional gas chromatography. A series of monocationic and dicationic ionic liquid (IL)-based stationary phases were evaluated as secondary columns in comprehensive two-dimensional gas chromatography (GC×GC) for the separation of aliphatic hydrocarbons from kerosene. In order to further understand the role that structural features of ILs play on the selectivity of nonpolar analytes, a series of dicationic IL-based stationary phases were evaluated using GC×GC. PIL-based sorbent coatings in solid-phase microextraction (SPME) will be discussed in their use for the extraction of a number of analytes within a variety of matrices. Finally, the ability to perform rapid DNA extraction from biological samples using custom-designed MILs will be presented. MIL solvents exhibit a number of advantages that allow them to be used in downstream DNA analysis including polymerase chain reaction (PCR).
KN09 – All you ever wanted to know about SFC mobile phases
Author: Caroline WestUniv Orléans, CNRS UMR 7311, ICOA, Pôle de Chimie, rue
de Chartres, BP 6759, F-45067 Orléans cedex 2, Franceemail: [email protected]
Abstract: Mobile phase composition in supercritical fluid chromatography (SFC) is flexible: it can be adjusted to the polarity of the analytes in changing the nature and proportion of co-solvent (alcohol, with or without water) mixed to carbon dioxide, and with the help of additives (acids, bases, salts…).
Some effects of mobile phase changes are rather well known: density variation, polarity variation and changes in the critical conditions.
But other effects of co-solvents and additives are somewhat unclear: “covering” of so-called “active sites” of the stationary phase, ion suppression, ion pairing, change of acidity of the mobile phase, chemical reactions?
In this presentation, I will discuss the different options based on old and new fundamental studies, and illustrate with examples.

23www.ISC2016.ie @isc2016 #isc2016 /isc2016
KEYNOTE ABSTRACTS
KN10 – Unique Carbon Adsorbents
Authors: Susan V. Olesik and Jiayi LiuDepartment of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 433210
Abstract: Carbon stationary phases are quite valuable for the separation of a broad range of analytes. For example, numerous liquid chromatographic-mass spectrometric separations of polymer bioanalytes are well documented. Precise control on the thickness of carbon adsorbents is necessary to obtain controlled retention. This control can be garnered both through synthetic methods in addition to self-assembly methods. Theory clearly shows that homogeneous chromatographic surfaces provide the highest efficiency and reproducibility. Carbon chromatographic surfaces have at least two distinct surface sites, basal and edge-plane. This talk will illustrate a range of new carbon adsorbents with controlled surface homogeneity and thickness. Fundamental studies on the differences in intermolecular interactions are also illustrated which will point to the optimal types of compounds best separated by either carbon type. These fundamental studies are fortified by chromatographic separations that illustrate the value of these ordered carbon materials.
KN11 - Bioanalysis – Reflections, Projections and the role of Separation Science
Author: Dr Derek Stevenson, visiting Senior LecturerUniversity of Surrey, Guildford, UK
Abstract: This lecture will review bioanalysis through the 60 years of the Chromatographic Society highlighting major developments as well as speculating on future needs. The analysis of drugs, metabolites, biomarkers and other trace organic compounds in biological samples has been one of the most difficult yet most important areas for separation science. Bioanalysis more broadly can involve different applications, for example pharmacokinetics, therapeutic drug monitoring, forensics, occupational and environmental monitoring and clinical biochemistry. Early methods to measure drugs included luminescent and colorimetric methods, immunoassays, radioderivatization, Thin Layer Chromatography (TLC) and Gas Chromatography (GC).
In the late 1960s/early 1970s High Performance Liquid Chromatography (HPLC) began to dominate. With most of these approaches sample extraction and concentration were also crucial as it was rarely possible to inject biological samples directly into instruments. Improvements in selectivity and sensitivity included the availability of a range of detectors as well as improvements in column technology. The desire to measure lower concentrations with higher sample throughput and automation along with the need to prove that results were valid has driven many developments. These include better detection (especially mass spectrometry), more efficient columns and more selective sample preparation.

www.ISC2016.ie @isc2016 #isc2016 /isc201624
KEYNOTE ABSTRACTS
Often advances in new or existing techniques show early promise (or receive much publicity) but these do not always come into routine use. This may be due to the rigorous validation requirements and the difficulty when actually dealing with biological samples.
Modern challenges still drive the need for advancement as more analytes are determined in a single run (metabolomics), large unstable biomolecules (proteins, antibodies) become more widely developed, lower detection levels are required, smaller amounts of sample are preferable.
The desire for more personalised health care means that the desire for devices capable of use in the home or local clinic is very strong. In the filed of bioanalysis further advances are required before this laudable goal can be achieved. The complexity of biological samples as well as appropriate validation guidelines means that in the foreseeable future separation method are still likely to dominate Bioanalysis.
KN12 - The Application RP and HILIC HPLC Hyphenated with HRMS/MS for Untargeted Metabolomics to Elucidate Nano Particle Induced Toxicity Mechanisms in A549 Human Lung Epithelial Cells
Authors: Roland Reischl1*, Christina Ranninger1, David Licha1, Matthew Boyles2, Albert Duschl2, Christian Huber1
1 Department of Molecular Biology, Division of Chemistry and Bioanalytics, Hellbrunnerstrasse 34, 5020 Salzburg, Austria
2 Department of Molecular Biology, Division of Allergy and Immunology, Hellbrunnerstrasse 34, 5020 Salzburg, Austria
email:*[email protected]
Abstract: Over the last decade the rise and availability of high resolution mass spectrometers delivering excellent mass accuracy has sparked the evolvement of ever more powerful omics methods. Proteomics and metabolomics both profited from the increase in sensitivity and in mass accuracy. Some metabolomics studies have been developed relying on the performance and selectivity of mass spectrometry as a standalone platform [1]. We analyzed A549 human lung epithelial cells, which had been treated with CuO nanoparticles and integrated information gathered by reversed phase HPLC and HILIC, MS and MS/MS in order to identify and relatively quantify as many metabolites as possible. As expected, we identified oxidative stress upon treatment with metal oxide nanoparticles [2, 3] and additionally we detected apoptotic cell death. The broad spectrum of chemically diverse metabolites identified allowed us to elucidate similarities and dissimilarities between apoptotic events occurring upon exposition

25www.ISC2016.ie @isc2016 #isc2016 /isc2016
KEYNOTE ABSTRACTS
of cells to CuO nanoparticles and apoptosis inducing agents such as camptothecin and staurosporine. On the course of our experiments we identified a potential biomarker occurring in all three apoptosis pathways. Furthermore we critically discuss the usefulness of chromatographic miniaturization to enhance the detectability of low abundant metabolites.References1 Han, J., et al., Metabolomics, 2008. 4: p. 128-140.2 Karlsson, H.L., et al., Toxicology Letters, 2009.
188(2): p. 112-118.3 Karlsson, H.L., et al., Chem. Res. Toxicol., 2008.
21: p. 1726-1732.
KN13 – Towards Hyperformance Two-Dimensional Liquid Chromatography
Authors: Peter Schoenmakers, Anna Baglai, Henrik Cornelisson van de Ven, Andrea Gargano and Bob Pirok,University of Amsterdam, Faculty of Science, Science Park 904, 1098 XH Amsterdam, The Netherlands
Abstract: Liquid chromatography (LC) is an immensely powerful separation technique for non-volatile samples. The combination of LC with mass spectrometry (LC-MS) can be used to characterize complex samples and to quantify selected analytes accurately. Yet, LC-MS still has its limitations. High-performance LC typically has a peak capacity of a few hundred, which implies that for any sample with more than a few dozen components peak overlap should be expected [1]. Usually, complex samples contain relevant analytes in divergent concentrations and major difficulties arise when low-abundant analytes are overshadowed by high-abundant ones.
Thus, we need to improve the separation power of LC.
Comprehensive two-dimensional liquid chromatography (LC×LC) provides additional separation power and additional selectivity. Peak capacities are typically increased from hundreds in high-resolution one-dimensional LC to thousands in LC×LC. By selecting two very different (“orthogonal”) retention mechanisms in LC×LC much additional selectivity can be provided and when the sample dimensionality (the number of structural features that distinguish the sample components) is relatively low structured chromatograms may be obtained.
Performing LC×LC is relatively straightforward, especially with the advent of modern dedicated instrumentation, but there are some challenges. These include dealing with the large amounts of data obtained in a correct and convenient manner and efficient method development. Also, detection sensitivity is hindered by the dissolution process in two successive LC columns. Solutions to the former two problems can be found in smart software programs. The latter problem can be addressed with active modulation [2]”ISSN” : “1520-6882”, “PMID” : “26709410”, “abstract” : “Online comprehensive two-dimensional liquid chromatography (LC \u00d7 LC.
In this lecture the principles of LC×LC will be briefly reviewed and contemporary developments will be illustrated with practical applications.References:1] J.M. Davis, J.C. Giddings, Statistical theory
of component overlap in multicomponent

www.ISC2016.ie @isc2016 #isc2016 /isc201626
KEYNOTE ABSTRACTS
chromatograms, Anal. Chem. 55 (1983) 418–424. doi:10.1021/ac00254a003.
[2] A.F.G. Gargano, M. Duffin, P. Navarro, P.J. Schoenmakers, Reducing Dilution and Analysis Time in Online Comprehensive Two-Dimensional Liquid Chromatography by Active Modulation., Anal. Chem. 88 (2016) 1785–1793. doi:10.1021/acs.analchem.5b04051.
KN14 – Investigations on the Metabolism of Non-Steroidal Anti-Inflammatory Drugs in Plants
Authors: Christian Klampfl*, Lisa Emhofer, Wolfgang Buchberger, Markus Himmelsbach
Abstract: The use of pharmaceuticals in human and in veterinary medicine has increased continuously. One negative side effect of this development is that pharmaceutically active ingredients (AI´s) can already be found in the aquatic system. Water contamination by AI´s occurs either due to incomplete uptake by the human body, so still active substances are excreted, drug containing gels and lotions, which are externally applied and washed off during personal hygiene, and unfortunately also by improper disposal of pharmaceuticals. Although contaminated municipal wastewater is treated in wastewater treatment plants, most of these substances are not completely removed. If water from these treatment plants is used for irrigation in agriculture, plants come into direct contact with these AI´s. Therefore uptake and metabolism by plants may occur representing a potential risk for consumers if edible plants are affected.
In the present work we studied the uptake and particularly the metabolism of non-steroidal anti-inflammatory drugs (NSAIDs) in plants. This class of drugs was selected as they are widely used and frequently found in the municipal waste water system up to the low mg L-1 range. For our experiments cress (lepidium sativum) and radish (raphanus sativus) were established as plant-model systems.
Cress was hydroponically grown with spiked tap water. After extraction the substances were separated on a RP-HPLC column and analyzed with high resolution mass spectrometry (QTOF and Orbitrap). For Diclofenac, one of the most frequently used NSAID´s, hydroxylation products as well as glucose and malonic acid conjugates could be detected in addition to the parent drug. Treatment of radish also led to drug uptake and formation of similar metabolites. Regarding the spatial distribution in the radish, the highest concentration was found in the roots, followed by the outer shell, while the inner part hardly contained any intact or metabolized AI.
KN15 - UHPLC and HRMS Methods for Bio-Therapeutic Protein Characterisation, Speed, Resolution and the PitfallsAuthor: Ken CookThermo Fisher Scientific, Waltham
Abstract: The Pharmaceutical industry has made a significant move towards the manufacture of Bio-Therapeutic proteins over small molecule therapeutics. The advantage in specificity and reduced side

27www.ISC2016.ie @isc2016 #isc2016 /isc2016
KEYNOTE ABSTRACTS
effects from molecules such as monoclonal antibodies [mAb] is very clear. These large proteins however are very different from the world of small molecule drugs and require different levels of characterisation with very different methodology. There are many types of protein bio therapeutics, mAb’s however remain the most popular due to the specificity and proven track record in the treatment of a broad range of diseases. The number of development candidates for monoclonal antibodies and related therapeutics is extensive and growing, creating the need for fast and efficient characterisation workflows during development and production of these products. This has led to a rapid improvement of UHPLC based methodologies. Ion exchange, size exclusion, Reverse phase intact protein, peptide mapping, hydrophilic interaction chromatography [HILIC] and hydrophobic interaction chromatography [HIC] methods have all been introduced into the characterisation arsenal very quickly. There is also a move towards global applicability of these methods to reduce the time it takes to develop optimised assays. To do all this well we need to fully understand these new methods and techniques. Which of these methods give the most useful information in terms of efficacy and safety? This all leads to the question are the methods presently being produced in the industry perfect or are they sub-optimal? Examples will be shown of some of the common pitfalls that lead to poor method development with protein characterisation and how UHPLC level analysis can be achieved for these large proteins with the correct approach. Multi-attribute methods will also be discussed and how the introduction of high resolution mass spectroscopy [HRMS] into the QC
environment can be implemented and what advantages it can bring to QC and PAT analysis.
KN16 - Application of capillary ion chromatography and capillary ion chromatography coupled with mass spectrometry in the production of high resolution inorganic and organic anion temporal profiles from Antarctic snow pit samples
Authors: Estrella Sanz Rodrigueza, Meredith Nationb,c, Andrew Moyb,c, Mark Curranb,c, Paul R. Haddada, Pavel N. Nesterenkoa, Brett Paulla,∗
a Australian Centre for Research on Separation Science, School of Physical Sciences, University of Tasmania, GPO Box 252-75, Hobart, Tasmania 7001, Australia.
b Australian Antarctic Division, 203 Channel Highway. Kingston Tasmania 7050. Australia.
c Antarctic Climate and Ecosystems Cooperative Research Centre, GPO Box 252-80, Hobart, Tasmania 7001, Australia.
* Corresponding author: Ph: +61 3 6226 6680; Fax: +61 3 6226 2858. email: [email protected]
Abstract: The high costs associated with collection of ice-cores from Antarctica demand scientists extract the absolute maximum data from these precious resources. Traditional analytical techniques are neither optimised nor indeed able to meet the demands of delivering high value multi-species data from sub-mL sample volumes, to provide higher temporal resolution in subsequent paleoclimatic records. To extract the most information from analytical data derived from these valuable ice cores, and/or low accumulation sites, the amount of sample required for each analysis must be drastically reduced. Capillary ion chromatography (cap-IC)

www.ISC2016.ie @isc2016 #isc2016 /isc201628
KEYNOTE ABSTRACTS
presents a new analytical capability to provide quantification of inorganic and organic ions based upon such sample volumes. In this presentation we will present results from a quantitative study on the simultaneous determination of organic and inorganic anions, including fluoride, methanesulfonate, chloride, sulphate and nitrate anions in Antarctic ice core and snow pit samples. The new method necessitated only 39 mL of injection volume to reach the analytical performances required. In this work, the capillary ion chromatograph was also coupled with mass spectrometry, and optimised for the identification and quantification of methansulfonate. The limit of detection for methanesulfonate was decreased to just 0.07 mg L-1 using this hyphenated technique, being the lowest reported until now in the literature for any ion chromatography based method. To validate the new method, a comparative study and statistical evaluation of the methanesulfonate results obtained for 101 snow pit samples from the Aurora Basin site, Antarctic, by three separate ion chromatography based methods, namely, standard ion chromatography, capillary ion chromatography and capillary ion chromatography coupled to mass spectrometry, was performed.
KN17 - LC-MS-Based Metabonomics, Metabolomics and Metabolic Phenotyping
Author: Ian WilsonImperial College of London, Division of Computational Systems Medicine, Department of Surgery and Cancer, Sir Alexander Building, Exhibition Road, South Kensington, London SW7 2AZ, United Kingdom.
Abstract: LC-MS-based global metabolite profiling ought to be easy – but there are many pitfalls for the unwary that can mean that the data produced from this type of analysis can be of less value than it should be. The tutorial will provide an overview of the strengths and weaknesses of the approach, and provide suggestions for practical solutions to problems wherever possible.
The areas to be covered will include study design, confounding factors, sample collection and storage, selection of chromatographic conditions, the need for (and effects of) sample preparation, how to ensure that the LC-MS system has the best chance of providing reproducible and reliable data, and how to demonstrate that this has been done. The topics of the “identification” of unknowns will be discussed, especially with reference to what actually constitutes identification and the use of “targeted vs non-targeted” methods (and what these terms actually mean) will be considered.
These topics will be illustrated with illustrative examples from studies in basic physiology, disease models and humans.

29www.ISC2016.ie @isc2016 #isc2016 /isc2016
KEYNOTE ABSTRACTS
KN18 – Analysis of bisphenol A in canned food: Tandem mass spectrometry fails, chromatography saves, high-resolution mass spectrometry elucidates
Authors: C. Czerwenka*, E. Dorn, H. Hinterwirth
Abstract: Bisphenol A is under increasing scrutiny as food contaminant due to its suspected endocrine disrupting activity. Therefore, selective and sensitive methods are required to detect this compound at low levels in complex food matrices, with liquid chromatography coupled to mass spectrometry being the method of choice.
In the course of the analysis of bisphenol A in canned food products using LC-MS/MS we noticed that the chromatograms of the selected reaction monitoring transitions of bisphenol A showed several peaks (up to ten). For all of these peaks a perfect co-elution of the two mass transitions was observed and the ratios of the transitions fully agreed with that of bisphenol A. Only the utilization of the retention time allowed for the correct identification of the target bisphenol A.
We then proceeded to elucidate the nature of the remaining peaks. The coating of food cans is often built upon bisphenol A diglycidyl ether (BADGE). During the curing process or in contact with food BADGE may undergo various reactions, including hydrolysis and hydrochlorination, leading to a substantial number of derivatives. Analyses by high-resolution (tandem) mass spectrometry provided useful information on the unknown peaks and showed that at least several of them
resulted from BADGE derivatives, all exhibiting the pseudomolecular ion of bisphenol A in their mass spectrum. Two compounds were unequivocally identified as BADGExHClxH2O and BADGEx2H2O by use of reference standards, whereas a bisphenol A monoglycidyl ether derivative and a proposed dimeric structure were substantiated by mass spectral data and their chromatographic behaviour.
It is concluded that BADGE-related compounds readily undergo in-source fragmentation, even under mild ionisation conditions, yielding bisphenol A ions. Thus, high chromatographic resolution is required to correctly identify bisphenol A in food products packaged in BADGE-coated cans.
KN19 – Microchip Electrophoresis-based Methods for Monitoring Oxidative Stress In Vivo and In Vitro Author: Susan M. LunteRalph N. Adams Institute for Bioanalytical Chemistry, Lawrence, KS USA
Abstract: Microchip electrophoresis is a powerful tool for the analysis of biological samples. In particular, its ability to perform fast, efficient separations makes it possible to monitor several compounds simultaneously with high temporal resolution. The small dimensions of the channels in the chip are compatible with the analysis of microdialysis samples and single cells. In this presentation, two applications of microchip electrophoresis (ME) for biochemical investigations will be presented. The first involves the development of ME-based methods for the detection of reactive

www.ISC2016.ie @isc2016 #isc2016 /isc201630
KEYNOTE ABSTRACTS
nitrogen and oxygen species (RNOS) in macrophages and immune cells. This includes direct amperometric detection of RNOS as well as the evaluation of fluorescent reagents used for specific species. ME allows detection of multiple substances in a single run, giving a better snapshot of the total RNOS production in the cells. The second application involves the combination of microdialysis with microchip electrophoresis for near real-time continuous monitoring of nitric oxide metabolites, catecholamines, and adenosine in awake, freely roaming animals. The goal of this application is to investigate the biochemical basis of neurodegenerative diseases caused by oxidative stress, such as epilepsy and traumatic brain injury.
KN20 - Using High Resolution LC-MS to Investigate Interplay Between Bioprocessing Parameters and Product QualityAuthors: Amy Farrell1, Kai Scheffler2, Jenny Ho3, Ken Cook3, Peter Mowlds3, Patrick Pankert2, Andrew Williamson3 and Jonathan Bones1
1 Comparability and Characterisation Laboratory, NIBRT – The National Institute for Bioprocessing Research and Training, Foster Avenue, Mount Merrion, Blackrock, Co. Dublin, Ireland.
2 Thermo Fisher Scientific, Im Steingrund 4-6, 63303 Dreieich, Germany
3 Thermo Fisher Scientific, Stafford House, 1 Boundary Park, Hemel Hempstead HP2 7GE, United Kingdom.
Abstract: Chinese Hamster Ovary (CHO) cells lines are the dominant expression system used in the biopharmaceutical industry for the production of therapeutic proteins. Despite numerous improvements to CHO bioprocessing based on empirical observations, there is a still a very poor fundamental understanding of the molecular
mechanisms governing biotherapeutic protein production from these important expression systems. Following serum free suspension batch culture, quantitative proteomics was performed on a Chinese Hamster Ovary (CHO) cell line that expressed an anti-Interleukin-8 IgG1 monoclonal antibody (mAb), to elucidate the cellular response to altered culture conditions of pH, temperature and dissolved oxygen. A multiplexing workflow using Tandem Mass Tags (TMT) and multi-dimensional liquid chromatography-mass spectrometry (LC-MS3) incorporating synchronous precursor selection (SPS) was applied for identification and quantification of proteins differentially expressed in response to culture conditions. In addition the multiplexing workflow was also utilised for the analysis of naïve CHO K1 cells following their exposure to spent culture media from the various production runs.
Furthermore, complete characterisation of the expressed mAb was carried out using advanced LC-MS methods including high resolution middle down MS; MS determination of sequence-level modifications, intact protein analysis of critical quality attributes, stable isotope based quantitative glycan analysis, hydrogen deuterium exchange MS for structural comparability analysis and surface plasmon resonance spectroscopy for determination of changes to the functional integrity of the expressed mAb. Combined, the findings herein provide a holistic insight into the effect of upstream parameters on the quality of therapeutic proteins and facilitate a greater understanding of the molecular mechanisms governing biopharmaceutical production systems, thereby creating a hypothesis for improved future cell line development using various engineering strategies.

31www.ISC2016.ie @isc2016 #isc2016 /isc2016
KEYNOTE ABSTRACTS
This research was supported by Science Foundation Ireland as part of a Starting Investigator Research Grant, grant reference: 11/SIRG/B107.
KN21 - Application of superficially porous chiral stationary phases for separation of enantiomers in liquid-phase techniquesAuthor: Bezhan ChankvetadzeInstitute of Physical and Analytical Chemistry, Tbilisi State University, I. Chavchavadze Ave 1, 0179 Tbilisi, Georgia
Abstract: The advantages of superficially porous silica (SPS) over fully porous silica (FPS) for achiral liquid phase chromatographic separations have been extensively documented. These advantages include significantly higher chromatographic efficiency, a marked shift to higher optimal flow rates and limited dependence of column performance on mobile phase linear velocity. The higher efficiency of columns made with SPS particles is probably due to the more uniform particle-size distribution of SPS particles compared to their fully porous analogues resulting in higher radial homogeneity of columns packed with SPS and to the shorter diffusion path available to analytes. These same characteristics are responsible for the limited dependence of column performance on the mobile phase linear velocity, minimizing both the eddy diffusion term (A term) and also the mass-transfer contribution (C term) to the height equivalent to a theoretical plate (HETP) as detailed in the van Deemter equation. Realizing the minimum HETP value at higher flow rate and maintaining such low HETP at even higher mobile phase flow rates makes SPS columns suited for high-speed separations.
Since polysaccharide esters and phenylcarbamates are recognized to be the most successful chiral selectors for the separation of enantiomers in liquid phase techniques such as high-performance liquid chromatography (HPLC), supercritical fluid chromatography (SFC), nano-chromatography and capillary electrochromatography (CEC), their combination with SPS seems logical for the preparation of highly efficient chiral stationary phases (CSP).
This presentation summarizes our recent studies on the preparation of polysaccharide-based CSPs for separation of enantiomers in HPLC, SFC, nano-LC and CEC. Various effects based on superficially porous structure of silica are reported and discussed in detail.
KN22 - Recent advances of capillary electrokinetic separation technologies and their applications in pharmaceutical and biochemical analyses Authors: C. Yan 1,*, Y. Wang 1, Y. Xue 1, B. Tang 1, S. Sun 2
1 School of Pharmacy, Shanghai Jiao Tong University, 2 R&D Department, Unimicro (Shanghai) Technologies,
Ltd., Shanghai, China
Content: Recent advances in two capillary electrokinetic separation technologies, namely pressurized capillary electrochromotography (pCEC) and quantitative capillary electrophoresis (qCE), will be reported.
Abstract: In order to overcome the problems and difficulties associated with bubbles formation in capillary

www.ISC2016.ie @isc2016 #isc2016 /isc201632
KEYNOTE ABSTRACTS
electrochromatography (CEC), a binary solvent delivery system consisting of two pumps is used to provide a pressurized flow that is complementary to the electroosmotic flow (EOF). The end result is pCEC. In such a system, quantitative sample injection can be realized through a rotary-type of nano-injector with a 10 nL sample loop. In addition, it is feasible to carry out a binary solvent gradient separation in pCEC, similar to that in HPLC, by programming the composition of mobile phase.
Another innovative qCE technique equipped with 10 nL sample injector was also developed which can perform quantitative analysis like in HPLC. Several type of detectors that are suitable for capillary-based pCEC and qCE techniques were developed, such as UV/Vis, LIF, ELSD detectors and MS interface. These analytical technologies were successfully used in many investigations including applications on pharmaceutical, biological, environmental analyses, as well as metabolomics.
KN23 - UHPLC-ESI-QTOF-MS/MS Based Lipidomics by Data-Independent Acquisition
Authors: Michael Laemmerhofer*, Jörg Schlotterbeck1, Bernhard Drotleff1
1 Institute of Pharmaceutical Sciences, University of Tuebingen, Tuebingen, Germany
Abstract: Lipidomics approaches are nowadays widely adopted to have a comprehensive view on lipids in biological samples as biomarkers of various diseases. We are particularly interested in biomarkers of oxidative stress in context of atherothrombosis and diabetes. For this
purpose comprehensive profiling by direct infusion ESI-MS/MS shotgun lipidomics or UHPLC-ESI-HR-MS/MS technologies with QqTOF or Orbitrap mass analyzers utilizing data-dependent acquisition (DDA) emerged as state-of-the-art. Thereby, precursor masses detected during a survey scan are fragmented and MS/MS spectra acquired which can be used for identification of distinctive features in samples. The advantage of such a scan mode is that MS/MS spectra can be unequivocally assigned to a particular precursor. Unfortunately, MS/MS triggers are often failed because it is a stochastic process fragmenting typically the most abundant precursors. The performance declines with complexity of samples and leads to incomplete analyte coverage which may be problematic in the course of biomarker identification. This problem can be overcome by data-independent acquisition (DIA). We use DIA by SWATH (Sequential Window Acquisition of all Theoretical Fragment-Ion Mass Spectra). In SWATH, precursor ions for simultaneous fragmentation are selected with intermediate Q1 mass window width (e.g. typically 10-100 Da) and the generated fragments readout by TOF analysis creating composite MS/MS spectra for each SWATH window. Data processing was done with commercial software PeakView, MasterView and MarkerView (Sciex), open source software MS-DIAL and statistical tools. In this presentation, the potential of SWATH in lipidomics will be elucidated and discussed on clinical examples.References: H. Hinterwirth et al., Anal. Chem., 85
8376-8384 (2013).

33www.ISC2016.ie @isc2016 #isc2016 /isc2016
KEYNOTE ABSTRACTS
KN24 – From separations to sensors for environmental monitoring
Author: Fiona Regan, DCU Water Institute, Dublin City University, Glasnevin,
Dublin 9, Ireland
Abstract: Growing concern about environmental contamination creates a greater demand for analytical methods that meet the needs of both compliance, investigative and trend monitoring. Coastal and freshwater pollution from nutrients leading to algal blooms and exposure of drinking water to metals and emerging contaminants means that improved analytical methods are required for bench and field. The growth of mass spectrometry with LC and GC separation means that measurement of environmental contaminants and their metabolites is now possible. However, these techniques are costly and are typically used for analysis of grab samples as part of a monitoring programme. There is a need to develop methods of measuring certain pollutants in real-time over prolonged periods of time, in areas of high risk so that environmental health can be protected. This paper will look at the development of analytical sensing platforms for nutrients, algal toxins and metals with the aim of real-time monitoring at concentrations relevant in the environment. It will also look at how generic technologies can be adapted for emerging chemicals of concern.
KN25 – Multiplatform metabolomics for nutrition related diseases
Authors: C. Barbas 1,*, A. Mastrangelo 1, D. Dudzik 1, A. García 1, L. F. Alguacil 1, P. Ramos 1
1 CEMBIO, Universidad San Pablo CEU, Boadilla del Monte. Madrid, Spain
Abstract: Non-targeted metabolomics techniques combined with multivariate statistics offer the ability to examine global changes in metabolites associated with physiological conditions. Thus, metabolomics is a powerful approach for examining disease-related metabolic changes and, accordingly, proves highly effective in identifying new biomarkers. We have applied a multiplatform (LC-QTOF/MS, GC-Q/MS and CE-TOF/MS) metabolomics approach to gain a better understanding of changes associated to nutrition related diseases at three stages of life, such as Gestational Diabetes Mellitus (GDM), insulin resistant (IR) prepubertal obese children and bariatric surgery effectiveness in adults.
Plasma fingerprints allowed for the discrimination of GDM pregnant women from controls [1]. In particular, lysoglycerophospholipids showed a close association with the glycemic state of the women. We provide evidence for the implication of these compounds in metabolic routes. In a second stage, we used the multiplatform metabolomics strategy and combined both untargeted and targeted (validation) approaches, to investigate the effects of insulin resistance in a specific population of obese children [2]. Finally in adulthood, we approached bariatric surgery (BS). Among metabolites able to predict effectiveness, bile acids and gut microbiota

www.ISC2016.ie @isc2016 #isc2016 /isc201634
KEYNOTE ABSTRACTS
by-products presented the most remarkable changes, reflecting the great influence of the microbial community on the surgery outcome. Moreover, we also investigated the correlation of metabolites with a psychological variable, food-craving, and its predictive capability.References: 1 D Dudzik, et al. Metabolic fingerprint of Gestational
Diabetes Mellitus.J Proteomics. 2014 103:57-71.2 A Mastrangelo, et al. Insulin resistance in
prepubertal obese children correlates with sex-dependent early onset metabolomic alterations. International Journal of Obesity (under final revision)
KN26 – Where the worlds of pharmaceutical and environmental analysis converge: Speciation analysis and bioimaging of gadolinium-based contrast agents
Uwe Karst*, Stefanie Fingerhut, Marvin Birka, David Clases and Michael SperlingUniversity of Münster, Institute of Inorganic and Analytical
Chemistry, Corrensstr. 30, 48149 Münster, Germanyemail: *[email protected]
Gadolinium-based magnetic resonance imaging (MRI) contrast agents have found increasing use in medical diagnostics in the last 25 years. The compounds are known to be tolerated well, and to exhibit only few side-effects due their fast excretion kinetics with a half-life of only two hours.
In 2006, these compounds started to be associated with a newly discovered disease, nephrogenic systemic fibrosis (NSF), which is only observed for dialysis patients. Furthermore, the disease has only been
described for contrast agents with linear ligands, but not for those with macrocyclic ligands. This indicates an influence of the lower kinetic stability of the contrast agents with linear ligands. While the general cause of the disease with a deposition of the gadolinium in the lower parts of the skin has been identified, many details of the pathogenesis are still unknown.
Very recently, small residues of the contrast agents have been discovered to remain in the human brain, and many related questions are currently under investigation. Even despite any current indication for respective pathogenic effects, scientists, manufacturers and regulatory agencies are concerned about these findings. To investigate this situation, liquid chromatography (LC) coupled to inductively coupled plasma-mass spectrometry (ICP-MS) and electrospray mass spectrometry (ESI-MS) are used, combined with spatially resolved analysis by laser ablation (LA)-ICP-MS imaging.
Similar hyphenated techniques are also used to determine residues of the contrast agents in wastewater, surface water and drinking water samples, as the compounds will pass wastewater treatment plants and waterworks almost quantitatively due to high polarity and very high chemical stability.
In this lecture, the development of the respective analytical methods and their application to address the challenges raised above are presented.

35www.ISC2016.ie @isc2016 #isc2016 /isc2016
KEYNOTE ABSTRACTS
KN27 - Potential of aptamers for the selective extraction of compounds at the trace level in complex samplesPichon Valérie 1,2
1 Department of Analytical and Bioanalytical Sciences and Miniaturization (LSABM), UMR CBI 8231 , ESPCI Paris, 10 rue Vauquelin, 75 005 Paris, France
2 Sorbonne Universités, UPMC , 4 place Jussieu, 75005 Paris, France
email: [email protected]
The evolution of the instrumentation in terms of separation and detection allowed a real improvement of the sensitivity and the analysis time. However, the analysis of ultra-traces from complex samples requires often a step of purification and of preconcentration before the chromatographic analysis.
To improve selectivity during sample pre-treatment, various selective tools inducing a molecular recognition mechanism during the extraction procedure can be developed. Indeed, sorbents constituted of immobilized antibodies, i.e. immunosorbents, have been largely applied to the selective extraction of various types of molecules, from small to large size molecule at the trace level in complex samples. Despite their high potential, their development is time-consuming and relatively expensive. These drawbacks have led to the development of molecularly imprinted polymers (MIPs) that are synthetic polymeric materials possessing specific cavities designed for a template molecule involving a retention mechanism based on molecular recognition. They present the advantage to be synthesized in a few days. In return, their application to real samples necessitates a careful optimization of the extraction procedure to reach the expected selectivity, this selective procedure
being very easy to develop when using antibodies.
More recently, as an alternative to both previous approaches, aptamers immobilized onto a solid support, i.e. oligosorbent, were proposed. Thanks to the high affinity and high selectivity of the interaction that offer some aptamers towards some target analytes, they also provide powerful techniques that enable a selective extraction and the concentration of a target analyte from liquid matrices in one step or the sample purification of extracts from solid matrices.
The development and the properties of these oligosorbents developed for different types of targets will be presented. After describing the immobilization procedures, different parameters characterizing the potential of aptamer-based support as extraction sorbents will be discussed. Indeed, close relations exist between extraction recoveries and the affinity and amounts of aptamers immobilized on the extraction device. In addition, analyte-aptamer interactions may be affected by matrix samples and by additives in the samples. This may also lower extraction recoveries and affect the stability and the possible reusability of the aptamer-based sorbent. All these different points will be illustrated by several examples.
The application of these sorbents to the treatment of complex samples such as food samples, environmental samples, and biological fluids will be presented and their potential will be compared with the potential of MIPs and immunosorbents. Their association with analytical devices, from conventional to miniaturized analytical systems, will be also discussed.

Oral Abstracts

37www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSPharmaceutical Analysis
Pharmaceutical Analysis
PA-003-001 - Use of a Triple Detection (UV-ELSD-MS) System for Mass Balance in the Forced Degradation of PharmaceuticalsP. Hong 1,*, S. M. McCarthy 1, P. R. McConville 1
1 Waters Corp, Milford, United States
Abstract: Forced degradation studies are typically performed using HPLC and UV detectors to understand the degradation pathway of pharmaceuticals and to insure all impurities are accounted. In these studies, performing mass balance or the conservation of mass is crucial. Given the range of impurities and their chemical and physical properties, mass balance studies can be challenging. These challenges may be due to co-elution of impurities and the active pharmaceutical ingredient (API), the presence of undetected components, or differences in response of the impurities to the API: all of which can lead to poor recoveries or a lack of understanding of the degradation pathway. 1
In this presentation we will evaluate mass balance using a triple detection system consisting of a photodiode array (PDA), evaporative light scattering detector (ELSD) and a mass detector. These studies will be performed in two parts. First, the relative response factors of impurities will be evaluated. Relative response, which is based on the ratio of UV peak area to concentration (mass), will be calculated using the slopes of the calibration curves. This well-established technique will then be compared to a method using the ratio of the response in UV to ELSD detection. The difference in linearity between the two
detection techniques will be addressed by converting the non-linear ELSD response to linear using standard mathematical principles. In the second part, the relative response factors will then be used to perform mass balance. Specifically, mass balance recoveries of the degraded samples will be determined using chromatographic software tools. The degradation pathway will then be confirmed through the identification of impurities and their by-products using a mass detector.References: 1. Baertschi SW, Pack BW, Hoaglund Hyzer CS,
Nussbaum MA. Assessing mass balance in pharmaceutical drug products: New insights into an old topic. TrAC Trends in Analytical Chemistry. 2013; 49:126-36.
Disclosure of Interest: None DeclaredKeywords: Evaporative light scattering detector, pharmaceutical, UPLC-MS
PA-003-002 - Characterization and Proteolytic Activity Studies of Botulinum Neurotoxin A in Pharmaceutical Products using High-resolution Tandem Mass Spectrometry
E. L. M. Wong 1,*, Y. C. Wong 1
1 Analytical and Advisory Services Division, Government
Laboratory, Hong Kong, Hong Kong, Hong Kong
Abstract: Botulinum neurotoxins (BoNT) are produced by the anaerobic bacterium Clostridium botulinum and are one of the most lethal known poisons (LD50 = 0.8 ug for a 70 kg human by inhalation). Despite its high lethal toxicity, BoNT have been used to treat spasms and other muscle

www.ISC2016.ie @isc2016 #isc2016 /isc201638
ORAL ABSTRACTS Pharmaceutical Analysis
problems. Most recently, BoNT serotype A (BoNT/A) gains its worldwide popularity in cosmetic surgery to prevent development of facial wrinkles. Clinical testing of BoNT/A is conventionally relied on in vivo mouse lethality assay, as stated in the pharmacopoeias, which requires huge amount of animals. In response to increased public pressure to the inhumane testing, sensitive replacement assays that can robustly measure BoNT/A in therapeutic or cosmetic products (containing ~0.1 ng or 50 MLD50 of BoNT/A) are of urgent need.
Identification of BoNT/A in pharmaceutical products using non-bioassay methods is always a challenge due to its complex structure, ultra-trace level and the interference from excipients (such as albumin and gelatin). This work reported a mass spectrometry study on BoNT/A in pharmaceutical preparations using non-bioassay method. BoNT/A in pharmaceutical matrix was isolated using magnetic beads immunoprecipitation followed by on-beads digestion and was characterized by peptide mass finger printing using high-resolution tandem mass spectrometry coupled with ion mobility and Q-ToF in MSE mode. The activity of the toxin was confirmed by its proteolytic activity towards specially designed synthetic SNAP-25 substrate. The specific cleaved peptide fragments obtained from SNAP-25 substrate correlated well with its proteolytic activity in linear range from 0.05 – 100 MLD50/uL (r2 > 0.99). Proteolytic activities of BoNT/A in different excipient matrices were also investigated. The method has been validated across few brands of BoNT/A pharmaceutical injections commonly available in the Asia Pacific and was found to produce reliable results.
Disclosure of Interest: None DeclaredKeywords: Botulinum neurotoxin A, High resolution mass spectrometry, Non-bioassay
PA-003-003 - Preliminary study of SFC critical method parameters for the determination of pharmaceutical compoundsA. Dispas 1,*, P. Lebrun 2, P.-Y. Sacré 1, P. Hubert 1
1 CIRM, University of Liège, Liège, 2Arlenda, Louvain-la-neuve, Belgium
Content: Nowadays, supercritical fluid chromatography is commonly presented as a promising alternative technique in the field of separation sciences. Nevertheless the selection of chromatographic conditions and sample preparation of pharmaceutical compounds remain a challenge and peak distortion was previously highlighted. The main objective of the present work was to evaluate the impact of different critical method parameters (CMPs), i.e. stationary phase, mobile phase composition and injection solvent nature. The experiments were performed considering two groups of antimalarial molecules: one group with neutral/apolar compounds and the other one with salt form of polar compounds. In this context, another objective was to propose a suitable sample solvent for quantitative analysis. During this study, design of experiments and desirability function approach enabled to highlight optimal chromatographic conditions in order to maximise peak capacity and to get acceptable value of symmetry factor. Regarding sample injection solvent composition, some counterintuitive results were observed: solvents closer to the mobile phase polarity did not provide best results in terms of peak symmetry. In

39www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSPharmaceutical Analysis
addition, acetonitrile and short aliphatic alcohols offered an interesting alternative as injection solvent: toxicity of solvents used is clearly reduced and better quantitative performances could be expected while keeping high peak capacity and symmetric sharp peaks. Finally, the quantitative performances were evaluated by the method validation for the quantitative determination of quinine sulfate in a pharmaceutical formulation. These better understandings on critical method parameters led SFC to be an even more promising technique in the field of the analysis of pharmaceutical compounds.References: 1. C. Hubert et al., J. Chromatogr. A 1395 (2015) 88-
982. C. Galea et al., J. Pharm. Biomed. Anal. 111
(2015) 333-3433. A. Dispas et al., J. Chromatogr. A 1353 (2014) 78-
88Disclosure of Interest: None DeclaredKeywords: critical method parameters (CMPs), Method validation, Supercritical fluid chromatography (SFC)

www.ISC2016.ie @isc2016 #isc2016 /isc201640
ORAL ABSTRACTS Column Technology Materials 1
Column Technology Materials 1
CTM1-004-001 - Advances In Fully Porous And Solid-Core Particle Technology
Kevin Wyndham* 1, Nicole Lawrence1, Beatrice Muriithi1, Bonnie Alden1, Jacob Fairchild1, Babajide Okandeji1, Daniel Walsh1, Cheryl Boissel1, Matthew Lauber1, James Cook1, Scott McCall1, Thomas Walter1
1 CBU, Waters, Milford, United States
Content: Solid-core stationary phases have had a significant impact in reversed-phase liquid chromatography, allowing for higher efficiencies and lower column pressures versus fully porous stationary phases of the same particle size. However, these efficiency gains are not always realized, as they are strongly dependant on the quality of packing, method, and instrumentation. One example of this has been the long held belief that solid-core packing materials could not be efficiently packed into 2.1 mm I.D. hardware and that users must accept a loss of efficiency when moving from larger to smaller I.D. columns. Higher system dispersions and lack of optimization of packed columns negatively impacts chromatographic resolution. In other areas fully-porous materials have clear advantages. First and foremost, the legacy and known quality of high performance columns gives users superior confidence. More tangible advantages include scalability of methods (e.g., direct, predictable scaling from UPLC to preparative scale), superior chemical resistance (e.g., pH 10-12), and sized based separations (e.g., SEC, GPC). It is evident from recent literature that both fully porous and solid-core materials have significant utility for contemporary
chromatographic separations. That stated - What advancements in solid-core materials are needed for further improvement in both separation performance and chemical stability? What is the future of solid-core stationary phases?
This presentation will explore the current state of solid-core and fully porous particle technologies and will contrast applications that show improvements in performance when used with solid-core or fully porous columns. Attention will be paid to optimization of solid-core columns for improvements in chemical stability and chromatographic performance. The evaluation of chemical stability of solid-core and fully porous particles, exposed to acidic and alkaline mobile phases will also be explored.Keywords: Hybrid, Reversed-Phase, Solid-Core
CTM1-004-002 - Analysis of Proteins including mAbs, and transfer of method using 2 µm analytical SEC column with dual functionality for HPLC and UHPLC
C. Benner 1,*, A. Chakrabarti 1
1 Tosoh Bioscience LLC, Philadelphia, United States
Content: Method transfer between HPLC and UHPLC is a very important and rather critical issue for chromatographers. Method transfer from HPLC to UHPLC is generally intended to reduce the run time, to increase the resolution with better column efficiency and also to reduce the solvent consumption. There are a few challenges in this regard. Changes in flow rate, solvent composition, mobile-phase pH, or column temperature

41www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSColumn Technology Materials 1
may be necessary to meet system suitability criteria while transferring a method. The end users without UHPLC instruments in the laboratory may also like to use the UHPLC column to take some of its advantage. Obviously the extra-column volumes in conventional HPLC need to be optimized, as these play a major role in the quality of the performance of a UHPLC column in HPLC system.
Here we will discuss the transfer of a method for the separation of proteins and monoclonal antibodies using a 5 µm particle size, 25 nm pore size analytical size exclusion chromatography conventional HPLC column (TSKgel G3000SWXL) to a new silica based, 2 µm particle Size, 25 nm pore size analytical size exclusion chromatography UHPLC column (TSKgel UP-SW3000) on a conventional HPLC instrument. We will also discuss the transfer of a method for the separation of proteins and monoclonal antibodies using the TSKgel UP-SW3000column from a conventional HPLC to a UHPLC instrument. We have shown that the new UHPLC column is compatible to both UHPLC and HPLC and can be interchangeably used with easy method transfer. Upon transferring the method from HPLC to UHPLC, the resolution between the peaks and the number of theoretical plates increased all over the range of separation including both LMW and HMW side of the monoclonal antibody. The experimental plan provided covers all the different variables for reproducibility issues and the overlay of the chromatograms clearly shows that the method is robust, easily transferable and reproducible. Disclosure of Interest: None DeclaredKeywords: Method Transfer, Monoclonal Antibody, UHPLC
CTM1-004-003 - Possibilities and limitations of computer-assisted method development in HILIC: a case studyE. Tyteca 1,*, S. Bieber 2, T. Letzel 2, G. Desmet 1
1 Chemical Engineering, Vrije Universiteit Brussel, Elsene, Belgium,
2 Technical University of Munich, Munich, Germany
Content: HILIC combines hydrophilic partitioning, H-bonding, electrostatic/ionic interactions, resulting in a non-linear dependency between ln(k) and the fraction of ACN φ and ln(φ). Also the salt concentration is known to have a significant influence in the modulation of the interactions.
The present study investigated the possibilities and limitations of computer-assisted method development in HILIC on a mixture of 14 isomeric hydroxyl- and aminobenzoic acids using a sulfobetaine (ZIC-HILIC) phase.
A limited number of isocratic runs were used to establish the Neue-Kuss parameters, enabling the retention prediction under any possible gradient profile. Errors < 2% were found, except for the compounds eluting at the very beginning of the gradient, which are too much influenced by the isocratic elution that precedes the actual start of the gradient (errors up to 10%).
Next to the observed k, also k at elution from the column was predicted and used to estimate the peak widths wp. Prediction of wp improved significantly using individual gradient N-values compared to employing one N for all compounds. Ngrad was calculated using the established relationship between Ngrad, Nobs, kel and kobs. The wp predictions were found to be least accurate for compounds

www.ISC2016.ie @isc2016 #isc2016 /isc201642
ORAL ABSTRACTS Column Technology Materials 1
eluting in the steepest region of the ln(k) vs φ relationship, where small errors in φel result in high kel errors.
Optimization was done via modeling of the Rs between each peak pair and constructing a Rs map, and the less model-dependent PEWS² strategy [1], using only an approximate modeling of the first and last peak to find the optimal elution window. Overall, the optimization was limited by the steep decrease in N (increase in wp) when increasing tgrad.
The separation was further improved by focusing on the most problematic zone in the chromatogram, as well as by applying a two-step gradient. The salt concentration was also optimized to improve the peak shapes.References: [1] E. Tyteca, Anal. Chem. 84 (2012)
7823-7830Disclosure of Interest: None DeclaredKeywords: Computer-assisted method development, HILIC, Retention modeling

43www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSFundamentals
Fundamentals
FS1-005-001 - Approaching Optimum Speed-Resolution Power Using Low-Density Mobile Phases. Application to Supercritical Fluid Chromatography
F. G. E. Gritti 1,*, M. Fogwill 1, S. Helmueller 1, J. A. Jarrell 1, M. Gilar 1
1 Core Research / Instrument / Fundamental, Waters Corporation, Milford, United States
Content: Across the wide bridge between conventional gas chromatography (GC) and liquid chromatography (LC), low-density mobile phases appear extremely attractive for the analysts: in contrast to GC, their solvation strength is easily adjustable by changing temperature and pressure while they provide higher separation speeds than classical LC eluents due to their lower viscosities [1].
However, low-density mobile phases have large Joule-Thomson (J-T) coefficients, which measure their temperature variation per unit increase of pressure under adiabatic conditions. For instance, the J-T coefficients of water (density 0.99 g/cm3) and carbon dioxide (density 0.29 g/cm3) at 60oC and 100 bar are -0.02 and 0.63 K/bar, respectively. During their decompression along the column, the temperature decrease of neat CO2 is thirty times larger than the temperature increase of water. Therefore, if the column is not properly thermally insulated when using low-density mobile phases, severe stationary radial temperature gradients are formed across the diameter of the column, which can no longer perform at its optimum efficiency.
In this presentation, experiments are investigated in order to approach the optimum efficiency of 3 mm x 150 mm columns packed with sub-2 µm particles using a low-density mobile phase such as neat CO2 at low outlet pressures (100 bar) and high temperatures (80oC). Such pressure/temperature conditions are extremely challenging in supercritical fluid chromatography (SFC) and lead to unacceptably low column efficiencies. The thermal insulation of the column using aerogel materials, the independent control of the temperatures of the eluent preheater and column oven, the design of ovens matching the axial temperature profile of the eluent, and the design of vacuum-based ovens are tested. Their performances are compared regarding the SFC separation of a series of alkylbenzenes and peptides.References: [1] J. C. Giddings, Unified Separation
Science, John Wiley & Sons, New York, (1991).Disclosure of Interest: None DeclaredKeywords: Column dispersion, Instrument technologies, Supercritical fluid chromatography (SFC)
FS1-005-002 - How to achieve and manipulate separations of polar and ionised compounds by hydrophilic interaction chromatography.
David McCalley, Centre for Research in Biosciences, University of the West
of England, Bristol BS16 1QY U.K.
It is now over 25 years since Alpert coined the term “hydrophilic interaction chromatography” (HILIC) to describe a technique in which polar and ionised

www.ISC2016.ie @isc2016 #isc2016 /isc201644
ORAL ABSTRACTS Fundamentals
compounds can be separated on a polar stationary phase in combination with an organic-rich mobile phase containing an appreciable amount of water. Examples of HILIC-like separations also exist in much earlier publications in liquid chromatography. However, the mechanism of the technique is still not entirely understood. It appears that partition of solutes between a water layer held on the surface of the column and the bulk mobile phase may be the major contributory mechanism. Evidence for the existence of the water layer has been obtained through experimental measurements and computer simulations. Adsorption and ionic retention/repulsion effects also contribute to the separation mechanism. A considerable number of different stationary phases are now commercially available, and the choice of mobile phase can also be confusing. Nevertheless, the practical implementation of HILIC methods is rather straightforward. In this tutorial lecture, the choice of stationary phases of appreciably different selectivity will be covered, together with how the composition of the mobile phase can influence the separation. Guidelines will be given as to the prediction of solutes that are suitable for HILIC through a consideration of their octanol water distribution coefficients, which can be obtained from a number of software packages. The complimentary nature of HILIC separations to reversed-phase will be stressed, particularly with regard to the retention of compounds which are insufficiently hydrophobic to be analysed by RP. While other advantages of HILIC will be reviewed, including those which result from the low mobile phase viscosity and its enhanced volatility, the disadvantages of HILIC will also be considered.
FS1-005-003 - Selection of Equivalent Columns according to Quality by Design principles
Molnár*, I., Rieger, H.J.Institute for Applied Chromatography, Berlin, GermanyMailing address: [email protected]
Content: HPLC methods help to control the quality of many chemical and pharmaceutical products.
They are usually developed after column selection by optimizing the properties of the mobile phase (tG, T, pH, ternary comp., etc.) according to Quality by Design (QbD) principles. If the original column is not available any longer, the separation will often change, if a different column under same conditions is used.
Therefore, regulatory agencies request applicants to submit one or two equivalent columns, which can replace the original column if it is not available anymore. If QbD principles were applied, no considerable change in selectivity and robustness of the method should be observed.
This presentation will discuss different ways to compare columns and describe how to select suitable equivalent columns. Different stationary phases were compared with the same Design of Experiments. By fixing the working point for critical resolution (Rs,crit > 1.5) the method robustness is calculated and documented for both columns and were found to be equivalent for 100 % success rate to achieve the selected Analytical Target Profile (ATP).
There is the possibility to establish a common working point or also different working points.

45www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSFundamentals
Furthermore it is also possible to work with both columns with different step gradients. If the separation methods are developed using modeling software, the established Common Design Space (CDS) allows small adjustments of the method for both columns without the need for revalidation, which make increased flexibility in routine Quality Control possible.

www.ISC2016.ie @isc2016 #isc2016 /isc201646
ORAL ABSTRACTS Mass Spectrometry
Mass Spectrometry
MS1-006-001 - Towards a Multipath Liquid Chromatography – Mass Spectrometry System for Simultaneous Small and Large Molecule Analysis from a Single Sample Injection
Kevin A. Schug* 1, Dananjaya Kalu Appulage1, Evelyn H. Wang1, Yehia Baghdady1, Yashaswini Nagarajan1, Frances Carroll2, Pete C. Combe3
1 Chemistry and Biochemistry, The University of Texas at Arlington, Arlington,
2 Restek Corporation, Bellefonte,3 Shimadzu Biotech, Columbia, United States
Content: Clinical samples obtained through invasive procedures are generally considered precious, especially when they are tied to some disease state. In order to gain the most information from a single injection of biological fluid, a multipath liquid chromatography (LC) – mass spectrometry (MS) has been conceptualized. Diluted biological fluid can be directly injected into the instrument. Small and large molecules are segregated with the use of restricted access media (RAM). Chromatography for each class of molecules is then developed simultaneously on individual LC paths, and the paths are recombined prior to MS detection. For this strategy to provide viable methods for trace quantitation of both classes from one injection, one needs a) efficient capture of target small molecules in the RAM, b) non-retention of target large molecules (proteins) in the RAM, c) efficient separations of each compound class (including refocusing/correction of band-broadening), and d) sensitive detection. Presented will be the systematic efforts
to date to realize such a system, which includes evaluations of trapping efficiency, automated method development and investigation of chromatographic variables, and demonstration of the ability to perform multiple reaction monitoring of intact proteins on a triple quadrupole – mass spectrometer. Currently, protein recovery is a major challenge to overcome.Acknowledgements: Research support from Restek Corporation and instrumentation support from Shimadzu Scientific Instruments is gratefully acknowledged.Keywords: Method development, Proteins, restricted access media
MS1-006-002 - Identification and Quantification of Aging Products in Lithium Ion Battery Electrolytes with LC-MSC. Schultz 1,*, S. Vedder 2, M. Winter 1, S. Nowak 1
1 University of Münster, MEET Battery Research Center, Münster,
2 Shimadzu Europa GmbH, Duisburg, Germany
Content: The demand of lithium-ion batteries (LIBs) offering high specific energy and high energy density increases, especially in the market of electric vehicles.[1] A challenge for the application of LIBs is the limited life time due to aging. To investigate aging mechanisms in LIBs, analytical methods for electrolyte investigation are important. Different organic carbonates like ethylene carbonate (EC) and dimethyl carbonate (DMC) are used as solvents for electrolytes. The lithium-ion conducting salt is mostly lithium hexafluorophosphate (LiPF6) which decomposes to PF5. This initiates decomposition of organic carbonates and leads to generation of several aging products.[2]
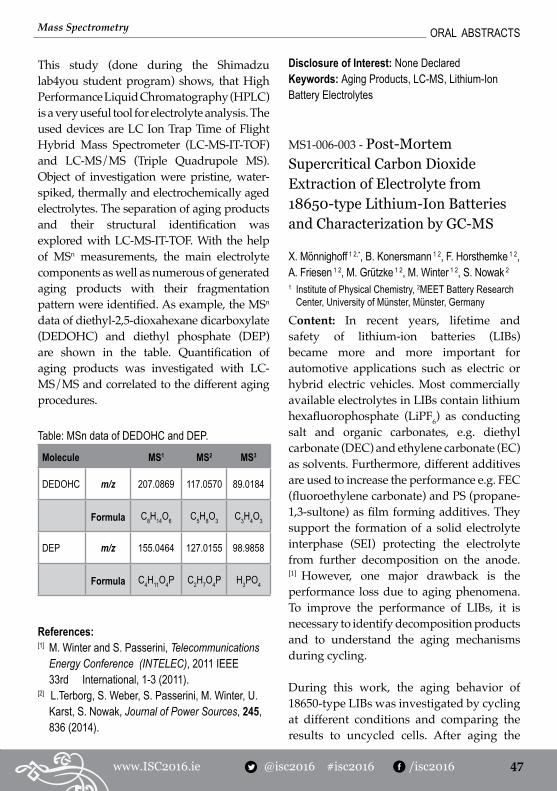
47www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSMass Spectrometry
This study (done during the Shimadzu lab4you student program) shows, that High Performance Liquid Chromatography (HPLC) is a very useful tool for electrolyte analysis. The used devices are LC Ion Trap Time of Flight Hybrid Mass Spectrometer (LC-MS-IT-TOF) and LC-MS/MS (Triple Quadrupole MS). Object of investigation were pristine, water-spiked, thermally and electrochemically aged electrolytes. The separation of aging products and their structural identification was explored with LC-MS-IT-TOF. With the help of MSn measurements, the main electrolyte components as well as numerous of generated aging products with their fragmentation pattern were identified. As example, the MSn data of diethyl-2,5-dioxahexane dicarboxylate (DEDOHC) and diethyl phosphate (DEP) are shown in the table. Quantification of aging products was investigated with LC-MS/MS and correlated to the different aging procedures.
Table: MSn data of DEDOHC and DEP.
Molecule MS1 MS2 MS3
DEDOHC m/z 207.0869 117.0570 89.0184
Formula C8H
14O
6C
5H
8O
3C
3H
4O
3
DEP m/z 155.0464 127.0155 98.9858
Formula C4H
11O
4P C
2H
7O
4P H
3PO
4
References: [1] M. Winter and S. Passerini, Telecommunications
Energy Conference (INTELEC), 2011 IEEE 33rd International, 1-3 (2011).
[2] L.Terborg, S. Weber, S. Passerini, M. Winter, U. Karst, S. Nowak, Journal of Power Sources, 245, 836 (2014).
Disclosure of Interest: None DeclaredKeywords: Aging Products, LC-MS, Lithium-Ion Battery Electrolytes
MS1-006-003 - Post-Mortem Supercritical Carbon Dioxide Extraction of Electrolyte from 18650-type Lithium-Ion Batteries and Characterization by GC-MS
X. Mönnighoff 1 2,*, B. Konersmann 1 2, F. Horsthemke 1 2, A. Friesen 1 2, M. Grützke 1 2, M. Winter 1 2, S. Nowak 2
1 Institute of Physical Chemistry, 2MEET Battery Research Center, University of Münster, Münster, Germany
Content: In recent years, lifetime and safety of lithium-ion batteries (LIBs) became more and more important for automotive applications such as electric or hybrid electric vehicles. Most commercially available electrolytes in LIBs contain lithium hexafluorophosphate (LiPF6) as conducting salt and organic carbonates, e.g. diethyl carbonate (DEC) and ethylene carbonate (EC) as solvents. Furthermore, different additives are used to increase the performance e.g. FEC (fluoroethylene carbonate) and PS (propane-1,3-sultone) as film forming additives. They support the formation of a solid electrolyte interphase (SEI) protecting the electrolyte from further decomposition on the anode.[1] However, one major drawback is the performance loss due to aging phenomena. To improve the performance of LIBs, it is necessary to identify decomposition products and to understand the aging mechanisms during cycling.
During this work, the aging behavior of 18650-type LIBs was investigated by cycling at different conditions and comparing the results to uncycled cells. After aging the

www.ISC2016.ie @isc2016 #isc2016 /isc201648
ORAL ABSTRACTS Mass Spectrometry
cells were opened and the electrolyte was extracted by supercritical fluid extraction (SFE) using CO2 as solvent and acetonitrile as co-solvent.[2] The decomposition products in the electrolyte were identified using gas chromatography (GC) with mass selective (MS) detection in electron ionization (EI), chemical ionization (CI) and negative chemical ionization (NCI) mode.
The fresh electrolyte contains three additives, FEC, PS and Succinonitrile. After aging at 20 °C FEC and PS were consumed and four well known decomposition products were detected. Furthermore, several new decomposition products were detected and identified.
Measurements on a GC-HR-MS/MS were performed to confirm the identified structures.References: 1. K. Xu, Chemical Reviews, 114 (11503-11618) (2014).2. M. Grützke, X. Mönnighoff, F. Horsthemke, V. Kraft,
M. Winter, S. Nowak, RSC Adv, 5 (43209) (2015).Disclosure of Interest:None DeclaredKeywords: GC-MS, GC-HR-MS, Lithium-Ion Battery Electrolytes, SFE
MS1-006-004 - How the mass spectrometry can be revealed as an innovative and powerful technique in the study of promising antiretroviral activity molecules
J. Ferey 1,*, D. Da Silva, C. Colas, B. Maunit1 University of Orleans, CNRS, ICOA, UMR 731, F-45067,
Orleans, France
Content: Nucleosides had led a considerable progress in the fight against viral infections such as herpes, hepatitis or AIDS. The
main objective of our studies is to screen new nucleoside analogues with higher activities and safety. In this context, our interest concerns the capacity of the new synthesized nucleoside analogues to be three times phosphorylated and able to stop the growing of viral DNA chain. In light of this, our study was focused on the development of a specific and sensitive analytical methodology based on an “on-line enzymatic assay” by UHPLC-HRMS for the monitoring of phosphorylation steps. In a first time, our work was focused to get quickly reliable data concerning the conversion of a monophosphate nucleotide into diphopshate and triphosphate nucleotide. An immobilization of the Thymidylate (TMPK) and Phosphoglycerate (PGK) kinases on silica beads was optimized first of all. The latter have been packed into a necessary bioreactor to perform a Frontal Affinity Chromatography (FAC) which enables the rapid and reliable screening of endogenous and new synthetic nucleotides. In the second step, a detailed research was realized to study the phosphorylation kinetic of each monophosphated nucleotide with the determination of the conversion rate. This kinetic study, using free enzymes (TMPK and PGK), was realized in Flow Injection Analysis Mass Spectrometry (FIA-MS). The activity of studied analogues used was classified according to the speed and the conversion rate. This methodology development allows improving significant advances in antiretroviral therapy by giving the possibility of testing in vitro the capacity of the new synthesized nucleoside analogues to be phosphorylated.Disclosure of Interest: None DeclaredKeywords: Enzymatic cascade, Frontal Affinity Chromatography, Immobilized Enzyme Reactor

49www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSInstrumentation/2D
Instrumentation/2D
I2D1-007-001
Method Development in Comprehensive 2D-LC – Column and solvent screening for finding the most orthogonal separation systems for RPLC×RPLC
Sonja Krieger*, Udo Huber1, Gerd Vanhoenacker2, Frank David2, Koen Sandra2, Pat Sandra2
1 Agilent Technologies, Waldbronn, Germany, 2 Research Institute for Chromatography, Kortrijk, Belgium
Content: Comprehensive two-dimensional liquid chromatography (comprehensive 2D-LC, LC×LC) offers high potential for the analysis of complex samples. It could be shown that the peak capacity can easily be increased by a factor of 3 to 4 using LC×LC compared to classical HPLC or UHPLC analysis, while maintaining analysis time1. The increased separation power of LC×LC can be reasoned by the peak capacity product rule2: Theoretically, the peak capacity of an LC×LC analysis is the product of the first and second dimension peak capacities. In practice, the peak capacity has to be corrected for limited orthogonality and undersampling of the first dimension effluent.
Despite the correlation of the separation mechanisms, the combination of two reversed-phase separations (RPLC×RPLC) is often used. When developing an RPLC×RPLC method, separation systems providing different selectivity should be selected.
For systematic development of an RPLC×RPLC method for a complex sample,
six different RP stationary phases were selected taking into account selectivity differences according to the Hydrophobic Subtraction Model. A one-dimensional screening was performed with the selected stationary phases and different mobile phases. For all possible combinations of separation systems, the correlation of retention factors of selected compounds was calculated. The combinations that provided the lowest correlations were tested in an LC×LC setup with full gradient in the second dimension and the fractional coverage of the resulting chromatograms was determined according to the Gilar-Stoll approach3. For the combination that provided the highest fractional coverage, a shifted gradient was designed for the second dimension separation. In this way, the fractional coverage could be greatly increased.References: 1. Vanhoenacker, et al., Agilent Technologies
Technical Overview (2015) 5991-6123EN.2. Giddings, Anal Chem 56 (1984) 1258A-1270A.3. Davis, et al., Anal Chem 80 (2008) 8122-8134.Keywords: Comprehensive 2D-LC, LC×LC, Method development
I2D1-007-002 - Comprehensive two-dimensional gas chromatography: from oil vacuum distillates to syngas
D. Thiebaut 1,*, E. Basset 2, M. Gallardo 2, Y. Kara 2, O. Guerrini 2, F. Hilaire 1, P. Sassiat 1, J. Vial 1
1 UMR CBI LSABM, ESPCI PARIS, PARIS, 2 ENGIE LAB CRIGEN’S, ENGIE, Research and
Technologies Division, St-Denis-la-Plaine, France
Content: The presentation will focus on GCxGC of samples to be converted in fuels. It will highlight the conditions of separations to be applied for successful elution of quite

www.ISC2016.ie @isc2016 #isc2016 /isc201650
ORAL ABSTRACTS Instrumentation/2D
heavy fractions such as vacuum distillates or process gas oils. For a higher resolution of hydrocarbon groups via the separation of saturated and unsaturated fractions, the benefits of the hyphenation of Supercritical fluid chromatography to GCxGC will also be recalled.
The presentation, more surprisingly, will also focus on supposed easier separation when gas samples are considered. Indeed, gasification of biomass associated to methanation is a new way to produce a green substitute of natural gas, called syngas. Syngas is composed of different chemical families covering a wide range of concentrations. Its complexity is quite new for Gas Industry and its purification is considered as one of the most important technological issues to be resolved in order to reach the level of purity required for industrial scale and commercial use. Thus, Gas chromatography (GC) and Comprehensive two-dimensional gas chromatography (GCxGC) associated to mass spectrometry (MS) will be compared for syngas detailed characterization. It will be shown that the complexity of various real syngas samples was high enough to justify the uncommon use of GCxGC for gas samples analysis because only partial resolution of the compounds could be obtained in GC-MS. Indeed, the implementation of GCxGC-MS enabled to detect or identify 2.5 times more compounds than in GC-MS. Thus, GCxGC can be used in routine analysis of syngas and other new energy-efficient gases (biogas, biomethane, etc.) for Enhanced Chemical, Safety and Performance Diagnostics for Gas and Bioenergy Industries.Disclosure of Interest: None DeclaredKeywords: Comprehensive 2D-GC, oil, Syngas
I2D1-007-003 - Introducing a New Generation Imaging Detection CE for CIEF and Application in Bio-Analysis
Gerardus P. Rozing*, Tiemin Huang1, 2, Martin Donker2
1 Advanced Electrophoresis Solutions, Cambridge, Canada,
2 Isogen Lifescience, De Meern, Netherlands
Content: Capillary IsoElectric Focusing (CIEF) has become an indispensable tool in life science research and in the development of new biopharmaceutical products. CIEF is the preferred method for study of post translational modifications, for protein degradation studies, variability in glycosylation, formulation of biopharma and for instance in profiling and authentication of food products.
Despite great efforts in study of fundamental aspects of CIEF, the method has remained cumbersome in execution, difficult to optimize and time consuming. Capillary isoelectric focusing with whole column imaging detection (iCIEF) was introduced on the bioanalysis market some years ago. The technique resolves these issues by providing faster method development, simpler practical operation, better reliability and robustness, and higher analytical throughput of CIEF.
Based on this technology, AES (Advanced Electrophoresis Solutions Ltd) has made some important breakthroughs such as high sensitivity imaging with 200 µm separation capillary, a modern optical system, and proprietary carrier ampholytes (CAs) and pI standards optimized for CIEF. AES has commercialized and released these improved technologies in the new CEInfinite instrument system.

51www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSInstrumentation/2D
High resolution, high sensitivity CIEF separations of therapeutic proteins such as antibody drug conjugates (ADC), monoclonal antibodies (mAbs), and fusion proteins with CEInfinite system have been demonstrated successfully and will be discussed.
In addition, the system allows high resolution preparative iCIEF. As an example, fractionation of hemoglobin control sample will be demonstrated. Fractions have been collected into individual vials, remixed with carrier ampholytes, and their purity checked with iCIEF.
Moreover the system allows to execute kinetic studies like determination of binding constants and the measurement of the hydrodynamic radius of proteins. Examples will be given.Keywords: Capillary Iso-Electric Focusing, iCIEF, Whole Column Image Detection
I2D1-007-004 - Polymer monolithic PLOT capillary columns for gas chromatography – Optimisation of layer thickness and morphologyZeliha Ates1, Katie Casey1, Ekaterina Nesterenko1, Shane Maguire1, David Collins1,2
1 National Centre for Sensor Research, Dublin City University, Glasnevin, Dublin 9, Ireland
2 School of Biotechnology, Dublin City University, Glasnevin, Dublin 9, Ireland
email: [email protected]
Recently, the potential of polymer monolithic porous layer open-tubular (PLOT) capillary columns was investigated as a solid adsorbent for gas phase separations, demonstrating that this type of phase is suitable for fast and efficient GC separations
[1]. This type of column possesses a covalently bound layer of organic polymer monolith which is formed inside a capillary through in-situ polymerisation, thus creating a long, open bore column with high flow through permeability. The primary advantage of this type of stationary phase over wall-coated open-tubular (WCOT) and other PLOT columns is that the layer is covalently attached to the capillary wall, resulting in a highly mechanically and chemically stable phase which exhibits little or no column bleed.
An in-depth study of the effect of layer thickness and morphology on separation performance was carried out. For this work, various PS-DVB monolithic porous layers were formed in-situ in 200 µm ID capillaries using a laminar flow polymerisation approach [2] with a C4D detection being used to monitor layer thickness [3]. The layer thickness was varied from 5 to 20 µm and an investigation into column load ability, efficiency, and optimisation of separation conditions was performed for test mixtures of common solvents and alkanes. The effect of layer porosity was further investigated for macro pore sizes between 3 and 8 µm. Initial results show that although increasing layer thickness improves column load ability, it has a profound effect on column efficiency. References: 1 Nesterenko, E.P.N., et al., RSC Adv. (2015), 5,
78902 Collins, D.A., et al., Chromatographia (2013), 76,
5813 Collins, D.A., et al., Analyst (2013), 138, 2540

www.ISC2016.ie @isc2016 #isc2016 /isc201652
ORAL ABSTRACTS Biomedical Analysis & Diagnostics
Biomedical Analysis & Diagnostics
BD1-008-001 - Proteomic Insights Into Aberrant and Perturbed MAPK Signaling Pathways:
I. M. Lazar*, J. Deng 1, N. Smith 1, F. Ikenishi 1
1 Virginia Tech, Blacksburg, United States
Content: In this study, our research was focused on evaluating the LC-MS technology for the discovery of mutated amino acids in proteins involved in cell-cycle signal transduction mechanisms. Proteins carrying such mutations represent useful disease markers or drug targets. The main objective was to identify the presence of mutations in SKBR3/HER2+ breast cancer cells and the associated pathways that are altered as a result of such mutations. We discuss the need for proper experimental design, the benefits of performing such analyses at both the genomic and proteomic levels, and the required performance characteristics of mass spectrometers for completing such studies. The breast cancer cells were cultured under conditions conducive to proliferation or apoptosis and analyzed by nano-LC-MS/MS. Raw MS data were processed with the Proteome Discoverer (FDRs<1) and further mined for biological content with bioinformatics tools enabled by KEGG, David and STRING. A total of ~6000 proteins were identified, representing a large number of cancer-relevant signaling pathways and biological processes. One of the most abundant categories was associated with disease mutations, encompassing ~10 % of all protein IDs. Point mutations, insertions and deletions were identified with our previously developed database of mutated peptides comprising ~900,000 entries [1]. We evaluated which pathways in
the database comprised the largest number of mutations, and which proteins were identifiable by our LC-MS protocols. The top ~400 proteins in the database (out of ~20,000 encoded by the human genome) carried ~15 % of all mutations (out of 900,000). LC-MS/MS analysis returned hundreds of mutations affecting processes involved in cancer cell signaling, proliferation, DNA damage repair and metastasis, providing encouraging results in support of future studies aimed at the development of novel personalized therapies.Acknowledgements: This work was supported in part by grant NSF/DBI-1255991 to IML.Disclosure of Interest: None DeclaredKeywords: Cancer, LC-MS, Proteomics
BD1-008-002 - The NIST mAb: A Reference Material to Advance Biopharmaceutical Characterization TechnologiesKaren Phinney, John Schiel, and Michael TarlovBiomolecular Measurement Division, National Institute of Standards and Technology, Gaithersburg, MD 20899 USA
Content: Monoclonal antibodies (mAbs) are the fasted growing class of therapeutics, and a wide variety of analytical techniques are used in their characterization. These analytical platforms continue to evolve, at the same time that new biopharmaceutical products are being introduced. Validation and acceptance of new characterization technologies would likely be enhanced by a widely available representative test material that is independent from product-specific reference standards. The National Institute of Standards and Technology (NIST) recently introduced a monoclonal

53www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSBiomedical Analysis & Diagnostics
antibody reference material (RM 8671) that is expected to provide a foundation for advances in analytical technology and facilitate the development of originator and follow-on biologics. The RM is intended for a variety of uses including, but not necessarily limited to establishing method or instrument performance and variability, comparing changing analytical methods, and assisting in method qualification. This presentation will describe characterization and certification of this reference material with a particular emphasis on separations-based techniques. Development of this reference material has included many industry collaborators.
BD1-008-003 - Chromatography-based evolution of theranostic peptides for targeted analysis of tumor markers and drug activation in living cells
Y. Huang 1, Y. Jin 1, R. Zhao 1,*
1 Institute of Chemistry,Chinese Academy of Sciences, Beijing, China
Content: Analytical tools for mapping and identification of cancer-related molecular profiles are urgently needed for sensitive, selective and accurate diagnosis and therapy. By taking advantage of high-performance affinity chromatography (HPAC) in rapid and high-throughput screening of biomolecular interaction, an efficient and directional strategy for functional evolution of cancer biomarker-specific peptides was established. With the combination of HPAC screening, RP-HPLC separation and mass spectrometry characterization, an optimal peptide AP2H was separated and identified
with high affinity towards an extracellular fragment of a tumor-related protein LAPTM4B. Capable of recognizing the expression level of LAPTM4B protein, AP2H guided the targeted analysis of cancerous cells. The high selectivity and sensitivity of the peptide further enabled tracing the translocation of LAPTM4B from plasma membrane to lysosomes. The employment of AP2H as an actively targeting moiety, peptide-stamped drug delivery systems were developed for photodynamic therapy and DNA-damaging of cancer cells. This work provides a versatile chromatography-based molecular evolution platform and an affinity peptide ligand for accurate mapping and targetable therapy of tumor in live biosystems.References: 1. R. Milkereit, A. Persaud, L. Vanoaica, A. Guetg, F.
Verrey, D. Rotin, Nat. Commun. 6, (7250) (2015).2. Z. Qian, X. Xu, J. F. Amacher, D. R. Madden,
E. Cormet-Boyaka, D. Pei, Angew. Chem. Int. Ed. 54, (1-6) (2015).
3. Y. Huang, F. Hu, R. Zhao, G. Zhang, H. Yang, D. Zhang, Chem.-Eur. J. 20, (158-164) (2014).
4. F. Hu, Y. Huang, G. Zhang, R. Zhao, H. Yang, D. Zhang, Anal. Chem. 86, (7987-7995) (2014).
5. Y. Jin, Y. Huang, H. Yang, G. Liu, Rui Zhao, Chem. Commun. 51, (14454-14457)(2015).
Acknowledgements: Financial supports from the National Natural Science Foundation of China, the Ministry of Science and Technology of China, and the Chinese Academy of Sciences are gratefully acknowledged.Disclosure of Interest: None DeclaredKeywords: affinity chromatography, peptide, tumor marker

www.ISC2016.ie @isc2016 #isc2016 /isc201654
ORAL ABSTRACTS Biomedical Analysis & Diagnostics
BD1-008-004 - Non-invasive profiling of human skin volatiles for chronic skin conditions
E. Duffy*, M. R. Jacobs, A. Morrin
Content: The human body emits hundreds of volatile compounds that can provide a valuable insight into the ongoing biochemical processes of an individual. These emissions are derived from glandular secretions and their interactions with bacterial populations on the skin. Studying volatile emissions from skin presents a non-invasive route towards monitoring pathophysiological status, and has been shown to aid in the early detection of diseases, such as melanoma1.
There can be considerable variation in the volatile fingerprints of individuals, with factors like diet, age, sex, skin type, genetic background and environmental conditions all influencing the gaseous emanations2-3. Thus, a greater understanding of the volatile fingerprint and its variation in different states of health and disease is required to identify clinically relevant compounds and to ultimately further the development of non-invasive diagnostics. We report the non-invasive profiling of skin volatiles using a HS-SPME method with GC-FID and GC-MS analysis.
Samples from a variety of participants were collected and analysed towards understanding variations in the unique signature. There was a particular focus on chronic skin conditions associated with a compromised skin barrier. Acute skin barrier disruption was simulated via tape stripping of the stratum corneum, and the resultant effect on the skin volatile
signature was characterised and compared to uncompromised skin. The analysis was then extended to participants with atopic dermatitis and other chronic skin conditions to highlight variations between different populations, and identify potential biomarkers for non-invasive health screening applications. References: 1. T. Abaffy et. al. Metabolomics 9 (998) (2013).2. V. Ruzsany et. al. J. Chromatogr. B 911 (84)
(2012).3. T. Abaffy et. al. J Cancer Sci. Ther. 3 (6) (2011).Acknowledgements: This research was funded by Science Foundation Ireland. Disclosure of Interest: None DeclaredKeywords: GC, Skin barrier, SPME

55www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSColumn Technology and Materials 2
Column Technology and Materials 2
CTM2-009-001 - Supercritical water as a flexible agent to treat fused silica capillaries for separation methods
P. Karásek 1, M. Horká 1, K. Šlais 1, J. Planeta 1, M. Roth 1,*
1 Institute of Analytical Chemistry of the CAS, v. v. i., Brno, Czech Republic
Content: Near- and supercritical water has been shown before to provide an efficient tool for pretreatment of fused silica capillaries as regards both their inner surface roughness and their internal diameter [1–3]. In the present contribution, an overview will be presented of the current options in the use of supercritical water to treat fused silica capillaries for applications in separation methods. Instrumental developments will also be briefly outlined. The sources and use of thermophysical properties of water to obtain hydrodynamic criteria including the Reynolds number and the Prandtl number will be discussed. These values are needed to estimate the hydrodynamic entry length and the thermal entrance length, the parameters that are useful to consider in the design of the etching apparatus. Finally, a recent example of application of supercritical water-treated fused silica capillary in electromigration separation will be given.References: 1. P. Karásek, J. Planeta, M. Roth: Anal. Chem. 85,
327-333 (2013).2. K. Šlais, M. Horká, P. Karásek, J. Planeta, M.
Roth: Anal. Chem. 85, 4296-4300 (2013).3. M. Horká, M. Tesařová, P. Karásek, F. Růžička,
V. Holá, M. Sittová, M. Roth: Anal. Chim. Acta 868, 67-72 (2015).
Acknowledgements: This contribution has been supported by the Czech Science Foundation (Project
No. 16-03749S) and by The Czech Academy of Sciences (Institutional Support RVO:68081715).Disclosure of Interest: None DeclaredKeywords: fused silica capillary, supercritical water, surface treatment
CTM2-009-002 - Preparation of organic monolithic columns in polytetrafluoroethylene tubes for reversed-phase liquid chromatography
M. Catalá-Icardo 1, S. Torres-Cartas 1, A. Calabuig-Belda 1, C. Gómez-Benito 1, E. J. Carrasco-Correa
2, E. F. Simó-Alfonso 2, G. Ramis-Ramos 2, J. M. Herrero-Martínez*
1 Research Institute for Integrated Management of Coastal Areas, Universitat Politécnica de València, Gandia (Valencia),
2 Analytical Chemistry, University of Valencia, Burjassot, Spain
Content: Monoliths are becoming very attractive stationary phases due to their advantageous hydrodynamic features and easy preparation and versatility. Up to now, most of the literature has been focused on enhancing column performance and on new column chemistries. However, scarce work has been done on increasing the column diameter and housing materials other than from the common fused-silica capillaries. In general, the fabrication of large-diameter monolithic columns involves some difficulties. One of these is caused by the monolith shrinkage during polymerization, which produced its detachment from the column wall, being this effect important in larger diameter tubes.
Polytetrafluoroethylene (PTFE) has been widely used in several analytical applications, due to its excellent properties

www.ISC2016.ie @isc2016 #isc2016 /isc201656
ORAL ABSTRACTS Column Technology and Materials 2
such as transparency to UV radiation, chemical inertness and others. However, its poor adhesion properties have caused considerable problems in several applications fields. In order to enhance its adhesiveness to other materials, several surface modification methods have been proposed. In this study, the use of a PTFE tubing as a column housing for microbore-LC using polymeric monoliths is investigated. To assure covalent attachment of the monolith to the inner wall of the PTFE tube, a chemical modification of this material was performed. For this purpose, two etchant reagents (a sodium naphthalene solution and H2O2/H2SO4 mixtures) were tried and compared. The obtained hydroxyl groups were then modified by methacryloylation to obtain a vinylized surface, and finally the polymerization was done. Special emphasis was also put on to reduce the unwanted effects of shrinking by using a proper mold and by selecting the adequate monomers to increase the flexibility of the polymer. The resulting monoliths were evaluated chromatographically with test mixtures and its capacity for the separation of proteins was also tested.Acknowledgements: Work supported by project CTQ2014-52765-R (MINECO of Spain and FEDER).Disclosure of Interest: None DeclaredKeywords: Polymeric monolith, PTFE tubing, microbore-LC
CTM2-009-003 - High-polar pyridinium ionic liquid phases for GC*GC separation complex mixtures of oxygen-containing compounds
M. Shashkov 1 2,*, V. Sidelnikov 2
1 Novosibirsk State University, 2Boreskov Institute of catalysis, NOvosibirsk, Russian Federation
Content: Comprehensive two dimensional gas chromatography (GC*GC) is a high-effective technique for analysis of complex multicomponent mixtures. An important advantage of GC*GC is that it allows analyzing of complex samples by separating the components in groups according to chemical classes.
In nowadays exist the problems which require as soon as possible high-polar columns (for one of the dimension) for achievement the high-resolution in GC*GC. Especially this is suitable for analysis the mixtures containing a lot of compound classes with different functionalities.
In our work we propose to use a high polar pyridinium ionic liquids (IL) as one of the column in GC*GC for separation complex multicomponent mixtures. Previously, it was shown that these columns show excellent results in the separation of polar compounds, particularly phenolic compounds [1]. And in this work we test columns with the different pyridinium ILs in pair with nonpolar HP-5 column. As the result we have found the best systems and conditions for separation certain objects in GC*GC. Among them products of flash pyrolysis wood (biooil), the products of pyrolysis of coal, essential oil of Pituranthos.

57www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSColumn Technology and Materials 2
We applied the system of nearest neighbor distances approach [2] for comparing different GC*
GC systems (different column pairs) and achieved the best resolution for our samples. The advantage this system is that the calculation harmonic mean Hs allow to find small differences in separation quality and to choose the best column pair and condition set.References: [1] Shashkov M.V. et al., J. Chromatogr. A 1309
(2012) 56[2] Nowik W. et al., Anal Chem. 85 (2013) 9459Disclosure of Interest: None DeclaredKeywords: gas chromatography, ionic liquids, resolution evaluation

www.ISC2016.ie @isc2016 #isc2016 /isc201658
ORAL ABSTRACTS Sample Preparation 1
Sample Preparation 1
SP1-010-001 - The electro membrane autosampler: Full automation of rapid sample extraction and analysis by LC-MS for high throughput applications
D. Fuchs*, N. J. Petersen 1
1 Department of Pharmacy, University of Copenhagen, Copenhagen, Denmark
Content: In the current work an autosampler with integrated electro membrane extraction (EME) was developed. The system fully automatically integrates rapid sample extraction by EME and sample analysis by LC-MS.
EME is a relatively new sample preparation technique using an electric field across a supported liquid membrane (SLM) to rapidly and selectively extract charged analytes from aqueous sample solutions. Up to now, most EME have been accomplished as off-line processes in which extracted analytes were manually transferred to the analysis method of choice. The newly developed EME-autosampler eliminates this manual sample handling step and can be used for consecutive extraction and analysis of a large sequence of samples without any manual sample handling.
For the setup a programmable CTC PAL autosampler was coupled to a LC-MS system via a 10-port-switching valve. Inside the autosampler’s syringe a 3 mm long porous hollow fiber, connected to fused silica capillaries was mounted. Electric potential was applied across the SLM by a programmable voltage sequencer. As sample was drawn by the autosampler’s syringe,
the sample passed the EME probe and analytes were extracted into the lumen of the hollow fiber. The extracted sample was then transported to the LC-MS system by switching a 10-port-valve that controlled the acceptor phase flow in the fused silica capillaries. The whole system was automatically controlled by the LC-MS software.
The EME-autosampler was successfully applied for fully automated extraction and analysis of large sequences of drug compounds in various sample matrices and proved good robustness, linearity and precision.
Due to its high degree of automation – the system integrates a whole analytical workflow of sample rapid extraction, separation and detection – the EME autosampler can potentially be used for all kinds of applications where fast and fully automated sample extraction and analysis is of interest.Disclosure of Interest: None DeclaredKeywords: automated sample extraction, autosampler, electro membrane extraction
SP1-010-002 - Towards the identification of individuals by odor-printing with original sampling approaches, multidimensional separation techniques and chemometrics
V. Cuzuel*, G. Cognon 1, I. Rivals 2, F. Heulard 1, D. Thiebaut 2, J. Vial 2
1 Institut de Recherche Criminelle de la Gendarmerie Nationale, Pontoise,
2 ESPCI Paris, PARIS, France

59www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSSample Preparation 1
Content: The characterization of human odor is of particular interest to forensics. Identification provided by dogs can be contested in courts. Developing a strategy using thermodesorption and comprehensive 2D gas chromatography coupled with mass spectrometry (TD-GC×GC-MS) to characterize the odorprint of individuals should be relevant to support information provided by dogs. Over the last decade, several studies have attempted to analyze substances released by human beings1. In this presentation, a strategy to characterize human odor using original sampling approaches, multidimensional separative techniques and chemometric analysis of data will be presented.
First, this communication will focus on techniques for human hand odor sampling. This crucial step was performed either by direct contact, an adsorbent being put directly on the skin of the subject during a predefined time, or indirectly with a home-made device based on air suction designed with an industrial partner. Both direct and indirect samplings were optimized using designs of experiments.
Then, TD-GC×GC-MS methods were developed using a mixture of 80 compounds representative of human hand odor. This technique allowed to perform efficient separations, to get more resolutive chromatograms, and therefore hopefully to collect enough information for further identification.
The last step is the implementation of advanced statistical data treatment for odor comparison and determination of target components. Our first results are promising since different people generated distinct
odoriferous profiles. For the approach to be used as a supportive evidence, further statistical data treatment have to be implemented 2 to compare samples and provide identification. The convergence of both frequentist and Bayesian approaches would bring more weight to evidence in courts of justice. References: 1. PA Prada, Rev Ciencias Forenses, 1(2), (81–7)
(2008)2. J Vial, J Chromatogr A, 1216(14) (2866–72) (2009)Acknowledgements: DIM Analytics, Région Ile-de-FranceDisclosure of Interest: None DeclaredKeywords: Comprehensive 2D-GC, forensic, odor-printing
SP1-010-003 - Magnetic Core-shell Molecularly Imprinted Nanomaterial for Selective Analysis of 2,4-Dichlorophenoxyacetic Acid in Real Samples
Y. Jin 1,*, L. Sheng 1, Y. Huang 1, R. Zhao 1
1 Institute of Chemistry, Chinese Academy of Sciences, beijing, China
Content: Molecularly imprinted polymers (MIPs), mimicking nature macromolecular recognition systems, are potential artificial alternatives to antibodies, enzymes and biological receptors. With extensive research in MIP these years, remarkable progress have been made in related areas, such as separations, biosensors, catalysts, biomedical materials. Fabrication of MIPs on magnetic nanoparticles gives access to smart materials with dual functions of target recognition and magnetic separation. In this study, the superparamagnetic core-shell MIP

www.ISC2016.ie @isc2016 #isc2016 /isc201660
ORAL ABSTRACTS Sample Preparation 1
nanoparticles were prepared via surface initiated reversible addition fragmentation chain transfer (si-RAFT) polymerization using 2,4-dichlorophenoxyacetic acid (2,4-D) as template. The living/controlled nature of si-RAFT polymerization reaction and optimum polymerization conditions allowed the construction of distinct core-shell structure with a 50 nm MIP layer, which favors fast mass transfer and rapid binding kinetics. The prepared MIPs revealed high selectivity and affinity towards 2,4-D. The target binding assays demonstrate the fast adsorption kinetics and high imprinting efficiency of Fe3O4@MIPs. The Fe3O4@MIPs nanoparticles coupled with RP-HPLC were successfully applied for the enrichment and analysis of 2,4-D from spiked tap water samples with recoveries ranging from 95.4-103.2%. This work provides a versatile approach for the fabrication of well-defined core-shell MIP for high fast and selective separation of target molecules.References: 1. C. Mahon, D. Fulton, Nat. Chem. 6 (665-672)
(2014).2. Y. He, Y. Huang, Y. Jin, X. Liu, G. Liu,
R. Zhao, ACS Appl. Mater. Interfaces 6 (9634−9642) (2014).
3. Y. Liu, Y. He, Y. Jin, Y. Huang, G. Liu, R. Rui, J. Chromatogr. A 1323 (11−17) (2014).
4. Y. Liu, Y. Huang, J. Liu, W. Wang, G. Liu, R. Zhao, J. Chromatogr. A 1246 (15−21) (2012).
5. J. Liu, W. Wang, Y. Xie, Y. Huang, Y. Liu, X. Liu, R. Zhao, G. Liu, Y. Chen, J. Mater. Chem. 21 (9232−9238) (2011).
Disclosure of Interest: None DeclaredKeywords: Core-shell particles, surface initiated reversible addition fragmentation chain transfer, target recognition

61www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSSupercritical Fluid Chromatography
Supercritical Fluid Chromatography
SFC-011-001 - SFC : what benefits for natural and cosmetic products analysis?
E. Lesellier, ICOA, UMR 7311, Université d’Orléans
Content: The numerous advantages of supercritical fluid chromatography with carbon dioxide and modifiers (SFC) vs liquid mobile phases are now well reported: safety, low cost, low viscosity, high eluting power, easy recycling of CO2 or collection of concentrated fractions. However, some specific points are yet often underestimated, such as the major role of the stationary phase choice with regards to the one of mobile phase conditions, or the dominant effect of modifier nature and percentage on retention over temperature and pressure changes.
Nevertheless, some of these advantages are of a great interest for complex mixtures, for instance, the extracts of natural products from plants (when looking on bioactive compounds), or the research of specific markers indicating adulteration or product origin. Because these extracts contain both several compound families, having different polarity, and numerous isomers, the selection of the suited stationary phase is the challenge to solve, or the coupling of numerous columns.
For cosmetic products, often water/oil emulsions, the issue is to quantify active compounds from a matrix containing a lot of lipids. The method development is based on the choice of the stationary phase from a restricted initial set of five columns having varied (orthogonal) properties, with an identical mobile phase, followed
by the final adjustment of mobile phase composition or other minor parameters. Varied detectors (UV; ELSD; MS) can be used either for calibration curves and/or for selective compound detection, depending on the concentration and the response of the studied compounds.
Several examples will be presented, with additional discussions on chromatographic system properties, method transfer from particle size to another one, and the use of gradient elution.
SFC-011-002 - Transferring solvent gradients in Supercritical Fluid Chromatography (SFC)
A. Tarafder 1,*, J. Hill 1
1 Waters Corporation, Milford, United States
Content: Solvent gradient chromatography (SGC), which varies solvent composition during method run, is used for separation of analytes with wide ranges of retention factors (k’) under isocratic conditions. Although SGC is normally associated with reversed-phase LC (RPLC), modern SFC widely uses SGC using CO2 as the principal mobile-phase (MP) component and an organic solvent as modifier.
Transferring gradients between systems is challenging in RPLC [1]. It can be more challenging in SFC due to MP compressibility. Reproduction of gradients is affected by several factors e.g. system volume, flow rates etc. The same factors also influence density variation in SFC system, imparting another dimension of variability in MP solvation power.
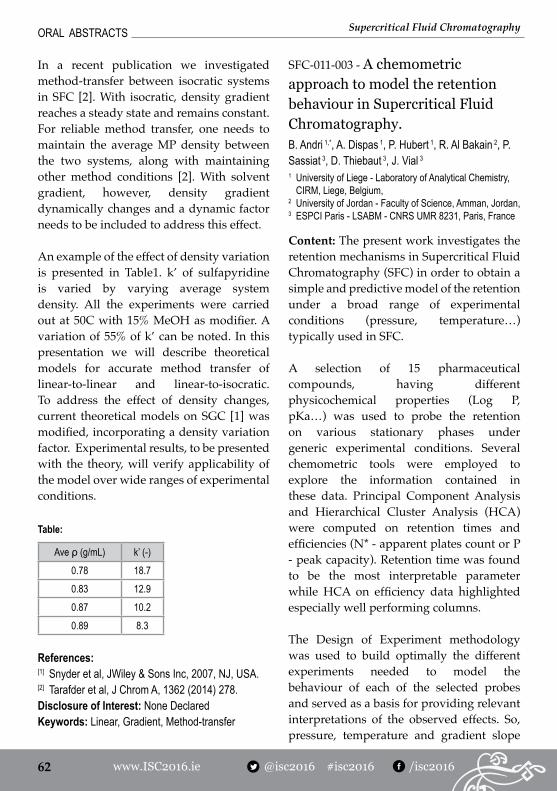
www.ISC2016.ie @isc2016 #isc2016 /isc201662
ORAL ABSTRACTS Supercritical Fluid Chromatography
In a recent publication we investigated method-transfer between isocratic systems in SFC [2]. With isocratic, density gradient reaches a steady state and remains constant. For reliable method transfer, one needs to maintain the average MP density between the two systems, along with maintaining other method conditions [2]. With solvent gradient, however, density gradient dynamically changes and a dynamic factor needs to be included to address this effect.
An example of the effect of density variation is presented in Table1. k’ of sulfapyridine is varied by varying average system density. All the experiments were carried out at 50C with 15% MeOH as modifier. A variation of 55% of k’ can be noted. In this presentation we will describe theoretical models for accurate method transfer of linear-to-linear and linear-to-isocratic. To address the effect of density changes, current theoretical models on SGC [1] was modified, incorporating a density variation factor. Experimental results, to be presented with the theory, will verify applicability of the model over wide ranges of experimental conditions.
Table:
Ave ρ (g/mL) k’ (-) 0.78 18.7
0.83 12.9
0.87 10.2
0.89 8.3
References: [1] Snyder et al, JWiley & Sons Inc, 2007, NJ, USA.[2] Tarafder et al, J Chrom A, 1362 (2014) 278.Disclosure of Interest: None DeclaredKeywords: Linear, Gradient, Method-transfer
SFC-011-003 - A chemometric approach to model the retention behaviour in Supercritical Fluid Chromatography.B. Andri 1,*, A. Dispas 1, P. Hubert 1, R. Al Bakain 2, P. Sassiat 3, D. Thiebaut 3, J. Vial 3
1 University of Liege - Laboratory of Analytical Chemistry, CIRM, Liege, Belgium,
2 University of Jordan - Faculty of Science, Amman, Jordan, 3 ESPCI Paris - LSABM - CNRS UMR 8231, Paris, France
Content: The present work investigates the retention mechanisms in Supercritical Fluid Chromatography (SFC) in order to obtain a simple and predictive model of the retention under a broad range of experimental conditions (pressure, temperature…) typically used in SFC.
A selection of 15 pharmaceutical compounds, having different physicochemical properties (Log P, pKa…) was used to probe the retention on various stationary phases under generic experimental conditions. Several chemometric tools were employed to explore the information contained in these data. Principal Component Analysis and Hierarchical Cluster Analysis (HCA) were computed on retention times and efficiencies (N* - apparent plates count or P - peak capacity). Retention time was found to be the most interpretable parameter while HCA on efficiency data highlighted especially well performing columns.
The Design of Experiment methodology was used to build optimally the different experiments needed to model the behaviour of each of the selected probes and served as a basis for providing relevant interpretations of the observed effects. So, pressure, temperature and gradient slope

63www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSSupercritical Fluid Chromatography
were systematically varied by means of a full factorial design. The fitting of an individual multi-linear regression model showed good agreement of the retention vs. the experimental conditions for the most efficient columns, proving the relevance of the approach.
Afterward, a global modelling of the retention, using the whole set of compounds, was also carried out with different input parameters, i.e. not only chromatographic conditions but also molecular descriptors, and various techniques. Partial Least Square regression allowed prediction of the retention in SFC under gradient elution mode in presence of methanol and ethanol.
Owing to the unavailability of truly predictive model of retention in SFC, this work should provide the basis for an efficient tool to favour and fasten the development of green and efficient separations.Disclosure of Interest: None DeclaredKeywords: Chemometrics, Chromatographic behaviour, Multivariate Analysis

www.ISC2016.ie @isc2016 #isc2016 /isc201664
ORAL ABSTRACTS Column Technology and Materials 3
Column Technology and Materials 3
CTM3-012-001 - A theoretical study on the advantage of core-shell particles with straight, purely radially-oriented mesoporesS. Deridder 1,*, M. Catani 2, A. Cavazzini 2, G. Desmet 1
1 Chemical Engineering Department, Vrije Universiteit Brussel, Brussels, Belgium,
2 Department of Chemistry , University of Ferrara, Ferrara,
Italy
Content: In the past 10 years, core-shell particles have revolutionized the speed and efficiency of chromatographic separations. Recently, Barber et al. [1] proposed a new type of particle that has the potential to make another leap in speed and efficiency. This material is of the core-shell type, but has all mesopores oriented purely radially. Because of this configuration, it can be expected that the longitudinal diffusion (B-term contribution) will be strongly suppressed, while the C-term mass transfer processes remain nearly unaffected. This was indeed confirmed in their study, showing unprecedented reduced plate heights of h=1.
To support these findings from a theoretical point of view and delimit the potential gain range of core-shell particles with radially-oriented mesopores (CS-ROM), computational fluid dynamics simulations were used to simulate the band broadening in ordered CS-ROM particle beds and the resulting reduced plate height curves were compared with previously obtained data for fully porous and conventional core-shell particles.
For the same zone retention factor (k”) and porous-zone diffusion coefficient (Dpz), the CS-ROM particles for example (k”=8
and Dpz/Dmol=0.5 case) produced a B-term which is 7 times lower than that of the fully porous and 6 times lower than that of the conventional core-shell particles. The CS-ROM particles displayed a minimal h-value that was 0.75 and 0.49 h-units smaller than the ordinary fully porous and core-shell particles respectively. Our study hence qualitatively confirms the experimental trends reported by Barber et al [1]. Covering a broad range of intra-particle diffusion coefficients and retention factors, our study allows for a comprehensive discussion of the gain margin one can expect from the radially oriented pores in core-shell particles compared to conventional core-shell and fully-porous particles.References: [1] T.-C. Wei, A. Mack, W. Chen, J. Liu, M. Dittmann,
X. Wang, W.E. Barber, J. Chromatogr. A 1440 (2016) 55-65.
Disclosure of Interest: None DeclaredKeywords: Core-shell particles, Radially oriented pores, Simulations
CTM3-012-002 - Photonic assisted fabrication of advanced liquid UTLC and modular SPE chromatographic platforms Paul F. O’Neill1,2,3,5, Ronan McCann1,2,3,5, Cian Hughes1,2,3,5, Komal Bagga1,2,3,5, Robert Groarke1,2,3,5, Apryll Stalcup1,2,4, Mercedes Vázquez1,2,3,4, Tashka Skrobisz1,2,3,5, Alessandro Scigliano1,2,3,5, Flora Klacsmann6, Yongfan Men6, Satyajyoti Senapati6, Hsueh-Chia Chang6, Dermot Diamond1,2,4, Dermot Brabazon1,2,3,5
1 Advanced Processing Technology Research Centre, Dublin City University, Dublin, Ireland;
2 National Centre for Sensor Research, Dublin City University, Ireland;
3 National Centre for Plasma Science and Technology,

65www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSColumn Technology and Materials 3
Dublin City University, Ireland; 4 School of Chemical Sciences, Dublin City University, Ireland; 5 School of Mechanical & Manufacturing Engineering,
Dublin City University, Ireland; 6 Centre for Microfluidic and Medical Diagnostic, University
of Notre Dame, United States
An overview of the most recent research and development of Ultra-Thin Layer Chromatography (UTLC) and modular Solid Phase Extraction (SPE) and detection will be presented as applied for chemical and biological sample speciation. Thinner stationary layers (<100 µm) used in UTLC result in shorter migration distances and quicker separations, as well as lower mobile phase consumption, when compared with conventional TLC [1]. To date, advances in UTLC have focused on metal-oxide or carbonaceous stationary phase sorbent layers deposited via advanced manufacturing techniques such as atomic layer deposition [2] and low-pressure chemical vapour deposition [3] or through the use of sol-gel deposition [4]. Polymers offer both cheap separation media and unique functionality however, to date, only polymer electrospinning has been examined as a process for UTLC plate fabrication [5]. In more recent work, we present a novel approach to the fabrication directly functionalized UTLC platforms utilising picosecond pulsed infrared laser to create networked microchannels.
Probe-functionalised Ion Exchange Membranes (IEMs) are a novel sensing method for the detection of nucleic acids in sample mixtures [6]. These label-free high-sensitivity biosensors are promising for on-site diagnostics due to their miniature form factor and potential for automation. However, one of the challenges in the development of an on-site
diagnostic platform is the miniaturisation of lab-based cell lysis and pre-treatment protocols for efficient extraction of nucleic acids from the lysed solution prior to sensing. In recent work, we report a novel pretreatment protocol for cell-lysis, volume reduction and buffer exchange in a microfluidic form factor to reduce conductivity of the sample for efficient electrochemical detection of bacterial and viral RNA for disease diagnosis. A 3D printed microfluidic Solid Phase Extraction (SPE) column was developed and used to capture negatively charged RNA molecules through a chaotropic binding mechanism. RNA molecules were eluted using DI water and volume fractions were analysed using quantitative reverse-transcription polymerase chain reaction (qRT-PCR) and compared with commercially available RNA purification methods. Development of this modular system is undertaken to improve reproducibility, and reduce assay time. The application of these systems for separation of environmental samples (herbicides and organic dyes), food samples, and point of care bio-diagnostics will be presented.References[1] S. K. Poole, J. Chromatogr. A 1218 (2648–60)
(2011).[2] J. Wannenmacher, J. Chromatogr. A 1318 (234–
43) (2013).[3] J. Song, Adv. Funct. Mater. 21 (1132–9) (2011).[4] H. E. Hauck, Chromatographia 57 (S313–5) (2003).[5] T. Lu, J. Chromatogr. B 912 (98–104) (2013).[6] S. Senapati, S. Basuray, Z. Slouka, L.-J. Cheng,
H.-C. Chang, Top Curr Chem (153-169) (2011).

www.ISC2016.ie @isc2016 #isc2016 /isc201666
ORAL ABSTRACTS Column Technology and Materials 3
CTM3-012-003 - Ion-exchange properties of synthetic diamond based phases
A. Koreshkova 1, A. Peristyy 1, P. N. Nesterenko*, B. Paull 1
1 ACROSS, University of Tasmania, Hobart, Australia
Content: Diamond based adsorbents have attracted significant attention as promising stationary phases for different modes of liquid chromatography [1]. This is due to low cost of microparticulated synthetic diamond and superior physico-chemical and mechanical properties of diamond. Diamond based stationary phases demonstrated remarkable stability under harsh separation conditions such as high temperatures and aggressive eluents, which can be used to achieve superior selectivities and efficiencies. However, the adsorption properties and chromatographic selectivity of diamond remain poorly characterised in liquid chromatography. This is due to complex surface chemistry of diamond, in particular, detonation nanodiamond.
Since it was recently shown that detonation nanodiamond has a property of amphiphilic polar adsorbent [2], it was particularly interesting to investigate its behaviour in ion chromatography (IC). In this work, we compared two most-commonly used types of synthetic diamond, namely, diamond synthesised at high pressure high temperature (HPHT) and microdispersed sintered detonation nanodiamond (MSDN) in terms of their performance in IC of common metal cations and inorganic anions. As expected, HPHT diamond column displayed classic weak cation-exchange selectivity series for alkali and alkaline earth metal cations with
no retention for inorganic anions. In contrast, several retention mechanisms and unusual ion-exchange selectivity were recorded for MSDN adsorbent. Surprisingly, MSDN column retains both anions and cations and has higher efficiency and performance at elevated temperatures. Additionally, adsorption isotherms are calculated using elution profile of chromatographic peaks of cations and anions and compared for both HPHT diamond and MSDN phases.References: 1. Anton Peristyy, Olga Fedyanina, Brett Paull, Pavel
N. Nesterenko, J.Chromatogr.A, 1357, 68-86, (2014).
2. Anton Peristyy, Brett Paull, Pavel N. Nesterenko, Adsorption-J. Intern. Adsorp. Soc., 22, 371-383, (2016).
Acknowledgements: This work was supported by grants DP110102046 and DP150101518 from the Australian Research Council.Disclosure of Interest: None DeclaredKeywords: ion chromatography, Ion-exchange, synthetic diamond

67www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSSample Preparation 2
Sample Preparation 2
SP2-013-001 - Parallel artificial liquid membrane extraction as an efficient tool for phospholipid removal from human plasmaK. S. Ask 1,*, S. Pedersen-Bjergaard 1, A. Gjelstad 1
1 School of Pharmacy, University of Oslo, Oslo, Norway
Content: LC-MS/MS is widely used for quantitative determination of drugs in plasma and other biological fluids. However, it has one major drawback; matrix components may cause ion suppression, leading to reduced accuracy and precision of the analytical method [1]. Endogenous phospholipids are highly abundant in plasma, and a significant source to matrix effects. One of the most efficient ways to avoid phospholipid-induced matrix effects is to remove the phospholipids by sample preparation before the analysis [1].
In 2013 a new approach to miniaturized liquid liquid extraction (LLE) was introduced, termed parallel artificial liquid membrane extraction (PALME) [2]. The technique is based on the use of supported liquid membranes (SLMs), where a porous membrane is impregnated with an organic solvent. The PALME equipment consists of two 96-well plates. The sample plate (at bottom) is housing the aqueous samples. The acceptor plate is housing the SLMs, and the aqueous acceptor solutions. Shaking the equipment, when assembled, leads to extraction of the analytes from the sample, through the SLM, and into the acceptor solution.
The aim of this study was to investigate the use of PALME for efficient removal of phospholipids from plasma.
PALME was performed from human plasma, using various organic solvents to create the SLM. After extractions of 120 minutes, the acceptor solutions were analysed by LC-MS/MS using in-source fragmentation, and monitoring the transition of m/z 184 → 184, which is characteristic for the most abundant phospholipids [3].
The experiments showed that the acceptor solutions from PALME were free from phospholipids. PALME can therefore be used as an efficient tool for phospholipid removal from human plasma.References: 1. Janusch, F., et al. Bioanalysis, 2013. 5(17):
p. 2101-14.2. Gjelstad, A., et al. Bioanalysis, 2013. 5(11):
p. 1377-1385.3. Little, J., et al. Journal of chromatography.
B, 2006. 833(2): p. 219-230.Disclosure of Interest: None DeclaredKeywords: Matrix effects, Parallel artificial liquid membrane extraction, Phospholipids
SP2-013-002 - Conception of Microporous Organogel Materials as Pesticide Sensor for Green Sfc and U-Hplc Separations.J.-C. Garrigues*, S. Franceschi, E. Perez
Content: Organogels are semi-solid systems in which an organic liquid is immobilized by self-assembled gelator fibers (1). The aim of this study was the elaboration of new porous organogels through the use of a particulate leaching method to introduce controlled micro-porosity into the material. To make an interconnected porous structure, we prepared sugar templates. The dried compact templates

www.ISC2016.ie @isc2016 #isc2016 /isc201668
ORAL ABSTRACTS Sample Preparation 2
were then soaked in octanol solutions containing 15 wt. % of HSA and heated over the T°gel. After cooling at room temperature, the sugar is eliminated by immersion in water. The advantage of this material as a pollutant sampler is that it behaves like a “porous liquid” without any diffusion barrier. These unique properties make these porous organogels ideal material to design sensors that can be used in air or water. In this study we decide to trap an insecticide largely used in veterinary treatment and often found in waste water: Fipronil® (2). In a first part, we will present the various stages of the organogel sensor manufacturing and the Fipronil® concentration capacity obtained with gelled octanol. The results show that 96% of the Fipronil® presents in an aqueous sample is trapped in 180 minutes, under agitation. In a second part, we will show the results obtained in the development of 2 analytical methods. A first method with U-HPLC separation using a water-ethanol gradient, on a C18 core-shell column allows the separation of Fipronil® in 0.7 minutes with quantification Limit (LOQ) of 2.82 nM. A second SFC method using CO2 and ethanol as co-solvent in gradient mode on a BEH-2EP column, allows the separation of Fipronil® in 2.18 minutes with a LOQ of 26.2 nM. The injection of the totality of the sensor in the analytical system reduces the sample preparation process to one step. With the use of microporous organogels, the LOQ decreased with a factor of 100 to 1000, compared to traditional LC and SFC/UV methods.References: 1 Chem Rev, 1997:97; 3133-31592 Environ Int 2014:74; 46–62.Disclosure of Interest: None DeclaredKeywords: Fipronil, pesticide sensor, porous organogels
SP2-013-003 - Coupling thin layer chromatography with MS, a setup for compound identification after sample preparation and chromatographic separation in one step
H. Griesinger 1,*, K. Matheis 2, M. Schulz 1, B. Fuchs
3, J. Schiller 3
1 Instrumental Analytics R&D, 2 Department of Bioanalytical Chemistry, Merck KGaA,
Darmstadt, 3 Institute of Medical Physics and Biophysics, University of
Leipzig, Leipzig, Germany
Content: Thin layer chromatography (TLC) allows sample preparation and chromatographic separation in one step. This is possible because of the high sample matrix tolerance of TLC. Thin layer chromatography can be coupled directly to mass spectrometry (MS) via various approaches [1]. As a result, the overall advantages of TLC to separate many samples in parallel without time-consuming sample preparation, are now combined with the powerful and versatile detection method mass spectrometry.
Several techniques for coupling TLC with MS are shown in the presentation. Examples include elution-based systems for direct solvent extraction from the plate, and the desorption-based approach MALDI (matrix-assisted laser desorption/ionization) which uses a laser beam for the spatial scanning of the plate.
The named hyphenation possibilities which can be applied to diverse types of mass spectrometers require distinct plate types. These TLC plates have to fulfil the demanding requirements of mass spectrometry concerning purity and sensitivity [2].

69www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSSample Preparation 2
All experiments were performed on newly developed HPTLC plates with a reduced separation layer thickness.
In this lecture we will present application examples with highly matrix-loaded samples in the areas of phytopharmaceuticals, food, cosmetic actives and consumer products. Data is shown for the:• determination of flavonoids• investigation of lipids in egg yolk• separation and identification of UV-filter
in suncream• analysis of methylisothiazolinone (MIT)
in wall paint
It is shown that the use of thinner TLC plates results in lower detection limits, increased sensitivity and improved S/N ratios. The sample matrices can be clearly separated from the target analytes. This results in highly unaffected mass spectra due to a very low level of ion suppression.References: [1] W. Schwack, J. Chromatogr. A. 1217 (6600-6609),
2010[2] G. Morlock, J. Liq. Chromatogr. Rel. Techn. 37
(2892-2914), 2014Disclosure of Interest: None DeclaredKeywords: mass spectrometry, matrix tolerance, thin layer chromatography

www.ISC2016.ie @isc2016 #isc2016 /isc201670
ORAL ABSTRACTS Bio- & Pharmaceutical Analysis
Bio- & Pharmaceutical Analysis
BP-014-001 - Hydrophilic Interaction Chromatography (HILIC) for the analytical characterization of protein biopharmaceuticalsV. D’Atri 1,*, S. Fekete 1, A. Periat 1, J.-L. Veuthey 1, D. Guillarme 1
1 School of Pharmaceutical Sciences, University of Geneva, Geneva, Switzerland
Content: Monoclonal Antibodies (mAbs) represent the largest and fastest growing category of biopharmaceutical proteins. They are manufactured through recombinant DNA technology, custom-designed to target chosen antigens (ranging from given substance to specific cell type), and employed for a wide range of applications, particularly in cancer, immune disorder, and infectious disease.
Antibody-Drug Conjugates (ADCs) are emerging as a promising class of mAbs-based pharmaceutical agents for targeted therapies in oncology. The strategic design of these immunoconjugates lies in the combination of the high specificity of mAbs with the potency of cytotoxic drug payloads, allowing a site-specific delivery of the toxic moieties to tumour cells. To date, two ADCs have been already approved by the FDA, but an increasingly number is currently in clinical trials.
A related issue to the analytical characterization of protein biopharmaceuticals arises from their wide structural micro-heterogeneity, due to their expression from living organisms. Although their structural characterization remains complex, it is mandatory due to the potential impact of micro-variants on safety. Several
methods might be applied for the analytical characterization of mAbs and ADCs, above them HPLC represents a gold-standard.
In this study, we compare the possibilities offered by reversed-phase liquid chromatography (RPLC) and hydrophilic interaction chromatography (HILIC) to evaluate whether HILIC might be used as an orthogonal approach to resolve structural isoforms, at the intact and middle-up levels of analysis. Up to now, HILIC was exclusively used for the separation of released glycans and for peptide mapping. Thanks to recent developments in column technology, now wide-pore HILIC phase is available and offers new possibilities for the characterization of mAb related products. Compatibility, suitability, and applications of coupling HILIC with electrospray ionisation mass spectrometry will also be discussed.Disclosure of Interest: None DeclaredKeywords: Hydrophilic interaction chromatography , Protein biopharmaceuticals
BP-014-002 - Recent Forays into ‘Mathematical Chromatography’ in Pharmaceutical Development – How Chromatogram Averaging and Subtraction Can Improve Performance
Christopher J. Welch*, Kerstin ZawatzkyMerck Research Laboratories, Rahway, NJ USA
Abstract: In this presentation we present recent examples of the use of chromatogram averaging and chromatogram subtraction to afford improved performance in pharmaceutical process research and development.

71www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSBio- & Pharmaceutical Analysis
Averaging of chromatograms can lead to enhancement of signal to noise ratio (S/N) in proportion to the square root of the number of measurements. Although the general principle has been known for decades, chromatogram averaging is almost never used in current pharmaceutical research. We describe the utility of this approach, showing it to be a simple and easily accessible method for boosting sensitivity for quantification of minor components and trace impurities, where current techniques deliver insufficient S/N.
Subtraction of chromatograms coming from two different samples collected under identical conditions can highlight small variations, serving as a useful tool for visualizing differences between experimental and control groups. While the basis for this general approach has been known for decades, the technique is seldom used in modern chromatographic analysis. We report an investigation into the application of subtractive chromatographic analysis in several areas of pharmaceutical research where detection of small differences between samples is important. Our investigation found that elimination of artifacts caused by peak misalignment was often necessary, especially for extremely sharp chromatographic peaks obtained in rapid injection MISER chromatography. Alignment of individual peaks prior to subtraction, combined with fast detector sampling rates, or data interpolation in cases where this is not possible, was found to afford convenient visualization of small differences (~1%) among samples, suggesting potential utility in high throughput screening of process adsorbents or other applications in pharmaceutical discovery and development.
BP-014-003 - New Modalities New Challenges
Nadim AkhtarAstraZeneca Macclesfield
In recent years the pharmaceutical industry has witnessed a major shift in the type of therapeutic agents, moving away from the traditional small molecule drugs. Small molecule drugs have dominated the industry for many years but as the trend is now moving toward biologics there is also an increased effort to identify novel synthetic platforms. Oligonucleotides, peptides, nanoparticles, dendrimers , conjugates are all examples of large synthetic therapeutic agents. Despite their inherent complexity the regulatory expectation is that small molecules ICH guidelines should be applied during the development of these products. This requirement has significant consequences for analytical science. Characterising these molecules is extremely challenging and quite often multiple orthogonal techniques are required to achieve the required resolution and specificity. This presentation will discuss the changing face of pharmaceutical development, challenges in developing novel synthetic platforms and the need to develop and adapt analytical technologies to allow successful commercialisation of such drugs.

www.ISC2016.ie @isc2016 #isc2016 /isc201672
ORAL ABSTRACTS Instrumentation & 2D HPLC
Instrumentation & 2D HPLC
I2D2-015-001 - Multiple Heart-Cutting 2D-LC-MS: Towards Real Time Determination of Related Impurities of Bio Pharmaceuticals in Salt Based Methods
P. Petersson 1,*, S. Buckenmaier 2, K. Haselmann 1
1 NovoNordisk A/S, Måløv, Denmark, 2 Agilent Technologies, Waldbronn, Germany
Content: The majority of the chromatographic methods used for determination of related impurities within the (bio) pharmaceutical industry are based on salt containing mobile phases. These often provide a superior chromatographic performance but are not compatible with mass spectrometry (MS). Peak tracking necessary for method development is therefore often based on peak areas and experience/intuition. In addition MS characterisation of impurities often requires time consuming fraction collection, often suffering from poor recovery or degradation of collected impurities.
Recent development of Multiple heart-cutting (MHC) two-dimensional liquid chromatography (2D-LC) provides a way to address these problems. A valve solution allows storage of cuts from the first dimension (1D) in 12 loops. Thereby the 1D and 2D dimension are decoupled from each other and, depending on the purpose, the 2D can be either a fast desalting gradient on a short column to allow MS analysis or a slow gradient on a long column with another selectivity to maximize separation power.
In this study we will show how MHC 2D-LC-MS can be used to obtain almost
real time MS data for bovine insulin related impurities present at low level. High quality MS spectra are obtained even for a 1D with a mobile phase containing high concentrations of sodium, sulphate and phosphate. Thereby MHC 2D-LC-MS offers a possibility to eliminate the guesswork currently associated with peak tracking in method development. Furthermore, in contrast to current workflow for characterization involving fraction collection, solvent reduction etc., MS determination is done directly in minutes instead of days reducing the risk for poor recovery and degradation.
Access to the type of MHC 2D-LC-MS instruments used in the current study will change the way purity methods are developed and impurities are characterized within the pharmaceutical industry.Disclosure of Interest: None DeclaredKeywords: 2D-LC-MS, multiple heart-cutting, related impurities
I2D2-015-002 - Improvement in the Determination of Mineral Oil Contamination in Vegetable Oils: Efficient Removal of Olefin Interferences by LC-LC-GC
M. Zoccali 1,*, G. Purcaro 1, L. Barp 2, M. Beccaria 1, D. Sciarrone 2, L. Mondello 1 2
1 Chromaleont srl, 2Scienze chimiche, biologiche, farmaceutiche ed ambientali, University of Messina, Messina, Italy
Content: Mineral oils are a complex mixture of compounds, derived from crude petroleum, mainly consisting of saturated (MOSH) and aromatic hydrocarbons

73www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSInstrumentation & 2D HPLC
(MOAH). MOAH can reach 10-30% of the MOSH content and are of potential concern for human health due to their carcinogenic effect. Vegetable oils can be contaminated with mineral oils from different sources. Liquid chromatography coupled to gas chromatography (LC-GC) with flame ionization detection (FID), represents the method of choice for the analysis of these two families. However, it is important to highlight that, the lack of mass spectrometry (MS) detection, due to the frequent controversies about the effective nature of the chromatographic hump, makes these determinations a hard task. Moreover, a correct quantification of the MOAH fraction can be affected by the presence of olefins, particularly squalene and its isomers, which can reach 5000 mg/kg in olive oils. In the present work, a novel on-line LC-GC method with a double detection [FID and triple quadrupole (QqQ) MS] was employed for the determination of hydrocarbon contamination in edible oils. Two different LC columns were coupled in series: a silica column to retain the bulk of the matrix (triglycerides) and to separate MOSH and MOAH, followed by a silver-ion one, which retains olefins allowed to obtain a MOAH hump free of interfering peaks. The QqQ MS system was used to evaluate the presence of hopanes as markers of petrogenic origin of the MOH contamination.Acknowledgements: The Project was funded by the Italian Ministry for the University and Research (MIUR) with a FIRB “Futuro in Ricerca” Project n. RBFR10GSJK “Tecniche Analitiche Avanzate per l’Analisi dei Contaminanti negli Alimenti”. The authors gratefully acknowledge Shimadzu and Sigma-Aldrich/Supelco Corporations for the continuous support.Disclosure of Interest: None DeclaredKeywords: hopanes, LC-GC, mineral oil
I2D2-015-003 - Multidimensional Gas Chromatographic Analysis of Trace Oxidised Species in Thermally Stressed Jet Fuels
R. Webster 1 2,*, D. J. Evans 2, P. J. Marriott 1
1 Australian Centre for Research on Separation Science, Monash University, Clayton,
2 Defence Science and Technology Group, Fishermans Bend, Australia
Content: In modern high performance jet aircraft, fuels are subjected to high thermal loads, particularly where fuel is used as a heat sink for cooling avionics and lubricants. When fuels are subjected to thermal stress, they may form insoluble deposits [1] and also result in an increased propensity towards poor fuel quality parameters. These negative effects can be attributed to the formation of oxidised species, despite only being present in trace amounts.
The thermal oxidative properties of alternative and synthetic fuels can be poor in comparison with those from conventional feedstocks [2]. However, the molecular mechanisms of the formation of oxidised compounds in these new fuel types have not been explored at a fundamental level and this knowledge is critical as their uptake increases in civilian and military aviation worldwide.
Matrix complexity makes it difficult to undertake detailed studies of oxidation products in thermally stressed fuels. However, application of advanced analytical techniques like multidimensional gas chromatography (MDGC) and comprehensive gas chromatography (GC×GC) provides an opportunity to overcome matrix complexity and study low concentrations of oxygenates amongst an

www.ISC2016.ie @isc2016 #isc2016 /isc201674
ORAL ABSTRACTS Instrumentation & 2D HPLC
forest of hydrocarbons. Here, MDGC has been used to analyse oxidised species in thermally stressed jet fuels [3], and quantify variation in concentration as a function of time over periods of thermal stressing [4]. In the absence of MDGC, the GC-MS approach alone does not provide adequate selectivity. References: 1. S. P. Heneghan & S. Zabarnick, Fuel, 73 (35-43)
(1994)2. R. L. Webster et al. Energy Fuels, 27 (889-897) (2012)3. R. L. Webster et al. Energy Fuels, 29 (2059-2066)
(2015)4. R. L. Webster et al. Energy Fuels, 29 (2067-2073
(2015)Disclosure of Interest: None DeclaredKeywords: aviation fuel, fuel oxidation, multidimensional gas chromatography
I2D2-015-004 - Liquid-liquid extraction can be turned into chromatography: the countercurrent chromatography techniqueA. Berthod 1,*, K. Faure 1
1 Institut of Analytical Sciences - CNRS, Villeurbanne, France
Content: Liquid-liquid extraction is commonly used in chemical purification. However, when you say that you are using a biphasic liquid system to perform countercurrent chromatography, people commonly tell you that you are doing liquid-liquid extraction. Nobody tells you that you are doing liquid-solid extraction when you describe HPLC or any chromatography working with a solid phase. If extraction is the chemical principle automatically repeated when doing chromatography, both purification techniques use very different equipments.
Countercurrent chromatography (CCC) is a preparative purification technique that works with a support-free liquid stationary phase. The major interest to use a liquid stationary phase is that the injected solutes can access the whole volume of the stationary phase and not just the surface of a solid stationary phase. The overloading problem of classical preparative chromatography is greatly reduced in CCC. The other advantages of a liquid stationary phase are presented. The major problem in CCC is to hold the support-free liquid stationary phase inside the CCC “column”. Centrifugal fields are used in most commercial CCC devices. They require rotating parts with gears or moving belts, rotary seals, motors making the CCC “columns” impressive devices that may frighten the new comers. Modern available equipments make CCC much more reliable with two different typres of columns that are presented. The use of the CCC technique to perform purifications will be illustrated by practical examples. It will be shown how a particular purification is optimized at the analytical scale with mL CCC columns and then, scaled-up to large CCC columns that can be as big as several liters. The biphasic liquid system being exactly the same in the different CCC columns, the scaling-up can be predictable. The solvent saving is outlined.References: A. Berthod, K. Faure, in Analytical Sep Sci, ISBN
978-3-527-33374-5, Wiley-VCH, Weinheim, Vol. 4, Ch. 3, pp. 1177-1206 (2015).
Disclosure of Interest: None DeclaredKeywords: biphasic liquid systems, preparative chromatography, support-free liquid stationary phase

75www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSForensic and Environmental Analysis 1
Forensic and Environmental Analysis 1
FE1-016-001 - Gradient retention time prediction for suspect screening of wastewaters using artificial neural networks and high resolution analysis data
L. P. Barron*, K. Munro 1, G. Mc Eneff 1, C. P. Martins
2, D. A. Cowan 1
1 Analytical & Environmental Sciences Division, King’s College London, London, United Kingdom,
2 Chromatography and Mass Spectrometry, Thermo Fisher Scientific, San Jose, CA, United States
Content: Characterising the breadth of emerging environmental contaminants is a dynamic challenge. Recent attention has focussed on liquid chromatography-high resolution mass spectrometry (LC-HRMS) for large and retrospectively mineable data capture. For preliminary identification of additional compounds, most works rely on HRMS data interpretation alone. Chromatographic data is often limited due to the unavailability or cost of reference standards for comparison. Prediction of retention is problematic especially for ionisable compounds under gradient conditions. Herein, a new reversed-phase retention time (tR) prediction model is presented for n=166 pharmaceuticals and illicit drugs in wastewater using artificial neural networks (ANNs). The predictive accuracy was <1.3 min for 75 % of all tR including n=30 blind cases. This enabled ~50 % of HRMS data to be rapidly discarded and compounds were identified at a rate of 73-83 % in effluent and influent, respectively. New drugs in wastewater influent are presented following a weeklong screening campaign at a major London sewage treatment works. Using an HRMS database of >1,400
compounds and the optimised ANN model allowed 30-40 additional compounds to be preliminarily identified on any one day. For example, mephedrone, a recently banned drug in the UK, was identified, confirmed and quantified (42-160 ng/L) in influent across the week [1]. Following this, the generalised performance of the ANN was investigated using 1,117 compound tR data in several complex matrices and across ten gradient reversed-phase LC-HRMS methods [2]. Multilayer perceptrons yielded the best correlations for 8/10 methods. Blind test predictions yielded an average absolute accuracy of 1.02 ±0.54 min for all methods. Finally, network dependency on molecular descriptors is critically discussed.References: [1] K. Munro, et al. J. Chromatogr. A, 1396, p 34-44
(2015)[2] L. Barron, et al. Talanta, 147, p 261-270 (2016)Acknowledgements: EPSRC (Ref:EP/J502029/1) and Thermo Fisher ScientificDisclosure of Interest: None DeclaredKeywords: High resolution mass spectrometry, retention time prediction, suspect screening
FE1-016-002 - Enantioseparation of various novel psychoactive drugs by HPLC
M. G. Schmid 1,*, M. Taschwer, J. Grascher, Y. Seidl1 Pharmaceutical Chemistry, Institute of Pharmaceutical
Sciences, Graz, Austria
Content: Besides consumation of well known illicit drugs, such as cocaine, heroine, MDMA and speed, recreational abuse of novel synthetic psychoactive drugs has become a challenging problem worldwide. Every year, dozens of new compounds enter the drug market and due to their similar

www.ISC2016.ie @isc2016 #isc2016 /isc201676
ORAL ABSTRACTS Forensic and Environmental Analysis 1
substitution patterns, full characterization is difficult.
Many of these compounds contain a stereogenic centre and as a consequence, pharmacological potency of the enantiomers might differ as known from various pharmaceutical drugs. Therefore, the development of analytical methods for chiral separation of new psychoactive substances is of big forensic interest.
This work gives an overview of different methods for enantioseparation of different drug compound classes including cathinones, amphetamines, benzofuries, thiophenes, phenidine and phenidate derivatives by HPLC. Most of these analytes were either purchased at various Internet shops or seized by Austrian Police, since they are hardly available at serious sources. Before serving as analytes, they underwent characterization by MS or NMR.
For successful enantioseparation, either commercially available chiral columns or chiral selectors as chiral phase additives on RP-columns were used. More than 40 new psychoactive compounds were resolved successfully. Obviously, all tested novel psychoactive drugs were traded as racemic mixtures.Disclosure of Interest: None DeclaredKeywords: Enantioseparation, HPLC, Novel psychoactive drugs
FE1-016-003 - Performances of the Continuous Flow Integrative Sampler (CFIS) for monitoring organic pollutants in surface and drinking watersA. Assoumani*
Content: Passive sampling has recently emerged as an efficient alternative to conventional active sampling for monitoring organic pollutants in water. The technique consists in exposing the passive sampler in water for period ranging from days to weeks, depending on the sampler, in order to accumulate the target pollutants. Average concentrations integrated on the exposure period are then determined from the mass of pollutants sequestered in the sampler. The concentration of target pollutants in the receiving phase allows reaching lower quantification limits than conventional sampling techniques such as spot sampling. Moreover, this technique reduces dramatically analytical costs, as it provides realistic average concentrations with a single analysis.
We have recently developed an automated, fully submersible, integrative sampling device to monitor both non-polar and polar organic pollutants as well as volatile organic compounds (VOC) in different types of water, the continuous flow integrative sampler (CFIS). The CFIS is capable of sampling a wide range of organic compounds because it is equipped with three different sorbents, each one specific to a range of compounds.
The performances of the CFIS were assessed and compared with spot sampling for monitoring PCB, PAH, hydrophobic pesticides, plasticizers,

77www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSForensic and Environmental Analysis 1
hormones, pharmaceuticals, BTEX and trihalomethanes in the Cecebre dam, and in the inflow and outflow waters of a drinking water treatment plant, located in A Coruña (NW Spain). The CFIS were exposed for 21 days, while spot samples were collected on day 1, day 7, day 14 and day 21 of the campaign.
Overall results showed very low concentrations of micropollutants at all sampling points, far below the regulated limits. The CFIS showed better sensitivity than spot sampling and the results of the two sampling techniques were in a good agreement. Thus, the CFIS proved to be an efficient, sensitive and cost-effective alternative for monitoring organic pollutants in surface waters.Disclosure of Interest: None DeclaredKeywords: Integrative sampling, Organic micropollutants, Water quality
FE1-016-004 - LC-MS/MS and SFC-MS/MS as alternative techniques to GC-MS/MS for the rapid screening of anabolic agents in urineV. DESFONTAINE 1,*, L. NOVÁKOVÁ 2, F. PONZETTO 3, R. NICOLI 3, M. SAUGY 3, J.-L. VEUTHEY 1, D. GUILLARME 11 Pharmaceutical Analytical Chemistry, School of
Pharmaceutical Sciences, University of Geneva, University of Lausanne, Geneva, Switzerland,
2 Department of Analytical Chemistry, Faculty of Pharmacy, Charles University in Prague, Hradec Králové, Czech Republic,
3 Swiss Laboratory for Doping Analyses, University Center of Legal Medicine Lausanne-Geneva, Epalinges, Switzerland
Content: The class of anabolic steroids is one of the most widely used by athletes to increase their performance. From an
analytical point of view, this is also one of the most challenging group of substances because: i) steroids share very similar backbone structures, leading to the presence of numerous isomers, ii) the World Anti-Doping Agency (WADA) has set very low minimum required performance levels (MRPL) for the detection of these compounds in urine. In consequence, the analytical methods used to detect these substances need to be highly selective and sensitive.
Historically, gas chromatography coupled to mass spectrometry (GC-MS or GC-MS/MS) has been used for the analysis of anabolic steroids in anti-doping laboratories. Its main issue is the time-consuming sample preparation, which generally includes a chemical derivatization. Because of these limitations, various analytical alternatives have emerged such as liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) which is certainly the most contemplated one. On the contrary, supercritical fluid chromatography (SFC) has been scarcely evaluated for doping control analysis up to now, despite the fact that steroids are good candidates for SFC.
In the present study, two methods (LC-MS/MS and SFC-MS/MS) were developed for the rapid screening of 43 anabolic agents in human urine. This included the development of a supported liquid extraction (SLE) procedure. The limits of detection (LOD) and matrix effects (ME) were compared with the reference GC-MS/MS method and several improvements were highlighted: i) the two developed methods were much faster than the reference one, ii) ME were very limited with the two developed methods (especially for SFC), iii)

www.ISC2016.ie @isc2016 #isc2016 /isc201678
ORAL ABSTRACTS Forensic and Environmental Analysis 1
LOD attained by LC-MS/MS and SFC-MS/MS were much lower (especially for LC-MS/MS with nearly all the target compounds detected at 0.1 ng/mL), iv) finally, both LC and SFC displayed complementary selectivity.Disclosure of Interest: None DeclaredKeywords: anabolic steroids, doping control, SFC-MS/MS

79www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSBiologics
Biologics
BIO-017-001 - Comprehensive on-line HICxRPLC-UV/MS for extensive characterization of cysteine-linked antibody-drug conjugatesM. Sarrut 1,*, S. Fekete 2, A. Beck 3, D. Guillarme 2, S. Heinisch 1
1 Institut des Sciences Analytiques, UMR CNRS 5280, Université de Lyon, 5 rue de la Doua, 69100 Villeurbanne, France, Villeurbanne, France,
2 School of Pharmaceutical Sciences, University of Geneva, University of Lausanne, Bd d’Yvoy 20, 1211 Geneva 4, Switzerland, Geneva, Switzerland,
3 Center of Immunology Pierre Fabre, 5 Avenue Napoléon III, BP 60497, 74160 Saint-Julien-en-Genevois, France, Saint-Julien-en-Genevois, France
Content: Antibody-Drug Conjugates (ADC) are a very promising class of human therapeutics. ADC technology combines the specificity of a monoclonal antibody (mAb) with a potent cytotoxic drug covalently bounded via a linker to the antibody, allowing increase in both safety and efficacy.
The goal of this work was to develop an on-line two-dimensional liquid chromatography method hyphenated to high resolution mass spectrometry in order to assess the drug loading distribution (i.e. drug-to-antibody ratio,DAR) while getting structural information on positional isomers. Hydrophobic interaction chromatography (HIC) and reversed phase liquid chromatography (RPLC) were used as first and second dimension respectively. To deal with the complexity of this coupling, many parameters had to be optimized including gradient conditions in both dimensions, injection solvent composition in 1D-HIC and the type of organic modifier in 2D-RPLC. Due to the presence of high salt concentration in 1D-mobile phase, the use
of a linear gradient bracketed by two water steps, followed by a proper desalting step at column outlet were necessary in the second dimension to prevent salt precipitation in the column and in MS source. Both peak capacity and sensitivity could be enhanced thanks to strong column focusing in RPLC allowing very large injection volume.
The optimized HICxRPLC-UV/MS method was applied to the analysis of a commercial cysteine-linked ADC (i.e. Brentuximab Vedotin, Adcetris®). Thus, in one single injection both the average DAR value was obtained and predominant positional isomers could be identified.
It is also shown that 2D-retention data can be very useful to assist ADC characterization through the identification of sub-units. In this way, the presence of odd DARs (1, 3 and 5) could be unambiguously demonstrated for both commercial and stressed ADC samples. It is also highlighted that MS might be conveniently replaced by UV-2D-colour plot to quickly assess the conformity of a given ADC batch.Disclosure of Interest: None DeclaredKeywords: Antibody-drug conjugates, HICxRPLC-UV/MS, On-line comprehensive 2D-LC
BIO-017-002 - Biosimilars Evolution and Demands on Separation Scientists
Lois Ann Beaver, (retired from the US Food and Drug Administration)
A perspective on the rapidly evolving field of Biopharmaceuticals and especially Biosimilars, (“copies” of biopharmaceutical

www.ISC2016.ie @isc2016 #isc2016 /isc201680
ORAL ABSTRACTS Biologics
products) will be presented. Scientific, legal and economic issues associated with production and marketing of biosimilars are undergoing development and change. In view of the increasing impact of these therapies on patients and providers the significance of advances and the key role of separation scientists will be demonstrated. Current regulatory guidance and the sources to follow for future information and counsel are addressed.
BIO-017-003 - Silver and platinum nanoparticles promote nucleation in crystallisation of the model protein lysozymeAleksandra Czekaja, b, Yehudi Blochb, Amir Khanc, Keith Linehand, Hugh Doyled and David Sheehana
a School of Biochemistry and Cell Biology, University College Cork, Cork, Ireland
b VIB Inflammation Research Centre, University of Gent, Belgium
c School of Biochemistry and Immunology, Trinity College Dublin, Dublin 2, Ireland
d Tyndall National Institute, University College Cork, Ireland
Content: Protein crystallization is a key step in determination of protein structure by X-ray diffraction, the principal approach to determining atomic-level protein structure and also has potential in protein purification in Biotechnology. Nucleation initiates crystal growth leading to ordered recruitment of proteins into the crystal. We synthesised platinum (average diameter 3.4± 0.6 nm) and silver (average diameter 5.4± 1.1 nm) nanoparticles as potential additives to a protein crystallization screen. We show that both nanoparticles gave crystals of the model protein lysozyme under more conditions than in their absence, the crystals grew faster and to a larger size. The crystals
obtained were of sufficient quality to determine three-dimensional structure by diffraction of X-rays from a synchrotron source and essentially identical structures were obtained for crystals grown in the presence of platinum or silver nanoparticles as for those previously-determined for nanoparticle-free crystals as determined by root mean square deviation. Neither the nanoparticles nor metals leached from them are present in the protein crystals, suggesting that nanoparticles aid the nucleation step of crystallization but thereafter do not become physically incorporated within the crystal. Nanoparticles are a promising novel additive category for sparse matrix sampling screening for protein crystals.Keywords: Protein; crystal; nanoparticles; silver; platinum; lysozyme
BIO-017-004 - Analytical approaches for the characterization and quantitation of monoclonal antibody oxidation variants using ion-pair reversed-phase HPLC-MS/MS
C. Regl 1 2,*, T. Wohlschlager 1 2, I. Forstenlehner 2 3, S. Ruzek 2 3, J. Holzmann 2 3, C. G. Huber 1 2
1 Department of Molecular Biology, Division of Chemistry and Bioanalytics,
2 Christian Doppler Laboratory for Innovative Tools for Biosimilar Characterization, University of Salzburg, Salzburg,
3 Analytical Characterization Biopharmaceuticals, Sandoz GmbH, Kundl, Austria
Content: Methionine oxidation induced by reactive oxygen due to inappropriate storage conditions, is a major shelf-life-limiting factor for biopharmaceuticals [1, 2]. Oxidation of methionine residues within the Fc region of IgG antibodies may not only lead to decreased bioactivity, but also

81www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSBiologics
cause faster plasma clearance [3]. Up to now analysis of monoclonal antibody (mAb) oxidation variants is carried out mainly via peptide mapping, requiring extensive sample preparation [1, 3]. In contrast, top-down or middle-down analysis would enable faster analysis, avoid artifacts occurring during sample preparation and provide information on individual proteoforms.
In order to develop such analytical workflows for the characterization of oxidized therapeutic antibodies, IgG1 antibody Rituximab was stressed by exposure to hydrogen peroxide. Limited proteolysis utilizing the IdeS enzyme under reducing conditions was applied in a middle-down approach to generate mAb Fc/2 fragments [4]. We achieved full chromatographic separation of single, double and non-oxidized Fc/2 species by ion-pair reversed-phase HPLC on a diphenyl column. Furthermore, oxidation site assignment was accomplished upon higher-energy collisional dissociation fragmentation in a quadrupole-Orbitrap mass spectrometer. Based on these results, we approach to quantify oxidation by UV-spectroscopy as well as full scan MS, as previously described [5]. Finally, possible approaches and limitations for oxidation analysis at the level of intact mAb will be discussed.References: [1] D. Houde et.al., Journal of Chromatography A,
2006. 1123: 189-198[2] J. Holzmann et.al., Anal Bioanal Chem, 2013. 405:
6667–6674 [3] J. Stracke et.al., 2014. mAbs 6:5: 1–14[4] Y. An et. al., 2014. mAbs 6:4: 879-893[5] I. Forstenlehner et. al., Anal. Chem. 2015, 87,
9336−9343
Disclosure of Interest: None DeclaredKeywords: middle-down, Monoclonal Antibody, Quantification

www.ISC2016.ie @isc2016 #isc2016 /isc201682
ORAL ABSTRACTS Ion Chromatography
Ion Chromatography
IC-018-001 - Preparation and Chromatographic Study of Novel Pellicular Stationary Phases for HILIC and Ion ChromatographyA. Zatirakha 1,*, A. Chernobrovkina 1, O. Shchukina 1, A. Uzhel 1, A. Smolenkov 1, O. Shpigun 1
1 Chemistry Department, Lomonosov Moscow State University, Moscow, Russian Federation
Content: Most of the recent developments in the field of stationary phase design are aimed at increasing selectivity and efficiency, which provides the possibility to solve a wider range of analytical problems with the particular chromatographic column. The best way to increase efficiency is to create a pellicular structure with a thin functional layer covering substrate particles; the structure of such layer and the way of its attachment to the substrate surface is responsible for separation selectivity.
In the present work two approaches for obtaining pellicular stationary phases for HILIC and ion chromatography based on silica and polystyrene-divinylbenzene substrates are proposed. In the first approach the substrate is modified with polyamine containing primary, secondary and/or tertiary amino groups, and selectivity variations are provided by choosing polyamine structure, its way of attachment, and, if necessary, the reagent for amino groups quaternization. The use of highly hydrophilic compounds for quaternization of attached polyamines results in obtaining anion exchange stationary phases that are suitable for the separation of 7 standard anions together with highly polarizable anions like thiosulphate, chlorate, bromate, iodide, and thiocyanate. Stationary phases
for HILIC obtained via such approach demonstrate good efficiency and selectivity for the separation of amino acids, water-soluble vitamins, and saccharides.
The second approach includes the creation of branched layers on the substrate surface using primary amines and diepoxides. The structure and the charge of the substitute in primary amine, as well as the location of the particular amine in the functional layer considerably influences selectivity, especially for separating weakly retained anions, which is demonstrated in the present work. The obtained anion exchangers provide the separation of up to 15 anions, including oxyhalides, anions of weakly retained organic acids and 7 standard inorganic anions.Disclosure of Interest: None DeclaredKeywords: hydrophilic interaction chromatography, ion chromatography, stationary phases
IC-018-002 - In vitro Study of the Reactions of Alkyl Phosphates from Lithium-Ion Battery Electrolyte in Human Whole Blood, Using simultaneous 2D-IC/ESI-MS/ICP-MS
J. Menzel 1 2,*, V. Kraft 1 2, M. Winter 1 2, S. Nowak 1
1 MEET Battery Research Center, 2Instiute of Physical Chemistry, University of Münster, Münster, Germany
Content: The lithium-ion battery (LIB) is a technology of great interest as it is used in every day portable electronics like mobile phones, cameras and computers. It is also a promising candidate for the use in electric vehicles (EVs). Still, the LIBs have issues with the capacity fading which is due to

83www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSIon Chromatography
aging processes inside the battery. One major aging mechanism is caused by reactions of conducting salt (LiPF6) with other battery components or protic impurities. One class of decomposition products are alkyl phosphates (APs). Their structures have been determined by ion chromatography (IC) coupled to electrospray ionization-mass spectrometry (ESI-MS).[1][2] Due to their structural similarity with the nerve agent sarin, they also have most likely toxic properties.
Yet, the toxicity of most of the APs has not been investigated. In order to gain information about the reactions of the compounds of aged electrolytes, a simultaneous coupling of two dimensional IC (2D IC) to ESI-MS and inductively coupled plasma-mass spectrometry (ICP-MS) was used for the quantification of the APs. In the ICP-MS chromatograms four compounds show distinct signals which are suitable for quantification. Their amount is in the low mmol/L range in thermally aged electrolytes.
During this work, blood samples were spiked with aged and unaged electrolyte and incubated at 36 °C. Afterwards the amount of the electrolyte components and aging products inside the blood samples were quantified in order to gain information about the reactivity of the substances with the blood constituents.
All components of pristine and aged electrolyte can be fully recovered from the blood plasma before incubation.References: [1] L. Terborg, S. Weber, F. Blaske, S. Passerini, M.
Winter, U. Karst, S. Nowak, Journal of Power Sources, 242, 832-837 (2013).
[2] V. Kraft, M. Grützke, W. Weber, M. Winter, S. Nowak, Journal of Chromatography A, 1354, 92-100 (2014).
Disclosure of Interest: None DeclaredKeywords: 2D IC/ICP-MS, in vitro study, Lithium-Ion Battery Electrolytes
IC-018-003 - Increasing Importance of Ion Chromatography for Pharmaceutical Analysis
J. Rohrer*
Content: A decade ago there were few United States Pharmacopeia (USP) monographs that used ion chromatography (IC). Today there are more than 50 official or proposed USP monographs that have an IC method and IC methods are more prevalent in other pharmacopeias. IC methods featuring either an anion-exchange or cation-exchange separation with either conductivity, electrochemical, or absorbance detection have been used in both drug substance and drug product monographs for assay as well as impurity tests. Some of the increased use of IC in USP monographs is a result of the USP’s monograph modernization efforts. IC is also one of the instrumental techniques allowed in the newly revised USP General Chapter 191 on identification tests. This talk will briefly describe IC and highlight the properties of IC that make it ideal for certain pharmaceutical analyses. Application examples from USP monographs that highlight these properties will be reviewed. The final portion of the presentation will detail the development of, and report the data for, three recent applications of IC for pharmaceutical analysis. The first application uses an anion-exchange separation and suppressed

www.ISC2016.ie @isc2016 #isc2016 /isc201684
Ion ChromatographyORAL ABSTRACTS
conductivity detection to assay the nitrite content of sodium nitrite as well as the amount of nitrate impurity at the specified limit. While the second application uses a cation-exchange separation with suppressed conductivity detection to assay the lithium content of lithium hydroxide as well as determine the amount of calcium impurity. These two examples are consistent with the USP’s monograph modernization efforts. The final example will show how anion-exchange chromatography with pulsed amperometric detection can be used to determine the sialic acid content of biopharmaceutical products.Disclosure of Interest: None DeclaredKeywords: Carbohydrate, Drug Product, USP

85www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSOmics 1
Omics 1
O1-019-001 - Investigation on the effect of combined cocaine and ethanol consumption through a Liquid Chromatography-Mass Spectrometry metabolomics approach
E. Sánchez-López 1,*, A. Marcos 2, E. Ambrosio 2, O. A. Mayboroda 3, A. L. Crego 1, M. L. Marina 1
1 Departamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcala, Alcala de Henares,
2 Departamento de Psicobiología, Universidad Nacional de Educación a Distancia, Madrid, Spain,
3 Center for Proteomics and Metabolomics, Leiden University Medical Center, Leiden, Netherlands
Content: Cocaine is the most commonly illicit psychostimulant drug used in Europe, and it is well known that it has deleterious effects on several metabolic pathways. More than 95 % of addicts mix cocaine use with ethanol intake, what produces an increased sense of euphoria compared to the use of these two drugs separately. Since research on the combined effect of these two addictive drugs is scarce, it is needed to further explore the consequences which could arise from this practice.
Metabolomics is a postgenomic discipline enabling a global, unbiased overview of the physiological/biochemical effects of the drug intake. In this work, we report the first metabolomic approach to study the effects of combined cocaine and ethanol exposure, where Liquid Chromatography coupled to high resolution Mass Spectrometry was employed to analyze plasma samples from rats exposed intravenously to these drugs.
Using a combination of the non-supervised (PCA) and supervised (PLS-
DA) multivariate analysis we explored and unraveled the metabolic differences between our experimental groups: cocaine alone, ethanol alone, cocaine plus ethanol, and the control group. A comparative analysis of the individual models and the first at all their VIP values has shown that every experiment intervention includes a subset of specific metabolites. Some of these metabolites were identified by comparison of retention times and MS/MS spectra of standards. The results demonstrated that the affected metabolic pathways were mainly those related to the metabolism of different amino acids.Acknowledgements: Authors thank financial support from Spanish Ministries of Economy and Competitiveness (project CTQ2013-48740-P) and Health, Social Services and Equality (Carlos III Health Institute, Spanish Network on Addictive Disorders, project RTA-RD12/028/0020, and National Plan on Drug Abuse, project PNSD-2012I057), the University of Alcalá (project CCG2014/EXP-059), and the UNED Plan for Research Promotion.Disclosure of Interest: None DeclaredKeywords: Cocaine and ethanol consumption, Liquid Chromatography Mass Spectrometry, Metabolomics
O1-019-002 - Highly specific separation and enrichment of phosphopeptides by electrospun titanium dioxide nanofibers
Y. Huang*, R. Zhao 1
1 Institute of Chemistry,Chinese Academy of Sciences, Beijing, China
Content: One-dimensional nanomaterials are attractive for separation and analysis of

www.ISC2016.ie @isc2016 #isc2016 /isc201686
ORAL ABSTRACTS Omics 1
low-content biological samples benefiting from their controllable composition, high surface area and uniformity. Herein, anatase TiO2 nanofibers with tunable dimensions were facially fabricated by electrospinning technique. The highly rough nanostructure provided large accessible surface area for the binding of phosphorylated species. After enrichment, the originally undetectable phosphopeptides in protein digest were efficiently captured, yielded a clean background and high signal-to-noise ratio in MS spectra. The selective binding capability towards both single- and multi-phosphorylated peptides were demonstrated by dephosphorylating assay and interference experiments using complex tryptic digests of different protein mixtures. The limit of detection for phosphopeptides was in the fmol range based on MALDI-TOF MS detection. Taking advantage of the high selectivity and sensitivity, the TiO2 nanofibers show great potential for the capture of phosphopeptides and identification of phosphorylation sites in disease-related proteins and complex biosamples.References: 1. H. Zhou, M. Ye, J. Dong, E. Corradini, A. Cristobal,
A. J. R. Heck, H. Zou, S. Mohammed, Nat. Protoc. 8 (461–480) (2013).
2. A. Greiner, J. H. Wendorff, Angew. Chem. Int. Ed. 46 (5670–5703) (2007).
3. J. Huang, F. Wang, M. Ye, H. Zou, J. Chromatogr. A 1372 (1–17) (2014).
4. Y. Huang, Q. Zhang, G. Liu, R. Zhao, Chem. Commun. 51 (6601–6604) (2015).
Acknowledgements: Financial supports from the National Natural Science Foundation of China and Chinese Academy of Sciences are acknowledged.Disclosure of Interest: None DeclaredKeywords: electrospinning, phosphopeptide, titanium dioxide nanofibers
O1-019-003 - Fermantanomics – Using NMR spectroscopy to monitor mammalian cell cultures
Kevin O’Sullivan1 Scott A. Bradley2 and Ian Sherlock1
1Eli Lilly S.A – Irish Branch, Kinsale, Ireland2Eli Lilly & Co., LTC-North, Indianapolis, United States of
America
Content: Using advanced analytical techniques like NMR it is possible to gain a global understanding of a culture state. Fermentanomics is a metabonomics technique for quantitating non-protein feed components and metabolites in mammalian cell cultures. The value of this methodology lies in its ability to provide insight into the nutrient levels needed to maintain productivity. The procedure has been applied to investigate the rate of change in nutrients and metabolites over time. This technique has also been used to support process optimisation and process changes. The advantages this approach offers over traditional analytical methodologies such as HPLC and mass spectrometry are summarised below.• Uniformly observes all organic species
in solution without the requirement for a chemical tag.
• Less method development in comparison to LC techniques.
• Minimal sample manipulation therefore more representative data sets.
• Detailed structural information on components as they appear so identification is possible without further experimentation.

87www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSFood and Health
Food and Health
FAH-020-001 - Development of an effective tool based on furanic profile to evaluate the quality and prevent frauds of sugarcane honey
P. Silva 1, J. Freitas 1, R. M. Perestrelo 1,*, C. L. Silva 1, F. Nunes 2, J. S. Câmara 1
1 CQM, Madeira University, Funchal, 2 CQ-VR, University of Trás-os-Montes and Alto Douro, Vila
Real, Portugal
Content: The sugarcane honey (SCH), known as ‘mel-de-cana’, is a black syrup produced in Madeira Island and widely used in traditional pastry and confectionery. The excellent quality and unique organoleptic properties of SCH arise from traditional production process and the use of sugarcane cultivars grown in this region. The SCH production is based only in thermal treatment of juice obtained by mechanical of fresh sugarcane stalks. However, in last years some products have been introduced in the market SCH that not respect the traditional processes. Thus, is imperative to develop tools that allow the evaluation of SCH quality as well as to guarantee its authenticity.
The purpose of this work was to develop a fast and sensitive analytical method to determine furanic derivatives (FDs) in SCH using a semi-automatic microextraction by packed sorbent (MEPS) procedure combined with ultra-high pressure liquid chromatography equipped with a photodiode array detection system (UPLC-PDA). Several chromatographic parameters (e.g. stationary phases, gradient conditions, eluents) were optimized to achieve the optimal FDs resolution. The analytical
method was validated according with IUPAC guidelines, and applied to quantify FDs in SCH samples.
As a result, the one-way ANOVA test showed significant differences on furanic profile of SCH groups. The principal components analysis (PCA) showed two separated clusters between certified and uncertified producers. From the results, it can be concluded that MEPS/UPLC-PDA can be used as a helpful tool to establish the authenticity and typicality of SCH. The simplicity and high extraction capacity of MEPS combined with the high sensitivity, reproducibility, and robustness of UPLC will be the main advantages of this approach.Acknowledgements: Fábrica Mel-de-Cana Ribeiro Sêco de V. Melim, Lda for the sample. ARDITI for the PhD Grant, FCT for the PhD grant (SFRH/BD/97039/2013), REDE/1508/RNEM/2015), HCV -New INDIGO/0003/2012; EU Project, QUI-Madeira-674.Disclosure of Interest: None DeclaredKeywords: sugarcane, MEPS, UPLC-PDA
FAH-020-002 - Determination of oxyhalides by ion chromatography-mass spectrometry
E. Gilchrist 1,*, D. Healy 2, V. Morris 2, J. D. Glennon 1
1 Department of Chemistry, University College Cork,2 Technical Function – R&D, PepsiCo, Cork, Ireland
Content: In recent years there has been increased interest in coupling ion chromatography (IC) to mass spectrometry (MS) in order to gather structural information at high sensitivity. The combination of IC-MS offers advantages over other analytical techniques in terms

www.ISC2016.ie @isc2016 #isc2016 /isc201688
ORAL ABSTRACTS Food and Health
of ICs ability to speciate, which can be essential in the identification of analytes, such as oxyhalides, as well as offering selectivity and sensitivity over traditional, non-specific detection modes such as conductivity and UV/Vis. Oxyhalides are commonly encountered in a range of analytical applications, from environmental to forensic to food and beverage quality, monitoring their concentrations in sample matrices related to these areas, such as water. This work aims to provide an outline to the application of IC-MS, with a focus on trace analysis of oxyhalides, along with the analytical considerations associated to enable sensitive analysis. Results from recent work comparing the use of IC-MS to typical IC with suppressed conductivity detection for common anions and oxyhalide analysis on a range of suitable columns will also be presented, looking at the effect on the retention of analytes, separation selectivity and efficiency.Disclosure of Interest: None DeclaredKeywords: ion chromatography, mass spectrometry
FAH-020-003 - Uncovering complex food fraud using metabonomic profiling
Kate Sidwick* 1, David Thompson
1
1
School of Physical and Geographical Sciences, Keele University, Newcastle-under-lyme, United Kingdom
Content: Food fraud is a challenge within the global food industry that affects the public on an everyday basis. This was apparent in 2013 when horse meat was discovered in processed meat products. The confidence in the meat industry has subsequently decreased, causing a need for reliable methods for meat
authentication to be developed. Methods for detecting food fraud usually involve searching for the presence or absence of a certain substance within the product to confirm its authenticity, but this can be easily adulterated. There are a number of specific frauds that are so complex, there are currently no methods for detection. The law states that all animals that are found to be dead on arrival to the slaughterhouse must be removed from the food chain. This project aims to use metabonomic methods to create a metabolic profile of the product, leading to a more reliable technique using a combination of markers, and their concentrations, to authenticate the product, and detect this subtle food fraud. This will involve using liquid chromatography-mass spectrometry based protocols, including established quality assurance methodology.

89www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSMicrofluidic Analyses
Microfluidic Analyses
MF-021-001 - Micro-plate array columns for fast gas chromatography
S. Jespers*, F. Lynen 1, G. Desmet 2
1 Separation Science Group, Universiteit Gent, Gent, 2 Department of Chemical Engineering, Vrije Universiteit
Brussel, Brussel, Belgium
Content: Since the first publication on a micro fabricated GC column (µGC column) in 1979 [1] the industrial and academic interest in µGC columns steadily increased. The advantages of these µGC columns are numerous, for example: lower gas consumption, faster analysis times, tunable in size and geometry, usable in hand held devices, etc. However, most µGC columns are still outperformed by their open tubular counterpart, specifically: the reduced plate height of µGC columns is substantially higher than this of capillary columns. This is the consequence of two effects. First, in order to provide enough sample capacity (while trying to keep the resistance to mass transfer low) the aspect ratio of the cross section of typical µGC columns is very high, which was shown by Poppe (2002) [2] to increase the dispersion dramatically. Second, coating of a channel with a square or rectangular cross section leads to pooling of the stationary phase in the corners; in these corners the stationary phase will thus be thicker than on the rest of the channel.
Recently, we designed a novel type of µGC column that has a 3 times lower reduced plate height compared to a capillary column. This is achieved by filling the channels with an array of radially elongated plates perpendicular to the direction of flow. These plates offer two big advantages: (1) they
allow the use of channels with square cross sections and (2) they reduce the observed axial dispersion. While many challenges still exist, the problem of pooling of the stationary phase being one, the preliminary results obtained with these µGC columns are very promising.References: [1] S.C., Terry, J.H., Jerman, J.B. Angell, A gas
chromatographic air analyzer fabricated on a silicon wafer, IEEE transactions on electron devices 26 (1880-1886) (1979).
[2] H., Poppe, Mass transfer in rectangular chromatographic channels, J. Chromatogr. A 948 (3-17) (2002).
Acknowledgements: The authors gratefully thank the research grant from the Research Foundation Flanders (FWO Vlaanderen).Disclosure of Interest: None DeclaredKeywords: chip, gas chromatography, micro fabricated
MF-021-002 - Performance comparison of capillary and microfluidic LC columns: efficiency versus peak capacity.M. Gilar*, T. S. McDonald 1, F. Gritti 1
1 Waters, Milford, United States
Content: We packed hundreds of capillary columns (cLC; 0.3 or 0.15 x 100 mm) and compared their performance to multiple titanium planar microfluidic (µLC). The efficiency N and peak capacity Pc (gradient elution) were monitored using a micro LC instrument with either 300 nL2 or 30 nL2 of extra column dispersion. The impact of this extra column dispersion on the observed efficiency and peak capacity is discussed. We show that for both cLC and µLC

www.ISC2016.ie @isc2016 #isc2016 /isc201690
ORAL ABSTRACTS Microfluidic Analyses
columns, testing in gradient mode is more practical than traditional isocratic testing. To exploit this bennefit, we developed a method for estimating a column’s efficiency from its gradient test. The measured peak capacity was converted to a column plate count using the theoretical relationship (Pc-1)=N0.5 × const., which we experimentally verified for 2.1 mm i.d. and 0.3 mm. i.d. columns. This simplified method requires the measurement of both peak capacity and efficiency for reference column 1 (wider i.d. for which both values can be easily obtained), while only measuring the peak capacity for column 2 (e.d. capillary column for which the isocratic efficiency is difficult to measure).
We applied this method for the efficiency determination of µLC columns. Planar titanium columns with straight channels have comparable efficiency/peak capacity to cLC columns of the same dimensions. However, a loss of efficiency and to a smaller degree a loss of peak capacity was observed for µLC devices that were machined with turns in the chromatographic channel. This is due to the so called race track effect causing on-column band broadening. The impact of turns in µLC column geometry increases with column i.d. The loss of performance was negligible for 150 µm i.d. devices, while 300 and 500 µm i.d. channels exhibited progressively larger loss. Turn tapering was used to correct for the race track effects. Experimental data confirm that µLC devices have to be carefully designed to achieve optimal performance.Disclosure of Interest: None DeclaredKeywords: Capillary scale, efficiency, Peak capacity
MF-021-003 - Characterization of efficiency of micro liquid chromatography columns by van Deemter and Kinetic Plot analysis
T. Hetzel 1 2,*, D. Loeker 2, T. Teutenberg 2, T. C. Schmidt 1
1 Instrumental Analytical Chemistry, University Duisburg-Essen, Essen,
2 Research Analysis, Institut für Energie- und Umwelttechnik e. V., Duisburg, Germany
Content: The efficiency of miniaturized liquid chromatography columns with an inner diameter between 200 and 300 µm has been investigated using a dedicated micro-LC system. Four different commercially available stationary phases differing in chromatographic support (fully porous, core-shell and monolithic) were compared applying classic van Deemter as well as kinetic plot analysis. It was found that the sub-2 µm fully porous as well as the 2.7 µm core-shell particle packed columns showed superior efficiency and similar values for the minimum reduced plate heights (hmin= 2.56-2.69) compared to normal bore columns. Moreover, the influence of column temperature and extra-column contribution on the achievable efficiency was examined in order to demonstrate the difference between apparent and intrinsic efficiency. It can be illustrated that 74% of the intrinsic efficiency could be reached using an alternative prototype detector. The results of the kinetic plot analysis indicate the superior performance of the sub-2 µm fully porous particle packed column for ultra-fast chromatography. In contrast, for high efficiency separations the monolithic column should be used if higher pressure capabilities are possible.

91www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSMicrofluidic Analyses
Acknowledgements: The authors would like to thank all column manufacturers for the donation of information and columns. We would like to especially thank Dr. Friederike Becker (YMC Europe GmbH), Dr. Stephan Altmaier (Merck KGaA) as well as Eike Logé and Dr. Peter Fischer (AB Sciex Deutschland GmbH).Disclosure of Interest: None DeclaredKeywords: extra-column volume, micro-bore columns, miniaturization

www.ISC2016.ie @isc2016 #isc2016 /isc201692
ORAL ABSTRACTS Process Analytical Technologies
Process Analytical Technologies
PAT-022-002 - PAT (Process Analytical Technology - Use to understand mechanisms and control
Dr. Norma M. ScullyEli Lilly and Company
Content: In situ analytics may be used throughout the pharmaceutical development lifecycle to support various aspects of the product control strategy. The application of these tools can vary depending on the product lifecycle and demands thereof. In early development phases, Process Analytical Technology (PAT) can enable process understanding, knowledge and scientific excellence. As the product moves through its lifecycle, PAT can play a key role in delivering enhanced process control, facilitating continuous improvement and right first time execution for supply products. Beyond product launch and into routine manufacturing, a robust control strategy is essential for long term manufacturability and CoPs (complex products and systems) reduction. Simplified PAT, or a correlation with manufacturing measurements (time, temperature, pressure, and flow) can be used to control the process. Novel PAT retained at this stage due to significant business/safety case.
Advanced process analytics tool can be exploited at each scale (lab, pilot, large scale) to gather data on each phase of each unit operation. Spectroscopic tools (e.g., near-infrared, mid-infrared, Raman, UV and NMR) are common place due to increased analytical specificity, which can provide both qualitative and quantitative trends.
Practical applications have demonstrated that these PAT tools can aid process understanding for unit operations such as crystalllisation and drying, therefore driving optimisation, and resulting in a leaner execution of supply. An on-line analytical method based on transmission near-infrared spectroscopy (NIRS) for the quantitative determination of water concentrations (in parts per million) was developed and applied to the manufacture of a pharmaceutical intermediat, from proof-of-concept phase to manufacturing implementation. A mechanistic approach was undertaken to understand the oxygen sensitivity of a Pd-catalyzed amination reaction used in the synthesis of an active pharmaceutical ingredient. FlowNMR and dissolved oxygen probes were used as process analytical technology alongside kinetic and unit operation models to better characterize the oxidative deactivation pathways of the catalyst.
PAT-022-003 - Process Chromatography for Extractable and Leachable Monitoring during the Upstream Bioprocess.
Christine Ta* 1, Noemi Dorival Garcia1, Sara Carillo1, Jonathan Bones1
1 Characterisation and Comparability Lab, NIBRT, Dublin, Ireland
Content: There is an increasing demand in the biopharmaceutical industry to move from stainless steel facilities to single-use or disposable, plastic bioprocessing technology. The many benefits of single-use technology (SUT) includes: reduced infrastructure and capital costs, reduced labour requirements

93www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSProcess Analytical Technologies
and the removal of cleaning and sterilisation procedures required in traditional facilities. Despite these economic benefits, SUT implementation is hampered by the risk of introducing extractable and leachable (E&L) compounds from plastic components into the bioprocess. These compounds pose a risk to patient safety and/or the biomanufacturing process, and therefore are a major concern for the biopharma industry. Recent studies show the detrimental effect of a leachable compound, bis(2,4-di-tert-butylphenyl)phosphate (bDtBPP) on CHO cell growth. This compound arises from the breakdown of Irgafos 168®, a widely used processing stabiliser in plastic bioprocessing containers. Therefore, a process analytical technology (PAT) method for the detection and monitoring of such compounds would be highly beneficial during the biomanufacturing process. Here we evaluate the PATROL UPLC Process Analysis system – a chromatography system capable of direct online sampling – as a PAT tool for E&L monitoring. In this study, an ultra-performance liquid chromatography method was developed for the separation and detection of bDtBPP and other extractable compounds related to Irgafos 168®. Both UV-VIS and fluorescence detection and C18 and CSH (charged surfaced hybrid) columns were evaluated. bDtBPP and related compounds were spiked in two cell culture media and monitored atline with a PATROL UPLC Process Analysis System. Notably, bDtBPP was still persistent in both cell culture media after 14 days. Cell culture medium stored in a disposable bioprocess bag was also analysed for bDtBPP. The effects of freeze-thaw cycles, incubation time and temperature on the E&L profile were also investigated.Keywords: extractables and leachables, UPLC, upstream monitoring

www.ISC2016.ie @isc2016 #isc2016 /isc201694
ORAL ABSTRACTS Column Technology and New Materials 4
Column Technology and New Materials 4
CTM4-023-001 - Significantly high retention and better selectivity of saccharides on a HILIC column, prepared by polymer-coatingT. Ikegami*
Content: The use of hydrophilic interaction chromatography (HILIC) is becoming popular for the separation of highly polar compounds. Thanks to great efforts by many researchers, it has been found that the retention mechanism in HILIC is essentially multimode composed by ion-exchange and hydrophilic partition [1,2]. The ratio of them varies depending on the chemical conditions in stationary and mobile phases. Retention of saccharides seems to be more difficult to improve, since it requires the development of more hydrophilic stationary phases, and ionic interaction does not help it.
Here, we reports on the preparation of novel HILIC stationary phase that possess significantly higher hydrophilic nature compared with commercially available HILIC columns. Silica particles (5 µm) were modified with 3-(methacrylamido)propyltriethoxysilane and functionalized by a radical polymerization of N-tetrazolylmethacrylamide (PTz) to prepare a HILIC type stationary phase. Polymerization was carried out at fixed monomer concentration (160 mg/mL), at 60°C for 3 hour, with varying initiator concentration. The PTz silica particles were packed into stainless steel columns and were characterized by a test scheme [3]. The retention factor of uridine (degree of hydrophilicity) was increased up to 8, as the initiator concentration reached over
10.4 mg/mL. The PTz column was found to provide approximately twice-great retention for saccharides than the other HILIC columns. In addition, selectivity for a glucose unit (k(maltose)/k(glucose)) on the PTz column was estimated as 2.24. This value was superior to those of commercially available HILIC columns.References: [1] G. Schuster and W. Lindner, J. Chromatogr. A,
1273, 73 (2013).[2] J. C. Heaton, et al., J. Chromatogr. A, 1347,
39 (2014).[3] Y. Kawachi et al., J. Chromatogr.
A, 1218, 5903 (2011).Acknowledgements: This work was supported by JSPS KAKENHI Grant Number 26410151.Disclosure of Interest: None DeclaredKeywords: HILIC saccharides
CTM4-023-002 - Microfluidic reactor for proteomics research
Kinga Meller, Bogusław Buszewski1
1 Nicolaus Copernicus University in Toru−, Faculty of Chemistry, Department of Environmental Chemistry and Bioanalytics, Gagarina 7, 87-100 Toru−, Poland e-mail: [email protected]
Content: Proteins are one of the most important compounds occurred in living organisms. Their play many biological functions including catalysis, molecules transport, biochemical processes regulation, immune functions etc. Proteins distribution, interactions, properties, structures, post-translational modifications (PTMs), and quantitative as well as qualitative determination are the subjects of the proteomic research [1]. Proteins digestion with specific enzyme is commonly used during their identification or sequencing

95www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSColumn Technology and New Materials 4
[2,3]. One of the relatively novel solutions making a progress on this field is application of Immobilized Enzyme Reactors (IMERs).
The main aim of presented study was the preparation of microfluidic reactor for fast and efficient proteolysis. For this purpose, trypsin was immobilized on monolithic support synthesized from 2-hydroxyethyl methacrylate and N,N’-methylene-bis-acrylamide. The mixture of 2-propanol, 1-decanol and water was used as a porogen solvent for polymerization of monolithic bed. Trypsin was immobilized on synthesized bed via support hydroxyl groups after their activation with 1,1’-carbonyldiimidazole. Two different immobilization strategies were tested and several microreactors were prepared to verify the reproducibility of elaborated methods. The IMERs activities were evaluated using nano-LC based on the degree of hydrolysis of low-molecular trypsin substrate. The prepared microreactors were also applied for protein digestion and identification using MALDI-TOF MS.References: [1] W.P. Blackstock, M. P. Weir, Proteomics:
quantitative and physical mapping of cellular proteins, Trends Biotechnol. 17 (1999) 121-127
[2] B. Thiede, W. Höhenwarter, A. Krah, J. Mattow, M. Schmid, F. Schmidt, P.R. Jungblut, Peptide mass Fingerprinting, Methods 35 (2005) 237–247.
[3] K. G. Standing, Peptide and protein de novo sequencing by mass spectrometry, Curr. Opin. Struct. Biol. 13 (2003) 595-601
Acknowledgments: This work was supported by the National Science Center in the frame of the project Maestro 6 (Cracow, Poland), no. NCN 2014/14/A/ST4/00641.
CTM4-023-003 - Multi-channel fused silica capillaries for the preparation of extraction devices and separation columns for HPLC.S. Currivan 1,*, N. Upadhyay 1, B. Paull 1 2
1 University of Tasmania, School of Physical Sciences, Australian Centre for Research on Separation Science,
2 University of Tasmania, School of Physical Sciences, ARC Centre of Excellence for Electromaterials Science, Hobart, Australia
Content: Photonic crystal fibres (PCFs) for optical fibre communications consist of a fused silica (FS) core, which is surrounded by an array of holes providing air insulation [1]. For microfluidics, the material consists of 126*4µm channels. The PCFs provide a higher surface-to-volume ratio when compared to single channel capillaries. The FS substrate can be easily modified for many applications, e.g. for the provision of anchor groups towards polymer attachment, or for immobilisation of specific groups, such as inorganic nanoparticles. The bioaffinity interaction between glycans and lectins (carbohydrate binding proteins) is well known, where highly specific bonding for different saccharides occurs, e.g. with the side chains of glycoproteins. Glycoproteins in biological fluids can act as biomarkers, and may provide diagnostic information for some disorders. Previous reports have shown the use of monolithic materials in pipette tips [2], with immobilised lectins for selective extraction of glycoproteins from complex mixtures, with subsequent separation in µHPLC. In this work, aminated FS capillaries were modified with a commercially available suspension of 15-20nm gold nanoparticles (GNPs). In a further step, the GNPs could be used as Au seeds for growth, which provided a ≤1µm gold layer upon the channel walls [3]. The

www.ISC2016.ie @isc2016 #isc2016 /isc201696
ORAL ABSTRACTS Column Technology and New Materials 4
Au surfaces supported the immobilisation of Erythrina cristagalli lectin, for selective glycan capture using the galactose affinity of the lectin. Glycan extraction could be performed offline, by means of a syringe pump with subsequent analysis by HPLC, or online, where the extraction capillary was placed ahead of a separation column under HPLC conditions. The performance of the capillary, and the efficiency of extraction was compared between the PCF array, and conventional capillaries of 25µm and 50µm ID. References:[1] .X, Yu, et al., Sensors and Actuators B 137 (462)
(2009)[2] .Alwael, et al., Analyst 136 (2619) (2011)[3] .K. Brown, et al., Langmuir 14, (726) (1998) Disclosure of Interest: None DeclaredKeywords: Bioaffinity, Microfluidic, Photonic Crystal Fibre

97www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSElectrodriven Separation Methods
Electrodriven Separation Methods
ED-024-001 - Interfaces and on-chip detection for two-dimensional coupling of isoelectric focusing with capillary electrophoresis – mass spectrometry
D. Sydes 1, P. A. Kler 2, H. Meyer 3, C. Huhn 1,*
1 Institute of Physical and Theoretical Chemistry, Eberhard Karls Universität Tübingen, Tübingen, Germany,
2 Centro de Investigación de Métodos Computacionales, Universidad Nacional del Litoral , Consejo Nacional de Investigaciones Científicas y Técnicas , Santa Fe, Argentina,
3 J&M Analytik AG, Aalen, Germany
Content: Multidimensional separation techniques recently gained significant attention. In comparison to chromatography the technical implementation for multidimensional electromigrative separation techniques is less advanced owing to the higher requirements essential for an efficient sample transfer between the two dimensions. Due to the small peak widths, mostly heart-cutting techniques are used today.
As an example we here present our technical innovations regarding the coupling of capillary isoelectric focusing with capillary electrophoresis – mass spectrometry (cIEF/CE-MS). Instead of increasing the peak capacity, the second dimension serves as a cleanup to remove ampholytes impairing the mass spectrometric detection from the proteins of interest.
The focus of our study is on the design and optimization of the microfluidic interface connecting up to four capillaries via a common intersection for both dimensions. This common intersection then houses the
sample plug at the start of the CE separation. In order to achieve an efficient analyte transfer, the monitoring of the common intersection is highly recommended. We here present the spatially resolved fluorescence monitoring of labeled proteins entering the chip interface using an array of optical fibers. For detection a hyperspectral camera which gives rise to a time dependent and wavelength-resolved image of the common intersection is implemented.
First results for cIEF/CE-MS with the hybrid capillary-microchip an on-chip spatially and wavelength-resolved monitoring of the analyte transfer are presented using model proteins.Disclosure of Interest: None DeclaredKeywords: microfluidics, multidimensional separation, on-chip detection
ED-024-002 - Understanding a Simple Approach to DNA Separation by Sequence using Capillary Zone Electrophoresis
L. B. Mcgown 1,*, J. Zhao 1, S. M. Cramer 2
1 Chemistry and Chemical Biology, 2 Chemical and Biological Engineering, Rensselaer
Polytechnic Institute, Troy, United States
Content: DNA analysis has widespread applicability in biology, astrobiology, medicine, ecology, biodefense, biotechnology and forensics. Most of these applications rely upon DNA separation by length, which is readily achieved using sieving gels in electrophoresis. Separation by sequence is a more complicated problem, generally requiring sufficient differences in native or induced conformation or differences in

www.ISC2016.ie @isc2016 #isc2016 /isc201698
ORAL ABSTRACTS Electrodriven Separation Methods
thermal or chemical stability of the strands that are hybridized prior to measurement. In many cases, applicability is limited detection of known target sequences. New approaches are needed that are analogous to separation by length in their simplicity and general applicability to any mixture of DNA. We have discovered such a technique that allows ssDNA to be separated by sequence using high concentrations of salts in simple buffer solutions. The approach does not require prior knowledge of the sequences or persistent conformational differences among the strands. This talk will focus on recent developments in our investigations of the basis of the separation in capillary zone electrophoresis. Separations were performed using both laser-induced fluorescence and UV absorption detection of labeled and unlabeled oligonucleotides.Acknowledgements: This work was supported by the United States National Institutes of Health under 1R21GM104693.Disclosure of Interest: None DeclaredKeywords: Capillary Zone Electrophoresis, DNA separation, DNA Sequence

99www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSLipidomics
Lipidomics
LP-025-001 - Liquid chromatography and mass spectrometry – tools to study differences in pea testa composition in relation to domestication and dormancy
Petr Bedná−* 1, Markéta Válková1, Monika Cechová1, Lukáš Ku−era1, Iveta Hradilová2, Petr Smýkal2, Aleš Soukup3, Anna Janská3
1 Regional Centre of Advanced Technologies and Materials, Department of Analytical Chemistry,
2 Department of Botany, Faculty of Science, Palacký University, Olomouc,
3 Department of Experimental Plant Biology, Faculty of Science, Charles University, Prague, Czech Republic
Content: Timing and efficiency of seed germination are key factors for plant life. In a population of wild seeds (unlike crop one) only a part of individuals starts to germinate in proper conditions. Fraction of seeds remaining inactive is expressed by dormancy. The study deals with development of methods distinguishing chemical differences of wild (dormant) pea genotypes from crop (non-dormant) ones in dry state for better understanding the process of imbibition and germination on molecular level. Liquid chromatography combined with high resolution tandem mass spectrometry (LC/HRTMS) and laser desorption/ionization mass spectrometry (LDI-HRTMS) were used.
Seeds of genotypes with different dormancy levels were studied. Crushed seed coats were extracted by solvents with different polarity and analyzed by LC/HRTMS. Surface or (micro)dissected parts of testa were studied by LDI-HRTMS.
Study of LC/HRTMS data treated by principal component analysis revealed signals with significant differences among dormant and non-dormant genotypes. Higher content of dimer and trimer of gallocatechin, isomers of quercetin and their hydroxylated forms were found in dormant genotypes indicating the significance of particular flavonoids. Chromatographic separation of isomeric markers was studied in detail on reverse and biphenyl stationary phases. Direct LDI-HRTMS analysis of testa outer surface revealed higher content of hydroxylated fatty acids in dormant genotypes presumably in hydrophobic cuticle hindering water penetration into seed. Strong signals of sugars were found in LDI-HRTMS spectra of seed coat (micro)dissections. Detailed interpretation of their differences in particular parts of seed testa (MS imaging) with respect to dormancy is now in progress. LC/HRTMS and LDI-HRTMS proved to work in complementary and synergistic way.Acknowledgements: Czech Science Foundation (14-11782S), Ministry of Education, Youth and Sports of the Czech Republic (LO1305) and Palacký University Olomouc (IGA_PrF_2016_016)Keywords: laser desorption/ionization mass spectrometry, liquid chromatography/mass spectrometry, seed dormancy
LP-025-002 - Addressing The Throughput Challenge Of Large Cohort Epidemiology Studies With Microbore Lc/Ms
Robert S. Plumb, Ian D. Wilson* 1
1Surgery and Cancer, Imperial College, London, United Kingdom

www.ISC2016.ie @isc2016 #isc2016 /isc2016100
ORAL ABSTRACTS Lipidomics
Content: The MRC-NIHR National Phenome Centre, Imperial College London, provides “high throughput, forensic quality, metabolic phenotyping to support large scale epidemiological studies & basic medical research into disease understanding or patient stratification”. Global life-styles changes lead to increases in obesity, diabetes, and mental health issues. This not only affects a person’s quality of life but also places increased strain on the healthcare systems to provide the right treatment & manage costs.
Metabolic Phenotyping offers a valuable and unique insight into the underlying biochemistry of diseases as & patients individual biochemistry “phenotype’, diet, health status, age and stress. To deliver this information the analytical data generated in processed via a variety of chemometric modelling and analysis methodologies to deliver the relevant biochemical information. These chemometric platforms employed vary from simple multivariant analysis to highly complex model based analysis and is presented in a format ready for interpretation by medics.
One of the key challeneges faced in metabolic phenotyping is addressing the large cohort size (2000-8000 samples) in a timely manner whilst maintaining analytical data quality. This presentation will discuss the development of a high throughput (1.5 min) high resolution assay employing sub 2µm particle HILIC and reversed-phase LC coupled to accurate mass MS to address this isses. The system developed employs short 1mm ID columns operated at very high mobile phase linear velocities, taking advatange of the extended van deemter curve of these small particles
to maintain separation performance. The system performance was compared to a standard system operating a 15 minute sepaation using both chromatographic parameters and multivariate statistics and found to deliver similar performance but with a 5 fold increase in throughput and 75% reduction in solvent consumption.Acknowledgements: Prof Jeremy NicholsonProf Ian WilsonDr Nicola GrayKeywords: biomarkers, epidemilology, Lc/MS

101www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSForensic and Environmental Analysis 2
Forensic and Environmental Analysis 2
FE2-026-001 - Liquid Chromatography –Mass Spectrometry (MS) for the Determination of Naturally Occurring Marine Toxins.Dr Ambrose Furey and Dr Mary Lehane, Mass Spectrometry Group, Cork Institute of Technology, Rossa Ave., Bishopstown, Cork.
Content: Marine algal borne toxins represent a serious threat to human health; these potent bioactive compounds have been associated with many severe poisonings and even deaths. These toxins can accumulate (becoming biomagnified) to very high levels in shellfish tissue due to the filter-feeding mechanism of these bivalve molluscs. Consequently most (but not all) poisoning incidents result from the consumption of shellfish.
Analytical determination of these natural toxins is challenging due to the fact that they represent a diverse range of compounds structurally (there are in fact many ‘families’ of marine toxins), by the lack of certified standard materials and also by the fact that the main toxins can undergo biotransformation in vivo in fish to produce toxic metabolites. Ireland exports over 80% of its shellfish abroad mainly to Europe, so it is imperative for the good of the industry that we can ensure the safety of our produce.
The Mass Spectrometry team in CIT specialises in the chromatographic isolation of natural toxins and their metabolites from complex matrices (fish tissue and algal material mainly) to generate reference materials, on the discovery of new toxins by studying the LC-MS data and on the development of robust and reliable quantitative methods for screening.
In this presentation we will focus on the in situ sampling of selected toxins in sea water using Solid Phase Adsorption Toxin Tracking (SPATT) technology, on LC-MS investigations into the chemical and structural nature of toxins and their analogues (many of these toxins are at too low a concentration and in too dirty a matrix to be able to easily obtain NMR spectra), on approaches to analysing isobaric isomers and on how to tackle recently emerging toxins in EU waters (toxins which have migrated from more tropical climes, including the dreaded Puffer fish toxin!).
FE2-026-002 - Complex separation of complexing agents in radioactive effluents and sludges
L. Sghaier 1,*, C. GAUTIER 1, C. Colin 1, J. Vial 2, D. Thiebaut 2, C. Mougel 1
1 Den - Service d’Etudes Analytiques et de Réactivité des Surfaces (SEARS), CEA, Université Paris-Saclay, Gif-sur-Yvette,
2 Department of Analytical, Bioanalytical Sciences and Miniaturization (LSABM), Institute of Chemistry, Biology and Innovation (CBI), ESPCI ParisTech, CNRS UMR 8231, PSL* Research University, Paris, France
Content: In France, the specifications of the National Radioactive Waste Management Agency (ANDRA) require to reliably quantify chelating agents present in nuclear wastes as a result of their use in decontamination processes. Indeed, these effluents are ultimately stored in dedicated waste disposal facilities and chelating agents may form complexes with radionuclides, which can facilitate their mobility in the environment. Four ligands must be identified in priority: ethylenediaminetetraacetic acid (EDTA), diethylenetriaminepentaacetic acid

www.ISC2016.ie @isc2016 #isc2016 /isc2016102
ORAL ABSTRACTS Forensic and Environmental Analysis 2
(DTPA), nitrilotriacetic acid (NTA), triethylenetetramine hexaacetic acid (TTHA).
At first sight, it seems easy to analyze only four compounds. But, when these molecules are chelating agents present in radioactive effluents characterized by high amounts of suspended solids, acidic pH, high content of heavy metals, high salt concentrations and an important radioactivity; when the study of the speciation of these analytes necessary to determine the major chelates is faced with the lack of stability constants in literature; when the sample treatment and separation steps must be compatible with a mass spectrometer detector; and when the standards used for the quantification are not always reliable [1], this analysis appears not so obvious.
Ion-pair chromatography with electrospray mass spectrometry detection [2] was chosen to perform the separation of these four compounds. Prior to that, a sample treatment was applied to convert the complexes into one major chelate form and thus achieve a reliable quantitative analysis. Mobile phase composition and mass spectrometer parameters were optimized, and the performances of the developed method in terms of linearity, repeatability and sensitivity were determined. The contribution of a core shell column was also assessed. After validation, this method was applied to real samples.References: [1] C, Gautier, Accred Qual Assur 21 (41-46) (2016)[2] L, du Bois de Maquillé, J. Chrom. A 1276 (20-25)
(2013)Disclosure of Interest: None DeclaredKeywords: EDTA, DTPA, NTA, TTHA, nuclear waste, speciation

103www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSOmics 2 & Food/Health
Omics 2 & Food/Health
O2FH-029-001 - High-performance thin-layer chromatography and mass spectrometry in the analysis of bioactives in plant extracts and food supplements
Irena Vovk, Vesna Glavnik, Alen Albreht, Breda Simonovska, Eva Kranjc, Samo SmrkeDepartment of Food Chemistry, National Institute of
Chemistry, Hajdrihova 19, SI-1000 Ljubljana, Sloveniaemail: [email protected]
Content: Today several medicinal plants and plant preparations are marketed as food supplements, although many of the bioactive ingredients (bioactives) have not yet been properly scientifically investigated. The endless possible combinations of the bioactive ingredients and excipients in different formulations such as hard/soft capsules, tablets, and liquids make the analysis of food supplements very difficult. Bioactive ingredients can be bioactive natural compounds, vitamins, minerals, plant extracts or even several plant extracts in one dietary supplement product, chemically modified compounds (e.g., encapsulated), etc.. Additional problems are the lack of available reference standards, standard reference materials (SRM), and marker compounds of particular plant materials, plant extracts or plant extract fractions, as well as a variety of chemical structures including isomeric compounds. Chromatographic techniques, especially their combined use and hyphenation to mass spectrometry and UV/vis spectrophotometry, are an indispensable tool in the research and development in this field.
We will present the state-of-the-art, the potential and the role of high-performance thin-layer chromatography (HPTLC) in all stages from the conception through to the final food supplement product, including the selection of the ingredients, selection of plant varieties, quality control of the raw materials and final products. The examples will be focused on the instrumental detection possibilities (densitometry, image analysis, mass spectrometry) in solving the analytical challenges (fast chemical fingerprinting, identification and characterization of biomarkers) related to different combinations of bioactive ingredients in multi-ingredient food supplements. We will show the importance of the detection reagents (stability and specificity) and the influence of the sorbent, pre-developing and developing solvents on ion suppression in HPTLC-MS and HPTLC-MS2 analyses. The potential of multidimensional planar chromatography (MPC) on two different sorbents and MPC-MS in fast analysis of proanthocyanidins in crude plant extracts will also be presented.
O2FH-029-002 - Standardising Analytical Metabolomics. Needs, ideas and the way ahead
G. Theodoridis*, H. Gika 1
1 Medicine, Aristotle University Thessaloniki, Thessaloniki, Greece
Content: Metabolomics is based on the use of high resolution spectroscopic and separation techniques: LC-MS, GC-MS and NMR spectroscopy. Despite noteworthy progress, metabolomics still presents major challenges to the analytical community: 1) high diversity of molecular structures

www.ISC2016.ie @isc2016 #isc2016 /isc2016104
ORAL ABSTRACTS Omics 2 & Food/Health
to be analysed, 2) huge differences in concentration ranges, 3) limitations in analytical response of different metabolites 4) need for multiple analytical platforms (NMR/LC-MS/GC-MS, HILIC/IPLC/RPLC), 5) ineffective data combination, 6) research effort fragmentation and slow rate of metabolite identification.
Poor standardisation in study design, sample collection/handling, chemical-statistical analysis, ineffective pathway analysis and fusion with other omics hinder expansion of metabolomics to epidemiology level.
These problems are largely analytically determined, so Analytical chemists should lead the quest for their resolution. Deep knowledge of Analytical Chemistry and high-quality in analytical practice is absolutely necessary; if this is not the case, poor outcome is close to certain despite the ever advancing instrumentation/software technology.
In the talk we will illustrate the needs for 1) standardisation of methods, 2) improving methods’ stability and 3) enforcing quality control (QC).
We will present results from our studies of the extraction of feces prior to comprehensive analysis by GC-MS and UHPLC-MS/MS. We also discuss the study of derivatisation conditions prior to GC-MS analysis. Finally we also present a roadmap/protocol for QC in untargeted UPLC-MS.Acknowledgements: Research has been co-financed by the European Union and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) - Research Funding
Program: Excellence II - Metabostandards. Investing in knowledge society through the European Social Fund. More information can be found at http://users.auth.gr/gkikae/aristeiaDisclosure of Interest: None DeclaredKeywords: harmonisation, metabolomics, quality control

105www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSTrace Element Analysis
Trace Element Analysis
TEA-030-001- Determination of Lithium and Transition Metals in Lithium-Ion Battery Components by Capillary Electrophoresis and Ion Chromatography
B. Vortmann 1,*, V. Kraft 1, C. Lürenbaum 1, S. Nowak
2, M. Winter 2
1 University of Münster, Münster, 2 University of Muenster, Muenster, Germany
Content: The lithium-ion battery (LIB) is one of the most important energy storage systems for portable electronic devices. One essential component of the LIB is the electrolyte, which contains lithium hexafluorophosphate as conducting salt and organic carbonates as solvents. The other important components are the electrodes, whereas lithiated layered transition metal oxides and graphite are the common used active materials. The determination of the total content of the active material components and the actual stoichiometry is very important for understanding of the material in its electrochemical application. The element amount was accessed so far by inductively coupled plasma - optical emission spectrometry (ICP-OES).
Table: Limits of detection and quantification for the ICP-OES, CE and IC system.
Element ICP-OES
µg/L CE
µg/LIC
µg/L LOD LOQ LOD LOQ LOD LOQ
Lithium 0.04 0.12 17.41 62.70 8.26 29.34
Cobalt 0.44 1.47 348.38 1282.96 - -
Manganese 0.07 0.24 42.09 244.07 - -
Nickel 0.55 1.85 838.04 2981.68 - -
The ion chromatography (IC) has increased more important in the field of battery research, especially in the area of electrolyte investigation, whereas the main focus is the identification of thermal aging and decomposition products [1]. The focus of the capillary electrophoresis (CE) method has been mainly in ionic liquids. Nevertheless, both methods were not applied for elemental determination of LIB components so far.
We have developed and optimized IC and CE methods for the determination of lithium and transition metals in battery components. The samples were separated by CE and IC and the element content was determined by UV/Vis spectroscopy and conductivity detection. The relative standard deviations, recovery rates, limits of detection and quantification were calculated and compared to those obtained with ICP-OES. These results present the CE and IC methods as good alternatives to the ICP-OES.
References: [1] V. Kraft, M. Grützke, W. Weber, M. Winter, S.
Nowak, J. Chromatogr. A 1354 (2014) 92-100.Disclosure of Interest: None DeclaredKeywords: Capillary electrophoresis, Lithium-Ion Battery, Transition metals

www.ISC2016.ie @isc2016 #isc2016 /isc2016106
ORAL ABSTRACTS Trace Element Analysis
TEA-030-002 - Applications of Ion Chromatography in Gas Analysis
Peter Adam*
Content: Studies on commercially available ion chromatographs have shown that this technology is a powerful tool for gas analysis if there is a complete dissociation of the analyt in the aqueous solvent. Design and function of an IC-system based on suppressed anion chromatography, as well as sampling methods for corrosive gases, will be outlined. Applications, focusing on the analysis of calibration gas mixtures with corrosive components, will be demonstrated.
Ion chromatographs can be calibrated with traceable salt standard solutions of high accuracy. Based on this, a detailed discussion of traceability and a model for the uncertainty calculation will be presented. Results of the analysis of gaseous primary reference material using an ion chromatograph will be reported.References: 1. J. Weiß, …Ionenchromatographie“, VCH
Verlagsgesellschaft, Weinheim (1995)2. D.T. Gjerde and J.S. Fritz, …Ion Chromatography“,
Dr. Alfred Hüthig Verlag, Heidelberg (1987)3. EURACHEM/CITAC Guide, …Quantifying
uncertainty in analytical measurement“, Third Edition, (2012)
4. J. Kragten, …Calculating standard deviations and confidence intervals with a universally applicable spreadsheet technique“, Analyst, 119, 2161-2166 (1994)
Keywords: calibration gas mixtures, gas analysis, traceability and uncertainty calculation

107www.ISC2016.ie @isc2016 #isc2016 /isc2016
ORAL ABSTRACTSSample Preparation 3
Sample Preparation 3
SP3-031-001 - Removal of phospholipids from human plasma samples by electromembrane extraction (EME)
L. Vårdal*, S. Pedersen-Bjergaard 1, A. Gjelstad 1
1 School of Pharmacy, University of Oslo, Oslo, Norway
Content: Electromembrane extraction (EME) was introduced in 2006 [1] as a microextraction technique based on electro kinetic migration of basic or acidic compounds across a supported liquid membrane (SLM).
In this project, EME was performed from plasma samples to investigate whether the extracts contain phospholipids. Phospholipids are fatty acids present in human plasma, which may interfere when plasma samples are analyzed with liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) by causing column contamination and matrix effects (ion suppression). Therefore, phospholipids should be removed before LC-MS/MS analysis.
EME was performed by filling a glass vial with 1 mL plasma sample (donor solution). Secondly, the SLM was prepared by dipping a porous hollow fiber of polypropylene into an organic solvent. Any excess of organic solvent was wiped off before the lumen of the fiber was filled with 25 µL of acceptor solution. Two electrodes were placed in the donor and acceptor solution, and then connected to a power supply. When simulating EME of positively charged compounds (bases), the negatively charged
electrode was placed in the acceptor solution. The whole setup was finally placed in an agitator, providing agitation of 900 rpm during the EME process. Extraction was carried out for five minutes.
To evaluate the amount of phospholipids in the EME extracts, LC-MS/MS analysis was performed on the acceptor solutions. The LC-MS/MS chromatograms were compared with the LC-MS/MS chromatogram obtained for a comparative protein precipitation (PP) extract, as this technique fails to remove phospholipids. The results were clear; no detectable peaks for phospholipids in the EME extracts compared to the PP chromatogram. To conclude; the present work has shown that EME extracts are free from phospholipids when operating with the most frequently used EME systems.References: 1. Gjelstad, A., K.E. Rasmussen, and S. Pedersen-
Bjergaard. Journal of Chromatography A, 2006. 1124(1): p. 29-34.
Disclosure of Interest: None DeclaredKeywords: Electromembrane extraction (EME), Removal of phospholipids, Sample clean-up
SP3-031-002 - Characterisation of volatiles and semi-volatiles in gin and whiskey using dispersive liquid-liquid microextraction with gas chromatography.
Damian Connolly*, Shelly Spoelders, Andrew Quigley, Wayne Cummins, Michael Breen.Department of Science, Waterford Institute of Technologyemail: [email protected]

www.ISC2016.ie @isc2016 #isc2016 /isc2016108
ORAL ABSTRACTS Sample Preparation 3
Gin is a widely consumed distilled beverage which includes various types and formulations. The most common type of gin is London Dry Gin, produced only by re-distillation of alcohol 96% (v/v) in the presence of juniper berries and other natural botanical ingredients including coriander seeds, cardamom seeds, orris root, angelica root, liquorice root, orange peel, lemon peel, and liquorice root. All of these ingredients are rich in essential oils, which contribute to the aroma of most gins, but the main flavour of distilled gin should come from the juniper berries. Alternative methods include vapour infusion in which the botanicals are held in a receptacle above the hot solvent during distillation. Since the presence of several volatile and semi-volatile compounds strongly contributes to gin flavour perception, detailed information about gin volatile composition is necessary for the characterization of distinct classes of products and as a basis for defining gin sensory quality. The most common analytical method for gin analysis is headspace solid phase micro-extraction (HS-SPME) followed by gas chromatography with mass spectrometric analysis (or direct immersion HS-GC-MS) which requires expensive laboratory equipment and consumables (SPME fibers). Here we report the development of dispersive liquid-liquid microextraction (DLLME) in which extraction of analytes takes place in a dispersion of the extracting solvent (chloroform, dichloromethane) made in water, in the presence of a third, bridging “dispersing solvent”. The dispersion is removed by centrifugation and the extracting solvent containing analytes is taken for analysis with a micro-syringe. We report the optimisation of a DLLME-GC-FID protocol for the analysis of
selected commercial gins as well as model gins produced in-house. Typical DLLME optimisation parameters such as solvent volume ratios, ionic strength and sample pH were studied using an internal standard to maximise extraction. Volatile and semi-volatile components were identified via Kovats Retention Indices using a mixed hydrocarbons standard, belonging mainly to the terpenoids family (monoterpenes, sesquiterpenes and their corresponding oxygenated compounds) including limonene, linalool, α-pinene, β-myrcene, γ-terpinene and verbenyl ethyl ether. The optimised DLLME protocol was used to (1): assay the batch-to-batch reproducibility of model gins, (2): to compare commercial gins and (3): to examine the influence of selected production methods on the generation of oxygenated terpenes and corresponding reduction in unwanted monoterpenes which is known to improve product stability. Finally, the method was also applied to the analysis of selected whiskeys and the evaluation of various accelerated whiskey maturation methodologies.

Tutorial Abstracts

www.ISC2016.ie @isc2016 #isc2016 /isc2016110
TUTORIAL ABSTRACTS
Tutorial Abstracts
TU01 - SFC without modifiers
Dr. Didier ThiébautUMR CBI 8231, CNRS - ESPCI-PARIS, Laboratoire
Sciences Analytiques, Bioanalytiques et Miniaturisation, PSL Research University, ESPCI-PARIS 10 rue Vauquelin, 75005 Paris - France,
Content: This presentation will remind the potential users of the SFC “hidden face”, SFC without modifiers, despite it has been developed for many years.
The history of SFC will be shortly presented as an introduction.
Then a first part will cover fundamentals on supercritical fluids properties and why they can be interesting in chromatography (SFC) and extraction (SFE). Their limitations will be presented too.
A second part will deal with the features of SFC without modifiers with a focus on the stationary phases to be used with neat carbon dioxide as the mobile phase and retention mechanism. The use of GC capillary columns will be commented.
The third part will be devoted to the apparatus with a focus on the wide choice of detectors that can be hyphenated to SFC and the easy coupling of SFC to GC or GCxGC.
The last part will discuss some applications of SFC without modifiers, mainly for the analysis of oil derived samples - for some SFC is a registered ASTM method -, including SFC-GCxGC examples.
TU02 - 60 years of chiral separations and thoughts about the future – a tutorial with historic perspective
Michael Laemmerhofer*
Content: In 2016, “The Chromatographic Society” celebrates its 60th anniversary. To account for its long tradition in the various fields of chromatographic science, this tutorial lecture attempts to highlight some important innovations in the field of chiral separations. In 1956, chromatographic chiral separation was essentially non-existent and the technology took off only with the advent of modern gas chromatography. It was pioneered by Gil-Av who demonstrated in 1966 a successful GC enantiomer separation of amino acids derivatives on a steel capillary coated with a chiral dipeptide selector. The modern column technology for enantioselective GC originates from seminal works in the late 70s and 80s. Cyclodextrins and amino acid derivatives attached to polysiloxane films coated on fused silica capillaries have become the state of art and are still the columns of broadest use. In LC, work by Davankov is sometimes referred to as the early start of modern chromatographic enantiomer separation. Yet, many of the modern chiral stationary phases used nowadays were invented in the 1980s. The success story in LC enantiomer separation was written by polysaccharide-coated silica supported CSPs invented by Okamoto. These phases have dominated for decades, and are still dominating applications in pharmaceutical industry, in analytical and preparative scale separations both in LC and SFC mode. They have been the enabling technologies in drug discovery like protein CSPs in early enantioselective bioanalysis. Nowadays, macrocylic

111www.ISC2016.ie @isc2016 #isc2016 /isc2016
TUTORIAL ABSTRACTS
antibiotics CSPs and chiral ion-exchangers are alternative CSPs particularly useful for polar ionic chiral solutes. This tutorial will spotlight important developments in the history of chiral separations and highlight studies which contributed essentially to the mechanistic understanding as well as advanced practical applications of this technology in pharmaceutical industries.References: 1. M. Lämmerhofer, J. Chromatogr. A, 1217, 814-856
(2010).
TU03 - Role of the Stationary Phase in HPLC
Attila FelingerDepartment of Analytical and Environmental Chemistry, University of Pécs, Ifjúság útja 6, H-7624 Pécs, Hungary
Content: Novel stationary phases in high-performance liquid chromatography have been developed continuously. During the recent years, new porous sub-2-micron packing materials, core-shell materials, monolith columns have been introduced to speed up separations keeping the efficiency high.
The novel phases guarantee fast mass transfer kinetics due to the shorter diffusion paths.
Pore sizes below 100 Å provide good efficiency for the separation of small molecules, but in bioseparations pores sizes larger than 300 Å are required. Besides pore size itself, the pore size distribution is another factor that influences the separation efficiency.
On the other hand, the developments of column technology, the smaller particles
that require larger pressure, bring along novel problems. The frictional heat limits the further increase of efficiency when sub-2-micron particles are employed.
The study of the mass transfer properties and thermodynamics of retention on modern fully porous and core-shell packing materials reveals the separation power of the latest generation of stationary phases. Concepts and results are presented focusing on the tools of stationary phase characterization.
TU04 - Two-Dimensional Liquid Chromatography (2D-LC) - Theory & Practice
Stephan BuckenmaierAgilent Technologies, Hewlett Packard Str 8, 76337 Waldbronn, Germany
Content: Two-dimensional liquid chromatography (2D-LC), already introduced a couple of decades ago, lately finds more extensive use in a variety of industries. Its role has been strengthened due to the advent of UHPLC based instrumentation, the availability of software facilitating 2D-LC operation and data analysis, a better understanding of limits and advantages of the technique, and the demonstration of the power of 2D-LC for applications where conventional 1D-(U)HPLC was simply not sufficient.
This tutorial provides theoretical concepts of 2D-LC and aims to introduce you to the motivation for employing this technique. You will be familiarized with different operational modes when comparing comprehensive, heart-cutting, and multiple heart-cutting (MHC) 2D-LC.

www.ISC2016.ie @isc2016 #isc2016 /isc2016112
TUTORIAL ABSTRACTS
Some emphasis will lie on MHC operation. This mode decouples the 2D from the 1D time scale and, unlike comprehensive mode, allows both dimensions to be operated under more optimal conditions. MHC is probably the best choice for samples of intermediate complexity, where the 2D-analysis of 5 to 20 compounds might be of interest.
In this context, strategies leading to successful 2D-LC methods will be discussed, in particular practical details on the selection of suitable separation modes, column types and instrumental parameters. Illustrative application examples such as the separation of pesticides in natural products, additives out of polymeric matrix, and impurity profiling in biopharmaceuticals will be shown to demonstrate the potential of MHC 2D-LC.
TU05 - Fundamental and Practical Aspects of Ultra-High-Performance Liquid Chromatography (UHPLC)
Monika Dittmann & Tony EdgeAgilent Technologies
Content: The increasing need for higher speed and resolution in analytical separations drives the trend towards moving from HPLC (ca. 400 bar max. pressure) to UHPLC (>1000 bar).
This tutorial will cover the basic principles of UHPLC and discuss various aspects that need to be considered to successfully apply this technology in method development and routine work.
In the first section the basic theory of mass
transfer in columns packed with different particle sizes and morphologies (fully porous, core-shell, monoliths) will be discussed. It will be demonstrated how the increase in operation pressure and/or temperature can result in enhanced resolution and/or speed of separation.
The second section will be dedicated to optimization strategies for UHPLC separations for different types of applications using the concept of kinetic plots. The kinetic plot (also known as Poppe plot) model uses column efficiency in combination with column permeability to determine the maximum achievable number of plates generated per unit time. This is a much more useful measure for comparing column performance than using column efficiency or plate number alone. The kinetic plot model will be explained in detail and examples for different particle sizes and morphologies will be shown.
The third section will focus on instrumental aspects of UHPLC such as dwell volumes and system dispersion, where the requirements are much more stringent as in HPLC instruments due to the smaller column diameters used in UHPLC. The impact of extra-column volume on the efficiency for small bore columns will be demonstrated for isocratic and gradient separations. Another topic is the transfer of methods between HPLC and UHPLC instruments where differences in system properties (dwell volume and mixing behavior) can lead to changes in retention times and selectivity.
The final section will investigate the effects of temperature on the chromatographic performance criteria. The effect of varying

113www.ISC2016.ie @isc2016 #isc2016 /isc2016
TUTORIAL ABSTRACTS
the temperature on the physical properties of the mobile phase will be discussed in relation to the mobile phase viscosity and also the dielectric constant. The session will then move into a discussion of the effects of temperature on the Gibbs free energy of binding. This allows for differences in selectivity for different compounds at different temperatures. The implications of this will be discussed in terms of method development. Finally, the effect that temperature has on the chromatographic efficiency will be discussed.
TU06 - Tutorial - The role of temperature in HPLC and how column thermostatting and mobile phase pre-conditioning details enable to take full advantage of it
Dr Frank SteinerThermo Fisher Scientific, Germering, Germany
Content: Column temperature and how to control it is of fundamental importance in liquid chromatography for both thermodynamic and kinetic reasons. Temperature variation allows for selectivity tuning in many applications and temperature increase enables faster separation at a given column pressure thanks to enhanced mass transfer and reduced eluent viscosity. Increasing the temperature also allows for reducing concentration of organic solvents.
The tutorial presents the thermodynamic and kinetic background of how temperature can be applied for method optimization. It provides valuable hints and rules of thumb and illustrates them with practical examples.
A special focus is on the instrumental pre-requisites of eluent pre-heating and column thermostatting. How to deal with significant frictional heat generated in columns at elevated pressures is an important aspect of this. While still air thermostatting generates higher efficiencies than forced air modes, there can be critical selectivity differences and changes in peak resolution between both thermostat types with some applications. The possibility to set an eluent pre-heater to a lower temperature than the column compartment to compensate for frictional heating effects in the column is discussed as a possible solution and illustrated with practical examples.
TU07 - Physical Origins of Band Broadening in Modern Chromatographic Columns Building Perspectives for Tomorrow’s Column Design
Fabrice GrittiWaters Corporation, Milford, MA, 01757, USA
Content: The tutorial is addressed to any audience interested in the separation performance of LC, UHPLC, and SFC columns from both a fundamental and practical viewpoints. The goals are three-fold:
Familiarize everyone with the basic fundamental and the relevant physical phenomena pertaining to broadening of a sample zone along modern chromatographic columns. They will include the dispersion contribution of the latest UHPLC instruments, longitudinal diffusion,

www.ISC2016.ie @isc2016 #isc2016 /isc2016114
TUTORIAL ABSTRACTS
sample diffusivity in the stationary phase, adsorption-desorption of the analyte onto the sorbent, eddy dispersion in the mobile phase, column thermal environment (and the nefarious effect of radial temperature gradients on column efficiency), and the axial heterogeneity of the column properties (temperature, eluent density, analyte retention).• Identify and anticipate from a minimum
of information (applied linear velocity, stationary phase size, and analyte diffusion) the dominant contribution to band broadening and the performance of any selected column, respectively. This approach applies to method transfer and to the optimization of separation performance for a given application.
• Build perspectives for the design of the next generation of instruments, columns, and particles that will enable the users to further improve LC and SFC separation performance in a near future.
TU08 - Potential of molecularly imprinted polymers as solid-phase extraction sorbent: synthesis, applications, on-line coupling (from LC to nanoLC).
Valérie Pichon1,2
1 Department of Analytical, Bioanalytical Sciences and Miniaturization (LSABM), UMR CBI 8231 ESPCI ParisTech/CNRS, PSL Research University, ESPCI ParisTech, Paris, France
2 UPMC, Sorbonne Universités, Paris, France
Content: The determination of compounds at very low levels of concentration is still a real analytical challenge in various applications fields. The evolution of the
instrumentation in terms of separation and detection allows a significant improvement of the sensitivity and the analysis time. However, the analysis of ultra-traces from complex matrices often requires a step of purification and preconcentration prior to chromatographic analysis. Therefore, extraction sorbents based on a molecular recognition mechanism can be developed and used for the selective extraction of a target molecule and its structural analogs, thus rendering their quantitative analysis more reliable and sensitive. For this, molecularly imprinted polymers (MIPs) have already shown a high potential for the selective extraction of target analytes from complex matrices.
The development of numerous MIPs (prepared by radical polymerization of organic monomers or by a Sol-Gel approach using organosilanes) to selectively extract a target analyte but also structurally related compounds from various complex matrices (real water, soils or biological fluids...) will be presented. Their potential for the selective trapping of organic compounds or metals from environmental matrices, foodstuff and biological fluids will be illustrated.
Most of the developments are achieved by introducing the MIP particles in disposable SPE cartridges. However, these selective tools are also particularly necessary when developing miniaturized devices because of the decrease of the resolution that results from the use of short length separation devices. In this context, miniaturized MIPs can be prepared by in-situ synthesis of an imprinted monolith in a capillary (I.D. 100 µm). The study of the repeatability of the synthesis of these miniaturized selective sorbents and their on-line coupling to nano-

115www.ISC2016.ie @isc2016 #isc2016 /isc2016
TUTORIAL ABSTRACTS
LC for the selective extraction of compounds from real samples will be presented.
TU09 - LC-MS-Based Metabonomics, Metabolomics and Metabolic Phenotyping
Ian WilsonImperial College of London, Division of Computational Systems Medicine, Department of Surgery and Cancer, Sir Alexander Building, Exhibition Road, South Kensington, London SW7 2AZ, United Kingdom.
Content: LC-MS-based global metabolite profiling ought to be easy – but there are many pitfalls for the unwary that can mean that the data produced from this type of analysis can be of less value than it should be. The tutorial will provide an overview of the strengths and weaknesses of the approach, and provide suggestions for practical solutions to problems wherever possible.
The areas to be covered will include study design, confounding factors, sample collection and storage, selection of chromatographic conditions, the need for (and effects of) sample preparation, how to ensure that the LC-MS system has the best chance of providing reproducible and reliable data, and how to demonstrate that this has been done. The topics of the “identification” of unknowns will be discussed, especially with reference to what actually constitutes identification and the use of “targeted vs non-targeted” methods (and what these terms actually mean) will be considered.
These topics will be illustrated with illustrative examples from studies in basic physiology, disease models and humans.
TU10 - Successful Gradient Reversed - Phase Liquid Chromatography - the Do’s Don’ts!
Course tutor Mel Euerby, Principal of Shimadzu’s Centre of Excellence in Liquid Chromatography and Visiting Professor at the University of Strathclyde and the Open University
Abstract: The 40-minute tutorial is aimed at junior chromatographers who wish to improve their understanding of gradient RP-LC (i.e. the operating parameters which control resolution) and learn how to perform it in a reproducible way. Attendees should be familiar with RP-LC instrumentation and basic chromatographic theory. The course will ONLY major on the minimum amount of theory that is required to perform gradient chromatography and will keep the number of equations down to a minimum!
The course will aim to cover the following topics:• Why use gradient LC? • Theoretical basis of gradient elution
chromatography • Isocratic and gradient scouting• Resolution equation• The importance of average k• Operating parameters (i.e. mobile phase
and instrumentation) which control resolution in gradient chromatography
• How to speed up your gradient chromatography
• Peak focussing• Practical tips for successful RP-
LC gradient chromatography – i.e. minimizing column bleed, ghost peaks and mobile phase effects
• What detectors can and cannot be used in gradient chromatography
• Introduction to retention modelling

Short Course Abstracts

117www.ISC2016.ie @isc2016 #isc2016 /isc2016
SHORT COURSE ABSTRACTS
SC01 - Short Course:
Development and Control of Robust HPLC Methods by Modeling
Imre Molnár, Hans-Jürgen Rieger, Molnár-Institute, Berlin, Germany
Abstract: Method Modeling became nowadays an important tool in analytical sciences. Since the introduction of QbD in analytical work, trial & error type of method development became obsolete. Robust HPLC methods are today required by regulatory agencies and communication about details of Robustness in HPLC methods need to be easily understandable for companies which produce products in Pharma, food and biotech area.
With modeling, method development timescale can be shortened by a factor of 5-10.
The short course is dealing with column selection using the Snyder-Dolan-Hydrophobicity Subtraction-Database and discusses 2- and 3-dimensional retention modeling, gradient elution and robustness issues in HPLC method modeling. Besides of several industrial case studies about pharmaceutical applications for small and large molecules we will talk about aspects of regulatory work and principles of quality by design (QbD) in HPLC method development. Printed Material will be provided. Laptops can be used for modeling.
SC01 Short Course Topics:
Development and Control of Robust HPLC Methods by Modeling 13.00 - 13.30 Introduction to UHPLC method development History of HPLC/UPLC Fundamentals of RPC, HILIC and other techniques
13.30 - 14.00 Isocratic work, Gradient elution, Critical Resolution, Principle of Equal Band Spacing Influence of column dimensions, L, ID, dp, Instrument parameter: Vd, Vext.col., tG, %B(start), %B(end)
14.00 - 14.30 Terms of Quality by Design: HPLC and Statistics Column selection and Characterization, Snyder-Dolan-HS-Database Characterization of Columns by the 3D-critical Resolution Cube’s
14.30 - 15.00 COFFEE BREAK
15.00 - 15.30 Design of Experiments (DoE) – Modeling UHPLC 1-D (OFAT): %B-, pH-, tG-modeling 2-D: tG-T, tG-pH-models
15.30 - 16.00 3-D-Extension to the Cube: mostefficientmodel pH-Cube: tG-T-pH-model, and the ternary Cube: tG-T-tC-model
16.00 - 16.30 Dealing with Multifactorial Variabilities: Robustness studies Instrument Variabilities Automated generation of the Cube
16.30 - 17.00 Case Studies of Modeling Separations in the Pharmaceutical Industry. Reducing analysis time from 60 min to 6 min Reducing analysis time from 160 min to 3 min Modeling methods for large molecules: Therapeutic Proteins, MAbs, ADC’s Knowledge Management Documentation

www.ISC2016.ie @isc2016 #isc2016 /isc2016118
SHORT COURSE ABSTRACTS
SC02 - Short Course:
Fundamental and Practical Aspects of Ultra-High-Performance Liquid Chromatography (UHPLC)
Monika Dittmann & Tony EdgeAgilent Technologies
Abstract: The increasing need for higher speed and resolution in analytical separations drives the trend towards moving from HPLC (ca. 400 bar max. pressure) to UHPLC (>1000 bar).
This tutorial will cover the basic principles of UHPLC and discuss various aspects that need to be considered to successfully apply this technology in method development and routine work.
In the first section the basic theory of mass transfer in columns packed with different particle sizes and morphologies (fully porous, core-shell, monoliths) will be discussed. It will be demonstrated how the increase in operation pressure and/or temperature can result in enhanced resolution and/or speed of separation.
The second section will be dedicated to optimization strategies for UHPLC separations for different types of applications using the concept of kinetic plots. The kinetic plot (also known as Poppe plot) model uses column efficiency in combination with column permeability to determine the maximum achievable number of plates generated per unit time. This is a much more useful measure for comparing column performance than using column efficiency or plate number alone. The kinetic plot model will be explained in
detail and examples for different particle sizes and morphologies will be shown.
The third section will focus on instrumental aspects of UHPLC such as dwell volumes and system dispersion, where the requirements are much more stringent as in HPLC instruments due to the smaller column diameters used in UHPLC. The impact of extra-column volume on the efficiency for small bore columns will be demonstrated for isocratic and gradient separations. Another topic is the transfer of methods between HPLC and UHPLC instruments where differences in system properties (dwell volume and mixing behavior) can lead to changes in retention times and selectivity.
The final section will investigate the effects of temperature on the chromatographic performance criteria. The effect of varying the temperature on the physical properties of the mobile phase will be discussed in relation to the mobile phase viscosity and also the dielectric constant. The session will then move into a discussion of the effects of temperature on the Gibbs free energy of binding. This allows for differences in selectivity for different compounds at different temperatures. The implications of this will be discussed in terms of method development. Finally, the effect that temperature has on the chromatographic efficiency will be discussed.

119www.ISC2016.ie @isc2016 #isc2016 /isc2016
SHORT COURSE ABSTRACTS
SC03 - Short Course:
SFC – Principles, Instrumentation, Method Development, and Applications
Abhijit Tarafdera, Isabelle Francoisb
a Waters Corporation, 34 Maple Street, Milford, MA 01757, USA
b Waters European Headquarter, 5 rue Jacques, Monod, 78280 Guyancourt, France
Abstract: SFC uses CO2 as the principal mobile phase solvent at pressures generally higher than 100 bar. Apart from this, from operational point of view, there is hardly any difference between SFC and RPLC. Regarding chromatographic capabilities, SFC has a more diverse scope with the types of analytes and selectivities. The recent availability of highly robust analytical SFC systems render the technique more attractive than ever before to a wide range of application areas.
The objective of this short-course is to introduce the students to the capabilities of modern SFC as an analytical instrument and separation device. We will describe in detail - (1) How SFC works (apparatus, detectors and coupling to MS) (2) How SFC compares with LC - focusing on where SFC has unique advantages (high flow rates, coupling achiral + chiral columns etc) and where not (e.g. analyzing large biomolecules), (3) How SFC instrumentation compares with LC, (4) Method development and optimization of (a) achiral SFC, and (b) chiral SFC (5) SFC scale-up and method-transfer, and (6) Sample applications.
The pre-requisite is experience with any chromatographic technique and the principles of chromatography. At the end of
the course it is expected that the student will be knowledgeable enough to start working with an SFC instrument and carry out an intended analysis.


Poster Abstracts

www.ISC2016.ie @isc2016 #isc2016 /isc2016122
POSTER ABSTRACTS Biopharmaceutical and Biologics
Biopharmaceutical and Biologics
P01-001-001 - Novel substituted arylcobalamin derivatives – their synthesis, chromatography and mass spectrometryG. Andryianau 1,*, G. Rotko 1, S. Wybraniec 1
1 Faculty of Engineering and Chemical Technology, Cracow University of Technology, Cracow, Poland
Content: Vitamin B12 is an essential cofactor for such important biochemical processes as several isomerization reactions as well as for methyl group transfer. In addition to this, cobalamin – dependent enzyme system plays a major role in uracil methylation reaction prior to DNA replication. Therefore, actively dividing tumor cells require large amounts of cobalamins. Only few microbial strains are able to synthesize corrinoid structures, so highly specialized uptake, transport and accumulation mechanisms were developed for these precious compounds. Elaboration of the idea of application axial – modified cobalamin derivatives for cancer diagnostic and treatment purposes led to the synthesis of several bioconjugates, containing cytotoxins or fluorescent indicators, recognizable by B12 transport proteins and enzymes. Main disadvantages of this class of compounds include their oxygen/light sensitivity and susceptibility to reductive cleavage of Co–CAliphatic bond catalyzed by CblC enzyme/glutathione system. Reported 4-ethylphenylcobalamin derivative don’t have these disadvantages, pointing arylcobalamins as promising new class of functional compound carriers.
Here, we report the synthesis of cobalamin derivatives, containing Co–CAr bonds by radical coupling of cob(II)alamin with aryl
radical generated in situ from aryldiazonium or diaryliodonium tetrafluoroborates in water solutions.
HPLC-DAD-MS control of described radical reactions showed them to be unselective, therefore prep. RP-HPLC isolation was required.
Numerous mass spectrometric experiments of pure compounds showed the presence of unusual triple-protonated dimeric forms of arylcobalamins in all cases. In addition to this, MS/MS fragmentation of title molecules was consequently studied with the use of collision cell – triple quadrupole system showing atypical fragmentation patterns. Disclosure of Interest: None DeclaredKeywords: Arylcobalamins, Drug carriers, Vitamin B12 derivatives
P01-001-048 - Analysis of deferoxamine and deferiprone in β-thalassemia patients’ plasma by capillary electrophoresisS.-M. Wu*
Content: Capillary electrophoresis (CE) is a widely used method which possesses some merits including high-resolution, low reagents consumption, and automation. The major disadvantages which affect CE sensitivity are narrow optical window and lower sample concentration. To achieve higher sensitivity for established methods, on-line sample stacking CE techniques were developed. Field-amplified sample injection (FASI) utilized the electrokinetic injection through different conductivity

123www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSBiopharmaceutical and Biologics
between background electrolyte (BGE) and sample zone to stack and enhance the sensitivity. Dynamic pH-junction (DypH) could stack the analytes in different buffer pHs. Large volume sample stacking (LVSS) could increase analytes by injecting large amount of samples. The sweeping strategy used micelles to gather up and separate analytes.
One capillary electrophoresis (CE) method was established for detecting deferoxamine (DFO) and deferiprone (DFR) in plasma. For β-thalassemia patients, DFO and DFR are major medicines to treat the iron overload caused by blood transfusion. This method was performed on an uncoated fused-silica capillary. After liquid-liquid extraction, the plasma samples were electrokinetically injected into capillary. The phosphate buffer was used as the rinse buffer. This method showed good linearity (r ≥ 0.9960). Precision and accuracy were evaluated by the results of RSD and RE of intra-batch and inter-batch analyses, and all of the absolute values were less than 6.12%. The limits of detection (LOD, S/N=3) were 200 ng/mL for DFO, and 25 ng/mL for DFR. This method was applied for clinical applications of five β-thalassemia patients.Disclosure of Interest: None Declared
P01-001-052 - How to ensure optimal performance when using SEC for the analysis of proteins
T. Edge 1,*, A. Coffey 1, L. Bevan 1, J. Davies 1
1 Agilent Technologies, Church Stretton, United Kingdom
Content: There has been a revolution in the pharma industry with the focus moving
away from traditional small molecules towards larger protein based or nucleic acid derived formulations. This has resulted in a relative surge in new separation technologies that can be readily applied to the analysis of these types of molecules. It has also seen a resurgence in some older technologies, such as size exclusion, which in particular has been applied to the analysis of the monoclonal antibodies, to glean structural information and the for the analysis of impurities that are generated during the manufacturing process.
Although size exclusion is a technique that has matured, it is technology that has not always been implemented in an effective manner. The effect that empirical parameters have on the performance of the separation mechanism for size exclusion will be discussed. The effect of flow rate will be invested, with the production of van Deemter1 curves that will demonstrate the differences between the analysis of small molecules and protein structures where a substantial differences in diffusion rates between these two forms of molecules exist. Another important parameter that can also affect the diffusion rate, is the temperature. A series of van’t Hoff2 plots will be constructed to demonstrate how the size exclusion process is affected by temperature, with the results being compared to the observations made using conventional reversed phase chromatography.
To perform effective size exclusion it is necessary to ensure that the compounds of interest do not interact with the stationary phase, other than in a size based mechanism. This will be investigated by altering the effect of the buffer concentration to demonstrate how secondary interactions can be minimised.

www.ISC2016.ie @isc2016 #isc2016 /isc2016124
POSTER ABSTRACTS Biopharmaceutical and Biologics
References:1. JJ van Deemter, FJ Zuiderweg and A Klinkenberg.
Chem. Eng. Sc. 5: 271–289, (1956)2. TL Chester, JW Coym, J. Chromatogr. A, 1003
(2003) 101–111Disclosure of Interest: None DeclaredKeywords: Monoclonal Antibody, SEC
P01-001-054 - Kynurenine in light of bone biomechanical properties in rats with experimental chronic kidney diseaseB. Kalaska 1,*, K. Pawlak 2, E. Oksztulska-Kolanek
2, B. Znorko 2, T. Domaniewski 2, A. Roszczenko 3, J. Rogalska 3, M. M. Brzoska 3, D. Pawlak 1
1 Department of Pharmacodynamics, 2Department of Monitored Pharmacotherapy, 3Department of Toxicology, Medical University Of Bialystok, Bialystok, Poland
Content: Many diseases can affect bone microarchitecture, e.g. chronic kidney disease [1]. We have previously demonstrated the significant disturbances in the peripheral kynurenine pathway in rats with renal insufficiency [2]. The aim of the present study was to understand the association between serum kynurenine concentration and biomechanical properties of bone in an experimental model of chronic renal failure in Wistar rats. We divided animals into two groups (sham-operated and subtotal nephrectomized rats [3]). Three months after surgery, serum samples were obtained for determination of kynurenine concentrations by HPLC according to Holmes [4]; and tibiae were collected for determination of biomechanical properties using three-point bending test. The subtotal nephrectomized rats presented higher serum concentrations of creatinine (0.63±0.09 vs. 0.39±0.04 mg/dL in the control group, p<0.001), urea nitrogen (74.4±11.3 vs. 40.0±2.6 mg/dL in
the control group, p<0.001), and kynurenine (2.50±0.77 vs. 1.51±0.34 µM/L in the control group, p<0.05). Kynurenine concentration correlated inversely with the biomechanical parameters of the tibia such as stiffness (r = -0.707, p<0.001), the resistance of the tibia diaphysis to deformation (r = -0.764, p<0.001) and fracture (r = -0.757, p<0.001). The observed associations between kynurenine concentrations and biomechanical properties of bone may develop new strategies for the diagnosis, treatment, and prevention of disturbances in bone structure in chronic kidney disease patients.References: [1] S, Moe, Kidney International, 2006;69:1945-1953.[2] D, Pawlak, Journal of Physiology and
Pharmacology, 2001;52:755-766. [3] J, Sviglerova, Physiological Research,
2010;59:81-88.[4] E, Holmes, Analytical Biochemistry, 1988;172:518-
525.Acknowledgements: The study was supported by National Science Centre Grant No. 2015/19/N/NZ4/01347.Disclosure of Interest: None DeclaredKeywords: Biomechanical testing, HPLC, Kynurenines
P01-001-055 Blood–brain barrier permeation: a microemulsion liquid chromatographic approach
X. Subirats 1,*, L. Muñoz-Pascual 1, M. Rosés 1
1 Department of Chemical Engineering and Analytical Chemistry, University of Barcelona, Barcelona, Spain
Content: Drugs designed to reach a pharmacological CNS target must be effectively transported across the blood-brain barrier (BBB), a thin monolayer

125www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSBiopharmaceutical and Biologics
of endothelial cells tightly attached together between the blood and the brain parenchyma. Because of the lipidic nature of the BBB, several physicochemical partition models have been studied as surrogates for the passive permeation of potential drug candidates across the BBB (octanol-water, alkane-water, PAMPA…). In the last years, biopartition chromatography (BPC) is gaining importance as noncellular systems for the estimation of biological properties in early stages of drug development. Microemulsions (ME) are suitable BPC mobile phases, because of their ease of formulation, stability and adjustability to a large number of compositions mimicking biological structures. In the present work, several microemulsion liquid chromatographic (MELC) systems have been characterized by means of the Abraham’s solvation parameter model, in order to assess their suitability as BBB permeability surrogates. In vivo data have been critically compared and evaluated, including rodent in situ brain perfusion determinations. The main parameters studied have been the passive blood-brain distribution (log BB or log PBBB, and log P0
BBB) and the passive permeability surface area product (PS, mL g-1 s-1) accounting for the rate of BBB penetration. In terms of similarity between BBB and MELC systems (dispersion forces arising from solute non-bonded electrons, dipolarity/polarizability, hydrogen-bond acidity and basicity, and molecular volume), the intrinsic passive permeability (log P0
BBB) is well correlated with the ME made of 3.3% SDS (w/v; surfactant) 0.8% heptane (w/v; oil phase) and 6.6% 1-butanol (w/v; co-surfactant) in 50 mM aqueous phosphate buffer, pH 7.4.References: J. Liu, J. Sun, Y. Wang, X. Liu, Y. Sun, H.
Xu, Z. He, J. Chromatogr. A, 1164 (2007) 129-138.
Disclosure of Interest: None DeclaredKeywords: Blood-Brain Barrier, Microemulsion Liquid Chromatography (MELC), Solvation Parameter Model
P01-001-056 - Characterization and Proteolytic Activity Studies of Botulinum Neurotoxin A in Pharmaceutical Products using High-resolution Tandem Mass Spectrometry
E. L. M. Wong*
Content: Botulinum neurotoxins (BoNT) are produced by the anaerobic bacterium Clostridium botulinum and are one of the most lethal known poisons (LD50 = 0.8 ug for a 70 kg human by inhalation). Despite its high lethal toxicity, BoNT have been used to treat spasms and other muscle problems. Most recently, BoNT serotype A (BoNT/A) gains its worldwide popularity in cosmetic surgery to prevent development of facial wrinkles. Clinical testing of BoNT/A is conventionally relied on in vivo mouse lethality assay, as stated in the pharmacopoeias, which requires huge amount of animals. In response to increased public pressure to the inhumane testing, sensitive replacement assays that can robustly measure BoNT/A in therapeutic or cosmetic products (containing ~0.1 ng or 50 MLD50 of BoNT/A) are of urgent need.
Identification of BoNT/A in pharmaceutical products using non-bioassay methods is always a challenge due to its complex structure, ultra-trace level and the interference from excipients (such as albumin and gelatin). This work reported

www.ISC2016.ie @isc2016 #isc2016 /isc2016126
POSTER ABSTRACTS Biopharmaceutical and Biologics
a mass spectrometry study on BoNT/A in pharmaceutical preparations using non-bioassay method. BoNT/A in pharmaceutical matrix was isolated using magnetic beads immunoprecipitation followed by on-beads digestion and was characterized by peptide mass finger printing using high-resolution tandem mass spectrometry coupled with ion mobility and Q-ToF in MSE mode. The activity of the toxin was confirmed by its proteolytic activity towards specially designed synthetic SNAP-25 substrate. The specific cleaved peptide fragments obtained from SNAP-25 substrate correlated well with its proteolytic activity in linear range from 0.05 – 100 MLD50/uL (r2 > 0.99). Proteolytic activities of BoNT/A in different excipient matrices were also investigated. The method has been validated across few brands of BoNT/A pharmaceutical injections commonly available in the Asia Pacific and was found to produce reliable results.Disclosure of Interest: None DeclaredKeywords: Botulinum neurotoxin A, High resolution mass spectrometry, pharmaceutical
P01-001-057 - High Resolution LC/MS Analysis of Therapeutic Oligonucleotides on a New Porous Polymer-based Reversed Phase Column
R. Van Ling*, S. Lin 1, J. Baek 1, J. Thayer 1, H. Wang
1, I. Birznieks 1, X. Liu 1
1Thermo Fisher Scientific, Sunnyvale, CA, United States
Content: Synthetic oligonucleotides (ONs) with different functionalities including antisense ONs, small interfering RNAs
(siRNAs), aptamers and immunostimulatory RNAs (isRNAs) are promising therapeutic agents due to their specificity, and well-established synthesis and modification technologies. Thorough characterization is required to demonstrate that efficacy and safety of these therapeutic ONs are ensured. This includes characterization of modifications to the base, sugar and backbone linkages, as these are commonly employed to decrease in vivo degradation and increase therapeutic efficacy. High performance LC/MS and LC/MS/MS are the preferred tools for these analyses, and are often used for more common ON purity assessments. Ion-pair reversed phase LC, with volatile mobile phase components, can be directly coupled to MS.
Here we introduce a new wide-pore polymeric reversed phase column and ion-pair methods for LC and LC/MS ON analysis. The new resin is stable to 110 °C and up to pH 14, and coupling the column to HRAM mass spectrometry successfully identified overlapping failure sequences, ONs with characteristic methylation, phosphorothioate diastereoisomers and 5’-phosphorylated DNAs. Short gradients allowed for the separation of mixed-base 21-mer and its n-1 failure sequence, and MS identified n-1 sequences where guanine, cytosine and thymine failed to couple during synthesis.Separation of diasteroisomers of several siRNA molecules with one or two phosphorothioate linkages was achieved, and MS analysis revealed identical mass values to within 9 ppm.
These results demonstrate the use of DNAPac RP and HRAM mass spectrometry for critical analyses of modified oligonucleotides.

127www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSBiopharmaceutical and Biologics
References: 1. Dias, N. et al. Molecular Cancer Therapeutics
(2002) 1, 347-355.2. Resnier, P. et al. Biomaterials (2013) 34, 6429-
6443.3. Jones, P.A. et al. Nature Reviews Genetics (2002)
3, 415-428.Disclosure of Interest: None DeclaredKeywords: Biopharmaceuticals, LC/MS, Oligonucleotides
P01-001-058 - Melatonin and its main metabolites in fish skin - quantitative determination by the liquid chromatography with FL and UV detection.
M. Gozdowska 1,*, E. Kulczykowska 1
1Institute of Oceanology of Polish Academy of Sciences, Sopot, Poland
Content: Melatonin (Mel; N-acetyl-5-methoxytryptamine) is present in many organisms: animals, plants and bacteria. In vertebrates, it acts as a regulator of circadian and seasonal rhythms, and immune and endocrine functions. Melatonin is also a potent endogenous antioxidant and free radical scavenger that reacts with several radicals generating various metabolites. Two of them are N1-acetyl-N2-formyl-5-methoxykynurenine (AFMK) and N1-acetyl-5-methoxykynurenine (AMK) which are even more potent than Mel itself.
Melatonin is metabolized through indolic and kynuric pathways to produce many biologically active metabolites including 6-hydroxymelatonin (6OH Mel), 5-methoxytryptamine (5MT), N1-acetyl-N2-formyl-5-methoxykynurenine (AFMK), N1-acetyl-5-methoxykynurenine (AMK).
In the skin, which is constantly exposed to unfavorable environmental factors, various defense mechanisms have been developed. Among them is the melatonergic system with melatonin and its metabolites (Slominski et al. 2007).
Herein, a simple, precise and selective assay for determination of Mel and its metabolites (6OH Mel, 5MT, AFMK and AMK) in fish skin is presented. The method is based on high performance liquid chromatography (HPLC-FL and DAD) analysis. The HPLC isocratic separation is achieved on a core-shall column (Kinetex, 2.6um, C18) preceded by metabolite enrichment on Strata-X on-line cartridge. The limit of detection/quantification for Mel, 6OH Mel, 5MT, AFMK and AMK are 0.2/1.2, 22/48, 18/38, 6/18 and 69/120 ng/mL, respectively. The assay is sensitive enough to determine Mel and its metabolites at physiological levels in fish skin.
Stability of all compounds examined here has been determined in different aqueous and organic solutions. An influence of selected factors as temperature, light, pH on stability of metabolites has also been tested.References: A Slominski et al., Trends Endocrin Met
19 (17-24) (2007)Acknowledgements: This study was supported by National Science Centre grant UMO-2012/07/B/NZ9/02144. All procedures were complied with Guidelines of the European Union Council (2010/63/EU) for the use and experimentation of laboratory animals and with the Guidelines of the local Ethics Committee on animal Experimentation.Disclosure of Interest: None DeclaredKeywords: melatonin, metabolites

www.ISC2016.ie @isc2016 #isc2016 /isc2016128
POSTER ABSTRACTS Biopharmaceutical and Biologics
P01-001-059 - An Analytical Platform for Early Stage Developability Assessment of Monoclonal Antibodies – How ‘Manufacturable’ is a mAb Candidate?
A. Trappe 1,*, R. Casey 1, J. Bones 1
1 Characterisation and Comparability Laboratory, National Institute of Bioprocessing Research & Training, Dublin, Ireland
Content: Development and commercialisation of new therapeutic monoclonal antibodies (mAbs) remains a long and expensive process. Although mAbs offer a higher degree of success due to their inherent specificity however many mAbs fail during development due to problems associated with expression, purification, formulation and stable storage of the mAb candidate1. Often, these stability issues are not detected until late in the drug discovery process, at which point the monetary costs and time required to remediate the problem may be prohibitive despite strong clinical indications of the mAb candidate. Therefore, the availability of highly informative analytical platforms to assess the ‘developability’ of a mAb candidate to derisk the scale up, manufacturing and formulation processes, at the earliest stage possible, is highly desirable.
We report the development of an analytical platform to derisk and reduce such late stage project attrition. CDR sequences from 6 monoclonal antibodies were cloned into a common human IgG1 framework and expressed using transient transfection in HEK 293 and CHO-S cells. Expressed mAbs were purified using a two-step process consisting of Protein A affinity chromatography and CaptoAdhere
polishing. Complete characterisation of the expressed mAbs was performed using top-down LC-MS for verification of experimental mass and PTM detection, Cation Exchange Chromatography (CEC), Hydrophobic Interaction Chromatography (HIC) and SEC-UV to determine the presence of acidic variants, oxidised mAbs and higher order structures. Bottom up peptide centred analysis was also performed to facilitate full sequence and PTM analysis using accurate mass LC-MS/MS. The expression of the mAbs using a common IgG1 framework under identical culture conditions facilitates an investigation into the role of the variable gene sequences on the modulation of product critical quality attributes.References: I. A Kaltashov, et al. Biotechnology
Advances 30 (1), 210-222.Disclosure of Interest: None DeclaredKeywords: LC-MS, Monoclonal Antibody
P01-001-060 - Comparison of Oenothein B And Sitosterols concentration in Epilobium Augustifolium Herb in Different Harvest Times
A. Gryszczyńska*, B. Opala 1, Z. Łowicki 1, A. Pietrowiak 1, M. Miklaś 1, M. Dreger 1, K. Wielgus 1, P. Mikołajczak 1 2
1 Institute Natural Fibres and Medicinal Plants, 2Department of Pharmacology, Poznań University of Medicinal Science, Poznań, Poland
Content: The Epilobium genus is rich in many bioactive compounds such as flavonol-3-O-glycosides, phenolic acids, steroids [1], oenothein B [2]. It should be mention that the following substances are also present: myricitrin, quercitrin, β-sitosterol [3], kaempferol [2].

129www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSBiopharmaceutical and Biologics
E. angustifolium seeds from Garden of Institute of Natural Fibers and Medicinal Plants as well as seeds bought in Rieger-Hofmann GmbH were used for induction of in vitro cultures. Seeds were sterilized with the use of 70% ethanol and ACE® solution (2:1) and washed three times in sterile water. Multi-shoots cultures were initiated from root’s explants according to Turker [4] in slight modification: MS [5] medium supplemented with BAP (0.1 mg/L), IAA (0.5 mg/L), vitamin C (0.1 g/L) and casein hydrolysate (0.5 g/L). Shoots were rooted on ½ MS medium with IAA (0.5 mg/L) with vitamin C. Rooted plants were transferred into soil substrate and perlite and acclimatized in the closed tunnels in the greenhouse conditions, then hardened and transferred into field. Raw material (herba Epilobii) was harvested in June (blooming phase) and in September.
HPLC-DAD (Agilent) was used to determinate of active compounds. A chromatography separation of sitosteroles were obtained on a column Zorbax Eclipse XDB-C8 (Agilent), 5µm x 4.6 x 150 mm. β-sitosterol, stigmasterol and cempesterol were detected at 208 nm. H2O: H3PO4 : THF (80:15:8 V/V) were used as eluents. Meanwhile, oenotheie B was separated on LiChrospher 100 RP-18e, 5µm x 4 x250 mm (Merck).Oenothein B was detected on 263 nm. To elution of active compound, mix of 2,5% CH3COOH and 2,5% CH3COOH:ACN (2:8 V/V) was used. Peaks were identified by the addition of standards.References: [1] Bazylko A. et al.:J Chromatogr A 2007,1173,146-
150[2] Kujawski R. et al. Herba Polonica 2011,57(4),33-44[3] Battinelli L. et al.: Il Farmaco 2001,56,345-348[4] Turker Aue et al.. Acta Physiol Plant 2008
30(4):421-426[5] Murashige T, Skoog F. Physiol Plantarum 1962
15(3):473-497Acknowledgements: The work was supported by the Polish National Centre for Research and Development, grant no PBS2/A8/23/2013.Disclosure of Interest: None DeclaredKeywords: EPILOBIUM AUGUSTIFOLIUM HERB, OENOTHEIN B
P01-001-061 - LC-MS proteomics workflow for the identification of Bioprocess Markers for Cell Stress and Cell Death during Perfusion Culture
S. Albrecht 1,*, C. Kaisermayer 2, A. Lindeberg 2, J. Bones 1
1 Characterisation and Comparability Laboratory, NIBRT, Dublin, 2BioMarin International Limited, Ringaskiddy, Cork, Ireland
Content: The maintenance of cell mass during perfusion culture is essential to achieve a steady production state during the manufacturing of biopharmaceuticals. Given the dynamic nature of perfusion systems reliable markers are required for monitoring cell death and general cell physiology1. However, the measurement of lactate-dehydrogenase (LDH) activity which is commonly used for the assessment of culture viability has limited reproducibility as the enzymatic activity does not always correlate with the enzyme expression levels. More informative cell death markers are therefore required.
Aiming at the identification of improved markers in spent media from perfusion culture we set out to investigate the cellular

www.ISC2016.ie @isc2016 #isc2016 /isc2016130
POSTER ABSTRACTS Biopharmaceutical and Biologics
biology of cell death. In the first instance a lab-scale model system is used in which apoptosis and necrosis is induced to CHO-K1 cultures in suspension. Tryptic digeststs of supernatant and cellular proteins are subjected to an LC-MS-based platform for screening and targeted proteomics analysis. The proteomics workflow includes high-pH low-pH reversed phase 2D-nanoLC-MSe for proteomics screening after addition of Hi3 for peptide quantitation2 followed by targeted QQQ-MS analysis of candidate viability markers.
Results from a model-apoptosis study using CHO-K1 are presented which demonstrate the suitability of the analytical platform for the in-depth identification and quantification of cellular and supernatant proteins related to cell death.References: 1. M.F. Clincke, C. Mölleryd, Y. Zhang, E. Lindskog,
K. Walsh, V. Chotteau, Biotechnol Prog, 2013, 29, 754–767.
2. A. Farrell, S. Mittermayr, B. Morrissey, N. Mc Loughlin, N. Navas Iglesias, I. W. Marison, J. Bones, Anal Chem, 2015, 87, 9186-9193.
Disclosure of Interest: None DeclaredKeywords: MRM, Nano-liquid chromatography-QTOF MS, Proteomics
P01-001-062 - Complete Characterisation of Monoclonal Antibody Following Production in Chinese Hamster Ovary Cells Under Conditions of Altered Bioprocessing
A. Farrell*, K. Scheffler, K. Cook, J. Bones
Content: Chinese hamster ovary (CHO) cell lines are an important expression system for therapeutic monoclonal antibodies (mAb) produced in mammalian cell lines. In this study various protein characterisation and comparability techniques are used to evaluate the impact of alteration of bioprocessing parameters (pH, temperature, dissolved oxygen) on the critical quality attributes of a humanised mAb secreted from CHO DP12 cell lines. Intact protein analysis of the mAb using liquid chromatography techniques were completed including determination of oxidised species using hydrophobic interaction chromatography; charge variant analysis using ion exchange chromatography and evaluation of aggregation profile by size-exclusion chromatography. Comparative analysis of N-glycans attached to the mAb was performed using 2AA-stable isotope labelling and subsequent UPLC-fluorescence-mass spectrometry analysis using a Waters Xevo G2 QToF. In addition, high resolution accurate mass LC-MS/MS based characterisation was employed to assess changes in the recombinant protein including peptide mapping of reduced and non-reduced mAb, and analysis of IdeS-digested mAb, using middle down LC-MS analysis on a Thermo Fisher Scientific Q Exactive HF hybrid quadrupole-Orbitrap mass spectrometer. Finally a Waters hydrogen deuterium exchange platform coupled to a Waters Synapt G2 HDMS mass spectrometer was used to evaluate conformation changes to the mAb in response to alterations in upstream bioprocessing conditions. Combined, these analyses revealed interesting insights into the effect of production of mAb in cell culture operated at the outer limits of the design space when compared to standard cell culture conditions.

131www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSBiopharmaceutical and Biologics
References: Millan Martin, S., et al., Comparative analysis of monoclonal antibody N-glycosylation using stable isotope lablling and UPLC-fluorescence-MS. Analyst, 2015. 140(5):p. 1442-7.
Acknowledgements: This research was supported by Science Foundation Ireland under grant reference 11/SIRG/B107.The authors gratefully acknowledge the contribution of the members of NIBRT Characterisation & Comparability Laboratory, NIBRT Training group, Dublin City University and Thermo Fisher Scientific.Disclosure of Interest: None DeclaredKeywords: Bioprocessing, Monoclonal Antibody, Protein Characterisation
P01-001-063 - Experimental study of arylcobalamin photolysis process
G. Andryianau 1,*, G. Rotko 1, A. Kumorkiewicz 1, S. Wybraniec 1
1 Faculty of Engineering and Chemical Technology, Cracow University of Technology, Cracow, Poland
Content: Intensive DNA replication requires large amounts of nucleotides and cobalamin-dependent enzyme system is responsible for uracil methylation reaction providing to thymidine. Therefore, tumors require abnormally high quantities of vitamin B12 [1]. Only few organisms can synthesize corrinoid structures, so during the evolution highly-specialized mechanisms of cobalamin management have been developed.
Axial-modified vitamin B12 aryl derivatives are considered to be promising class of drugs carriers. The application of irradiation for selective drug release directly inside the tumor cells is one of the conceptions providing to decrease of the cancer treatment toxicity. This is the reason of importance of the study of Co–CAr bond photodissociation process.
Here we report the study of arylcobalamin photolysis process. The solutions of substituted arylcobalamins were exposed to monochromatic light (531,6 nm) and photolysis progress was monitored by UV-Vis. For analysis of “cobalamin core” photodissociation products HPLC-DAD-MS system was applied. Volatile compounds were analyzed using GC-FID. Numerous photochemical experiments showed that there is no strong dependence between Hammet constant of the substituents in aryl moiety and photolysis rate constants. Aquacobalamin is the main product of photolysis in all cases. Additionally, we showed that the mixtures of appropriate benzenes and phenols are formed during the photochemical reaction.
Radical mechanism of arylcobalamins photolysis was proposed. Homolytic Co–CAr bond cleavage probably is the first stage of the process. Recombination of aryl radicals with hydrogen atoms from the environment and cob(II)alamin oxidation is the second one. The reaction of aryl radical with dioxygen followed by recombination with cob(II)alamin may be considered as concurrent reaction. Arylperoxocobalamin product may decompose to hydroxocobalamin and appropriate phenol.References: [1] Z. Schneider, A. Stroinski,
Comprehensive B-12, de Gruyter: Berlin, 1987Disclosure of Interest: None DeclaredKeywords: Arylcobalamins, Photolysis, Vitamin B12 derivatives

www.ISC2016.ie @isc2016 #isc2016 /isc2016132
POSTER ABSTRACTS Biopharmaceutical and Biologics
P01-001-064 - Innovator and biosimilar comparison using HPLC coupled to Time-of-Flight Mass spectrometry: A peptide mapping study
R. Gudihal 1, S. Krieger 2, U. Huber 2,*
1 Agilent Technologies India Pvt Ltd, Bangalore, India, 2 Agilent Technologies, Waldbronn, Germany
Content: Monoclonal antibodies (mAbs) are one of the rapidly growing classes of biomolecules for treatment of various cancers. Biosimilar mAbs, which are the replica of the licensed innovator product in the market are also gaining lot of attention due to patient expiry(1). LC/MS characterization to show comparability between innovator and biosimilar, is proving to be vital. Amino acid sequence similarity is of crucial importance between biosimilar mAbs with the innovators. Hence peptide mapping is one of the critical steps to show similarity in the sequence and modifications between the innovator and biosimilar pair. Peptide mapping of innovator and biosimilar mAbs was generated using trypsin protease followed by analysis using HPLC system coupled to quadrupole time-of-flight (Q-TOF) Mass Spectrometry. Mass spectrum was acquired in positive ion mode with both in MS only mode and in MS/MS mode.
Similar total ion chromatogram profile for both biosimilar and innovator mAbs was observed for Peptide mapping using LCMS. Minor differences was observed between the innovator and biosimilar peptide maps. Differential Analysis of peaks showed a peak around 14.4 min is prominently found in biosimilar, but not in innovator sample. This peak corresponded to a C- terminal sequence (SLSLSPGK) which was further confirmed
by MS/MS. The higher abundance of this C-terminal peptide was assigned to charge variants (lysine addition, basic variants) seen in the biosimilar mAb(2). Similarly in the innovator peptide map, the C-terminal peptide without lysine (SLSLSPG) was identified with higher abundance around 18.7 min. Peptide maps were also used to quantify the extent of oxidation(3)and deamidation as these are the two most commonly occurring modifications seen during storage, formulation and sample handling. References: 1. Biosimilar, bio better and next generation
therapeutic antibodies, mAbs. 2011 Mar-Apr; 3(2): 107
2. Agilent publication number 5991-5557EN3. Agilent publication number 5990-8769ENDisclosure of Interest: None DeclaredKeywords: monoclonal antibodies, Peptide mapping, Time of Flight
P01-001-066 - Quantitative examination of extracellular profile of neurotransmitters in median raphe nuclei associated with optogenic photo stimulation
M. Baranyi*, D. Zelena 1, B. Sperlagh 2
1 Institute of Experimental Medicine, Hungarian Academy of Sciences, Budapest, Hungary,
2 Molecular Pharmcology, Institute of Experimental Medicine, Hungarian Academy of Sciences, Budapest, Hungary
Content: The median raphe nucleus (MRN) is a major 5-hydroxytriptamine (5-HT) containing cell group of the brainstem it can control the activity of ascending serotonergic systems and the release of

133www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSBiopharmaceutical and Biologics
5-HT in terminal areas of the forebrain. Several intrinsic and extrinsic factors regulate 5-HT release in MRN neurons and their malfunctions may lead pathologically altered behavior. In vivo microdialysis technique was used for sampling of MRN of anaesthetized transgenic mice that were microinjected with light-sensitive mutant adenoviral vector (rAAV2/9-synapsin-ChR2/eGFP). Blue light illumination using 20 Hz frequencies and theta burst stimulations protocol at 5 min interval was used. Depolarizing effect of KCl in same interval used as positive control. Dynamic changes of extracellular levels of monoamine and amino acid neurotransmitters were analyzed using two column switching methods [1] with same HPLC instrument. Speed of analysis of monoamines and their metabolites was predominated by the retention behavior of norepinephrine, the nonideal behavior of matrix components, and the loss in signal of 5-HT. This method was optimized to meet the requirements for detection sensitivity and minimizing the size of collected fractions, which determines temporal resolution in microdialysis. The amino acid neurotransmitters glutamate (Glu) and γ-aminobutyric acid (GABA) were analyzed after dansyl derivatization procedure. The detection limit for Glu and GABA was 10 nmol/L (0.10 fmol in 25 µL); the monoamine neurotransmitters had a detection limit between 55 and 95 pmol/L (0.15-0.45 fmol in 25 µL) in standard solutions. This is the first study to use LC electrochemical and fluorescence detection to analyze monoamines and amino acid neurotransmitters in the same microdialysates from mouse MRN.References: [1] M. Baranyi, E. Milusheva, E. S. Vizi
and B. Sperlagh, Journal of Chromatography A 2006, 1120, 13-20;
Disclosure of Interest: None DeclaredKeywords: Electrochemical and fluorescence detection, median raphe nucleus, optogenetics
P01-001-067 - Non-Toxic Antibody Drug Conjugate Mimic Characterization to Enable LC-MS Method Development Without RiskA. Fridström*, K. Ray 1, J. Turner 1, N. Caffarelli 2, J. Dapron 1, B. Gau 1
1 Millipore Sigma, St Louis, 2Millipore Sigma, St.Louis, United States
Content: Characterizing the distribution of drugs per antibody is paramount to understanding ADC efficacy, process control, and to obtaining regulatory approval. However, a significant roadblock to the development of ADC analysis methods is their hazardous nature. Here we show how analytical methods, in particular MS-based methods, may be optimized using a non-toxic surrogate of the ADC, or “ADC-mimic”, with highly similar physicochemical properties to commercial cysteine-conjugated ADCs.
Methods: A cysteine-fluorophore conjugate was prepared by coupling purified monoclonal antibody to dansyl-cadaverine fluorophore via an LC-SMCC crosslinker in a two-step reaction. Samples were deglycosylated with PNGase F and analyzed by native SEC-MS. Reduced SEC-MS was accomplished after further DTT treatment and less energetic source conditions. For peptide mapping, samples were reduced, alkylated, and trypsin proteolyzed prior to injection onto a Thermo QE-Plus LC-MS/MS platform with a 75 µm x 25 cm PicoFrit column packed with 2.7 µm, 160Å, fused-core C18 particles.

www.ISC2016.ie @isc2016 #isc2016 /isc2016134
POSTER ABSTRACTS Biopharmaceutical and Biologics
ADC-mimic samples were characterized by peptide mapping and SEC-MS analyses in conjunction with a commercial cysteine-conjugated ADC Adcetris (Seattle Genetics). The DAR determined by reduced SEC-MS of the ADC-mimic heavy and light chains matched the native SEC-MS of the full ADC-mimic, and comports with spectrophotometric DAR measurements. Tryptic peptide analysis showed 95% coverage of the mimic. The Cys-conjugated dansyl-cadaverine drug linker imparts significant hydrophobicity to peptides, comparable to the hydrophobicity shift of Cys-linked MMAE in the commercial ADC. Finally, reversed phase protein LC-MS analysis of the fully reduced and IdeS-treated ADC-mimic is compared to Adcetris in terms of chromatography and spectrometry characteristics.References: To be added
Disclosure of Interest: None DeclaredKeywords: Antibody Drug Conjugate, cytotoxicity, LC-MS
P01-001-068 - Should I stay or should I go? - Antioxidant efficiency of Beech (Fagus sylvatica L.) leaf polyphenols in terms of adaptation to climate change
T. Hofmann*, E. Nebehaj 1, L. Albert 1
1 University of West Hungary, Faculty of Forestry, Institute of Chemistry, Sopron, Hungary
Content: Temperate zone forests are heavily affected by climate change. Certain species are especially vulnerable and threatened by retreat or possible extinction. One of these species is beech (Fagus sylvatica L.), common
across Europe with high economic and ecological value. Future task for forestry is to find varieties, which can adapt to changing climatic conditions due to their efficient chemical defense mechanisms. Antioxidant (AO) leaf polyphenols (LPP) represent one of these defense mechanisms [1]. By understanding the AO efficiency of LPPs, varieties with a high potential to withstand future climatic anomalies and to grow into healthy forests in Hungary, can be selected. In this work the HPLC-MS/MS identification and quantitative evaluation of 56 LPP compounds from beech leaves was carried out. The AO capacity was assayed by FRAP and ABTS methods. Investigated beech varieties (Pidkamin/UA, Farchau/D, Torup/S, Bánokszentgyörgy, Magyaregregy/HU, Gråsten/DK) were grown at Bucsuta/H in the framework of an IUFRO project. Correlation analysis was performed between concentrations of LPP compounds, AO capacities, Ellenberg’s climate quotient (EQ) and average stem diameter. Results: most efficient LPPs ((+)-catechin, (-)-epicatechin, coniferin derivative, procyanidins (3), quercetin glycosides(3)) were found in the highest levels in the leaves of the most vulnerable varieties (Gråsten, Torup), with the poorest growing results. Highest FRAP and ABTS levels could also be measured in these samples. The varieties originating from warmer regions (high EQ) had lower concentations of the most powerful LPPs, indicating good adaptability. These LPPs could be markers of climatic adaptation and used for future research of selecting varieties of beech afforestation in Hungary. Research was supported by the János Bolyai Research Scholarship of the Hungarian

135www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSBiopharmaceutical and Biologics
Academy of Sciences and by the VKSZ_12-1-2013-0034 Agrárklíma.2 project.References: [1] Cadahía E et al. Phytochemical
Analysis 26, 171-182 (2015)Disclosure of Interest: None DeclaredKeywords: Beech leaf polyphenols, Climate change, HPLC-MS/MS

www.ISC2016.ie @isc2016 #isc2016 /isc2016136
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
Column Technology and New Materials as Separation Media
P01-001-007 - New Approaches to Stationary Phases on Superficially Porous Particles
L. A. Colon*, Z. Xue, A. C. Borges-Muñoz, J. Ezzo, J. Velez-González, K. M. Tirado-González
Content: The ever-evolving area of column technology continues to propel new advances in HPLC, enabling fast, efficient, and selective separations of complex sample mixtures. Our research group is investigating new approaches to column chemistries for use as stationary phases for HPLC. In one example, we have generated a thin layer of nanodiamonds on silica nanoparticles and are exploring the chromatographic behavior of these materials. We have also attached benzyl groups at the surface of the superficially porous silica material which have been carbonized in placed to form a dark carbonaceous layer on the silica surface. These materials have been characterized spectroscopically, packed into columns, and tested chromatographically. This presentation will focus on the silica modification approaches to produce carbonaceous surface on the silica particles, their characterization, and the preliminary chromatographic performance of the materials under HPLC conditions.Disclosure of Interest: None DeclaredKeywords: carbonaceous stationary phases, superficially porous particles
P01-001-021 - Bioinspired and Biocompatible Nanogels: Self-Tuning Fluids to Process, Sort, Select, and Characterize Biopharmaceutical Glycosylation
L. Holland*
Content: Smart nanomaterials are an innovative alternative to support flexible and reprogrammable sample processing and tunable microscale bioseparations. The morphology of phospholipid nanogels is temperature dependent, rendering the viscosity thermally responsive. These material features are leveraged to integrate sample processing and complex separations in a single platform. By harnessing a smart material, the on-board fluid is programmed to concentrate, process, steer, and then separate targeted analyte. The reversible nanogel can be directed to different microfluidic channels using thermoelectric modules to spatially control the temperature in a microfluidic chip with a 2-second switching time. This is significant to chemical separations because generic microscale channels generate a sophisticated multifunctional device that can be programmed, erased, and reprogrammed repeatedly. These materials extend the range and precision of biomolecular size discrimination and support separation-based enzyme and lectin processing. The nanophase is key to this approach because it preserves the protein activity, supports catalytic processing using nanoliter volumes of enzymes and enhances electrophoretic separation of the products. The analyte resolution of biomolecules separated by these nanophases are exceptionally high, yielding separation efficiency up

137www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
to 2 million theoretical plates. A critical advantage of self-assembled nanophases for efficient separations of biomolecules is the ease with which they are modified and then introduced, patterned, and replaced in the capillary. The applicability of this technology will be demonstrated with glycan separations relevant to next generation pharmaceuticals.Acknowledgements: This work is supported by the National Institutes of Health through R01GM114330Disclosure of Interest: None DeclaredKeywords: antibody therapeutics, glycosylation, microscale sequencing
P01-001-028 - Column Selectivity Choices in HPLC Method Development
S. Luke 1,*, W. Long 2, A. Mack 2, J. Link 2
1 Agilent Technologies, Church Stretton, United Kingdom, 2 Agilent Technologies, Wilmington, United States
Content: Modern superficially porous particle (SPP) based columns were introduced in 2006, and since then, many bonded phases have been introduced from a variatey of column manufacturers, allowing easy transition from totally porous to superficially porous particles. These columns deliver high efficiency as well as very long lifetimes at a fraction of the pressure of totally porous sub-2 micron particles.
Short highly efficient SPP columns can be used to replace a longer 5 micron columns with the resulting separation often having more resolution and efficiency while requiring significantly less time. End-capped C18 SPP columns such are an excellent first choice in separations because they typically
result in successful separations. However, additional bonded phases are available if additional options are needed
When performing method development, selectivity is the most powerful parameter to adjust, influencing resolution far more than efficiency or rention factor, and can be driven by column bonded phase changes, or modifications in the mobile phase composition or pH.
In many cases an alternative column to a C18 should be chosen early in the method development cycle based on the type of compounds to be analyzed. For example separations involving closely related compounds such as isomers can be quickly separated using a pentafluorophenyl (PFP) column while highly polar compounds can be retained and separated using an Aq or HILIC column.
Examples of successful and unsuccessful separations will be shown as well as a discussion of using Tanaka and Hydrophobic Subtraction models to help explain the differences in bonded phase performance.Disclosure of Interest: None DeclaredKeywords: Column characterization and comparison, HPLC, Selectivity

www.ISC2016.ie @isc2016 #isc2016 /isc2016138
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
P01-001-029 - Method Transfer and Column Scalability with Superficially Porous Particles
S. Luke 1,*, A. Mack 2, W. Long 2, J. Link 2
1 Agilent Technologies, Church Stretton, United Kingdom, 2 Agilent Technologies, Wilmington, United States
Content: Superficially porous particles offer improved efficiency and performance over similarly sized traditional totally porous particles. This is primarily due to a shorter mass transfer distance and substantially narrower particle size distribution of the particles in the column. Higher efficiency leads to improved resolution and possible time savings with superficially porous particles, hence their growing popularity for HPLC analyses. Columns using superficially porous particles are currently available in a a number of particle sizes, pore sizes and stationary phase chemistries to meet most analysts’ needs. This work will address method transfer and scalability from totally porous to superficially porous particles, as well as among different varieties of superficially porous particles. It will also discuss when to use which particle and column configurations. Overcoming common barriers of method transfer with high efficiency columns, including instrument configuration, method settings, and proper connections will also be addressed. Example applications are transferred from traditional totally porous particle HPLC columns to more modern superficially porous particle columns. Methods are geometrically scaled, as column volumes are adjusted, with the intent to preserve chromatographic integrity. As methods are adjusted to higher efficiency columns with smaller column volumes, instrument hardware and software is also appropriately
adjusted. Some example applications include compendial USP methods, which are transferred to superficially porous particle HPLC columns from their prescribed totally porous particle columns. Results are compared against the permissible adjustments found in USP General Chapter 621.Disclosure of Interest: None DeclaredKeywords: Core-shell particles, HPLC, Method Transfer
P01-001-031 - Laser fabricated cyclic olefin polymer-based ultra thin layer chromatography platforms
R. McCann 1 2 3,*, K. Bagga 2 3, R. Groarke 2 3, A. Stalcup 2 4, M. Vázquez 2 3 4, D. Brabazon 1 2 3
1 National Centre for Plasma Science and Technology, 2 Irish Separation Science Cluster, 3 Advanced Processing Technology Research Centre, 4 School of Chemical Sciences, Dublin City University,
Dublin, Ireland
Content: Although less common in analytical environments than their liquid chromatographic counterparts, planar chromatographic techniques such as thin-layer chromatography (TLC) remains widely used. More recently, ultra-thin layer chromatography (UTLC) has become a topic of interest in planar chromatography. The thinner stationary layers (<100 µm) used in UTLC result in shorter migration distances and quicker separations, as well as lower mobile phase consumption, when compared with conventional TLC [1]. To date, advances in UTLC have focused on metal-oxide or carbonaceous stationary phase sorbent layers deposited via advanced manufacturing techniques such as atomic layer deposition [2] and

139www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
low-pressure chemical vapour deposition [3] or through the use of sol-gel deposition [4]. While these methods are successful in the fabrication of UTLC platforms, they are unsuited for low-cost device fabrication. In contrast, laser processing of polymers is well suited towards lower-cost fabrication and rapid device prototyping. Polymers offer both cheap separation media and unique functionality however, to date, only polymer electrospinning has been examined as a process for UTLC plate fabrication [5].
Here we present a novel approach to the fabrication of UTLC platforms utilising a picosecond pulsed infrared laser to create networked microchannels (which behaves as the functional stationary phase layer) directly fabricated on a 188 µm thick cyclic olefin polymer (COP) polymer substrate. Structural characterisation was done using scanning electron microscopy and 3D optical profilometry. The produced UTLC plates were applied to a range of organic dye separations demonstrating the capabilities of these laser fabricated UTLC plates.References: 1. S. K. Poole, J. Chromatogr. A 1218 (2648–60)
(2011).2. J. Wannenmacher, J. Chromatogr. A 1318 (234–
43) (2013).3. J. Song, Adv. Funct. Mater. 21 (1132–9) (2011).4. H. E. Hauck, Chromatographia 57 (S313–5) (2003).5. T. Lu, J. Chromatogr. B 912 (98–104) (2013).Disclosure of Interest: None DeclaredKeywords: cyclic olefin polymer, laser fabrication, ultra-thin layer chromatography
P01-001-036 - Characterization of zwitterionic HILIC columns by means of the Abraham solvation parameter modelM. Roses 1, X. Subirats 1,*, M. Ferrer 1
1 Analytical Chemistry, Universitat de Barcelona, Barcelona, Spain
Content: Nowadays hydrophilic interaction chromatography (HILIC) is a continuously growing technique in applications related to the separation of polar or ionic biological molecules of medical interest. In present work, with the aim of better understanding the fundamentals of HILIC retention, three zwitterionic columns have been studied (SeQuant® ZIC®-pHILIC, ZIC®-cHILIC, and ZIC®-HILIC), which present differences in the nature of the stationary phase or in its support.
The columns have been characterized according to the Abraham’s solvation parameter model at different pH values with different mobile phase compositions with acetonitrile (HILIC-MeCN) and methanol (HILIC-MeOH) modifiers. This solvation model describes the chromatographic retention in a particular system according to the sum of specific interactions related to dispersion forces arising from solute non-bonded electrons, dipolarity/polarizability, hydrogen-bond acidity and basicity, and molecular volume.
The results obtained have been compared to those of common reversed phase (RP) and immobilized artificial membranes (IAM) columns.
An increase in solute volume strongly increases retention in RP and IAM, but strongly decreases retention in HILIC-MeCN and HILIC-MeOH.

www.ISC2016.ie @isc2016 #isc2016 /isc2016140
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
An increase in solute hydrogen bond basicity strongly increases retention in HILIC-MeCN, but decreases retention in RP and IAM (strongly) and in HILIC-MeOH (moderately).
An increase in solute hydrogen bond acidity produces a small increase of retention in HILIC-MeCN, HILIC-MeOH and IAM, and a small decrease in RP.
An increase in solute dipolarity produces a moderate increase of retention in HILIC-MeOH, almost no effect in HILIC-MeCN and a small decrease of retention in RP and IAM.
An increase in solute polarizability leads to a weak or moderate increase of retention in all columns.References: E. Lázaro, C. Ràfols, M.H. Abraham, M.
Rosés, J. Med. Chem. 49 (2006) 4861.Disclosure of Interest: None DeclaredKeywords: Column characterization and comparison, HILIC, Solvation Parameter Model
P01-001-051 - Switchable Chromatography using Carbon Dioxide Modified Solvents
X. Yuan 1,*, E. G. Kim 1, P. G. Jessop 1, R. D. Oleschuk 1
1 Department of Chemistry, Queen’s University, Kingston, Canada
Content: In this work, we collaborate with chemists in the green chemistry field, aiming at the development of CO2-responsive separation media and aqueous/organic mobile phases.
During the past decade, there has been significant interest in using CO2 as a
dynamic chemical stimulus. Researchers demonstrated the extraordinary ability of CO2, for the reversible hydrophobicity switch of materials such as solvents, surfactants, or solutes [1-3]. An example of the CO2-switchable chemistry is that, tertiary amine groups can be protonated, becoming hydrophilic, when CO2 is dissolved, providing protons and bicarbonate ions. Removing CO2 by heating or purging with nitrogen/argon leads to the deprotonation of tertiary amine groups switching them to a hydrophobic form.
To investigate the chromatographic behavior of CO2 switchable groups, three chromatographic columns were examined using aromatic analytes with neutral or ionizable substituents. When aqueous mobile phase is purged with CO2, at 1 atm, the retention of neutral compounds showed a significant decrease, resulting from a hydrophobicity switch of the stationary phase. Furthermore, the retention of ionizable analytes showed different selectivity.
By comparing the differential effects of organic solvent composition, CO2 concentration, and pH on the interaction between analytes and the switchable stationary phase, we demonstrate the capability of CO2 in the presence of water as an efficient stimulus for chemical separation. This may significantly reduce the amount of organic solvent required to separate compounds of interest and will provide users with another dimension of chromatographic control.References: (1) Boniface, K. J.; Dykeman, R. R.; Cormier, A.; Wang,
H.-B.; Mercer, S. M.; Liu, G.; Cunningham, M. F.; Jessop, P. G. Green Chem. 2016, 18, 208-213.
(2) Scott, L. M.; Robert, T.; Harjani, J. R.; Jessop, P. G. RSC Advances 2012, 2, 4925-4925.
(3) Jessop, P. G.; Heldebrant, D. J.; Li, X.; Eckert, C.

141www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
A.; Liotta, C. L.Nature 2005, 436, 1102.Disclosure of Interest: None DeclaredKeywords: CO2-switchable Chromatography
P01-001-069 - Analysis of primary proteic amino acids using serially-coupled RPLC columns
J. R. Torres-Lapasió*, T. Alvarez-Segura 1, C. Camacho-Molinero 1, M. C. García-Alvarez-Coque 1
1 Analytical Chemistry, University of Valencia, Burjassot (Valencia), Spain
Content: Along the years, a lot of effort has been invested in the development of reversed-phase liquid chromatographic (RPLC) methods to analyse amino acids. Some reports deal with the HPLC separation of the o-phthalaldehyde/N-acetyl-L-cysteine derivatives of the 19 primary proteic amino acids using gradient elution. These derivatives allow sensitive procedures for amino acids using fluorescence detection. However, the amino acid derivatives are frequently resolved only in excessively long analysis times, even using multi-linear or multi-isocratic gradients. If the analysis time is reduced, significant overlapping occurs for several compounds. Single columns in RPLC are thus insufficient to solve complex separations, due to their limited functionality.
A relatively simple solution to resolve the amino acid derivatives in sufficiently short analysis times is the serial connection of two or more columns with different stationary phases. Each combination performs as a totally new column. The full potential of this approach requires, however, the development of a powerful interpretive optimisation strategy. For this purpose, a unique predictor system was developed
in our laboratory, which implemented the different strategies with single and serially coupled columns, in both isocratic [1], and gradient elution [2]. An outstanding agreement has been found in all cases between experimental and predicted chromatograms.
In this study, we considered five types of stationary phases with different selectivity (C18, pentafluorophenyl-C18, C4, cyano and phenyl), considering the nature, length and order of the coupled columns, and multi-linear gradient profiles. The mixture of amino acid derivatives was successfully resolved in practical times, in spite of the extremely poor performance obtained when the columns were used isolately.References: [1] C. Ortiz-Bolsico et al., J. Chromatogr. A, 1281, 94
(2013). [2] C. Ortiz-Bolsico et al., J. Chromatogr. A, 1373, 51
(2014).Disclosure of Interest: None DeclaredKeywords: Amino acids, Analysis time, Serially coupled columns
P01-001-070 - Study of a new Chiral Bioactive and Cholesteric GC Column
O. Ferroukhi*, N. Mermat 1, M. H. Guermouche 1
1 Faculte De Chimie, University Of Sciences And
Technologie Of Algiers Algeria, Algiers, Algeria
Content: A new chiral bioactive mesogenic (CBM) material used as stationary phase in capillary GC has been tested. Ibuprofen has been used as chiral selector covalently immobilised on a liquid crystal (LC) cholesteric material.

www.ISC2016.ie @isc2016 #isc2016 /isc2016142
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
It was demonstrated that the linked group influence significantly the specific intermolecular interactions between the LC used as astationary phase and a chromatographed analyte [1,2].
Due to the enantioselective recognition, the reactivity of the bioactive part of the phase and the geometrical discrimination induced by the presence of the liquid crystal state, the separation of this column can be improved compared to conventional phases.
Synthesis and characterization of the new CBM stationary phase has been realized. The filling of capillary column by the dynamic method was carried out. The polarity test was performed to select the chemical families to separate.
In order to determine the separation capabilities of the material, thermal and analytical studies of the new column by inverse GC were carried out by injecting several compounds of different chemical families. The results obtained are satisfactory witch highly encourage the pursuing of the work in order to analyze more complexes mixtures of enantiomers to understand the phenomena governing the interactions necessary for the separation of molecules and evaluate discrimination against couples of enantiomers.References: [1] O Ferroukhi , A Addoun , S Guermouche , J P
Bayle , M H Guermouche, Chromatographia 78 (1251-1261) (2015).
[2] A Addoun , O Ferroukhi , M Dahmane , S Guermouche , J P Bayle , M H Guermouche, Chromatographia 79 (1367-1377) (2014).
Disclosure of Interest: None DeclaredKeywords: Chiral stationary phase , Gas Chromatography, liquid crystal
P01-001-071 - Possibilities of current sub-3 µm SEC columns for the characterization of biopharmaceuticals aggregationA. GOYON 1,*, J.-L. VEUTHEY 1, A. BECK 2, D. GUILLARME 1, S. FEKETE 1
1 School of Pharmaceutical Sciences, University of Geneva, Geneva, Switzerland,
2 Center of Immunology Pierre Fabre, Saint-Julien-en-Genevois, France
Content: Monoclonal antibodies (mAbs) and antibody-drug conjugates (ADCs) have attracted some attention in recent years as therapeutic proteins with a high specificity and affinity for target antigens [1]. However, the complexity of protein structure might cause potential modifications which need to be characterized. Proteins aggregation is a phenomenon that may reduce drug efficacy, and could potentially have detrimental impact on drug safety. Quantification of proteins aggregation is therefore an important issue in the pharmaceutical industry widely assessed by size exclusion chromatography (SEC) [2]. While conventional SEC is typically performed using 6-8 mm id × 300 mm columns packed with 5–10 µm particles operated at relatively low column pressures, a new class of SEC columns packed with sub-3 µm particles have been recently launched by several suppliers. The introduction of ultra-high pressure size-exclusion chromatography columns offers a new opportunity to improve both the resolution and throughput of SEC analysis. The aim of this study was to perform an evaluation of the practical possibilities and limitations of four new commercially available sub-3 µm SEC columns. Measurements were performed with several different model proteins, mAbs and ADCs to assess the achievable plate

143www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
counts, analysis time and resolution. Baseline resolution of the monomer and dimer peaks was obtained at about Rs = 2.0 within less than ten minutes. The inertness of the stationary phases was also systematically evaluated and showed the predominance of secondary hydrophobic interactions over secondary ionic interactions. Plate height (H) versus mobile phase linear velocity (ui) curves, kinetic plots and calibration curves were also constructed and compared. References: [1] AM Scott, JD Wolchok, LJ Old, Antibody therapy of
cancer, Nature Rev. Cancer 12 (278–287) (2012).[2] S. Fekete, D. Guillarme, Ultra-high-performance
liquid chromatography for the characterization of proteins, TrACS 63 (76-84) (2014).
Disclosure of Interest: None DeclaredKeywords: Antibody-drug conjugate, protein aggregate, size exclusion chromatography
P01-001-072 - Stationary liquid phase based on ionic liquids for the analysis of enantiomers.
M. Shashkov*, V. N. Sidelnikov
Content: Chemical processes involving optically active substances is wide and developing area of the modern chemistry. Investigations in this area are important for the development of fundamental and applied aspects of modern biology, medicine, pharmacology and related fields. For the analysis of volatile chiral compounds (or substances that are easily converted to volatile state) is the most effective method of is a gas chromatography (GC) on with a column loaded by chiral stationary liquid phases (SLP). Currently GC analysis of the enantiomers is performed mainly
with two types of columns – columns with cyclodextrine-based SLP and valine-based phases. However, these phases have a number of limitations and do not cover the ever-expanding range of tasks for chiral analysis. Therefore is the search for new SLP, which would expand the range of capabilities of modern chiral gas chromatography is an interesting task.
One of the promising materials that can be used as chiral SLP – are ionic liquids (IL). ILs have a number of considerable advantages in comparison with traditional phases. The main one is they are high-polar, and at the same time, thermostable [1]. Therefore, the use of IL allows to solve a number of problems associated with traditional chiral SLP – low thermostability, sensitivity to oxidation and a limited selectivity range. In our work, we study a number of approaches to the design of chiral IL SLP. The first approach involves the use of achiral IL and the chiral selector as an additive to SLP. Used as a selector permethylated β-cyclodextrin we included one in a number of imidazolium and pyridinium IL. This phases showed selectivity toward a number of chiral analytes
Another approach, used in this work assume an application of pure chiral IL as SPL. The example this approach is an IL with dication based on the structural unit of tartaric acid or anion tartaric acid.References: [1] Anderson J, Armstrong D.W. Anal.
Chem 2005, 77(19):6453-62Disclosure of Interest: None DeclaredKeywords: enantiomers and related substances, gas chromatography, ionic liquids

www.ISC2016.ie @isc2016 #isc2016 /isc2016144
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
P01-001-073 - Pellicular anion exchangers for ion chromatography with negatively charged functionalities in ion exchange layer
A. Uzhel 1,*, O. Shchukina 1, A. Zatirakha 1, A. Smolenkov 1, O. Shpigun 1
1 Chemistry department, Moscow State University, Moscow, Russian Federation
Content: Polystyrene-divinylbenzene (PS-DVB) and ethylvinylbenzene-divinylbenzene copolymers are of major interest for the preparation of the stationary phases for ion chromatography due to their mechanical and chemical stability, and method of their modification to the large extent determines chromatographic properties of obtained anion exchangers. Most of the modern polymeric anion exchangers have pellicular structure, where anion exchange layer is located on the substrate surface and diffusion of exchanging ions into the pores of the particle is limited. It results in the faster mass transfer and thus provides higher column efficiencies. For the modern latex-agglomerated and polyelectrolyte anion exchangers the diffusion of anions is additionally restricted because of the repulsion from the negatively charged sulfonic groups covering the substrate surface.
In this work negatively charged functionalities were introduced into the structure of ion exchange layer of chemically derivatized pellicular anion exchangers for preventing the diffusion of analytes into the pores of PS-DVB substrate and reducing non-ionic interactions of polarizable anions with polymeric surface. It was found that the type, number, and location of negatively
charged groups in the functional layer had a considerable impact on chromatographic performance of the obtained anion exchangers.
The proposed approach resulted in the drastic increase of selectivity, especially for weakly retained anions and anions of organic acids. The obtained anion exchangers are compatible with hydroxide solutions as eluents and provide the separation of 14 anions in 30 minutes in gradient elution mode including 7 standard inorganic anions (F-, Cl-, NO2
-, Br-, NO3-, SO4
2-, PO43-), anions
of weakly retained organic acids (HCOO-, CH3COO-, C2H4COO-, CH2(OH)COO-), and oxyhalides (ClO2
-, BrO3-, ClO3
-). The mixture of 7 standard anions can be resolved in only 7-9 minutes using gradient elution with hydroxide eluent.Disclosure of Interest: None DeclaredKeywords: Ion Chromatography, Pellicular anion exchangers , Polystyrene-divinylbenzene
P01-001-074 - Novel anion exchangers with covalently bonded hydrophilic polymeric functional layers for ion chromatographyO. Shchukina*, A. Uzhel 1, A. Zatirakha 1, A. Smolenkov 1, O. Shpigun 1
1 Moscow State University Chemistry Department, Moscow, Russian Federation
Content: Polystyrene-divinylbenzene (PS-DVB) copolymers are the best substrates for obtaining anion exchangers for suppressed ion chromatography (IC) due to their high chemical stability, but non-ionic interactions taking place between polarizable anions and aromatic rings of PS-DVB can be a reason of poor efficiency for such anions. This problem

145www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
can be solved by shielding polymer surface with functionalized polyamine attached to the substrate surface via hydrophilic linkers thus creating pellicular structure.
In the present work new anion exchangers obtained via covalent attachment of polyetheleneimine to the PS-DVB surface with hydrophilic linkers followed by the quaternization with different hydrophilic reagents are presented.
The synthesis included preliminary covering of PS-DVB substrate with the secondary aminogroups, which served as the anchors for the attachment of the functional layer. Diepoxides with different structure were used as a hydrophilic linkers for the covalent attachment of aminopolymer, which was further alkylated with glycydol and 1,4-butanediol diglycidyl ether (1,4-BDDGE).
Stationary phase modified with glycydol provides the separation of 11 anions including 7 standard anions (fluoride, chloride, nitrite, bromide, nitrate, phosphate and sulphate), formiate, chlorite, iodide and thiocyanate in 26 minutes using gradient elution with hydroxide solutions. Moreover, such anion exchanger enables efficient and selective separation of highly polarizable anions including thiosulphate, chlorate, bromate, iodide, and perchlorate in 20 minutes. Anion exchanger having the layer of polyamine quaternized with 1,4-BDDGE provides the separation of the anions of weakly retained organic acids, which also can be separated from fluoride and chloride. The obtained anion exchangers can be successfully used for solving wide variety of analytical tasks, including the analysis of water, air and soil extracts.
Disclosure of Interest: None DeclaredKeywords: Anion exchangers, ion chromatography, Pellicular anion exchangers
P01-001-075 - HILIC, Polar, and Shape Selectivity of the FluoroPhenyl Phase
T. Kahler*, H. Majer 1, F. Carroll 1, S.-H. Liang 1, S. Lupo1 Restek Corporation, Bellefonte, United States
Content: The FluoroPhenyl stationary phase has long been marketed as a phase that offers alternative, or orthogonal, selectivity to a C18. The FluoroPhenyl phase offers unique selectivity by incorporating strongly electronegative fluorine atoms on a phenyl ring. In addition to the traditional reversed-phase dispersive interactions, this phase also exhibits shape selective, polar, cation-exchange and even HILIC retention mechanisms which aid in selectivity of specific analytes.
In this presentation we aim to demonstrate the useful and alternate retention of the FluoroPhenyl stationary phase. We chose several relevant target analytes which we plan to use to exemplify the unique retention characteristics of the FluoroPhenyl phase when used in either HILIC or reversed-phase mode. All of these analytes have been pursued due to either poor retention, poor resolution, or both on a traditional C18 phase.References: C Santasania, D. Bell, Mechanisms of
Interaction Responsible for Alternative Selectivity of Fluorinated Stationary Phases, LCGC North America, Vol 34, Issue 2, Pg 92-105 Feb 01 2016
Disclosure of Interest: None DeclaredKeywords: FluoroPhenyl, HILIC, Selectivity

www.ISC2016.ie @isc2016 #isc2016 /isc2016146
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
P01-001-076 - Chemically stable merged inorganic/organic silica material with full scalability in reversed phase chromatographyFredrik Limé, Therése Tran and Robert FredrikssonAkzoNobel/Kromasil, SE-445 80 Bohus, Sweden
Content: Classic silica materials for reversed phase chromatography are often limited to pH between 2 and 8 due to breakage of the siloxane bonds at low pH or dissolution of silica at high pH. Since inorganic/organic silica materials offer the opportunity to work in a wider pH range due to its organosilane reinforcement, analytical, medicinal and drug discovery laboratories adopt these phases when analyzing and purifying mixtures that require harsh pH conditions. The merged silica materials are useful in analytical laboratories where there is a need for repeated injections without shift in retention times both at low and high pH and serves as fast screening columns and in multi-purpose purifications. This new organosilane reinforced silica creates an interfacial gradient in the surface that maintains the superior mechanical stability, pore size and pore structure. The reinforced silica material was further derivatized with C8, C18 and phenyl hexyl followed by end-capping to completely shield the silica matrix from silianol interactions. To use the same chemistry for all particle sizes (1.8, 2.5, 5 and 10 µm) allows for scaling the chromatography all the way from time saving UHPLC up to semi prep and preparative chromatography.References: N/ADisclosure of Interest: None DeclaredKeywords: HPLC, Hybrid, stationary phase
P01-001-077 - Characterization of polyaniline-coated stationary phases using linear solvation energy relationship in HILIC mode by capillary liquid chromatography
L. Taraba 1,*, T. Křížek 1, O. Hodek 1, P. Coufal 1
1 Department of Analytical Chemistry, Charles University in Prague, Faculty of Science, Prague 2, Czech Republic
Content: Polyaniline-coating-modified stationary phases have been investigated using linear solvation energy relationship (LSER) approach in hydrophilic liquid chromatography (HILIC) mode in order to estimate their dominant solute-retention-related interactions. Polyaniline, an easily synthesized highly stable conducting polymer, was polymerized in situ on the surface of original bare and octadecyl-modified silica gel spherical particles in the form of thin coating from aniline hydrochloride solution; ammonium persulfate was used as the oxidation agent [1]. Optical microscopy and scanning electron microscopy (SEM) techniques were used for surface characterization of the newly prepared stationary phases; specific surface areas of these materials were assessed by Brunauer-Emmett-Teller nitrogen adsorption method (N2-BET). The sorbents were consequently slurry-packed into capillary columns [2] and examined employing LSER chemometric method in HILIC mode [3]. According to obtained results, surface modification of the original materials was successful and brought so intended mix-mode separation mechanisms. In conclusion, this is the very first systematic chromatographic characterization of these complex stationary phases with a promising application potential.

147www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
References: [1] J. Stejskal, O. Quadrat, I. Sapurina, J. Zemek,
A. Drelinkiewicz, M. Hasik, I. K−ivka, J. Prokeš, European Polymer Journal 38 (631-637) (2002).
[2] M. Franc, J. Vojta, J. Sobotníková, P. Coufal, Z. Bosáková, Chemical Papers 68 (22-28) (2014).
[3] G. Schuster, W. Lindner, Journal of Chromatography A 1273 (73-94) (2013).
Acknowledgements: The study was financially supported by the Charles University in Prague (projects GAUK227233 and SVV260317).Disclosure of Interest: None DeclaredKeywords: HILIC, linear solvation energy relationship, polyaniline-coated stationary phase
P01-001-078 - Separation potential of novel sulfobutylether-β-cyclodextrin stationary phase
H. Procházková 1,*, G. Kučerová 1, K. Kalíková 1, E. Tesařová 1
1 Department of Physical and Macromolecular Chemistry, Charles University in Prague, Prague 2, Czech Republic
Content: Cyclodextrin-based stationary phases (SPs) are frequently used for separation of various isomers [1,2]. In this work we used sulfobutylether-β-cyclodextrin (SBE-β-CD) for preparation of novel SP. SBE-β-CD-based SP was prepared by dynamic coating procedure. Negatively charged SBE-β-CD was coated on oppositely charged ion exchange SP surface. Forming of ionic interactions was expected in order to modify the original SP surface.
Separation potential of newly prepared SP was tested using different analytes, i.e. amino acid and (non)chiral dipeptide derivatives of Tyr, Phe and Trp. Analyses were performed in RP-HPLC and hydrophilic interaction liquid chromatography. Mobile phases composed of organic modifier methanol and
four different aqueous parts: (a) deionized water; (b) aqueous solution of formic acid, pH 2.1; (c) ammonium acetate buffer, pH 4.7 and (d) ammonium acetate buffer, pH 8.8; in different volume ratios.
SBE-β-CD coated SP proved to be suitable for separation of chiral and also nonchiral analytes. Out of 23 chiral analytes, 9 were baseline and 7 were partially enantioseparated. Out of 9 mixtures of achiral dipeptides, 8 baseline separations were observed. Prepared SP was stable for more than 500 injections.References: [1] D.W. Armstrong, W. Demond, J. Chromatogr. Sci.
22 (411-415) (1984).[2] Y. Xiao, S.-C. Ng, T.T.Y. Tan, Y. Wang, J.
Chromatogr. A 1269 (52-68) (2012).Acknowledgements: The authors gratefully acknowledge financial support of the Grant Agency of the Charles University, Grant No. 790314 and the Grant Agency of the Czech Republic, Grant No. P206/14-19278P, and 16-05942S.Disclosure of Interest: None DeclaredKeywords: (enantio)separations, dynamic coating, sulfobutylether-β-cyclodextrin
P01-001-079 - Straightforward tips and tricks and troubleshooting for improved HPLC performance
S. Forster*, S. Altmaier, A. Piper, M. Schulz
Content: The result of a quantitative or qualitative chromatographic separation depends on a large variety of factors – including all HPLC system components and their setup, the choice of connectors and tubing, the selection of the chromatographic column as well as of all solvents and

www.ISC2016.ie @isc2016 #isc2016 /isc2016148
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
additives applied. HPLC performance as a combination of plate count and peak symmetry as well as sensitivity is influenced by several factors:• System setup, care and maintenance• HPLC column care & use• Mobile phase preparation
System setup and settings include the influence of dwell volume and dead volume including pump technology, tubing and detector cell, the detector response time and autosampler settings (mainly underdosing issues but also autosampler washing solution and sample solvent effects). System maintenance involves performing of system suitability tests, proper system flushing and mobile phase composition as well as regular maintenance (valves, seals). HPLC column care and use combines topics such as the choice of suitable connectors and fittings and a match of both column backbone and housing with eluent composition. In addition, column care (equilibration, cleaning, regeneration) as well as possible column bleeding issues are relevant. Mobile phase preparation is an important parameter in terms of solvent and additive properties (purity, viscosity, absorbance) as well as final composition and temperature (match with stationary phase properties necessary).
In this poster the influence of aforementioned topics and issues on overall chromatographic performance is discussed. Recommendations are given for the choice and setup of HPLC system components as well as the selection of solvents, tubing, connectors and HPLC columns. Problems and handling pitfalls will be described and tips and tricks as well as troubleshooting will be provided in order to optimize system and results and to gain maximum
performance.References: no external references used, data from
Merck internal application scientistsDisclosure of Interest: None DeclaredKeywords: column care, HPLC, system optimization
P01-001-080 - Hybrid monolithic materials synthetized from tetramethoxysilane and 1,2-bis(trimethoxysilyl)ethane and their use in capillary liquid chromatography
J. Planeta 1,*, P. Karásek 1, D. Moravcová 1, M. Roth 1
1 Department of fluid phase separations, Institute of Analytical Chemistry of the CAS, v. v. i., Brno, Czech Republic
Content: Over the recent decades, monolithic silica columns have been studied intensively as a separation medium in high-performance liquid chromatography (HPLC).
The common route for silica monolith synthesis is the acid hydrolysis of tetramethoxysilane (TMOS) in the presence of polyethylene glycol (PEG) and urea under acidic conditions at elevated temperature. This method works greatly for polymerization in capillaries up to 100 µm i.d. [1, 2], but in larger diameters, monolith cracks and the loss of contact with the capillary wall because of the monolith shrinking severely decrease chromatographic performance.
One of possible solutions of this problem can be synthesis of hybrid silica material, which is less rigid than pure silica due to the presence of organic moieties.

149www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
In the present work, we will demonstrate the synthesis of monolithic columns from mixture of TMOS and bis(trimethoxysilyl)ethane (BTME). We optimized the TMOS-BTME ratio and then synthetized monolithic materials with different ratios of PEG and silica precursors. All materials were examined by SEM and the sizes of skeletons and macro pores were calculated. The prepared capillary columns were evaluated by HPLC and their separation efficiencies, permeabilities and total porosities were calculated.References: 1. Planeta J., Moravcová D., Roth M.,
Karásek P., Kahle V., Journal of Chromatography A, 1217 (2010) 5737–5740
2. Moravcová D., Planeta J., Kahle V., Roth M. Journal of Chromatography A, 1270 (2012) 178– 185
Disclosure of Interest:None DeclaredKeywords: capillary column, monolith, silica
P01-001-081 - Customization of interior detection segment of fused silica capillary for C4D detection by supercritical waterP. Karásek 1,*, P. J. Barwa 1 2, P. Kubáň 1, M. Roth 1, J. Planeta 1
1 Institute of Analytical Chemistry of the CAS, v.v.i, 2 Department of Chemistry, Masaryk University, Brno,
Czech Republic
Content: Water, the most environment-compatible medium, is a unique solvent with high values of polarity and dielectric constant. Water has relatively high boiling point due to intermolecular interactions based on highly hydrogen-bonded structure. Increasing the temperature above the boiling point leads to acceleration of thermal motion of water molecules and
to dismemberment of hydrogen-bonded lattice. This results in a drastic change in water physical properties; above its critical point (374°C, 221 bar), supercritical water (SCW) becomes a non-polar and very aggressive medium which can dissolve silicon dioxide [1].
The purpose of this work is a reproducible manufacturing of in-capillary detection cell by etching with SCW and with predefined cell parameters (length, inner diameter, volume). The cells were tested by capillary electrophoresis with a referenced contactless conductivity detector (C4D). To prevent higher baseline conductivity of larger detection cells, the cell signal was compensated by selection of proper electrolyte concentration in the reference detector channel. The sensitivity such system was tested at different separation conditions. Major benefit of this approach is that the SCW is locally generated right in the location of produced cell. In comparison with traditional methods, based on HF mixtures, the other parts of capillary, notably the separation part, are not touched with an aggressive medium and the surface is not polluted with any residual chemical species.
The capillaries made by this way could be used in high-selectivity and high-efficiency biomolecular separations.References: 1. P.Karasek, J.Planeta, M. Roth: Anal.
Chem 85, 327-333 (2013).Acknowledgements: This contribution has been supported by the Czech Science Foundation (Project No. 16-03749S, 16-09135S) and by The Czech Academy of Sciences (Institutional Support RVO:68081715).Disclosure of Interest:None DeclaredKeywords: bubble cell, supercritical water, surface treatment

www.ISC2016.ie @isc2016 #isc2016 /isc2016150
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
P01-001-082 - Next Generation High Performance Thin Layer Chromatography (HP-TLC) Using Additively Manufactured Acrylate Based Polymer Stationary Phases
R. Groarke 1 2,*, T. Skrobisz 2, C. Hughes 1 2, R. McCann 1 2, A. Scigliano 3, D. Brabazon 1 2
1 Advanced Processing Technology Research Centre, 2 School of Mechanical and Manufacturing Engineering,
DCU, Dublin, Ireland, 3 Department of Mechanical and Aerospace Engineering,
Sapienza University of Rome, Rome, Italy
Content: Thin Layer Chromatography has long been used to facilitate the separation and visualization of separated chemical components in mixtures. This technique has been used as a rapid and facile way to check for reaction product purity and to separate dye mixtures 1 and pesticides 2. Silica, alumina as well as cellulose and silica-C18 are the traditional, commercially available media used in such applications. Polymer based TLC stationary phases have thus far been limited to electro-spun fiber based media 3. 3D Printing or Additive Manufacturing has been applied for many products as a means of rapid prototyping since its development and commercialization in the 1990s 4.
In this work, 3D printing was employed to fabricate a novel dual phase acrylate polymer based TLC plate with dimensionally well-defined micro-structured features which can be utilised as a separation platform requiring low solvent and sample volumes.
As a proof of concept, separations of a standard dye test mixture, and a food dye solution were carried out on each of these plates using a variety of mobile phases to
optimize the separation conditions. Rapid prototyping of the plates is possible with 25 plates (50mm x20mm x 1mm) produced in 51 mins. Results presented here include separation times and resolution. These plates are discussed in terms of their potential as a cost effective separation and detection alternative to other possible detection methods including UV-vis, MS, HPLC and other commercial TLC alternatives.References: (1) Wang, F.; Wu, H.; Zhu, Q.; Huang, K.; Wei, Y.; Liu,
C.; Zhai, Y.; Yang, Z.; Weng, S.; Xu, Y.; Noda, I.; Wu, J. Anal. Methods 2013, 5 (16), 4138–4144.
(2) Patel, R. B.; Gopani, M. C.; Patel, M. R. Chromatographia 2013, 76 (19-20), 1225–1231.
(3) Clark, J. E.; Olesik, S. V. Anal. Chem. 2009, 81 (10), 4121–4129.
(4) Sachs, E. M.; Haggerty, S.; Michael, J.; Williams, P. A. US Patent Number 5,204,055: Three-Dimensional Printing Techniques. 5,204,055, 1993.
Disclosure of Interest:None DeclaredKeywords: microfluidics, Polymeric materials, thin layer chromatography
P01-001-083 - Core-Shell Stationary Phases For The Separation Of Chiral Pharmaceuticals With Capillary Electrochromatography S. Declerck 1,*, B. Chankvetadze 2, Y. Vander Heyden
1, D. Mangelings 1
1 Analytical Chemistry and Pharmaceutical Technology (FABI), Vrije Universiteit Brussel, Brussels, Belgium,
2 Institute of Physical and Analytical Chemistry, School of Exact and Natural Sciences, Tbilisi State University, Tbilisi, Georgia
Content: Capillary electrochromatography (CEC) is a hybrid technique that combines the selectivity of HPLC and the high

151www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
separation efficiency of CE and has proven to be a suitable technique to develop chiral separations (Declerck et al., 2016).
Stationary phases with core-shell particles are merely used in high-performance liquid chromatography (HPLC) up till now. There are many advantages of core-shell materials compared to porous particles, such as a shorter diffusion path-length which results in a lower contribution of the mass transfer to the separation efficiency. Another important advantage is a more uniform particle-size distribution, resulting in a lower longitudinal diffusion. These advantages result in higher column efficiencies and faster elution times (Hayes et al., 2014).
Combined with CEC as separation technique, core-shell phases should theoretically produce an even higher efficiency than in HPLC.
This study compares results on two core-shell chiral stationary phases, coated with 2% (w/w) cellulose tris(3,5-dimethylphenylcarbamate), with different nominal particle and pore sizes with these on their two porous counterparts.
The test set of this study consists of 20 chiral pharmaceutical compounds (10 basic, 5 acidic and 5 neutral). In addition, van Deemter curves will be generated, by applying different voltages between 3-30kV, evaluate the column performances. References: R. Hayes; A. Ahmed; T. Edge; H. Zhang. (2014).
Core shell particles: Preparation, fundamentals and applications in high performance liquid chromatography. Journal of Chromatography A, doi:10.1016/j.chroma.2014.05.010.
S. Declerck; Y. Vander Heyden; D. Mangelings (2016). Enantioseparations of pharmaceuticals with capillary electrochromatography: A review. Journal of Pharmaceutical and Biomedical Analysis, doi:10.1016/j.jpba.2016.04.024.
Disclosure of Interest:None DeclaredKeywords: Capillary electrochromatography, Core-shell particles
P01-001-084 - Synthesis and characterization of molecular imprinted polymers for polyphenols M. Biesaga*, A. Ciric 1, K. Pedzich 2
1 Faculty of Science, University of Kragujevac, Kragujevac, Serbia,
2 Department of Chemistry, University of Warsaw, Warsaw, Poland
Content: In the current work, a molecularly imprinted polymers (MIP) have been synthesised and used to enable the extraction of a naturally-occurring antioxidant from complex media. Molecularly imprinted polymers (MIP) for solid phase extraction of polyphenols have been successfully prepared by a thermal polymerization method using caffeic acid or quercetin as templates, methacrylate acid or 4-vinylpyridine as functional monomer, ethylene glycol dimethacrylate as crosslinker and acetonitrile was used as porogen. Systematic investigations of the influence of monomer, cross-linker, porogen, as well as polymerization conditions on the properties of the MIPs were carried out. The binding parameters was calculated directly using the Langmuir-Freundlich fitting coefficients that yield total number of binding sites, mean binding affinity and heterogeneity. The sorption kinetic data was described by pseudo

www.ISC2016.ie @isc2016 #isc2016 /isc2016152
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
second-order models. The good fit obtained using the pseudo second-order model indicating the proposed chemical reaction mechanisms, supporting the assumption that the adsorption rate was controlled by physico-chemical interactions. Chemical and morphological characterization of the MIP was investigated by SEM (scanning electron microscopy) which confirmed a high degree of polymerization. The caffeic acid MIPs were evaluated according to their selective recognition properties for caffeic acid, structurally related compounds (chlorogenic, p-coumaric, ferulic, sinapic acid). The synthesized MIPs exhibited the high selectivity towards caffeic acid in the presence of huge amount of chlorogenic acid. Finally, the MIP was successfully applied to the clean-up and preconcentration of caffeic acid from coffee samples.References: 1. L.J. Schwartz, B. Danylec, S.J.
Harris, R.I. Boysen, M.T.W. Hearn, Journal of Chromatography A, 1218 2189-2195 2011
Acknowledgements: The authors would like to thank Erasmus Mundus Scholarship SIGMA AGILE 2SIG1401575 for partially supporting this projectDisclosure of Interest:None DeclaredKeywords: molecular imprinted polymer, polyphenols, preconcentration
P01-001-085 - Bonding Strategies for Monodisperse Core Shell and Fully Porous Silica Particles synthesised by modified Stober Type ProcessJ. P. Hanrahan*, T. F. O’Mahony, J. M. Tobin, J. J. Hogan
Content: HPLC is one of the most important and most widely used analytical tools by analytical chemists in the world. It
is estimated that between 70 and 80% of all HPLC separations use reversed phase liquid chromatography (RPLC or RP-HPLC) where the separation mechanism is dependent upon interactions between the analyte and the surface of the non-polar stationary phase packings in conjunction with largely aqueous polar mobile phases. [1] Classic HPLC packings are based on silica supports due to the particles being mechanically strong and therefore can withstand high pressures without compression. The surface of silica is polar as it contains many hydroxyl groups and in turn must be functionalized for non-polar separations. Due to the surface modifications and by easy manipulating the pore size, pore volume and surface area of the silica; the packing can be optimized for different analyte separations, e.g. small molecular entities or proteins.
The aim of this work was to use improve the existing bonding process utilized by Glantreo Ltd in an attempt to increase the bonding densities of organosilanes onto the silica surface of SOLAS™ and EIROSHELL™ in order to produce a reverse phase with a more homogeneous C18 coverage compared with standard commercial products . A design of experiments involving different prebonding treatments, different solvents and acid scavengers was conducted in an attempt to increase the ligand coverage on the silica surface. These bonded phases were then packed into narrow bore columns (2.1 x 50mm) and their efficiency was tested using a simple two compound separation to determine the number of theoretical plates.
References: [1] J.O. Omamogho, J.P. Hanrahan, J. Tobin, J.D. Glennon (2011) ‘Structural variation of solid core and thickness of porous shell of 1.7 mu core-shell silica particles on chromatographic performance:

153www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
Narrow bore columns’. Journal of Chromatography A, 1218 (15):1942-1953
Disclosure of Interest:None DeclaredKeywords: Core Shell, Fully Porous Particle, Stober
P01-001-086 - Polymeric microspheres with modified by RAFT polymers surface and their application as column packings for HPLCM. Grochowicz 1,*, B. Gawdzik 1
1 Faculty of Chemistry, Department of Polymer Chemistry, Maria Curie Sklodowska University, Lublin, Poland
Content: Permanently porous copolymers in the shape of microspheres are important materials used as column packings in different chromatographic methods. Methacrylate copolymers synthesized from monomers possessing polar functional groups are less hydrophobic than the traditional ST-DVB stationary phases. However, the radical polymerization of highly polar monomer is awkward and usually leads to the product with an insufficient amount of polar groups. Surface modification of microspheres aimed at achieving their shell-functionalization is an interesting tool for modifying their properties. In general, two different approaches can be mentioned, the ‘grafting to’ and the ‘grafting from’ approach.
The aim of this study was to obtain uniform polymeric microspheres of glycidyl methacrylate (GMA) and 1,4-dimethacryloyloxybenzene (1,4DMH) using seed polymerization. Poly(GMA-co-1,4DMH) microspheres which were subjected to the further chemical modification possessed diameters about 8,61 µm, the surface area
(SBET) equal 236 m2/g, the pore volume Vp = 0.25 cm3/g, and the pore diameters were in the range of mesopores. They were reacted with sodium cyclopentadienide which is a derivative of the most reactive dienes in the Diels–Alder (DA) reactions. The chemical modification of such prepared microspheres was carried out with hetero-DA reaction. As a dienophile, the RAFT poly(geranyl methacrylate) linear chains were used. The ‘grafting to’ approach was chosen for grafting polymeric chains with well-defined properties on the surface of the microspheres.
Polymeric beads were synthesized with the intention to use them as polymeric stationary phases in high performance liquid chromatography (HPLC). To evaluate the chromatographic properties of porous packing the Smith method was applied [1]. The retention indices of five sets of homologous compounds and the selectivity test compounds were measured.References: 1. RM, Smith, DR, Garside, J.
Chromatogr. 407 (1987) 19Disclosure of Interest:None DeclaredKeywords: HPLC packings, Polymeric materials, Surface modification
P01-001-087 - Application of polymeric high internal phase emulsion (polyHIPE) coated stationary phase columns in open tubular capillary electrochromatography
S. Choudhury 1,*, D. Connolly 2, B. White 1
1 School of Chemical Sciences, Dublin City University, Dublin,2 Department of Science, Pharmaceutical and Molecular
Biotechnology Research Centre (PMBRC), Waterford Institute of Technology, Waterford, Ireland

www.ISC2016.ie @isc2016 #isc2016 /isc2016154
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
Content: The application of polymeric high internal phase emulsion (polyHIPE) capillary coatings for open tubular analytical separation columns is demonstrated here for the first time. Multiple polystyrene-co-divinylbenzene (PS-co-DVB) polyHIPE layers of average total depth 1.73 µm are coated on internal capillary surfaces, creating open tubular columns (20 cm coated and 32.5 cm effective length). Utilising these columns for open tubular capillary electrochromatography (CEC), ethylbenzene and pentylbenzene are separated. While the overall separation capacity of the columns produced is low, the polyHIPE coatings improve analyte peak shape, decrease total run time and improve peak symmetries relative to comparable unmodified open tubular columns. In addition, use of these novel polyHIPE columns leads to use of 30% less organic modifier, potentially improving the shelf life for open tubular columns typically utilised in CEC.
The work presented here builds on previous work demonstrating reversed phase liquid chromatographic (RP-HPLC) separation of small molecules using a polystyrene-co-divinylbenzene (PS-co-DVB) polyHIPE stationary phases housed within 1.0 mm i.d. silcosteel columns [1]. In that work, a 90% PS-co-DVB polyHIPE was covalently attached to the walls of the column housing by prior wall modification with 3-(trimethoxysilyl) propyl methacrylate and could withstand operating backpressures in excess of 200 bar at a flow rate of 1.2 mL/min. Unfortunately peak widths did not approach those obtainable with commercial columns, with baseline widths of approx. 2 minutes for ethylbenzene and over 8 minutes for pentylbenzene observed.By utilizing the polyHIPE materials in
small molecule separation in capillary electrochromatography (CEC) in the work shown here, their chromatographic performance capabilities have been demonstrably enhancedReferences: 1. S. Choudhury, L. Fitzhenry, B. White,
D. Connolly, Materials 9 (212) (2016)Acknowledgements: Funding from Irish Research Council for S. Choudhury (Grant number RS/2012/24) is gratefully acknowledged.Disclosure of Interest:None DeclaredKeywords: Capillary electrochromatography, open tubular column, polyHIPE
P01-001-088 - Synthesis and characterisation of bonded aminopropyl silica intermediate using supercritical carbon dioxide and toluene as reaction solvent.B. A. Ashu-Arrah 1,*, J. D. Glennon 1
1Chemistry, UCC, Cork, Ireland
Content: Bonded aminopropyl silica (APS) intermediate was prepared when porous silica particles (Exsil-pure, 3µm) were reacted with 3-aminopropyltriethoxysilane (3-APTES) and N N- dimethylaminopropyltrimethoxysilane (DMAPTMS) using supercritical carbon dioxide (sc-CO2) and toluene as reaction solvent. Covalent bonding of APS intermediate was confirmed using elemental microanalysis (CHN), thermogravimetric analysis (TGA), zeta potential (ξ), diffuse reflectance infrared fourier transform (DRIFT) spectroscopy and solid-state nuclear magnetic resonance (CP/MAS NMR) spectroscopy. The authors present results which demonstrate that APS can be prepared successfully under

155www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
sc-CO2 conditions of 100˚C/414 bar in a substantial reduced time of 3h compared to a temperature of 110˚C in 24h using conventional organic solvent such as toluene. Moreover, bonded APS generated under supercritical CO2 technology avoid extensive purification steps hence no generation of waste organic solvent [1] leading to “green chemistry” alternative and cost reduction. The surface coverage of APS (evaluated from %C obtained from elemental analysis) prepared with APTES (%C: 8.03, 5.26 µmol/m-2) and DMAPTES (%C: 5.12, 4.58 µmol/m2) is somewhat higher when compared to organic based reactions under reflux with APTES (%C: 7.33, 4.71µmol/m2) and DMAPTMS (%C: 4.93, 4.38 µmol/m2) at a temperature of 110˚C in 24h This increase is attributed to the fact that inaccessible silanol groups on silica surface are accessible in sc-CO2 resulting in extensive hydrolysis and silanols condensation or cross- linkage [1, 2].References: [1] B.A. Ashu-Arrah, J.D. Glennon, and K. Albert, J.
Chromatogr. A 1236 (2012) 42-50.[2] B. McCool, and C.P. Tripp, J. Phys. Chem. B 109
(2005) 8914-8919.Acknowledgements: The authors wish to acknowledge financial support from Science Foundation Ireland (SFI) for the Irish Separation Science Cluster (ISSC) http://www.separationscience.ie/ under their Strategic Research Clauster (SRC) programme. Acknowledgement also goes to the outstanding collaboration with the Universität Tubingen, Institüt fur Organische Chemie, D-72076 Tubingen, Germany, in particular Emeritus Professor Albert and Miss Helen Yeman for assistance with acquisition of 29Si and 13C CP/MAS NMR spectra.Disclosure of Interest:None DeclaredKeywords: Chemical functionalisation, Supercritical CO2, Green Chemistry, Aminopropyl bonded silica
(APS), bi-functional bonding, Surface coverage/conversion efficiency.
P01-001-089 - An N-alkylbetaine bonded phase for the separation of polar molecules using hydrophilic interaction liquid chromatographyV. K. Langsi 1,*, B. A. Ashu-Arrah 2, A. Buzid 2, J. D. Glennon 2
1 Innovative Chromatography Group, Irish Separation Science Cluster (ISSC), Department of Chemistry and the Analytical, Biological and Chemistry Research Facility (ABCRF),
2 Innovative Chromatography Group, Irish Separation Science Cluster (ISSC), Department of Chemistry and the Analytical, Biological and Chemistry Research Facility (ABCRF), University College Cork, Cork, Ireland
Content: An N-alkyl betaine stationary phase was successfully prepared using ExsilTM pure silica (3.0 µm, 223 m2/g, 120 Å) by silanisation with N,N-dimethylaminopropyltrimethoxysilane (DMAPTMS)1 followed by quarterisation of the N,N-dimethylaminopropyl intermediate with sodium chloroacetate. Characterisation of the bonded particles by elemental analysis, TGA and spectrometric DRIFT analysis confirmed the presence of covalent bonding and N-alkylbetaine functionality. The bonded material was slurry packed in a 4.6 ID x 150 mm column and labelled as Nab-HILIC phase. The column was chromatographically evaluated using the Tanaka test mixture and the results revealed close selectivity properties with a commercially available ZIC®-HILIC column (4.6 ID x 150 mm) having a sulfobetaine functionality.
Further investigations of the effect of changes in the mobile phase pH and buffer concentration were performed to decipher

www.ISC2016.ie @isc2016 #isc2016 /isc2016156
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
the different HILIC retention mechanisms when separating acidic, basic, neutral or zwitterionic polar solutes.References: 1. V. K. Langsi, B. A. Ashu-Arrah, J. D.
Glennon, J. Chromatogr. 1402 (2015) 17-26.Acknowledgements: The authors would like to thank Science Foundation Ireland for research funding under the Strategic Research Cluster programme (Grant Number 08/SRC/B1412)Disclosure of Interest:None DeclaredKeywords: N-alkylbetaine, quarterisation, selectivity
P01-001-090 - Non-bonded 1.7 µm thin-shell silica particles for fast determination of uric acid and creatinine in human urine by HILIC methodV. K. Langsi 1,*, J. D. Glennon 1
1 Innovative Chromatography Group, Irish Separation Science Cluster (ISSC), Department of Chemistry and the Analytical, Biological and Chemistry Research Facility (ABCRF), University College Cork, Cork, Ireland
Content: Non-bonded 1.7 µm thin-shell silica particles (1.5 µm solid core and 100 nm thin shell layer) was synthesised by a method developed in our laboratory [1]. Particle size distribution analysis of the thin shell particles (labelled as TS1.7-100) showed the particles were monodispersed with polydispersity index < 0.2, BET characterisation gave pore size 88 Å and specific surface area of 78 m2/g. The non-bonded thin shell particles were slurry packed in a 4.6 ID x 100 mm column and HILIC chromatography carried out using a mobile phase of 70 % ACN, flow rate 1.25 ml/min with UV detection at 254 nm. A common indication associated with kidney disease are elevated levels of creatinine and uric acid and this research reports
the separation and analysis of these renal markers within 1 minute with hypoxanthine as internal standard on the TS1.7-100 column under HILIC chromatographic conditions. Calibration plots of peak area ratio of uric acid and creatinine to that of hypoxanthine against concentrations of uric acid and creatinine yielded correlation coefficients > 0.999. The method was applied to spiked urine samples and recoveries of uric acid and creatinine were between 86.7– 92.1% and 92.7– 94.3% respectively, with the limit of detection (LoD) evaluated at 0.03 µg/ml for uric acid and 0.05 µg/ml for creatinine. References: 1. V. K. Langsi, B. A. Ashu-Arrah, J. D.
Glennon, J. Chromatogr. 1402 (2015) 17-26Acknowledgements: The authors would like to thank Science Foundation Ireland for research funding under the Strategic Research Cluster programme (Grant number 08/SRC/B1412)Disclosure of Interest:None DeclaredKeywords: characterisation, recovery, Thin shell particles
P01-001-091 - Synthesis and characterization of porous polymeric microspheres with pendant polar groupsM. Maciejewska*, J. Osypiuk-Tomasik
Content: One of the significant trends in modern chromatography is searching for new stationary phases in order to improve the mass transfer, allow fast separation and provide column stability in the whole range of pH. The adsorbents based on porous polymers can meet these requirements. Additionally, they can provide a huge variety of functional groups available on the surface. The polymeric adsorbents with suitable functional groups can be obtained

157www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
from numerous types of monomers as well as by modification of copolymers that contain reactive group [1]. The aim of this work was to create porous cross-linked microspheres with pendant polar groups that can be used as polymeric stationary phases in high performance liquid chromatography (HPLC). To achieve this goal, in the first step the copolymers of poly(2,3-epoxypropyl methacrylate-co-trimethylolpropane trimethacrylate) were synthesized by suspension-emulsion polymerization. Next, the porous methacrylate network was modified by subsequent reactions with diethylenetriamine, 1-vinyl-2-pyrrolidone and 1-(2-hydroxyethlyl)-2-pyrrolidone, respectively. As a result permanently porous polar copolymers in the form of microspheres were obtained. Their porous structure was evaluated on the basis of nitrogen adsorption–desorption measurements at 77 K. The surface area of the parent copolymers was in the range from 86 to 333 m2/g depending on the synthesis parameters. After the process of modification it slightly decreased. The pore volume was about 0.65 cm3/g and the pore diameters was in the range typical for mesopores. The highly developed internal structure along with polar character of surface make the obtained copolymers promising materials for HPLC applications. References: [1]. M, Maciejewska, D, Ko−odynska,.
Mat. Chem. Phys. 149 (2015) 43.Acknowledgements: The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/ under REA grant agreement no. PIRSES-GA-2013-612484.Disclosure of Interest:None DeclaredKeywords: permanently porous microspheres, polar groups, Polymeric materials
P01-001-092 - Development of hybrid organic/inorganic monoliths containing mesoporous silica nanoparticles for capillary electrochromatographyA. Weller 1, E. J. Carrasco-Correa 1, P. Amorós 2, J. M. Herrero-Martínez*
1 Analytical Chemistry, University of Valencia, Burjassot, 2 Institut de Ciencia dels Materials, University of Valencia,
Valencia, Spain
Content: Polymethacrylate-based monolithic columns are the most widespread and the best characterized columns. Thus, these polymeric beds present several advantages as adjustable polarity, control for pore characteristics and stability under extreme pH conditions. In spite of these good features, these macroporous polymeric monoliths have relatively low surfaces areas which results in weak retention for chromatographic applications. In order to overcome this limitation, one of the several strategies relies in the incorporation of nanoparticles (NPs) to the monoliths, which have emerged as a promising way of increasing the surface area.
In the last years, mesoporous silica NPs have gained a lot of attention due to their favourable properties. In particular, these materials consist of an amorphous silicon matrix with mesopore sizes ranging from 2-50 nm and large surface areas (ca. 1000 m2/g). For this reason, their potential is to enhance the performance of organic monoliths could be of significant value in separation techniques.
In this work hybrid organic–inorganic monolithic stationary phases were prepared by incorporation of two mesoporous silica NPs (MCM-41 or UVM-7) for capillary

www.ISC2016.ie @isc2016 #isc2016 /isc2016158
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
electrochromatography (CEC) applications. The NPs were dispersed in a polymerization mixture containing butyl methacrylate and ethylene glycol dimethacrylate as monomers. The stability of the dispersions was investigated by UV-Vis measurements at several contents of silica NPs. Using short UV-polymerization times, polymeric beds with homogenously dispersed NPs were prepared. The resulting hybrid monolithic columns were characterized using scanning electron microscopy and its CEC performance of these novel stationary phases was evaluated by using alkylbenzenes and benzoic acid derivatives as test solutes. The prepared columns led to an increase in the retention of all the analytes compared to the parent monolith. The reproducibility of the resulting monolithic columns showed values below 11.2%..Acknowledgements: Project CTQ2014-52765-R (MINECO of Spain and FEDER)Disclosure of Interest:None DeclaredKeywords: capillary electrochromatography, hybrid organic-inorganic monolithic columns, silica nanoparticles
P01-001-093 - Development of novel through-porous particle as a separation medium for HPLC
N. Ishizuka 1,*, R. Takahashi 2, H. Kanezaki 2, M. Miyashita 2, K. Tsutsui 2, T. Yukiyama 2, M. Kobayashi 2, N. Kuroda 2, N. Kuriyama 2
1 Emaus Kyoto Inc., 2R&D, YMC Co., Ltd, Kyoto, Japan
Content: ‘Through-porous particle’ is a novel porous material, having co-continuous structure with through-pores and skeletons like a monolithic structure[1]. It is expected to be used as a
chromatographic medium, an adsorption medium and a support in solid-phase synthesis, etc. In this study, we succeeded in preparation of novel porous silica and polymer particles with co-continuous structure. These particles synthesized using emulsion polycondensation accompanied by gelation and phase separation based on spinodal decomposition, have well-controlled through-pores in the micrometer range. Furthermore, the particle size can be controlled in the range of ca. 10 ~ 100 µm. We assessed chromatographic properties obtained with a column packed with the through-porous silica. As a result, this column gave a low operating pressure compared to the conventional porous silica particles. We will report chromatographic properties obtained with a column packed with the through-porous silica particles in reversed phase HPLC.References: [1] H. Minakuchi, K. Nakanishi, N.
Soga, N. Ishizuka, N. Tanaka, Anal. Chem., 68, 3498-3501, 1996
Disclosure of Interest:None DeclaredKeywords: monolith, particle, porous
P01-001-094 - Embedded ester and phosphodiester stationary bonded phases for liquid chromatography purposesK. Krzemińska 1,*, S. Bocian 1, B. Buszewski 1
1 Chair of Environmental Chemistry & Bioanalytics, Nicolaus Copernicus University in Torun, Torun, Poland
Content: In recent years there is a noticeable increasing interest in development of chemically bonded stationary phases which are stable under highly aqueous conditions [1, 2]. There are known two approaches to obtain such type of packing materials. It

159www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
is possible to incorporate polar group to silica support or endcapped synthesized stationary phases. Chemically bonded stationary phases (CBPs) contained diol ligands are known since many years. The main reason for using this type of chromatographic material is its ability to form hydrogen bonding between the analyte and the stationary phase surface. Furthermore, it is possible the formation of ester bonds with carboxylic acids and their derivatives. Responsible for these capabilities are present in the structure hydroxyl groups. For this reason diol stationary phase can be used as starting material for the synthesis of a series of chromatographic packings. Polar embedded ester and phospodiester bonded stationary phases are stable under highly aqueous conditions. Attachment of various functional groups affects the change in retention and selectivity in comparison with classically CBPs for RPLC. These packing materials offer improved peak shape for basic analytes, an alternative selectivity profile for determination of a large group of substances with a polar nature Such chemically bonded stationary phase surface exhibits structural similarity to natural biological membrane. For this reason the proposed stationary phases may constitute a model system to study effects of the substance with biological membrane.References: [1] Sz. Bocian, A. Nowaczyk, B. Buszewski, Analytical
and Bioanalytical Chemistry 404 731 – 740 (2012)[2] N. Furusawa, American Journal of Analytical
Chemistry 3 295-299 (2012)Acknowledgements: This work was supported by Ministry of Science and Higher Education, Grant no. NCN 2013/09/D/ST4/03807 for period 2014–2017.Disclosure of Interest: None DeclaredKeywords: aqueous mobile phase, ester - bonded stationary phases, phosphodiester stationary phases
P01-001-095 - Study of the retention and selectivity of the amino acids- and peptide-silica stationary phases with different sequences of immobilized ligands
M. Skoczylas 1,*, S. Bocian 1, B. Buszewski 1
1 Chair of Environmental Chemistry and Bioanalytics, Nicolaus Copernicus University in Toru−, Torun, Poland
Content: Selectivity is a primary parameter in terms of chromatographic techniques. Consequently, the development of new chromatographic packings as well expansion of application existing materials are significant parts of the chromatographic investigation. The interesting sources of new stationary phases are biological systems and compounds contained in them [1].
The amino acids- and peptide-silica stationary phases are noteworthy materials characterized by the specific properties and the multitude of combination of their structures. Thus, immobilized on the silica surface suitable amino acids groups or their sequences allow obtaining stationary phases of different hydrophobicity and polarity. The optical properties of this type of packings can further classified them as chiral materials capable to separate enantiomers [2]. Additionally, immobilization of amino acids enables receiving zwitterionic stationary phases, as alternative materials for analysis of ionized analytes [3].
The aim of the research was the application of a new way in peptide-silica phases preparation, bases on the fundamental principles of Merrifield method. In this work, different types (regarding type and length of immobilized amino acids sequences) of home-made adsorbents have been studied. The investigated stationary phases were modified by the following amino acids:

www.ISC2016.ie @isc2016 #isc2016 /isc2016160
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
glycine, alanine, phenylalanine, leucine, and aspartic acid. The study of the retention and selectivity of the model compounds allowed the recognition and comprehension of the retention mechanism. Consequently, the classification of the peptide-materials according to their chromatographic properties was performed.References: [1] S. Ray, M. Takafuji, H. Ihara, Analyst
137, 4907-4909 (2012).[2] M. Xue, H. Huang, Y. Ke, C. Chu, Y. Jin, X. Liang,
J. Chromatogr. A 1216, 8623-8629 (2009).[3] A. Shen, Z. Guo, X. Cai, X. Xue, X. Liang, J.
Chromatogr. A 1228, 175-182 (2012).Acknowledgements: This work was supported by grants from the Ministry of Science and Higher Education “Iuventus Plus” no. IP2014 003673.Disclosure of Interest:None DeclaredKeywords: Column characterization and comparison, peptide-silica stationary phase, synthesis methodology
P01-001-096 - Separation of amino acids and small peptides by silica-based monolithic capillary columns
D. Moravcová 1,*, J. Planeta 1, M. Roth 1
1 Institute of Analytical Chemistry of the CAS, v. v. i., Brno, Czech Republic
Content: Separation of amino acids and small peptides by silica-based monolithic capillary columns (0.1mm x 150mm) is presented. All columns were prepared by acidic hydrolysis of tetramethoxysilane in the presence of polyethylene glycol and urea [1] and modified to RP (C18-) or HILIC (zwitterionic-, amino-) stationary phases. Effects of buffer type and concentration as well as content of organic solvent in mobile phase on separation of selected analytes were evaluated. Due
to hydrophilic nature of compounds of interest, gradient RP separation required pre-column derivatization of sample by orthopthaldialdehyde to increase analyte retention and selectivity of separation. On the other hand, both HILIC columns were capable to separate a sample mixture of native amino acids and peptides under isocratic separation conditions, e.g., zwitterionic column modified by [2-(methacryloyloxy)ethyl]-dimethyl-(3-sulfopropyl)-ammonium hydroxide allowed separation of nine amino acids (Phe, Trp, Met, Tyr, Pro, Lys, Ala, Gly, Ser) in a time up to 5 min employing 75% v/v of acetonitrile with 25% v/v of 25 mM ammonium formate, pH = 4.5, as mobile phase.References: 1. D. Moravcová, J. Planeta, V. Kahle,
M. Roth, Journal of Chromatography A 1270 (178– 185) (2012).
Disclosure of Interest:None DeclaredKeywords: Amino acids, Monolithic silica column, Peptides
P01-001-097 - Conjoint Immobilized Metal-ion Affinity Chromatography (IMAC) Approach: A novel pre-fractionation for plasma proteomics.
K. Karkra 1,*, K. K. R. Tetala 1, V. M.A 1
1 CBST, Centre for Bioseparation Technology, VIT University, vellore, India
Content: One of the major challenges in the proteomic analysis of complex samples such as plasma is the masking of low abundant proteins, potential biomarkers, by high abundant proteins such as Albumin, IgG, etc.1 In order to overcome this drawback various enrichment &pre-fractionation strategies like SEC, IEX, Affinity Chromatography etc. are used either independently or in tandem

161www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
mode.2 However, multiplexing them into a single system is challenging due to different binding and elution conditions.
Here, we evaluate the potential of novel sequential conjoint immobilized metal affinity chromatography (IMAC) to pre-fractionate proteins in their native state from a complex biological sample such as human Plasma prior to LC-MS analysis. The study was performed using CIM (Convective Interactive Media) Disk (BIA Separation) containing 4 different transition metal ions placed in the sequential order (Co (II) → Zn (II)→Ni (II)→Cu (II)) chelated with Iminodiacetic Acid (IDA) in a single housing. The rational to place the metal ions in the above-mentioned sequential order is due to their demand for histidine surface topography.3 A mixture of 5 model proteins (Fibrinogen, Transferrin, IgG, HSA, and lysozyme) was subjected to pre-fractionation using the conjoint IMAC system. Fibrinogen, Transferrin and IgG with “His-Clusters” were retained by Co(II) and/or Zn (II) only. HSA with multiple “His” was retained by Ni (II), whereas Lysozyme with a single surface exposed “His” was retained by Cu (II) only. Using this pre-fractionation strategy, we were able to reveal ~ 2.5 times more plasma proteins (both crude and Alb-IgG depleted plasma) in comparison to without pre-fractionation (LC-MS data).References: 1. N. L. Anderson and G. A. Norman., Molecular &
cellular proteomics 1 (845-867) (2002).2. Y. Shi, X. Rong, H. Csaba, A. W. James.,
J.Chromatogr.A,1053,(27-36) (2004).3. E. S. Hemdan, Y. J. Zhao, E. Sulkowski, J.
Porath., Proceedings of the National Academy of Sciences, 86, (1811-1815) (1989).
Acknowledgements: We would like to acknowledge
Department of Science & Technology (DST) India for financial support. The first author acknowledges ICMR, India for Junior Research Fellowship (JRF). BIA Separation, Slovenia is deeply acknowledged for their generous support of CIM disks.Disclosure of Interest:None DeclaredKeywords: pre-fractionation, IMAC , Plasma
P01-001-098 - Analytical characterization of stationary phases packed with pearl cellulose particlesT. Hetzel 1,*, J. Leonhardt 1, D. Loeker 1, T. Teutenberg
1, J. Bohrisch 2
1 Research analysis, Institut für Energie- und Umwelttechnik e. V., Duisburg, 2Fraunhofer Institute for Applied Polymer Research, Potsdam-Golm, Germany
Content: Reducing toxic organic solvents for liquid chromatographic separations is still a topic of huge interest. One approach to perform liquid separations with an aqueous mobile phase is high temperature liquid chromatography [1]. However, long-term stability of stationary phases as well as on-column analyte degradations has to be considered. An alternative to high temperature elution could be stationary phases whose hydrophobicity can be influenced by slightly elevated temperature or pH. Here, modified pearl cellulose particles could be an interesting option. Therefore, an unmodified and two different modified stationary phases were used. Particles grafted with Nacryloylpyrrolidine were expected to be temperature sensitive whereas diisopropylaminoethyl moieties containing material should have pH-sensitive characteristics. The columns were investigated using the Neue-test, van´t Hoff analysis and linear solvent strength model. An inverse trend of the van´t Hoff plot was

www.ISC2016.ie @isc2016 #isc2016 /isc2016162
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
observed using the temperature-sensitive column and a switching point of separation behavior at 52 °C could be obtained. Based on the observed retention factors it could be illustrated that there is a change of the retention mechanism for pH-sensitive stationary phase at a pH-value of 8. Due to the possibility to perform an aqueous elution at low temperatures, the results indicate a high applicability especially in the field of isotope-ratio mass spectrometry, solid phase extraction as well as for biological applications.References: [1] T. Teutenberg, High-Temperature
Liquid Chromatography : A User’s Guide for Method Development The Royal Society of Chemistry, 2010.
Disclosure of Interest:None DeclaredKeywords: pH and temperature sensitive stationary phases, van’t Hoff analysis, water-only mobile phase
P01-001-099 - Development of organic monolithic stationary phases with stepped functionality for capillary liquid chromatographyE. Morcom 1, E. J. Carrasco-Correa 1, G. Ramis-Ramos 1, J. M. Herrero-Martínez*
1 Analytical Chemistry, University of Valencia, Burjassot, Spain
Content: The development of stationary phases for high-performance liquid chromatography with stepped functionality constitutes a challenge from a technical point of view. However, the synthesis of stationary phases with different selectivities may yield mixed separation mechanisms that are not possible with a single column, and a potential increase in chromatographic performance. In addition, the use of stepped functionality columns could lead to avoid
the employment of gradient mobile phases. Using traditional packed columns, the formation of above multi-function columns could be particularly difficult.
On the other hand, the use of polymer monoliths for the production of stepped functionality columns has received considerable attention in recent years. Due to the ease preparation and surface modification of these polymers, stepped stationary phases have been synthetized in situ using UV grafting [1-3]. However, the application of these novel materials to chromatographic separation of several types of solutes has not been studied in detail.
In this work, dual functionality monolithic stationary phases for capillary liquid chromatography have been developed. For this purpose, glycidyl methacrylate-based monoliths were first prepared and then modified with ammonia. Both types of monoliths (parent and amine-modified monoliths) were modified using photografting to obtain a hydrophobic zone by using a single-step approach (benzophenone and lauryl methacrylate). These stepped stationary phases were chromatographically characterized by using alkyl benzenes, phenol derivatives and inorganic anions as test solutes. A comparison with monolithic columns synthetized with a single one functionality was also done.References: 1. S. Currivan, Michrochem. J., 111
(32-39) (2013)2. S. Currivan, Analyst, 137 (2559-2566) (2012)3. V. Pucci, J. Sep. Sci., 27 (779-778) (2004)Acknowledgements: Project CTQ2014-52765-R (MINECO of Spain and FEDER)Disclosure of Interest:None DeclaredKeywords: capillary liquid chromatography, monolithic columns, stepped stationary phases

163www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
P01-001-100 - Incorporation of ZIF-8-derived nanoporous carbons in methacrylate polymeric monoliths for capillary electrochromatographyE. J. Carrasco Correa 1, A. Martínez-Vilata 1, J. M. Herrero-Martínez*, F. Maya 2, V. Cerdà 2, C. Palomino
2, G. Turnes-Palomino 2, F. Svec 3
1 Analytical Chemistry, University of Valencia, Burjassot, 2 Chemistry, University of Balearic Island, Palma de
Mallorca, Spain, 3 E.O. Lawrence National Laboratoru, The Molecular
Foundry, Berkeley, CA, United States
Content: Metal-organic frameworks have gained particular attention in recent years as a novel class of porous materials mainly due to their designable framework structures modularly built from transition-metal clusters and organic ligands. Inspired by their diverse structures, high surface area, and large pore volume, MOFs have been considered as an alternative precursor to construct nanoporous carbons.
On the other hand, organic monolithic stationary phases, have attracted in the last decade increasing attention due to their good features. Nevertheless, these monoliths have low surface areas due to absence of a dedicated mesoporous structure being accompanied by weak retention in separation techniques. In order to overcome this limitation, one of the strategies relies in the incorporation of nanoparticles to the monoliths.
In this study, composites of zeolitic imidazole framework-8 (ZIF-8)-organic polymeric monoliths were prepared for its use in capillary electrochromatography. For this purpose, ZIF-8-derived with different crystal size were first synthesized by simple carbonization of ZIF-8 materials and then
incorporated to methacrylate polymerization mixtures, followed by UV initiation. The effect of crystal size and content of prepared ZIF-8-derived materials on the performance of the hybrid monolithic columns was investigated. The resulting composites were characterized using optical and scanning electron microscopy. Using short UV-polymerization times, polymeric beds with homogenously dispersed incorporated ZIF-8-derived materials were obtained. The chromatographic performance of these composites was evaluated using polycyclic aromatic hydrocarbons and non-steroidal anti-inflammatory drugs as test solutes. The hybrid ZIF-8-organic polymers led to an increase in the retention of all the analytes.References: .Acknowledgements: Project CTQ2014-52765-R (MINECO of Spain and FEDER)Disclosure of Interest:None DeclaredKeywords: capillary electrochromatography, organic polymeric monoliths, zeolitic imidazole framework-8
P01-001-101 - Considerations when Designing a Bonded Phase for Polar Analytes
B. A. Alden 1,*, T. H. Walter 1, J. N. Fairchild 1, B. Okandeji 1, J. Cook 1, D. P. Walsh 1
1 Consumables Group, Waters Corporation, Milford, United States
Content: Due to its reproducibility and broad applicability, reversed-phase chromatography is a widely used method of analysis for a variety of compounds. However, one difficult issue still exists, the low retention of polar compounds. In many cases, to achieve sufficient retention, highly aqueous (>90% water) mobile phases are required. Traditional reversed-phase materials with high surface
www.ISC2016.ie @isc2016 #isc2016 /isc2016164
POSTER ABSTRACTS Column Technology and New Materials as Separation Media
concentrations of hydrophobic (C18) groups do not give reproducible results under these conditions due to the lack of penetration of the highly aqueous mobile phase into the pores of the stationary phase (1). We have specifically designed bonded phases for the retention of polar analytes and compatibility with highly aqueous mobile phases. We have investigated the parameters of pore diameter and bonded phase surface concentration and will demonstrate mitigation of retention loss due to lack of mobile phase penetration into the pores.Table: Effect of C
18 Surface Concentration on
Retention Loss
Material Surface Conc. % Retention Loss
A 2.3 18
B 2.0 8
C 1.8 5
References: (1) T. H. Walter, P. Iraneta and M. Capparella J. Chrom. A 1075, 2005, 177.
Disclosure of Interest:None DeclaredKeywords: aqueous mobile phase, Dewetting, Polar compounds
P01-001-102 - Solid-Core Particles for Chromatographic Separations under High pH Conditions
N. L. Lawrence*, K. D. Wyndham, J. A. Wilson, K. H. Glose, J. N. Fairchild, C. A. Boissel, M. A. DeLoffi, S. J. Shiner, B. Muriithi 1
1 Waters Corporation, Milford, United States
Content: The use of solid-core (i.e., superficially porous) particles for chromatographic separations has been increasing in popularity over the past decade due their ability to provide significant enhancements in efficiency and speed while lowering backpressure. Since the majority of these commercially available
materials are silica based, they are limited in application due to restrictions in pH.
We have recently explored modifying the surface of these solid-core silica particles using a hybrid organic/inorganic surrounding material in an effort to broaden their chromatographic utility by expanding the useable pH range. In this poster, we will present the characterization and chromatographic performance of these novel packing materials. In addition, we will compare the results with competitor solid-core materials, marketed for high pH applications, as well as conventional fully porous particles. References: N/ADisclosure of Interest:None DeclaredKeywords: Chromatography, Solid-Core
P01-001-104 - Automation of Equivalent Column Selection in HPLC
I. Molnar*, I. Santoso
Content: HPLC methods help to control the quality of many chemical and pharmaceutical products.
Methods are usually developed by optimizing the properties of the mobile phase with a given column. If the original column cannot be used any longer, the separation will often change if another column is used.
Therefore, one might want to have one or two equivalent columns which can replace the original column without changes in selectivity and robustness of the method.

165www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSColumn Technology and New Materials as Separation Media
This presentation will show a new way to compare columns and to select suitable replacement columns. The presented procedure will even be able to evaluate the possibility to use different columns with the same method, and to evaluate the robustness of the common method at different columns.
Three stationary phases, Kinetex C18, Cortex C18 and HALO C18 were compared. By fixing the working point, Kinetex C18 and Cortex C18 were found to be equivalent for 100 % success rate (success criteria: critical resolution Rs,crit >1.5).References: R. Kormany, I. Molnar, H.J. Rieger, J.
Pharm. Biomed. Analysis 80 (2013) 79-88Disclosure of Interest:None DeclaredKeywords: Chromatography Modeling, Column Selection
P01-001-105 - Comparison of various Aryl and Alkyl modified Sorbents in RP Chromatography
H. Riering 1,*, N. Bilmann 1, G. Cozzupoli 1
1 Macherey-Nagel GmbH & Co KG, Dueren, Germany
Content: Alkyl, especially octadecyl modified silicas are the workhorses in RP chromatography. Their retention behavior can be explained mainly by hydrophobic interactions. For difficult separations this may be insufficient. Therefore, stationary phases with alternative separation mechanisms are still of interest for HPLC. Aromatic ligands on the sorbent are particularly suitable for this [1], because they are chemically inert and exhibit as hydrocarbons sufficient hydrophobicity. But due to the π,π-interactions their selectivity is different compared with alkyl modified silicas [2,3]. In this poster we compare the selectivities of
various aryl modified silicas with each other and with alkyl modified sorbents (octyl and octadecyl). Phenylpropyl, phenylhexyl, biphenyl and biphenylpropyl are used as aromatic groups. In addition, a bifunctional phase with phenyl and octadecyl groups is investigated. Numerous test substances are used to characterize van der Waals and π,π-interactions and the results are discussed.References: 1. F. Chan, L.S. Yeung, R. LoBrutto, Y.V. Kazakevich
J. Chromatogr. A 1069 (2005) 217 - 2242 M.R. Euerby, P. Petersson, W. Campbell, W. Roe
J. Chromatogr. A 1154 (2007) 138 – 1513 K. Croes, A. Steffens, D.H. Marchand, L.R. Snyder
J. Chromatogr. A 1098 (2005) 123 -130Disclosure of Interest:None DeclaredKeywords: aryl modified silica, RP chromatography, selectivity

www.ISC2016.ie @isc2016 #isc2016 /isc2016166
POSTER ABSTRACTS Electrodriven Separation Methods
Electrodriven Separation Methods
P01-001-011 - Definition of guidelines to facilitate analytical inter-instrumental method transfer of capillary electrophoretic separations
B. De Cock 1,*, D. Mangelings 1, Y. Vander Heyden 1
1 FABI, Vrije Universiteit Brussel, Brussel, Belgium
Content: The increased interest in the separation of small peptides, proteins and chiral molecules leads to an increased demand for appropriate analytical methodologies. As a result of its separation mechanism, capillary electrophoresis (CE) is frequently used in those analyses. The more common application of CE methods therefore entails more frequent analytical method transfers (AMT). Due to the lower precision and robustness of CE methods, the instrumental differences and the larger impact of multiple parameters on the separation, the AMT of CE methods is more challenging compared to HPLC methods. In this project AMT guidelines were defined which should facilitate a successful AMT rate of CE methods, both between laboratories and instruments.
In a first step, the repeatability of a chiral CE method was increased by application of a constant current. Using a constant current also generated more analogous results during inter-instrumental AMT. In a second step settings that were different between instruments were selected as robustness-test parameters. The information from the robustness test and the earlier performed precision study were applied to achieve both a successful inter-laboratory and
inter-instrumental transfer. The developed guidelines were evaluated with a more complex separation of angiotensin II and derivatives. However loss of baseline separation occurred during this AMT and an update of the developed guidelines was needed through equalization of the electrical resistances on both instruments to overcome separation-efficiency differences. In a third step, the differences concerning the detector and data-handling specifications were analysed by means of a second robustness test using the initial chiral separation as test case, and the results were incorporated in the guidelines. Finally, the occasional differences in effective length that occur with short end injection methods were studied using a separation of two strategic metals as example.Acknowledgements: Disclosure of Interest:None DeclaredKeywords: Analytical method transfer, Capillary electrophoresis, Interinstrumental differences
P01-001-016 - Capillary electrophoresis-laser induced fluorescence and microscale thermophoresis: miniaturization tools for human neutrophil elastase kinetic study
S. Fayad 1,*, R. Nehmé 1, P. Morin 1
1 ICOA, University of Orléans - CNRS, FR 2708, UMR 7311, Orléans, France
Content: Human neutrophil elastase (HNE) is a main actor in the development of chronic obstructive pulmonary diseases and in the degradation of the extracellular matrix causing then skin aging. In order to assay HNE kinetics, a novel online

167www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSElectrodriven Separation Methods
capillary electrophoresis (CE) assay has been developed for the determination of the maximum velocity (Vmax) and of the Michaelis–Menten constant (Km) of HNE regarding several new potential substrates. This assay is based on short-end injection to decrease analysis time, on transverse diffusion of laminar flow profiles (TDLFP) for in-capillary reactant mixing, and on UV or laser-induced fluorescence (LIF) detection [1]. The developed assay was then optimized to monitor HNE inhibition. Two natural well known inhibitors from the triterpene family, ursolic acid and oleanolic acid, were tested to validate the developed assay. Since the solubility of pentacyclic triterpenes in aqueous media where the enzymatic reaction will take place is limited, the effect of DMSO and ethanol on HNE was studied using microscale thermophoresis (MST). IC50 values were determined to rank HNE inhibitors relative to each other. Furthermore, MST technique was used for evaluating HNE interaction with the ursolic acid (Ki determination). Up to 16 capillaries were automatically processed to obtain in one titration experiment the dissociation constant for the HNE-ursolic acid complex. The complementarity between CE and MST was thus evaluated [2].References: [1) S. Fayad, R. Nehmé, P. Lafite, P. Morin, Assaying
Human Neutrophil Elastase activity by capillary zone electrophoresis combined with laser-induced fluorescence (CZE-LIF), J. Chromatogr. A 1419 (2015) 116-124.
[2] S. Fayad, R. Nehmé, B. Claude, P. Morin, Human neutrophil elastase inhibition studied by capillary electrophoresis with laser induced fluorescence detection (LIF) and microscale thermophoresis (MST) J. Chromatogr. A 1431 (2016) 215–223.
Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis-laser induced
fluorescence, Kinetic study, Transverse diffusion of laminar flow profiles
P01-001-024 - Electrophoretic separation of linear synthetic polymers in non-aqueous medium containing ionic surfactantsS. Kitagawa 1,*, T. Yamamura 1, N. Fukai 1, H. Ohtani 1
1 Nagoya Institute of Technology, Nagoya, Japan
Content: In the field of polymer science, liquid chromatographic methods are generally used for characterizing linear polymers. Electrophoresis is an alternative separation method that is the standard for biopolymers; however, electrophoresis has not been applied to the analysis of synthetic polymer except for a few cases. In this study, we developed a method for separating water-insoluble and neutral synthetic polymers using electrophoresis in non-aqueous medium containing ionic surfactant. In non-aqueous capillary electrophoresis using ionic surfactant, a mixture of polymers (polystyrene, polybutadiene, polycarbonate, and polymethylmethacrylate) were successfully separated. It was found that the selectivity toward the polymer species depended on both the kind of surfactants and the composition of electrophoretic medium. Interestingly, electrophoretic mobility of the polymers was independent of the molecular weight in the proposed method. Using the method, poly(styrene-co-methylmethacrylate)s with different compositions were also successfully separated.References: 1. T. Yamamura, S. Kitagawa, H.
Ohtani, J. Chromatogr. A, 2015, 1393, 122-127.Acknowledgements: This work was supported by JSPS KAKENHI Grant Number 15K13723.

www.ISC2016.ie @isc2016 #isc2016 /isc2016168
POSTER ABSTRACTS Electrodriven Separation Methods
Disclosure of Interest:None DeclaredKeywords: ionic surfactants, linear synthetic polymers, non-aqueous electrophoresis
P01-001-106 - Large volume sample injection and transient sweeping via dynamic chelation for analysis of heavy metal ions in cosmetics Y.-L. Chen*, K.-L. Chen
Content: Due to toxicity of heavy metal ions, international limited provisions of metal impurities in cosmetics were set up to prevent overexposure of heavy metal ions. Large volume sample injection-transient sweeping via dynamic chelation (LVSI-tSDC) was established by capillary electrophoresis to analyze the limited impurities lead (Pb), cadmium (Cd) and mercury (Hg) in cosmetics (1). The mechanism of LVSI-tSDC was large volume of metal ions were swept by the UV absorbing chelating agents electrokinetically injected into the capillary transiently to increase the detection sensitivity. Sample solution diluted by 25 mM ammonium acetate (pH 6.0) was injected into the capillary by pressure, 3.5 psi 99.9 s. Then the EDTA was injected by -25 kV applied for 1 min from EDTA buffer and metal ions were swept and stacked simultaneously. Finally, separation was performed at -20 kV by using separation buffer. The sensitivities relative to capillary zone electrophoresis were improved 33, 50 and 100-fold of Pb, Cd and Hg respectively. The correlation coefficients greater than 0.998 found that this method has good linearity. The relative standard deviation and relative error were less than 8.7% with high precision and accuracy. The recovery value of homemade lotion simulating the
real sample matrix was 93-104% indicated that sample matrix does not affect the quantitative results. Ultimately, commercial cosmetics were selected to demonstrate the feasibility of the method to determine Pb, Cd and Hg.References: (1) K. Isoo, S. Terabe, Analysis of metal
ions by sweeping via dynamic complexation and cation-selective exhaustive injection in capillary electrophoresis, Anal. Chem. 75 ( 6789-6798) (2003)
Acknowledgements: We gratefully acknowledge the support of the Ministry of Science and Technology of Taiwan (MOST 104-2113-M-037-007) and partially by Kaohsiung Medical University “Aim for the Top Universities Grant” (No. KMU-TP104PR05) for funding this work.Disclosure of Interest:None DeclaredKeywords: large volume sample injection-transient sweeping via dynamic chelation, on-line preconcentration technique, lead, cadmium, mercury, cosmetics.
P01-001-107 - Effect of human serum albumin glycation on its interaction with the antidiabetic drugs studied by capillary electrophoresis – frontal analysis
L. Michalcová 1, Z. Glatz 1,*
1 Department of Biochemistry, Faculty of Science, Masaryk University, Brno, Czech Republic
Content: Binding of drugs to plasma proteins, moslty to serum albumin is one of key issues that influences drug disposition. However it can be affected by numerous factors including the endogenous protein modifications. As a result, the knowledges about interaction between drugs and modified protein are very important for

169www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSElectrodriven Separation Methods
the pharmacotherapy. The main objective of this study was to compare the binding constants (Kb) as a numerical expression of the binding strength of normal (HSA) and modified - glycated (gHSA) serum albumins with the antidiabetic drugs - tolbutamide, chlorpropamide, acetohexamide, carbutamide.
Capillary electrophoresis - frontal analysis (CE-FA) was chosen for this purpose since it is characterized by very low sample consumption, the high sample throughput with the possibility of automation and a short analysis time. Besides CE-FA measurements can be performed with the binding partners in their native states without immobilization or labelling under physiological conditions [1]. CE-FA was performed with the external calibration. The binding parameters were determined by nonlinear regression. The Kb values obtained exhibited a good repeatability. They decreased in the sequence acetohexamide > tolbutamide > chlorpropamide > carbutamide for both albumin forms. Although the Kb exhibited the same sequence for HSA as well as for gHSA, the glycated form showed lower values of Kb for these drugs. The Kb for acetohexamide-HSA and tolbutamide-HSA are in agreement with the literature [2-5]. The Kb of carbutamide, chrorpropamide with HSA and information related to interactions with gHSA are not adequately described in the literature.References: [1] L. Michalcová and Z. Glatz, J. Sep. Sci. 38 (325-
331) (2015).[2] E. Lazaro, et al., J. Med. Chem. 51 (2009-
2017) (2008).[3] K. S. Joseph, et al., J. Pharm. Biomed. Anal. 54
(426-432) (2011).[4] H. Koyama, et al., Biopharm. Drug Dispos. 18
(791-801) (1997).[5] K. S. Joseph, et al., J. Chromatogr. B 878 (2775-
2781) (2010).Acknowledgements: This work was supported by grant No. P206/12/G014 from the Czech Science Foundation.Disclosure of Interest:None DeclaredKeywords: capillary electrophoresis – frontal analysis, drug-protein interaction, human serum albumin glycation
P01-001-108 - Chiral separation of radezolid using charge single isomer derivatives of cyclodextrins by capillary electrokinetic chromatography
K. Michalska 1,*, E. Gruba 1, J. Cielecka-Piontek 2
1 Department of Antibiotics and Microbiology, National Medicines Institute, Warsaw,
2 Department of Pharmaceutical Chemistry, Poznan Univeristy of Medicinal Sciences, Poznan, Poland
Content: A method for the enantioseparation of radezolid, the next analogue after linezolid and tedizolid of a truly new class of antibacterial agents – the oxazolidinones, was developed based on capillary electrokinetic chromatography using cyclodextrin as a chiral pseudophase (CD-cEKC). During the experiment the behaviour of radezolid together with linezolid and its precursor were observed.
The single isomer S-radezolid possesses one chiral center at C5 of oxazolidinone ring, which is associated with antibacterial activity of the drug. The stereochemistry of the enantiomers of S- and R-radezolid as well as behaviour of selected complex formation were established using electronic circular dichroism (UV-CD).

www.ISC2016.ie @isc2016 #isc2016 /isc2016170
POSTER ABSTRACTS Electrodriven Separation Methods
The charge of radezolid is pH-dependent. At low pH is cationic, due to protonation of amino group at aliphatic bridge between phenyl and triazole rings, at higher pH are mix forms, while at pH above 10.0 the anionic form dominates. During the development of the method the anionic single-isomer cyclodextrins, from a hydrophilic, through moderately hydrophobic, up to a hydrophobic: HS-β-CD, HDAS-β-CD, ODAS-γ-CD as well as HDMS-β-CD respectively, were tested. Experiments were performed at 2.5, 6.6, 8.2 and 10.0 pH.
Cyclodextrins, which carry the acetyl and methyl group at the C2 and C3 positions, with seven residues of glucopyranose units, HDAS-β-CD and HDMS-β-CD provided the partly and baseline separation of enantiomers however required higher temperature in the first case, and acetonitrile addition in the second case, respectively. During the experiments, different organic solvents were tested. The best results for separation of enantiomers were obtained with 38 mM HDMS-β-CD in 50 mM phosphate buffer (pH 6.6) with addition of acetonitrile (60:40, v/v), 27ºC, normal polarity, 28 kV. Finally, the apparent binding constants for each enantiomer - HDMS-β-CD pair were calculated.References: K. Michalska, E. Gruba, J. Cielecka-
Piontek, E. Bednarek, J Pharm Biomed Anal 120 (2016) 402–412
Disclosure of Interest:None DeclaredKeywords: CD-cEKC, cyclodextrin, Enantioseparation
P01-001-109 - Separation of racemic drugs with brand-new cyclodextrin derivatives : Single-isomer Carboxymethylated CyclodextrinsE. Varga 1,*, G. Benkovics 1, J. Szemán 1, L. Szente 1
1Cyclolab Ltd., Budapest, Hungary
Content: It is widely recognized today that chirality is an important modulator of the effects and properties of chiral substances in a variety of fields such as pharmacology, agrochemistry, food chemistry, environmental chemistry, etc. The phenomenon of chirality exists in all biological systems. Therefore, analytical methods for the determination of single enantiomers in different natural and industrial samples are required. Cyclodextrins (CDs) play an important role as chiral selectors in capillary electrophoresis (CE) and other chromatographic techniques[1].
Carboxymethyl cyclodextrins (CM CDs) represent a group of negatively charged CD derivatives which have been already successfully used for the separation of enantiomers of compounds in capillary electrophoresis .
The newly synthesized single isomer CM CDs, namely heptakis-(2,3-di-O-methyl-6-carboxymethyl)-β-CD and the octakis-(2,3-di-O-methyl-6-carboxymethyl)-γ-CD that are fully carboxymethylated at the 6-carbon positions and contain methoxy groups at the 2- and 3-carbon positions, were utilized as additives to the background electrolyte in CE and tested for the enantioseparation of several biologically significant molecules. Their enantioselectivities were compared with the analogous members of the

171www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSElectrodriven Separation Methods
commonly used persubstituted single-isomer CD family, 2,3-dimethyl-6-sulfato CDs.References: 1 Escuder-Gilaberta, L. Martín-
Bioscaa, Y., Medina-Hernándeza, M.J., Sagrado, S. Cyclodextrins in capillary electrophoresis: Recent developments and new trends. J. Chromatogr. A., 29, (2014) 2–23.
Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis, Enantiomer separation, Single Isomer Cyclodextrin Derivative
P01-001-110 - Separation of Carbon Dots by Capillary Electrophoresis
K. M. Tirado-González 1,*, Z. Xue 1, L. A. Colón 1
1 Chemistry , University at Buffalo, The State University of New York, Buffalo, United States
Content: Carbon dots (C-dots) have attracted substantial interest within the scientific community because of their intrinsic properties, such as photoluminescence, water solubility, chemical stability, and low toxicity. It has been recently shown that after dialysis and filtration, these materials can still exist as a mixture with different surface functionalities, sizes, and surface charge. Therefore, high resolution separations are necessary to fully characterize C-dots after their initial synthesis. We have used capillary electrophoresis (CE) with laser induced fluorescence (LIF) detection to monitor C-dots prepared under different reaction conditions in an effort to establish conditions that produce less heterogeneous C-dots samples. The C-dots have been prepared using the bottom-up approach using citric acid (CA) and diethylenetriamine (DETA) as the molecular precursors. Photoluminescence and electrophoretic methods were used to
characterize the mixtures. Electrophoretic separation of the species contained in the mixture prepared with different CA:DETA ratios showed that the maximum number of resolved components in the sample was achieved using a CE run electrolyte at a pH of 5. At low ratios of amine to citric acid, the predominant species is negatively charged; however, at equal ratio or higher the predominant species is neutral. This work will show how CE-LIF has provided insight in the understanding and characterization of C-dots mixture.References: 1. J.C., Vinci; I.M., Ferrer; S.J., Seedhouse; A.K.,
Bourdon, J.M., Reynard; B.A., Foster; F.V., Bright; L.A. J. Phys. Chem. Lett., 4 (239-243)(2013).
2. H., Ding; S.-B., Yu; J.-S., Wei; H.-M., Xiong. ACS Nano, 10 (484-491)(2016).
3. S.N., Baker; G.A., Baker. Angew. Chem., Int. Ed., 49 (6726-6744)(2010).
4. J.S., Baker; L.A., Colón. J. Chromatogr. A. 1216 (9048-9054)(2009).
Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis - laser induced fluorescence, Carbon nanomaterials
P01-001-111 - Electrophoretic Separations of Oligosaccharides Coupled with On-Line Fluorescent LabellingZ. Nováková 1,*, P. Česla 1, H. Fischer 1
1University of Pardubice, Department of Analytical Chemistry, Pardubice, Czech Republic
Content: Carbohydrates, either alone or as constituents of glycoproteins and glycolipids, are mediators of several events, such as intra and extracellular recognition, differentiation, proliferation or even signal transduction [1].

www.ISC2016.ie @isc2016 #isc2016 /isc2016172
POSTER ABSTRACTS Electrodriven Separation Methods
Oligosaccharides play a significant role in posttranslational modifications of proteins. The changes in protein glycosylation may serve as an indicator of some diseases [2]. Carbohydrate derivatization is required for most of the analytical procedures. The determination of glycans (without treatment) presented in organisms is very difficult. Detection of intact carbohydrates can be utilized by spectroscopic measurement at 195 nm, amperometric or refractive index detection, all of which are associated with low sensitivities [3]. Derivatization with UV-absorbing or fluorescent molecules significantly enhances the detection sensitivity of high-performance liquid chromatography (HPLC), capillary electrophoresis (CE) and polyacrylamide gel electrophoresis (PAGE). It is necessary to insert an optically active substance into the analyzed molecule. For example, maltooligosaccharides (maltose to maltoheptaose) can be labelled with 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) and used for their characterization.
In present work, conditions in non-coated and coated capillaries were optimized. Further, we had examined and compared various fluorescent derivatization agents in terms of resolution, migration time, and sensitivity of detection. Finally, we have developed the method for on-line labelling of oligosaccharides using procedure for fluorescent derivatization.References: [1] F. N. Lamari, R. Kuhn, N. K. Karamanos / J.
Chromatogr. B 793 15-36 (2003).[2] P. Smejkal, M.C. Breadmore, R.M. Guijt, J. Grym,
Foret, Bek, F., M. Macka, Anal. Chim. Acta 755, 115-120 (2012).
[3] F .N. Lamari, M. Militsopoulou, T.N. Mitropoulou, A. Hjerpe, N.K. Karamanos, Biomed. Chromatogr.
16 (2002) 96.Acknowledgements: The work was financially supported by the Czech Science Foundation (1406319S).Disclosure of Interest:None DeclaredKeywords: Electrophoretic separation, fluorescent labelling, Oligosaccharides
P01-001-113 - Improving CZE data with cobalt complexes as internal standards: a cisplatin/GMP case studyH. U. Holtkamp 1,*, S. J. Morrow 2, M. Kubanik 1, C. G. Hartinger 1
1 School of Chemical Sciences, 2University of Auckland, Auckland, New Zealand
Content: To facilitate metal-based anticancer drug development towards clinical trials and subsequent commercialisation, preliminary studies are required that can characterise the hydrolytic stability of these complexes and their binding affinity and reactivity with biological binding partners. Capillary electrophoresis (CE) is an ideal technique for these studies as CE is capable of the separation of charged metal-containing species formed under physiological conditions.1 CE separations are also prone to run-by-run variations resulting in imprecision in both the migration times and peak areas, making peak and kinetic trend identification difficult and error prone.2 This study aimed to develop suitable standards for CE separations that can be well detected by both UV and mass spectrometric (MS) detectors. The CoIII complexes [Co(en)3]Cl3, [Co(acac)3] and K[Co(EDTA)] were considered for their stability, accessibility, low cost, range of charge states and the low detection limit for Co in inductively coupled plasma (ICP)-MS.3 These CoIII

173www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSElectrodriven Separation Methods
chelate complexes were evaluated as internal standards in a model system using the anticancer drug cisplatin and guanosine 5’-monophosphate in various analytical backgrounds. It was found that by employing the Co complexes resulted in significantly improved data sets. References: (1) Holtkamp, H.; Grabmann, G.; Hartinger, C. G.
Electrophoresis 2015, 37 (7-8), 959-972.(2) Michalke, B. J. Anal. At. Spectrom. 1999, 14 (9),
1297-1302.(3) Pröfrock, D.; Prange, A. Appl. Spectrosc. 2012, 66
(8), 843-868.
Acknowledgements: We thank the organizations and foundations that have supported our research efforts in this area, especially the University of Auckland (University of Auckland Doctoral Scholarship to H.H. and M.K.), the India-New Zealand Education Council, Education New Zealand, the India-New Zealand Research Institute, the Royal Society of New Zealand, and COST CM1105. We thank Auckland Science Analytical Services of the University of Auckland for access to their facilities. We are grateful to Prof. Gordon Miskelly for useful discussions.
Disclosure of Interest:None DeclaredKeywords: Capillary Electrophoresis, Internal Standards, Metal-based anticancer complexes
P01-001-114 - Preconcentration and separation of all proteinogenic amino acids by hyphenation of isotachophoresis and capillary electrophoresis mass spectrometry
T. Melzer*, P. Rath 1, P. Kler 2, C. Huhn 1
1 Institute for Physical and Theoretical Chemistry, Eberhard Karls Universität Tübingen, Tübingen, Germany,
2 Centro de Investigacion de Metodos Computacionales,
(UNL-CONICET), Santa Fe, Argentina
Content: Isotachophoresis (ITP) is an electromigrative separation technique which is often applied for preconcentration of analytes and removal of sample matrix. Its application is generally restricted to analytes of high to intermediate effective electrophoretic mobility and thus often not applicable for weak acids and bases as the mobility window is impaired by H+ or OH- at very low or high pH values required to sufficiently charge the analytes. Thus, the preconcentration of all proteinogenic amino acids is impossible in aqueous ITP as some analytes are excluded from the isotachophoretic stack and migrate zone-electrophoretically in the terminating electrolyte.[1]
To enable an ITP preconcentration of all proteinogenic amino acids as cations, a non-aqueous electrolyte system using the aprotic solvent dimethylsulfoxide (DMSO) was developed by Kler et al.[1]. Mass spectrometry (MS) and capacitively coupled contactless conductivity detection (C4D) were applied to monitor the ITP processes. By ITP-MS, different capillary inner diameters and changes in the electrolyte system were examined.
In this study, we use this ITP system as the first dimension in ITP/CE-MS implemented in a modular setup for on-line column-coupling of ITP and CE-MS using a microfluidic glass interface with intermediate C4D detection. We here show the possibilities and limitations for the effective ITP preconcentration of proteinogenic amino acids before CE-MS analysis varying injection parameters, capillary inner diameters and lengths in the first dimension. Matrix effects and matrix

www.ISC2016.ie @isc2016 #isc2016 /isc2016174
POSTER ABSTRACTS Electrodriven Separation Methods
removal is investigated using high salt conditions.References: [1] P. Kler, C. Huhn, “Non-aqueous
electrolytes for isotachophoresis of weak bases and its application to the comprehensive preconcentration of the 20 proteinogenic amino acids in column-coupling ITP/CE–MS”, Analytical and Bioanalytical Chemistry, 406 (2014) 7163-7174.
Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis, Electromigrative separation, Isotachophoresis
P01-001-115 - Comparison of electromigrative enrichment possibilities: Capillary isotachophoresis, capillary electrophoresis and free-flow electrophoresis
T. Melzer*, G. Weber 1, A.-J. Wicht 2, C. Huhn 2
1 FFE Service GmbH, Munich, 2 Institute for Physical and Theoretical Chemistry, Eberhard
Karls Universität Tübingen, Tübingen, Germany
Content: Sample preparation is crucial for a large number of different analytical approaches. Especially trace analysis demands for effective sample preconcentration or enrichment. For extremely complex samples, such as in environmental and bioanalysis, prefractionation for a reduction of the complexity is required. Furthermore, matrix effects impairing the measurements are to be avoided by separating analytes from the matrix.
When it comes to polar and ionogenic analytes, electromigrative separation methods are of high interest as they are
applicable to analyze substances which would elute in the dead volume of modern RPLC. Here, methods such as capillary electrophoresis (CE), capillary isotachophoresis (ITP) and free-flow electrophoresis (FFE)[1] are of interest.
In this study, a method to concentrate different pharmaceuticals of environmental concern by CE was developed. In this case, the analytes were forced to migrate from a large volume at the inlet of a commercial CE instrument to the outlet with a smaller volume by applying an electric field. E.g. we used 3 mL at the inlet and 100 µL at the outlet vial to preconcentrate various cations in acetic acid. Anionic or cationic compounds can be separated from neutrals according to the chosen pH. Optimization of the process mainly included measures to effectively sample the large volume. Enrichment was characterized by LC-MS.
Anionic ITP was used in a (semi)preparative mode for enrichment and fractionation. High throughput was reached using free-flow isotachophoresis (FFITP). While the volumes in capillary based CE and ITP are in the low µL range, FFITP proved able to handle up to 30 mL/h to yield a preconcentration factor of 15 for 8 anion fractions. Analytes with pI values up to 8.5 could be included in the mobility window. As a real sample, river water was enriched and fractionated by FFITP and subsequently analyzed by LC-MS.References: [1] FFE Service GmbH, “Technology”,
http://www.ffeservice.com/technology, (accessed 25th May 2015)
Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis, Enrichment, Free-flow isotachophoresis

175www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSElectrodriven Separation Methods
P02-002-001 - Improvement of a SDS-CE method for the control of protein enrichment and depletion from biological samples to enhance automation and quantification
M. Meixner 1,*, C. Huhn 1
1 Chemistry Department – Institute for Physical and Theoretical Chemistry, University Tuebingen, Tuebingen, Germany
Content: Low abundance proteins in blood are known to be reliable biomarkers for diseases like e.g. cancer and Alzheimer’s disease. Identification and quantification is still difficult and cumbersome due to the large number of proteins as well as their large concentration range which comprises 12 orders of magnitude. Therefore, my work focuses on the development of a robust and quick technique for a reduction of the concentration range combining depletion and enrichment using a hexapeptide ligand library commercialized as ProteoMiner-beads1.
Currently, the enrichment/depletion experiments are mostly followed by SDS-PAGE experiments, which is rather laborious. In this study, we use capillary sieving electrophoresis with sodium dodecyl sulfate with UV-detection (SDS-CE-UV), a promising alternative for the classic slab gel electrophoresis. The SDS-CE method was greatly enhanced using a preinjection plug made of SDS solutions in various concentrations, which greatly improves the baseline and separation efficiency for a separation buffer made of 100 mM Tris-CHES and 0.1% SDS and polyethylene oxide (Mr = 600000 Da)2. With the optimized method, analysis times of 28 min, including preparation of the
capillary, were reached; the capillary-based system allowed automated analysis of model protein mixtures using commercially available standard chemicals instead of cost-intensive kits.
We here show that no further sample processing is necessary for eluates from the hexapeptide ligand library so that a fast and effective direct quantitative analysis is possible.References: 1. C. Huhn, L.R. Ruhaak, M. Wuhrer, A.M. Deelder,
Journal of Proteomics 2012, 75(5), 1515-1528.2. T. Kaneta, D. Yamamoto, T. Imasaka,
Electrophoresis 2009, 30(21), 3780-3785.Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis, capillary sieving electrophoresis, low abundance proteins enrichment
P02-002-002 - Contactless conductivity detection for assaying pancreatic lipase activity and inhibition by capillary electrophoresisS. Fayad*, J. Vilmen 1, R. Nehme 1, P. Morin 1
1 Institut de chimie organique et analytique (ICOA), Orleans, France
Content: Pancreatic Lipase is one of the main digestive enzymes secreted from the pancreas that hydrolyzes dietary fat molecules in the human digestive system. It converts triglyceride substrates found in ingested oils to monoglycerides and free fatty acids [1]. It has vast importance for our health, not just in regard to the commonly recognized diseases of the fat metabolism such as overweight and underweight, but also for skin problems, autoimmune diseases and cancer [2].

www.ISC2016.ie @isc2016 #isc2016 /isc2016176
POSTER ABSTRACTS Electrodriven Separation Methods
In this study, capillary electrophoresis (CE) was used for the first time for evaluating pancreatic lipase kinetics (maximum velocity Vmax and Michaelis–Menten constant Km) toward paranitrophenylbutyrate (PNPB). The affinity of standard inhibitor (epigallocatechine gallate) was also assessed with the determination of the half maximal inhibitory concentration (IC50). The pre-capillary approach was chosen since it is very simple to conduct. The products were detected and quantified with a contactless capacitively coupled conductivity detector (C4D) allowing the evaluation of lipase kinetics. This study shows, that capillary electrophoresis with contactless conductivity detection can be very useful for monitoring lipase activity. Comparing to the conventional spectrophotometric method, the CE method developed here is simple, automated, economic, rapid and robust. Results very well compared with those reported in literature using the conventional method.References: [1] K. Satouchi, T. Mori, S. Matsushita,
Characterization of inhibitor protein for lipase in soybean seeds, Agricultural and Biological Chemistry 38 (2014) 97-101.
[2] C.Y. Jeong, Y.D. Han, J.H. Yoon, H.C. Yoon, Bioelectrocatalytic sensor for triglycerides in human skin sebum based on enzymatic cascade reaction of lipase, glycerol kinase and glycerophosphate oxidase, Journal of Biotechnology 175 (2014) 7–14.
Disclosure of Interest:None DeclaredKeywords: Contactless conductivity detection , Capillary electrophoresis, Kinetic constants

177www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
Food and Health, Food Safety and Authenticity
P01-001-005 - Synthesis of molecular imprinted polymers for the selective extraction of organophosphorus in vegetable oilsS. Boulanouar Al Massati 1,*, S. Mezzache 2, A. Combès 1, V. Pichon 1 3
1 Department of Analytical, Bioanalytical Sciences and Miniaturization (LSABM), UMR CBI 8231 ESPCI Paris /CNRS, PSL Research University , ESPCI ParisTech, Paris,
2 Chimie analytique, L’Oréal Recherche avancée, AULNAY SOUS BOIS, 3UPMC, Sorbonne Universités, Paris, France
Content: The increasing use of pesticides for agricultural purposes cause serious risks to the human health. Their determination at very low concentration levels in vegetable oils constitutes a real analytical challenge. Hence, their analysis requires often an extraction and a purification step followed by chromatographic analysis. Therefore, extraction sorbents providing a molecular recognition mechanism can be developed and applied to the selective extraction in order to obtain more sensitive and reliable quantitative analysis. Aim of this work was to develop these sorbents for organophophorus (OPPs) that can be found in vegetable oils. Even if molecularly imprinted polymers (MIPs) have already been developed for the extract of two OPPs in a vegetable oil, purpose of this work was to develop a MIP able to selectively trap numerous OPPs belonging to a wide range of physico-chemicals properties.
MIPs for OPPs were synthesized by the radical polymerization of organic monomers but also by the Sol-Gel approach using organosilanes. Different conditions of
synthesis were screened in order to study the creation of specific cavities able to trap up to 12 studied OPPs. The evaluation of the selectivity brought by the resulting MIPs was done by studying the retention of the OPPs in pure media at first time. The organic solvents involved in the SPE procedure were also selected in order to favor the development of selective interactions and to remove non-specific ones. These non-specific interactions were estimated by the retention of OPPs in non-imprinted polymer
These sorbents were applied to the selective extraction of OPPs from different vegetable oils to evaluate their potential in terms of ability to remove matrix components. Liquid chromatography (LC) coupled to mass spectrometry (MS) was used to obtain a limit of quantification, which were lower than maximum residual limits (MRLs) established for each OPPs.Disclosure of Interest:None DeclaredKeywords: moleculary imprinted polymers , organophosphorus, vegetable oils
P01-001-014 - Sensitive micronutrient analysis in spices and monofloral honeys by capillary electrophoresis coupled with LIF detectorP. Hashemi 1, G. Gezek 1, H. Kaygusuz 1, F. Tezcan 1, F. B. Erim*
1 Chemistry, Istanbul Technical University, Istanbul, Turkey
Content: During the last decade capillary electrophoresis as a separation technique has gained a great popularity in food analysis. Low costs, low sample and reagent consumption, ease of automation, high resolution, and less analyzing time are

www.ISC2016.ie @isc2016 #isc2016 /isc2016178
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
some of the superiorities of this technique. Hyphenation with a laser induced fluorescence detector has led CE to reach higher sensitivities. Our work is the first study that gives the vitamin B2 level in saffron, salvia species and monofloral honeys, using a trustable, easy, and sensitive capillary electrophoretic method and therefore provides the missing information on the nutritional value of these precious food. The limit of detection (LOD) and limit of quantification (LOQ) values of vitamin B2 for the method were 4.15 and 13 nM, respectively. Moreover, vitamin B2 is shown to be one of the main components to control honey authenticity of 14 monofloral honeys and 6 honeydew honey samples from different botanical origins and different regions of Anatolia, using principal component analysis.Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis-laser induced fluorescence, Saffron, Vitamin B2
P01-001-015 - Impact of different forage types (grass, grass/clover and TMR) on the sensory quality and volatile profile of bovine milk in Ireland
H. Faulkner 1,*, T. F. O’callaghan 1, S. Mcauliffe 2, D. Hennessy 2, C. Stanton 3, M. G. O’sullivan 4, J. P. Kerry 4, K. N. Kilcawley 5
1 Department of Food Biosciences, Teagasc Food Research Centre, Moorepark, Fermoy, Co. Cork, Ireland., Teagasc/UCC,
2 Animal & Grassland Research and Innovation Centre, Teagasc Moorepark, Fermoy, Co. Cork, Ireland, Teagasc,
3 a Department of Food Biosciences, Teagasc Food Research Centre, Moorepark, Fermoy, Co. Cork, Ireland., Teagasc/UCC,
4 School of Food and Nutritional Sciences, University College Cork, Ireland, UCC,
5 Department of Food Biosciences, Teagasc Food Research Centre, Moorepark, Fermoy, Co. Cork, Ireland., Teagasc, Cork, Ireland
* Corresponding author:e-mail: [email protected] (K.Kilcawley)
Content: No scientific studies have been undertaken to evaluate sensorial differences of bovine milk produced from different feeding systems in an Irish context. This study evaluated the sensorial and volatile properties of milk produced from 54 Friesian cows on three separate diets; grass only (G), grass/clover (GC) and total mixed rations (TMR) over a single season. The feed (G, GC & TMR) and milk was sampled in duplicate at each stage of lactation. Milk was pasteurised (HTST) and homogenised prior to sensory evaluation, using acceptance (hedonic) and Ranking Descriptive Analysis (RDA). Grass fed pasture produced the best preferred milk and also rated higher for a number of key attributes (Creamy Flavour, Dairy Sweet Flavour, Colour). Some negative sensory attributes were associated with GC and TMR milks. Volatile profile analysis by gas chromatography mass spectrometry could distinguish each milk type and major differences in the volatile components of the G, GC and the TMR forage samples were also evident. A direct correlation between forage and some volatile components was evident. Some potential biomarkers were identified that could be used distinguish milk from grass based feeding systems.Disclosure of Interest:None DeclaredKeywords: GC/MS, SPME, Volatiles

179www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
P01-001-022 - Determination of Boswellic resins origin and authenticity using two complementary methodologies: TLC-MALDI/TOF and GC-MS
Z. Jemmali 1,*, A. Chartier 1, D. Da Silva 1, E. Destandau 1, C. Elfakir 1
1 Institut de Chimie Organique et Analytique (ICOA), UMR 7311, Univ. Orleans, CNRS, Orleans, France
Content: Boswellia resins are hydrocarbon secretions of frankincense trees distributed in India (B. serrata), Arabia (B. sacra) and Africa (B. carterii and B. neglecta) and are composed mainly of terpenoids. Well known for their health benefits, they are used in drug composition and as food supplements. Their commercial usage demands a rapid identification of the resins to certify their origin and ensure quality control.
The chemical characterization of this material is a complex task due to the wide array of polarity and volatility of its constituents. Thus, in our study, a chemical differentiation of four Boswellia resins has been achieved.
First, thin layer chromatography (TLC) was developed to investigate quickly the resin composition in major markers. To have a clear assignment of the obtained spots, matrix assisted laser desorption / ionization and time of flight mass spectrometry (MALDI-TOF) was directly combined to TLC. This methodology remains challenging given the low molecular weight of terpenoids thus several parameters were optimized (nature of matrix, ionization mode,…). This hyphenated technique gives a clear identification of TLC spots with significant spatial resolution for boswellic acids and
their O-acetates and allows to distinguish resins from their Indian, Arabian or African origin.
In order to have more information about their minor markers composition, a gas chromatography-mass spectrometry (GC-MS) method was developed for a simultaneous determination of mono- to triterpenoids in resins. The method was validated in terms of detection limits (100 µg.L−1 - 200 µg.L−1), linearity and repeatability. Qualitative and quantitative GC analysis showed significant differences between the two closely related African resins for monoterpenoids (α-thujene, verbenol,…), diterpenoids (incensole and serratol) and triterpenoid distribution.
The complementary of these two methodologies gives a powerful approach for fast and reliable differentiation of Boswellia resins.Disclosure of Interest:None DeclaredKeywords: GC-MS, Natural resins, TLC-MALDI/TOF
P01-001-023 - Innovative Screening for Non-Intentionally Added Substances in Food Contact MaterialsC. Kanakaki 1,*, N. Steiner-Reischütz 2, M. Pyerin 2, V. Osorio Piniella 1
1OFI, Wr. Neustadt, 2OFI, Vienna, Austria
Content: Current guidelines of the European Regulation (EU) 10/2011 for the first time mention the obligation of the manufacturers of food contact materials (FCM) or articles made of plastic to evaluate their products with respect to non-intentionally added

www.ISC2016.ie @isc2016 #isc2016 /isc2016180
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
substances (NIAS). NIAS are substances which may be present e.g. due to impurities in raw materials used, or they are created during processing, such as reaction and degradation products. They can cause, especially if they are low molecular weight, volatile components, sensory impairments of the packaged foods or even lead to health risks.
In our research project, we are evaluating different automated sample pre-concentration techniques and their combination with gas chromatographic mass spectrometric methods for the identification and quantification of NIAS. Targeting the establishment of an analytical basis for the optimization of packaging materials and their processing conditions (i.e. sterilization), we analyze the original materials as well as their products from specific migration studies, so that we can significantly contribute not only in the production of organoleptic neutral materials, but also in the enhancement of product safety.
To facilitate this purpose, we selected to investigate the dynamic headspace (DHS) and the solid phase micro-extraction (SPME) technique, coupled to a gas chromatographic-mass spectrometric (GC-MS) system. The basic principles of these techniques will be briefly discussed in our contribution, together with the optimization steps of the developed methods and a number of application examples.Acknowledgements: We would like to acknowledge Prof. Erich Leitner and his research group at the Technical University of Graz for the technical support and co-operation. Financial support of this study, provided by the Austrian Research Promotion Agency (FFG, Project ‘‘CoinSenses’’, Project Number
415403), is also gratefully acknowledged.Disclosure of Interest:None DeclaredKeywords: dynamic headspace, non-intentionally added substances, solid phase micro-extraction
P01-001-030 - A Novel Superior Gas Chromatographic Method for Free Fatty Acid Analysis of Dairy ProductsD. T. Mannion*
Content: Quantification of free fatty acids (FFA) in dairy products by gas chromatography flame ionization detection is an important requirement for quality, research, nutritional, authenticity and legislative purposes. Current methods are time consuming, labour intensive and environmentally unfriendly. This study involved the development of an alternative novel derivitisation method using butylation, incorporating automation, reducing solvent usage that is applicable for a wide range of dairy products. This method was validated and compared against pre-existing widely used methods for the quantification of FFA in representative dairy products varying in lipid composition and degree of lipolysis. Significant limitations were evident with the existing methods; accumulative column phase deterioration, irreversible FFA absorption, loss of polyunsaturated free fatty acids, periodic emergence of interfering artefact peaks and issues with quantification of highly volatile butyric acid. The novel butyl derivitisation method overcame all the major issues associated with existing methods. In addition the derivitisation step was automated, reducing solvent usage and increasing sample throughput and accuracy, with limits of

181www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
quantification and detection comparable to pre-existing methods. Overall the novel butyl derivitisation method is more suitable for research and industrial applications for the quantification of FFA in dairy products.Disclosure of Interest:None DeclaredKeywords: Dairy, derivitization, gas chromatography
P02-002-004 - Establishment of sugarcane honey authenticity using the highthrouput potential of chromatography
P. Silva 1, J. Freitas 1, R. M. Perestrelo 1,*, C. L. Silva 1, F. Nunes 2, J. S. Câmara 1
1 CQM, Madeira University, Funchal, 2CQ-VR, University of Trás-os-Montes and Alto Douro, Vila Real, Portugal
Content: The food authenticity has become a global problem, increasing the importance of markers identification to guarantee the authenticity and typicality of products and consumer safety. The sugarcane honey (SCH) produced in Madeira Island, known as ‘mel-de-cana’, is widely recognized for its excellent quality and sui generis organoleptic properties [1]. The production of good quality treacle are affected by many factors, as the sugarcane cultivars, agricultural treatments, soil and climatic conditions, as well by manufacturing process, packing and storage.
The purpose of the work is to establish the SCH volatile profile produced at Madeira Island by a certified producer, in order to establish the authenticity and typicality of the product in order to avoid frauds expressed by the commercialization of adulterated SCH. Solid-phase microextraction in headspace mode (HS-
SPME) combined with gas chromatography-mass spectrometry (GC-MS) methodology was applied to establish the volatomics profile of SCH. The HS-SPME extraction was optimized for stationary phase (PA, PDMS, CAR/PDMS, PDMS/DVB and DVB/CAR/PDMS), temperature (30, 40, 50, 60, 70 and 80 ºC) and time (30, 45, 60 and 75 min), and validated according with IUPAC guidelines. The optimal conditions for HS-SPME extraction were obtained using a DVB/CAR/PDMS at 50 ºC for 60 min. Different volatomics profiles for certified and uncertified samples can be recognized. After analysis of profiles were identified 80 volatile compounds belonging distinct chemical classes, mainly aldehydes, alcohols, ketones and furans. The application of the discriminant analysis (PCA) showed two separated clusters for the samples from certified and uncertified producers. From the results, it can be concluded that GC-MS combined with HS-SPME revealed as a very appropriate sampling technique to establish the authenticity and typicality of SCH.References: [1] Ruiz-Matute, A.I., et al., J Food
Compost Anal.. 2010, 23, 260-263.Acknowledgements: Fábrica Mel-de-Cana Ribeiro Sêco de V. Melim, Lda for the sample. ARDITI for the PhD Grant, FCT for the PhD grant (SFRH/BD/97039/2013), REDE/1508/RNEM/2015), HCV -New INDIGO/0003/2012; EU Project, QUI-Madeira-674.Disclosure of Interest:None DeclaredKeywords: sugarcane, HS-SPME, GC-qMS

www.ISC2016.ie @isc2016 #isc2016 /isc2016182
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
P02-002-005 - Chromatographic data as useful tool to evaluate the impact of storage time and temperature on furanic derivatives evolution in fortified wines
R. M. Perestrelo*, E. Rodriguez 1, P. Pedro 1, J. S. Câmara 1
1CQM, Madeira University, Funchal, Portugal
Content: Furanic derivatives (FDs) are a group of compounds formed during non-enzymatic browning reactions, such as caramelization, which involves the degradation of sugars, and the Maillard reaction, involving Amadori rearrangement. Furan and its derivatives are of high concern to human health due to their potential toxicity. Some furan derivatives can be used as potential wine aging markers [1].
The main goal of this work was to evaluate the effect of some winemaking procedures, namely the storage time (fortnight sampling during 5 months) and heating temperatures (30, 45 and 55 ºC) on FDs evolution (e.g. 2-furfural, 5-hydroxymethyl-2-furfural, 1-(2-furanyl)-ethanone, 5-methyl-2-furfural) in fortified wines, through semi-automatic microextraction by packed sorbent (MEPS) combined with reversed-phase ultra-high pressure liquid chromatography (RP-UPLC) equipped with a photodiode array (PDA) detection system. The MEPS/UPLC-PDA performance was assessed and good linearity was obtained with a regression coefficient (r2) higher than 0.992, and low detection (LOD) and quantification limits (LOQ) were achieved for both synthetic wine models. The recovery for the target FDs in dry/medium dry wines ranged from 74 to 97 %, whereas for sweet/medium sweet wines from 84 to 99 %.
With regard to the FDs, a tendency was observed as its abundance increased with storage time and temperature in both sweet/medium sweet and dry/medium dry wines. Moreover, it was noticed that sweet/medium sweet wines have a greater FDs abundance than dry/medium dry wines. According to the obtained data, it is possible to conclude that target FDs can be used as a potential wine aging markers and the developed methodology allows a quick, high-throughput and efficient FDs analysis.References: 1. Perestrelo, R.; Barros, A.S.; Câmara,
J.S.; Rocha, S.M. Journal of Agricultural and Food Chemistry, 59 (2011), pp 3186-3204.
Acknowledgements: The authors acknowledge the FCT for the PhD grant (SFRH/BD/97039/2013), REDE/1508/RNEM/2015), QUI-Madeira-674.Disclosure of Interest:None DeclaredKeywords: Fortified wine, HS-SPME, GC-qMS
P02-002-006 - Determination of sulfonamides in animal tissues by modified QuEChERS and liquid chromatography tandem mass spectrometry
M.-R. Fuh 1,*, C.-H. Wen, S.-L. Lin1 Department of Chemistry, Soochow University, Taipei,
Taiwan, Province of China
Content: Veterinary drugs such as sulfonamides have been widely applied for treating various infections in livestock. However, the occurrence of sulfonamide residues in edible animal tissues might cause potential adverse effects, such as allergy and thyroid follicular tumor, on human health. Due to the health concerns, maximum residue limits (MRLs) have been set to regulate the residual sulfonamides

183www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
found in food products by many countries. For example, the European Union has set a MRL at 100 ng g-1 for total sulfonamides in edible animal tissues.
In this study, the salting-out solvent extraction and dispersive solid-phase extraction (dSPE) clean-up steps in QuEChERS (quick, easy, cheap, effective, rugged, and safe) method were optimized to reduce matrix effect and efficiently extract 9 target sulfonamides from a variety of edible animal tissues (beef, pork, chicken, pork liver and beef tripe). The extracted sulfonamides were then analyzed using liquid chromatography tandem mass spectrometry (LC-MS/MS). Good extraction recoveries (74.0 - 100.3% in five different sources of animal tissues; n= 3) with minor matrix effect (≦ 10%) were obtained using the proposed method. Limits of detection were estimated at 0.02-0.08 ng g-1 and good linearity ranged from 0.125-12.5 ng g-1 were determined. In addition, intra-day/inter-day precision (1.0-10.5% / 0.4-8.0%) and accuracy (95.2-107.1% / 97.8-102.1%) were measured. The applicability of the newly developed method for food safety was demonstrated by determining the occurrence of target sulfonamides in various edible animal tissues.References: 1. R.P. Lopes, É.E. de Freitas Passos, J.F. de
Alkimim Filho, E.A. Vargas, D.V. Augusti, R. Augusti, Food Control 28 (2012) 192-198.
2. H. Abdallah, A. Arnaudguihem, F. Jaber, R. Lobinski, J. Chromatogr. A 1355 (2014) 61-72.
Disclosure of Interest:None DeclaredKeywords: LC-MS, sulfonamide
P02-002-007 - Optimizing the Separation of 14C-labeled Compounds for Two Detectors: Mass Spectrometry and 14C-Radio-Detection
M. Speitling*
Content: During the registration process of plant protecting compounds several studies are performed for which 14C-labeled compounds are used. By this, the metabolization of the active ingredient (AI) is investigated both quantitatively and qualitatively. The data can be generated on one LC-system equipped with two detectors: the quantitative results from a 14Cradiodetector, the qualitative information from a mass spectrometer. The impact of the variation of the flow path of the chromatographic system and of the cell volume of the radio detector on the peak parameters in both channels is shown.References: Cuyckens F, Koppen V, Kembuegler R,
Leclercq L., J Chromatogr A., 1209(1-2), 128-135, 2008
Disclosure of Interest:None DeclaredKeywords: 14C radiodetection, LC-MS
P02-002-008 - Separation, identification and mutagenicty screening of photo-degradation products of Tartrazine (E102) dye in a commercial soft drink
K. Yamjala*, M. Subramania Nainar 1
1Pharmaceutical Analysis, JSS University, Udhagamandalam, India
Content: The present study deals with the separation and identification of the potential

www.ISC2016.ie @isc2016 #isc2016 /isc2016184
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
photo-degradation products of tartrazine (E102) dye present in commercial soft drink. Six beverages (containing E102 dye 20 ppm, citric acid 0.1%, sucrose 8%, fructose 7%, nata de coco 25% and cantaloupe juice 25% as indicated on label) were subjected to natural sunlight irradiation during the month of September ( ̴21.k lux and >11 UV light index) of Ooty, India. The results indicated that colour of the beverage changed gradually from lemon yellow to pale yellow and then to colourless as clearly evidenced by naked eye observation. After the exposure the bottles at t0, t1, t2 and t3 were opened, filtered through 0.22 µm nylon membrane filter and subjected to UHPLC-MS/MS analysis. In order to understand the degradation kinetics of E102, model solutions were also subjected to sun irradiation along with beverages and the half-life times were calculated. The photo-degradation products of E102 dye in beverage were identified by non-target screening analysis (Since the most abundant product ion in the mass spectra of E102 was the sulphonate radical anion with an m/z of 79.9570) by UHPLC-Q-TOF. Six degradation products were identified and their structures were elucidated on the basis of the elemental composition and accurate mass data using a software tool. The mutagenicity of the degradation products was evaluated through the invitro bacterial reverse mutation assay using five strains of bacteria both in presence and absence of metabolic activation system (S9). Statistically, Dunnett’s test is used an aid to evaluate the mutagenicity results. References: Gosetti F, Gianotti V, Angioi S, Polati
S, Marengo E, Gennaro M (2004) Oxidative degradation of food dye E133 Brilliant Blue FCF: liquid chromatography–electrospray mass spectrometry identification of the degradation
pathway. J. Chromatogr. A 1054: 379-387.Acknowledgements: This work was supported by ICMR, New Delhi (3/1/2/32/2014-Nut).Disclosure of Interest:None DeclaredKeywords: Mutagenicity, Photo-degradation products, Tartrazine dye
P02-002-009 - Development Of The Chiral Separation of L-AND D- β-N-METHYLAMINO-ALANINE Using Response Surface DesignA. Combes 1,*, J. vial 1, V. Pichon 1 2
1 LSABM - UMR CBI 8231, ESPCI - Paris/CNRS, 2 Sorbonne universite, UPMC, 10 rue vauquelin 75005
Paris, France
Content: Recently the cyanotoxin β-methylamino-L-alanine (BMAA) has received renewed attention as an environmental risk factor for sporadic cases of a neurodegenerative disease, the amyotrophic lateral sclerosis1. BMAA has been described as present at different concentration levels in various strains of cyanobacteria. Although no biosynthetic pathway has been investigated, the hypothesis is that the toxin is produced by the cyanobacteria themselves. Nevertheless, the detected BMAA could also be issued from others metabolites. Discrimination between the L and D enantiomers of BMAA may allow to provide evidence in favor of one of these hypotheses.
Therefore, the chiral separation of the L- and D-BMAA and the L- and D-2,4 diaminobutyric acid, one of its natural isomer, was performed on a zwitterionic chiral stationary phase Chiralpak ZWIX (+). The objective was to maximize resolution during the separation step and sensitivity during MS detection in MRM mode.

185www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
The most common approach consists in optimizing each factor separately, thus requiring numerous experiments. Therefore, a screening design was used to determine the most important factors affecting the separation in order to reduce the number of experiments for the subsequent optimization design of experiment. In a first step, six different parameters have been studied with a 26-2 factorial fractional design. The influence of these parameters on the resolution and on the sensitivity in the LC-MS/MS analysis was assessed. According to the results obtained with the screening design, a response surface design (central composite design with 4 factors and 5 levels for each factors -6 center points- divided in 3 blocks of experiments) was implemented for the four major factors influencing the resolution: the amount of water, the methanol/acetonitrile ratio, the concentration of salt and the acid/base ratio in the mobile phase.References: 1 -Gil J. et al., Rev neurology 163 (2007) 1021-302 -Murch S.J., et al., PNAS 101 (2004) 12228-31Disclosure of Interest:None DeclaredKeywords: Zwiterionnic chiral separation, L- and D-BMAA, Screening design, Design of experiments
P02-002-010 - Authentic Turkish Bee Pollen: Analysis of Organic Acids by Capillary Electrophoresis
Z. Kalaycıoğlu 1,*, F. B. Erim 1
1Chemistry, Istanbul Technical University, Istanbul, Turkey
Content: Honeybee-collected pollen is a natural food product packed by honeybees into pellets and later harvested as food for humans. Bee pollen is recognized as potent
antibiotic, antineoplasic, and antioxidant agent due to its high carbohydrate, lipid, protein, vitamin, and amino acid contents [1]. Recent investigations have drawn attention to the importance of organic acid profiles for the description of functional food quality such as freshness and purity. However, there has been no analytical study for the analysis of individual organic acids in bee pollen samples. The purpose of this study was to analyze the organic acid content of 10 different bee pollens collected from Turkey. A capillary electrophoresis method, which was recently applied by Tezcan et al. for the analysis of the organic acids in some authentic Turkish honeys, was used here for the analysis of organic acids in the bee pollen samples with some modifications [2]. The optimal separation buffer was 5 mM PDC (2,6-pyridinedicarboxylic acid), 0.1 mM CTAB (N-Cetyl-N,N,N-trimethylammonium bromide) at pH 5.50. Gluconic acid was the predominant organic acid while oxalic, tartaric, malic, citric, acetic, and lactic acids were determined at minor concentrations.References: [1] S. Bogdanov, Bee Product Science, p. 3, (2014).[2] F. Tezcan, S. Kolaylı, H. −ahin, E. Ulusoy, F.B.
Erim, Journal of Food and Nutrition Research, 33-40, (2011).
Disclosure of Interest:None DeclaredKeywords: Bee pollen, Capillary electrophoresis, Organic acids

www.ISC2016.ie @isc2016 #isc2016 /isc2016186
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
P02-002-011 - Quantitation of various capsaicinoids in different chili varieties by ultra-high performance liquid chromatographyA. Schmidt*, G. Fiechter 1, E.-M. Fritz 1, H. K. Mayer 1
1 Food Chemistry Laboratory, University of Natural Resources and Life Sciences Vienna - BOKU, Vienna, Austria
Content: The reason for the pungent, burning or stinging sensation caused by irritants such as capsaicin can be explained on a molecular level by the interaction with special receptors which are also activated by thermal stimuli; hence the common perception of “heat” while eating chili containing foods. Besides “classical” sensorial/organoleptic assays for quantifying “hotness”, nowadays analytics mainly emphasizes HPLC methods focusing on the determination of individual capsaicinoids rather than on overall pungency. Thus, the aim of this work was to establish a rapid UHPLC method for the determination of capsaicin and dihydrocapsaicin as being the most powerful pungent capsaicinoids found in chili fruits.
Based on an implemented HPLC-UV method, elution was adapted to UHPLC and further optimized resulting in a significantly shortened separation time (1.7 vs. 20 min), hence presented a most feasible approach for high-throughput pungency profiling of chili containing foods. Using UV and fluorescence detection, the established method was validated for the analysis of both capsaicinoids regarding linearity, precision, recovery and suitability of the sample preparation procedure. Compared to UV, fluorescence detection showed superior sensitivity with LODs ranging
around 1 ng/mL, thus proved suitable for analyzing even very low pungent chili products. Moreover, to give an estimate on the massive variability between chilies, typical varieties were characterized for their intrinsic pungency with total capsaicinoid amounts ranging from 200 ng/g for Jalapeño to 28000 ng/g for Bhut Jolokia corresponding to “mild” 3000 or “hot” 450000 Scoville heat units, respectively [1].References: [1] N. Kozuke, J-S. Han, E. Kozuke,
S-J. Lee, J-A. Kim, K-R. Lee, C. E. Levin and M. Friedman (2005). Analysis of Eight Capsaicinoids in Peppers and Pepper-Containing Foods by High-Performance Liquid Chromatography and Liquid Chromatography-Mass Spectrometry. J. Agric. Food Chem., 53, 9172-9181
Disclosure of Interest:None DeclaredKeywords: Capsaicinoids , Chili , Ultra-high Performance Liquid Chromatography
P02-002-012 - A novel ultra-high performance liquid chromatography method for the rapid assessment of the native vitamin B6 spectra in bovine milk
A. Schmidt*, M. Schreiner 1, H. K. Mayer 1
1 Food Chemistry Laboratory, University of Natural Resources and Life Sciences Vienna - BOKU, Vienna, Austria
Content: Vitamin B6 is an essential food component and its intake is necessary for maintaining many functions of the human organism. Although being part of the human diet for ages, its adequate quantitation is still challenging today. It can occur in up to seven different vitamers, namely: pyridoxine, pyridoxal, pyridoxamine as well as their mono phosphate analogues

187www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
and 4-pyridoxic acid. As those vitamers do not exhibit the same stability during thermal food processing, the investigation of the native spectra is of great importance.
In this work, we present an ultra-high performance liquid chromatography (UHPLC) method for milk in order to study the native spectra of B6 vitamers. It features a simplified sample preparation, which together with UHPLC technology enables high-throughput routine analysis. Separation of six relevant B6 vitamers was achieved within 5.8 minutes, by a total runtime of 11 min per analysis [1]. Furthermore, the method is suitable for the quantification of vitamin B2 and its phosphate analogue flavin mononucleotide; both are of great relevance in bovine milk. As thermal induced interconversions as well as inactivation of B6 vitamers in milk have been observed [2], it is vital to assess the full native vitamer spectra in order to estimate possible losses and alterations of vitamin B6 in bovine milk during processing.References: [1] R. Gatti and M. G. Gioia (2005). Liquid
chromatographic determination with fluorescence detection of B6 vitamers and riboflavin in milk and pharmaceuticals. Analytica Chimica Acta, 538, 135-141
[2] A. V. Pingali and P. R. Trumbo (1992). Effect of Sterilization of Milk on Vitamin B-6 Composition and Bioavailability. Journal of Agricultural and Food Chemistry, 40, 1860-1863
Disclosure of Interest:None DeclaredKeywords: Bovine Milk, Ultra-high Performance Liquid Chromatography , Vitamin B6
P02-002-013 - Non-targeted characterization of Physalis alkekengi L. pyhsalins by TLC–MS
E. Kranjc 1,*, I. Vovk 1, A. Albreht 1, V. Glavnik 1, M. Križman 1, N. Ferant 2
1 Laboratory for Food Chemistry, National Institute of Chemistry, Ljubljana, 2Institute of Hop Research and Brewing, −alec, Slovenia
Content: Physalis alkekengi L. is of great medicinal value in traditional Chinese medicine due to its anti-inflammatory, antipiretic, diuretic, antimicotic, antitumor, antibacterial and immunomodulatory effects. Physalins, steroidal compounds possessing a unique 13,14-seco-16,24-cycloergostane skeleton characteristic for genus Physalis, are known for its highly oxygenated, complex, fused-ring system. These secondary metabolites represent pharmacologically potent constituents of P. alkekengi L., therefore their characterization has attracted much attention in recent years.
Until now analyses used for determination of physalins were mainly performed by HPLC–UV [1] or HPLC–MSn techniques [2]. In our study several TLC methods for a preliminary screening of physalins from P. alkekengi L. were developed. Since very few physalin standards are commercially available (only physalin L was obtainable), an optimization of chromatographic parameters was based on the resolution of compounds assumed to belong to the physalin class of compounds present in P. alkekengi L. orange husk extracts. Hyphenation of TLC with MS enabled detection of physalins in a non-targeted approach. Furthermore, classification of different physalin types based on their MS/MS fragmentation patterns was attempted. Our TLC(–MSn) separation method based

www.ISC2016.ie @isc2016 #isc2016 /isc2016188
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
on silica gel layer was extended to various silica gel bonded stationary phases; reversed-phase as well as hydrophilic layers were tested for separation of P. alkekengi L. physalins. Due to the high complexity of investigated P. alkekengi L. extracts, grafted two-dimensional-TLC (2D-TLC) was also applied. Preliminary screening of physalins in extracts of different parts of P. alkekengi L. harvested in different stages of maturity was performed.References: 1. B. Xu, C. Ju, H. Guan, L. Xu, N. Xu, B. Wang, J.
Med. Plants Res. 6 (2012), 2438-2442.2. C. Huang, Q. Xu, C. Chen, C. Song, Y. Xu, Y.
Xiang, Y. Feng, Q. Ouyang, Y. Zhang, H. Jiang, J. Chromatogr. A 1361 (2014) 139–152.
Disclosure of Interest:None DeclaredKeywords: physalins, Physalis alkekengi L., TLC-MS
P02-002-014 - High-Troughput Methodoloy For The Determination of 5-NITROIMIDAZOLE residues in Fish Roe Samples by UHPLC-MS/MS
M. Hernández-Mesa 1,*, C. Cruces-Blanco 1, A. M. García-Campaña 1
1 Department of Analytical Chemistry, University of Granada, Granada, Spain
Content: 5-nitroimidazoles (5-NDZs) are a wide-spectrum antibiotic class used for treating infections due to anaerobic protozoan and bacteria, however, their use in veterinary medicine has been restricted within European countries according to Regulation (EU) No 37/2010 [1]. Alerts about their presence in animal products destined to human consumption are still
notified by the Rapid System of Food and Feed (RASFF) portal, so analytical methods are required for 5-NDZ determination in order to ensure food safety. A novel multiresidue method has been proposed for the determination of twelve 5-NDZs in fish roe samples by UHPLC-MS/MS. A salting-out assisted liquid-liquid procedure has been proposed for the extraction of the analytes. Moreover, the variables that affected the separation such as the mobile phase composition and flow rate as well as the parameters involved in the ionization and fragmentation for MS detection were studied in detail. Finally, the separation was accomplished in a C18 Zorbax Eclipse Plus (50 mm × 2.1 mm, 1.8 µm) column using a mobile phase consisted of 0.025% (v/v) formic acid aqueous solution (eluent A) and MeOH (eluent B). Mobile phase was supplied at 0.5 mL/min and column temperature was set to 25°C. Samples were injected in mobile phase (5/95 (v/v) MeOH:0.025% (v/v) formic acid aqueous solution), and an injection volume of 17.5 µL was considered. The proposed method was characterized in terms of linearity (R2 ≥0.9996), extraction efficiency (≥71.4%), repeatability (≤9.8%), reproducibility (≤13.9%) and trueness (≥72.3%). Decision limit (CCα) and detection capability (CCβ) values between 0.3-1.5 and 0.5-2.5 µg/kg, respectively, were obtained for all 5-NDZ compounds. The reached results accomplish with the requirements of Regulation (2002/657/EC) and the recommendations of European Union Reference Laboratories for the determination of these substances.References: [1] Commission Regulation (EU) No 37/2010 of 22
December 2009, Off. J. Eur. Union L15 (2010) 1–72.Acknowledgements: The “Junta de Andalucía” has supported this work (Excellence Project Ref: P12-

189www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
AGR-1647). MHM thanks to the Plan Propio of the University of Granada for a post-doctoral contract.Disclosure of Interest:None DeclaredKeywords: 5-nitroimidazoles, Fish roe, UHPLC-MS/MS
P02-002-015 - ATHEROGENIC TRANS - Fatty Acid Content In Selected Dietary Fats In The Greek MarketE. G. Barmpounis*, D. Goulousi 1, M. Tzatzarakis 2, E. Vakonaki 2, A. Tsatsakis 2, A. Rizos 2
1 University of Crete, Herkalion, 2University of Crete, Heraklion, Greece
Content: Trans fatty acids (TFA) are strongly correlated with an increased risk of cardiovascular and other chronic diseases. Consumption of TFA increases the plasma cholesterol concentration of low-density lipoprotein (LDL) and decreases high-density lipoprotein (HDL). The aim of the current study was to determine the levels of TFA in a number of animal and vegetable fats on the Greek market using a gas chromatography-mass spectrometry based protocol.
Materials and Methods: Thirty two (32) samples of margarines and butters were collected and categorized in three groups: margarines (n=18), animal origin (n=9) and blends (n=5). The analytical procedure consisted of the esterification of TFA by methanolic-KOH solution followed by mass spectrometric analysis with a Shimadzu GCMS-QP 2010 .
Results & Discussion: All of the analysed samples contained TFA. Specifically, they tested positive for oleic acid (t-C18:1), linoleic acid (t-C18:2) and a-linoleic acid (t-C18:3).
Conclusion: TFA’s levels were lower than 1% in the majority of the samples and much lower than those reported (0.1-19%) in previous published studies over the past decades. Moreover, our results are in agreement with those reported in more recent study where the TFA’s levels of Greek market’s margarines are in a range of 0.16%>0.97%. Considering that the strong evidence for the harmful effects of industrially produced trans fatty acids, it is feasible to eliminate them from the food supply for a more healthy diet for the entire population in any given country.References: Trans Fatty Acids and Cardiovascular Disease1. Dariush Mozaffarian, M.D., M.P.H., Martijn B.
Katan, Ph.D., Alberto Ascherio, M.D., Dr.P.H., Meir J. Stampfer, M.D., Dr.P.H., and Walter C. Willett, M.D., Dr.P.H.
N Engl J Med 2006; 354:1601-1613April 13, 2006Disclosure of Interest:None Declared
P02-002-016 - Determination of tropane alkaloids atropine and scopolamine by UHPLC-MS/MS in cereal products E. Vysatova 1,*, R. Stepan 1, P. Cuhra 1, M. Kubik 1
1 Czech Agriculture and Food Inspection Authority (CAFIA), Prague 5, Czech Republic
Content: Atropine (racemic mixture of optical isomers (S)- hyoscyamine and (R)- hyoscyamine) and scopolamine are secondary metabolites which naturally occur in plants of the Datura genus and Hyoscyamus genus belongs to the Solanaceae family. Datura stramonium and Hyoscyamus niger (commonly known as Jimson weed and black henbane, resp.), which contain hallucinogens that represent a serious

www.ISC2016.ie @isc2016 #isc2016 /isc2016190
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
danger to humans and animals, are widely distributed in temperate and tropical regions. Seeds of these plants can be found as impurities in sorghum, millet, linseed, soybean, sunflower and buckwheat and products thereof. All parts of the Datura genus and Hyoscyamus genus contain dangerous levels of tropane alkaloids which are classified as anticholinergics. The deadly symptoms are caused by disruption of the parasympathetic nervous system’s ability to regulate breathing and heart rate. Therefore, a reliable method for the determination of above mentioned tropane alkaloids content is of high importance.
The present work was aimed to optimize and validate a rapid and sensitive method for determination of atropine and scopolamine by UHPLC-MS/MS. Tropane alkaloids were extracted from sample using mixture of methanol/water/formic acid and injected without clean-up to UHPLC-MS/MS. Separation were performed using Waters XBridge C18 chromatographic column.
During the validation process basic parameters such as repeatability, recovery, linearity, limit of detection (LOD), limit of quantification (LOQ) and reporting limit were evaluated for both analytes. Excellent parameters enable application of the presented method to processed cereal-based foods including baby foods for infants and young children where maximum limits 1 ppb were set by the Commission Regulation (EC) No 1881/2006 for both tropane alkaloids.References: COMMISSION REGULATION (EC) No
1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs, OJ L 364, 20.12.2006, p. 5
Disclosure of Interest:None DeclaredKeywords: Tropane alkaloids, UHPLC-MS/MS
P02-002-017 - Characterisation of volatiles and semi-volatiles in gin using dispersive liquid-liquid microextraction with gas chromatography
D. Connolly 1,*, S. Spoelders 1, M. Breen 1, W. Cummins 1
1 Pharmaceutical and Molecular Biotechnology Research Centre, Waterford Institute of Technology, Waterford, Ireland
Content: Gin is produced by re-distillation of alcohol 96% (v/v) in the presence of juniper berries and other natural botanical ingredients including coriander seeds, orris root, angelica root and lemon peel. All of these ingredients are rich in essential oils, which contribute to the aroma of most gins. Detailed information about gin volatile composition is necessary as a basis for defining gin sensory quality. The most common analytical method for gin analysis is headspace solid phase micro-extraction (HS-SPME) followed by gas chromatography with mass spectrometric detection. Here we report the development of dispersive liquid-liquid microextraction (DLLME) in which extraction of analytes takes place in a dispersion of the extracting solvent in water, in the presence of a third, bridging “dispersing solvent”. The dispersion is disrupted by centrifugation prior to analysis. We report the optimisation of a DLLME-GC-FID protocol for the analysis of selected commercial and laboratory gins. Volatile and semi-volatile components were identified via Linear Retention Indices using a mixed hydrocarbons standard, belonging mainly to the terpenoids family (monoterpenes, sesquiterpenes and their corresponding oxygenated compounds) including limonene, linalool, α-pinene,

191www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
β-myrcene and γ-terpinene. The optimised DLLME protocol was used to (1): assay the batch-to-batch reproducibility of model gins, (2): to compare commercial gins and (3): to examine the influence of selected production methods on the terpene composition of gin. References: Vichi, S., Riu-Aumatell, M., Mora-Pons,
M., Buxaderas, S., & Lopez-Tamames, E. (2005).Characterization of volatiles in different dry gins. Journal of Agricultural and Food Chemistry, 53, 10154–10160.Vichi, S., Riu-Aumatell, M., Mora-
1: S. Vichi, M. Riu-Aumatell, M. Mora-Pons, S. Buxaderas, E. Lopez-Tamames, E. Journal of Agricultural and Food Chemistry, 53, 10154–10160 (2005)
Disclosure of Interest:None Declared
Keywords: Alcoholic beverage, gas chromatography, Gin
P02-002-018 - Determination of CARBADOX, OLAQUINDOX, 1-DESOXYCARBADOX, 4-DESOXYCARBADOX, and DI Desoxycarbadox in Porc Muscle and Liver
M. Funeva-Peycheva 1, T. Yankovska-Stefanova 1, N. Stoilova 1, S. Andriana 2,*
1 VMP Analysis Department, Central Laboratory of Veterinary Control and Ecology (CLVCE), Bulgarian Food Safety Agency, 1528 Sofia,
2 Analytical Chemistry dep, University of Chemical technology and metallurgy, Sofia, Bulgaria
Content: Carbadox, olaquindox and their metabolites have been widely applied as veterinary medicines, for improving the feed efficiency, increasing the rate of weight, and gain in aquacultures [1,2].
The path of carbadox metabolism is via carcinogenic mono-and desoxy compounds to quinoxaline-2-carboxilic acid (QCA), which is not carcinogenic. QCA is a marker residue for Carbadox determination. In vivo olaquindox metabolizes at the methyl-3-quinoxaline-2-carboxylic acid, a marker residue for olaquindox. The analytical methods recently reported in the literature are based on HPLC [3,4].
The aim of the present study is to compare separation efficiency of several chromatographic columns and to develop optimal gradient elution program for separation and determination of carbadox, olaquindox and their metabolites by liquid chromatography coupled with electrospray ionization–tandem mass spectrometry. The separation efficiency of newly available chromatographic columns was studied. The composition of the mobile phase (ammonium acetate buffer and acetonitrile) and the gradient elution programs were optimized to be effective and to ensure optimum separation of the analytes on each of studied chromatographic columns. Multiple reaction monitoring was used for selective determination of compounds. An analysis time up to 5 min for all studied substances was achieved on Kinetex PFP and Zorbax Eclipse PH columns. Although the separation time on Synergi and Waters columns was higher than other studied columns (up to 10 min), it is short enough to ensure good separation at low reagent and analysis time consumption. The developed chromatographic method showed detection limits as low as 10 ppb below recommended concentration limits for carbadox, olaquindox and their metabolites.References: 1. W. Jiang et al., J. Agricult. Food Chem.

www.ISC2016.ie @isc2016 #isc2016 /isc2016192
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
doi/10.1021/jf40374972. L. Cheng et al, Anal. Methods 4 (217-221)(2012)3. J. J. Nasr et al, Chromatographia 76 (523-528)
(2013)4. L. Cheng et al, Anal. Methods 4 (217-221 (2012)Disclosure of Interest:None DeclaredKeywords: carbadox, LC–MS/MS, olaquindox
P02-002-019 - Authentication of whiskey and wine using dispersive liquid-liquid microextraction with gas chromatographic and liquid chromatographic analysis.
D. Connolly 1,*, S. Spoelders 1, M. Breen 1, W. Cummins 1
1 Pharmaceutical and Molecular Biotechnology Research Centre, Waterford Institute of Technology, Waterford, Ireland
Content: Dispersive liquid-liquid microextraction (DLLME) involves the injection of a mixture of a water-immiscible extraction solvent into an aqueous sample in the presence of a third “dispersion” or bridging solvent. The emulsion thus formed comprises many droplets of extraction solvent into which the analyte partitions; the huge increase in interfacial area relative to conventional solvent extraction results in almost instantaneous extraction and very high enrichment factors (often several hundred-fold). Typical extraction solvents are usually denser than water allowing their recovery by centrifugation of the emulsion, with usual dispersion solvents being acetone, methanol or acetonitrile. Hydroalcoholic beverages such as wine and whiskey are ideally suited to facile DLLME since they already contain suitably high levels of ethanol as a dispersive solvent. Here we therefore report the use of DLLME
for the extraction of key congeners from whiskey and wine prior to GC and HPLC analysis. Whiskey is produced by the fermentation of a barley-derived wort, its distillation to increase the alcohol content, followed by maturation in oak barrels for 3 years (minimum, but often up to 18 years). Typically bottled at 40% ethanol by volume, whiskey is comprised of a complex mix of hundreds of compounds including higher alcohols, esters, organic acids, phenols, aldehydes and ketones. We report the use of DLLME with GC and HPLC to distinguish selected Irish and Scottish whiskeys as well as the authentication of wine based upon grape variety and geographical origin.References: 1: M. Rezaee, Y. Yamini, M. Faraji. J.
Chromatogr. A 1217, 2342, 2010Disclosure of Interest: None DeclaredKeywords: Liquid-Liquid microextraction, Whiskey, Wine
P02-002-020 - Analysis of natural blue dyes and products of their degradation in historic fabrics by liquid chromatography mass spectrometry
M. Ganeczko 1, B. Witkowski 1, T. Gierczak 1, A. Laudy
2, M. Biesaga 1,*
1 Department of Chemistry, University of Warsaw, 2Museum of King Jan III’s Palace at Wilanów, Warsaw, Poland
Content: Indigo is a blue dye, which is known since antique. The natural indigo was obtained in Europe from two plants Isatis tinctoria and Indigofera tinctoria [1-2]. After years of exposition to the different environmental factors, as a result of degradation processes, the natural indigo dyed textiles fade.

193www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
The aim of this project is examination of the impact of solar radiation on natural blue dyes degradation processes in the silk samples obtained from the royal chambers from Museum of King Jan III’s Palace at Wilanów, Poland. To do this, the continuance of blue natural dyes from the samples of patterned velour of the upholstery and wall fabrics were analyzed. Each sample came from another part of textiles and only a few of these were from the places exposed to UV radiation.
The high performance liquid chromatography coupled by tandem mass spectrometry (LC/MS/MS) was used to identification of indigotin, main chemical compound responsible for blue color, and other natural dyes and their degradation products in the textiles samples. The comparison of results obtained from historical fabrics with results obtained for standards and model samples is the basis for the identification of originally used dye. The dyes extraction from the textiles is an important step before the LC/MS/MS analysis. Dimethyl sulfoxide (DMSO) was used as a solvent to extract the vat dyes, like indigo.
The analyses show that solar radiation is not the only one factor affecting natural dyes degradation process. However, textiles not exposed to the UV radiation were characterized with the highest content of indigotin.References: 1. M.P. Colombini,F. Modungo, Organic Mass
Spectrometry in Art and Archaeology, John Willey & Sons, 2009.
2. J.H. Hofenk van Graaff, W.G.T. Bommel, The Colourfull Past: Origins, Chemistry and Identyfication of Natural dyestuffs, Archetype
Publication, London 2004.Acknowledgements: The research was supported by the POIS.11.01.00-00.068/14-00 from day 03.10.2014 r.Disclosure of Interest: None DeclaredKeywords: dyes, historic fabrics, indygo
P02-002-021 - In-silico prediction and wet lab validation of Arisaema tortuosum (Wall.) Schott extracts: a comparative antioxidant and antibacterial studyK. Kant 1,*, U. R. Lal 1, M. Ghosh 1
1 Pharmaceutical Sciences & Technology, Birla Institute of Technology, Mesra, Ranchi, Jharkhand, India
Content: Arisaema tortuosum (Wall.) Schott (ATWS); (Araceae), a famous folklore medicine of Asian region & used commonly against a variety of disorders since ancient times. In the present investigation, we have computationally predicted the reported principal bioactive metabolites of ATWS for possible antioxidant and antibacterial biological spectrum using molecular docking & prediction of activity spectra for substances (PASS-online) tools and further ATWS tubers & leaves extracts were validated experimentally. The phytoconstituents of ATWS showed best hits glide score against poly (ADP-ribose) polymerase (PARP); quercetin: -11.76 (antioxidant) & dihydrofolate reductase; (DHFR); rutin: -11.69 (antibacterial) receptors, respectively. Pass spectrum results of top screened too showed promising antioxidant and antibacterial potential. Overall, ethyl acetate tubers (B3) fraction (ABTS; IC50: 67.46 µg/ml, DPPH; IC50: 87.08 µg/ml) was resulted in more promising antioxidant activity, with a remarkable amount of FRAP (195.96 µg/mg), TPC (0.112 µg/mg) and TFC (12.5

www.ISC2016.ie @isc2016 #isc2016 /isc2016194
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
µg/mg). In addition, leaves extract and their fractions have shown varying level of minimum inhibitory concentration (MIC)/zone of inhibition (ZOI)-hexane (A1): 78.13×10-3 mg/ml/9±0.4mm (S.aureus), ethanolic (A): 1250×10-3 mg/ml/11±0.7mm (B.subtilis), chloroform (A2): 2500×10-3 mg/ml/11±0.8mm (E. coli) & ethanolic (A): 1250×10-3 mg/ml/11±0.4mm (S. typhi), respectively. Finally, the B3 fraction showed greater antioxidant activity whereas leaves varying fractions (A, A1 & A2) resulted in good potential against bacterial strains. The results indicate that the tubers and leaves part of the plant can be used as natural antioxidants and preservatives in food for promoting health benefits.References: 1. K, Kant, M, Walia, VK, Agnihotri, V, Pathania, B,
Singh, 3 (324-329) (2013).2. TM, Joska, AC, Anderson, 10 (3435-3443) (2006).Acknowledgements: The authors thankfully acknowledge the funding agency UGC, New Delhi, India (UGC-BSR fellowship) for their financial support. The authors also grateful to Head, Department of Pharmaceutical Sciences & Technology, BIT Mesra, Ranchi (Jharkhand), India for providing needed facility during the research investigations.Disclosure of Interest:None DeclaredKeywords: Arisaema tortuosum, In-silico, Antioxidant, Antibacterial
P02-002-022 - Determination of steviol glycosides using HPLC-MS/MS
H. Pastell 1,*, K. Kahila 2, M. Raatikainen 1
1 Research and Laboratory Department, Finnish Food Safety Authority Evira, 2Department of Food and Environmental Sciences, University of Helsinki, Helsinki, Finland
Content: Steviol glycosides, used as sweetener (E 960), are extracted from Stevia rebaudiana Bertoni –plant leaves. According to Commission regulation (EU) No 231/2012 (1), at least 75 % of steviol glycosides in E 960 consists of stevioside (Stv) and/or rebaudioside A (Reb A). Furthermore, 95 % of the final product should comprise of Stv, Reb A, B, C, D, E and F, steviolbioside (Stvb), rubusoside (Rub) and dulcoside (Dul). High performance liquid chromatography (HPLC) in combination with UV or mass spectrometry (MS) has been used in the analysis of steviol glycosides. Limiting factor in the method development has been a poor resolution between the two major components, Stv and Reb A. The aim of this study was to develop a HPLC-MS/MS method for determination of three most abundant steviol glycosides, Stv, Reb A and Reb C. The objective was also to validate the method for juices.
Using ZIC-HILIC column (H2O:ACN 15:85 (v/v), 10 mM ammonium acetate, 0.5 ml/min, 28oC), a good resolution between Stv and Reb A was achieved. Altogether six steviol glycosides were identified and separated in the following order: Rub, Stvb, Reb B, Stv, Reb C, Reb A. The steviol glycosides were identified based on their retention time and characteristic MS transitions. Stv, Reb C and Reb A were quantified using one quantitation and two verification ions.

195www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
Juice, sweetened with steviol glycosides, was diluted and purified using solid phase extraction by C18 cartridge. Juice matrix induced ion suppression in MS, but the quantification was successful using standard curves and digitoxin as an internal standard. The developed HPLC-MS/MS method demonstrated good performance in terms of trueness, accuracy, repeatability and reproducibility for juice. The limit of quantification (LOQ) for the individual steviol glycosides was 0.1 mg/l, while the permitted maximum levels range from 20 to 3300 mg/kg.References: 1. Commission regulation (EU) No
231/2012, Official Journal of European Union L83/1 (2012).
Disclosure of Interest:None DeclaredKeywords: Food additive, HPLC-MS/MS, Steviol glycosides
P02-002-023 - Determination of nitroimidazoles in feed by LC-MS/MS
K. Mitrowska*, A. Posyniak 1
1 Department of Pharmacology and Toxicology, National Veterinary Research Institute (PIWet), Pulawy, Poland
Content: Nitroimidazoles are antibacterial and antiprotozoal drugs used formerly for the treatment of diseases in food producing animals but have been classified as suspect mutagens and carcinogens and are not allowed to use in food producing animals within European Union. When nitroimidazoles in food of animal origin are detected, an investigation should be initiated at the source farm which includes additional sampling of feed. To analyse these samples a multiresidue method for the determination of seven nitroimidazoles
in feed was developed. Feed samples were extracted with acetonitrile and cleaned-up on strong cation-exchange (SCX) solid phase extraction (SPE) cartridges. The obtained extracts were evaporated to dryness, reconstituted in 0.1% formic acid and injected onto a LC-MS/MS. The separation of analytes was performed on a C18 column using a mobile phase of 0.1% formic acid in acetonitrile and 0.1% formic acid in water with gradient elution. The MS/MS was operated in MRM mode with positive electrospray ionization. The whole procedure was evaluated according to the European Union requirements [1] and all validation criteria were in the required ranges. The method can easily detect and confirm metronidazole, dimetridazole, ronidazole, ipronidazole and their hydroxy metabolites below the recommended concentration level of 3 µg/kg.References: 1. Commission Decision 2002/657/EC.
OJ L 221, 8-36.Acknowledgements: The research was funded by KNOW (Leading National Research Centre) Scientific Consortium “Healthy Animal - Safe Food” allocated on the basis of the decision of the Ministry of Science and Higher Education No. 05-1/KNOW2/2015.Disclosure of Interest:None DeclaredKeywords: feed, LC-MS/MS, nitroimidazoles
P02-002-024 - A method for the determination of malachite green and leucomalachite green in feed by LC-MS/MSK. Mitrowska*, A. Posyniak 1
1 Department of Pharmacology and Toxicology, National Veterinary Research Institute (PIWet), Pulawy, Poland
Content: Malachite green (MG) is a synthetic triphenylmathane dye which has a broad

www.ISC2016.ie @isc2016 #isc2016 /isc2016196
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
spectrum of fungicidal and ectoparasiticidal activities. MG has been used as an effective compound in the treatment of external fungal and protozoan infections of fish but it has never been registered as a veterinary drug for use in food producing animals within European Union because of its potential carcinogenicity, mutagenicity and teratogenicity in mammals. In living organisms, MG is extensively metabolised to the reduced, colourless compound leucomalachite green (LGM). When malachite green or leucomalachite green in fish are detected, an investigation should be initiated at the source farm which includes additional sampling of feed. To analyse these samples a method for the determination of malachite green or leucomalachite green in feed was developed. The extraction of a feed sample was performed with acetonitrile-acetate buffer mixture followed by portioning with dichloromethane. The organic phase was cleaned up on a strong cation-exchange (SCX) solid phase extraction (SPE) column. The obtained extract was evaporated to dryness, reconstituted in a mixture of acetate buffer, acetonitrile and ascorbic acid and injected onto a LC-MS/MS. Chromatographic separation was achieved on a phenyl–hexyl column using a mobile phase of acetonitrile and acetate buffer with isocratic elution. The MS/MS was operated in MRM mode with positive electrospray ionization. The whole method was validated according to the European Union requirements [1] and all performance criteria were in the required ranges. The method can easily detect and confirm malachite green and leucomalachite green below the minimum required performance limit (MRPL) of 2 µg/kg.References: 1. Commission Decision 2002/657/EC.
OJ L 221, 8-36.
Acknowledgements: The research was funded by KNOW (Leading National Research Centre) Scientific Consortium “Healthy Animal - Safe Food” allocated on the basis of the decision of the Ministry of Science and Higher Education No. 05-1/KNOW2/2015.Disclosure of Interest:None DeclaredKeywords: feed, LC-MS/MS, malachite green
P02-002-025 - Analysis of acrylamide from water according to DIN EN ISO 38413-6
H. R. Wollseifen*, T. Kretschmer, H. Riering, M. Roedel
Content: Polyacrylamides are often used in the water industry as coagulant for water clarification. Residual amounts of acrylamide monomers in drinking water, included in the purification treatments, are observed. In animal tests carcinogenic and mutagenic properties have been observed. Subsequently amounts of acrylamide in drinking water are limited by the german law [1]. Hence the interest in highly sensitive analysis for acrylamide has increased. The most important german guidelines for analysis of acrylamide are described in DIN EN ISO 38413-6 method [2]. This determination method provides a solid-phase extraction and high perormance liquid chromatographic method by mass spectrometric detection for acrylamide.
The first part of this work deals with a methodology for sample preparation of water analysis including a solid-phase extraction (SPE) method. SPE is carried out successfully on an activated carbon with methanolic elution. The activated

197www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
carbon phase is highly porous and suited to the DIN specification of a specific surface higher than 1000 m2/g. The recovery is compared with the requirements of DIN EN ISO 38413-6. The second part of this work points out the optimal high performance liquid chromatographic conditions on NUCLEODUR®C18 Gravity for acrylamide. The identification of acrylamide in this work was carried out by ESI mass spectrometry.References: [1] Verordnung über die Qualität von Wasser für den
menschlichen Gebrauch, (Trinkwasserverordnung - TrinkwV 2001)
[2] Determination of acrylamide – Methode using high performance liquid chromatography with mass spectrometric detection (HPLC-MS/MS)
Disclosure of Interest:None DeclaredKeywords: acrylamide, activated carbon, water analysis, HPLC-MSMS, SPE
P02-002-026 - Olive seeds as sustainable sources of antihyperlipidemic peptides
I. M. Prados Nieto 1,*, M. L. Marina 1, M. C. García 1
1 Departamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcalá, Alcalá de Henares, Spain
Content: Exploitation and efficient use of natural resources by minimizing waste generation are priorities for the United Nations Organization and the European Commission. Moreover, population aging and the increasing incidence of certain diseases have led to the demand for foods containing ingredients with beneficial effects on health. The valorization of food industry byproducts could respond to both demands. Processing of olives (Olea europaea) generates large amounts of residues, mainly
comprised by the stones. Protein content in olive stones is concentrated in the seed and supposes more than 20% [1].
Hyperlipidaemia is a group of metabolic disorders characterized by an elevated level of triglycerides and cholesterol in blood and it has increased significantly in recent years. Antihyperlipidemic peptides have not been much studied. The aim of this work was to explore the antihyperlipidemic capabilities of peptides obtained from olive stones.
Olive seed peptides were obtained from defatted seeds by the extraction of proteins followed by hydrolysis with Alcalase [1]. Four different parameters yielding information on different mechanisms to reduce absorption of lipids were evaluated: the ability to reduce cholesterol micellar solubility, the bile acid binding capacity, and the capacity to inhibit cholesterol esterase and pancreatic lipase enzymes. Hydrolysates were fractionated by ultrafiltration observing that most hipocholesterolemic peptides showed the highest molecular weights (> 5 kDa). The highest ability to reduce triglyceride absorption was observed in peptides below 5 kDa. A further fractionation of these peptides by semipreparative RP-HPLC was also carried out.References: [1] C. Esteve, M.L. Marina, M.C.
García, Food Chem. 167 (2015) 272.Acknowledgements: Authors thank the Spanish Ministry of Economy and Competitiveness (AGL2012-36362), the Centro para el Desarrollo Tecnológico Industrial (ITC-20151193), the Comunidad of Madrid (Spain) and FEDER program (S2013/ABI-3028). We also thank FAROLIVA S.L. (Spain) for the kind donation of olive stones.Disclosure of Interest:None Declared

www.ISC2016.ie @isc2016 #isc2016 /isc2016198
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
P02-002-027 - Developed and validation of a LC-MS/MS method for the determination of five dye residues in aquaculture products
A. Macaluso*, V. Giaccone 1, G. Polizzotto 2, A. Alongi 1, A. Vella 1, V. Ferrantelli 1
1 Area Chimica e Tecnologie Alimentari, Istituto Zooprofilattico Sperimentale della Sicilia “A. Mirri”, 2Dipartimento STEBICEF, Università di Palermo, Palermo, Italy
Content: A method for the quantitative determination of the triphenylmethane dyes, malachite green (MG), leuco malachite green (LMG), brilliant green (BG), crystal violet (CV) and leuco crystal violet (LCV) in fish muscle was developed and validated. MG is used in aquaculture as fungicide. LMG is the reduced metabolite of MG and accumulates in fatty tissues of the fish.
Both and CV have been bannen due to their carcinogenic activity by FDA and UE. BG was evaluated for the similar chemical structure and the possible similar toxicity. The analytes were extracted from the matrix using a LC/LC extraction with a mixture of acetonitrile and citrate buffer; the clean up was performed using the quechers tecnique and the purified sample was injected in a LC-MS/MS instrument with electrospray ionization source for the determination. An internal calibration was used. In accordance with the Commission Decision 2002/657/EC the method has been validated in line with the minimum required performance limit MRPL of 2 µg/Kg. The validation parameters such as instrumental linearity, specificity, precision (repeatability and reproducibility), recovery, decision limit (ccα), detection capabilities (ccβ) and ruggedness were evaluated with good results for the suitability of the method.
A method for the quantitative determination of the triphenylmethane dyes, malachite green, leuco malachite green, brilliant green, crystal violet and leuco crystal violet in fish muscle was developed and validated. The analytes were extracted from the matrix using a LC/LC extraction with acetonitrile and citrate buffer; the clean up was performed using the quechers tecnique and the purified sample was injected in a LC-MS/MS (ESI) for the determination. In accordance with the Decision 2002/657/EC the method has been validated in line with the minimum required performance limit MRPL of 2 µg/Kg. The validation parameters were evaluated with good results for the suitability of the method.References: J. S. Diana, BioScience 59, 27-
38 (2009)Disclosure of Interest:None DeclaredKeywords: acquaculture, dye, LC-MS/MS
P02-002-028 - Evaluation of angiotensin converting enzyme inhibitory capacity and citotoxycity of peptides from peach and olive seeds
R. Vásquez-Villanueva 1,*, M. L. Marina 1, M. C. García 1
1 Departamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcalá, Alcalá de Henares, Spain
Content: Hypertension is a major risk factor linked to cardiovascular diseases that leads to premature mortality. Angiotensin converting enzyme (ACE) plays an important role in the renin-angiotensin system, which is the main blood pressure regulator [1]. ACE catalyzes the conversion

199www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
of angiotensin I to the potent vasoconstrictor angiotensin II and deactivates the vasodilator bradykinin. Synthetic ACE inhibitors are widely used for hypertension treatment but they can cause undesirable side effects. Some natural peptides have shown ACE inhibitor properties without provoking adverse effects.
Previous researches demonstrated that some seeds from Prunus genus and Olea Europaea contain high protein contents and that enzymatic hydrolysis of proteins results in the release of peptides with ACE-inhibitory capacity [2, 3]. Furthermore, more active peptides were concentrated in the fraction with peptides below 3 kDa. This work proposes to further study these peptides to select highly antihypertensive peptides and to study their cytotoxic effects.
Peach and olive seeds proteins were extracted and hydrolysed with Thermolysin. Peptides in the fraction below 3 kDa after ultrafiltration were further fractionated by RP-HPLC. Eight different subfractions were collected and their ACE inhibition capacity was determined. One subfraction from the olive seeds and two subfractions from the peach seeds exhibited potent ACE-inhibitory capacity. Peptides in these fractions were identified by HILIC- and RP-HPLC-ESI-Q-ToF and by de novo sequencing. Some identified peptides were synthetized and characterized. Moreover, toxicity of fractions and peptides was evaluated using three epithelial cell lines. References: [1] P. Puchalska, et al. Crit. Rev. Food Sci. Nutr. 55
(2015) 521-551.[2] R. Vásquez-Villanueva, et al. J. Funct. Foods 18
(2015) 137-146.[3] C. Esteve, et al. Food Chem. 167 (2015) 272-280.
Acknowledgements: Authors thank the Spanish Ministry of Economy and Competitiveness (AGL2012-36362), the Centro para el Desarrollo Tecnológico Industrial (ITC-20151193), the Comunidad of Madrid (Spain) and FEDER program (S2013/ABI-3028). R.V.V. thanks the University of Alcalá for her pre-doctoral contract.Disclosure of Interest:None Declared
P02-002-029 - Comprehensive profiling of amino acid degradation products by LC-QTOFMS and establishment of a spectrum libraryS. Neubauer 1,*, M. Laemmerhofer 1
1 Institute of Pharmaceutical Sciences, Eberhard-Karls-University Tuebingen, Tuebingen, Germany
Content: This work deals with the degradation of amino acids, since they are widely in food supplements. Test solutions of the proteinogenic amino acids are exposed to stress conditions, precisely to elevated temperature, UV-irradiation and contact to oxygen. Comprehensive impurity profiling is achieved by complementary separation (reversed phase liquid chromatography – RPLC and hydrophilic interaction liquid chromatography - HILIC) in combination with a quadrupole time of flight mass spectrometer (LC-ESI-QTOFMS) operated in both, positive and negative ionization mode.
Data extraction using a target list based on degradation products found in literature as well as non-targeted peak finding reveals the degradation profiles of the proteinogenic amino acids after stress testing conducted at elevated temperature and varying the exposure to oxygen or to UV irradiation. The sum formulae of unknowns are determined by accurate mass and isotopic resolution.

www.ISC2016.ie @isc2016 #isc2016 /isc2016200
Food and Health, Food Safety and AuthenticityPOSTER ABSTRACTS
Furthermore a spectrum library of amino acid degradation products is established comprising retention time (RPLC and HILIC), accurate mass, isotope pattern and fragmentation spectra. Finally the chemical structures obtained after spectra interpretation and search in chemical databases are included.References: S. Schiesel, M. Laemmerhofer, W.
Lindner, Journal of Chromatography A 2012, 1259, 100-110.
Disclosure of Interest: None DeclaredKeywords: amino acid, Degradation products, LC-MS
P02-002-030 - Pyrrolizidine alkaloids in honey: Analytical and Toxicological and regulatory considerationsA. A. Sheehan*, C. T. Griffin, M. Danaher, A. Furey 1
1 Department Physical Sciences, Cork institute of Technology, Cork, Ireland
Content: Pyrrolizidine alkaloids (PAs) are secondary plant metabolites found primarily in the plant families Boraginaceae, Asteraceae and Fabaceae, an estimated 6,000 plant species or 3% of all flowering plants [1]. Over 600 PAs have been identified todate [2] several of which are known to be highly hepatotoxic and carcinogenic. PAs have been associated with a number of livestock diseases and documented cases of human PA poisioning through food date back to 1920 (Wilmot and Robertson). Currently there are no EU regulatory limits for PAs and their N-oxides.The European Food Safety Authority (EFSA) have issued guidelines not to exceed an intake of 0.007 µg kg-1 body weight day-1 based on a safety assessment of PAs.
Liquid chromatography coupled to mass spectrometry has advanced the analytical capabilities of regulatory authorities to quantitatively profile PAs in food. Focusing specifically on honey, a simple acidic extraction of samples followed by solid phase extraction (SCX) with subsequent LC-MS/MS [3] analysis resulted in the identification of a number of PA toxins in honey samples sold in Irish supermarkets. The PA positive samples were 24% (n=369) with Lycopsamine (m/z 300) and echimidine (m/z 398) being the predominant PAs found in these samples.
An LTQ Orbitrap mass spectrometer is a hybrid instrument that combines linear ion trap (LIT) mass spectrometry (MS) with high-resolution Fourier Transform(FT) MS and this was exploited to perform simultaneous ultra-high-resolution full-scan MS analysis and collision-induced dissociation (CID) tandem mass spectrometry (MS/MS). PA positive honey samples were investigated using the LTQ Orbitrap mass spectrometer to confirm the results generated from LC-MS/MSReferences: [1] LW Smith , CC Culvenor., J Nat Prod. 1981 Mar-
Apr;44(2):129-52[2] L Cramer, T Beuerle., Planta
Med. 2012 Dec;78(18):1976-82.[3] CT Griffin, J O’Mahoney, M Danaher, A Furey.,
Food Anal Methods 2015 8:18-31Acknowledgements: Funded under the Food for health research initiative (FHRI) by the Irish Dept. of Agriculture, Fisharies and Food (DAFF) and the health research board (HRB) 2008 - 2012Cork Institute of Technology, Risam scholarship research grant 2014-2017.Disclosure of Interest:None DeclaredKeywords: Honey, Liquid Chromatogrpahy, Pyrrolizidine alkaloid

201www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
P02-002-031 - Analysis of polar ionic pesticides by ion-exchange chromatography tandem mass spectrometry: a possible solution to a problematic analysis?J. Beck 1, S. Adams 2, J. Guest 2, R. Fussell 3, F. Schoutsen 4, J. Weiss 5,*
1 Thermo Scientific, San Jose, United States, 2 FERA, York, 3 Thermo Scientific, Hemel Hempstead, United Kingdom, 4 Thermo Scientific, Dreieich, Germany, 5 Thermo Scientific, Sunnyvale, United States
Content: Polar ionic pesticides, such as glyphosate, perchlorate, chlorate and the like, often occur as residues in food, but are not always included in pesticide monitoring programs, simply because they are not ‘amenable’ to generic multi-residue methods. The introduction of the Quick Polar Pesticides (QuPPe) Method by the European Reference Laboratory for single residue methods (EURL-SRM) has enabled more laboratories to conduct analysis for at least some of the polar pesticides. Still, the absence of a liquid partitioning step, or clean-up step, results in ‘dirty extracts’ containing high concentrations of matrix co-extractives. Thus, the separation and accurate quantification of analytes in QuPPe extracts is challenging. Analysts attempt to mitigate these issues by analyzing a single extract a number of times, using different chromatographic columns and conditions. These separation conditions are often less than ideal and the large amounts of co-extractives often contaminate the low capacity columns to cause variation in retention time and a decrease in the ruggedness of the method.
The application of high resolution ion-exchange chromatography with high
capacity columns, coupled to a triple quadrupole mass spectrometer can overcome the issues experienced with other chromatographic techniques. Using the IC-MS/MS approach for direct analysis of QuPPe extracts, low limits of quantification (typically < 5 ng/g) , and associated repeatability (typically < 20%) have been achieved for chlorate, perchlorate, glufosinate, N-acetyl glufosinate, 3-MPPA, glyphosate, AMPA, Fosetyl-Al, phosphonic acid, ethephon, HEPA and more, in a single analysis. Further details on separation, quantification and validation in various matrices will be presented.References: EU Reference Laboratory for pesticides requiring
Single Residue Methods (EURL-SRM) Version 8.1, March 2015
CVUA Stuttgart, Schaflandstr. 3/2, DE 70736 Fellbach, Germany
Website: www.eurl-pesticides.eu,[email protected] of Interest:None DeclaredKeywords: IC-MS/MS, ionic pesticides, Polar pesticides

www.ISC2016.ie @isc2016 #isc2016 /isc2016202
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
P02-002-032 - Enantiomeric determination of citrulline in food supplements by Electrokinetic Chromatography
R. Pérez-Míguez 1,*, M. L. Marina 1, M. Castro-Puyana 1
1 Departamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcalá, Alcalá de Henares, Spain
Content: Citrulline is a non-protein amino acid that can be synthesized during L-arginine metabolism. This compound is frequently used in dietary supplements due to its favorable effects on the metabolism of corporal fatty excess. L-Citrulline is involved in urea cycle reducing ammonia levels and playing an important role in NO cycle, whereas its D-enantiomer has demonstrated to have different in vivo/in vitro behavior [1, 2]. Since the use of D-enantiomers in the elaboration of dietary supplements is not allowed by regulatory agencies [3], the development of estereoselective methodologies presents a high interest.
The aim of this work was to carry out the enantiomeric determination of citrulline in food supplements by an Electrokinetic Chromatography methodology based on the use of a formate buffer at acidic pH and sulfated γ-CD as chiral selector. Moreover, the effects of the storage time on citrulline racemization were also studied. Citrulline enantiomers were separated in less than 20 min (Rs 2.7). L-citrulline was quantified in different dietary supplements while D-citrulline was not detected in any of the samples analyzed. No racemization process took place in those samples submitted to a long storage time.References: [1] E. Curis et al., Amino acids, 29
(2005) 177-205.
[2] L. A. Van Geldre et al., Eur. J. Pharmacol., 455 (2002) 149-160.
[3] Commission Decision 2001/15/EC. Official Journal of the European Communities, L 52, 2001, pp. 19-25.
Acknowledgements: Authors thank the Spanish Ministry of Economy and Competitiveness (CTQ2013-48740-P) and the Comunidad of Madrid (Spain) and European funding from FEDER program (S2013/ABI-3028, AVANSECAL-CM). M.C.P. also thanks the Ministry of Economy and Competitiveness (Spain) for her “Ramón y Cajal” research contract (RYC-2013-12688). R.P.M. thanks the University of Alcalá for her pre-doctoral contract.
Disclosure of Interest:None Declared
P02-002-033 - Chemical characterization of eggplant polyphenols by using liquid chromatography coupled with diode array and mass spectrometry
P. Šilarová*, V. Brighenti 1, M. Steinbach 2, L. −eslová 3, S. Benvenuti 1, L. Boulekbache-Makhlouf 4, F. Pellati 1
1 University of Modena and Reggio Emilia, Modena, Italy, 2 Justus-Liebig University Giessen, Giessen, Germany, 3 University of Pardubice, Pardubice, Czech Republic, 4 Université A. Mira de Bejaia, Bejaia, Algeria
Content: Solanum melongena L. (eggplant) is among the best vegetables in terms of antioxidant content, which is linked to various health benefits. Phytonutrients contained in eggplant include polyphenolic compounds, such as phenolic acids and flavonoids. The different cooking processes may affect the quali- and quantitative profile of polyphenols in eggplant. In the light of this, the present study was addressed at the development of a new analytical method for the characterization

203www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
of the polyphenolic compounds contained in eggplant extracts obtained from both fresh and cooked samples. The quali- and quantitative analysis of eggplant polyphenols was carried out by means of a new RP-HPLC method coupled with both UV/DAD and ESI-MS2 detection, based on the use of a fused-core stationary phase. The application of the fused-core technology allowed us to obtain an improvement of the HPLC performance in comparison with that of conventional particulate stationary phases. As regards the MS detection, a QTOF mass analyzer coupled with an ESI source was employed and the system was operated both in the positive and negative ion mode. The identification of the main compounds in the eggplant extracts was achieved by comparing the UV/DAD spectra and the MS fragmentation data of the chromatographic peaks with those described in the literature and further confirmed by available reference standards. The samples considered in this study consisted of extracts obtained from fresh, fried, baked and grilled eggplant and prepared using aqueous solution of different organic solvents. The change in the polyphenolic composition among the samples was monitored by comparing their chromatographic profiles.References: Lynda Arkoub-Djermoune, Lila
Boulekbache-Makhlouf, Sabrina Zeghichi-Hamri, Salima Bellili, Farid Boukhalfa and Khodir Madani, Influence of the thermal processing on the physicochemical propreties and the antioxidant activity of a solanaceae vegetable: eggplant, Journal of Food Quality, 00, (00-00), 2016
Disclosure of Interest:None DeclaredKeywords: Eggplant, HPLC-MS, Polyphenols
P02-002-034 - Shampoos and hair products deformulation using spectroscopic and advanced chromatographic toolsN. Longieras*
Content: Shampoos and hair products are complex formulation with multiple components and various chemical elements. Competitive benchmark from the analytical point of view is very demanding.
In the past last years, we developed approaches for shampoo products deformulation. Bulk characterization using NMR, vibrational spectroscopy, X-ray fluorescence are followed by chromatography. Different separation modes (liquid, gas, ions) allow most of the identification. Unknowns are characterized using liquid chromatography and intelligent detectors (FTIR, High Resolution MS, NMR). Mixture of polymer are separated by adsorption chromatography (LAC) and size-exclusion chromatography (SEC) to obtain information on composition and molar mass distribution.References: H. Pasch, B. Trathnigg,
Multidimensional HPLC of Polymers, Springer (2013)
Disclosure of Interest: None Declared
P02-002-035 - Fast and Cost-effective Sugar Analysis Using HPAE-PAD
H. Yang*, J. Weiss, G. Ellison
Content: Mono- and disaccharide sugar determinations are often used in the food and beverage industry to comply

www.ISC2016.ie @isc2016 #isc2016 /isc2016204
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
with the mandatory nutrition declaration (EU regulation No.1169/2011), to ensure the quality of a formulated product, to maintain or select for desired sweetness, and to characterize and confirm the source of the carbohydrates. Carbohydrates have poor chromophores and are hard to detect by absorbance detectors without lengthy and costly derivitization. However, High Performance Anionic-exchange Pulsed Amperometric Detection (HPAE-PAD) is a well-established method that does direct detections without derivitization.
Here demonstrates the use of HPAE-PAD for direct determination of the carbohydrates in food samples. With the new Thermo Scientific™ Dionex™ CarboPac™ SA10-4µm column, the sugar analysis is completed in less than 10 minutes. The new Thermo Scientific™ Dionex™ Integrion™ HPIC™ system introduces more easy-of-use features for this application such as the Thermo Scientific™ Dionex™ Reagent-Free™ IC Eluent Generation (RFIC™-EG) and Thermo Scientific™ Dionex™ IC PEEK Viper™ fitting.References: 1. 21 CFR 101.9 - NUTRITION LABELING
OF FOOD http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=101.9 (accessed Feb 1, 2016)Thermhtml?matchDim=Y&keyword=technical+ note+21&activeTab=DOC&prd Refine=0&continueSearch=true (accessed Feb 1, 2016)
2. Thermo Scientific Dionex Technical Note 20. http://www. thermoscientific.com/ en/search-results.html?matchDim=Y&keyword= technical+note+20 &activeTab=DOC&prd Refine= 0& continueSearch=true (accessed Feb 1, 2016)
3. Thermo Scientific Dionex Technical Note 21. http://www.thermoscientific.com / en/
search-results.html?matchDim=Y& keyword =t echnical+note+21&activeTab=DOC&prd Refine= 0&continueSearch=true (accessed Feb 1, 2016)
4. Thermo Scientific Dionex Application Brief 127. http://www.thermoscientific.com /en/search-results.html?matchDim=Y& keyword =ab127& activeTab=DOC&prd Refine= 0&continue Search=true (accessed Feb 2, 2016)
Disclosure of Interest: None DeclaredKeywords: carbohydrate, HPAE-PAD, sugar
P02-002-036 - Metabolite profiling of chlorogenic acids after ingestion of coffee by high performance liquid chromatography with tandem mass spectrometry detection
M. Gwiazdon 1,*, M. Biesaga 1
1 Department of Chemistry, University of Warsaw, Warsaw, Poland
Content: Coffee is a well known beverage containing large amounts of chlorogenic acids, which might play antioxidant role in human body [1]. Chlorogenic acids are the family of compounds containing ester bonds between quinic acid and hydroxycinnamic acids’ derivatives such as ferulic acid (feruoylquininc acids) and caffeic acid (caffeoylquinic). However, the biological properties of phenolic acids, depend on their bioavailability and metabolism in human body. The aim of these studies was to identify the metabolites of coffee chlorogenic acids, mainly 5-O-caffeoylquinic acid in urine samples of healthy volunteers. The study was conducted in two days cycles during which volunteers were on polyphenol-free diet. A cup of coffee was consumed after 24 hours of diet. Urine samples were collected after different times from coffee consumption for

205www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
a total period of 24 hours. All samples were analyzed using high performance liquid chromatography tandem mass spectrometry (LC-MS/MS). 12 metabolites were identified in urine samples after coffee ingestion. The main compounds identified in the samples were sulfate and glucuronide conjugates of ferulic acid, caffeic acid, dihydroferulic acid and dihydrocaffeic acids. Only trace amount of unchanged 5-O-caffeoylquinic acid were detected. Short time to reach maximum of metabolites concentrations (Cmax) for sulfate derivatives of ferulic acid and caffeic acid indicates their absorption in small intestine. Reduced forms of phenolic acids: dihydroferulic and dihydrocaffeic acids and their derivatives were detected later in urine samples than non-reduced, which indicates their absorption in the large intestine. Highly significant increase in the excretion of studied acids after coffee ingestion relative to the levels before coffee consumption can be observed. This indicates that chlorogenic acids are intensively metabolized in human digestive track.References: [1] Stalmach A., Mullen W., Nagai C., Crozier A.,
Brasil J Plant Physiol 18 (253–262) (2006)Acknowledgements: The research was carried out with the approval of the ethics committee, KEIB – 5/2016Disclosure of Interest: None DeclaredKeywords: coffee, metabolites, polyphenols
P02-002-037 - Urinary excretion of hesperidin and naringin after ingestion of orange juice
M. Gwiazdon 1,*, I. Ostrowska 1, M. Biesaga 1
1 Department of Chemistry, University of Warsaw, Warsaw, Poland
Content: Epidemiological studies show strong correlation between diet rich in plants and food of plant origin and reducing the risk of some chronic diseases such as cardiovascular diseases, diabetes and cancer [1,2]. The dietary flavonoids are present in plants primarily in the glycosides form in which they are not able to cross the cellular membranes. Due to this fact, the key task of polyphenols absorption is their hydrolysis to aglycones. The aim of these studies was to describe the metabolism of two flavonoids glycosides: naringin and hesperidin in human urine samples. The study was carried out in two days cycles during which healthy volunteers were on polyphenol-free diet. A cup of orange juice was consumed after 24 hours. Urine samples were collected in different times during next 24 hours. Measurements were carried out using high performance liquid chromatography with mass spectrometry detection (LC-MS/MS). Preliminary studies demonstrate that only trace amounts of unchanged glycosides were found in urine samples. This indicates the hydrolisis of glycosides to their aglycones in human digestive track. However, similar to glycosides, only small amounts of free aglycones were found in samples. The main metabolites presented in urine are glucuronide and diglucuronide conjugates of hesperetin and naringenin. Maximum concentration (Cmax) of glycosides can be detected in urine samples after about 2-4 hours after orange juice consumption. Time to reach Cmax for

www.ISC2016.ie @isc2016 #isc2016 /isc2016206
POSTER ABSTRACTS Food and Health, Food Safety and Authenticity
glycosides was significantly shorter than for aglycones. The area under the curve (AUC(0-
24)) for glucuronide and diglucuronide derivatives are much more higher than for free glycosides and aglycones. This indicates higher bioavailability of conjugated forms that unconjugated. These results show that orange juice flavonoids are intensively metabolized in human digestive track. References: [1] Sies, H., Archives of Biochemistry and Biophysics
501 (2-5) (2010) [2] Petti, S; Scully, C., Journal of Dentistry, 37 (413-
423) (2009)Acknowledgements: The research was carried out with the approval of the ethics committee, KEIB – 5/2016Disclosure of Interest: None DeclaredKeywords: flavonoids, metabolites, orange juice
P02-002-038 - Sensitive Detection of Organophosphorus Pesticides in Medicinal Plants Using the acetylcholinesterase reaction system based on quantum dots
J. WEI 1,*, P. LI 1
1 Institute of Chinese Medical Sciences, University of Macau, Macau, Macao
Content: A simple, rapid, and sensitive method based on the acetylcholinesterase (AChE) reaction system and core-shell quantum dots (QDs) has been developed for the detection of organophosphorus pesticides. It is well-known that acetylcholinesterase (AChE) can hydrolyze acetylcholine chloride (ACh) to choline, which can be oxidized to betaine with the concomitant generation of hydrogen peroxide (H2O2) in the presence of choline
oxidase (ChOx). The fluorescence of QDs could be effectively quenched by enzyme-generated H2O2. In the presence of pesticides, the activity of AChE is inhibited, leading to the decrease of the generated H2O2, and hence the fluorescence of QDs increases. By measuring the increased fluorescence, the inhibition efficiency of pesticide to AChE activity was evaluated. Under the optimized conditions, there is a good linear relationship between the inhibition efficiency and the concentration of dichlorvos (DDVP) in the range of 0.11−4.4 mg kg−1. The detection limit of DDVP was lower than the maximum residue limits regulated by the European Union and the United States Food and Drug Administration. To evaluate the matrix effect and the applicability of the proposed method for the analysis of real samples with complex matrices, slope ratios of calibration in the Panax ginseng matrix and in neat solvent (Phosphate buffered saline, PBS) were compared. According to the results, the matrix-matched calibration curves were required for accurate quantification. The excellent performance of the proposed biosensor shows that this method is suitable for the determination of trace pesticide residue in real samples with complex matrices.References: [1] Cédric Mongin, Sofia Garakyaraghi, Natalia
Razgoniaeva, Mikhail Zamkov, Felix N. Castellano, Science. 351(369-372)(2016).
[2] Yinhui Yi, Jianhui Deng, Youyu Zhang, Haitao Lia, Shouzhuo Yao, Chemical Communications. 49(612-614)(2013).
[3] Gemma Aragay, Flavio Pino, Arben Merkoçi, Chemical reviews. 112(5317-5338)(2012).
Acknowledgements: This research was supported by the National Natural Science Foundation of China (31160065), the Macau Science and Technology Development Fund (052/2012/A2 and 007/2014/

207www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFood and Health, Food Safety and Authenticity
AMJ), and the Research Committee of the University of Macau (MYRG109-ICMS13-LP and MYRG2014-00089-ICMS-QRCM).Disclosure of Interest: None DeclaredKeywords: biosensor, nanomaterials, pesticides

www.ISC2016.ie @isc2016 #isc2016 /isc2016208
POSTER ABSTRACTS Forensic and Environmental Analysis
Forensic and Environmental Analysis
P01-001-008 - Development of complementary analytical strategies for the characterization of the degradation of solvents used during CO2 capture process.
L. Cuccia 1 2 3,*, J. Dugay 3, D. Bontemps 2, V. Bellosta
4, M. Louis-Louisy 2, J. Vial 3
1 Agence de l’environnement et de la Maîtrise de l’Energie, Angers,
2 Departement Mécanique des Fluides Energie et Environnement, EDF R&D, Chatou,
3 Department of Analytical, Bioanalytical Sciences and Miniaturization, ESPCI ParisTech - LSABM, 4Department of Organic Chemistry, ESPCI ParisTech, Paris, France
Content: In 2010, 25% of the CO2 emissions were caused by electricity and energy production1. One of the most promising technologies to limit these is post-combustion CO2 capture using amine solvents. The main drawback is amine degradation resulting in the formation of degradation compounds in both liquid and gas phase i.e. the treated flue gas. In this study, the degradation of an innovative blend composed of 1-methylpiperazine (30% w/w), piperazine (10% w/w) and water (60% w/w) was studied. To our knowledge, no study has even described the degradation of this blend. In this project, both liquid and gaseous matrices were studied.
In the objective to identify utmost compounds, complementary analytical methods were developed. Regarding the liquid phase of the solvent, Ionic Chromatography (IC), liquid injection gas chromatography coupled to mass spectrometry (GC-MS), and Liquid
Chromatography (LC-MS) were used. A quantification method of the 2 main amines was developed and validated in IC, and allowed the monitoring of the stability of the blend during the process. GC-MS allowed the identification of close to 15 compounds; among them, 2 alkylpyrazines (pyrazine; 2-methylpyrazine) and 7 piperazine derivatives (e.g. 1,4-diformylpiperazine; 1-formylpiperazine). The development of a LC-MS method will allow the identification of compounds with higher molecular weight and/or not volatile, not analysable by GC-MS. Regarding the gas phase, a solid phase sampling method using adsorbent tubes filled with Tenax TA® was used to preconcentrate the compounds emitted with the treated flue gas. Tubes were then thermodesorbed and analyzed by GC-MS. Up to now, 25 compounds were identified; among them 9 alkylpyrazines (e.g. 2-vinylpyrazine) and 7 piperazine derivatives (1-piperazineethanamine; trimethylpiperazine). Quantitative methods will be proposed to quantify targeted compounds in both matrices. To confirm the formation of these compounds, reaction mechanism will be proposed. References: 1. IPCC, 2014Disclosure of Interest:None DeclaredKeywords: CO2 capture, Degradation products

209www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSForensic and Environmental Analysis
P01-001-034 - “DSCAC”: A Unique Chromatographic Method for the Determination of Sugar Alcohols, Carbohydrates and Polysaccharides of both the Fructan- & Starch-type
M. Raessler*
Content: Sugar alcohols, carbohydrates and polysaccharides, such as polyfructans and starch, are important plant components the composition of which may markedly vary with season, light availability and status of plant growth. Carbohydrate distribution also reflects changes of photosynthetic activity and abiotic stress phenomena, such as drought. In addition to plant physiology, the exact identification and reliable quantification of both polysaccharides and carbohydrates in plant material is of growing importance for the calculation of more sophisticated carbon balances of numerous biogeochemical processes.
To tackle this goal, we will present a unique method based on HPLC-PAD, assigned as “DSCAC”: dual-step chromatographic analysis of carbohydrates. It allows the exact and straightforward determination of the sugar alcohols, carbohydrates and polysaccharides relevant to the calculation of what is called non-structural carbohydrates (NSC). It can be applied to both the fructan- and starch-based plant species after appropriate sample preparation.Disclosure of Interest:None DeclaredKeywords: Polysaccharides
P01-001-038 - Simultaneous determination of y-hydroxybutyric acid, ibotenic acid and psilocybin in saliva samples by capillary electrophoresis
P. Saar-Reismaa*
Content: The abuse of fast metabolizing compounds such as y-hydroxybutyric acid (GHB)1, ibotenic acid (IBO)2, and psilocybin (PY)3 is a part of the world-wide problem of using psychoactive substances. Therefore, this study has been conducted to simultaneously determine GHB, IBO and PY in saliva samples by capillary zone electrophoresis with contactless conductivity detection (CE-C4D). The saliva samples were spiked with GHB, IBO, PY and then precipitated by adding acetonitrile (ACN) with a ratio of 1:2 (saliva:ACN) and centrifuged at 500 rpm during 5 minutes. The separation was carried out using an optimized background electrolyte (BGE) of 17.8 mM L-arginine, 9.6 mM succinic acid and 0.002% hexadimethrine bromide, adjusted to pH 7.3 by adding an appropriate amount of 1M sodium hydroxide. The instrument detection limit in saliva was 0.5 mg/L for GHB, 0.7 mg/L for IBO and 1.5 mg/L for PY. Good repeatability, reproducibility and high selectivity were achieved. The proposed CE-C4D method could also be used on-site for the detection of GHB, IBO and PY.References: 1. H. R. Sumnall, K. Woolfall, S. Edwards, J. C. Cole,
and C. M. Beynon, Drug Alcohol Depend., 92 (286–290), (2008)
2. L. Satora, D. Pach, B. Butryn, P. Hydzik, and B. Balicka-−lusarczyk, Toxicon, 45 (941–943), (2005)
3. R. M. Hallock, A. Dean, Z. a. Knecht, J. Spencer,

www.ISC2016.ie @isc2016 #isc2016 /isc2016210
POSTER ABSTRACTS Forensic and Environmental Analysis
and E. C. Taverna, Drug Alcohol Depend., 130 (245–248), (2013)
Acknowledgements: This work has been supported by Internal Security Fund.Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis, psychoactive substances, saliva
P02-002-039 - Determination of artificial sweeteners in environmental water by electrode-assisted liquid phase microextraction coupled with LC-MS/MS
Y.-H. Lin 1, C.-Y. Chen 1, M.-R. Lee 1,*
1 Chemistry, National Chung Hsing University, Taichung City, Taiwan, Province of China
Content: Artificial sweeteners are food additive containing few or no-calories and widely used as sugar substitutes in beverages and foods all over the world. In general, the artificial sweeteners are safe for human consumption, however, the environmental distribution and ecotoxicological impact of artificial sweeteners are limited. Therefore, development of a sensitive analytical method to determine the trace artificial sweeteners in environmental aqueous samples is necessary and important in environmental safety. In this study, an electrode-assisted liquid phase microextraction coupled to liquid chromatography-tandem mass spectrometry (EA-LPME-LC-MS/MS) method was developed for determination of trace artificial sweeteners in environmental aqueous samples. The anodic electrode was connected to the fiber of hollow fiber liquid phase microextraction (HF-LPME) and the voltage of 100 V was applied to the anodic
electrode during extraction. The linearity range of the developed method was 0.05-50 µg/L for acesulfame potassium, 0.625-125 µg/L for sodium saccharin, 0.25-100 µg/L for sodium cyclamate, 10-500 µg/L for sucralose, 1-100 µg/L for aspartame, 1.25-250 µg/L for dulcin, and 0.0625-25 µg/L for neotame. The limits of detection (LODs) and limits of quantitation (LOQs) of seven artificial sweeteners were between 0.1-167 ng/L and 0.4-466 ng/L, respectively. The proposed method was tested by analyzing of artificial sweeteners in environmental waters of Taichung area. Acesulfame potassium and sodium cyclamate were detected in the real environmental water samples with concentration of 1-5.2 µg/L.References: 1. Z. Gan, H. Sun, R. Wang, B. Feng, Journal of
Chromatography A, 1274 (87-96)(2013).2. J.Y. Gui, W. Sun, C.L. Zhang, Y.T. Zhang, L.
Zhang, F. Liu, Chinese Journal of Analytical Chemistry, 44 (361-366) (2016).
3. M.G. Kokotou, N.S. Thomaidis, Analytical Methods, 5 (3825-3833) (2013).
4. M. Wu, Y. Qian, J.M. Boyd, S.E. Hrudey, X.C. Le, X.F. Li, Journal of Chromatography A 1359 (156-161) (2014).
Disclosure of Interest:None DeclaredKeywords: Artificial sweeteners, Electrode-assisted liquid phase microextraction, LC-MS/MS
P02-002-040 - Tracing Pollutants In Groundwater By Passive Sampling And Gas Chromatography Mass SpectrometryP. Auersperger 1,*, K. Lah 1, A. Vrbec 1, V. K. Zidar 1, A. Koroša 2, N. Mali 2
1 JP Vodovod-Kanalizacija d.o.o., 2Geološki zavod Slovenije, Ljubljana, Slovenia

211www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSForensic and Environmental Analysis
Content: Rigorous EU legislation on natural waters pushed analytical methods to new frontiers1-4. Monitoring programmes for natural waters are still mostly based on a collection of spot samples. More than a decade ago passive sampling was introduced as an attractive alternative to the sampling of natural waters. In the year 2012, ISO 56637-23 was published as the first standard for passive sampling of surface waters, followed by ASTM D7929–14 on the passive sampling of groundwater in 2014. Passive sampling monitoring is less expensive, but a validation procedure including sampling uncertainty evaluation is still a challenge. A real case on the use of the qualitative passive sampling method for the detection of a wide range of organic compounds in groundwater by gas chromatography mass spectrometry will be presented with data from more than 40 sampling points in the Ljubljansko polje and Ljubljansko barje aquifers. Additional validation of qualitative passive sampling procedure will be obtained from the comparison of passive sampling results with those from quantitative monitoring at the same sampling points in the aquifer. References: 1. P.C.Lindholm, J.S.Knuutinen, H.S.J.Ahkola,
S.H.Herve, 2014, Analysis of Trace Pharmaceuticals and Related Compounds in Municipal Wastewaters by Preconcentration, Chromatography, Derivatization, and Separation Methods, BioResources 9(2), 3688-3732.
2. H. Brielmann,F.Humer,M.Kralik,J.Grath,M.Clara,S.Weiß,S.Kulcsar,S.Scharf,S.Voerkelius, Identifying anthropogenic nitrogen sources in ground- and surface water, Presented on International Symposium on Isotope Hydrology: Revisiting Foundations and Exploring Frontiers,Vienna 11.-15. May 2015.
3. Lab and field screening of 5 selected passive
samplers for the measurement of VOC fluxes in groundwater, J. Agric. Sci. Appl. 3 (2), 2014, 30-38.
4. CITYCHLOR Interreg IVB-NWE project report, INERIS reference DRC-13-102468-03494A, 2013, Groundwater quality measurement with passive samplers - Code of best practices, 66.
Acknowledgements: This work was financially supported by JP Vodovod – Kanalizacija d.o.o, Ljubljana which the authors greatly appreciate. Disclosure of Interest:None DeclaredKeywords: groundwater, unknown organic pollutants, passive sampling, GC-MS
P02-002-041 - Developing a portable Ion Chromatography System for Freshwater Analysis
E. Murray 1 2,*, B. Moore 1, A. Morrin 2
1 R&D Department, TE Laboratories Ltd, Carlow, 2Chemical Sciences, Dublin City University, Dublin, Ireland
Content: Water is a precondition for human, animal and plant life as well as an indispensable resource for the economy. The protection of water resources is therefore one of the cornerstones of environmental protection. Despite this fact, 80% of the world’s population is still exposed to high levels of threat to water security [1]. High costs and non-representative pollution estimates are associated with manual sampling and off-line analysis. To overcome these disadvantages and to facilitate early detection of pollution events, in-situ analysis is necessary.
When considering the monitoring of anionic pollutants in water, ion chromatography (IC) is the gold standard. However, current commercially available benchtop systems have limited deployment capabilities. To achieve efficient in-situ monitoring,

www.ISC2016.ie @isc2016 #isc2016 /isc2016212
POSTER ABSTRACTS Forensic and Environmental Analysis
portable, automated analytical systems which remotely communicate results to the user are required. This research aims to achieve this by developing a field deployable IC system for in-situ monitoring of anions in fresh water matrices. The system will be low cost and will employ monolithic columns and 3D printed components.
Here, we report the separation and analysis of freshwater anions using monolithic silica columns which have been modified using a range of cationic surfactants and a number of various eluents. Both indirect UV and conductivity detection mechanisms have been investigated and are reported. The current set up and future design of the system are displayed and the applications of the system are also discussed.References: [1] Vörösmarty, C.J.; McIntyre, P.B.; Gessner, M.O.;
Dudgeon, D.; Prusevich, A.; Green, P.; Glidden, S.; Bunn, S.E.; Suillivan, C.A.; Reidy, C. and Davies, P.M. Nature, 2010, 467 (7315), 555-561.
Acknowledgements: This research has been funded through the Irish Research Council’s Employment Based PhD Programme. Disclosure of Interest:None DeclaredKeywords: In Situ Monitoring, Ion Chromatography, Water Quality
P02-002-042 - Characterisation of chemical waste from illegal amphetamine synthesis to support forensic assessment of clandestine drug laboratories
F. M. Hauser 1,*, T. Rößler 1, D. Weigel 1, R. Zimmermann 2, M. Pütz 1
1 KT 34 - Toxicology, Bundeskriminalamt, Wiesbaden,
2Institute of Chemistry, University of Rostock, Rostock, Germany
Content: The illegal synthesis of amphetamines is of great forensic relevance but it is difficult to detect this hidden activity. Synthesis waste is often dumped into the sewage system or into the open country and has a serious impact on the environment [1]. The following work describes the chemical and physical characterisation of waste produced during amphetamine synthesis following the Leuckart route. It is part of the EU project ‘microMole’ which investigates the possibility to identify clandestine labs by means of their disposed waste [2]. Defined amphetamine synthesis waste was produced following instructions found in a clandestine lab. Physical characteristics were determined showing conductivities between 30 and 130 mS/cm, pH values above 11 and densities around 1.2 g/mL. A solid phase extraction (SPE) method was developed to extract organic compounds from the waste. A gas chromatography/mass spectrometry (GC/MS) method going from 60 up to 300 °C within 34 minutes was developed to identify the most common organic compounds present in the waste. Out of these, benzylmethylketone, N-formylamphetamine, amphetamine and 4-methyl-5-phenylpyrimidine were further investigated. Their solubility was determined using high performance liquid chromatography (HPLC) coupled to a diode array detector (DAD) showing values around 10 g/L. Finally a capillary electrophoresis method was developed to quantify ions like ammonium and formate in the waste. Details on the composition of clandestine lab waste can be used forensically to make estimations about production scale, the number of production cycles and characteristics of clandestine labs.

213www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSForensic and Environmental Analysis
Differences in the waste from different synthesis steps allow to identify which synthesis steps took place.References: [1] A. Janusz, K.P. Kirkbride, T.L. Scott, R. Naidu,
M.V. Perkins and M. Megharaj, Forensic Sci. Int., 2003, 134, 62-71.
[2] EU-project microMole, Horizon 2020, FCT-05-2014, agreement number 653626, Web: http://micromole.eu/ (accessed April 2016)
Disclosure of Interest:None DeclaredKeywords: amphetamine laboratories, forensic science, synthesis waste
P02-002-043 - Analysis of pharmaceutical drugs residues in South Africa by gas chromatography-mass spectrometry
B. P. Gumbi*, P. Ndungu, B. Moodley 1, G. Birungi1 School of Chemistry and Physics, University of KwaZulu-
Natal, Durban, South Africa
Content: Environmental concentration data for pharmaceuticals in surface water in the African continent is currently limited. Due to limited methods permitting detection of pharmaceuticals at low concentration in complex matrices. This paper presents method development and application for simultaneous detection and quantification of acidic pharmaceutical drugs residues (salicylic acid, acetylsalicylic acid, nalidixic acid, ibuprofen, phenacetin, naproxen, ketoprofen, meclofenamic acid and diclofenac) in surface water using gas chromatography-mass spectrometry, associated with solid phase micro-extraction for the sample preparation. For most of the acidic drugs, recovery was in the range 70
to 120% and the percent standard deviation was below 15% for the entire method, with limits of detection ranging from 0.041 to 1.614 ppb. The developed method was applied in the analysis of acidic drugs in Umgeni River system, KwaZulu-Natal South Africa. Five of nine analysed acidic drugs were detected and quantified, their concentration in Umgeni River system ranged from 0.0200 to ppb. References: K. Jindal, M. Narayanam and S. Singh,
J. Pharm. Biomed. Anal., 2015, 108, 86-96.M. Saravanan, J. H. Hur, N. Arul and M. Ramesh,
Environ. Toxicol. Pharmacol., 2014, 38, 948-958.G. Shanmugam, S. Sampath, K. K. Selvaraj, D. G.
J. Larsson and B. R. Ramaswamy, Environ. Sci. Pollut. Res., 2014, 21, 921-931.
Z. Li, X. Xiang, M. Li, Y. Ma, J. Wang and X. Liu, Ecotox. Environ. Safe., 2015, 119, 74-80.
Q. Qin, X. J. Chen and J. Zhuang, Crit. Rev. Environ. Sci. Technol., 2015, 45, 1379-1408.
M. Chen, Q. Yi, J. Hong, L. Zhang, K. Lin and D. Yuan, Analytical Methods, 2015, 7, 1896-1905.
K. Kümmerer, Chemosphere, 2009, 75, 417-434.Acknowledgements: National research foundation of South Africa and Umiversity of KwaZulu-Natal.Disclosure of Interest:None Declared
P02-002-044 - Determination of Hormones in Drinking Water by LC-MS-MS using a Poroshell HPH ColumnR. Fu*, A. Zhai, S. Luke
Content: A method for the simultaneous determination of the seven hormones in finished water was developed and validated according to the EPA 539 method [1]. The analytes were extracted and cleaned by Agilent SPEC 47mm C18AR disc solid

www.ISC2016.ie @isc2016 #isc2016 /isc2016214
POSTER ABSTRACTS Forensic and Environmental Analysis
phase extraction (SPE), separated on Agilent Poroshell HPH-C18, 4 µm and 2.7 µm HPLC columns under a basic mobile phase with a high pH of 10.5 and quantified by liquid chromatography coupled to electrospray ionization tandem mass spectrometry (LC/MS/MS). The dynamic calibration range for the analytes was obtained from 0.5 to 70 ng/L. Limit of detection (LODs) for the method analytes fortified into reagent water ranged from 0.03 to 0.7 ng/L. The overall recoveries ranged from 82.6% to 105.6%, with RSD values between 2.7% and 6.0%.
The new Agilent Poroshell HPH-C18 columns packed with superficially porous materials based on the Poroshell 120 family was used for this method. The column is designed to be stable in high pH mobile phases by integrating organic into the porous outer silica layer, thus resisting dissolution under extreme high pH and high temperature conditions. The lifetime test showed column has a superior stability under a high pH mobile phase. References: 1, EPA Method 539 – Determination of Hormones
in Drinking Water by Solid Phase Extraction (SPE) and Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry (LC-ESI-MS/MS), EPA Document No. 815-B-10-001, November 2010.
2, Ralph Hindle. Improved Analysis of Trace Hormones in Drinking Water by LC/MS/MS (EPA 539) using the Agilent 6460 Triple Quadrupole LC/MS; Application note, Agilent Technologies, Inc. Publication number 5991-2473EN, 2013.
Disclosure of Interest:None DeclaredKeywords: drinking water, Hormones, poroshell
P02-002-045 - HPLC-MS for analysis of biophenols in the extracts from heather flowers (Calluna vulgaris)
P. Dróżdż 1,*, K. Pyrzyńska 2
1 Laboratory of the Natural Environment Chemistry, Forest Research Institute, Sękocin, 2Department of Chemistry, University of Warsaw, Warsaw, Poland
Content: Calluna vulgaris is a dominant species of forest shrub growing in Poland. It can be also found in most parts of Europe, North America, Asia and New Zealand. Traditionally, flower and herbal material are used to treat urinary tract disorders and as an antiseptic, antirheumatic and choleretic remedy [1].
The objective of this study was to determine the content of some biophenols in selected heather collected from forest and to compare them with cultivated plant. Aerial parts of Calluna vulgaris were collected from natural environmental localities of central Poland in order to have uniform climatic conditions and altitude.
Chromatographic analysis was performed using a high performance liquid chromatography system (Shimadzu) equipment with a binary pump, degasser, autosampler and connected to 3200 QTRAP Mass spectrometer.
Chromatographic separations were evaluated on a KinetexTM C-18 column (100 x 2.1 mm, 2,6 µm). Gradient elution was used: 8 mM formic acid (pH 2,8) and acetonitrile. Compounds were identified by comparing retention time and m/z values obtained by MS and MS2 with the mass spectra from standards tested under the same conditions.
Extract of heather flowers is a rich source

215www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSForensic and Environmental Analysis
of biophenols. Chlorogenic acid had been isolated as the major phenolic acid in the extract of heather flowers. Moreover, catechins as well as quercetin and quercetrin were found in the significant amounts. The presence of tricetin (5,7,30,40,50-pentahydroxy flavone) with potent anti-inflammatory and anticancer activities was not reported yet in the extract of heather flowers.
Phytochemistry of heather extracts strongly depend on the used extractant. The highest content of biophenols was found in 60% ethanol, the lowest, especially for quercetin and rutin, was obtained when water was used.References: 1. M. Monschein, J. I. Nira, O. Kunert, F. Bucar,
Phytochemistry Review, 9, (205-215), (2010)Acknowledgements: The authors would like to thank Forest Research Institute for financial support given to the project ‘The possibility of utilisation non-wood forest products (NWFPs) as a source of antioxidants in beneficial for health supplements’, topic no. 260801.Disclosure of Interest:None DeclaredKeywords: biophenols, heather (Calluna vulgaris), HPLC-MS/MS
P02-002-046 - Chromatographic techniques and quality control assurance in the forest monitoring programme in Poland P. Dróżdż 1,*, Z. Cieśla 1
1 Laboratory of the Natural Environment Chemistry, Forest Research Institute, S−kocin, Poland
Content: The national forest monitoring programme is a comprehensive programme for monitoring forest ecosystems, which
is now implemented using a three-level structure of permanent observation plots. In 1992, the Forest Research Institute established the central laboratory (now the Laboratory of the Natural Environment Chemistry), entrusting it with all the chemical surveys in the forest monitoring programme. Measurements are carried out in accordance with a uniform methodology used in the national and European forest monitoring programme [1].
12 Intensive Monitoring plots were set up, where on a monthly basis samples of ambient air, deposition and soil solutions are tested. Chemical surveys cover the concentrations of NO2 and SO2 in ambient air and the chemical composition of open field, throughfall, stemflow deposition samples and soil solutions at 25 and 50 cm depth.
The concentrations of selected anions: Cl-, NO3
-, PO43-, SO4
2- in water samples were determined by ion chromatography system (DIONEX ICS-1000) according to the PN-EN ISO 10304-1:2009. The analysis of NO2 and SO2 in ambient air samples were conducted using liquid chromatograph (Shimadzu LC-20AD) according to modified Amaya-Krochmal passive sample method.
To ensure the accuracy of analytical results the laboratory has quality control procedures with different statistical techniques. Quality control includes regular use of certified reference materials, analyses of duplicate samples and participation in laboratory intercomparisons or proficiency-testing programmes. The precision and bias of the analytical methods are monitored with the use of control charts.References: 1. ICP Forests, Manual on methods and criteria for

www.ISC2016.ie @isc2016 #isc2016 /isc2016216
POSTER ABSTRACTS Forensic and Environmental Analysis
harmonized sampling, assessment, monitoring and analysis of the effects of air pollution on forests, (14-15) (2010)
Disclosure of Interest:None DeclaredKeywords: forest monitoring programme, ion chromatography, quality control
P02-002-047 - Investigating the pH influence on the UV degradation of sulfamethoxazole
K. Eckey*, U. Karst 1
1 Institute of Inorganic and Analytical Chemistry, University of Muenster, Muenster, Germany
Content: The removal of micropollutants from surface waters is a topic of growing awareness. Not all xenobiotics are sufficiently removed by municipal wastewater treatment plants. As a result, additional disinfection steps such as ozonation or direct photolysis utilizing UV light are investigated. Many of these procedures are already well elucidated regarding degradation efficiency or energy costs but only few studies deal with the identification of formed transformation products and their toxicity.
In the present study, the photolytic degradation of the frequently used antibiotic sulfamethoxazole (SMX) using polychromatic UV light is investigated focusing on the identification and characterization of formed transformation products via high performance liquid chromatography coupled to electrospray ionization mass spectrometry (HPLC-ESI-MS). The oxidative degradation of SMX using direct photolysis with UV light has been achieved with a high-pressure mercury lamp (250 W) emitting light in the range of 220-500 nm. The SMX
solutions were adjusted to pH 3 and pH 8 using a phosphate buffer to study the pH dependency of product generation.
The photolytic degradation of the antibiotic sulfamethoxazole in aqueous solutions has been investigated to characterize transformation products. In total, more than 20 different mass-to-charge ratios could be found, only ten of which have been published before. It was shown that the pH value has a great impact on how SMX is degraded and which transformation products are formed. Most obvious is the greatly reduced degradation time of SMX at pH 3. To degrade SMX to a similar level, the photolysis needs 18 min at pH 8 whereas it takes only 4 min at pH 3. Additionally, the main transformation product varies depending on the used pH value. While the photolysis of SMX at pH 3 mainly leads to an isomerization of SMX itself, at pH 8, a broader variety of main products is formed.References: 1. K. Eckey, Master thesis, University of
Münster 2015Disclosure of Interest: None DeclaredKeywords: Advanced oxidation processes, Sulfamethoxazole, UV degradation
P02-002-048 - Analysis of designer benzodiazepines by non-aqueous capillary electrophoresis - tandem mass spectrometry using ionic-polymer coated capillary
M. Svidrnoch 1,*, P. Ondra 2, V. Maier 1
1 Analytical Chemistry, Palacky University in Olomouc, Faculty of Science, 2Forensic Medicine and Medical Law, Faculty Hospital, Olomouc, Czech Republic

217www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSForensic and Environmental Analysis
Content: Designer benzodiazepines (BZD) represent a group of semilegal compounds. Even if some of them have been scheduled, new derivatives have been synthesized [1,2]. Capillary electrophoresis with tandem mass spectrometry (CE-MS/MS) is considered to be a powerful technique, namely due to fast analyses, ease of parameters changing and high versatility. Despite the fact that CE-MS/MS covers a very wide range of utilization, some compounds with similar physico-chemical properties and structural relation are difficult to be completely separated. An interesting solution is the use of non-aquous buffers which could be also compatible with the MS detection. The non-aqueous CE (NACE) enables to shift the borders of the conventional CE and its limitations. The NACE-MS/MS was used to investigate the separation and detection conditions of nine BZDs and main parameters were evaluated to reach suffcient separation conditions. Different buffers based on ammonium acetate and trifluoroacetic acid (TFA) in methanol, acetonitrile and their mixtures were studied in terms of separation selectivity. The separations were performed in cationically coated capillary with a reversed EOF. The capillary was prepared by a successive multiple ionic-polymer coating (SMIL) using polybrene and dextran sulfate, which showed very good repeatability of the analyses and led to full separation of BZDs. Another important parameters such as composotion of the buffer were optimized. Also the parameters of the MS/MS detection were optimized to gain the highest signal intensity. The BZDs were detected in single/multiple reaction monitoring modes (SRM/MRM) which provided valuable structural information and allowed identification of each studied BZD.
References: 1. B. Moosmann, L.M. Huppertz, M. Hutter, A.
Buchwald, S. Ferlaino, V. Auwarter, J. Mass Spectrom. 48 (1150-1159), 2013.
2. L.M. Huppertz, P. Bisel, F. Westphal, F. Franz, V. Auwarter, B. Moosnann, Forensic Toxicol. 33 (388-395), 2015.
Acknowledgements: The authors gratefully acknowledge the support of the project LO1305 of the Ministry of Education, Youth and Sports of the Czech Republic. Disclosure of Interest:None DeclaredKeywords: Benzodiazepine, Capillary electrophoresis, mass spectrometry
P02-002-049 - Comparison of two methods for extraction of cardiovascular drugs from waters
A. Klancar 1, J. Trontelj*, Z. Temova 1, A. Kristl 1, R. Roskar 1
1 Faculty of Pharmacy University of Ljubljana, Ljubljana, Slovenia
Content: Cardiovascular diseases (CVD) have nowadays the dominance among chronic diseases (1). Once the disease state is epidemiologically dominant, the use of suitable medications is expanded and pharmaceuticals released in the environment may pose an ecological burden. Our aim was to compare two different extraction methods for 8 representative pharmaceuticals used in CVD from water samples.
Methods Two solid phase extraction (SPE) methods were tested. First was carried out by a manual SPE using Strata-X and the second by SPE-DEX® semi-automated system using HLB discs. Experiments were performed in a synthetic mixture of pharmaceuticals (atorvastatin, bisoprolol, clofibric acid, gemfibrozil, glibenclamide,

www.ISC2016.ie @isc2016 #isc2016 /isc2016218
POSTER ABSTRACTS Forensic and Environmental Analysis
metoprolol, rosuvastatin and verapamil) in Milli-Q water. Extraction of 250 mL water was carried out and subsequently analysed by LC-MS/MS Agilent 6460 triple quadrupole (Agilent Technologies).
The linearity was established on five calibration standards in the concentration range from 1 to 1000 ng/L at pH 7. The recovery and imprecision (n=5) for each analyte were examined at 1 µg/L.
Results The determined recoveries from both methods were comparable, mostly higher than 80% with imprecision 1-8%. The determination coefficients were higher than 0.999. Limits of detection were 1-5 and 5-10 ng/L for manual and automated SPE, respectively. By and large, SPE-DEX method enables faster procedure with a lesser probability of blocking, thus it was chosen for further analysis of wastewater samples.
Finally, optimised SPE-DEX method was applied to five wastewater effluent 24-hours composite samples. Four of target analytes were detected in all samples; in the highest concentrations bisoprolol and rosuvastatin (~ 0.4 µg/L), while clofibric acid was not detected.References: 1. Institute of Medicine (US)
Committee on Preventing the Global Epidemic of Cardiovascular Disease.
Acknowledgements: This work was supported by the LIFE+ project: LIFE13ENV/SI/000466.Disclosure of Interest:None DeclaredKeywords: cardiovascular drugs, LC-MS/MS, Solid phase extraction
P02-002-050 - Assessment of phenolic composition of the forest underbrush
N. Tiso 1,*, K. Bimbiraitė-Survilienė 1, A. S. Maruška
1, V. Kaškonienė 1, R. Daubaras 2, L. Česonienė
2, V. Stakėnas 3, M. Muraškienė 3, V. Tamutis 4, R. Rimgailė-Voicik 5, M. Zych 6
1 Biology, 2 Botanical Garden, Vytautas Magnus University, Kaunas,
3Forestry institute, Lithuanian Research Centre for Agriculture and Forestry, Girionys,
4 Biology and Plant Biotechnology, Aleksandras Stulginskis University, Kaunas,
5 Botany and Genetics, Vilnius University, Vilnius, Lithuania, 6 Botanical Garden, University of Warsaw, Warsaw, Poland
Content: Clear-cutting is a cutting technique widely used worldwide. The functioning and biodiversity of forest ecosystem are deeply related to its structural complexity [1,2]. The variation of the biodiversity and particularly of the chemical composition of the plants before and after the wood-cutting results useful for a better understanding of the modifications that the forest management technique used generates in the forest structural complexity. The research presented is focused on the screening and evaluation of the phenolic composition of the forest ecosystem before the cutting. It includes the phenolic composition analysis of several plants of the underbrush (Vaccinium myrtillus, Lycopodium clavatum, Calluna vulgaris, etc.) and the forest background, performed by Ultraviolet-visible spectroscopic methods and by High Performance Liquid Chromatography analysis. References: 1. M.R. Roberts, F.S. Gilliam, Ecological Applications
5(4), (969-977), (1995).2. J. Bengtsson, S.G. Nilsson, A. Franc, P. Menozzi,
Forest Ecology and Management 132, (39-50), (2000).

219www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSForensic and Environmental Analysis
Acknowledgements: Acknowledgement : This project is financed by Research Council of Lithuania, National Science Program: “Agro- forest and water ecosystems sustainability”, grant No SIT-1/2015.Disclosure of Interest:None DeclaredKeywords: forest ecosystem, High Performance Liquid Chromatography, Phenolic composition
P02-002-051 - Determination of the four major surfactant classes in detergents by RP- HPLC using serially connected UV and evaporative light-scattering detection
J. M. Herrero-Martínez*, A. Escrig-Domenech 1, E. F. Simó-Alfonso 1, G. Ramis-Ramos 1
1 Analytical Chemistry, University of Valencia, Burjassot, Spain
Content: An HPLC procedure capable of determining, the four major surfactant classes present in household detergents - linear alkyl benzenesulphonates (LAS), alkyl ether sulphates (AES), fatty alcohol ethoxylates (FAE) and oleins (soaps, fatty acid salts) - has been developed. On reverse phase (RP) eluting conditions, and for all modifications in the mobile phase assayed, the anionic surfactants LAS and AES, coelute, at lower migration times. Whereas, non-ionic surfactants (fatty alcohol ethoxylates) and fatty acid salts (soaps) appears separated at higher migration times. To evaluate the content of LAS and AES in real samples, two detectors, UV and ELSD detectors, connected in series were selected. Only LAS provides peaks using the UV detector, then the LAS concentration can be obtained using this detector, and the AES concentration can be determined by the subtraction of the LAS concentration from
the sum of the LAS and AES concentrations predicted from the ELSD chromatogram. However, a quadratic adjust is necessary to construct calibration curves, which produces elevated errors in the AES prediction. Two solutions were successfully tried in this work. First, to linearly correct the predicted AES concentrations making use of the LAS concentrations obtained from the UV chromatogram. Second, the addition of a known amount of AES to the samples before analysis to modify the LAS/AES ratio and therefore use the calibration curve for the lowest LAS/AES ratio where the calibration curves showed highest accuracies. These two approaches may be of interest in any other cases in which coeluting species are calibrated using UV and ELSD. The procedure has been applied to the quantification of the four most frequently found surfactant classes in detergents with success. Acknowledgements: Project CTQ2014-52765-R (MINECO of Spain and FEDER) and Quimicas Oro S.A. (Spain). A. E-D thanks the MINECO for an FPU grant for PhD studies.Disclosure of Interest:None DeclaredKeywords: Detergents, HPLC-UV-ELSD, Surfactant determination
P02-002-052 - Separation of Organophosphate Nerve Agents by Capillary Electrophoresis and Microchip Capillary ElectrophoresisX. Cao 1,*, Y. Wang 1, W. Messina 1, A. Hogan 1, E. Moore 1
1 University College Cork, UCC, University College Cork, UCC, Cork, Ireland
Content: Generic Integrated Forensic Toolbox (GIFT) is an FP7 funded project that

www.ISC2016.ie @isc2016 #isc2016 /isc2016220
POSTER ABSTRACTS Forensic and Environmental Analysis
focuses on developing a forensic toolbox for investigating CBRN incidents, and our aim within this project is to develop miniaturised chemical analysis devices for chemical agents’ detection. Organophosphate (OP) compounds are very toxic substances and commonly used as chemical warfare agents and pesticides. Due to their toxicity and requirements of rapid warning and field deployment, innovative analytical tools are urgently needed. Capillary electrophoresis (CE) is one of the most powerful techniques for pharmaceutical, biomedical and environmental analysis due to its high separation efficiency, low solvent consumption and short analysis time. UV is the most commonly used detection technique for CE, while electrochemical detection has emerged as a popular method coupling to CE, due to its high sensitivity, easy operation, compact size and low cost. In this study, methyl paraoxon (MPX), ethyl paraoxon (EPX), methyl parathion (MPT), fenitrothion (FT) and ethyl parathion (EPT) were baseline separated by Micellar Electrokinetic Capillary Chromatography (MECK) and Microchip Capillary Electrophoresis (MCE), with a diode array detector. The short-end injection was used in MECK, so that the separation was achieved in an 8.5 cm capillary within 2.5 minutes. Also a faster separation was performed by MCE (channel length 2.5 cm) which is only 45 seconds. The effects of the SDS concentration, buffer pH, buffer type, separation voltage were investigated. The future work will focus on coupling microchip with contactless conductivity detector, which is much smaller in size and lower in cost.References: 1. J. Zhang, J. Konecny, Z. Glatz, J. Hoogmartens,
A. Van Schepdael, Curr Anal Chem. 3(3)(197-217)(2007)
2. J. Wang, M.P. Chatrathi, A. Mulchandani, W.
Chen, Anal Chem. 73(8)(1804-1808)(2001)3. P. Gozel, E. Gassmann, H. Michelsen, R.N. Zare,
Anal. Chem. 59(44)(1987)4. A Wang, Y. Fang, Electrophoresis 21(1281-1290)
(2000)Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis, Microchip capillary electrophoresis, Organophosphate nerve agents
P02-002-053 - Experimental design as a tool to optimization of pesticides extraction method and quantification by GC-MS
M. I. Paez*, J. M. Martinez 1
1 Department of Chemistry, Universidad del Valle, Cali, Colombia
Content: The present study is focused on the development of an analytical method for the simultaneous analysis of pesticides, the quick, easy, cheap, effective, rugged and safe (QuEChERS) method was one of the most effective preparation methods when dealing with multiple pesticides for its high efficiency of extraction of analytes with a wide polarity range, as well as its simplicity, low cost and amenability to high throughput.
QuEChERS methodology for the extraction of fourteen organochlorine and organophosphate pesticides in soil samples was assessed. The extraction of the pesticides by the selected methodology was evaluated by two experimental designs: Taguchi orthogonal design to determine most influential factors was used and fractional factorial design (25-1) to determine the optimal preparative conditions was used. The quantification was carried out by gas chromatography coupled to a quadrupole ion trap (GC-ITQ) in tandem mode (MS2).

221www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSForensic and Environmental Analysis
Results showed that pH, sonication and use of cleaning adsorbents are irrelevant factors in the preparation process of the sample. Humidity, sample:solvent ratio, and mass of MgSO4 and NaCl, as partition salts, play an important role in the process. The final method obtained consisted in the hydration of 10 g of sample with 5 ml of type I water, stirring for 1 min., adding 10 ml of acetonitrile (ACN) as extractor solvent, salting out with 2 g of MgSO4 and 2 g NaCl, finally, the extract is cleaning out with 100 mg of Octadecylsilane (C18), 10 mg of graphitized carbon (GCB) and 600 mg of MgSO4 per ml of extract. This method allowed extracting, quantitatively, pesticides from soils, with recoveries between 70-120%, RSD less than 10% and detection limits between 4.9 - 23.5 mg / kg.References: Xiao-Qin L., Yun-Fei L., Wen-Ting M.,
Dong-Xiang L., Sun H., Ling T., Guo-Xiang S., J. of Chromatography B, 1015–1016 (2016) 1–12
Disclosure of Interest:None DeclaredKeywords: FRACTIONAL FACTORIAL, GC-ITQ, QuEChERS method
P02-002-054 - Environmental Risk Assessment Of Exposure To Pesticides And Pharmaceutical Compounds In Cauca River Basin (Valle Del Cauca, Colombia)
J. M. Martinez*, M. I. Paez Melo 1, M. Peña 2
1 1Department of Chemistry, 2 Cinara Institute, Universidad del Valle, Cali, Colombia
Content: Different industrial, domestic and agricultural activities in the Valle del Cauca affect the water quality of the Cauca River, one of the most important rivers in Colombia. Among the different compounds that are discharged into the river basin
are e.g pesticides, pharmaceuticals and other organic and inorganic compounds; such compounds may be classified as toxic or hazardous to the extent that its concentration prevails and the time of exposure of individuals increases. The aim of this work was to determine pesticides and pharmaceutical compounds in different points of Cacua River and some of its tributaries, water and sediment samples were taken..
The presence of pesticide residues in sediments collected from some points of the upper basin were determinated; Chlorpyrifos, 4,4-DDE, Endrin, 4,4-DDD, were identified. With regard to the presence of drug compounds, synthetic hormones, analgesics, antipyretics, hypolipemiant agents and antibiotics were found more often in the sample points; bisphenol A, gemfibrozil, naproxen and Iopromid were most prevalent compounds in the water.
The hazard coefficient (HQ) of each group of compounds was calculated. Regarding the pesticides in sediments, chlorpyrifos detected in Virginia and Mediacanoa represents a moderate ecological risk, since the HQ calculated in both places were among 3 and 4; for 4,4-DDE and 4,4-DDD, which are organochlorine pesticides banned, their presence represents according to the data, a low ecological risk (coefficients were among 0.5 and 0.6).For pharmaceuticals, the HQ obtained was low for most compounds detected; for oxacillin, sulfametaxol, the amoxicillin and 4-iso-nonylphenol, quotients higher than 0.1 were obteined, suffesting a low risk but with potential adverse effects. References: EPA Guidelines for Exposure
Assessment 1992, 57, 4–20.

www.ISC2016.ie @isc2016 #isc2016 /isc2016222
POSTER ABSTRACTS Forensic and Environmental Analysis
Warren, N.; Allan, I. J.; et al.,Pesticides and other micro-organic contaminants in freshwater sedimentary environments—a review. Applied Geochemistry 2003, 18, 159–194Disclosure of Interest:None DeclaredKeywords: Risk Assessment, pesticides, pharmaceutical compounds, exposure
P02-002-055 - A novel CE microchip with micro pillars column & double-L injection design and use of Capacitance Coupled Contactless Conductivity detection technolog
Y. Wang 1,*, W. Messina 1, E. Moore 1
1LSI, Tyndall National Institute, Cork, Ireland
IntroductionApplication with FP-7 Project GIFTDesign featuresC4D electrodes design
Content: This novel capillary electrophoresis microchip or known as µTAS (micro total analysis system) was designed to separate complex aqueous based compounds, similar as commercial CE & microchip (capillary electrophoresis) system, but more compact. This system is potentially made for mobile/portable chemical analysis equipment. Un-doped silicon wafer & ultra-thin borofloat glass wafers been used as the main components with Tyndall’s semi-conductor fabrication processes. Double-L injection feature, micro pillars column, bypass separation channel & hybrid-referenced C4D electrodes design to deliver a high SNR (signal to noise ratio), easy-separate, durable and reusable µTAS for CE use.
References: 1) K Tsougeni, K Ellinas etc. A microfabricated cyclo-
olefin polymer microcolumn used for reversed-phase chromatography.
2) Benjamin J. Hindson,a Dora M. Gutierrez etc. Development of an automated DNA purification module using a microfabricated pillar chip.
3) Chien Fat Chau and Tracy Melvin. Design and fabrication of a quasi-ordered nanoporous silicon membrane suitable for thermally induced drug release.
4) Junshan Liu, Fei Xu etc. A polydimethylsiloxane electrophoresis microchip with a thickness controllable insulating layer for capacitatively coupled contactless conductivity detection
5) Wendell Karlos Tomazelli Coltro etc. Renato Sousa Lima etc. Capacitively coupled contactless conductivity detection on microfluidic systems—ten years of development.
6) José Geraldo Alves Brito-Neto1, José Alberto Fracassi da Silva etc. Understanding Capacitively Coupled Contactless Conductivity Detection in Capillary and Microchip Electrophoresis. Part 2. Peak Shape, Stray Capacitance, Noise, and Actual Electronics.
7) Che-Hsin Lin, Ruey-Jen Yang etc. Double-L injection technique for high performance capillary electrophoresis detection in microfluidic chips
Acknowledgements: This research was supported by European Commission FP-7 project GIFT. Thanks to my supervisor Dr. Eric Moore & co-supervisor Dr. Walter Messina from who provided tutorials and expertise that greatly assisted the research.I would also like to show our gratitude to the Tyndall National Institute, University College Cork for sharing their pearls of wisdom with us during the course of this research.Lastly, I would thanks my research partner, Xi Cao, Anna Hogan, Patricia Vazquez and so on…and all the members of LSI, Tyndall and Chemistry Dpt, UCC.Disclosure of Interest:None DeclaredKeywords: micro fabricated, Microchip capillary electrophoresis, microfluidics

223www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSForensic and Environmental Analysis
P02-002-056 - Operational Environment Characterisation for the Development of Future Chemical DetectorsA. Harvey 1,*, R. Lidster 1, S. Lowe 1, C. Burnell 1
1 Chemical Detection & Protection, Dstl, Salisbury, United Kingdom
Content: A wide range of chemical detectors are available for the detection of chemical warfare agents (CWA), toxic industrial chemicals (TICs) and explosives. Depending on the technique used and its application, these detectors can be prone to false alarms due to interferent chemicals present in the environmental background.To address this problem and to allow detector performance assessment, an understanding of materials commonly encountered within different military operational environments is required.Whole air sampling by means of evacuated Entech MiniCans™ provides an attractive method for air and vapour sample collection as it provides an unbiased, comprehensive sample without the need for power, pumps or user skill. These cans are analysed by Thermal Desorption Gas Chromatography (TD-GC-MS) and the individual chemical components identified. Various military environments have been characterised in this way with two priority aims in mind: (1) to populate a database of commonly found chemicals in various operational environments for developing future detection capabilities, and (2) to specify and develop standardised test mixes for realistic testing and evaluation of chemical detectors.The environmental sampling campaign is ongoing with plans to carry out testing in more environments. Disclosure of Interest: None Declared
P02-002-116 - Application of Advanced Analytical Characterisation Techniques to support Process Investigations
Author: Ian Sherlock, Ken Desmond, Kevin F O’Sullivan and Barry O’Sullivan
Content: The Eli Lilly, Kinsale site has evolved from a traditional synthetic chemistry based pharmaceutical manufacturing facility to one that also supports biopharmaceutical manufacturing. In addition to this, process development and clinical trial manufacturing are also performed on site. As the complexity of the site has increased a broad range of advanced forensic analytical techniques such as scanning electron microscopy with energy dispersive spectroscopy (SEM-EDS), stereoscopic microscopy and fourier transform infrared microscopy (FTIR-microscopy) have been established. These forensic analytical techniques have supported a broad spectrum of process investigations. Case studies are presented that highlight the benefit of having this analytical capability.

www.ISC2016.ie @isc2016 #isc2016 /isc2016224
POSTER ABSTRACTS Forensic and Environmental Analysis
Fundamentals of (U)HPLC and GC Separations
P01-001-017- Measurement of resolution and optimisation of chromatographic conditions in the absence of standards
M. C. García-Alvarez-Coque*, J. R. Torres-Lapasió 1, T. Alvarez-Segura 1, J. A. Navarro-Huerta 1
1 Analytical Chemistry, University of Valencia, Burjassot (Valencia), Spain
Content: Getting useful chemical information from samples containing many compounds is still a challenge to analysts. The highest complexity corresponds to situations for which there is no prior knowledge about the chemical composition. Not only there are no standards that could be used to forecast separation conditions of optimal selectivity, but the identity of peaks in chromatograms taken in different conditions can be ambiguous. Moreover, peak size differences affect seriously the separation (a minor peak in a longer elution time can become undetectable), and the resolution level has to be outlined in a probabilistic basis. Most chromatographic objective functions (COFs) described in the literature to measure the separation performance cannot be applied to this type of sample.
In this work, an unsupervised measurement is described to overcome these situations, based on the concept of peak prominence (area fraction exceeding the line joining the valleys delimiting each peak). This measurement is based on the overall signal and does not require a comprehensive knowledge of the individual signals of each compound. This makes it suitable
for the evaluation of the resolution of chromatograms of high complexity. Peak prominences allow the selection of the same best experimental conditions found by a conventional COF with a full knowledge of the underlying number of compounds and their individual signals. MATLAB functions have been developed for the automatic processing of raw chromatograms of high complexity, such as those obtained from extracts of medicinal herbs. The developed COF has been applied to the optimisation of the extraction conditions. Also, it is useful for the selection of the gradient conditions providing maximal information content. This is based on the construction of partial models from peak tracking of the major peaks between independent training experiments, and the comparison of the local peak capacity between them and the actual measurement.Disclosure of Interest:None DeclaredKeywords: Chromatographic objective function, Method development optimisation, Unsupervised resolution measurement
P01-001-042 - Effect Of Pre- And Post-Column Band Broadening On The Performance Of High-Speed Chromatography Columns Under Isocratic And Gradient Conditions
K. Vanderlinden*, K. Broeckhoven 1, Y. Vanderheyden
1, G. Desmet 1
1 VUB, Elsene, Belgium
Content: The development of extremely efficient columns have spurred a new interest in the problem of extra-column band broadening (ECBB). The small peak

225www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSForensic and Environmental Analysis
volumes associated with a decrease in column i.d. and an increase in efficiency make the reduction of ECBB a critical aspect of modern UHPLC instruments.
We report on the results of an experimental and theoretical study of the effect of the ECBB on the performance of small-bore columns filled with the smallest commercially available particles (dp = 1.3µm). Emphasis is on the difference between the effect of ECBB under gradient and isocratic conditions, as well as on the ability to model and predict the ECBB effects.
We investigated the attribution to band broadening of different parts of the instrument in isocratic mode, where the POISe method can be used to (partly) eliminate pre-column band broadening. Since in gradient elution the pre-column band broadening can also be neglected, the contributions of post-column volumes to ECBB in isocratic and gradient mode were compared and modeled. Whereas it is frequently assumed that gradient separations suffer less from ECBB than isocratic separations, we found that gradient separations are very susceptible to ECBB as well. Even more, the steeper the gradient, the more pronounced the ECBB losses become.
We found that it is possible to use a set of column and extra-column data measured under isocratic conditions to get a fair prediction of the influence of the extra-column contribution to the peak width under gradient conditions. This was achieved using well-established band broadening expressions, including the full transient expression (more complete than the Taylor-Aris) and by treating different
connected tubing pieces as one single piece with the same total length and an equivalent diameter.References: K. Vanderlinden, K. Broeckhoven, Y.
Vanderheyden, G. Desmet, J. Chromatogr. A JCA-16-167R1 (2016).
Acknowledgements: The authors wish to thank Dr.Tivadar Farkas (Phenomenex) for providing the columns.Disclosure of Interest:None DeclaredKeywords: extra-column band broadening, gradient elution
P01-001-116 - Evaluation of alternative solvents in normal phase chromatography and lipid classes analysis
N. Prache 1 2,*, P. Sassiat 2, S. Abreu 1, D. Thiebaut 2, P. Chaminade 1
1 Lip(Sys)², Faculty of Pharmacy – Paris Sud University, Paris Saclay University , Chatenay-Malabry,
2 UMR 8231Chimie Biologie Innovation CNRS – ESPCI, LSABM, PSL University, ESPCI Paris, Paris, France
Content: Green analytical chemistry development represents one of the main issues of the 21th century. Many investigators in analytical chemistry are actually involved in the development of methods that prevent irreversible damage to humans and environment in particular, by using “green” alternative solvents.
In the domain of lipid analysis, structural diversity as well as difference in solubility of these compounds is leading to work with a very large range of solvent polarity to separate lipids by classes. Normal-phase chromatography and more specifically, adsorption chromatography, allow elution of compounds in order of increasing

www.ISC2016.ie @isc2016 #isc2016 /isc2016226
POSTER ABSTRACTS Fundamentals of (U)HPLC and GC Separations
polarity. Most of ternary elution systems are inspired of elution scheme of Christie [1]. Hydrocarbons are used to help retention of hydrophobic compounds. Water-alcohol mixture is used to elute phospholipids. Miscibility between the two phases requires the use of solvent of medium polarity as chloroform. Organic solvents stated above although well performing are raising different problems due to their original source, i.e. fossil hydrocarbons, volatility and toxicity for humans and environment. One of the ways to avoid such solvents is the substitution with alternative solvents, as proposed by various actors in green chemistry.
In this presentation, it is shown that separation methods could be developed with solvents alternative to n-heptane (hexamethyldisiloxane) and chloroform (cyclopentyl methyl ether, 2-methyltetrahydrofuran and isoamyl acetate). Binary gradients composed by one or both alternative solvents were carried out to evaluate their chromatographic selectivity for the analysis of major neutral lipid classes. Alternative solvents offer an appropriate selectivity in terms of separation of lipid classes compared with n-heptane/chloroform gradient. Finally, ten classes of lipids were separated by using ternary gradient developed with alternative solvents.References: 1. William W. Christie, Journal of Lipid Research, 26
(507-512) (1985)Disclosure of Interest:None DeclaredKeywords: Alternative solvents, Evaporative light scattering detector, Normal phase liquid chromatography
P02-002-057 - Approach to evaluate the dispersion parameters in HPLC based on the information obtained from a set of compoundsM. C. García-Alvarez-Coque*, M. D. Peris-Díaz 1, T. Alvarez-Segura 1, J. J. Baeza Baeza 1
1 Analytical Chemistry, University of Valencia, Burjassot (Valencia), Spain
Content: The study of column performance is of great interest to characterise stationary phases, develop new materials and increase the resolution of complex samples. Traditionally, this study has been performed through the elution of a probe compound at different flow rates and the representation of Van Deemter plots, which relate the plate height of the column to the linear mobile phase velocity. The interest of these plots is that they can reveal the different contributions to band broadening. The disadvantages of this approach are the need of measuring the extra-column variance, the use of a unique compound which limits the conclusions of the approach, and the increase of the uncertainty due to the manipulation of two peak parameters (position and variance) to obtain the theoretical plate height.
In this work, three complementary approaches are proposed and compared that avoid these disadvantages. One of the approaches is based on the direct treatment of peak variances of several compounds according to a linear relationship with the squared retention time, including the extra-column variance as a fitting parameter. In another approach, the peak variance (expressed in volume units), for each substance, is fitted against the flow rate, and the model parameters are further related to the retention factors. The third

227www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFundamentals of (U)HPLC and GC Separations
methodology is based on a combination of both approaches [1].
The behaviour of several sulphonamides and β-blockers, eluted from C18 columns of different types using acetonitrile-water mixtures was studied. The results show that it is possible to characterise the columns with greater reliability using the above approaches, without previous knowledge of the extra-column variance. Also, they allow the inspection of the whole experimental range of the columns, based on the retention behaviour of several compounds eluted at different retention times. References: [1] J.J. Baeza Baeza, C. Ortiz Bolsico, M.C. García
Álvarez-Coque, Chromatography 2, 625 (2015).Disclosure of Interest: None DeclaredKeywords: Column dispersion, Peak variance, Van Deemter plots
P02-002-058 - Modified Gaussian functions to model the peak shape and asymmetry of chromatographic peaksJ. R. Torres-Lapasió*, J. J. Baeza-Baeza 1 1, J. A. Navarro-Huerta 1, M. D. Peris-Díaz 1, M. C. García-Alvarez-Coque 1
1 Analytical Chemistry, University of Valencia, Burjassot (Valencia), Spain
Content: Chromatographic peaks often show some degree of skewness. This has given rise to the proposal of many theoretical, semiempirical and empirical functions for their description. An appropriate fitting of experimental peaks is needed in the measurement of peak properties, the deconvolution of partially resolved peaks, the evaluation of peak overlapping, and
the prediction of peak profiles at different experimental conditions, which is needed to carry out an accurate interpretive optimisation of the resolution.
To be practical, the peak function should make explicit the properties usually monitored (i.e., retention time, efficiency, asymmetry, and area or height). In this regard, the modified Gaussian functions (MGFs) have demonstrated good performance. MGFs constitute a family of peak functions that interpret the deviations from ideality as a change in the standard deviation or variance with time. Among the proposed MGFs, the polynomially modified Gaussians (PMGs), which assume a change in the standard deviation with time, has awakened special interest [1]. Linear PMG has the drawback of the uncontrolled growth of the predicted signal outside the elution region, which departs from the experimental baseline. To solve this problem, several MGFs have been developed for peaks of different skewness, which have shown been flexible in the simulation of tailing and fronting peaks, being able to describe skewed chromatographic peaks, with errors below 2–3%.
In this work, the peaks of several compounds (sulphonamides and β-blockers) showing a broad range of peak properties, eluted from columns of different type under diverse experimental conditions, were fitted using MGFs of diverse complexity. The capability of the MGFs to fit the experimental peaks and describe their asymmetry is evaluated. Finally, their convenience to develop different tasks and the relationships among them is discussed.References: [1] J.R. Torres, J.J. Baeza, M.C. García,
Anal. Chem. 69, 3822 (1997).

www.ISC2016.ie @isc2016 #isc2016 /isc2016228
POSTER ABSTRACTS Fundamentals of (U)HPLC and GC Separations
Disclosure of Interest:None DeclaredKeywords: Asymmetry, Modified Gaussian, Peak models
P02-002-059 - High performance liquid chromatography at -196ºC and its retention control
T. Motono 1,*, S. Kitagawa 1, H. Ohtani 1
1 Department of Materials Science and Engineering, Graduate School of Engineering, Nagoya Institute of Technology, Nagoya, Japan
Content: High performance liquid chromatography (HPLC) is a general analytical method widely used in various research fields, and nearly all HPLC separations are performed at ambient temperatures. Temperature is an important parameter for HPLC, and its expansion has a potential to achieve separations that are difficult at ambient temperature. However, a lower limit of separation temperature in HPLC depends on freezing a mobile phase. In this study, HPLC at -196ºC using a mixture of liquefied gases, which has a sufficiently low freezing (melting) point, as the mobile phase was developed.
HPLC separation of low molecular weight alkanes at -196ºC was successfully achieved with liquid nitrogen mobile phase and bare-silica stationary phase. The HPLC separation at -196ºC is the lowest temperature ever reported in the literature. The addition of methane to the liquid nitrogen mobile phase suppressed the retention of analytes, so that the retention in ultra-low temperature HPLC can be controlled by the concentration of methane in N2/CH4 mobile phase, as similar to the retention in general HPLC. Moreover, the retention behaviors with N2/He, N2/Ar, and N2/CH4/C2H6 mobile
phase were also investigated. Furthermore, a change in selectivity of n- and iso-isomer and significant enhancement of π-electron/dipole interaction between alkene and stationary phase were found as specific behaviors in HPLC at -196ºC.References: 1. T. Motono, S. Kitagawa, H. Ohtani,
Jornal of Separation Science (38, 2381-2386) (2015)
Acknowledgements: This work was supported by JSPS KAKENHI Grant Number 23655065 and Shimadzu Science Foundation.Disclosure of Interest:None DeclaredKeywords: -196ºC, HPLC, liquefied gas
P02-002-060 - Influence of solvent on the optical rotation of alpha-benidipine enantiomers
O. Kurka 1,*, L. Kucera 1, P. Bednar 1
1 Palacky University, Fac. of Science (RCPTM, KACh), Olomouc, Czech Republic
Content: Rotation of polarized light is a phenomenon observed in chiral compounds. Such compounds play an important role especially in the pharmaceutical industry. There, routine checks are performed to ensure sufficient quality of produced drugs. Separation techniques are widely used to detect impurities in the samples. For this purpose, online optical rotation detector can be suitable as it detects both presence and the optical rotation of an optically active compound. However the influence of the solvent (i.e. mobile phase) must also be taken into consideration. Different studies show that change of the solvent can influence intensity of the observed optical rotation for example by its suitability for hydrogen bonding [1] and can even change the sign of the rotation [2].

229www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFundamentals of (U)HPLC and GC Separations
In our study we deal with benidipine – a chiral compound used as a calcium channel blocker for the treatment of hypertension (high blood pressure) [3]. Racemate of alpha-benidipine was separated into two enantiomers on Lux Cellulose-1 column and isolated enantiomers were analyzed in FIA (flow injection analysis) mode using a optical rotation detector in different solvents used commonly as mobile phases. The results show that low-molecular alcohols (i.e. methanol, ethanol, isopropanol) provide similar response in the detector. Acetonitrile amplifies the signal/noise ratio significantly compared to aforementioned alcohols. Effects of water and hexane are currently being studied. Achieved results can be used to enhance response of benidipine enantiomers in the optical rotation detector by using a suitable solvent as a mobile phase or a post-column additive.References: [1] A. T. Fischer, R. N. Compton, R. M. Pagni, J.
Phys. Chem. A 110 (2006), 7067.[2] Y. Kumata, J. Furukawa, T. Fueno, B. Chem. Soc.
Jpn. 43 (1970), 3663.[3] K. Terada, K. Nakao, K. Okabe, K. Kitamura, H.
Kuriyama, Br. J. Pharmac. 92 (1987), 615.Acknowledgements: The authors thank to Grant Agency of the Czech Republic project (13-01809S), Ministry of Education, Youth and Sports of the Czech Republic (LO1305), and Palacky University Olomouc (project IGA_PrF_2016_016) for financial support.Disclosure of Interest:None DeclaredKeywords: chiral, optical, rotation
P02-002-061 - The effect of buffer concentration and cation type in the mobile phase on retention in HILIC
K. Kalíková*, P. Kozlík 1, E. Tesa−ová 2
1 Analytical Chemistry, 2Physical and Macromolecular Chemistry, Faculty of Science, Charles University in Prague, Prague, Czech Republic
Content: Hydrophilic interaction liquid chromatography (HILIC) is usually employed to separate polar compounds that are unretained or weakly retained in RP-HPLC [1,2]. HILIC mechanism is believed to be complex. Both the partition and adsorption mechanisms may contribute to the overall retention in HILIC [3]. Significant contribution of ion exchange in the retention/separation mechanism of some amino acids was shown [2].
In this work the effect of buffer concentration and cation type in the mobile phase (MP) on retention of dipeptides and amino acids was tested. Two HILIC columns, i.e. TSK gel Amide and Kromasil 60-5SIL were used. MP composed of acetonitrile/buffer 80/20 and 90/10 (v/v). Various buffers differing in cation type were used: LiOH/HAc; NH4
+/HAc; TRIS/HAc; TEA/HAc. The cation concentrations in buffers ranged from 5 to 50 mM. pH values of all buffers was the same, i.e. 4.0.
Obtained results showed that the retention of tested polar compounds is very influenced by concentration and type of cation in MP. This behavior was observed for both tested columns in different extent. The highest retention of dipeptides on amide-based column was achieved using TRIS in MP. Interestingly, using TEA/HAc buffer the

www.ISC2016.ie @isc2016 #isc2016 /isc2016230
POSTER ABSTRACTS Fundamentals of (U)HPLC and GC Separations
retention of compounds decreased with increasing concentration of TEA. None of other buffers showed similar behavior.References:[1] A. J. Alpert, J. Chromatogr. 449 (177-196) (1990).[2] M. Dousa, J. Srbek, Z. Stransky, P. Gibala, L.
Novakova, J. Sep. Sci. 37 (739-747) (2014).[3] B. Buszewski, S. Noga, Anal. Bioanal. Chem. 402
(231-247) (2012).Acknowledgements: The authors gratefully acknowledge the financial support of the Grant Agency of the Czech Republic, Grant No. P206/14-19278P and 16-05942S.Disclosure of Interest:None DeclaredKeywords: buffer type, HILIC, retention
P02-002-062 - Nano-liquid chromatography for analysis of plant extracts
L. Kucera 1,*, M. Cechová 1, S. Fanali 2, Z. Aturki 2, P. Bednář 1
1 Regional Centre of Advanced Technologies and Materials, Department of Analytical Chemistry, Palacky University, Fac. of Science (RCPTM, KACh), Olomouc, Czech Republic,
2 Institute of Chemical Methodologies, Italian National Research Council, Monterotondo, Italy
Content: One of the main trends in modern analytical chemistry is miniaturization of analytical devices. Miniaturized version of LC (nano and capillary LC) found its place in different areas, mostly due to their higher separation efficiency and resolution, lower sample and solvent consumption and in particular higher analysis speed, making these techniques environmentally friendly and cost-effective. The laboratory-made prepared capillary column packed with fully porous particles Chromosphere C18, dp = 3 mm, core–shell particles Kinetex C18, dp = 2.6
mm (100 mm i.d.) and Chromolith CapRod (100 mm i.d.) were used for the separation of diastereomers of (+)-catechin-ethyl-malvidin-3-glucoside. They belong among bridged anthocyanin dyes originating in mixtures of flavanols and anthocyanins in the presence of acetaldehyde (e.g. in wine products). Those dyes were prepared in our laboratory by mixing of their precursors (i.e. (+)-catechin, malvidin-3-glucoside and acetaldehyde), isolated by semipreparative chromatography and their identity was confirmed by nuclear magnetic resonance prior to nanoLC experiments. Fully porous columns Chromosphere C18 provide the best chromatographic performance in less than 7 minutes using mobile phase water:acetonitrile (80:20) acidified with trifluoroacetic acid (0.1%, v/v/v) with flow rate 360 nL min−1. The developed chromatographic method was compared with conventional high-performance liquid chromatography (HPLC) equipped by the same stationary phase (Chromosphere C18, 3 mm, 100 mm × 4.6 mm). The nanoLC provides higher separation efficiency compared to HPLC [1]. At present the nanoLC chromatography is tested for analysis of complex plant extracts including microsamples of pea seed coat tissues during study of seed dormancy.References: [1] L. Kučera, S. Fanali, Z. Aturki, T.
Pospíšil, P. Bednář, J. Chromatogr A 1428 (2016), 126-133.
Acknowledgements: Authors gratefully acknowledge the support of Czech Science Foundation (14-11782S), Ministry of Education, Youth and Sports (LO1305) and Palacký University Olomouc (IGA_PrF_2016_016) for financial supportDisclosure of Interest:None DeclaredKeywords: Capillary column, Flavonoids, Nano-liquid chromatography

231www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFundamentals of (U)HPLC and GC Separations
P02-002-063 - Choose your Adventure. Development, optimization and validation of a generic UPLC method
J. Jokovic*, F. Kuivenhoven, M. Verberne, E. Baltussen, T. Noij
Content: When the challenge is to analyse thousands of different and varying samples on a daily basis, rapid and straightforward method development is a must. For the majority of substances, a partition coefficient and/or dissociation constant can be predicted, which are an important starting point for method development. A generic method development procedure was developed which is based on the use of these predicted parameters to select starting conditions for a reversed phase C18 column which can be further optimized by varying mobile phase composition or by adding mobile phase modifiers. If retention cannot be accomplished, alternative columns for e.g. polar compounds such as HSS T3 or HILIC can be used. Following these guiding principles, a generic procedure that can help analytical chemists establish methods in just a few steps was developed. Like the book “Choose your own Adventure”, the analytical chemist can choose his or her own Method. Various applications will be shown to demonstrate that when a suitable method is chosen according to these principles, it can be readily validated and successfully applied for the sample analysis.References: 1. Veronika R. Meyer, Practical High-Performance
Liquid Chromatography, 5th eddition, 20102. Daniel C. Harris, Quantitative Chemical Analysis,
8th edditiona, 2010Disclosure of Interest:None DeclaredKeywords: Generic, Method development optimisation, UPLC
P02-002-064 - Regularities of quinoline derivatives sorption on carbon surface under conditions of reversed-phase liquid chromatography
N. A. Nekrasova*
Content: The problem of determination of quantitative relationships between structure and properties of compounds, such as chromatographic retention, does not lose relevance. There are many approaches to solving it, which are based on using of molecular structure descriptors and parameters of the studying system [1]. An interesting question is studying of planar and non-planar sorbates retention on sorbents with flat surface. Thereby, the aim of our study was investigating of influence of physicochemical and structural parameters of quinoline and tetrahydroquinoline derivatives on their chromatographic retention on porous graphitized carbon. The main sorption sites of investigated compounds are quinoline and tetrahydroquinoline fragments, which differ in the basicity of the nitrogen and by geometrical shape: quinoline has planar structure while tetrahydroquinoline has deviations from planarity. These explain the lower values of tetrahydroquinolines retention compared with quinolines. Increasing of such parameters as polarizability, lipophilicity, surface area, area of projection of the molecules on plane leads to linear increasing of retention due to the strengthening of sorbates dispersion attraction to the sorbent. The dipole moment growth leads to increasing of induction interactions with graphite π-electron system and retention increasing. In addition, the

www.ISC2016.ie @isc2016 #isc2016 /isc2016232
POSTER ABSTRACTS Fundamentals of (U)HPLC and GC Separations
preferred orientation of molecules with respect to the sorbent surface was obtained and adsorption and solvation energies of the complexes were calculated. Adsorption energy for quinoline derivatives is much higher than for tetrahydroquinolines, due to the greater extension of the aromatic system and possibility to approach closer to the sorbent surface. Among others, we obtained six two-parameter equations. Generally, the obtained correlations allow to predict sorption behavior of structurally similar compounds under conditions of reversed phase liquid chromatography.References: 1. R. Kaliszan, Chem. Rev., 107 (3212-
3246)(2007).Acknowledgements: The work was supported by the Russian Government (grant −14.B25.31.0005).Disclosure of Interest:None DeclaredKeywords: porous graphitized carbon, quinoline derivatives, Reversed-Phase High Performance Liquid Chromatography
P02-002-065 - Programmable At Column Dilution for Peak Shape Management
R. W. Andrews*, P. C. Beals 1, S. Cormier 1
1 RDE, Waters Corp, Milford, United States
Content: With the introduction of sub 2 micron particles and superficially porous particles, the enhanced chromatographic efficiency allows the reduction of column length and volume while preserving resolution. Smaller columns require a reduction of injection volume in order to preserve column efficiency and ensure minimal peak asymmetry. Chromatographs which have been optimized for UPLC and UHPLC feature have reduced pre-column
dispersion which means that the sample which arrives at the head of the column with minimal dilution with mobile phase. This reduces sample focusing for early eluting peaks; it can also cause peak fronting and, in extreme cases, peak splitting. We describe a method to introduce so-called at column dilution by segmenting the desired injection volume and bracketing those segments with weak solvent. This effectively decreases the solvent strength of the sample diluent, increases the retention factor, and ensures sample focusing. The net result is a reduction of peak asymmetry and chromatographic efficiency. The method has been applied to conventional HPLC separations such as the USP monograph method for aspirin and to HPLC and UPLC gradient separations. Enhanced mass sensitivity for UPLC separations run on 1 mm ID columns is demonstrated when at column dilution is employed to ensure sample focusing.References: noneDisclosure of Interest:None DeclaredKeywords: dilution, peak shape, programmable
P02-002-067 - Quantitative analysis of cyclohexanohemicucurbiturils
M. Fomitšenko*, R. Aav 1
1 Tallinn University of Technology, Tallinn, Estonia
Content: Host-guest chemistry is an attractive field in science. One representatives of host molecules are cucurbiturils (CB), which have many family members applications even in drug delivery. We have synthesised first enantiomerically pure CB family members - (all-R,R)-cyclohexanohemicucurbiturils1,2 - that also have chiral recognition ability.

233www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFundamentals of (U)HPLC and GC Separations
The synthesis of any cucurbituril family member is relatively simple, but since it is a polymerization reaction, it causes a mixture of homologues, isomers and oligomers, so analysis and separation of this mixture is challenging.
Many analytical methods are developed to understand and describe host-guest chemistry of CBs but at the same time there is very limited number of examples published for simultaneous screening and quantifying several homologues or isomers of CB themselves.3,4
Herein we describe a chromatographic technique to analyse crude mixture of reaction to follow macrocycle formation5
and calculate the yields of products. All synthesised macrocyclic homologues and isomers are separated on C-18 reversed phase column and detected with UV detection at 210 nm where even different oligomers are distinguishable. Calibration of macrocyclic products is achieved by establishing their purity by quantitative 1H-NMR. Unlike the biotin[6]uril and biotin4 UV response did not correlate with the number of chromophores in case of cyclohexanohemicucurbiturils. Therefore UV spectrum of each compound was measured and molar extinction coefficients on 210 nm were determined. It was observed that UV intensity of macrocycles were magnitude larger compared to linear oligomers, which reflect the sensitivity of UV absorption to spacial geometry. References: 1. R, Aav, Organic Letters, 15, (3786), (2013)2. M, Fomitsneko, Supramolecular Chemistry, 26,
(698), (2014)3. V, Lewin, European Journal of Organic Chemistry,
(3857), (2013)
4. M, Lisberg, Journal of American Chemical Society, 137, (4948), (2015)
5. E, Prigorchenko, Chemical Communication, 51, (10921), (2015)
Disclosure of Interest: None DeclaredKeywords: cucurbituril, Quantitative HPLC analysis, UV absorption
P02-002-068 - Solvation processes on phenyl-bonded stationary phases - the influence of the polar functional groupsS. Bocian 1,*, I. Goryńska, M. Matyska 2, J. Pesek 2, B. Buszewski 1
1 Chair of Environmental Chemistry and Bioanalytics, Nicolaus Copernicus University in Toru−, Toru−, Poland, 2San Jose State University, San Jose, United States
Content: When the mobile phase is in contact with the hydrophobic surface of the stationary phase, molecules of the higher hydrophobicity are preferentially adsorbed on the surface. The composition of the mobile phase at the interface is different from its bulk composition. Thus, the acting stationary phase is a combination of three components: bonded ligands, adsorbed solvent molecules and residual silanols [1].
Summarizing, the solvent from binary mobile phase interacts preferentially with the functional groups on the surface of the stationary phase. For understanding the distribution of the solvent between the stationary phase and a bulk binary solution the excess adsorption of the organic modifier has to be described
A series of phenyl-bonded stationary phases with incorporated polar functional groups were subjected into adsorption investigation. Measurement of acetonitrile

www.ISC2016.ie @isc2016 #isc2016 /isc2016234
POSTER ABSTRACTS Fundamentals of (U)HPLC and GC Separations
and methanol was measured using minor disturbance method. It was observed that adsorption of organic solvent strongly depends on the presence of polar functional groups in the bonded phases. Additionally, relative adsorption of acetonitrile and methanol confirms earlier observations, that presence of amine and amide groups in the stationary phase changes the relative elution strength of organic solvents.References: [1] Sz. Bocian, P. Vajda, A. Felinger, B.
Buszewski, Anal. Chem. 2009 (81) 6334–6346Acknowledgements: This work was supported by Ministry of Science and Higher Education, Grant no. NCN 2013/09/D/ST4/03807 for period 2014–2017.Disclosure of Interest:None DeclaredKeywords: mobile phase, solvation processes, stationary phase
P02-002-069 - Optimizing Selectivity and Efficiency in Critical UHPLC Methods by Separating Eluent Pre-heating from Column Thermostatting
F. Steiner 1,*, S. Patzelt 1, M. De Pra 1, M. M. Martin 1
1 Thermo Fisher Scientific, Germering, Germany
Content: It is commonly considered best practice in HPLC for effective retention control and highest possible efficiency to achieve a homogeneous temperature distribution across the entire separation column. UHPLC methods at pressures up to 1500 bar can generate substantial frictional heat and thus impact on the effective local temperature in columns. Depending on the type of thermostat such frictional heat will cause either radial or longitudinal temperature gradients or both. These
effects can have significant implications for method optimization, because temperature gradients and effective temperatures also change with column length, flow rate, and mobile phase viscosity. The influence on the chromatogram can be substantial if the selectivity of critical peak pairs depends on temperature.
We applied an instrument with pressure capability up to 1500 bar under heat generating conditions in columns of 2.1 mm i.d. packed with particle sizes between 1.5 µm and 2.2 µm. The applied thermostat was able to control the degree of heat dissipation by varying the fan speed to alter air circulation as well as setting the mobile phase pre-heater at a temperature independent of the compartment temperature.
This setup enabled thermal balance studies and effective ways to develop temperature based kinetic and thermodynamic method optimization. While still air thermostatting generated the highest theoretical peak capacities, we sometimes observed critical selectivity differences and changes in peak resolution between both thermostatting modes. Setting the pre-heater to a lower temperature than the column compartment to compensate for pronounced frictional heating effects in still air mode turned out to be a key solution for holistic optimization on efficiency and selectivity of such methods.References: Lucie Nováková, Jean Luc Veuthey,
Davy Guillarme, Journal of Chromatography A, Volume 1218, Issue 44 (7971–7981) (2011).
Disclosure of Interest:None DeclaredKeywords: active and independent pre-heater, frictional heating, still air vs. forced air thermostat

235www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSFundamentals of (U)HPLC and GC Separations
P02-002-070 - Thermodynamics of the Adsorption of α-Phenylcarboxylic Acid Enantiomers on a Chiral Stationary Phase with a Grafted Antibiotic Eremomycin.
E. N. Reshetova*
Content: Chiral stationary phases (CSPs) based on macrocyclic antibiotics grafted onto the surface of silica gel suggested by Armstrong 20 years ago became widespread in analysis of optically active compounds due to the multiple functional groups and chiral centers that ensure successful separation of the enantiomers of compounds of different classes [1, 2]. One of such adsorbents is CSP “Nautilus-E” with a grafted antibiotic eremomycin introduced in chromatographic practice relatively recently [3].
The effect of the pH and ionic strength of aqueous–ethanol mobile phases on the retention and adsorption thermodynamics of the optical isomers of several α-phenylcarboxylic acids on a CSP “Nautilus-E” was studied. It was shown that the sterical structure of substituents on the chiral atoms of the acids affect the mechanism of retention. It was revealed that the “Nautilus-E” adsorbent was most selective to acids, the adsorption of which is an enthalpy-controlled process. The dissociation constants of α-phenylcarboxylic acids have been determined by chromatographic method in aqueous–ethanol solution. A statistical analysis of the phenomenon of enthalpy-entropy compensation was performed. It was shown that the corresponding points of the investigated adsorbates are arranged on
the compensation dependences according to spatial structural characteristics of the molecules.References: 1. D.W. Armstrong, Y. Tang, S. Chen, et al. Anal.
Chem. 66 (1473) (1994).2. S.M. Staroverov, M.A. Kuznetsov, P.N.
Nesterenko, et al. J. Chromatogr. A, 1108 (263) (2006).
Acknowledgements: This work was supported by the Russian Foundation for Basic Research, project no. 15-03-01159-А and by a comprehensive program of the Ural Branch of RAS, project no. 15-3-3-36.Disclosure of Interest:None DeclaredKeywords: Adsorption, Enantiomers, Eremomycin

www.ISC2016.ie @isc2016 #isc2016 /isc2016236
POSTER ABSTRACTS Ion Chromatography and Hyphenation with ICP-MS
Ion Chromatography and Hyphenation with ICP-MS
P01-001-018 - Investigation of capillary ion chromatography for nuclear prospects
C. Gautier*, C. Rey, D. Roussignol, E. Machon, J. Randon, M. Crozet, C. Rivier
Content: The recent development of commercial capillary ion chromatography (IC) systems provides a major opportunity to increase the reactivity of laboratories and to reduce the amount of radioactive effluents produced from the chromatographic analyses in the nuclear field. As the column replacement in a radioactive environment is tedious, the retention behavior of IC columns was investigated at capillary scale to anticipate this operation.
In this framework, the evolution of retention factors was studied on several ion exchange columns (known as AS10, AS15, AS11-HC-4µm for anions and CS12A for cations) implemented on commercial capillary ion chromatography systems so as to achieve a better characterization of their retention behaviors. A linear decrease of retention was measured as a function of the operating time of the capillary columns for all anions whereas no decline was observed for cations. In particular, the retention factor of sulphate exhibited a monthly decrease between 2.5 % and 5 % when using AS15 columns at 30 °C in isocratic mode. Comparisons made with conventional scale showed that this drift was more pronounced at capillary scale. It was also proved that the reduction of the column temperature enabled to limit the retention decline. From this work, we suggest to apply a column temperature of
30 °C when performing the analyses and a low column temperature (maximum 20 °C) when no measurement is done on the Cap-IC device in order to preserve the long-term column retention capabilities. Despite the decrease of retention factors, the analytical performance of capillary ion chromatography was validated with an interlaboratory comparison exercise, which makes this technology convenient for applications to radioactive samples.
This work is of prime interest for nuclear analysts but also for IC users who intend to increase the flexibility and the reactivity of their laboratories.Disclosure of Interest:None DeclaredKeywords: Capillary scale, ion chromatography, Radioactive effluents
P02-002-071 - Trace element determinations of clinical samples by High Resolution ICP-SFMS, with speciation applications by IC-ICP-SFMS
G. Ring*, J. O’Mullane, A. Furey 1
1 Physical Sciences, Cork Institute of Technology, Co. Cork, Ireland
Content: The determination of trace elements and metals is an unavoidable consideration for many scientific industries, including Pharmaceutical, Environmental, Food, Nuclear, Semiconductor and Clinical. While trace metals are necessary for cellular development, growth, and reproduction, the determination of metal profiles can provide important information that reflects the overall health and wellbeing of patients. It is possible to generate such profiles across

237www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSIon Chromatography and Hyphenation with ICP-MS
a variety of clinical matrices. By examining metallic element concentrations, clinicians can link certain metals with medical disorders/diseases or evaluate the efficacy of metallic-alloy implants by monitoring biological outputs from orthopaedic patients.
ICP-SFMS is one of the greatest analytical tools at the disposal of clinicians for such investigations. The instrument combines the extremely high temperatures of an ICP flame with the resolving power of a magnetic sector Mass Spectrometer, making it compatible for analyses that incorporate both inorganic and organic sample matrices.
Some trace elements have an additional level of analytical intrigue, given that their risk of toxicity and bioavailability are dependent upon their species (oxidation state/chemical form). In order to quantify individual chemical species, clinicians must deploy a hyphenated analytical technique known as IC-ICP-MS. As metal speciation analyses for clinical samples is on the increase , the development of analytical methods that allow for the reliable identification and quantification of analytes (and their respective species) that are clearly resolved from interferences is essential.References: 1. G Ring, Clinical Biochemistry, 2016;
(In Press) http://dx.doi.org/10.1016/j.clinbiochem.2016.01.001
2. J Delafiori, G Ring, Talanta (153), 2016; (In Press)Acknowledgements: The author acknowledges the funding obtained for the project titled “Development of a novel elemental point-of-use device for Orthopaedic applications and its validation using High Resolution Inductively Coupled Plasma Mass Spectrometry (ICP HRMS)” [Irish Research
Council (IRC), EMBARK Initiative, Government of Ireland Postgraduate Scholarship Scheme 2014 (Gavin Ring, Ref. GOIPG/2014/1406)]. The author also acknowledges the funding of a High Resolution Sector Field ICP-MS (HR-ICP-SFMS) under the Science Foundation Ireland (SFI) Research Infrastructure Call 2012 [Proposal #12/RI/2335 (2)].Disclosure of Interest:None DeclaredKeywords: Clinical Analysis, ICP-SFMS, Trace Element
P02-002-072 - Using Ion Chromatography with High Resolution Orbitrap Mass Spectrometry for Metabolomic Profiling of Three Representative Food Diet Samples
J. Weiss 1,*, T. T. Christison 2, G. Ellison 3, J. T. Wang
4, L. T. Lopez 2
Technical Director, Chromatography Sales, Thermo Fisher Scientific, Dreieich, Germany,
2 Ion Chromatography, Thermo Fisher Scientific, Sunnyvale, United States,
3 Chromatography Sales, Thermo Fisher Scientific, Hemel Hempstead, United Kingdom,
4 Metabolomics Marketing, Thermo Fisher Scientific, San Jose, United States
Content: Analysis of small polar metabolites is critical to understanding many of the metabolic disturbances as a result of disease, lifestyle, and diet. In this experimental design, UC Davis Center of Metabolomics generated three food samples representing three different diets: low animal protein (Davis), high fish and vegetables (California), and high beef, high sugar, and high fat (USA). Recently it has been shown that ion chromatography (IC) when combined with HRAM MS can provide superior separations and sensitivity for polar ionic species as compared to

www.ISC2016.ie @isc2016 #isc2016 /isc2016238
POSTER ABSTRACTS Ion Chromatography and Hyphenation with ICP-MS
other LC methods. These results have been demonstrated using a capillary IC and replicated using a higher throughput IC system.
Over 1028 compounds were detected in the food samples, of which about 180 small polar compounds were assigned using IC-HRAM via software Compound Discoverer v2.0. Compound Discoverer utilizes mzCloud and ChemSpider to identify compounds, based on MS/MS and accurate mass, respectively. Succinate, as an example, was confirms by comparing the MS/MS spectra with mzCloud library, and we displayed the results found in each sample and diet.
Many of the small polar compounds found by IC were central carbon metabolism compounds, i.e., TCA metabolites. As an illustration of IC-HRAM analysis coverage, these compounds and their differences were summarized in the cycle. Monophosphate and diphosphate sugars and nucleotides, such as AMP were typically difficult to resolve by HILIC and RP chromatography methods, they were also well resolved and easily detected by IC-HRAM. In this study these samples were analyzed using IC-Orbitrap MS and processed using Compound discoverer 2.0.References: 1. J. Wang, T. Christison, etal. Anal Chem., 86 (10),
(5116-5124), (2014).2. S. Hu, J. Wang, etal. Anal Chem., 87 (12), (6371-
6379), (2015),Acknowledgements: We wish to acknowledge Dr. Oliver Fiehn, Ms Arpana Vaniya, and the West Coast Metabolomics Center in Davis, CA, USA for organizing the metabolomics project and for providing the food diet samples.Disclosure of Interest:None Declared
Keywords: High resolution mass spectrometry, ion chromatography, metabolomics
P02-002-073 - Validation of a screening method for polyphoshates identification in fish using ion chromatography with suppressed conductivity detection.
F. Longo*, R. Baccelliere 1, D. Colangelo 1, E. Romualdi 1, R. C. Bonanni 1 1, B. Neri 1
1 Chemistry, Istituto Zooprofilattico Sperimentale del Lazio e della Toscana M.Aleandri, Rome, Italy
Content: A srceening method was developed and validated for determination of polyphosphates ( diphosphates, triphosphates and terapolyphosphates) in fish products by ion chromathography with suppressed conductivity detection. The chromatographic separation was carried out with an anion exchange colum eluted with a sodium hydroxide gradient. The method was validated according to Regulation 882/2004/EC and parameters for screening methods (linearity, specificity and limit of detection) were tested and resulted conforme with the European Directive. The rang of linearity was from 10 mg L-1 to 100 mg L-1 with R2 >0.995. To test method specificity, 20 independent blank samples of different species of fish were analyzed and limit of detection was set to 200 mg Kg-1 . This method is used for fingerprint of sodium hexamethaphosphate with chain lenghts to about 20 phosphates units, compound used as an additive in foodstuffs, in particular meat, processed cheeses and fish products. The European Regulation N° 1333/2008 and its Annex II N° 1129/2011 established a maximum limit permitted for polyphosphates in frozen fishes and did not

239www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSIon Chromatography and Hyphenation with ICP-MS
authorize their use in not-frozen fishes. The method is rapid and simple and it allows the identification of polyphosphates added in foods in which they are forbidden.References: M. Iammarino, A. Di Taranto, Eur Food
Res Technol 235 (409-417) (2012)Disclosure of Interest:None Declared

www.ISC2016.ie @isc2016 #isc2016 /isc2016240
POSTER ABSTRACTS Mass Spectrometry (LC-MS, CE-MS, GC-MS)
Mass Spectrometry (LC-MS, CE-MS, GC-MS)
P01-001-027 - Analysis of organophosphates as aging products of lithium-ion battery electrolytes by liquid chromatography tandem mass spectrometry (LC-MS/MS)
V. Kraft 1,*, M. Winter 1, S. Nowak 1
1University of Münster, Muenster, Germany
Content: Solutions of LiPF6 dissolved in different organic carbonates are used as state-of-the-art electrolytes for lithium-ion batteries. The batteries have excellent properties with regard to cell performance, cycling stability and safety when used at defined operational conditions. The electrolytes are thermally and electrochemically non-stable at elevated temperatures and potentials leading to severe decomposition of LiPF6 and the formation of organophosphates. Some of them like dimethyl fluorophosphate (DMFP) and diethyl fluorophosphate (DEFP) are known in literature as highly toxic species due to their irreversible reaction with the mammal enzyme acetylcholine esterase. The investigation of the aging products is important for the understanding of reaction mechanisms of the decomposition as well as toxicological aspects.
In our previously published work, identi-fication and quantification using external calibration of some organophosphates by LCMS/MS in thermally and electrochemically treated LiPF6based electrolytes was performed.[1] The presented study focused on development of a highly
sensitive quantification method to study organophosphates in electrochemically aged electrolytes. Therefore, MS parameters needed for multiple reaction monitoring (MRM) transitions were optimized for several organophosphates both commercially available and synthesized. Furthermore, the chromatographic behavior of the analytes on reversed-phased columns was investigated. For the quantification, external calibration and standard addition methods were applied and compared. In an electrochemically aged electrolyte treated at 5.5 V vs. Li/Li+, the concentrations of DMFP and DEFP were determined at 1162.4 ppm and 611.6 ppm, respectively. The studies included the investigation of the suppression effects caused by the sample matrix in the LC-MS/MS setup. References: [1] V. Kraft, W. Weber, B. Streipert, R.
Wagner, C. Schultz, M. Winter, S. Nowak, Rsc Adv, 6, 8-17, 2016.
Disclosure of Interest:None DeclaredKeywords: LC-MS/MS, lithium-ion battery electrolytes, organophosphates
P01-001-033 - The Decomposition of Ionic Liquids Monitored by Capillary Electrophoresis Hyphenated to a High-Resolution Mass Spectrometer
M. Pyschik 1 2,*, M. Winter 1 2, S. Nowak 2
1 University of Muenster, Institute of Physical Chemistry, 2 University of Muenster, MEET Battery Research Center,
Muenster, Germany
Content: Nowadays, ionic liquids (ILs) are in the center of attention for different chemical applications, for example, as catalysts for chemical reactions or as

241www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMass Spectrometry (LC-MS, CE-MS, GC-MS)
electrolyte in lithium-ion batteries. The application fields are widely spread. For this reason, it is necessary to know in detail the decomposition products of ILs. So far, the degradation of ILs were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) or theoretical calculations. Only few studies deal with the investigation of decomposition products via chromatographic methods, like Zhou et al., which used high-performance liquid chromatography (RP-HPLC) with a monolithic column for the determination of BF4
- in ILs [1].
We have developed and optimized new capillary electrophoresis (CE) and ion chromatography (IC) methods. The IC system was hyphenated to a triple quadrupole mass spectrometer [2, 3] and the CE to a quadrupole time-of-flight (Q-TOF) mass spectrometer. Because of the hyphenations, it was possible to identify cationic decomposition products of pyrrolidinium- and imidazolium- based ILs and anionic decomposition products of trifluoromethanesulfonyl imide (TFSI) and fluorosulfonyl imide (FSI) [2, 3]. Comparing the IC and CE methods with each other, it can be said that the CE method is the more usefully method for the investigation of ILs. With the CE method, it was possible to separate the decomposition products well from each other and quantify the certain decomposition products.References: [1] S. Zhou, H. Yu, L. Yang and L. Ai, J. Chrom. A
1206 (200-203) (2008).[2] M. Pyschik, V. Kraft, S. Passerini, M. Winter
and S. Nowak, Electrochim. Acta 130 (426-430) (2014).
[3] M. Pyschik, C. Schultz, S. Passerini, M. Winter
and S. Nowak, Electrochim. Acta 176 (1143-1152) (2015).
Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis, ionic liquids, quadrupole time-of-flight mass spectrometer
P02-002-074 - Liquid Chromatography Mass Spectrometry Analysis Of Sodium Sulfo Palmityl Methyl Ester Surfactant A. Asselah 1 2,*, A. Tazerouti 1
1 Organic chemistry, University of science and technology, Cité El Alia- Bab Ezzouar,
2 Environment Engineering , faculty of Engineering science- University of M’Hamad Bougara, Cité de l’indépendance- Boumerdes, Algeria
Content: A sodium sulfo palmityl methyl ester surfactant (C16-MES) synthesized by photochemical process [1-2] was analyzed by liquid chromatography electrospray ionization tandem mass spectrometry ( LC-MS/MS-ESI) to confirm it chemical composition determined. The LC-MS/MS analysis by ESI of (C16-MES) diluted in ethanol revealed the presence of two peaks in LC/MS spectrum. The first one corresponded to sodium sulfo palmityl methyl ester salt giving the pseudo molecular ion [M-H]-. The second peak corresponded to the disalt is retained longer than the monosalt. The spectra reveal the presence of the characteristic fragments for this surfactant indicating the sulfonate group at m/z 80 and others fragments corresponding for the loss of sodium or methyl group to molecular ion. These results confirm the structure of the product and agree the literature data [3] who report that MES surfactants are composed in their structure of mono and disalt.

www.ISC2016.ie @isc2016 #isc2016 /isc2016242
POSTER ABSTRACTS Mass Spectrometry (LC-MS, CE-MS, GC-MS)
References: [1] ASSELAH A., TAZEROUTI A., J of Surf and
Deterg, 17 (6):1151-1160, 2014[2] AZIRA H., ASSASI N., and A.TAZEROUTI, J of
Surf and Detergs, 6 : 55-59, 2003[3] HOLMBERG K., Novel surfactants: preparation,
applications, and biodegradability, Marcel Dekker, New York, pp 425–466, 2003
Disclosure of Interest:None DeclaredKeywords: liquid chromatography, methyl ester sulfonates, mass spectrometry
P02-002-075 - Separation of Quorum Sensing Signalling Molecules of Pseudomonas aeruginosa by HILIC Coupled with Q-TOF MSA. Buzid 1 2,*, V. K. Langsi 1 2, E. Ó Muimhneacháin 1, F. J. Reen 3, F. O’Gara 3 4, G. P. McGlacken 1, J. D. Glennon 1 2
1 Department of Chemistry and Analytical & Biological Chemistry Research Facility (ABCRF), University College Cork,
2 Innovative Chromatography Group, Irish Separation Science Cluster (ISSC),
3 BIOMERIT Research Centre, School of Microbiology, University College Cork, Cork, Ireland,
4 School of Biomedical Sciences, CHIRI, Curtin University, Perth, WA, Australia
Content: Pseudomonas aeruginosa, a gram-negative opportunistic pathogen, is capable of surviving in a wide range of natural environments. It is an antibiotic-resistant human pathogen associated with hospital acquired infections and causes acute pneumonia and chronic lung infections in cystic fibrosis (CF) patients. 2-heptyl-3-hydroxy-4-quinolone (PQS) and its precursor 2-heptyl-4-quinolone (HHQ) are two important signal molecules produced by P. aeruginosa while pyocyanin (PYO) is a redox active toxin involved in virulence and pathogenesis. Early detection is possible
through this molecular signature analysis, and is crucial in the clinical management of this pathogen, as established infections enter a biofilm lifestyle that is refractory to conventional antibiotic therapies. Herein, the chromatographic separation and determination of the three biomarkers is reported using hydrophilic interaction liquid chromatography (HILIC) coupled to quadrupole-time-of-flight mass spectrometry (Q-TOF-MS) detection. The method is applied for the simultaneous detection of PYO, HHQ and PQS in microbial culture and in biological fluids, including P. aeruginosa spiked CF sputum samples. The performance of the method is compared to a sensitive electrochemical approach using a boron-doped diamond (BDD) electrode previously reported from this laboratory.References: Molecular signature of Pseudomonas aeruginosa
with simultaneous nanomolar detection of quorum sensing signaling molecules at a boron-doped diamond electrode. Scientific Report, 2016.
Disclosure of Interest:None DeclaredKeywords: cystic fibrosis , mass spectrometry, Pseudomonas aeruginosa
P02-002-076 - TLC-MS: coupling thin layer chromatography with mass spectrometry - a practical approach for matrix-loaded samplesH. Griesinger 1,*, E. Reuss 2, M. Oberle 1, K. Matheis
2, M. Schulz 1
1 Instrumental Analytics R&D, 2 Department of Bioanalytical Chemistry, Merck KGaA,
Darmstadt, Germany
Content: A straightforward way to couple thin layer chromatography (TLC) with mass

243www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMass Spectrometry (LC-MS, CE-MS, GC-MS)
spectrometry (MS) is the TLC-MS Interface from Camag [1]. It is an elution-based, semi-automatic system to extract zones from the TLC plate and transfer them online into the MS. It is suitable for all thin layer materials and every eluent that can be sprayed in the ion source. The interface can be connected to any kind of LC-coupled mass spectrometer.
We show how the TLC-MS interface can be used for the development of TLC-MS applications in the areas of food & beverage, pharmaceutical ingredients, cosmetic actives and peptide & protein analysis:• separation and identification of insulin
species• investigation of UV-filters in suncream• analysis of steroids• determination of caffeine in energy
drinks
All experiments were performed on newly developed HPTLC plates with a reduced separation layer thickness. After chromatographic separation the analytes were extracted with acetonitrile/water (95:5, v/v) and transferred online into the MS with a flow rate of 0.2 ml/min. The ionization technique was electrospray ionization (ESI) in the positive mode.References: [1] H. Luftmann, A simple device for the
extraction of TLC spots: direct coupling with an electrospray mass spectrometer, Anal. Bioanal. Chem., 378, 964-968, 2004
Disclosure of Interest:None DeclaredKeywords: mass spectrometry, thin layer chromatography, TLC-MS interface
P02-002-077 - MS Spectra Database of Flavours and Fragrances coupled with Linear Retention Index Approach: Reliable Identification of Complex Essential Oil Samples
M. Utczas 1, M. Zoccali 1,*, G. Micalizzi 1, G. Purcaro 1, L. Mondello 1 2
1 Chromaleont srl, 2 Dipartimento di Scienze Chimiche, Biologiche,
Farmaceutiche ed Ambientali, University of Messina, Messina, Italy
Content: Gas chromatography coupled to mass spectrometry (GC-MS) is a widely used analytical method for a detailed characterization of any kind of complex sample, like essential oils. A commercial spectra database can help in the identification of the unknown substances, but in many cases misidentification can occur due to a high spectral similarity. Conventional mass spectral search combined with the Linear Retention Indices (LRI) approach can provide a great possibility to boost the identification of “challenging” molecules, occurring in essential oils. To the right use of the LRI information it is important to know exactly the GC column stationary phase and the chromatographic conditions. A GC-MS database devoted to the flavour and fragrance field, namely FFNSC library, collecting around 3500 spectra derived from pure chemicals, essential oils and perfumes, has been built-up. The quadrupole MS spectrum of each compound is registered in the database, along with structural information, IUPAC name, CAS number, and the experimental value of LRI. The latter have been obtained utilizing three different types of reference standards (n-alkanes, fatty acid methyl esters and fatty acid ethyl esters) and three different stationary phases (1% and 5% diphenyl-95%

www.ISC2016.ie @isc2016 #isc2016 /isc2016244
POSTER ABSTRACTS Mass Spectrometry (LC-MS, CE-MS, GC-MS)
polymethylsiloxane, polyethyleneglycol). To maximize the potentiality of such a database, the use of a post-run software, CromatoPlus Spectra, was exploited. This software allows to import data coming from different systems and to carry out the identification with the simultaneous support of the spectral similarity and the LRI filter. It has been proven that a LRI filter in the ± 5 range allows to perform a highly reliable identification of complex essential oil samples.Disclosure of Interest:None DeclaredKeywords: Gas chromatography , Linear Retention Indices , mass spectrometry
P02-002-078 - Tips and Tricks for enhanced sensitivity in LC-MS laboratories
H. Griesinger 1,*, S. Altmaier 1, A. Domanov 2, M. Schulz 1
1 Instrumental Analytics R&D, Merck KGaA, Darmstadt, Germany,
2 Lab Water Applications, Millipore S.A.S., St. Quentin, France
Content: The performance of LC-MS instruments regarding sensitivity and resolution has improved tremendously in recent years. In parallel, regulatory requirements regarding the limits of detection of various analytes have become increasingly challenging.
The two most important prerequisites in LC-MS analyses are the use of purest solvent and additive quality available as well as the establishment of a workflow avoiding contamination of setup and sample during processing [1]. This is, because any impurity or contamination will negatively affect sensitivity (signal-to-
noise ratio) and hence increase the limit of detection: By increasing background noise, by signal suppression or adduct formation as well as by increasing the complexity of a mass spectrum or chromatogram (formation of ghost peaks). All solvents and additives (buffers, acids, bases, salts) have to be of MS-grade quality and need to be volatile. Especially organic solvents can leach contaminants out of container surfaces (e.g. stabilizers or plasticizers). Normal glass is dissolved by ultrapure water and silica and alkali is released, and mainly the latter ions form adducts with analytes. Other sources of contamination are solvent containers cleaned in a dishwasher, unsuitable HPLC columns and tubing, contaminated system filters or frits as well as solvent compositions leading to microbial growth or buffer precipitation. These issues not only compromise analyses, complicate data interpretation and add the risk of repeating experiments, but also decrease column life time and maintenance intervals of analytical instruments.
In this poster handling pitfalls in a typical LC-MS workflow will be discussed. Aforementioned problems will be described in detail and tips and tricks will be provided in order to enable or maintain high-quality and high-sensitivity LC-MS analyses.References: [1] B.O. Keller, Interferences and contaminants
encountered in modern mass spectrometry, Anal. Chim. Acta, 627, 71-81, 2008
Disclosure of Interest:None DeclaredKeywords: LC-MS, sensitivity, tips & tricks

245www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMass Spectrometry (LC-MS, CE-MS, GC-MS)
P02-002-079 - Liquid Chromatography/Coordination Ionspray-Mass Spectrometry for the Analysis of Liquid CrystalsT. Berg*, T. Elseberg, P. Leonhard, C. Enders, M. Sperling, U. Karst
Content: Many flat panel displays in electronic devices are based on the liquid crystal display (LCD) technology. Mixtures of between ten and twenty small organic molecules (liquid crystals) are important for the image buildup.[1] For the correct function of LCDs, quality control of the single components and mixtures is important during the whole production process. Here, molecular information of the low polar liquid crystals mostly is obtained by atmospheric pressure chemical ionisation-mass spectrometry (APCIMS). However, the analysis of nonpolar compounds is still challenging due to the poor ionisation efficiency of the analytes. Therefore, coordination ionspray-mass spectrometry (CISMS) is demonstrated as an alternative analytical technique for the investigation of rather polar liquid crystals. Application of CIS-MS leads to the formation of charged complexes by adding cations or anions to the analyte solution. Consequently, an increased polarity and thereby an improved detectability of the compounds results. Multicomponent mixtures can be analysed by previous liquid chromatographic (LC) separation.
A liquid crystal mixture containing eleven compounds was analysed by means of LC/CIS-MS using reversed phase chromatography. After LC separation, a solution of silver tetrafluoroborate in acetonitrile was added to the eluate. The
formed complexes of the analytes with the silver(I) ions were detected after electrospray ionisation in the positive ionisation mode.
Six of eleven compounds could be detected as their silver adducts by LC/CIS-MS. Monomer adducts [M+Ag]+, acetonitrile adducts [M+Ag+ACN]+ and dimers [2M+Ag]+ were obtained. All complex forming analytes contained two or more aromatic rings in their structures.References: 1. D. Pauluth, K. Tarumi. Journal of
Materials Chemistry 14(8) (1219-1227) (2004).Acknowledgements: S. Fartowski and D. Gemeinder, Merck KGaA, Darmstadt.Disclosure of Interest:None DeclaredKeywords: Coordination Ionspray-MS, Liquid Crystals, Mass Spectrometry
P02-002-080 - Design of Experiments to set up variables in a gas chromatography coupled to mass spectrometry (GC-MS) to determine persistent organic pollutants
N. Castro*, M. I. Páez 1
1Chemistry, Universidad de Valle, Cali, Colombia
Content: The persistent organic pollutants (POP) is a group of chemicals, that can be stable in the environment, they can travel long-range and these can accumulate in animals, human and food chains, therefore, they can produce Eco-toxic effects in environmental and humans1. Analytical methods that can determine them are highly required together with an instrumental technique that ables to detect and quantify at trace level. Usually GC/MS is applied, which has numerous variables that require

www.ISC2016.ie @isc2016 #isc2016 /isc2016246
POSTER ABSTRACTS Mass Spectrometry (LC-MS, CE-MS, GC-MS)
to be optimized 2. In this research, Design of Experiments (DOE) were applied to set up six variables of the GC/MS to determined organochlorine pesticides and polychlorinated biphenyls. These were inlet temperature (° C), splitless time (s), pulse pressure (KPa), time pressure, final temperature of oven program (° C) and transfer line temperature (° C). 2-Level fractional factorial design (FF) was carried out to check if these variables have any influence on the signal/noise response for each compound, then, central composite design (CCD) was used to optimized the variables that affected the response.
The FF results showed that the factors inlet temperature(XA), splitless time(XB) and transfer line temperature(XC) are the most affect the pollutant responses. The optimization phase obtained a function relating to the values of the three variables with the response. The empirical model was applied to predict responses of pollutants according to different values of the studied variables. The results showed that optimal value is the best for majority of pollutants and these were XA: 303 °C, XB:1,17s and XC:237°C. They were evaluated and compared with other instrumental conditions using calibration curves and their slopes certified that the optimal values are better than the other conditions.References: 1. K,Valsaraj, J. Phys. Chem. Lett.1, (1694–1700)
(2010)2. M,Callao, Trends Anal. Chem. 62, (86–92) (2014)Disclosure of Interest:None DeclaredKeywords: Design of Experiments, gas chromatography, Persistent Organic Pollutants
P02-002-081 - Peptidomic approaches for the identification of antihyperlipidemic peptides inolive seeds and study of their cytotoxicityI. M. Prados Nieto 1,*, R. Vásquez-Villanueva 1, M. C. García 1, M. L. Marina 1
1 Departamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcalá, Alcalá de Henares, Spain
Content: Olive seed proteins are a sustainable source of bioactive peptides [1]. Indeed, this byproduct presents a high protein content and peptides with antihypertensive, antioxidant and antihyperlipidemic capabilities are hide within proteins sequences. Hyperlipidemia can be due to a high level of cholesterol (hypercholesterolemia), triglycerides (hypertriglyceridemia) or both. Antihyperlipidemic peptides in foods and food byproducts have been scarcely studied. We have demonstrated that olive seeds contain antihyperlipidemic peptides and we have obtained fractions concentrating peptides exerting the highest capacity to absorb cholesterol and triglycerides. The aim of this work was to identify by HPLC-Q-TOF-MS and de novo identification the sequences of the peptides present in these fractions and to study theircytotoxicity.
Olive seed peptides were obtained by a procedure previously described [1] comprising a first extraction of proteins followed by digestion with Alcalase enzyme. This hydrolyzate was fractionated by ultrafiltration and fractions were subfractionated by semipreparative RP-HPLC to obtain fractions showing the highest ability to reduce cholesterol and triglycerides. Identification was carried out by HPLC-Q-TOF-MS and de novo sequencing

247www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMass Spectrometry (LC-MS, CE-MS, GC-MS)
using PEAKS software. Moreover, two different chromatographic modes were employed for the comprehensive identification of peptides, a fused-core reversed-phase HPLC column and a fused-core column in the hydrophilic interaction chromatographic mode. Cytotoxicity of most active fractions was evaluated using the colorimetric MTT assay and three different kinds of cells (HK2, HeLa and HT-29).References: [1] C. Esteve, M.L. Marina, M.C.
García, Food Chem.167 (2015)272-280.Acknowledgements: Authors thank the Spanish Ministry of Economy and Competitiveness (AGL2012-36362), the Centro para el Desarrollo Tecnológico Industrial (ITC-20151193), the Comunidad of Madrid (Spain) and FEDER program (S2013/ABI-3028). We also thank FAROLIVA S.L. (Spain) for the kind donation of olive stones.Disclosure of Interest:None Declared
P02-002-082 - Development and validation of HPLC-MS/MS method for the simultaneous determination of seventeen metabolites in human urine
A. Yumba Mpanga 1,*, D. Siluk 1, J. Jacyna 1, O. Szerkus 1, R. Bujak 1, M. Markuszewski 2, M. Matuszewski 2, R. Kaliszan 1, M. J. Markuszewski 1
1 Department of Biopharmaceutics and Pharmacodynamics,
2 Department of Urology, Medical University of Gdansk, Gdansk, Poland
Content: Seventeen endogenous metabolites were find to be statistically significantly different in the concentration levels in a previous non-targeted analysis of urine from bladder cancer patients and healthy
volunteers. In order to confirm those differences, an HPLC-MS-MS-ESI (+/-) method for the simultaneous determination of those metabolites in urine was developed and validated following FDA Guidelines [1]. Afterwards, the method was used to compare metabolites levels in 40 patients with bladder cancer and 40 healthy volunteers.
The LC-MS analysis were conducted using a gradient elution of mobile phase consisted of 0.06 % acetic acid in water and 0.06 % acetic acid in acetonitrile on an 150 mm x 4.6 mm x 2.7 µm Ascentis Express C-18 column (Supelco Analytical, USA). The validation was performed with the use of blank urine obtained by three-fold purification of a pooled urine on a solid phase extraction (Agilent Technologies, USA). The validation was ensured in terms of linearity, the limit of detection (LOD), the limit of quantification (LOQ), accuracy, precision, recovery, matrix effect and stability. The analysis of the data obtained by the investigation of biological samples was provided by the use of U-Mann Whitney test.
The LOD and the LOQ was in the range of 0.07-13.76 ng/ml and 0.22-45.85 ng/ml, respectively. The concentration range of compounds was found to be between 2.5 and 12500 ng/ml. Only one compound showed a significant matrix effect (61 %). The method presented recovery and precision values within the ranges proposed by FDA [1].
In this study, a targeted analysis of 17 metabolites present in human urine was conducted in order to confirm the potential of those metabolites as biomarkers in bladder cancer detection.

www.ISC2016.ie @isc2016 #isc2016 /isc2016248
POSTER ABSTRACTS Mass Spectrometry (LC-MS, CE-MS, GC-MS)
References: 1. FDA, Guidance for Industry – Bioanalytical Method Validation, May 2001.
Acknowledgements: The project was supported by the National Science Center granted on the basis of the decision number 2012/07/N/NZ75/00081 and 2012/05/B/NZ7/03293.Disclosure of Interest:None DeclaredKeywords: biomarkers, HPLC-MS/MS, Method validation
P02-002-083 - Quantification of Steroid Hormones in Plasma using a Surrogate Analyte Approach and HPLC-HR-MS/MS with SWATH-Acquisition
B. Drotleff 1,*, M. Hallschmid 2, M. Laemmerhofer 1
1 Institute of Pharmaceutical Sciences, 2 Institute for Medical Psychology and Behavioural
Neurobiology, Ebernhard-Karls-University Tuebingen, Tuebingen, Germany
Content: Quantification of low level steroids still faces many analytical challenges. Although a wide variety of sensitive immunoassays is available, reliable determination with mass spectrometric methods is commonly demanded due to known disadvantages of immunoassays, such as matrix interferences and cross reactivity1. Another main issue is the lack of true blank plasma for steroids, so that suitable alternative solutions for correct calibration and quantification are needed. To overcome this problem, a surrogate analyte approach2 was applied for the target compounds testosterone and estradiol. Here, calibration is done via 13C-labeled isotopic analogues that are spiked into pooled plasma.
In order to achieve calibration ranges that cover total endogenous testosterone and
estradiol plasma levels of women and men, high-end triple-quadrupole mass analyzers are usually utilized. By comprehensive instrument optimization and solid phase extraction of samples, these ranges could be satisfyingly reached with a high resolution tandem quadrupole/timeofflight (QqTOF) instrument. In combination with a five minute gradient elution on a core-shell reversed-phase column, limits of quantification of 10 ng/mL could be achieved for both analytes in positive mode.
With this method total testosterone and total estradiol levels of male patients, who received estradiol treatment via a transdermal patch, were determined. The used SWATHacquisition allows combined generation of targeted data for absolute quantification as well as untargeted data for metabolite or steroid profiling. This technique also showed sufficient selectivity and superior sensitivity compared to MRMHR.References: 1 M. D. Krasowski et al., BMC Clinical Pathology,
14:33, 20142 W. Li, L. H. Cohen, Anal. Chem., 75:5854-5859,
2003Disclosure of Interest:None DeclaredKeywords: LC–MS/MS, Steroid hormones, SWATH

249www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMass Spectrometry (LC-MS, CE-MS, GC-MS)
P02-002-084 - A survey of the presence of toxins in water reserviors in Singapore which threaten human healthC. Porojan*, F. Abbas, M. Mowe, S. M. Mitrovic, R. P. Lim, D. C. J. Yeo, A. Furey
Content: Microcystins (MC) are a group of toxins produced by harmful algal blooms. These toxins can cause serious poisoning in humans and animals and chronic exposure can cause liver cancer. Toxic algae have been found in the fresh water reservoirs of Singapore so human exposure is a possibility. In order to conduct a comprehensive hazard analysis it was imperative to determine the species of toxin present and to profile the toxin compounds produced and their concentrations. To this end a survey of 17 resrvoirs was undertaken where samples of water and algae were collected and monitored regularly over a 12 month period. The survey found a number of MC toxin producing cyanobacteria in Singapore’s reserviors but for the duration of this particular study the toxin levels that were found were relatively low in comparison to the WHO provisional MC-LR guideline limit of 1.0 µg/L (one sample in 200 exceeded the WHO limit).Acknowledgements: Funding support for this study was provided by a research grant from the Public Utilities Board of Singapore[AF1] . The Higher Education Authority (Programme for Research in Third-Level Institutions, Cycle 4 (PRTLI IV) National Collaboration Programme on Environment and Climate Changes: Impacts and Responses is also acknowledged for the capital investment in LC-MS.[AF1]Reference Number??Disclosure of Interest:None DeclaredKeywords: Singapore reservoirs, cyanobacterial blooms, microcystin toxins, LC-MS/MS
P02-002-085 - The application of Capillary Electrospray Ionization with negative ion electrospray ionization to the analysis of plant metabolites S. J. Lock, J. Thorn 1,*, W. Smith 1
1 MARKET DEVELOPMENT, SCIEX SEPARATIONS, WARRINGTON, United Kingdom
Content: CESI is the integration of capillary electrophoresis (CE) and electrospray ionization (ESI) into a single process in a single device. CESI-MS operates at low nL/min flow rates offering several advantages including increased ionization efficiency and a reduction in ion suppression. In this study we describe use of CESI-MS in negative mode for the analysis of anionic plant metabolites including low molecular weight organic acids. These compounds are generally small polar compounds typically not retained on traditional liquid chromatography columns making LC-separations difficult.
Using a bare fused silica OptiMS Cartridge and 10% acetic acid as a background electrolyte, low nanoM/10 µL concentration of plant metabolites were detected. These metabolites included low molecular weight organic acids including succinate and malate. In addition, separation of the isobaric phosphorylated disaccharides of Sucrose-6-Phosphate and Trehalose-6-Phosphate was possible and the partial separation of phosphorylated monosaccharides was observed in the same separation. This study investigated both separation reproducibility and sensitivity for numerous plant metabolites difficult to analyze by other means.References: 1. M. C. Gulersonmez, S. Lock, T.
Hankemeier and R. Ramautar. Electrophoresis,

www.ISC2016.ie @isc2016 #isc2016 /isc2016250
POSTER ABSTRACTS Mass Spectrometry (LC-MS, CE-MS, GC-MS)
1-8, 2015.Disclosure of Interest:None DeclaredKeywords: CE-MS/MS, Metabolites, negative electrospray
P02-002-086 - The application of Capillary Electrospray Ionization to the analysis of Intact Proteins
S. J. Lock, J. Thorn*, W. Smith 1
1 Technical Support, Scie, Darmstadt, Germany
Content: CESI is the integration of capillary electrophoresis (CE) and electrospray ionization (ESI) into a single process in a single device. CESI-MS operates at low nL/min flow rates offering several advantages including increased ionization efficiency and a reduction in ion suppression. In this study we describe use of CESI-MS in positive mode for the analysis of mixture of proteins of various sizes and different isoelectric points (pI). The proteins ranged in size from 4.5KDa to 148KDa and from acidic to basic proteins.
The aim of the study was to investigate the conditions affecting protein separation with a view to highlighting some of the factors that affect CESI-MS of intact proteins including biopharmaceuticals. In this work we used a neutral OptiMS Cartridge to reduce wall effects on protein separations and acidic background electrolytes. The study investigates the effect of mass and charge of proteins on CE separations with CESI-MS and the effects of salt concentration in the sample, separation pressure and voltage as well as background electrolyte buffer on the separations achieved. Throughout this study a QTOF mass spectrometer was used to detect the proteins in positive electrospray TOF only mode.
References: 1. J-M, Busnel., et.al. Anal. Chem. 9476-9483, 2010.
Disclosure of Interest:None DeclaredKeywords: CE-MS/MS, Intact protein
P02-002-087 - Does column bleed affect the determination of unknowns in LC/MS?
R. Clarke 1, L. Pereira 2,*, D. Thompson 1
1 University of Keele, Keele, 2Thermo Fisher Scientific, Runcorn, United Kingdom
Content: The retention and separation of polar molecules is often performed on columns which have a degree of polarity in the stationary phase. These columns cover a broad separation space meaning that the separation scientist is well supported in developing separation methodologies for polar compounds. However, these stationary phases often also suffer from a degree of column bleed which is not significant when using LC-UV, but, when using untargeted LC-MS analysis can become a limiting factor. It is therefore important to understand the impact that column bleed can have on both targeted and untargeted analysis.
The work presented here investigates first the effect of column bleed when running targeted samples, focussing on the amount of suppression effects that are observed. The LC/MS analysis is performed on commercially available columns which are known to bleed and then compared to same analysis on a material which has significantly reduced bleed, using full scan initially to determine the relative amounts of bleed coming from both type of columns when running under typical analysis conditions. The comparison is

251www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMass Spectrometry (LC-MS, CE-MS, GC-MS)
also made in SRM mode looking at signal-to-noise ratio and sensitivity. The second stage of the work presented looks at effect of column bleed on the determination of unknown markers. Food samples were used as the sample sets and comparisons were made between the different columns in the determination of a specific fingerprint structure. This information was used to determine if the column bleed has an effect on the determination of unknowns.References: L.PereiraDisclosure of Interest:None DeclaredKeywords: background ions, determination of unknowns, untargeted analysis
P02-002-088 - Towards Optimized Holistic Performance and Throughput in UHPLC-MS
F. Steiner 1, M. Krajewski 1,*, M. Ruehl 1, R. Swart 1
1 Thermo Fisher Scientific, Germering, Germany
Content: Most mass spectrometry (MS) based applications depend on automated sample introduction and effective chromatographic compound separation by liquid chromatography (LC) to maximize the qualitative and quantitative performance. The ever-growing demand for LC-MS systems and the wide range of LC-MS applications in different markets and user segments require systematic and profound examinations to identify the LC-MS combinations with optimum holistic system performance. In this study, we evaluate the impact of three different UHPLC systems on retention time (RT) reproducibility, peak properties, and sample throughput to ensure optimal MS performance and capacity utilization. We used binary high
pressure gradient (HPG) and quaternary low pressure gradient (LPG) systems with different pressure specifications, gradient slopes, column formats at different flow rates, and varied gradient delay volumes (GDVs) by varying mixer volumes. LC-MS analysis of small molecules and in particular pesticides was the main application example used in these studies.References: Mona I. Churchwell, Nathan C.
Twaddle, Larry R. Meeker, Daniel R. Doerge, Journal of Chromatography B, Volume 825, Issue 2 (134-143) (2005)
Disclosure of Interest:None DeclaredKeywords: gradient slope, LC-MS throughput, mixer volume
P02-002-089 - A novel, simple, and direct method based on UPLC-MS for the simultaneous quantitation of various fatty acids in olive oils of different origins
Z. A. Alothman*, S. Wabaidur 1
1 King Saud University, Riyadh, Saudi Arabia
Content: A simple and direct method using UPLC-MS has been established for the simultaneous quantitation of a few saturated and unsaturated fatty acids in olive oils from various countries. Many methods previously reported involves sample preparation [1-6]. But in the current method No sample pretreatment techniques were employed such as extraction or derivatization for the analysis of target acids from oil samples, as the oil samples were just diluted, filtered and then directly injected to the instrument. The chromatographic separations of all target fatty acids were achieved on a Hypersil Gold C18 column of

www.ISC2016.ie @isc2016 #isc2016 /isc2016252
POSTER ABSTRACTS Mass Spectrometry (LC-MS, CE-MS, GC-MS)
particle size 1.9 µm, 50 × 2.1 mm I.D, while the gradient elution using a binary mobile phase mixture of acetonitrile and water at a flow rate of 1.5 mL/min was adopted for achieving optimum separations. The recoveries of the fatty acids were obtained higher than 89% with good validation parameters; linearity (r2>0.992), detection limit between 0.09 and 0.24 µg/ml, run to run and day to day precisions with percent relative standard deviation lower than 2.4% at both low (1 µg/ml) and medium (10 µg/ml) concentration levels. The total content of fatty acids in each individual oils was found in the range of 472.63 to 7751.20 µg/ml of olive oil. The obtained validation parameters confirm that the proposed analytical method is rapid, sensitive, reproducible and simple and it could be applied for the successful evaluation of fatty acids in various oils and other matrices.References: [1] E. Mariotti, M. Mascini, Food Chem. 73 (2001)
235.[2] A.A. Ismail, F.R. van de Voort, G. Emo, J.
Sedman, J. Am. Oil Chem. Soc. 70 (1993) 335.[3] Z. Güler, A.C. Gürsoy-Balcı, Food Chem. 127
(2011) 1065–1071.[4] D.M. Pereira, P. Valentão, N. Teixeira, P.B.
Andrade, Food Chem. 141 (2013) 2412–2417.[5] T.C. Sindhu Kanya, L. Jaganmohan
Rao, M.C. Shamanthaka Sastry, Food Chem. 101 (2007) 1569–1574.
Disclosure of Interest:None Declared
P02-002-090 - The effect of pH on the partitioning of polychlorinated biphenyls (PCBs) between sediment grain sizes and waterG. C. Adeyinka*, B. Moodley 1
1 Chemistry and Physics, University of KwaZulu-Natal., Durban, South Africa
Content: The sorption of organic pollutants within the environmental media is one of the significant factors affecting the distribution, transportation and fate of the pollutant as well as the remediation of polluted water within the aquatic ecosystem1. The aim of this research was to investigate the effects of pH on the phase partitioning of organic pollutants such as PCB congeners between the aqueous phase and various modelled sediment grain sizes. A model sample of sediment was sieved into five grain sizes, <75 µm, 100 µm, 200 µm, 300 µm and 425 µm, respectively. Laboratory batch experiments was used to evaluate the adsorption as well as the adsorption ratio of the soil/sediment (Kd) within the different sediment grain sizes2,3. The results showed that soil grain size of <75 µm showed the most adsorption of PCB congeners, in the range of 79.90-89.97 % while a sediment grain size of 300 µm showed lowest adsorption between 32.60-64.14 %. This could be attributed to the fact that sediment grain size (75 µm,) with high surface area adsorbed more of the analytes compared to other sediment grain sizes. The effect of pH on the sorption capacity of the sediment grain sizes were also studied. A pH of 6.5 showed the highest percentage adsorption. This was due to the non-polar PCBs preferring to partition to the sediment at near neutral pH.References: 1 Morel, F.M.M., and Gschwend, P.M (1987), In
Aquatic Surface Chemistry: Chemical Process at

253www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMass Spectrometry (LC-MS, CE-MS, GC-MS)
the Particle – Water Interface; Stumm, W., Ed.; Wiley: New York, pp 405-422.
2 US EPA, 2000. Organization for Economic Corporation and Development (OECD 106) guideline for the testing of chemicals. Adsorption - Desorption Using a Batch Equilibrium Method.
3 Karickhoff, S. W., D. S. Brown, and T. A. Scott. 1979. “Sorption of Hydrophobic Pollutants on Natural Sediments.” Water Research, 13:231-248.
Disclosure of Interest:None DeclaredKeywords: Aqueous Phase, Partitioning, pH
P02-002-091 - Kinetic study of hyaluronidase by capillary electrophoresis - high resolution mass spectrometryS. Fayad*, R. Nehmé 1, C. Colas 1, M. Langmajerova
2, G. Zdenek 2, P. Morin 1
1 Institut de chimie organique et analytique (ICOA), Orleans, France,
2 Department of Biochemistry, Faculty of Science, Masaryk University, Brno, Czech Republic
Content: Hyaluronic acid (HA) is a key molecule for skin health due to its unique capacity in retaining water. Hyaluronidase is one of the endotype glycosidases that hydrolyzes the endo-N-acetylhexosaminic bonds of hyaluronic acid (HA).
As for elastases and collagenases, hyaluronidase is implicated in skin aging and in tumor suppression. Therefore, for cosmetic as well as therapeutic applications, it is essential to develop efficient and robust methods to study the activity of hyaluronidase and to identify novel inhibitor molecules. Capillary electrophoresis (CE) is a versatile technique that permits the analysis of small amounts of reactants in a short analysis time [1]. It has been widely used for enzymatic assays
and the determination of kinetic constants (Km, Vmax and IC50) [2]. In this study, CE-UV/MS was used for the first time to assess hyaluronidase kinetics. To identify the different products of this enzymatic reaction, CE was coupled to high resolution mass spectrometry (HRMS). Sheath liquid nature and flow, capillary positioning in the electrospray ionisation source (ESI), the needle voltage and the ionization mode were optimized. Kinetic constants (km and Vmax) and IC50 of a standard inhibitor, epigallocatechin gallate (EGCG) [3], were determined by CE-UV and CE-HRMS. The results obtained were cross compared and compared to the literature in order to validate the developed assays. This assay will be used for screening natural inhibitors.References: [1] S. Fayad, R. Nehmé, P. Lafite, P. Morin, J.
Chromatogr. A 1419 (2015) 116-124.[2] R. Nehmé and P. Morin, Electrophoresis, 2015.[3] W.D. Ratnasooriya, W.P.K.M. Abeysekera, Asian
Pac J Trop Biomed, 4 (2014) 959-963.Disclosure of Interest:None DeclaredKeywords: Capillary electrophoresis, High resolution mass spectrometry, Kinetic constants
P02-002-092 - Identification of Skin Whitening Ingredients in Misbranded Cosmetics Products using UHPLC with PDA and Mass Detection
M. Twohig 1, C. Stumpf 1, J. A. Cooper 2, H. Boiteux 3,*
1 Waters Corporation, 34 Maple Street, Milford, MA 01757, United States,
2 Waters Corporation, Stamford Avenue, Altrincham Road, Wilmslow, SK9 4AX, United Kingdom,
3 Waters S.A.S. , BP 608, 78056 Saint-Quentin, En Yvelines Cedex , France

www.ISC2016.ie @isc2016 #isc2016 /isc2016254
POSTER ABSTRACTS Mass Spectrometry (LC-MS, CE-MS, GC-MS)
Content: Skin whitening products are used to lighten and produce a more even skin tone, and are mainly designed for use on the face and neck. The use of pharmaceutical active ingredients such as corticosteroids is prohibited in cosmetics due to the potential side effects.1,2 Further, in the EU, hydroquinone, is prohibited in cosmetics while several other countries either ban or limit the amount allowed in cosmetic products. Despite the legal regulations on these components in cosmetics they can still be found in formulated cosmetic products. In this study, skin whitening products were obtained from online vendors to assess their composition. The samples were extracted and analysed using UHPLC with PDA and mass detection.
UHPLC separation of eight whitening agents ranging in polarity was performed at a flow-rate of 0.80 mL/min using 0.1% formic acid in water as the aqueous mobile phase and acetonitrile as the organic modifier. A C18 column designed for providing exceptional retention for both polar and non-polar analytes even at 100% aqueous conditions was used for the analysis. The column was maintained at a temperature of 35 0C and had dimensions of 3.0 x 100 mm and a particle size of 2.7 µm. Detection was by photodiode array (PDA) and mass detection.
Several samples in this study were found to contain prohibited skin whitening agents. In some cases packaging labels omitted the presence of the skin lightening agent on the enclosed product information thus increasing the likelihood of improper long-term use and adverse side effects to consumers.
References: 1. B. Desmedt, P. Courselle, J.O. De Beer, V.
Rogiers, E. Deconinck, K. De Paepe. (2014), Illegal cosmetics on the EU market: a threat to human health? Arch Toxicol 88:1765-1766.
2. B. Desmedt, E. Van Hoeck, V. Rogers, P. Courselle, J.O De Beer, K. De Paepe, E. Deconinck. (2014), Characterisation of suspected illegal skin whitening cosmetics. J. Pharm. Biomed. Anal. 90, 85-91.
Disclosure of Interest:None DeclaredKeywords: Cosmetics, skin whitening, corticosteroids, hydroquinone, label claim, misbranded
P02-002-093 - Bi-platform metabolomics analysis of individual plasma and brain samples using UPLC-qToF-MS and GC-MS: a pilot study in Alzheimer’s disease.
A. A. Ebshiana 1,*
1 Institute of Pharmaceutical Science -Faculty of Life Sciences & Medicine, King’s College London, London, United Kingdom
Content: Metabolomic analysis of biological fluids and tissues has become an increasingly routine tool in the biological toolbox. Owing to the wide range of concentrations at which metabolites are present and their diverse phsyio-chemical properties no single platform is capable of comprehensive coverage. A number of studies have looked to do this by analyzing samples on multiple platforms [1-2], and whilst this does increase coverage it also increases the amount of sample required. Here we describe a metabolomics strategy for the analysis of brain tissue and blood plasma to optimize coverage from limited sample volumes. This was achieved

255www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMass Spectrometry (LC-MS, CE-MS, GC-MS)
by coupling a split phase in-vial dual extraction (IVDE) with high resolution LC-MS and GC-MS, and powerful multivariate statistical analysis. The non-aqueous phase was analyzed by a combination of reversed phase RP-LC-MS and a 49 minute GC-MS gradient, whilst the aqueous phase was analyzed using hydrophilic liquid interaction chromatography HILIC-LC-MS and a 33 minute GC-MS gradient. This experiment showed that the applied strategy is both highly sensitive, measuring 19027 metabolite features in plasma samples, and highly reproducible with 42.3% of features with CV’s below 15% and 79.1% below 30%. To assess the analytical performance of the method developed it was applied to a small pilot experiment of 40 plasma samples (20control Vs 20 Alzheimer’s disease(AD). The method was shown to be capable of discriminating healthy controls from AD, Also were able to identify a number of previously identified AD biomarkers and new potential AD biomarkers as being associated with disease pathology. This study has demonstrated the utility of combining a split phase IVDE with LC-MS and GC-MS analysis to create a comprehensive metabolomics strategy from only 20ml of plasma and 3mg of brain tissue. References: [1]. O.Yanes,et al,Analytical
Chemistry.,2011,83,2152-2161.[2]. N.Psychogios.,et al,PLoS One.,2011,6,e16957,
DOI:10.1371/journal.pone.0016957.Disclosure of Interest:None DeclaredKeywords: GC/MS, LC-MS/MS, metabolomics
P02-002-094 - Characterization of a newly identified lipid class in vernix caseosa by chromatographic and mass-spectrometry techniques E. Háková*
Content: Vernix caseosa is a multicomponent mixture, which is consisted of water (80 %) and proteins and lipids roughly in the same proportion (10 % each). This unique human material starts to be formed in the third trimester of a pregnancy and is present on the skin of newborns after a delivery1,2. Lipids of vernix caseosa are characterized by high complexity. In many lipid classes are presented thousands of molecular species which differ in the length, branching and presence of double bonds in carbon chains of lipids. A separation and identification of these lipids need very effective chromatographic techniques and modern spectral analytical methods. In this work we discovered a new, so far undescribed minor lipid class in vernix caseosa. The total lipid extract was gained by the extraction according to Bligh/Dayer. Thin layer chromatography (TLC) was used as a first separation step and the separation of main lipid classes was achieved. TLC is not efficient enough for a separating lipid classes which have very similar polarity. Therefore, high-performance liquid chromatography in normal-phase in coupling with mass spectrometry (NP-HPLC-MS) method was developed. This method made it possible to identify a new species of acylceramides in vernix caseosa. Its structure corresponds to amino alcohol linked to a non-hydroxylated fatty acid (FA) via an amide bond and another non-hydroxylated FA via an ester bond. This general structure was deduced from derivatization reactions focused on

www.ISC2016.ie @isc2016 #isc2016 /isc2016256
POSTER ABSTRACTS Mass Spectrometry (LC-MS, CE-MS, GC-MS)
hydrolysis of the ester and amide bonds, FA analysis and measurement by nuclear magnetic resonance. Reversed-phase HPLC-MS was used for characterization of the molecular species within this lipid class. Fragmentation experiments provided information about the FAs, amino alcohol chain length and double bond. References: 1. Hoat S. B: Neonatal Skin, Structure
and Function. 2nd ed. New York, Marcel Dekker, 2003.2. Pickens W. L., Warner R. R., Boissy Y. L., Boissy R. E., Hoath S. B.: J Invest Dermatol. 115 (2000), 875-881.
Acknowledgements: This work was supported by the Grant Agency of Charles University in Prague (Project No. 1182216), Czech Science Foundation (Project No. P206/12/0750) and Charles University in Prague (Project No. SVV260317). Disclosure of Interest:None Declared

257www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMetabolomics, Glycomics, Lipidomics and Proteomics
Metabolomics, Glycomics, Lipidomics and Proteomics
P01-001-039 - Volatomic signature of human breast cancer cell lines by solid-phase microextraction combined with gas chromatography-mass spectrometry
C. L. Silva 1,*, H. Tomás 2, J. S. Câmara 2
1 CQM-Centro Química da Madeira, Universidade da Madeira,
2 CQM-Centro de Química da Madeira, Faculdade de Ciências Exatas e da Engenharia, Universidade da Madeira, Campus da Penteada, 9020-105 Funchal, Portugal
Content: Breast cancer (BC) remains as the most prevalent oncologic pathology in women having huge psychological, economic and social impact in our society and the current diagnostic tools present limited sensitivity and specificity. Nowadays, there is no single screening test that is totally reliable and a number of tests can be combined to help an early breast cancer detection. Metabolome analysis emerge as a powerful tool of information about the biological processes that occur in the organism using several biofluids as a mean to discover new biomarkers or disease diagnosis.
The aim of this work was to identify volatile organic metabolites (VOMs) being emitted or metabolized by human mammary epithelial cells (HMEC) when compared with breast cancer type (MCF-7, T-47D, MDA-MB-231) as a mean to better understand the origin of metabolites from cells. Their establishment relied on gas chromatography with mass spectrometric
detection (GC–qMS) and solid-phase microextraction in headspace mode (HS-SPME).
It was possible to identify 46 VOMs in cell lines analyzed, belonging to several chemical families, namely alkanes, aldehydes, ketones, acids, alcohols, among others. From these, 2-pentanone, 2-heptanone, 3-methyl-3-buten-1-ol, 1,2,4-trimethylbenzene, ethyl acetate, ethyl propanoate, and 2-methyl butanoate were detected only on cell lines headspace. Multivariate statistical methods were then used to verify the volatomic differences between oncologic and normal cell lines in order to find related volatile metabolites that could be associated with breast cancer, providing comprehensive information on volatile metabolites as potential cancer biomarkers.
The characterization of these volatiles using GC-qMS approach is feasible and will contribute to increase our knowledge about the cancer etiology and consequently improve the diagnostic tools.Acknowledgements: Catarina Silva to FCT for the PhD grant (SFRH/BD/97039/2013), REDE/1508/RNEM/2015), HCV -New INDIGO/0003/2012 ; EU Project, QUI-Madeira-674.Disclosure of Interest:None DeclaredKeywords: breast cancer cell lines, solid-phase microextraction, volatile metabolites

www.ISC2016.ie @isc2016 #isc2016 /isc2016258
POSTER ABSTRACTS Metabolomics, Glycomics, Lipidomics and Proteomics
P01-001-044 - Analysis of N-Glycans Derived from Glycoproteins - HPAE-PAD versus Mixed-Mode Liquid ChromatographyJ. P. Weiss*
Content: N-Glycans bound to proteins modulate many biological properties. Understanding protein glycosylation is an important step in unraveling the function of glycans in glycoproteins. For the chromatographic separation of carbohydrates numerous LC techniques have been developed in the past. For many years, the highest resolution separations of carbohydrates were achieved using anion-exchange chromatography (HPAE) at high pH. Pulsed amperometric detection (PAD) is commonly used for detection to provide a sensitive and selective tool for determining a wide variety of carbohydrates. Introduced in 1983, HPAE-PAD has become a widely accepted method for analyzing carbohydrate moieties derived from glycoproteins.
For more than three decades mass spectrometry (MS) has been a primary technique in carbohydrate structural analysis. Numerous publications have described oligosaccharide sequence, branching pattern, and linkage information deduced from ESI mass spectra. A prerequisite for combining anion-exchange chromatography of carbohydrates based on hydroxide eluents with ESI-MS is the use of a membrane desalter, which is placed between the column and the ESI interface to eliminate time-consuming dialysis steps.
As an alternative to HPAE-PAD, some uniquely designed stationary phases including weak anion-exchange-HILIC
mixed-mode based phases have been developed enabling researchers to study diverse situations including quantitative, linkage, and qualitative structural analysis in a single platform. In addition these stationary phases also provide a unique way to separate both native and fluorescently labeled N-glycans based on charge, size, and polarity. In addition, this analysis and characterization of N-glycans will be using an optimized LC-MS workflow based on these stationary phases coupled online with a benchtop Orbitrap mass spectrometer.
It is the scope of this presentation to compare both liquid chromatographic techniques in terms of resolution and analysis times.Disclosure of Interest:None DeclaredKeywords: High-performance anion-exchange chromatography, Integrated pulsed amperometric detection, Weak anion-exchange/HILIC mixed-mode chromatography
P02-002-095 - Profiling urinary volatile metabolites as a non-invasive and powerful strategy to detect potential lung cancer-specific biomarkers
P. Silva 1, C. L. Silva 2,*, M. Caldeira 3, F. Aveiro 4, J. Câmara 5
1 CQM-Universidade da Madeira, Universidade da Madeira, Campus da Penteada, 9020-105, Funchal,
2 CQM-Centro Química da Madeira, Universidade da Madeira, Campus da Penteada, 9020-105 Funchal,
3 CQM-Centro de Química da Madeira, Universidade da Madeira, Campus da Penteada, 9020-105, Funchal,
4 Unidade de Hematoncologia, Hospital Dr. Nélio Mendonça, Av. Luís Camões, 9000, Funchal,
5 CQM-Centro de Química da Madeira, Faculdade de Ciências Exatas e de Engenharia, Universidade da Madeira, Campus da Penteada, 9020-105 Funchal, Portugal

259www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMetabolomics, Glycomics, Lipidomics and Proteomics
Content: Lung cancer is the leading cause of cancer-related deaths throughout most of the world. The most common methods currently used to diagnose lung cancer can occasionally miss tumors because they depend on tumor size. In contrast, urine testing has long been recognized as a medical technique that allows diagnosis of disease by linking specific volatile organic metabolites (VOMs) in urine to medical conditions. Metabolomics helps to understand metabolic state of biological systems with comprehensive insight by detecting the metabolome.
Solid-phase microextraction (SPME) using CAR/PDMS sorbent at 50 oC during 60 min was selected as a powerful and efficient strategy to isolate urinary VOMs, combined with gas chromatography-mass spectrometry and used to obtain metabolomic information patterns of 25 samples from lung cancer patients and 20 samples from healthy individuals. A total of 92 VOMs were identified in urine samples from healthy subjects and lung cancer patients. The analysis of variance (ANOVA) was performed on both groups to evaluate if there were differences between the groups for each volatile compound. Results showed that positive rates of 38 metabolites among the total of 92 detected were found to be different with statistical difference (p <0.05). The application of partial least squares discriminant analysis (PLS-DA) showed the existence of two separated clusters for the healthy subjects and lung cancer patients.
It can be concluded that SPME coupled to GC-qMS is a very appropriate sampling technique to distinguish the urinary volatile metabolomic profile of healthy subjects from lung cancer patients. The data in this study
suggests that diagnostically useful VOMs are produced in patients with lung cancer and excreted into urine, thus providing support for a possible early diagnostic tool.References: C. Silva, M. Passos, J. Câmara, British Journal of Cancer 105(1894-1904)(2011)Acknowledgements: C.Silva to FCT for the PhD grant (SFRH/BD/97039/2013), (REDE/1508/RNEM/2015), HCV -New INDIGO/0003/2012; EU Project, QUI-Madeira-674; Hematology-Oncology Unit at Hospital Dr. Nélio Mendonça.Disclosure of Interest:None DeclaredKeywords: lung cancer biomarkers, volatile metabolites, HS-SPME, GC-qMS
P02-002-096 - A metabolomic approach as a powerful strategy for identification of urinary volatile metabolites as potential breast cancer biomarkers
C. L. Silva 1,*, H. Tomás 2, J. S. Câmara 2
1 CQM-Centro Química da Madeira, Universidade da Madeira,
2 CQM-Centro de Química da Madeira, Faculdade de Ciências Exatas e da Engenharia, Universidade da Madeira, Campus da Penteada, 9020-105 Funchal, Portugal
Content: Despite of global efforts to limit the incident of this disease, cancer continues to be the major source of morbidity and the second most common cause of mortality worldwide. Breast cancer (BC) is the most prevalent pathology in women having huge psychological, economic and social impact in our society and the current diagnostic tools present limited sensitivity and specificity. Hence, new and more efficient diagnostic approaches are necessary to invert this

www.ISC2016.ie @isc2016 #isc2016 /isc2016260
POSTER ABSTRACTS Metabolomics, Glycomics, Lipidomics and Proteomics
scenario. Although various compounds from blood, saliva and urine, including proteins, tumor antigens, anti-tumor antibodies, cell type-specific peptides, metabolic and epigenetic products such as hyper-methylated DNA, RNA, and the expression of specific genes, have been explored as biomarkers, most of them continue to fail in reaching clinical use. The cancer cells metabolism is necessarily different from their normal counterparts, being certain molecules differentially expressed in cancer cells. Many of these metabolites are volatiles and could be used to establish a cancer signature to differentiate health subjects from BC patients. The characterization of these volatiles using GC-qMS approach is feasible and will contribute to increase our knowledge about the cancer etiology and consequently improve the diagnostic tools.
In this study, we tested the potentialities of a powerful and solvent-free miniaturized high-throughput analytical methodology based on dynamic solid phase microextraction in headspace mode (dHS-SPME) in combination with GC-qMS. This technique was used for the metabolomic analysis of urine samples that were obtained from clinically diagnosed BC patients and from healthy controls, to provide comprehensive information on volatile metabolites as potential cancer biomarkers.References: C. L. Silva, M Passos, J.S. Câmara, Talanta 89,
360-368Acknowledgements: Catarina L. Silva acknowledge the Portuguese Foundation for Science and Technology (FCT) for the PhD grant (SFRH/BD/97039/2013). The authors also acknowledge the Portuguese Foundation for Science and Technology (FCT) through the MS Portuguese Networks (REDE/1508/RNEM/2015), HCV - New INDIGO EU
Project (FCT reference: New-INDIGO/0003/2012) and Pluriannual base funding (QUI-Madeira-674).Disclosure of Interest:None DeclaredKeywords: VOMs, Urine; cancer biomarkers; HS-SPME, GC-qMS
P02-002-097 - Application of combinatorial peptide ligand libraries to the identification of plum and peach seeds proteinsE. González-García 1,*, M. L. Marina 1, M. C. García 1, P. G. Righetti 2, E. Fasoli 2
1 Departamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcalá, Alcalá de Henares, Spain,
2 Department of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Milan, Italy
Content: Plum (Prunus domestica L.) and peach (Prunus persica (L.) Batsch) seeds have attracted the attention of researchers for their high protein content. Moreover, it has been demonstrated that proteins present in this waste material are source of bioactive peptides [1-3]. Nevertheless, there is still no study on the identification of proteins present in plum and peach seeds.
Vegetable samples preparation in proteomics analysis is hindered by the presence of high-abundance proteins which mask the detection of low-abundance ones. Combinatorial peptide ligand libraries (CPLLs) are exceptional tools to reduce proteins dynamic concentration range. The aim of this work was to comprehensively identify proteins in plum and peach seeds by applying the CPLL technology and to evaluate the generation of bioactive peptides, derived from proteins previously recognized.

261www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMetabolomics, Glycomics, Lipidomics and Proteomics
Milled and defatted seeds were treated with native and denaturing extraction buffers. The extracted proteins were precipitated and the pellet was resuspended in native buffer, adjusting the pH to 7.5 or 2.2 to optimum CPLLs incubation. Proteins were eluted using a boiling SDS solution and separated by SDS-PAGE for a final identification by nLC-MS/MS. Comparison of MS data, against a general vegetable database (Uniprot_Viridiplantae) and a specific one for Prunus genus, enabled the identification of 146 and 98 proteins in plum and peach seeds, respectively. It was possible to identify 21 specific Prunus proteins, several histones and seed storage proteins. In addition, 21 and 14, previously reported bioactive peptides [1-3] in plum and peach seeds, respectively, were found within the sequence of identified proteins. These peptides have shown antioxidant and antihypertensive bioactivities.References: [1] E. González-García, et al., J. Funct. Food. 11
(2014) 428-437.[2] R. Vásquez-Villanueva, et al., J. Funct. Food. 18
(2015) 137-146.[3] R. Vásquez-Villanueva, et al., J. Chromatogr. A
1428 (2015) 185-192.Acknowledgements: Authors thank the Spanish Ministry of Economy and Competitiveness (AGL2012-36362) and the Comunidad of Madrid (Spain) and FEDER program (S2013/ABI-3028). E.G.-G. thanks the University of Alcalá for her pre-doctoral contract and mobility grant.Disclosure of Interest:None Declared
P02-002-098 - Development of LC-QTOF-MS method for human lung tissue fingerprinting with application to non-small cell lung cancer.
M. Ciborowski 1,*, K. Pietrowska 1, J. Kisluk 2, P. Samczuk 1, T. Kowalczyk 1, M. Kozlowski 3, J. Niklinski 2, A. Kretowski 1
1 Clinical Research Centre, 2 Department of Clinical Molecular Biology, 3 Department of Thoracic Surgery, Medical University of
Bialystok, Bialystok, Poland
Content: Histologic subtypes of non–small cell lung cancer (NSCLC) include adenocarcinoma, squamous cell carcinoma (SCC), and large-cell carcinoma1. Molecularly targeted agents have surprisingly failed in treatment of SCC patients2. Moreover, even the most careful examination of biopsies by expert pulmonary pathologists leave about 10-30% of NSCLC as not specified3. Novel pathways for targeted therapy in case of SCC and additional methods for NSCLC subtyping are needed. Following analytical platforms were proposed for lung tissue fingerprinting: CE-MS, 1H-NMR, and GC-MS. LC-MS was only used for lipidomics as a complementary approach for multiplatform rat lung tissue fingerprinting4. LC-MS has never been proposed as stand-alone technique for human lung tissue fingerprinting. In this study different solvents for tissue homogenization (100% and 50% methanol) and metabolites extraction (water/methanol/MTBE or water/methanol/chloroform) were tested. Obtained extracts were analyzed on LC-QTOF-MS using different C18 and C8 columns. Homogenization with 50% methanol and extraction with water/methanol/MTBE

www.ISC2016.ie @isc2016 #isc2016 /isc2016262
POSTER ABSTRACTS Metabolomics, Glycomics, Lipidomics and Proteomics
(10%/74%/16%) was selected as the most effective. Such prepared extracts gave the best results when analyzed with Zorbax C8 column (~1800 metabolic features RSD<20% after blank extraction). This method was applied to human NSCLC lung tissues. Multivariate analysis of obtained fingerprints showed separation not only between cancer and adjacent control tissue (OPLS-DA model, R2=0.993 and Q2=0.926), but also between NSCLC subtypes (PLS-DA model, R2=0.885 and Q2=0.351). Developed method is a promising tool for diagnosis and better understanding of NSCLC different subtypes.References: 1) DN. Hayes, et al. J Clin Oncol 24 (5079-90)
(2006).2) CJ. Langer, et al. J Clin Oncol 28 (5311-20)
(2010).3) G. Rossi, et al. Int J Surg Path 21 (326-36) (2013).4) S. Naz, et al. Anal Chem 85 (10941−8) (2013).Acknowledgements: N/NCN/OP/15/001/1196 and STRATEGMED2/266484/2/NCBR/2015Disclosure of Interest:None DeclaredKeywords: LC-MS, lung cancer biomarkers, metabolomics
P02-002-099 - Comparison of performance of three trypsin columns compatible with HPLC apparatus T. Šlechtová 1,*, M. Gilar 2, K. Kalíková 1, E. Tesařová 1
1 Department of Physical and Macromolecular Chemistry, Charles University in Prague, Faculty of Science, Prague, Czech Republic,
2 Waters Corporation, Milford, United States
Content: Trypsin is the most widely used enzyme in proteomic research for its high specificity. Although the in-solution
digestion is still frequently used, it is burdened with several drawbacks, such as long digestion time, autolysis, intolerance to high temperatures and organic solvents [1,2]. To overcome these shortcomings, immobilization of trypsin on solid support is convenient, since it ceases autolysis, allows use of high enzyme concentration and is compatible with modern LC-MS instruments [3]. In this work, three trypsin columns were tested for their catalytic activity and compared using Nα-benzoyl-L-arginine 4-nitroanilide hydrochloride as a substrate. Two commercial trypsin columns, Perfinity and Poroszyme, and one column prototype from the University of North Carolina at Chapel Hill were used. For this purpose, various pH of buffers, flow rates and temperatures were used. Relative standard deviation values were determined to describe repeatability of digestions and the whole separation process. Activities of columns differed the most using buffer pH 9.0, when the highest digestion activity was possessed by the column prototype and the lowest by Poroszyme column at all conditions tested.References: [1] Siepen, J. A.; Keevil, E. J.; Knight, D.; Hubbard, S.
J. J. Proteome Res. 2007, 6, 399−408.[2] Yen, C. Y.; Russell, S.; Mendoza, A. M.; Meyer-
Arendt, K.; Sun, S.; Cios, K. J.; Ahn, N. G.; Resing, K. A. Anal. Chem. 2006, 78, 1071−1084.
[3] Ma, J.; Zhang, L.; Liang, Z.; Zhang, W.; Zhang, Y. Anal. Chim. Acta 2009, 632, 1–8.
Acknowledgements: The Grant Agency of the Charles University in Prague, project No. 254214, and the Grant Agency of the Czech Republic, project No. 16-05942S, are gratefully acknowledged for the financial support. The authors also express their gratitude to the Waters Corporation for technical support and to Jim Jorgenson from the University of North Carolina for the gift of trypsin column prototype.

263www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMetabolomics, Glycomics, Lipidomics and Proteomics
Disclosure of Interest:None DeclaredKeywords: digestion efficiency, HPLC, trypsin columns
P02-002-100 - LC-QTOF-MS serum metabolic fingerprinting after exposure of rats to quinolinic acid
B. Kalaska 1,*, M. Ciborowski 2, T. Domaniewski 3, U. Czyzewska 4, J. Godzien 5, W. Miltyk 4, A. Kretowski 2, D. Pawlak 1
1 Department of Pharmacodynamics, 2 Clinical Research Centre, 3 Department of Monitored Pharmacotherapy, 4 Department of Pharmaceutical Analysis, Medical
University of Bialystok, Bialystok, Poland, 5 Centre for Metabolomics and Bioanalysis, San Pablo
CEU University, Boadilla del Monte, Spain
Content: Quinolinic acid (QUIN), one of the end metabolites in the kynurenine pathway, plays an important role in the pathogenesis of several neurological diseases. Serum QUIN concentration rises in patients with renal dysfunction, liver cirrhosis, sepsis and many other inflammatory diseases [1,2]. We intraperitoneally implanted osmotic minipumps containing QUIN (0.3 and 1 mg/day) into rats for 28 days, and then evaluated the physiological and toxicological variables and performed LC-QTOF-MS serum metabolic fingerprinting. Preparation of serum samples for metabolic fingerprinting and chromatographic conditions were the same as described previously [3]. Data were collected in positive and negative ESI ion modes in separate runs on a QTOF operated between m/z 50 to 1000. Metabolic profile changes were analyzed with partial least squares-discriminate analysis (PLS-DA). QUIN significantly decreased the serum concentrations of phenylalanine, valine, tyrosine, tryptophan, pantothenic acid,
pivaloylcarnitine, total cholesterol, and glucose (by 35, 25, 35, 29, 44, 43, 21, and 33%, respectively); increased the serum concentrations of pentadecanoic amide, palmitic amide, oleamide, stearamide, spermine, spermidine, sphingosine, and deoxy-prostaglandin (by 35, 33, 41, 38, 108, 87, 83, and 41%, respectively); and caused alterations in phospholipids. This study represents the first report of comprehensive metabolites analysis after chronic intraperitoneal administration of QUIN. Further studies could develop new therapeutics for patients with disorders accompanied by increased serum level of QUIN.References: [1] Y, Chen, International Journal of Tryptophan
Research, 2009;2:1-19.[2] G, Guillemin, FEBS Journal 2012;279:1356-1365.[3] M, Ciborowski, Journal of Proteome Research,
2012;11:6231-6241.Acknowledgements: The study was supported by Leading National Research Center Grant No. 31/KNOW/2013.Disclosure of Interest:None DeclaredKeywords: Metabolic fingerprinting, Metabolomics, Quinolinic acid
P02-002-101 - Retention behavior of lipids in reversed-phase ultrahigh-performance liquid chromatography coupled with electrospray ionization mass spectrometry
M. Ovčačíková 1,*, M. Lísa 1, E. Cífková 1 2, M. Holčapek 1 2
1 Department of Analytical Chemistry, 2 Analytical Chemistry, University of Pardubice, Pardubice,
Czech Republic

www.ISC2016.ie @isc2016 #isc2016 /isc2016264
POSTER ABSTRACTS Metabolomics, Glycomics, Lipidomics and Proteomics
Content: Lipids fulfill multiple essential roles within all eukaryotic cells in living organisms [1]. The dysregulation of the lipid metabolism contributes to numerous serious human diseases, such as obesity, diabetes, cardiovascular diseases and cancer. Therefore, they are investigated as possible biomarkers of these diseases [2]. Lipidomics is a systematic study which includes detailed identification of individual cellular lipid species. Reversed-phase ultrahigh-performance liquid chromatography (RP-UHPLC) method using two C18 columns coupled in series is developed with the goal to separate and identify a high number of lipid species in biological samples. The identification is performed by the coupling with high-resolution tandem mass spectrometry (MS/MS) using quadrupole – time-of-flight (QTOF) instrument. Electrospray ionization (ESI) full scan and tandem mass spectra are measured in both polarity modes with the mass accuracy better than 5 ppm, which provides a high confidence of lipid identification. 416 lipid species covering 14 polar and nonpolar lipid classes from 5 lipid categories are identified in total lipid extracts of human plasma, human urine and porcine brain. The general dependences of relative retention times on relative carbon number (CN) or relative double bond (DB) number are constructed and fit with the second degree polynomial regression. The regular retention patterns in homologous lipid series provide additional identification point for UHPLC/MS lipidomic analysis, which increases the confidence of lipid identification. References: [1] G. van Meer, D. R. Voelker, G. W. Feigenson,
Membrane lipids: where they are and how they behave, Nat. Rev. Mol. Cell Biology 9 (2008) 112-124.
[2] E. Cífková, M. Holčapek, M. Lísa, D. Vrána, B. Melichar, V. Študent, Lipidomic differentiation between human kidney tumors and surrounding normal tissues using HILIC-HPLC/ESI–MS and multivariate data analysis, J. Chromatogr. B 1000 (2015) 14-21.
Acknowledgements: This work was supported by the ERC CZ grant project LL1302 sponsored by the Ministry of Education, Youth and Sports of the Czech Republic. Disclosure of Interest:None DeclaredKeywords: Lipidomics, Mass spectrometry, Ultrahigh-performance liquid chromatography
P02-002-102 - Comparison of on-capillary derivatization of amino acids using different strategies of reactant mixing for capillary electrophoresis coupled with LIF
A. Cela*, A. Madr 1, M. Sulcova 1, Z. Glatz 1
1 Department of Biochemistry, Faculty of Science, Masaryk University, Brno, Czech Republic
Content: Amino acids are very important compounds for living organisms. They exhibit low or no native fluorescence, thus derivatization is essential for their highly sensitive determination by laser induced fluorescence detection. Derivatization reaction is often time consuming, laborious and derivatization dyes are quite expensive. In capillary electrophoresis, derivatization can be done precapillary, oncapillary or postcapillary. In the case of oncapillary arrangement, the inner space of the separation capillary is used both for separation and for derivatization. This approach offers many advantages, such as requirement of smaller volumes of sample and other reagents and possibility of automation. Moreover, the time between a reaction and detection of

265www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMetabolomics, Glycomics, Lipidomics and Proteomics
derivatives is strictly defined and automation reduces manual steps thus on-capillary derivatization being more precise. Mixing of the reactants in narrow capillaries can be performed by longitudinal diffusion (atinlet), by transverse diffusion of laminar flow profiles (TDLFP), or by electrophoretically mediated microanalysis (EMMA) [1].
Two methods for on-capillary derivatization of amino acids with naphthalene2,3dicarboxaldehyde/sodium cyanide were optimized using mixing of reactants both by TDLFP [2] and EMMA. Both mixing strategies were compared in terms of efficiency of mixing of reactants, sensitivity, repeatability and applicability to various biological samples. The EMMA based method finally combined with the on-line preconcentration technique - sweeping, provides up to 10× higher sensitivity and proved to be more robust to the sample composition compared with TDLFP.References: [1] Z. Glatz, Electrophoresis, 36, 744-763, 2015, doi:
10.1002/elps.201400449.[2] A. Celá, A. Mádr, T. Dědová, M. Pelcová,
M. Ješeta, J. Žáková, I. Crha, Z. Glatz, Electrophoresis, in press, 2016, doi: 10.1002/elps.201500587.
Acknowledgements: Financial support granted by the Czech Science Foundation (Projects No. P206/12/G014) is highly acknowledged.Disclosure of Interest:None DeclaredKeywords: Amino acids, Capillary electrophoresis - laser induced fluorescence, On-capillary derivatization
P02-002-103 - HILIC-HPLC/ESI-MS analysis of gangliosides in biological samples
R. Hájek 1,*, R. Jirásko 1, M. Lísa 1, E. Cífková 1, M. Holčapek 1
1 Analytical Chemistry, University of Pardubice, Pardubice, Czech Republic
Content: Gangliosides are a class of sphingolipids [1] characterized by ceramide lipid chain attached to the oligosaccharide moiety that varies in the complexity based on the level of sialylation (N˗acetylneuraminic acid, NeuAc) and uncharged sugars (GlcCer, Gal, GlcNAc, GalNAc, Fuc). These lipids are abundant in the central nervous system and play important roles in many physiological processes in cells, such as memory control, cell signaling, neuronal recovery, neuronal protection or apoptosis. Gangliosides are also related to some serious disease such as cancer, Alzheimer disease, Tay-Sachs disease and Fabry disease. Gangliosides are extracted using chloroform, methanol and water. Aqueous phase containing gangliosides is further purified on C18 solid-phase extraction column to remove undesired salts before LC˗MS. The main goal of this study is the development of analytical method for ganglioside analysis based on the hydrophilic interaction liquid chromatography coupled to negative-ion electrospray ionization mass spectrometry (HILIC/ESI-MS). The optimization of chromatographic parameters including the buffer type, its concentration and pH value is performed to achieve the optimal HILIC separation with regard to the best sensitivity and chromatographic resolution of studied compounds. The best results are obtained on porous shell particle column Ascentis (150 x 2.1 mm, 3 µm). Individual lipid species were identified based on accurate

www.ISC2016.ie @isc2016 #isc2016 /isc2016266
POSTER ABSTRACTS Metabolomics, Glycomics, Lipidomics and Proteomics
m/z values in the negative-ion mode using high-resolution tandem mass spectra. The final HILIC/ESI-MS method is applied for the lipidomic analysis of human kidney, lungs, plasma, urine, erythrocytes and porcine brain. More than fifty gangliosides from GM1, FucGM1, GM2, GM3, GD1, GD3 and GT1 classes and other phospholipids (e.g., sulfatides) are identified in biological samples.References: [1] http://www.lipidmaps.org/,
downloaded in April 2016.Acknowledgements: This work was supported by ERC CZ project No. LL1302 (MSMT, Czech Republic).Disclosure of Interest:None DeclaredKeywords: gangliosides, HPLC, mass spectrometry
P02-002-104 - Evaluation of ion mobility/ TOF mass spectrometry with multiple LC method parameters for enhanced detection in metabolic profiling
P. Rainville*, J. Castro-Perez 1, R. Plumb 2, J. Langridge 3
1 Waters Corporation, Milford, United States, 2 Imperial College London, London, 3 Waters Corporation, Wimslow, United Kingdom
Content: The aim of this study was to evaluate ion mobility spectrometry (IMS) coupled with rapid high resolution LC/MS and determine the impact upon metabolome and lipidome coverage. Metabolic profiling can provide a comprehensive and quantitative view of the metabolite variation in biological systems. The total number of metabolites can vary from 600 for simple yeast cells to over 200,000 metabolites for higher order organisms. Due to this complexity it is very challenging to establish analytical
profiling methodologies that can isolate, identify, and quantify analytes in a high throughput environment. IMS offers an extra dimension of separation that when coupled with LC/MS increases the overall peak capacity for metabolic profiling.
Human plasma and urine obtained from breast cancer, Type I diabetes and healthy subjects was used as metabolite test samples in the study. Three column lengths were utilized in the study (150, 75, and 30 mm), LC gradient conditions were: Scaled for each of the column length and therefore gradient duration by maintaining constant linear velocity, or increased of the mobile phase linear velocities and reduction of the gradient duration accordingly.
Results from these experiments showed that IMS significantly increased feature detection with fast analysis on short columns. Results will be discussed with respect to the dynamic range of detection, mass measurement accuracy of detected features. Finally, we will show that the increase in the number of features that could be detected by utilizing IMS allowed for the use of shorter gradients and columns thus increasing the throughput of analysis without reducing metabolic coverage. References: 1. Paglia. G. et al, Ion mobility
derved cross sections to support metabolomics applications, Analytical Chemistry, 2014, 86, p 3985-3993.
Disclosure of Interest:None DeclaredKeywords: ion mobility mass spectrometry, Metabolic fingerprinting, UPLC-HRMS

267www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMetabolomics, Glycomics, Lipidomics and Proteomics
P02-002-105 - Application of UHPSFC/ESI-MS method for the lipidomic analysis of kidney cancer tissue samplesM. Khalikova 1,*, M. Lísa 1, E. Cífková 1, D. Vrána 2, B. Melichar 2, V. Študent 3, M. Holčapek 1
1 Department of Analytical Chemistry, University of Pardubice, Pardubice,
2 Department of Oncology, Palacky University, Medical School and Teaching Hospital,
3 Department of Urology, Palacky University, Faculty of Medicine and Dentistry, Olomouc, Czech Republic
Content: Kidney cancer is the twelfth most common cancer in the world (joint position with pancreatic cancer), with 338 thousand new cases diagnosed in 2012 [1]. The determination of lipidomic differences is crucial for the improvement of cancer early diagnostic since the changes in lipids profiles are associated with cancer development and may be correlated to the disease stage. The ultrahigh-performance supercritical fluid chromatography (UHPSFC) technique brings benefits for the separation and analysis of polar and nonpolar lipid classes [2]. Optimized UHPSFC – electrospray ionization mass spectrometry (ESI-MS) method is applied for the characterization of changes in lipid profiles of kidney cancer tissue samples. 422 lipid species are identified and quantified in kidney tissues from 10 different lipid classes including cholesterylesters (CE), triacylglycerols (TG), diacylglycerols (DG), monoacylglycerols (MG), phosphatidylethanolamines (PE), lysophosphatidylethanolamines (LPE), ceramides (Cer), phosphatidylcholines (PC), lysophosphatidylcholines (LPC) and sphingomyelins (SM). The quantitation of lipids using UHPSFC/ESI-MS is based on peak abundances of corresponding ions in lipid class mass spectra after the isotopic correction related to the peak abundance of
one internal standard per each lipid class. We apply multivariate data analysis using unsupervised principal component analysis and supervised orthogonal partial least square analysis to group and identify significant differences in lipid species by comparing lipid profiles of cancer and health tissues. Up-regulated nonpolar lipid classes, such as CE and TG, containing long chain fatty acyls, are observed in tumor samples and show a clear association with kidney tumor tissues. References: 1. Peng Li et al. Eur. Urol. 67, 1134–1141, 20152. Lísa M., Holčapek M., Anal. Chem. 87, 7187-
7195, 2015Acknowledgements: This work was supported by ERCCZ project No.LL1302 sponsored by the Ministry of Education, Youth and Sports of the Czech Republic.Disclosure of Interest:None Declared
P02-002-106 - Comprehensive Serum N-Glycome Characterization Utilizing 2-Dimensional UPLC and LC-MS/MS analysis with DMT-MM Derivatisation
J. Smith 1,*, V. Hilborne 2, S. Mittermayr 1, C. Clarke 1, E. McCarthy 2, J. Bones 1
1 National Institute of Bioprocessing Research and Training (NIBRT), Dublin, Ireland,
2 Northeastern University, Boston, United States
Content: Glycosylation is the most common protein post-translational modification with changes in the serum N-glycome having been shown to be associated with a wide variety of diseases1. However, a general consensus as to the number of N-glycans present in the serum N-glycome does not exist2,3. Here we have performed a complete

www.ISC2016.ie @isc2016 #isc2016 /isc2016268
POSTER ABSTRACTS Metabolomics, Glycomics, Lipidomics and Proteomics
characterization of the serum N-glycome to fully identify the glycans present. To perform our analysis, human serum was first depleted of IgG and albumin and pooled. N-Glycans were released using a FASP based sample prep method and PNGase F digestion, with subsequent 2-aminobenzamide (2-AB) derivatisation. Weak anion exchange (WAX) fractionation was performed on the labelled glycans to separate them by the number of sialic acids present. To comprehensively identify the glycan structures, exoglycosidase digestions, in addition to subsequent hydrophilic interaction liquid chromatography (HILIC)-UPLC fluorescence fractionation was performed on the WAX fractionated glycans. All the peaks from the HILIC fractionation runs were collected, with each fraction containing between 55 - 70 individual peaks and over 300 individual peaks in total. The collected peaks were analysed using LC-MS/MS with a portion of each peak first derivatized with 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) to aid in sialic acid linkage and positional isomerism annotation4. Glycan identifications were then applied to glycan profiles of patient gastric cancer (GC) serum samples taken before (Pre-Op) and after (Post-Op) surgery.References: (1) Dwek, R. A. Chem Rev 1996, 96. 683-720.(2) Bereman, M. S.; Williams, T. I.; Muddiman, D. C.
Anal Chem 2009, 81. 1130-6.(3) Saldova, R.; Asadi Shehni, A.; Haakensen, V.
D.; Steinfeld, I.; Hilliard, M.; Kifer, I.; Helland, A.; Yakhini, Z.; Borresen-Dale, A. L.; Rudd, P. M. J Proteome Res 2014, 13. 2314-27.
(4) Tousi, F.; Bones, J.; Hancock, W. S.; Hincapie, M. Anal Chem 2013, 85. 8421-8.
Disclosure of Interest:None DeclaredKeywords: Glycomics, Mass Spectrometry, UPLC
P02-002-107 - Optimisation of Square Wave Voltammetric conditions for the rapid detection of Guaiacol using a Boron Doped Diamond electrode
P. E. Hayes 1,*, L. Reddy 2, V. Morris 2, J. D. Glennon 3
1 Innovative Chromatography Group, Irish Separation Science Cluster (ISSC), Department of Chemistry and the Analytical and Biological Chemistry Research Facility (ABCRF), University College Cork, Cork,
2 PepsiCo, Technical Function - R&D, Little Island, Cork, 3 Innovative Chromatography Group, Irish Separation
Science Cluster (ISSC), Department of Chemistry and the Analytical and Biological Chemistry Research Fcaility (ABCRF), University College Cork, Cork, Ireland
Content: Alicyclobacillus spp. are thermophilic and acidophilic spore forming bacteria associated with spoilage in fruit juices and beverages. As Alicyclobacillus spp. can grow at low pH levels and survive the pasteurisation process, it causes spoilage through the formation of a medicinal or antiseptic like off odour. Guaiacol is accepted as the predominant metabolite associated with spoilage, and is formed as a result of microbial metabolism through the nonoxidative decarboxylation of vanillic acid. Previous techniques applied for its detection include HPLC-ECD1 and HS-GC-MS2. The aim of this study was to apply a bare boron doped diamond electrode for the detection of guaiacol thus providing manufacturers with a rapid method for its detection. Under cyclic voltammetric conditions, guaiacol exhibited quasi-reversible behaviour with one oxidation peak at + 1.076 V and a smaller reduction peak at – 0.144 V. The effect of supporting electrolyte, pH, scan rate and square wave voltammetric parameters (frequency, amplitude and scan increment) on the current response of guaiacol were examined

269www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMetabolomics, Glycomics, Lipidomics and Proteomics
to determine the optimum detection conditions. Under optimum conditions (0.1 M phosphate buffer, pH 2 and an accumulation step of 60 s) this method provides a simple and rapid technique for guaiacol detection.References: 1. M. Takahashi, S. Sakamaki, A. Fujita, Biosci.
Biotechnol. Biochem, 77, (595-600), (2013)2. R.C. Witthuhn, Y. Smit, M. Cameron, P. Venter,
Food Control, 30, (700-704), (2013)Disclosure of Interest:None DeclaredKeywords: Alicyclobacillus spp., Boron doped diamond electrode, Guaiacol
P02-002-108 - Liquid chromatography of conformational isomers of an adduct between monoamine oxidase A and an irreversible inhibitorA. Albreht 1,*, R. R. Ramsay 2
1 Laboratory for Food Chemistry, National Institute of Chemistry, Ljubljana, Slovenia,
2 Biomedical Sciences Research Complex, University of St Andrews, St Andrews, United Kingdom
Content: In liquid or gas chromatography, the ideal behaviour of an analyte is usually represented by a symmetric, narrow, and sharp peak. In reality, deviations from this form are often observed such as peak tailing/fronting, peak broadening, splitting, shouldering, and so forth. These phenomena are frequently the result of improper sample preparation, unstable chromatographic conditions or some kind of instrumentation failure. However, there are instances when chromatographic abnormalities come into play due to intrinsic properties of an analyte.1
An example of such an occurrence is an analyte having a high interconversion barrier between two conformational isomers. In this study, we report on a peculiar chromatographic behaviour of an adduct, which is formed by irreversible inhibition of monoamine oxidase A with ASS234 followed by subsequent proteolytic digestion. Under different HPLC-MS conditions we observed various scenarios, a single peak and multiple resolved peaks being the two extremes. Combined with the MS data, these results were finally used for the estimation of the interconversion activation energy barrier and aided us in the structural elucidation of the adduct.References: 1 O. Trapp, Anal. Chem. 2006, 78, 189-198.Disclosure of Interest:None DeclaredKeywords: conformational isomers, Liquid Chromatography Mass Spectrometry, Structural similarity
P02-002-109 - LC-HRMS Applied to Study of Metabolomic Profile Modification in Milk after Enrofloxacin Administration A. Junza 1,*, J. Saurina 2, D. Barron Bueno 1, C. Minguillón 1
1 Food and Nutrition Torribera Campus, 2 Analytical Chemistry, Universitat de Barcelona,
Barcelona, Spain
Content: LC-HRMS was used in the search and identification of metabolites and transformation products (TPs) in raw milk from medicated cows. Enrofloxacin (ENR) was administered as a pharmacological treatment to ill cows for three days. Samples were collected daily during seven days (from the first day of treatment and until four

www.ISC2016.ie @isc2016 #isc2016 /isc2016270
POSTER ABSTRACTS Metabolomics, Glycomics, Lipidomics and Proteomics
days after). A conventional extraction step by solid phase extraction (SPE) previous to LC-HRMS (operating in full scan MS mode) was used as a sample treatment.
Original raw data, consisting of m/z values, were taken throughout the entire chromatogram and were compared with those obtained from blank milk samples. For each sample, the significant MS peaks, characterized by their retention time and m/z value, have been subsequently taken to construct a data set focused on the differentiation of positive and blank samples. Twenty-six different compounds, present only in positive samples, were identified and structurally elucidated on the basis of their high resolution MS/MS spectra. Some of these compounds resulted to be related to ENR structure, while others were assigned as di- or tripeptide derivatives. Also, the identification of discriminant components among compounds and samples has been studied by Principal Component Analysis (PCA). Thus, on the basis of the presence of metabolites and their concentration profile evolution with time, samples were classified in three different groups corresponding to days 1-2, day 3 and days 4-7. A tentative metabolic pathway was proposed to rationalize the metabolic formation of the newly identified compounds.References: A. Junza, S. Barbosa, M. R. Codony,
A. Jubert, J. Barbosa, D. Barrón. J. Agric. Food Chem. 2014, 62, 2008−2021
A. Junza, J. Saurina, D. Barrón, C. Minguillón. Metabolic profile modifications in milk after Enrofloxacin administration studied by Liquid Chromatography coupled with High Resolution Mass Spectrometry. J. Chromatogr. A, 2016, under revision.
Acknowledgements: The authors are grateful to the Ministerio de Economia y Competitividad (Project CTQ2013-44077-R) for finantial support.
Disclosure of Interest:None DeclaredKeywords: enrofloxacin, metabolomics, milk
P02-002-111 - Retention time indexing in reverse phase liquid chromatography for enhancing metabolite identification in LC-MS based metabolomics. A cross-lab trial
M. Witting 1, N. Sillner 1, D. Spaggiari 2, S. Rudaz
2, J. Lintelmann 1, J. P. Schnitzler 1, O. Begou 3, G. Theodoridis 3, H. G. Gika 3,*, P. Schmitt-Kopplin 1, M. Quilliam 4
1 Helmholtz Center, Munich, Germany, 2 Geneva University, Geneva, Switzerland, 3 Aristotle University, Thessaloniki, Greece, 4 Institute for Marine Biosciences, National Research
Council, Halifax, Canada
Content: Metabolite identification is still the major bottleneck in non-targeted metabolomics. Different levels of identifications have been proposed by the Metabolomics Society Identification Task group (Sumner et al 2007, plus paper with refined levels). The highest level of identification can be only achieved by comparison with an authentic standard using two independent properties such as retention times and MS/MS spectra. Having reference standards for all possible metabolites in a single laboratory is nearly impossible and not feasible. Several public repositories storing MS/MS spectra have been created, also covering various instrumentations including MS analyzers (e.g., Q-ToF, IT or ICR) (MassBank, Metlin etc.).
MS/MS spectra represent only one part of an accurate identification and can be ambiguous in case of isomeric compounds,

271www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMetabolomics, Glycomics, Lipidomics and Proteomics
wrong collision energy queries, etc. Retention times could provide valuable information in LC-MS based metabolomics but are comparable only over a certain range. Even when using the same column and mobile phase chemistries and column dimensions, retention times can vary between labs due to different LC instrumentation. The concept of retention time indexing (RTI), already widely used in GC-MS, can help simplify the process by converting retention times to the dimensionless retention index, which is only dependent on stationary phase chemistry and mobile phase composition.
Here we present our first preliminary results from a ring-trial using a novel RTI system based on a homologous series of substances purposely designed for LC-MS and several metabolite standards measured in 5 different laboratories using different LC-MS systems.References: M .Quilliam et al., 42nd Symposium
of HPLC 2015, Geneva, Switzerland / poster presentation, “Anew retention index system for LC-MS/MS”
Disclosure of Interest:None DeclaredKeywords: Liquid Chromatography Mass Spectrometry, Metabolomics, retention index
P02-002-112 - Analysis of Siamese crocodile (Crocodylus siamensis) Eggshell Matrix Proteins
I. Miksik 1,*, S. Pataridis 1, A. Eckhardt 1, D. Sedmera 1 2
1 Institute of Physiology ASCR, Praha 4, 2 Institute of Anatomy, First Faculty of Medicine, Charles
University in Prague, Praha 2, Czech Republic
Content: Electrophoretic and nanoLC-MS methods were developed for the analysis of proteins in an Crocodylus eggshell matrix.
This research continued on our analysis of proteins from avian eggshell [1].
Eggs of Crocodylus siamensis originated from breeding at Crocodile Zoo Protivín (Czech Republic). The eggshell was decalcified by various methods (EDTA or acetic acid) and two main layers (cuticle and palisade) were analyzed. From these layers we used both soluble as well as insoluble parts. Final analysis of presented proteins was made by nLC-MS (QTOF) or combination of polyacrylamide gel electrophoresis and nLC-MS (QTOF). Proteins were identified by correlating tandem mass spectra to the selected database set for Crocodylia extracted from NCBI database and using the MASCOT searching engine when semi-trypsin was chosen as the enzyme parameter. We also tried to search avian database.
More then 70 proteins were determined at the whole eggshell matrix when the most dominant proteins were thyroglobulin, IgGFc-binding protein, calumenin, lysyl oxidase, ovostatin, mucin-5AC, ectonucleotide pyrophosphatase/phosphodiesterase, vitellogenin 1 and apolipoprotein B-100. Cuticle contained lower amount of proteins (cca 35) and relative distribution of proteins was slightly different between both layers.
Conclusion: It is a first attempt to describe proteome of reptile eggshell. In comparison to proteins determined in avian eggshell we observed substantial differences (only a few proteins were determined at both animal classes, as vitellogenin 1 and 2) however not all proteome of Crocodile is described yet.References: 1. Ivan Mikšík, Pavla Sedláková,
Katerina Lacinová, Statis Pataridis, Adam Eckhardt: Determination of insoluble avian

www.ISC2016.ie @isc2016 #isc2016 /isc2016272
POSTER ABSTRACTS Metabolomics, Glycomics, Lipidomics and Proteomics
eggshell matrix proteins, Analytical and Bioanalytical Chemistry, 397: 205–214, 2010.
Acknowledgements: The financial supports by the Czech Science Foundation (GA CR) (15-01948S – IM, SP, AE and 16-02972S - DS) are gratefully acknowledged.Disclosure of Interest:None DeclaredKeywords: Crocodile, Eggshell, Proteomics
P02-002-113 - Application of GC-QqQ/MS and LC-TOF/MS for validation of tentative urine biomarker candidates of prostate cancer R. Bujak 1, W. Struck-Lewicka 1, M. Patejko 1,*, M. Kordalewska 1, M. Markuszewski 2, M. Matuszewski 2, R. Kaliszan 1, M. Markuszewski 1
1 Department of Biopharmaceutics and Pharmacodynamics,
2 Department of Urology, Medical University of Gdańsk, Gdańsk, Poland
Content: According to worldwide statistics, prostate cancer (CaP) is one of the most common and lethal type of cancer among men. The pathogenesis of CaP is still unclear and not fully explained. As early diagnosis of the disease increases chances for implementation of efficient pharmacotherapy, there is a need of specific and noninvasive diagnostic methods evaluation. Metabolomics approach seems to be useful tool in searching for biomarkers of CaP. The aim of this study was to perform intra-laboratory validation of previously identified potential markers of prostate cancer [1]. Urine samples from CaP patients (n=39) and healthy volunteers (n=43) were analyzed with the use of two analytical platforms: gas chromatography coupled with triple quadrupole mass spectrometry (GC-QqQ/MS) and liquid chromatography hyphenated with time-
of-flight mass spectrometry (LC-TOF/MS). Samples were prepared using previously optimized procedure. Obtained datasets were processed with deconvolution, alignment, filtering and normalization steps. Afterwards, uni- (t-test, U Mann-Whitney test depending on data distribution) and multivariate statistical analysis (PCA, OPLS-DA) were performed with the use of SIMCA P+ and Matlab software. Statistically significant metabolites were selected according to adjusted p value (FDR p value < 0.05) and variable importance into projection (VIP value > 1). In comparison to previous results, the most reproducible metabolites, which contributed the most into group classification, were selected in independent set of samples. The obtained results provided a list of compounds that could be considered as validated potential biomarker candidates of prostate cancer. Nevertheless, quantitative targeted metabolomics approach applied on larger set of samples is recommended for confirmation of the diagnostic potential of the proposed metabolites.References: 1. W. Struck-Lewicka, et al., JPBA
111(2015), 351-361Acknowledgements: The project was supported by National Science Centre grant no. 2014/13/D/NZ7/00368Disclosure of Interest:None DeclaredKeywords: biomarkers, metabolomics, Prostate cancer

273www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMetabolomics, Glycomics, Lipidomics and Proteomics
P02-002-114 - LC-QTOF metabolomics reveals biochemical changes during Klebsiella pneumoniae biofilm eradication by greater omentumJ. Teul 1,*, S. Deja 2, K. Siemionow 1, A. Ząbek 3, P. Młynarz 3, P. Barć 4, A. Junka 5, D. Smutnicka 5, M. Bartoszewicz 5, W. Miltyk 1
1 Department of Pharmaceutical Analysis, Medical University of Bialystok, Bialystok,
2 Faculty of Chemistry, Opole University, Opole, 3Department of Chemistry, Wroclaw University of Technology,
4 Department of Vascular, General and Transplantation Surgery,
5 Department of Pharmaceutical Microbiology and Parasitology, Wroclaw Medical University, Wroclaw, Poland
Content: One of the serious problems in hospital treatment are nocosomial infections. Many of them are associated on bacteria biofilm formation causing an acute inflammation in patient’s organs especially in case of implant grafting. Bacterial biofilm is difficult to eliminate with standard doses of antibiotics and new practices are being explored [1]. One of them is application of greater omentum for implant coverage. Greater omentum is an organ of exceptional versatility, angiogenic and germicidal abilities [2]. The aim of the study was to extend present knowledge of effectiveness of microbial biofilm eradication by greater omentum and evaluate metabolic changes in rat urine after implant grafting using LC-MS (QTOF) metabolomics. Metabolomics is a global analysis and gives an opportunity to uncover information concealed in a metabolic profile and apply it to diagnose and monitor various pathological states [3].
In this study 4 groups of rats were subjected to implantation of biomaterial (sterile or covered with bacteria biofilm). The
biomaterial was introduced surgically into peritoneum or greater omentum. Urine samples were collected within 30 days. Metabolomic profiles of urine were registered by LC-MS. Multivariate analysis was applied to build statistical models that enabled to observe significant differences in generated profiles among studied cases. Discriminant variables were found and corresponding metabolites were identified. Results were correlated with the number of bacteria left on a removed implant at the time when the experiment was stopped for each rat.
LC-MS metabolomics enabled to observe and identify metabolic changes connected to the place of implant grafting and determine if metabolic changes after a surgery are connected with bacteria biofilm eradication on the surface of implant.References: [1]. PS Stewart,JW Costerton, Lancet, 358(9276):135-
8, 2001.[2]. J Vernik, AK Singh, Int Artif Organs, 30:95-9, 2007.[3]. J Teul et all, J Proteome Res, 8:5580-9, 2009.Acknowledgements: This work was founded by National Science Centre in Poland (DEC-2012/07/N/NZ4/02528). J.Teul acknowledges the financial support from Foundation for Polish Science (FNP).Disclosure of Interest:None DeclaredKeywords: biofilm eradication, greater omentum, metabolomics

www.ISC2016.ie @isc2016 #isc2016 /isc2016274
POSTER ABSTRACTS Metabolomics, Glycomics, Lipidomics and Proteomics
P02-002-115 - Investigation of the derivatization conditions for GC-MS metabolomics of biological samplesG. Moros 1, C. A. Chatziioannou 1, G. Theodoridis, N. Raikos 2, H. GIKA 2,*
1 Chemistry, 2 Medicine, Aristotle University Thessaloniki, Thessaloniki,
Greece
Content: GC-MS provides a powerful tool for clinical analysis. Metabolomics applications represent an upcoming field where significant efforts are directed. Metabolomics analysis necessitate two step derivatization to alter metabolites in foms more amenable to chromatographic separation and mass spectrometric detection. Derivatization is a very important step of the experiment process. Different derivatization have been reported for GC-MS metabolomics. However still no universally accepted protocol is established. The scope of this work is to examine derivatisation reaction conditions including selection of derivatization agent.
Different derivatization agents have been reported in the literature. Two-step derivatization procedure is the method most often used; methoxymation followed by silylation. In the present study, frequently used silylation agents (MSTFA, BSTFA with 1% TMCS and MSTFA with 1% TMCS) were tested for the derivatisaion of blood plasma following a standardised experimental protocol. Optimization of the methoxymation reaction was performed examining different conditions when processing the same standard extract, consisting of 60 different compounds belonging to several chemical classes (endogenous metabolites). Completion
of the methoxymation reaction depends on reaction time and temperature. Both parameters were studied using conditions commonly found in literature. A further experiment was performed in the same standard extract testing the time and the temperature of silylation reaction. Data were extracted with XCMS, AMDIS and Chemstation software. After extensive statistical analysis the optimum results for biological matrixes were given using MSTFA as silylation agent. Also methoxymation at room temperature followed by silylation at high temperature leads to efficient derivatization of most metabolites.References: 1. H. Kanani, PK Chrysanthopoulos,
M Klapa. Standardizing GC-MS metabolomics. J Chromatogr B, 871 (191-20) (2008).
Disclosure of Interest:None DeclaredKeywords: derivatization, GC-MS, silylation
P03-003-001 - Towards Increasing Metabolite Coverage By Combination Of Sample Preparation, Hilic And Reversed-Phase Lc/MsA. T. Faccio 1 2,*, M. F. M. Tavares 3, C. Barbas 2, F. J. Rupérez 2
1 Química Fundamental Instituto de Química - , Universidade de São Paulo, Sao Paulo, Brazil,
2 CEMBIO, Universidad CEU-San Pablo, Boadilla del Monte, Spain,
3 Química Fundamental Instituto de Química -, Universidade de São Paulo, Sao Paulo, Brazil
Content: Metabolomics (both targeted and untarged profiling) aim to achieve a comprehensive analysis of all metabolites (small molecules, intermediates and products of metabolism) in biological systems such as biofluids, tissues or cells

275www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMetabolomics, Glycomics, Lipidomics and Proteomics
from an organism under specific conditions (disease vs control, different diets, different treatments, etc.). Difficulties related to the large variability of physicochemical properties and range of concentrations of all the possible metabolites within a biological sample are well known. Among the analytical platforms used for metabolomics studies, LC-MS is the most popular, due to its robustness and reliability, as well as the high sensitivity and dynamic range of MS. Moreover, the versatility of the available combinations of modes (RP vs. HILIC) and mobile phases allows obtaining complementary results with only one instrument. However, despite many attempts and publications, HILIC chromatography is far from having the robustness that is necessary to compare profiles.
The aim of the present work was to evaluate different procedures for sample (plasma) treatment and analysis, to obtain the maximum metabolite coverage, with the highest reproducibility. One-phase and two-phases extraction procedures were compared, according to the extraction efficiency and simultaneous compatibility with both HILIC and reversed phase LC. We will show results of the comparison of different sample treatments, different chromatographic conditions (columns, mobile phases, gradients), as well as different detection modes, in two different LC/MS instruments. The differences between conditions were evaluated according to the number of molecular features, the number of putatively identified metabolites, the coefficient of variance of intensities for all features, as well as the coefficient of variance of the retention times and the shape of peaks.
References: Disclosure of Interest:None DeclaredKeywords: HILIC, Metabolites, metabolomics
P03-003-002 - Microfluidic reactor for proteomics research
B. Buszewski 1,*, K. Meller 1
1 Nicolaus Copernicus University in Toruń, Faculty of Chemistry, Chair of Environmental Chemistry and Bioanalytics, Torun, Poland
Content: Proteins are one of the most important compounds occurred in living organisms. Their play many biological functions including catalysis, molecules transport, biochemical processes regulation, immune functions etc. Proteins distribution, interactions, properties, structures, post-translational modifications (PTMs), and quantitative as well as qualitative determination are the subjects of the proteomic research [1]. Proteins digestion with specific enzyme is commonly used during their identification or sequencing [2,3]. One of the relatively novel solutions making a progress on this field is application of Immobilized Enzyme Reactors (IMERs).
The main aim of presented study was the preparation of microfluidic reactor for fast and efficient proteolysis. For this purpose, trypsin was immobilized on monolithic support synthesized from 2-hydroxyethyl methacrylate and N,N’-methylene-bis-acrylamide. The mixture of 2-propanol, 1-decanol and water was used as a porogen solvent for polymerization of monolithic bed. Trypsin was immobilized on synthesized bed via support hydroxyl groups after their activation with 1,1’-carbonyldiimidazole. Two different immobilization strategies were tested and several microreactors

www.ISC2016.ie @isc2016 #isc2016 /isc2016276
POSTER ABSTRACTS Metabolomics, Glycomics, Lipidomics and Proteomics
were prepared to verify the reproducibility of elaborated methods. The IMERs activities were evaluated using nano-LC based on the degree of hydrolysis of low-molecular trypsin substrate. The prepared microreactors were also applied for protein digestion and identification using MALDI-TOF MS.References: [1] W.P. Blackstock, M. P. Weir, Trends Biotechnol. 17
(1999) 121-127[2] B. Thiede, W. Höhenwarter, A. Krah, J. Mattow,
M. Schmid, F. Schmidt, P.R. Jungblut, Methods 35 (2005) 237–247.
[3] K. G. Standing, Curr. Opin. Struct. Biol. 13 (2003) 595-601
Acknowledgements: This work was supported by the National Science Center in the frame of the project Maestro 6 (Cracow, Poland), no. NCN 2014/14/A/ST4/00641.Disclosure of Interest:None DeclaredKeywords: monolithic support, 2-hydroxyethyl methacrylate-co-N,N’-methylene-bis-acrylamide, Immobilized Enzyme Reactor (IMER)

277www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMicrofluidic Separation Devices
Microfluidic Separation Devices
P03-003-003 - On-chip integration of high pressure chip LC and droplet microfluidics
R. Gerhardt 1,*, D. Belder 1
1 Institute of Analytical Chemistry, University of Leipzig, Leipzig, Germany
Content: We present the first integration of droplet-based microfluidics with chip-based liquid chromatography in a full-body glass chip. Segmentation of the eluent flow is achieved by introducing the immiscible oil phase immediately following separation. By coupling the two functionalities analytes are encapsulated into numerous discrete volumes which are isolated by the surrounding oil phase. Glass was chosen as chip material for numerous reasons. First, chromatographic separations could be performed under high pressure conditions (up to 300 bar). Furthermore, excellent optical properties and chemical inertness are appealing as well. Monolithic integration of high pressure chip-based LC with subsequent droplet generation was achieved by using a T-junction design. Four polycyclic aromatic hydrocarbons were separated under reversed-phased conditions and compartmentalized into a sequence of droplets (about 50 droplets per peak) using 80% acetonitrile as the mobile phase. Fluorescence spectroscopy was applied as detection technique. Detection of the separation at a later position, i.e. further downstream, resulted in an equal chromatogram, proving that the chromatographic resolution is preserved within the droplets. Generation of the segmented flow had no disadvantageous effects on the separation performance. Further downstream processing such
as derivatization, additional analysis, extraction of the droplet content or storage is easily realizable.References: S. Thuermann, L. Mauritz, C. Heck, D.
Belder, Journal of Chromatography A, 1370, 33 (2014).
Disclosure of Interest:None DeclaredKeywords: chip LC, droplet microfluidics
P03-003-004 - Development of 3D printed modular microfluidic elements for pre-treatment of RNA samplesP. F. O’Neill 1,*, F. Klacsmann 2, Y. Men 2, S. Senapati 2, H.-C. Chang 2, D. Diamond 3, D. Brabazon 1
1 Advanced Processing Technology Research Centre, Dublin City University, Dublin, Ireland,
2 Centre for Microfluidics and Medical Diagnostics, University of Notre Dame, Notre Dame, United States,
3 National Centre for Sensor Research, Dublin City University, Dublin, Ireland
Content: Probe-functionalised Ion Exchange Membranes (IEMs) are a novel sensing method for the detection of nucleic acids in sample mixtures [1]. These label-free high-sensitivity biosensors are promising for on-site diagnostics due to their miniature form factor and potential for automation. However, one of the challenges in the development of an on-site diagnostic platform is the miniaturisation of lab-based cell lysis and pre-treatment protocols for efficient extraction of nucleic acids from the lysed solution prior to sensing. This poster reports a novel pretreatment protocol for cell-lysis, volume reduction and buffer exchange in a microfluidic form factor to reduce conductivity of the sample for efficient electrochemical detection of bacterial and viral RNA for disease diagnosis. A 3D

www.ISC2016.ie @isc2016 #isc2016 /isc2016278
POSTER ABSTRACTS Microfluidic Separation Devices
printed microfluidic Solid Phase Extraction (SPE) column was developed and used to capture negatively charged RNA molecules through a chaotropic binding mechanism. The column was fabricated with integrated luer-lock fittings for connection to pumping and sensing peripherals in a single step using a UV curable photopolymer. SiO2 particles were then packed against the integrated packing weir and the device was sealed using a standard luer-lock fitting and flushed with RNase Zap and DI H2O to remove RNase enzymes. The SPE column was subsequently loaded with the lysed cell solution and the column was washed using a buffer optimised to reduce the conductivity of the eluted sample while retaining the nucleic acids on the column. Finally, RNA molecules were eluted using DI water and volume fractions were analysed using quantitative reverse-transcription polymerase chain reaction (qRT-PCR) and compared with commercially available RNA purification methods. Further development of this modular system will be undertaken to improve reproducibility, and reduce assay time.References: 1. S. Senapati, S. Basuray, Z. Slouka,
L.-J. Cheng, H.-C. Chang, Top Curr Chem (153-169) (2011).
Acknowledgements: Financial support from Science Foundation Ireland (Grant Nos. 12/IA/1576 and 12/RC/2289) and the Naughton Foundation (Naughton Graduate Fellowship) is gratefully acknowledged.Disclosure of Interest:None DeclaredKeywords: 3D printing, RNA, Solid phase extraction

279www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMolecular Diagnostics, Clinical and Biomedical Analysis
Molecular Diagnostics, Clinical and Biomedical Analysis
P01-001-013 - Detection of bacteriophages’ lytic enzymes associated with their bacterial hosts’ protein profiles analysisA. A. Elshayeb*
Content: Protein chromatograpgy can be assesed by bioinformatics or in silico biology analysis programs.
Objective: To identify the relationship between bacteriophages and bacteria by the protein profiles analysis.
Methods: The proteins of the E.coli phage bacteria were separated by (SDS-PAGE) to estimate their molecular weights; the migration of each band was compared to the standards of known weights in the molecular ladder.
Results: The proteins of the phage showed four major bands with MW of 46, 35, 24 and 14 kDa. Meanwhile, the bacteria showed clear nine bands with MW ranged between 96 and 24 kDa. This was analysed by the programme ImageJ136b which showed molecular masses of 47, 34 and 16 kDa, The analysed data showed the protein bands distance migration and plot area of the phage and the bacteria, the results in size from 47.7 to 34.3 kDa resembles 43.3% of total phage protein which formed the capsid head and the coil, while the MW mass of 22.5 formed the tail proteins,. The lytic enzymes MW was ranged between 18-14 kDa according to the type of the enzyme. We found that the band of 34 kDa was the common shared peak between bacteriophage and allied bacterium.
Conclusion: Protein separation by SDS-PAGE by the support of bioinformatics and data mining ensures the proteins molecular weight and functions. During phage assembly, lytic enzymes 47 – 34 kDa are built in the capsid head and the lysozyme 14kDa in the tail, they facilitate the enzymatic decaye for bacterial host. This is related to the lytic cycle of the bacteriophages and their phenomenon in utilizing the bacterial DNA in proteins manufacturing during their multiplication inside bacteriaReferences: Ayman, A., Elshayeb, Abdalazim, A.,
Ali, Marmar A. Elsiddig and Adil, A., EL-Hussien. (2016). Detection of bacteriophages’ lytic enzymes associated with their bacterial hosts’ protein profiles analysis. University of Khartoum.
Acknowledgements: The authors would like to thank Mrs. Nada Eltahir, DNA Fingerprint laboratory SudanDisclosure of Interest:None DeclaredKeywords: Bacteriophage, Bioinformatics, Protein Profile
P03-003-006 - Stress and gut health: fast and efficient mannitol determination by UPLC-MS for the assessment of intestinal permeability
J. J. Pastor 1,*, A. Mereu 1, G. Tedó 1, G. Rimbach 2, I. R. Ipharraguerre 1 2
1 Feed Additives Innovation, Lucta S.A., Montornes del Valles (Barcelona), Spain,
2 Institute of Human Nutrition and Food Science, University of Kiel, Kiel, Germany
Content: In pigs, weaning stress results in long-term gut barrier dysfunction and thereby impaired animal performance. These consequences are partly mediated by

www.ISC2016.ie @isc2016 #isc2016 /isc2016280
POSTER ABSTRACTS Microfluidic Separation Devices
the release of corticotrophin releasing factor (CRF), which induces degranulation of intestinal mast cells (MC) and the subsequent release of cytokines and proteases that disrupt normal barrier function. The objective of this study was to investigate whether blocking MC degranulation at weaning contributes to increase long-term intestinal integrity and animal performance. To this end, recovery of intestinal permeability markers cobalt-EDTA (Co) and mannitol from plasma samples was assessed 35 d after weaning in piglets either treated or not with the MC stabilizer cromolyn. For mannitol determination different methodologies are available, including enzymatic analysis and gas chromatography. Main drawbacks associated to them are limited accuracy, time consuming sample preparation and poor sensitivity. In this work, a new UPLC methodology for mannitol quantification was developed, based on HILIC chromatography with exact MS detection. The methodology is characterized by a simple sample preparation procedure, low amounts of sample necessary (25 µL) and fast gradient conditions (5 min). Mannitol can be determined with a linearity range from 2.9 to 290 ng/mL, high precision (CV < 10% intra and inter-day) and accuracy (114 and 104% for lower and higher concentrations, respectively). Results showed that piglets treated with cromolyn had a significant decrease in plasma Co concentration (42.3 vs 129.0 ± 29.5 µg/mL; P < 0.048) and mannitol (7.1 vs 8.6 ± 0.5 µg/mL, P < 0.067). Therefore, attenuation of weaning-induced MC degranulation seems to improve intestinal permeability long after weaning. Furthermore, the use of mannitol as described in this study represents a practical tool for assessing gut permeability in vivo.
References: A. J. Moeser; Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G173
Disclosure of Interest:None DeclaredKeywords: Intestinal Permeability, Mannitol, UPLC-MS
P03-003-007 - Application of LC-MS/MS method for free mycophenolic acid measurement in plasma ultrafiltrate samples from lupus nephritis patients
T. Pawinski*, P. Luszczynska 1, A. Dobrzanska 1, M. Krajewska 2, M. Hurkacz 3
1 Department of Drug Chemistry, Medical University of Warsaw, Warsaw,
2 Department and Clinic of Nephrology and Transplantation Meducine,
3 Department of Clinical Pharmacology, Wroclaw Medical University, Wroclaw, Poland
Content: Background: Mycophenolic acid (MPA) is an immunosuppressive drug used in the form of two prodrugs, either mycophenolate mofetil (MMF) or enteric-coated mycophenolate sodium (EC-MPS). It was reported that free MPA measurements might prove useful in predicting patients’ outcome in the course of therapeutic drug monitoring (TDM), especially in case of patients with hypoalbuminemia, kidney or liver diseases [1]. Therefore, the aim of this preliminary research was to apply newly developed LC-MS/MS method for the measurement of free MPA in plasma ultrafiltrate samples from lupus nephritis patients.
Methods: Samples were collected at three time points (C0, C0.5, C2) from eleven lupus nephritis patients on MMF therapy cared for in the Clinic of Nephrology and

281www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMolecular Diagnostics, Clinical and Biomedical Analysis
Transplantation Medicine (Wroclaw Medical University). Free MPA concentrations in plasma ultrafiltrate were measured in the Department of Drug Chemistry (Medical University of Warsaw) using LC-MS/MS method with MPA-d3 as an isotope-labelled internal standard. Zorbax Eclipse XDB-C18 column and a gradient mobile phase of water:methanol (with 0.1% formic acid and 2.5 mM ammonium acetate) were used. The transitions of [M+NH4]+ adduct ions were m/z 338.3→207.0 for MPA and m/z 341.3→210.1 for MPA-d3.
Results: A total of sixteen abbreviated pharmacokinetic profiles were obtained. Free MPA C0, C0.5, C2 concentrations in ultrafiltrate were between 5.31 and 165.30 ng/mL, 20.54 and 251.10 ng/mL, 19.36 and 137.50 ng/mL, respectively. Free MPA trough concentrations in ultrafiltrate were significantly correlated (r=0.8694, p=0.0110) with total MPA trough plasma concentrations additionally measured for seven samples using HPLC-UV method.
Conclusions: Free MPA concentrations were determined with success in plasma ultrafiltrate samples from lupus nephritis patients with the use of LC-MS/MS method what confirms its potential applicability in pharmacokinetic and TDM studies.References: 1. Shaw L.M. et al.: Ther. Drug Monit.
2001, 23, 305-315.Disclosure of Interest:None DeclaredKeywords: free mycophenolic acid, LC-MS/MS, lupus nephritis
P03-003-008 - LC-MS/MS methods for determination of coenzymes Q10, Q10H2 and Q9 in rat plasma and tissuesD. Iaroshenko 1,*, A. Grigoriev 1, A. Sidorova 1
1 CSU “Analytical Spectrometry”, Bioanalytical Laboratory, Peter the Great Saint-Petersburg Polytechnic University, Saint-Petersburg, Russian Federation
Content: A lot of studies have been conducted to elucidate the coenzymes despite this there are some gaps in the pharmacokinetics of these analytes. New drug formulations are developing, preclinical and clinical trials are carrying out. The pharmacokinetics is one of main part of these studies. To meet the demands of new tasks a sensitive and robust method of determination of coenzymes and their metabolites in complex biological matrix are required.
The aim of our study was a development and validation of the methods for simultaneous LC-MS/MS determination of Q10, Q10H2 and Q9 in rat plasma, liver and heart. Most of previous trials were performed by HPLC with UV-detection. The MS/MS-quantification of coenzymes in physiological fluids and tissues are poorly described in the literature.
Because the standard of reduced form of Q10 is unstable (storage under inert atmosphere at -86°C, light and temperature sensitive) we developed special effective procedure for obtaining the Q10H2 from the Q10 by NaBH4 in lab. MRM for Q10, Q10H2 and Q9 were 880.8→197, 882.8→197 and 812.8→197 respectively. Due to the instability of analytes at room temperature a sample preparation should be done at shorter period of time with minimum steps. Therefore a

www.ISC2016.ie @isc2016 #isc2016 /isc2016282
POSTER ABSTRACTS Molecular Diagnostics, Clinical and Biomedical Analysis
protein precipitation by acetonitrile has been utilized for extracting of coenzymes from rat plasma. The isotopic-labeled ubiquinone-10 (Q10-d6) was used as internal standard. This procedure was reproducible with high accuracy (96-105%) and precision (CV=3.6-9.4%) in concentration range 1-60 µg/mL. The extraction from liver and heart tissues was more difficult issue and especial procedure of homogenization and liquid-liquid extraction by hexane has been developed. The recovery of Q10, Q10H2 and Q9 were 88±3%, 70±3% and 83±5% respectively for tissues and 78±6, 60±3, 78±4% respectively for plasma. The developed methods were validated in according with EMA requirements [1] and were applied to pre-clinical trials.References: [1] EMA. 21 July 2011.Disclosure of Interest: None DeclaredKeywords: Coenzyme, HPLC-MS/MS, Validation
P03-003-009 - A UPLC-HRMS Investigation of Medication Adherence Based on the Analysis of Dried Blood Spot SamplesD. Bernieh 1,*, G. Lawson 1, S. Tanna 1
1 Leicester School of Pharmacy, De Montfort University, Leicester, United Kingdom
Content: There is evidence that ~50% of patients prescribed cardiovascular drugs do not adhere to their prescribed regimen. The combination of UPLC separation capabilities with high resolution mass spectrometry (HRMS) was used to identify and quantify a range of cardiovascular (CVD) drugs in a single dried blood spot (DBS), collected from volunteers by a simple finger prick to assess medication adherence.
Methods: An HRMS method [1] was developed and validated for the quantification of 9 commonly UK prescribed heart medicines in DBS samples. Chromatographic analyses was performed on a Zorbax Eclipse Plus C18 HD 2.1x100mm, 1.8µm using gradient elution with a run time of 2.5 min. Identification was carried out using electrospray ionization (positive) in conjunction with high resolution (ToF) MS on an Agilent 6530A QTOF mass spectrometer. The method was applied to DBS samples from volunteers previously administered one or more of the target drugs: atenolol, atorvastatin, bisoprolol, diltiazem, doxazosin, losartan, ramipril, simvastatin and valsartan.
Results: The method validation showed good linearity; the accuracy (relative error) and precision (coefficient of variation) values were within the pre-defined limits of ≤15% at all concentrations. Drug recoveries from spiked blood spots were 57% for simvastatin and ≥82% for the other target drugs. Results of DBS samples taken from 14 volunteers prescribed with one or more of the CVD drugs were within expected levels with respect to published pharmacokinetic data.
Conclusions: The developed DBS based LC-HRMS method has the potential to assess medication adherence and to allow re-interrogation of data collected for heart disease patients. This method has potential to save health service providers money and will allow health practitioners to personalize and optimize drug treatments for their patients.References: 1. S. Tanna et al, J Bioanal Biomed
2015, 7:1, p1-5, http://dx.doi.org/10.4172/1948-593X.1000115

283www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMolecular Diagnostics, Clinical and Biomedical Analysis
Disclosure of Interest:None DeclaredKeywords: Dried blood spot (DBS), Liquid chromatography- high resolution mass spectrometry (LC-HRMS), Medication adherence
P03-003-010 - Diagnostic approach for diabetes mellitus using Dried Saliva Spot (DSS) by UPLC-MS/MS
T. Toyo’oka*, M. Numako 1, T. Takayama 1, K. Todoroki 1, H. Mizuno 1, J. Z. Min 1
1 School of Pharmaceutical Sciences, University of Shizuoka, Shizuoka , Japan
Content: Diabetes Mellitus is one of the interesting diseases due to its increasing worldwide prevalence, not only in the European-American, but also Asian countries. The diagnosis during the pre-disease state is important to avoid the crisis and/or complications with other disorders. L-Lactic acid (LA), which is one of the biologically-active carboxylic acids, ubiquitously occurs in living organisms. Its optical isomer, D-LA, also appears in living systems, even though its amount is relatively low compared to L-LA. Based on this background, we have developed a new methodology using UPLC-MS/MS for the simultaneous determination of L- and D-LA. The proposed method was applied to the simultaneous determination of L- and D-LA in the saliva of diabetic patients and healthy volunteers. The ratio of D-LA to L-LA in diabetic saliva was significantly higher than that in the saliva of the healthy persons. Although the saliva sampling is very easy, the transport of the sample to the analytical facility may be complicated for the general public. This paper proposes the dried saliva spot (DSS) as a convenient sampling technique for bioanalysis. The DL-LA in the DSS was labeled with a chiral
reagent (DMT-3(S)-Apy) for carboxylic acids and determined by UPLC-ESI-MS/MS. The limits of detection (S/N=3) for the DSS analysis were in on an amol level (approximately 30 amol). Because a good stability, recovery, accuracy and precision of the DL-LA for the DSS method was also obtained from the proposed procedure, the DSS method was applied to the determination of the D- and L-isomers of LA of diabetic patients, and prediabetic and healthy persons. The D/L-LA ratio by the present DSS method and the HbA1c value in blood were well correlated to the diabetic states.References: T. Takayama, T. Kuwabara, T. Maeda, I.
Noge, Y. Kitagawa, K. Inoue, K. Todoroki, J.Z. Min, T. Toyo’oka, Anal. Bioanal. Chem., 407, 1003-1014 (2015).
Disclosure of Interest:None DeclaredKeywords: Diabetes mellitus, DL-Lactic acid, Dried saliva spot
P03-003-011 - Simultaneous determination of nifedipine and lidocaine in human plasma by HPLC-MS/MSA. Nikitina*, A. Grigoriev, A. Sidorova
Content: Nifedipine is a calcium channel blocker and can be used in treatment of hypertension, angina pectoris; lidocaine is a local anesthetic. The combination of these drugs in ointment can be used as anti-haemorrhoidal medicine. The method of the simultaneous determination of nifedipine and lidocaine was developed, validated and successfully applied to a pharmacokinetic trial on 12 healthy volunteers. The nifedipine photodestruction was one of

www.ISC2016.ie @isc2016 #isc2016 /isc2016284
POSTER ABSTRACTS Molecular Diagnostics, Clinical and Biomedical Analysis
the main challenge in the development of this study. All the actions with nifedipine solutions and plasma samples were performed under sodium lamp illumination or in the darkness (using aluminum foil). The advantage of the reported method is rapid one step sample preparation for both nifedipine and lidocaine. The analytes were extracted from human plasma by liquid-liquid extraction using methyl tert-butyl ether with recoveries 90 ± 5% for nifedipine, 78 ± 3% for lidocaine. Chromatographic separation of the analytes was performed on an YMC-Triart C18 HPLC column (100×2.0 mm; S5 µm 12 nm). The mobile phase was methanol:water, 60:40, (v/v), and contained 0.15% acetic acid. The multiply reaction monitoring (MRM) experiments were performed in a positive ion mode with fragmentation transitions m/z 347.1→315.1 for nifedipine, m/z 235.2→86.1 for lidocaine. The LLOQ for nifedipine and lidocaine were 0.5 and 1 ng/mL, respectively. The linearty of the method has been validated in the concentration ranges of 0.5 to 50 ng/mL for nifedipine and 1.0 to 500 ng/mL for lidocaine. The reported method was validated according EMA requirements [1], tested on real plasma samples and can be applied for clinical research.References: [1] Guideline on Bioanalytical Method
Validation. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). 21 July 2011.
Disclosure of Interest:None DeclaredKeywords: HPLC-MS/MS, Lidocaine, Nifedipine
P03-003-012 - Development and validation of HPLC-UV method for determination of A77 1726 in plasma of patients with rheumatoid arthritis.
T. Pawinski*, I. Szlaska 1, M. Lobacz 1, B. Kwiatkowska 2
1 Department of Drug Chemistry, Medical University of Warsaw,
2 National Institute of Geriatrics, Rheumatology and Rehabilitation, Warsaw, Poland
Content: Background: Leflunomide (commercial name Arava) is leading drug for the treatment of rheumatoid arthritis. After oral administration, leflunomide is rapidly converted into its active metabolite A77 1726. Pharmacokinetics of A77 1726 significantly differs in particular patients. Moreover, there is a high incidence of adverse events during therapy [1].
Objective: The aim of this study was to develop a rapid and sensitive HPLC-UV method for determination of A77 1726 in human plasma.
Methods: Sample preparation based on a simple protein precipitation procedure. 0.5 mL plasma sample containing A77 1726 and clorazepate dipotassium salt as an internal standard were deproteinized by addition of 0.05 mL 0.2M ZnSO4 and 0.25 mL acetonitrile. After thorough mixing and shaking for 1 min , the samples were centrifuged for 5 min ( 3000 rpm) at 5oC. 20 µl of each samples were injected into Shimadzu HPLC system. Isocratic HPLC analysis was achieved on Nova-Pak C18 (150 mm × 3.9 mm ; 4 µm) column. The mobile phase was consisted of acetonitrile and 1/15M phosphate buffer adjusted to

285www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMolecular Diagnostics, Clinical and Biomedical Analysis
pH 3.5 (33:67 v/v) and was delivered at a flow rate 1.0 mL/min. Spectrophotometric detection was performed at the wavelength of 293 nm [2].
Results: All validation parameters met the acceptance criteria. The calibration curve was linear within the range of 0.5 – 100 µg/mL with a correlation coefficient 0.9985. The within-run and between-run accuracy ranged 89.7 – 112.4 % and 87.2 – 109.4 %, respectively, while the within-run and between-run precision ranged 1.25-6.1% and 1.6 – 7.2 %, respectively.
Conclusions: The HPLC-UV method for determination of A77 1726 can be recommended in further studies on pharmacokinetics of leflunomide. The method might be applied in therapeutic drug monitoring of A77 1726 during treatment of patients with rheumatoid arthritis.References: 1. E. van Roon et al., Ann Rheum Dis, 64, 569-574
(2005)2. V. Chan et al., Journal of Chromatography B, 803,
331-335 (2004)Disclosure of Interest:None DeclaredKeywords: A77 1726, HPLC-UV, leflunomide
P03-003-013 - The Analysis of C3-Epimers of 25-Hydroxyvitamin D in Serum by LC/MS/MS
T. W. Kahler, H. Majer 1,*, F. Carroll 1, S.-H. Liang 1, S. Lupo1 Restek Corporation, Bellefonte, United States
Content: Vitamin D analysis has increased dramatically in clinical practice due to its association with multiple human diseases
and the prevalence of vitamin D deficiency worldwide. Vitamin D exists in two forms, vitamin D2 and vitamin D3; each undergoes metabolism to form 25-hydroxyvitamin D2 [25(OH)D2] and 25-hydroxyvitamin D3 [25(OH)D3] which are used as the biomarkers for the assessment of vitamin D status. The epimeric forms of 25(OH)D, 3-epi-25-hydroxyvitamin D2 and D3, have been identified and may contribute to a large portion of the total 25(OH)D concentration, particularly in infant populations. Studies have shown that the C3 epimers have much lower bioactivity than the primary metabolites; therefore, a specific quantitation of these epimers is necessary for a proper clinical assessment of vitamin D status. Since these epimers are isobaric, chromatographic separation is necessary for accurate quantitation. In this study, the Raptor™ FluoroPhenyl column was used for chromatographic separation of 25(OH)D and their C3-epimers. The established chromatographic method was able to accurately quantitate the 3-epi-25-hydroxyvitamin D2 and D3 metabolites in fortified beagle serum.
It was demonstrated that the Raptor™ FluoroPhenyl column can provide unique selectivity for accurate and differential quantitation of 25-hydroxyvitamin D and C3-epimers in serum. The chromatographic analysis was performed using 0.1% formic acid in water and methanol as mobile phases with a 7-minute analysis time. The analytical method is applicable to the clinical analysis of total 25-hydorxyvitamin D concentration and provides the option to report the C3-epimer concentrations separately. References: D. Bailey, et al., Analytical
Measurement and Clinical Relevance of Vitamin D3 C3-epimer, Clinical Biochemistry, Vol 46, Issue

www.ISC2016.ie @isc2016 #isc2016 /isc2016286
POSTER ABSTRACTS Molecular Diagnostics, Clinical and Biomedical Analysis
3, Pg. 190, Feb 2013Disclosure of Interest:None DeclaredKeywords: Epimer, FluoroPhenyl, Vitamin D
P03-003-014 - A Multi-Class Drug and Metabolite Screen of 231 Analytes by LC-MS/MS
T. W. Kahler, H. Majer 1,*, F. Carroll 1, S.-H. Liang 1, S. Lupo1 Restek Corporation, Bellefonte, United States
Content: The use of pain management drugs has been steadily increasing. As a result, labs are seeing an increase in patient samples that must be screened for a wide variety of drugs to prevent drug abuse and to ensure patient safety and adherence to their medication regiment. Therapeutic drug monitoring can be challenging due to the low cut-off levels, potential matrix interferences and isobaric drug compounds. To address these challenges, many drug testing facilities are turning to liquid chromatography coupled with mass spectrometry (LC-MS/MS) for its increased speed, sensitivity, and specificity. In this example, a method was developed for a multi-class drug and metabolite screen containing 231 compounds.
There are many challenges one must consider when developing a large screening assay. Experiments performed included mobile phase considerations, sample diluent, isomer resolution, drug interferences, and instrumentation capabilities. During mobile phase investigations, it was found that methanol provided the best retention of early eluting analytes, such as morphine, oxymorphone, nicotine and norcotinine. Sample diluent was optimized to improve the peak shape of the early eluting
compounds. Scan rates and retention time windows were optimized to insure enough data points per peak were collected. In this example, a method was developed for a multi-class drug and metabolite screen containing 231 compounds.References: J. Jones, S. Mogali, S. Comer, Polydrug abuse: a
review of opioid and benzodiazepine combination use, Drug Alcohol Depend, 125 (1-2) (2012) 8-18
U. Garg, Chromatography in therapeutic drug monitoring of nonnarcotic analgesics and anti-inflammatory drugs, in: A. Dasgupta (Ed.), Advances in chromatographic techniques for therapeutic drug monitoring, Taylor & Francis Group, Boca Raton, 2010, 385-396.
Disclosure of Interest:None DeclaredKeywords: drug monitoring, Multi-class drugs, screening methods
P03-003-015 - Application of TLC with densitometry for pterins analysis
A. Waligóra 1,*, K. Tyrpień-Goldder 1, S. Waligóra 1
1 School of Medicine with the Division of Dentistry in Zabrze, Medical University of Silesia, Zabrze, Poland
Content: Pterins are nitrogen heterocyclic compounds characterized by 2-amino-4-hydroxypyridine core. They occur in human body fluids e.g. serum, urine and play an important biological function. Among them neopterin (NEO) and biopterin (BIO) are formed during the metabolic pathway of 5,6,7,8- tetrahydrobiopterin (BH4). Any changes in the mechanism of BH4 metabolism disrupt synthesis of neopterin and biopterin leading to various diseases. Therefore, measuring the amount of NEO and BIO has currently diagnostic, monitoring and prognostic value. The determination of pterins is complicated and challenging

287www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMolecular Diagnostics, Clinical and Biomedical Analysis
due to their physicochemical properties. That is the reason, why pterins are usually determined by immunoassay. This work shows how successfully determine both neopterin and biopterin as well as their reduced forms by two-dimensional thin layer chromatography with densitometry, after their separation from urine by solid phase extraction (SPE).References: 1. P Kośliński, P Jarzemski,
M Markuszewski, R Kaliszan, J Pharm Biomed Anal, 91 (37-45) (2014)
2. A Jimenez Giron, E Martin Tornero, MC Hurtado Sanchez, I Duran Meras, A Espinosa-Mansilla, Talanta, 101 (465-472) (2012)
3. F Canada-Canada, A Espinosa-Mansilla, A Munoz de la Pena, A Mancha de Llanos, Anal Chim Acta, 648 (113-122) (2009)
4. A Mancha de Llanos, A Espinosa-Mansilla, F Canada-Canada, A Munoz de la Pena, J Sep Sci 34 (1283-1292) (2011)
5. E Martin Tornero, I Duran Meras, A Espinosa-Mansilla, Talanta, 128 (319-326) (2014)
6. R Dobson, Mult Scler Relat Disord 1 (76-80) (2012)
7. L Yanchun, H Zhidong, J Med Coll PLA 26 (45-51) (2011)
8. R Sucher , K Schroecksnadel, G Weiss , R Magreiter , D Fuchs, G Brandacher, Cancer Lett 287 (13-22) (2010)
9 H Tomsikova, P Solich, L Novakova, J Pharmaceutical Biomed 95 (265-272) (2014)
10. M Ribeiro de Castro , G Seno di Marco, DY Arita, LC Texeira, AB Pereira, DE Casarini, J Biochem Biophys Methods 59 (275-283) (2004)
Acknowledgements: These result were supported by KNW-2-012/N/5/N.Disclosure of Interest:None DeclaredKeywords: biopterin, neopterin, TLC-densitometry
P03-003-016 - Diagnostic efficacy of HPLC-ED measurements of plasma free metanephrines and methoxytyramine for detection of pheochromocytomas and paragangliomas
D. Nieć 1,*, P. K. Kunicki 1, M. Pęczkowska 2, A. Prejbisz 2, A. Januszewicz 2
1 Department of Medical Biology, 2 Department of Hypertension, Institute of Cardiology,
Warsaw, Poland
Content: Aim: The aim of the study was to assess measurements of plasma free normetanephrine (NMN), metanephrine (MN) and methoxytyramine (MTY) by high performance liquid chromatography with electrochemical detection (HPLC-ED) as a biochemical assay for detection of catecholamine-producing tumors: pheochromocytomas and paragangliomas (PPGL).
Patients and methods: The study population consisted of 464 patients (217/247, M/F) admitted on a routine basis to the Institute of Cardiology in Warsaw including 30 patients with confirmed PPGL. All the patients underwent biochemical testing using HPLC-ED measurements of free NMN, MN and MTY in plasma. Following solid phase extraction, separation and detection of the analytes of interest was achieved on an HPLC column (C18, 150x4.6 mm, 3 µm, 100 Å) and coulometric electrodes [1]. The method was assessed in terms of diagnostic sensitivity and specificity, an area under the receiver operating characteristic (ROC) curve and positive and negative predictive values (PPV and NPV).

www.ISC2016.ie @isc2016 #isc2016 /isc2016288
POSTER ABSTRACTS Molecular Diagnostics, Clinical and Biomedical Analysis
Results: Among all patients, levels of NMN, MN and MTY ranged: 15.2-10305.6, 10.9-2231.7 and 10.3-3031.9 pg/ml, respectively. Median concentrations of NMN, MN, and MTY were, accordingly, 11.4, 2.1 and 1.6-fold higher for patients with than without PPGL. The assay was characterized by diagnostic sensitivity of 100% and diagnostic specificity of 95.8%. The area under the ROC curve was 0.981. The NPV was at 100% irrespective of the pretest probability of PPGL. The PPV was 62.5%, however, when the concentration of any of the analytes of interest was more than two times the respective upper cut-off of reference interval, the PPV was 100% at all pretest probabilities of the disease.
Conclusion: HPLC-ED measurements of plasma free NMN, MN and MTY proved to be an efficient biochemical assay in diagnosing of PPGL, allowing the detection of all diseased patients in the studied population.References: 1. D. Nieć, PK. Kunicki. J Chromatogr
B Analyt Technol Biomed Life Sci. 1002 (63-70) (2015)
Disclosure of Interest:None DeclaredKeywords: diagnostic efficacy, HPLC-ED, plasma free metanephrines and methoxytyramine
P03-003-017 - Internal standarization used in HPLC-UV method for MPA determination in plasma – is it always needed and beneficial ?P. K. Kunicki 1,*, A. Wróbel 1, E. Kowalska 1
1 Clinical Pharmacology Unit, Department of Medical Biology, Institute of Cardiology, Warsaw, Poland
Content: The aim of the work was to compare mycophenolic acid (MPA) plasma concentrations calculated with or without
internal standard (IS, fenbufen) on both: MPA spiked drug-free plasma and samples from heart transplant patients.
MPA concentration was measured by a validated HPLC-UV method based on protein precipitation with acetonitrile. A total number of 1202 patients samples as well as 45 low (~1.2 µg/mL) and 45 high (~25.0 µg/mL) MPA spiked samples were evaluated. MPA concentration results were calculated using two equations: one involving IS, and the other one based only on MPA peak area measurement.
The chromatograms of 15 patients samples (1.25%) revealing significant interference with IS were excluded from further analyses. For remaining 1187 patients samples, MPA concentration calculated using IS ranged: 0.16-9.49; mean: 1.99±1.24 µg/mL, whereas calculated without IS ranged: 0.16-9.54; mean: 1.98±1.22 µg/mL. Bland-Altman test revealed mean bias of -0.011 µg/mL (95% CI: -0.017; -0.005) comprising -0.14% (95% CI: -0.39; +0.11). Passing-Bablok regression was: y=0.986x+0.014 (95% CI for slope: 0.981; 0.992 and for intercept: +0.007; +0.021). Regardless of very close (r2=0.993, p<0.0001) correlation, the statistical analyses (Wilcoxon test, p=0.0154) confirmed the significant difference between both ways of calculating MPA results. For spiked ones, MPA concentrations calculated using IS were: 1.23±0.07 and 25.24±1.05 µg/mL; whereas calculated without IS were: 1.21±0.05 and 25.14±0.69 µg/mL providing higher variability when using IS (RSD of 5.91 vs 4.55 % and 4.18 vs 2.73 %, for low and high MPA level, respectively).
Statistically significant difference between the measurements calculated using both

289www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMolecular Diagnostics, Clinical and Biomedical Analysis
algorithms is clinically irrelevant when related to MPA therapeutic range. Our HPLC method provided reliable results without the necessity of using IS. Moreover, for a few patients samples, the IS chosen for the method was useless because of interference observed.References: noneDisclosure of Interest:None DeclaredKeywords: HPLC-UV, internal standard, mycophenolic acid
P03-003-018 - Chlorinated decarboxy-betanins and their chromatographic properties
G. Andryianau 1,*, S. Wybraniec 1, K. Starzak 1, G. Rotko 1, Z. Pietrzkowski 2
1 Faculty of Engineering and Chemical Technology, Cracow University of Technology, Cracow, Poland,
2 Applied BioClinical Labs., Irvine, CA, United States
Content: Betacyanins and decarboxylated betacyanins are glucosylated derivatives of the betanidin & decarboxylated betanidin chromophore, respectively. All the compounds belong to betalains, water-soluble plant pigments commonly used as food colorants.
Recently, the first identification of chlorinated betacyanins that can be generated by in vivo chlorination of the ingested pigments as a result of inflammatory processes has been reported [1]. It explains preliminarily a possible function of betalains in inhibiting the effects of chlorination in inflamed tissues and increasing the comfort of joints in individuals as had been observed previously [2, 3].
Strong antioxidative activity as well as low toxicity point betalains as a promising class of compounds for use in health-protection applications. In addition, they can presumably serve as indicators of hypochlorite in inflammed tissues because their chlorination can be confirmed by the applied analytical methods.
A betalain chlorination study was carried out in the Cl2/HClO/ClO- system and the chlorination products were identified by LC-DAD-ESI-MS/MS. The characteristic isotopic profiles confirm the presence of one chlorine atom per molecule. Fragmentation experiments resulted a subtraction of glucose unit and mono-chloro-decarboxy-betanidin/isobetanidin ions formation confirming that the chlorination takes place at the aglycone unit.
The experiments indicate that not only betacyanins but also decarboxylated betacyanins are potent scavengers of hypochlorous acid, a powerful oxidant produced by human neutrophils. Such scavengers may provide protective activity against hypochlorous acid-induced inflammatory processes.References: [1] S. Wybraniec, K. Starzak, Z. Pietrzkowski, J.
Agric. Food Chem., 2016, 64, 2865−2874[2] Z. Pietrzkowski, B. Nemzer, T. Michałowski, S.
Wybraniec, New Med., 2010, 1, 12−17[3] Z. Pietrzkowski, R. Argumedo, C. Shu, B. Nemzer,
S. Wybraniec, T. Reyes-Izquierdo, Nutr. Diet. Suppl., 2014, 6, 9−13
Disclosure of Interest:None DeclaredKeywords: chlorination, decarboxy-betanins, inflammation

www.ISC2016.ie @isc2016 #isc2016 /isc2016290
POSTER ABSTRACTS Molecular Diagnostics, Clinical and Biomedical Analysis
P03-003-019 - High speed LC-MS/MS determination of multiple steroids using the Nexera MX system for multiplex analysisP. Jochems*, A. Grüning 1
1Shimadzu Europa GmbH, Duisburg, Germany
Content: Steroids are crucial for normal biological activity in the human body. Steroid analysis has also had important impact on the research of disorders such as congenital adrenal hyperplasia (CAH), Cushing’s disease and polycystic ovarian disease.
Traditional methods to measure steroids have been immunoassays, however, these assays lack specificity, are affected by matrix interferences and could take up to a week to be completed. Recently, liquid chromatography mass spectrometry (LC-MS/MS) has become the industry standard in evaluating steroids due to the specificity, precision and sensitivity of the system. A highly sensitive analysis method for simultaneous determination of 15 steroids by UHPLC-MS/MS was developed to support research or clinical studies.
Since high sample throughput is of utmost importance this method was applied to the Shimadzu Nexera MX multiplex LCMS system to reduce duration of the whole analysis time. With its Dual Stream Technology (MX-DST) it uses two separate analysis systems to perform overlapping sample injections. Therefore it is possible to either inject two batches of samples at the same time or to inject one batch of samples using the two separate streams of the Nexera MX system. This study also considers the influence on the results when using the two
separate streams for one batch compared to the use of only one stream for one batch.References: NoneDisclosure of Interest:None DeclaredKeywords: LC-MS/MS, multiplexing, Steroid hormones
P03-003-020 - Fast Analysis of six Insulin Variants and Analogs using a Fused Core Particle HPLC Column with UV and MS Detection Spectral Detection
A. Fridström 1,*, H. Brandes 2, C. Muraco 3, S. Shollenberger 3
1 Sigma-Aldrich GMBH now part of Merck, St Gallen, Switzerland,
2 Analytical Biomacromolecule Separations, 3MilliporeSigma, a subsidiary of Merck KGaA, Darmstadt, Germany, Bellefonte, United States
Content: Purpose Insulin is an approximately 6000 Da peptide hormone used to regulate the glucose concentration in blood. There are several different forms of insulin that are both natural and recombinant; the recombinant forms have been engineered to improve pharmacokinetic properties. A quick and reliable chromatographic technique was developed to identify and detect a mixture of insulin variants in an unknown sample.
Methods: A mixture of six insulin variants (insulin-bovine, insulin-human, insulin-porcine, insulin-lispro, insulin-Asp, and insulin-glargine, 100 µg/mL, 0.1% aqueous trifluoroacetic acid) was analyzed chromatographically on a BIOshell™ A160 Peptide C18 HPLC column using UV and MS Detection. All analyses were performed

291www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSMolecular Diagnostics, Clinical and Biomedical Analysis
in triplicate and the elution order of the insulin variants was confirmed by mass spectral analysis and injections of reference standards.
Conclusion: Baseline resolution of the six insulin variants was achieved by reversed-phase HPLC on a (BIOshell™) A160 Peptide C18 column. The analytical method was optimized for quick and efficient analyses, which is a benefit for larger, pharmaceutical quality control labs that are examining insulin formulations. References: 1. Kirkland, J.J., US Patent 3,505,785. April 14, 1970. 2. Schuster, S.A., Wagner, B.M., Boyes, B.E.,
Kirkland, J.J. J Chrom Sci 48 (566 - 571) (2010).Disclosure of Interest:None DeclaredKeywords: Fast U/HPLC Analysis, Insulin, Peptide Hormone
P03-003-021 - Quantification of 24R,25-dihydroxyvitamin D3, 25-hydroxyvitamin D3 and its 3-epimer, and 25-hydroxyvitamin D2 in serum by an LC-MS/MS method using LLE
K. G. Dowling*, K. D. Cashman 1 2
1 Food and Nutritional Sciences, 2Department of Medicine, University College Cork, Cork, Ireland
Content: Serum concentrations of 25-hydroxyvitamin D (25(OH)D) are considered the best indication of vitamin D status. Two minor vitamin D metabolites are common interferences encountered in 25(OH)D assays. The first is 3-epi-25-hydroxyvitamin D3 (3-epi-25(OH)D3), which if not chromatographically resolved from 25-hydroxyvitamin D3 (25(OH)D3),
can overestimate 25(OH)D concentrations in LC-MS/MS assays. The second is 24R,25-dihydroxyvitamin D3 (24R,25(OH)2D3), which can cross-react with 25(OH)D immunoassays.(1) Our aim was to develop an LC-MS method capable of detecting 24R,25(OH)2D3 in serum without the use of a derivatisation agent.(2) We report an isotope dilution LC-MS/MS method, with electrospray ionization in the positive mode, which can simultaneously detect 24R,25(OH)2D3, 25(OH)D3, 3-epi-25(OH)D3, and 25-hydroxyvitamin D2. The method employs a cost effective liquid-liquid extraction (LLE) using only 150 µL of sera and a total run time of 10 minutes. Accuracy of the method was assessed by measuring NIST Standard Reference Materials 2973 and 972a, Level 1-4 in triplicate and comparing to assigned target values. Pooled sera at three different concentration levels of 25(OH)D3 were used to assess method precision. Sera previously analysed by immunoassay were re-analysed by our method and compared.
References: 1 Carter GD, Jones JC, Shannon J, Williams EL,
Jones G, Kaufmann M, et al. 25-Hydroxyvitamin D assays: Potential interference from other circulating vitamin D metabolites. J Steroid Biochem Mol Biol. 2015 Dec 21. PubMed PMID: 26718874. Epub 2016/01/01. Eng.
2. Muller MJ, Volmer DA. Mass spectrometric profiling of vitamin D metabolites beyond 25-hydroxyvitamin D. Clin Chem. 2015 Aug;61(8):1033-48. PubMed PMID: 26130585.
Acknowledgements: Supported by funding from the European Commission: ODIN (Food-based solutions for optimal vitamin D nutrition and health through the life cycle; Contract code 613977).Disclosure of Interest:None DeclaredKeywords: LC-MS/MS, Vitamin D metabolites
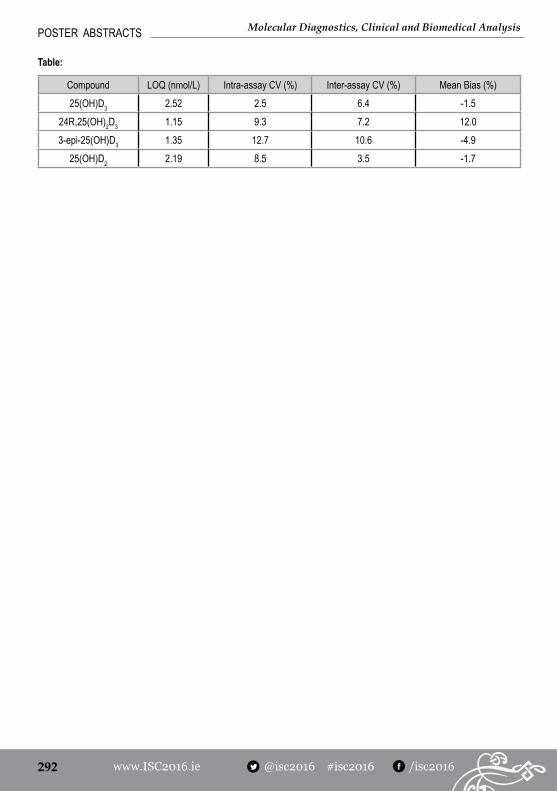
www.ISC2016.ie @isc2016 #isc2016 /isc2016292
POSTER ABSTRACTS Molecular Diagnostics, Clinical and Biomedical Analysis
Table:
Compound LOQ (nmol/L) Intra-assay CV (%) Inter-assay CV (%) Mean Bias (%)
25(OH)D3
2.52 2.5 6.4 -1.5
24R,25(OH)2D
31.15 9.3 7.2 12.0
3-epi-25(OH)D3
1.35 12.7 10.6 -4.9
25(OH)D2
2.19 8.5 3.5 -1.7

293www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSNew Instrumentations and Multidimensional Separations
New Instrumentations and Multidimensional Separations
P01-001-006 - Offline 2D LC with hybrid high resolution mass spectrometry for phospholipid analysis in lipidomic studies: heart cutting vs comprehensive
T. H. Chappuis 1,*, A. Solgadi 2, J. Vial 1, P. Sassiat 1, A. Combès 1, P. Chaminade 3, D. Thiébaut 1
1 Analytical and Bioanalytical Sciences and Miniaturization Laboratory, UMR CNRS CBI 8231,, ESPCI Paris, PSL Research University, Paris,
2 UMS IPSIT, 3 Lip(Sys)²- Chimie Analytique Pharmaceutique (FKA
EA4041 Groupe de Chimie Analytique de Paris-Sud), Université Paris-Sud, Université Paris-Saclay, Châtenay-Malabry, France
Content: We used offline bidimensional liquid chromatography with hybrid high resolution mass spectrometry to study phospholipids in Pecten Maximus. To bring orthogonality to the separation system, normal phase liquid chromatography in the first dimension was coupled to reversed phase liquid chromatography in the second dimension The first dimension allowed for the separation of 10 phospholipid classes out of which three, phosphatidic acid, phosphatidic ethanolamine, and phosphatidic choline, were studied in detail in the second dimension. Solvent incompatibility between the two dimensions was resolved by evaporating the first dimension mobile phase and resolubilizing the extract in the second dimension mobile phase with good repeatability (RSD < 10 %). This layout was applied to standards and to a scallop extract (Pecten Maximus) with an heart cutting coupling and allowed for the separation of hundreds of molecular species of phosphatidic acid, phosphatidic
ethanolamine, and phosphatidic choline. Hybrid high resolution mass spectrometry provided elementary composition and structural information using data dependent MSn experiments. A higher sampling rate, i.e. a comprehensive coupling, induced higher performances in peak detection and structure determination of PL molecular species with up to 27 % more observed peaks and 50 % more determined structures. This was allowed by a simplification in the obtained chromatograms in the second dimension originating from a higher separation power with a comprehensive approach. The higher separation power allowed for a higher ionization yield and a simplification of the complexity of the samples entering the MS. It explains why we were able to detect very low concentrated PLs, to distinguish phospholipids differentiated by only the position of double bonds on a fatty acid, and to describe novel unknown phospholipid structuresDisclosure of Interest:None DeclaredKeywords: Hybrid High resolution mass spectrometry, Lipidomic, Multidimensionnal liquid chromatography
P01-001-035 - Characterising the Response Behaviour of the Barrier-Ionisation Discharge Detector for Gas Chromatography (GC-BID)E. Rosenberg 1,*, M. Antoniadou 1 2, G. A. Zachariadis 2
1 Institute of Chemical Technologies and Analytics, Vienna University of Technology, Vienna, Austria,
2 Department of Chemistry, Laboratory of Analytical Chemistry, Aristotle University Thessaloniki, Thessaloniki, Greece
Content: The dielectric-barrier-discharge plasma has already been used for long for

www.ISC2016.ie @isc2016 #isc2016 /isc2016294
POSTER ABSTRACTS New Instrumentations and Multidimensional Separations
spectroscopy [1], but only relatively recently as a gas chromatographic detector. In the commercial instrument, the dielectric barrier discharge serves as an ionization source that produces a concentration-proportional electron current, similarly to the GC-FID. The acronym GC-BID (GC with dielectric barrier discharge-ionization detection) coined for this system [2] emphasizes the analogy to the flame ionization detector (FID).
In this study, the performance of the BID as a GC detector was characterized and compared to GC-FID. Interesting characteristics and differences compared to the GC-FID system were found: While it is claimed that the BID’s sensitivity is equal to or exceeds that of the FID, it was demonstrated that this not necessarily is the case, but depends largely on the operation conditions (e.g. detector gas flow rate and purity), and to an even higher extent on the analyte.
This is because in contrast to the FID, where practically complete atomization and oxidation of the analyte is achieved, the BID is a low-thermal energy excitation and ionization source that does not lead to a complete destruction of the analyte molecules. Instead, singly charged molecular ions are formed, leading to a concentration- rather than a mass-dependent response. As a consequence, the advantage of superior sensitivity compared with the FID is quickly lost when the analyte’s molecular weight increases. This and other important characteristics of the BID as GC detector will be presented and discussed in detail, providing for the first time a comprehensive characterization and an objective comparison of this detector with the FID.
References: 1. Y.-L. Yu, Y.-T. Zhuang, J.-H. Wang, Anal. Methods,
2015, 7, 1660-1666.2. K. Shinada, S. Horiike, S. Uchiyama, R. Takechi,
T. Nishimoto, Shimadzu Review SR13_001E (2013) 5pp.
Acknowledgements: Shimadzu Austria is greatfully acknowledged for the support of this study by the loan of a GC-BID system.Disclosure of Interest:None DeclaredKeywords: barrier ionisation discharge detector, gas chromatographic detectors, response factor
P01-001-045 - Manipulating Chromatographic Selectivity Using Automated Control of Mobile Phase pHT. E. Wheat, A. B. Dlugasch 1, P. R. McConville 1,*
1 Waters, Milford, United States
Content: Chromatographic resolution is best maximized by manipulations that change the selectivity of the separation conditions. For reversed-phase methods, numerous manipulations can affect selectivity, including for example, strong solvent, bonded phase, temperature, as well as other parameters. While mobile phase pH is recognized as a powerful tool for altering selectivity, particularly for ioniozable analytes, this parameter is seldom used in separation development protocols since the preparation of buffered mobile phases is time consuming and labor-intensive. In common practice, only widely separated pH conditions are used to maximize retention. For groups of related compounds, however, small changes in pH can be more effective because the analytes behave as though partially charged. We describe here the automated screening

295www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSNew Instrumentations and Multidimensional Separations
of this separation variable in a software controlled system. A quarternary solvent pump is used with specific algorithms to permit programming directly in units of pH and buffer concentration. Buffered mobile phases are prepared on demand from concentrated stocks using the solvent proportioning capabilities of the UPLC pump. A series of small incremental changes in pH can be evaluated to identify the point of maximum selectivity. As expected, the effective pH ranges center around the pK’s of the analyte molecules. The approach is particularly useful for structurally related molecules that differ slightly in pK. In the present study, an alternative protocol using continuous gradients of pH has been tested. The software embodying the control algorithms can be used to generate gradients of pH. Buffer systems have been developed for the reversed phase application that give stable, predictable, and reproducible gradients over the required pH range while maintaining compatibility with MS detection. The utility of this approach will be shown with several sample mixtures representing pharmaceuticals, foods, and industrial chemicals.Disclosure of Interest:None DeclaredKeywords: Method development optimisation, Mobile Phase pH, Selectivity
P01-001-046 - Optimizing the Combination of Multi-Segment, Multi-Dimensional Separations with Photodiode Array and Mass Spectrometry Detection
A. B. Dlugasch 1,*, T. E. Wheat 1, P. R. McConville 1
1 Waters, Milford, United States
Content: Multi-dimensional separations can provide a solution to the fundamental limits on chromatographic resolution, particularly when the separation steps use orthogonal mechanisms for selectivity. This approach can be particularly powerful when combined with information-rich detection. The detection becomes more useful when the multiple detectors respond to different properties of the analytes. For the system to provide the greatest resolution and information, both the chromatographic steps and the detection must be chemically compatible and must not impose time constraints. We describe an instrument configuration to approach these objectives. The system includes two UPLC gradient pumps, and a pair of valves that are used to divert one or more segments of a first dimension chromatogram to a holding facility, either a chromatographic trap or an empty loop. The system includes the capacity to hold multiple segments before beginning the second stage of analysis. Each trap cartridge is eluted in turn onto a second column and into the detectors. In this arrangement, maximum resolution is possible in both dimensions because the cycles and timing of the two separations are independent. At-column dilution is available at each chromatographic step to establish compatibility between the successive chromatographic modes and to

www.ISC2016.ie @isc2016 #isc2016 /isc2016296
POSTER ABSTRACTS New Instrumentations and Multidimensional Separations
ensure optimum detectability. The system function is illustrated with separation of small molecules and with monoclonal antibodies. The small molecules were separated using low pH reversed-phase followed by high pH reversed-phase with a set of C18 trap cartridges as the holding facility. The monoclonal antibodies were separated using ion exchange followed by SEC. Between dimensions, the protein peaks were isolated in lengths of empty tubing to ensure good recovery of the proteins. This system reliably combines multiple modes of chromatography and detection.Disclosure of Interest:None DeclaredKeywords: Multi-Dimensional Chromatography, Orthogonal Detection, Proteins
P03-003-023 - Analysis of Volatile Defensive Secretions of True Bugs by Two-Dimensional Gas Chromatography-Mass SpectrometryM. Havlíková 1,*, J. Krajicek 1, Z. Bosakova 1, R. Cabala 1
1 Department of Analytical Chemistry, Charles University in Prague, Prague 2, Czech Republic
Content: All the insects are potential prey for some predator, and during the evolution many defense strategies of insects have been developed. The true bugs (Hemiptera: Heteroptera) use a multimodal protection against their predators. The most widely used is combination of optical and chemical signaling [1]. True bugs have very well-developed scent glands which produce about hundred diverse and partly strongly smelling compounds when the bug is irritated by their predators.
A new non-lethal method has been developed for defense secretion sampling. Samples were then analysed by gas chromatography with mass spectrometric detection (GC-MS). For the irritation of the bugs was used the pressing the bugs with the plunger of a syringe and the preconcentration step was performed by sorption on a solid phase microextraction (SPME). The composite divinylbenzene/carboxene/polydimethylsiloxane (DVB/CAR/PDMS) fiber was shown to be the most suitable for SPME sorption. The main advantage of the developed method is that it requires only a few individuals who remain alive, and their secretion is gathered comprehensively. Because the secretion is very complex mixture of compounds, it is a problem to determinate all macro, as well as micro components by simple GC-MS analysis. Therefore, new GCxGC method has been developed using commercial cryogenic modulator (ZOEX, USA) [2]. For the first dimension nonpolar SLB-5ms column was used whereas polar column Supelcowax 10 was applied for the second dimension. The newly developed method was tested on analysis of secretions produced by adult males and females and also larvae of true bugs (Graphosoma lineatum etc.).References: [1] C. Rowe, C. Halpin, Behav. Ecol. Sociobiol. 67
(1425-1439) (2013)[2] M. Adahchour, J. Beens, R.J.J. Vreuls,
U.A.T. Brinkman, Trends in Analytical Chemistry 25 (540-553) (2006)
Acknowledgements: The study was financially supported by the Charles University in Prague, project GA UK No 760216.Disclosure of Interest:None DeclaredKeywords: solid phase micro-extraction, true bugs, two-dimensional gas chromatography

297www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSNew Instrumentations and Multidimensional Separations
P03-003-024 - Making Analytically Incompatible Approaches Compatible
F. T. Van Beek 1 2,*, B. Pirok 1 2, B. Wouters 1 2, T. O’Sullivan 3, W. Genuit 4, R. A. Peters 1 5, M. Blom 6, P. J. Schoenmakers 1
1 Analytical Chemistry Group, University of Amsterdam, 2 TI-COAST, Amsterdam, 3 Global Innovation and Research, Heineken, Zoeterwoude, 4 PTD/ TASE, Shell Global Solutions, Amsterdam, 5Coating
Resins, DSM, Waalwijk, 6 Micronit Microfluidics, Enschede, Netherlands
Content: Comprehensive two-dimensional liquid chromatography (LC×LC) is an essential technique for the separation of highly complex samples. Successful coupling of LC systems for comprehensive analysis requires a modulator, which transfers an analyte solution in the first-dimension solvent to the second-dimension. The modulator links two processes with different time scales and should be small and repeatedly useful [1]. These overall characteristics make LC×LC modulators suitable for coupling many other seemingly incompatible devices. The potential of modulators can be further enhanced by allowing fast, controlled chemical reactions to take place. A modulation reactor may contain immobilized enzymes, microbes or target materials under investigation. Combining modulators and modulation reactors yields a myriad of possibilities.
The research project “Making Analytically Incompatible Approaches Compatible” (MAnIAC) aims for a generic approach to couple two vast “orthogonal” domains, i.e. chemistry (chemical reactions and processes) and physics (modern methods of instrumental analysis). Our strategy is based on the use of physical modulators and
chemical modulation reactors, which can be coupled to separation techniques (liquid chromatography, gas chromatography, etc) and/or mass spectrometry (including high-resolution mass spectrometry and ion-mobility spectrometry), as well as to other detection methods, such as NMR.
The processes to be studied include the degradation of various types of natural and synthetic particles (such as milled grains, synthetic resins) and complex molecules, such as lignins, as well as microbe metabolism in relation to the beer-brewing using a microbial modulation reactor. The key objective of the project is creating a generic MAnIAC toolbox useful for various industrial applications.References: 1. Georges Guiochon, et al., Journal of
Chromatography A (190-168)(2008).Disclosure of Interest: None DeclaredKeywords: LCxLC, MAnIAC, Modulation reactor
P03-003-025 - QbD Based Method Development on 1290 Infinity II instrument through Empower 3 Software combined with a seamless method transfer to HPLC using ISET
V. Azhakaprakalam 1, S. Krieger 2,*
1 Agilent Technologies, Bangalore, India, 2 Agilent Technologies, Waldbronn , Germany
Content: Quality by Design (QbD) based approach incorporates robustness to the method during method development. Transferring a method from UHPLC to HPLC conditions without compromising the Critical Method Attributes (CMAs) is challenging. A method which has been developed on a UHPLC system even on

www.ISC2016.ie @isc2016 #isc2016 /isc2016298
POSTER ABSTRACTS New Instrumentations and Multidimensional Separations
conventional columns may not provide the same resolution after its transfer to a HPLC system due to differences in the systems delay volumes and gradient mixing behaviors. To overcome these issues Intelligent System Emulation Technology in combination with QbD Method Development and Validation Software were used to transfer UHPLC methods to HPLC methods within the QbD principles. Empower 3 software was used to control 1290 Infinity II instrument using Instrument control framework (ICF). Screening of sub 2 µm columns, organic solvents and pH were performed. The best conditions will be further optimized to achieve a robust UHPLC method and design space will be generated. Method will be transferred to an HPLC method using Method Translator. The 1290 Infinity system, a UHPLC system will be operated as HPLC (1260 System) using ISET emulation mode and final HPLC method will be verified. The best answer after screening and optimization in UHPLC system will be generated. The final robust method will be transferred to HPLC method and emulated as 1260 infinity method. The CMA performance requirements should have met in all conditions. The robust HPLC method was replicated on a 1260 infinity system. The RSDs of API Area, RT and resolution should be within the acceptance limit.References: 1. Vinayak AK, Syed Salman Lateef, Automated
QbD Based method development and validation of oxidative degraded atorvastatin, publication number : 5991-4944EN, 2014
2. Vinayak AK, QbD based method development on an Agilent 1290 infinity UHPLC System Combined with a Seamless Method Transfer to HPLC Using Intelligent System Emulation technology, publication number : 5991-5701EN, 2015
Disclosure of Interest: None DeclaredKeywords: Method Transfer, QbD, UHPLC Method developent
P03-003-026 - Sequential injection chromatography as a tool for automation of on-line hyphenated extraction and chromatographic methods
P. Chocholouš 1,*, L. Zelená 1, P. Svoboda 1, D. Šatínský 1, H. Sklenářová 1, P. Solich 1
1 Department of Analytical chemistry, Faculty of Pharmacy in Hradec Králové, Charles University in Prague, Hradec Králové, Czech Republic
Content: Sequential Injection Chromatography (SIC) belongs among newer and quickly developing liquid chromatography methods. The pioneering work of the SIC technique was presented in 2003 by Šatínský et al. [1]. The SIC with flexible manifold setup enables to handle liquids in various modes and bidirectional flow.
Different approaches are used for development of fully automated sample analysis based on on-line two-dimensional method (SPE and liquid chromatography). The SPE step is performed by various disposable Lab-On-Valve Bead Injection or reusable MEPS or lab packed column. The chromatographic step is performed by short (30 or 50 mm) commercial monolithic or fused-core particle packed column. The SIC enables to perform all the steps of sample handling, sample pre-treatment, on-line transfer of the sample from SPE column to chromatographic column and separation. Based on the analytes and sample matrix both columns formats and

299www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSNew Instrumentations and Multidimensional Separations
both columns chemistries are selected – for mutual compatibility, selectivity, and high performance analysis. Up to date 2D SIC methods were used for determination of pharmaceuticals in environmental samples (water) and biological samples (urine) [2,3].
Further development led into a new automated on-line method with the broaden potential. Key characteristics of the SIC method compared with column switching methods developed on the HPLC will be discussed.References: 1. D. Šatínský, P. Solich, P. Chocholouš, R. Karlíček,
Anal. Chim. Acta 499 (2003) 205–214.2. A.D. Batista, P. Chocholouš, D. Šatínský, P.
Solich, F.R.P. Rocha, Talanta 133 (2015) 142-149.3. I. Šrámková, P. Chocholouš, H. Sklenářová, D.
Šatínský, Talanta 143 (2015), 132-137.Acknowledgements: The authors gratefully acknowledge the financial support of the Ministry of Education of the Czech Republic, project no. SVV 260292, Grant Agency of the Czech Republic, project no. 15-10781S/P206.Disclosure of Interest: None DeclaredKeywords: on-line hyphenation of extraction and separation, sequential injection chromatography
P03-003-027 - Small ScalePurification of Constituents from Complex Natural Products
R. Cleary*, J.-A. Jablonski
Content: Extracts from natural product samples can be complex, often containing a large number of diverse compounds. Increased separation performance of sub 2um column chemistries provide a tool that produces sharp, narrow and
more concentrated peaks. When there is a need to collect narrow peaks from thee complex mixtures, traditional preparative HPLC fraction collection instrumentation is not suitable for the collection of such narrow peaks. This poster will discuss the fundamentals and practice of small volume fraction collection, which are required to provide optimal operation with small particle chemistries.References: Tim S Bungi, Journal o Natural
Producs, J. Nat. Prod., 2008, 71 (6), pp 1095–1098
Disclosure of Interest: None DeclaredKeywords: Isolation, Purification
P03-003-028 - UV imaging detection with 10 and 25 micron ID capillaries for CE, nanoLC and open tubular liquid chromatographyD. Goodall 1,*, T. Allen 1, A. Chapman 1
1Paraytec Ltd, York, United Kingdom
Content: This poster presents results obtained with an ActiPix D200 system (Paraytec, UK) demonstrating the versatility of UV imaging detection with narrow bore columns in capillary electrophoresis, nanoLC and open tubular chromatography. Whilst detection path length across columns with ID down to 10 µm is low, addition of time displaced snapshots allows peaks to be monitored during travel across the full 11 mm length without sacrificing the 5.5 µm spatial resolution of the imager.
Our new generation detector can be run with USB3 data output at frame rates of 36 fps with 5.5 µm resolution for the whole 11x11 mm chip, ideal for monitoring sharp

www.ISC2016.ie @isc2016 #isc2016 /isc2016300
POSTER ABSTRACTS New Instrumentations and Multidimensional Separations
features. For single capillary applications as used here, the narrower area imaged allows for increase in frame rate. Multi-wavelength imaging is achieved using up to 4 light sources pulsed in sequence (typically xenon lamp/filter at 190 or 214 nm, LEDs at 255, 280 and 525 nm). The measured RMS noise is 7 µAU (1x16 binning / 100 fps / 1 s time constant / 255 nm / 75 µm ID capillary).
In CE, Joule heating effects are small using narrow bore columns, allowing high peak efficiencies to be realised. We will show CE separation of choride isotopes, 35Cl- and 37Cl-, using direct detection at 190 nm in a 25 µm ID/ 150 µm OD capillary. Such work has only previously been possible using indirect detection [1]. In a second application, separation of serum proteins in a 25/150 capillary gives peaks with far higher efficiency than previously described [2].
Using a cylindrical focusing lens, results for 10 micron ID capillaries show detection of peptides and proteins in post-column transfer lines for nanoLC with satisfactory sensitivity without loss of peak quality. Work is also being carried out in open tubular and PLOT columns [3] with imaging multiple windows along the column length.References: 1. C.A. Lucy & T.L. McDonald, Anal. Chem., 67, 1074
(1995).2. C. Chartier et al., Clin. Biochem., 44, 1473 (2011).3. Q. Liu et al., Anal. Chem., 79, 6174 (2007). Acknowledgements: Disclosure of Interest: None DeclaredKeywords: Capillary electrophoresis, Nano-liquid chromatography, UV absorption
P03-003-029 - Analysis of Lignin Oil by two-Dimensional Comprehensive Gas Chromatography by comparison of a non-Polar/Polar and a Polar/non-Polar column set.
J. H. Marsman*, E. Heeres 1
1 Chemical Engineering, University Groningen, Groningen, Netherlands
Content: Lignin is a solid product from biomass (wood) and is generated during paper production. By hydro-deoxygenation (hydrogenation at elevated temperature) of lignin the liquid lignin oil is formed.
The analysis of this complex multicomponent lignin oil was performed by 2-D comprehensive Gas Chromatography (GCxGC) by two different column sets.
For the GCxGC analysis a non-polar/polar and a polar/non-polar column system was tested to find out the best profiles for characterization of the various classes of chemicals.
Test mixtures with different types (functional group) of chemicals were used for classification purposes.
The results of separation efficiency and identification op components on both column systems will be shown. Finally the both column sets will be evaluated to choose the most appropriate column set for this application.

301www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSNew Instrumentations and Multidimensional Separations
Table 1: Column specifications
No. Column Stationary phaseGeometric dimension:
Length(m)xID.(mm) x filmthickness (df = µm)
1 Rxi- 5Sil MS Aryl-PDMS /Phenyl 95/5 30 x 0.25 x 0.25
2 Rtx-1701 PDMS-cyanopropyl-phenyl 86/14 30 x 0.25 x 0.25
* PDMS= poly dimethylsiloxane;
References: M. Windt, D. Meier, J.H. Marsman, H. J.Heeres, S. de Koning J. of Anal. Appl. Pyrol. 85 (2009) 38.
1. J.H. Marsman, J. Wildschut, P. Evers, S. de Koning, H.J. Heeres, J. Chromatogr. A 1188 (2008) 17.
2. T. Sfetsas, C. Michailof, A. Lappas, O. Li, B. Kneale, J. Chromatogr. A 1218 (2011) 3317.
Disclosure of Interest: None Declared
P03-003-030 - Longitudinal thermal modulation in online two-dimensional liquid chromatography
M. J. Egeness 1,*, J. P. Foley 1 2, R. A. Shellie 1 3, H. J. Cortes 4, E. F. Hilder 5, M. C. Breadmore 1
1 ACROSS, University of Tasmania, Hobart, Australia, 2 Department of Chemistry, Drexel University, Philadelphia,
United States, 3 Trajan Scientific and Medical, Ringwood, Australia, 4 HJ Cortes Consulting, LLC, Midland, United States, 5 Future Industries Institute, University of South Australia,
Adelaide, Australia
Content: We have developed a longitudinally modulated resistively heated modulator for LC x LC. The modulator relies on a trap column packed with porous graphitic carbon to retain solutes eluted from the first dimension separation column. Focused bands are mobilized into the second dimension separation column by rapidly and substantially changing the partition coefficient of retained solutes using high temperature. To this end, a resistively heated sleeve is fitted over the trap column
and moved along the column length by aid of a mechanically actuated arm. The longitudinally resistively heated modulator presented achieves LC x LC separations without recourse to switching valves. References: (1) B., Gu; H., Cortes; J., Luong; M.,
Pursch; P., Eckerle; R., Mustacich. Anal. Chem.(1488–1495) (2009)
(2) P.J., Marriott; R.M., Kinghorn. Anal. Chem. (2582-2588) (1997)
(3) P.J., Marriott; R.M., Kinghorn. J.High Res.Chromatogr. (620-622) (1998)
(4) P.J., Marriott; R.M., Kinghorn. J.High Res.Chromatogr. (235-238) (1999)
Disclosure of Interest: None DeclaredKeywords: Thermal modulation, Two-dimensional liquid chromatography

www.ISC2016.ie @isc2016 #isc2016 /isc2016302
POSTER ABSTRACTS Pharmaceutical Analysis
Pharmaceutical Analysis
P03-003-031 - Exploitation of chiral uHPLC using chiral mobile phase additives: achiral/chiral assay and related substances for brompheniramine and chlorpheniramine
D. Meston*, W. J. Lough 1
1 Sunderland Pharmacy School, The University of Sunderland, Sunderland, United Kingdom
Content: Despite the advantages of chiral uHPLC, it will be a while before it is readily accessible with a wide range of chiral stationary phases (CSP) commercially available as in conventional chiral HPLC1. However, it has been found that a convenient route in to chiral uHPLC is to use achiral uHPLC columns with chiral mobile phase additives (CMPA). Despite apprehension regarding slower mass transfer that may be involved in chiral interactions, it was found that the beneficial flat van Deemter C term characteristic of uHPLC was obtained and CMPA consumption was not the issue it is in conventional scale LC. While this makes uHPLC with CMPA viable, the question remained, as to whether it could easily be applied to typical pharmaceutical R&D analytical problems.
Using β-cyclodextrin (β–CD) as the CMPA and a 100 mm x 2.1 mm I.D. column containing 1.9 µm silica particles (Hypersil Gold) it was possible to not only resolve the enantiomers of brompheniramine from each other but also from related substances, chlorpheniramine and pheniramine. Further, similar separations could be achieved for the much more challenging example of chlorpheniramine enantiomers and related
substances. Critically, it was possible to baseline resolve chlorpheniramine enantiomers from those of its BP impurity C in a systematic fashion, fine tuning via [CMPA] with eventual mobile phase [acetonitrile – (water - triethylammonium acetate - acetic acid (100:0.89:0.21, v/v/v) (10:90, v/v)] containing 6.81 mg ml-1 β-CD.
These examples demonstrated clearly the practicality of CMPA as an alternative to CSP for chiral uHPLC. Given the low CMPA consumption and the potential for systematic adjustment of achiral as well as chiral selectivity, perhaps CMPA use in uHPLC may not just be a stopgap until the full development of CSP for uHPLC, but have a promising long term future, irrespective of incompatibility with MS.References: 1. W.J.Lough, Chapter 7 in (2012). «Drug
Stereochemistry: Analytical Methods and Pharmacology pp 95-122.
Disclosure of Interest: None DeclaredKeywords: chiral mobile phase additives, enantiomers and related substances, uHPLC
P03-003-032 - Development of a New UHPLC Method for Separation of Eight Phytosterols using UV/CAD detectionJ. Fibigr 1,*, I. Lhotská 1, D. Šatínský 1, P. Solich 1
1 Department of Analytical Chemistry, Faculty of Pharmacy in Hradec Králové, Charles University in Prague, Hradec Králové, Czech Republic
Content: Plant stanols and sterols, generally known as phytosterols, are natural steroid compounds with ability of decreasing the low density lipoprotein cholesterol [1]. Structures of phytosterols vary only

303www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
in carbon side chains and in presence or absence of a double bond, which make their separation quite complicated. In this study a new ultra-high performance liquid chromatography (UHPLC) method for separation of eight phytosterols (ergosterol, brassicasterol, fucosterol, campesterol, stigmasterol, β-sitosterol, campestanol, and stigmastanol) was developed using fused-core column Ascentis Express Phenyl-hexyl (100 × 2.1 mm), particle size 2.7 µm, with mobile phase acetonitrile/pure water according to the gradient program at a flow rate of 1.0 mL min−1 and at temperature 50 °C. The phytosterols have very low response in UV spectrum provided by photodiode array (PDA) detector, even at limiting wavelength 190 nm, and stigmastanol has no response at all, due to the lack of chromophore in its structure. That was verified by comparison of chromatograms acquired from PDA detector and from Charged Aerosol detector (Corona) during analysis of standard solution. For these reasons a Charged Aerosol detector was chosen to obtain better chromatogram of separation of all peaks. Detection of any nonvolatile or semivolatile analyte with or without a chromophore, gradient compatibility and very good sensitivity are main advantages of this detector. The Charged Aerosol detector was operated with nitrogen of purity 99.99% at 40.0 psi gas flow. A 2-µL volume of standard solution, which was prepared by dissolving standards of all analysed phytosterols in 100% methanol with use of ultrasonic bath, was directly injected into the UHPLC system.References: [1] C.T. Srigley, E.A. Haile, Quantification of plant
sterols/stanols in foods and dietary supplements containing added phytosterols, J. Food Compos. Anal. 40 (2015) 163–176.
Acknowledgements: The authors are grateful to the Charles University, grant project no. GAUK 181216 and they would like to acknowledge financial support of the project of specific research, project no. SVV 260 292.Disclosure of Interest: None DeclaredKeywords: Charged Aerosol detection, Phytosterols separation, UHPLC
P03-003-033 - Analytical study of biologically active rhenium complexes with aromatic alcohol ligandsD. Kaliba 1,*, I. Jelínek 1, M. Štícha 2
1 Department of Analytical Chemistry, 2 Department of Chemistry, Charles University in Prague,
Prague 2, Czech Republic
Content: Nuclear medicine is based on application of the source of ionizing radiation into the patient body. Technetium 99mTc is the most widely used radiopharmacum in the present nuclear medicine. Close resemblance of rhenium (186Re and 188Re) and technetium is evident from their positions in periodic table. Physical properties of complexes with the same ligand (size, lipophilicity etc.) are usually close. However, technetium and rhenium analogues possess several chemical differences; therefore, the behavior of Re complexes cannot be simply predicted on the basis of known technetium analogues.
Rhenium complexes with aromatic alcohol ligands 1,2-dihydroxybenzene and 1,2,3-trihydroxybenzene were prepared by modified procedure. Depending on actual experimental conditions (addition of triethylamine as a base, heating on oil bath), different rhenium complexes were obtained as major reaction products. The structures of prepared complexes and their intermediates

www.ISC2016.ie @isc2016 #isc2016 /isc2016304
POSTER ABSTRACTS Pharmaceutical Analysis
were estimated by ESI-MS [1]. Quantitative data describing the composition of complex forming reaction mixture (necessary for future medical application) were obtained by HPLC/UV and CZE/UV [2,3].
All components of a reaction mixture containing major bis(1,2,3-trihydroxybenzene)oxorhenium were successfully separated by HPLC/UV (column Zorbax SAX); mobile phase acetonitrile:ammonium formiate (80:20, pH=7.47). Capillary zone electrophoresis was carried out in nonmodified silica capillary (50 cm total length, 50 µm I.D.) with UV/Vis detection. Aqueous borate and phosphate buffers with different pH values as well as nonaqueous buffers containing acetonitrile and perchloric or acetic acids were successfully used as BGE. References: [1] M.Sticha, I.Jelinek, J.Polakova, D.Kaliba,
Analytical Letters 48 (2329-2342) (2015)[2] M.Koudelkova, V.Jedinakova-Krizova, J Chrom A
990 (317-323) (2003)[3] M.Kohlickova-Koudelkova, V.Jedinakova-Krizova,
Z.Deyl, Electrophoresis 23 (245-248) (2002)Acknowledgements: This research was carried out within the framework of the project of the Specific University Research (SVV 260317).Disclosure of Interest: None DeclaredKeywords: antitumor agents, Capillary Zone Electrophoresis, mass spectrometry
P03-003-034 - Sensitive Analysis of Esters of Aryl Sulfonic Acids using Mass and UV Detection in Genotoxic Impurities MonitoringM. Maziarz 1, M. Wrona 1, C. Henry 1,*, S. M. McCarthy 1
1Waters Corporation, Milford, United States
Content: Sulfonic acids such as methanesulfonic acid (mesylate), benzenesulfonic acids (besylates) and p-toluenesulfonic acid (tosylates) are commonly used as counter ions to form Active Pharmaceutical Ingredients (API) salts and can interact with residual alcohols to generate alkyl esters, which are considered potential genotoxic impurities1. The genotoxic compounds have the potential to react with DNA, consequently produce a carcinogenic response and tumor. It is therefore essential to have reliable and highly sensitive methods for their accurate determination in both drug substance and drug product.
In this work, we present the development of a robust and quick dual detection UPLC method for the analysis of esters of benzenesulfonic and p-toluenesulfonic acids. We will utilize both the UV and mass detection with an ACQUITY QDa detector for fast and accurate monitoring of genotoxic impurities. During the study, we will evaluate the impact of different parameters on the chromatographic separation of the sample components. In addition to the chromatographic parameters, we will investigate the effect of different factors that can affect the quality of MS data and sensitivity of the MS method. These factors include mobile phase, sample diluent and acquisition parameters of the MS detector. Then, we will demonstrate the

305www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
linearity, sensitivity and specificity of the method achievable with both UV and mass detection. To show that the analytes can be accurately measured in the presence of the sample matrix, we will spike the amlodipine besylate API with the analytes and calculate recovery. Overall, we will show that by employing mass detection we improve sensitivity and specificity of the method, which is essential for quick and accurate determination of low level genotoxic impurities in pharmaceutical samples.References: 1. Liu D.Q., Sun M., Kord A.S., Recent Advances
in Trance Analysis of Pharmaceutical Genetoxic Analysis, Journal of Pharmaceutical and Biomedical Analysis, 2010, 999-1014
Acknowledgements: Margaret Maziarz, Mark Wrona, Sean M. McCarthy, Janet HamondDisclosure of Interest: None DeclaredKeywords: potential genotoxic impurities, UPLC, mass detection
P03-003-035 - Design and development of new class of water compatible molecularly imprinted solid phase sorbents for the extraction of carbamazepine scaffolds
P. Kadhirvel*, A. Combes 1, V. Pichon 1
1 LSABM, ESPCI, Paris, France
Content: Carbamazepine (CBZ) is an anticonvulsant and mood-stabilizing drug primarily used in the treatment of epilepsy. It is one of the most frequently detected pharmaceutical residues in water supplies so far. The estimated global consumption of CBZ is exceeding 1000 tons and trace
amounts up to a concentration of 610 ng L-1
have been found in groundwater, emanating from its poor elimination by wastewater treatment plants.
Molecularly imprinted polymerization (MIP) is a versatile approach successfully employed to selectively capture a single molecule of interest in complex matrices, even at trace levels1. One of the most intriguing aspects of MIP is the unique possibility to obtain a predetermined selectivity, which is developed during the imprinting process (polymerization) in presence of a “print molecule”. Subsequent removal of the “print molecule” from the polymer matrix creates the confined 3-dimensional highly selective binding cavities, which are complementary in shape and to its functional groups of the “print molecule”.
Research articles, published so far, regarding the successful and exclusive targeting of CBZ through MIPs are based on the implementation of carbamazepine as a “print molecule”. Nonetheless, these MIPs suffer from CBZ leakage during analysis because of the struggle based in the complete removal of CBZ after polymerization, thus, complicating its quantification.
Herein, we aim to synthesize MIPs based on the carbamazepine structural analogues to avoid leakage and with the aim of simplifying the quantification process. Through rational choice of new classes of functional monomers and cross linkers, both employed synergistically to obtain highly selective MIPs for binding carbamazepine scaffolds including some of its metabolites and through further investigation of appropriate solvents in the polymerization

www.ISC2016.ie @isc2016 #isc2016 /isc2016306
POSTER ABSTRACTS Pharmaceutical Analysis
process, granting usage of these MIPs in aqueous media.References: 1. George V, Lars I A, Ralf M, Klaus M, Letters to
Nature, 361 (645-647) (1993).Disclosure of Interest: None DeclaredKeywords: carbamazepine, imprinted sorbents,
P03-003-036 - Controlled cycloporine A release from poly(D,L-lactic acid) nanofibers with poly(ethylene glycol) of various molecular weight studied by HPLC- MS/MS
Z. Hampejsova 1,*, Z. Bosáková 1, J. Širc 2, P. Kozlík 1
1 Department of Analytical Chemistry, Charles University in Prague, Prague 2, 2Institute of Macromolecular Chemistry of the Academy of Sciences, Prague 6, Czech Republic
Content: Cyclosporine A (CsA) is an immunosuppressive drug used to prevent the suppression of immune reaction after organ transplantations. Its local delivery (via biocompatible nanofibers) has a potential to reduce a number of dose-dependent side effects. It was shown that properties of drug-loaded nanofibers and drug release profile can be modified by varying composition of a polymer solution for nanofibers preparation; especially by addition of amphiphilic polymer poly(ethylene glycol) (PEG). [1,2]
Poly(D,L-lactic acid) nanofibers with incorporated immunosuppressant CsA and with addition of amphiphilic poly(ethylene glycol) (PEG) were prepared by needle-less electrospinning technology. Four types of nanofibrous carriers varying in the presence and molecular weight of PEG (6, 20 and 35 kDa) were extracted for 7 days in phosphate
buffered saline at 37 °C. Release kinetics of CsA were studied by newly developed and validated HPLC-MS/MS method based on Phenyl-Hexyl stationary phase in the combination with mobile phase containing the mixture of methanol and 1mM ammonium acetate, pH 4.5.
Nanofibers containing PEGs showed significantly higher released amounts of CsA compared to the nanofibers containing pure CsA. The release times were also extended - CsA was releasing even after 144 hours of the experiment and the release profiles, obtained for nanofibers with PEGs, were reproducible and well-balanced. CsA released amounts increased and release profiles prolonged with decreasing molecular weight of the added PEGs.
The obtained results indicate that the addition of PEG with particular molecular weight can be promising way to control CsA release from nanofibrous carriers for various medical applications.References: 1. J. Hrib, Belstein Journal of Nanotechnology,
(1939-1945) (2015)2. T. Steele, Acta Biomaterilia, (1973-1983) (2011)Acknowledgements: This work was supported by the Charles University in Prague (project no. 307115 and project SVV), by the Ministry of Education, Youth and Sports of CR within the National Sustainability Program II (Project BIOCEV-FAR) and by the project ‘‘BIOCEV’’ (CZ.1.05/1.1.00/02.0109).Disclosure of Interest: None DeclaredKeywords: cyclosporine A, HPLC-MS/MS, poly(D,L-lactic acid) nanofibers

307www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
P03-003-037 - Chiral Separation of 2-Hydroxyglutaric Acid on Cinchonan Carbamate based Weak Chiral Anion Exchangers by HPLC-Charged Aerosol Detection
C. Calderón 1,*, J. Horak 1, M. Laemmerhofer 1
1 Institute of Pharmaceutical Sciences, Eberhard Karls Universitaet Tuebingen, Tuebingen, Germany
Content: D- and L-2-Hydroxyglutaric acid (D- and L-2-HG, respectively) are metabolites which are present in the human body in relative low concentrations. Abnormal higher concentrations of these metabolites have been linked to some diseases like D- and L-2-Hydroxyglutaric aciduria, gliomas, glioblastomas and acute myelogenous leukemia (AML), which make their identification and analysis crucially important for diagnostic purposes. Chiral stationary phases (CSP) based on tert-butylcarbamoyl-quinine and -quinidine (Chiralpak QN-AX and QD-AX), and the corresponding zwitterionic derivatives (Chiralpak ZWIX(+) and Chiralpak ZWIX(-)) were employed in a weak anion-exchange mechanism to perform the enantiomer separation of D- and L-2-HG without derivatization.
Most promising separation was obtained with QD-AX CSP and therefore optimization of eluent, flow rate, additives, and temperature, required for the baseline separation of solutes was performed. A baseline separation with a resolution value of 2.0 was possible with run time < 20 min and MS-compatible conditions. Employment of the pseudo-enantiomeric QN-AX CSP allowed the inversion of the elution order of D- and L-2-HG, which represents an important advantage when
the minor enantiomer must be analysed in the presence of the major one.References: 1. Kranendijk, M.; Struys, E. A.; Salomons, G. S.;
Van der Knaap, M. S.; Jakobs, C. J. Inherit. Metab. Dis. 2012, 35 (4), 571–587.
2. Dang, L.; White, D. W.; Gross, S.; et al. Nature 2009, 462 (7274), 739–744.
3. Cheng, Q.-Y.; Xiong, J.; Huang, W.; Ma, Q.; Ci, W.; Feng, Y.-Q.; Yuan, B.-F. Sci. Rep. 2015, 5 (October), 15217.
4. Ianni, F.; Pataj, Z.; Gross, H.; Sardella, R.; Natalini, B.; Lindner, W.; Lämmerhofer, M. J. Chromatogr. A 2014, 1363, 101–108.
5. Lämmerhofer, M.; Lindner, W. Adv. Chromatogr. 2008, 46, 1–107.
6. Franco, P.; Zhang, T.; Gargano, A.; Mahut, M.; Lammerhofer, M.; Lindner, W.; Points, K. E. Y. LC GC Eur. 2012, 25 (11), 600–610.
Acknowledgements: We are grateful for financial support by the “Struktur- und Innovationsfonds für die Forschung (SI-BW)” by the regional government of Baden-Württemberg. C.C. gratefully acknowledges financial support from the German Academic Exchange Programme (DAAD).Disclosure of Interest: None DeclaredKeywords: 2-Hydroxyglutaric acid, Chiral ion-exchangers, Cinchona alkaloid
P03-003-038 - Increased Efficiency of Method Scouting Procedure Using SFC/HPLC Switching System for Chiral Drug Separation
Y. Watabe 1,*, Y. Hayakawa 1, S.-I. Kawano 1, T. Hattori 1, H. Terada 1, T. Uchikata 1, Y. Funada 1
1Shimadzu Corporation, Kyoto, Japan
Content: Nowadays most commercial low molecular drugs are chiral compounds. One

www.ISC2016.ie @isc2016 #isc2016 /isc2016308
POSTER ABSTRACTS Pharmaceutical Analysis
of the most expectant application fields for super critical fluid chromatography (SFC) is chiral separation of low molecular weight drugs that have stereoisomers. In chiral separation, the first and important step to establish analytical condition is “column scouting” because there is no universal stationary phase like the C18 in the case of reversed phase chromatography. Newly designed SFC system can go with column scouting device that can equip up to twelve columns and it affords increased productivity with dedicated supporting software. On the other hand, A configuration with additional single solvent delivery pump to SFC affords HPLC/SFC switching function within a single instrument and the switching can be performed automatically even in a single batch analysis using dedicated controlling software. Here, we report ultra-high efficiency and flexibility of real method development including column scouting for omeprazole, one of chiral drugs for gastric acid inhibitor, in a single sequence.References: T. Uchikata, et al, J. Chromatogr.A 1250
(69-75) (2012)Disclosure of Interest: None DeclaredKeywords: Chiral drug, method scouting, SFC
P03-003-039 - Development of matrix effect-free MISPE-UHPLC-MS/MS method for determination of lovastatin in food samplesP. Svoboda 1,*, L. Zelená 1, L. Matysová 1, L. Nováková 1
1Department of Analytical Chemistry, Faculty of Pharmacy in Hradec Králové, Charles University in Prague, Hradec Králové, Czech Republic
Content: Lovastatin represents the first marketed cholesterol-lowering drug
which mechanism of action is based on inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase. This statin molecule is produced by several fungal species that were isolated from food samples such as Pu-erh tea, Red yeast rice and Oyster mushroom. Statins occur in lactone (prodrug) and open hydroxy-acid (active) form1. Interconversion between them is observed both in vivo and in vitro. Therefore, both forms were monitored simultaneously during method development and also lovastatin determination.
The analyte of interest was determined using UHPLC-MS/MS method. Food samples such as mushrooms, tea leaves and fermented rice represent complex matrices that are prone to matrix effects. This undesirable phenomenon is mostly caused by components of the complex samples2. To get rid of these components a sample preparation step based on molecularly imprinted solid-phase extraction (MISPE) was employed. Molecularly imprinted polymers (MIPs) enable to retain the target analyte selectively, purify the complex matrices3 and thus to reduce or fully eliminate the matrix effects. MIP sorbent was synthesized using simvastatin as a template, methacrylic acid as a functional monomer and ethylene glycol dimethacrylate as a cross-linker. MIP material was fully characterized by experiments involving evaluation of the selectivity and repeatability of the MISPE procedure, repeatability of the synthesis of the MIP material, determination of the capacity and evaluation of the matrix effects.
The resulting matrix effect-free MISPE-UHPLC-MS/MS method was employed for determination of lovastatin in different mushroom samples, different kinds of Pu-erh teas and dietary supplements containing

309www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
red yeast rice extract.References: 1. D.-J. Yang, L. S. Hwang, J. Chromatogr. A 1119
(2006) 277-284.2. F. Gosetti, E. Mazzucco, D. Zampieri, M. C.
Gennaro, J. Chromatogr. A 1217 (2010) 3929-3937.
3. V. Pichon, J. Chromatogr. A 1152 (2007) 41-53. Acknowledgements: The authors gratefully acknowledge the financial support of the Charles University in Prague; GA UK project no. 274216 and SVV 260 292.Disclosure of Interest:None DeclaredKeywords: lovastatin, matrix effects, molecularly imprinted polymers
P03-003-040 - Rivaroxaban under hydrolytic stress: identification of degradation products by UHPLC-ESI-MS/MS and in vitro toxicity assessmentN. R. Wingert 1,*, M. D. Arbo 2, B. da Costa 2, L. F. Altknecht 2, S. C. Garcia 2, M. Steppe 1
1 Laboratory of Pharmaceutical Quality Control, 2 Laboratory of Toxicology (LATOX), Federal University of
Rio Grande do Sul, Porto Alegre, Brazil
Content: Evaluation of synthesis and degradation impurities is substantial for guarantee drug safety and successful pharmaceutical therapy. Rivaroxaban (RIV) is a long-term use oral anticoagulant. The aim of present work was to evaluate RIV behaviour under hydrolytic stress conditions in order to verify the formation of degradation products (DPs) and assess whether those compounds presented toxic effect to human cells. RIV was exposed to acid (1.0 M HCl) and alkaline (0.1 M NaOH) media up to 8 h, for each condition one main additional peak was detected on chromatograms. Analyses were performed
in UPLC coupled to quadrupole time-of-flight MS. ESI was applied in positive mode, and C18 column used for separation of compounds1. RIV molecular íon [M+H]+ (m/z 436.07) was fragmented under 20 kV, best energetic condition to obtain clear and reproducible fragmentation pattern. With support from UPLC separation and specific detection by MS, DP-1 and DP-2 were successfully elucidated as 5- Chloro- N- [[(5S) - 3- [4- [(2- hydroxyacetyl) (2- hydroxyethyl) amino] phenyl] - 2- oxo- 5- oxazolidinyl] methyl] -2- thiophenecarboxamide and 5- Chloro- N- [3- [[4- [(2- hydroxyacetyl) (2- hydroxyethyl) amino] phenyl] imino] propyl] -2- thiophenecarboxamide, respectively. In vitro tests were applied to assess toxic effect of RIV exposed to hydrolysis compared to RIV intact drug. MTT reduction and neutral red uptake assays were performed with human hepatoma HepG2 cell line. A direct relationship between increased amount of alkaline degradation product (DP-2) and higher cytotoxic potential was found. Elucidation of RIV DPs enables major access and knowledge regarding drug stability, increasing tools for quality control and safer treatments. Results obtained through viability assays support the concerns on risks associated with acute toxicity of pharmaceutical samples containing DPs as impurities.References: 1. N.R. Wingert, M.A.G et al. Curr. Anal.
Chem. 11 (124-129) (2015).Acknowledgements: PPGCF and UFRGS-Brazil; CAPES.Disclosure of Interest:None DeclaredKeywords: chemical elucidation, cytotoxicity, UPLC-MS/MS

www.ISC2016.ie @isc2016 #isc2016 /isc2016310
POSTER ABSTRACTS Pharmaceutical Analysis
P03-003-041 - Transferring a GPC method to a more efficient SEC method for Zoladex Co-polymer with Advanced Polymer Chromatography U(H)PLC system with RI detection
C. Henry*, J. Bowden 1, M. Wrona 2, A. Boughey 3, R. Ladd 4
1 Quality Control, AstraZeneca, Macclesfield, United Kingdom,
2 Waters Corporation, Milford, United States, 3 AstraZeneca, Macclesfield, 4 Waters Corporation, Wilmslow, United Kingdom
Content: We will present the workflow, optimization and steps towards validation of a compound which is routinely analysed by quality control departments using a registered Pharmacopeial Gel Permeation Chromatography (GPC) coupled with Refractive index RI detection method for molecular weight average.
GPC is a well established method for the characterization of polymers but has limitations associated with low resolution due to large particle size and long equilibration times due to the relatively unstable packing material in traditional GPC columns.
Within this body of work we present a faster, more efficient method for determination of the molecular weight average of the co-polymer.
The method was developed using a low dispersion Waters Advanced Polymer Chromatography (APC) system using sub 3µm Acquity APC columns reducing run time by over three-fold and improving resolution. Columns evaluated employed
hybrid silica technology which was found to greatly improve method robustness due to the rigid silica based packing’s lack of susceptibility to the shrinking/swelling inherent with traditional gel-based columns. The advantages of this technology are reduced equilibration time and more consistent chromatography.
We will also demonstrate efficiency savings in instrument time of approximately 70% with column equilibration time savings of over 80% while detailing the corresponding solvent usage savings using this method and technology.
This will be achieved whilst comfortably satisfying the existing system suitability criteria laid down in the GPC method currently used by Astra Zeneca.
The methodology developed provides a robust and reliable alternative to a challenging legacy GPC method.References: 1. USP General Chapter, <621> Chromatography,
USP36-NF31, The United States Pharmacopeia Convention, official December 1, 2013
2. Astra Zeneca standard operating method “Copolymer molecular weight and polydispersity”.Disclosure of Interest:None Declared
Keywords: Polymer analysis, QC, UHPLC

311www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
P03-003-042 - Optimization and speeding up a stability indicating HPLC method for an antibiotic suspension formula by application of Core-Shell stationary phases
M. I. Elian*
Content: The effectiveness of using core-shell stationary phases for acceleration of analysis with preservation of method performance criteria was demonstrated. This was illustrated as two steps for optimization and acceleration of a separation method for amoxicillin, sodium benzoate and saccharin sodium in a suspension dosage form. At first, the separation was optimized using a conventional Agilent HC C18(2) 150mm×4.6mm, 5µm column. Effects of methanol concentration, column temperature and pH of mobile phase buffer were evaluated regarding separation. Mobile phase buffer pH was found to have the most significant effect for bringing the retention times of all compounds in a practical range. Profiles of retention time as a function of pH enabled selection of a buffer the most optimum pH value. The optimized mobile phase consisted of methanol:sodium acetate buffer at pH 6.5; (5:95 v/v) at flow rate of 2ml/minutes. The separation was achieved within 8 minutes. At the second step, the stationary phase was downscaled to Kinetex C18, 50mm×3.0mm, 2.6µm column with core-shell particles on an optimized HPLC system for lower extra-column volume. The optimized method enabled separation within about 1.2 minute with a flow rate of 1 ml/min and UV detection wavelength of 215nm. The two methods were validated according to ICH Q2 (R1) guidelines regarding assay of active
ingredient and preservative. Selectivity regarding to degradation products and other formula ingredients was demonstrated by photodiode array detector. Robustness was confirmed for the two methods and factors affecting separation were identified for establishment of resolution solution acceptance criteria [1] for resolution between amoxicillin and sodium benzoate peaks and resolution between sodium benzoate and saccharin sodium peaks.References: 1. Y, Vander Heyden, A, Nijhuis, J,
Smeyers-Verbeke, B, Vandeginste & D. Massart, Journal of pharmaceutical and biomedical analysis, 24, (723-753) (2001).
Disclosure of Interest: None DeclaredKeywords: Core-Shell stationary phases, Method optimization, Robustness testing
P03-003-043 - Sulfobutylether-β-cyclodextrin as mobile phase additive for amino acids and dipeptides separationG. Kucerova 1,*, H. Prochazkova 1, K. Kalikova 1, E. Tesarova 1
1Department of Physical and Macromolecular Chemistry, Charles University in Prague, Prague 2, Czech Republic
Content: Cyclodextrins (CDs) still represents important tool for analysis of structurally different compounds. They are used either as chiral stationary phase (SP) or mobile phase (MP) additives [1-3]. In this work we focused on separation potential of sulfobutylether-β-CD (SBE-β-CD) used as MP additive. Analytes of interest were chosen with the respect to their nature. Our focus lies particularly in amino acids Tyr, Trp, Phe and their derivatives as well as dipeptides derived from the mentioned amino acids. Dipeptide isomers form

www.ISC2016.ie @isc2016 #isc2016 /isc2016312
POSTER ABSTRACTS Pharmaceutical Analysis
unique group of analytes examined in this study. Dipeptides used for mixtures have the same molecular formula but the sequence of amino acids constituting the dipeptide is reversed. Hereby, we show that SBE-β-CD could be also successfully used for separation of these compounds.
XTerra® RP C18 column (dimensions: 150 × 4.6 mm i.d.; silica particle size 5 µm) was used for measurements with SBE-β-CD present in MP. MPs composed of organic modifier (methanol) and three different aqueous parts: (i) aqueous solution of formic acid, pH 2.1, (ii) ammonium acetate buffer, pH 4.7, and (iii) ammonium acetate buffer, pH 8.8 in various volume ratios.
The following results were obtained. At first, this chromatographic system provided very low separation ability towards chiral analytes, just one compound was baseline resolved (DL-Trp butylester) and two partially (Gly-DL-Trp and Val-Tyr). On the other hand, situation was opposite when dipeptide isomers mixtures were employed. The percentage success of the RP C18 SP with SBE-β-CD as MP additive was more than 70%. Seven mixtures of dipeptide isomers were baseline separated (RS > 1.5). One mixture of dipeptide isomers was separated partially (RS = 1.38).References: [1] D.W. Armstrong, W. Demond, J. Chromatogr. Sci.
22 (411-415) (1984).[2] Y. Xiao, S.-C. Ng, T.T.Y. Tan, Y. Wang, J.
Chromatogr. A 1269 (52-68) (2012).[3] J. Zhou, J. Tang, W. Tang, Anal. Chem. 65 (22-29)
(2015).Acknowledgements: The authors gratefully acknowledge financial support of the Grant Agency of the Charles University, Grant No. 790314 and the Grant Agency of the Czech Republic, Grant No.
P206/14-19278P, and 16-05942S.Disclosure of Interest:None DeclaredKeywords: (enantio)separations, mobile phase additive, sulfobutylether-beta-cyclodextrin
P03-003-044 - Novel method for β-secretase kinetic and inhibition assays with unlabelled substrates using capillary electrophoresis
J. Schejbal 1,*, R. Řemínek 1, Z. Glatz 1
1 Department of Biochemistry, Faculty of Science, Masaryk University, Brno, Czech Republic
Content: Since its discovery in 1999, β-secretase 1 (BACE1) has drown much attention of both academia and pharmaceutical industry [1]. Its key role in amyloid plaque creation, the hallmark of Alzheimer’s disease, led to the amyloid hypothesis, which states that specific BACE1 inhibition would halt amyloid plaque production and consequently slow down or stop the progression of Alzheimer’s disease. Nowadays Förster resonance energy transfer (FRET) based assays are performed almost exclusively to study BACE1 kinetics and for inhibitor screening. Regrettably substrates for FRET assays are fluorescently labelled on both ends which leads to numerous drawback including low solubility, high variability of results depending on assay conditions and proneness to false positivity and negativity [2].
In our work we present novel method for BACE1 kinetic and inhibition assays using unlabelled decapeptide, which contains so called Swedish mutation of amyloid precursor protein, as substrate using capillary electrophoresis coupled with mass spectrometry. Thanks to low sample

313www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
consumption and high sensitivity the enzyme reaction can be carried out in as small volume as only 100 µl in just 30 min. These features together with deploying unlabelled substrate ensure low cost per analysis. What is more, our results show high difference between kinetic parameters of unlabelled substrate in comparison with kinetic parameters of fluorescently labelled substrates published in literature by two orders of magnitude even though the incubation conditions used were comparable. On the other hand determined inhibition parameters of two reported inhibitors show excellent agreement with literature data. These results were confirmed by comprehensive kinetic study of BACE1 carried out using number of detection techniques.References: [1] S.L. Cole, R. Vassar, Molecular neurodegeneration
2 (22-46) (2007)[2] F. Mancini, A. De Simone, V. Andrisano, Analytical
and bioanalytical chemistry 400 (1979-1996) (2011)Acknowledgements: This work was supported by grant No. GA16-06106S from the Czech Science Foundation.Disclosure of Interest:None DeclaredKeywords: Beta-secretase, Capillary electrophoresis, Enzyme assay
P03-003-046 - Investigation of various chromatographic approaches for resolution of foridone and its oxidized formT. Upmanis 1,*, A. Kreicberga 1, H. Kazoka 1
1 Latvian Institute of Organic Synthesis, Riga, Latvia
Content: A systematic search for new calcium antagonists among
1,4-dihydropyridines (DHPs), performed at the Latvian Institute of Organic Synthesis (Riga), resulted in the synthesis of 2,6-dimethyl-3,5-dimethoxycarbonyl-4 - ( 2 ’ - d i f l u o ro m e t h o x y p h e n y l ) - 1 , 4 -dihydropyridine known as foridone, which possesses antihypertensive and antianginal properties.
1,4-DHPs belong to the class of nitrogen-containing heterocycles having a 6-member ring. One of the typical reaction of the 1,4-DHP ring is aromatization to a pyridine ring. Therefore it is important to have analytical method for oxidized form control in 1,4-DHP pharmaceutical forms.
It is well known that reversed-phase (RP) high performance liquid chromatography is widely used method for separation and qualitative and quantitative detection of chemical compounds, especially in pharmacological studies. Unfortunately poor resolution of foridone and its oxidized form was observed on classical C18 column under RP mode [1].
In this study various chromatographic approaches has been investigated with the aim to improve resolution of foridone and its oxidized form. The first set of experiments was related to selectivity changes due to the stationary phase itself. Largest selectivity effect was found under RP mode when a classical stationary phase was compared to one with an embedded polar group. The mobile phase organic modifier (methanol vs acetonitrile) produced the second largest selectivity changes. Good resolution was achieved under normal-phase high performance liquid chromatography mode on bare silica and cyano stationary phases. In continuation the suitability of

www.ISC2016.ie @isc2016 #isc2016 /isc2016314
POSTER ABSTRACTS Pharmaceutical Analysis
supercritical fluid chromatography was also investigated.References: 1. I.A. Barene, V.V. Kastrone, V.G.
Mishneva, G.Ya. Duburs, Pharm.Chem.J. 31 (389-390) (1997)
Acknowledgements: The authors thank Laboratory of membrane active compounds and β-diketones of the Latvian Institute of Organic Synthesis for kind donation of foridone and its oxidized form samples, and a company Waters for the ACQUITY UPC2 demo-system. Disclosure of Interest:None Declared
P03-003-047 - Development of chiral separation strategies to analyze amino acids and small peptides
A. Ceuterick 1,*, Y. Vander Heyden 1, D. Mangelings 1
1 Department of Analytical Chemistry and Pharmaceutical Technology, Vrije Universiteit Brussel (VUB), Brussel, Belgium
Content: Because of their unique properties, peptides are interesting compounds for biopharmaceutical drug development. Their building blocks, natural amino acids, with the exception of glycine, possess one or two asymmetric centers, resulting in two or four stereoisomers. The enantiomeric and diastereomeric purities of peptides are important quality aspects since they influence the biological and chemical activity. Chiral method development is often a trial-and-error approach. Therefore, generic chiral separation strategies that rapidly give an idea about the enantioselectivity of a system in a limited number of experiments are useful for enantioselective method development. The goal of this project is to define generic chiral separation strategies applicable to amino acids and peptides with diverse
characteristics. Techniques such as capillary electrophoresis (CE), normal phase liquid chromatography (NPLC), reversed phase liquid chromatography (RPLC), polar organic solvents chromatography (POSC) and supercritical fluid chromatography (SFC) will be considered in this project. In a first study, the applicability of earlier defined chiral strategies for small molecules will be evaluated for their usefulness in peptide and amino acid analysis. These earlier defined strategies used in total nine polysaccharide-based chiral stationary phases for the NPLC, RPLC and POSC modes (1). In CE highly sulfated cyclodextrins were used as chiral selectors (2). In a next step, other polysaccharide CSP’s and other types of chiral selectors will be evaluated to be possibly included in an updated strategy. The same applies to other types of cyclodextrines and different selectors for CE.References: 1. AA. Younes, J. Pharm. Biomed. Anal., 75 (74-85)
(2013)2. N. Matthijs, J. Pharm. Biomed. Anal., 27(515-529)
(2002)Disclosure of Interest:None DeclaredKeywords: Chiral separations, Peptides, Polysaccharides
P03-003-048 - Comparison of four different methodologies for drug-protein interaction studies
H. Nevídalová*, L. Michalcová, Z. Glatz
Content: Pharmacokinetics and pharmacodynamics behaviour of a drug is influenced by its binding on human serum albumin (HSA) [1]. Therefore the evaluation

315www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
of analytical methods for analysis of these interactions with a small amount of sample solution is a highly desirable [2].
In this work, four methodologies differing in their basis – capillary electrophoresis - frontal analysis (CEFA), equilibrium dialysis (ED), circular dichroism (CD) and isothermal titration calorimetry (ITC) were used for the determination of diclofenac-HSA binding constant (Kb) as a criterion of the interaction strength. Integrating benefit of these methodologies is the possibility to measure interaction of binding partners in their native states without immobilization or labelling under physiological conditions. ED is considered as referent method for such studies, but it suffers from drawbacks, such as long equilibrium time and nonspecific adsorption. CD approach can determine only intermediate affinity interactions and requires purified samples, but structural information on drug-protein complex can be obtained. Large sample consumption and problem with estimation of very high and very low affinity processes are disadvantages of ITC, on the other hand the thermodynamics parameters can be acquired. Protein adsorption on the capillary wall may occur with CEFA, but there are several advantages as low sample consumption, high resolution and the possibility for studying low and high affinity interactions.
The main objective of this study was to determine Kb and to compare these methodological approaches. Obtained results are in a range of (1.22 - 7.91) · 104 L.mol-1 and these values are comparable with values from literature [3].References: [1] S., Schmidt, et al., J. Pharm. Sci. 99 (1107-1122)
(2010).
[2] K., Vuignier, et al., Anal. Bioanal. Chem. 398 (53-66) (2010).
[3] L., Michalcová, Z., Glatz, J. Sep. Sci. 38 (325-331) (2015).
Acknowledgements: This work was supported by grant No. P206/12/G014 from the Czech Science Foundation.Disclosure of Interest:None DeclaredKeywords: binding constant, human serum albumin, diclofenac
P03-003-049 - Development of UHPLC-MS/MS method for a pilot bioactivation study of sobuzoxane
P. Reimerová 1,*, N. Čalkovská 1, T. Hergeselová 1, J. Škoda 1, J. Roh 1, P. Kovaříková 1
1Charles University, Faculty of Pharmacy in Hradec Králové, Hradec Králové, Czech Republic
Content: Pro-drug sobuzoxane was synthetized to improve bioavailability of anticancer agent ICRF-154. Furthermore, thanks to the structural similarity of ICRF-154 to a cardioprotective drug dexrazoxane, it is also investigated for its protective effect against antracycline-induced cardiotoxicity1.
Although sobuzoxane is clinically approved drug there is no modern analytical method for simultaneous analysis of sobuzoxane and ICRF-154 in a biological material that fits for a bioactivation study.
The aim of our study was to develop a UHPLC-MS/MS method for simultaneous assay of sobuzoxane and ICRF-154 in NVCM (rat neonatal ventricular cardiomyocytes) and cell media and to utilize it in a pilot bioactivation experiment.

www.ISC2016.ie @isc2016 #isc2016 /isc2016316
POSTER ABSTRACTS Pharmaceutical Analysis
Analyses were performed on Nexera UHPLC system coupled with LCMS-8030 triple quadrupole mass spectrometer with ESI ion source (Shimadzu). Zorbax SB-AQ chromatographic column (3 x 100 mm, 1.8 µm, Agilent) with mobile phase composed of ammonium formate and methanol in a gradient mode provided the best separation. Different internal standards were tested to reflect the distinct retention behavior of the analytes. Ice-cold methanol, acetonitrile or these solvents with addition of 0.1% formic acid were assessed as precipitation and dilution media for treatment of NVCM and cell media. Linearity was evaluated over concentrations of 10 – 150 µM. The method was then utilized for analysis of samples taken from a pilot bioactivation study of sobuzoxane in vitro. After incubation with sobuzoxane (100 µM) the NVCM cells were flushed with ice-cold PBS and precipitated. Cell medium was simply diluted.References: 1. M. Sterba, O. Popelova, A. Vavrova, E. Jirkovsky,
P. Kovarikova, V. Gersl, T. Simunek, Antioxid. Redox Signal. 18 (899-929) (2013)
Acknowledgements: This work was supported by Charles University in Prague (projects GAUK 344615 and SVV 260291).Disclosure of Interest:None DeclaredKeywords: bioactivation, ICRF-154, sobuzoxane
P03-003-050 - Development of a Capillary Electrophoresis-Mass Spectrometry Method for the Analysis of 5-Nitroimizale Antibiotics in Urine SamplesM. Hernández-Mesa 1,*, A. M. García-Campaña 1, C. Cruces-Blanco 1
1 Department of Analytical Chemistry, University of
Granada, Granada, Spain
Content: 5-nitroimidazoles (5-NDZs) are antimicrobials employed in clinical medicine for treating infections due to protozoan such as Trichomonas vaginalis, Entamoeba histolytica or Giardia lamblia, or bacterial pathogens such as Helicobacter pylori, Clostridium difficile, Gardnerella vaginalis and Bacteroides fragilis. Although 5-NDZ determination has been traditionally carried out by liquid chromatography [1], capillary electrophoresis (CE) has been proved to be a powerful technique in clinical analysis. In this work CE with tandem mass spectrometry (CE-MS/MS) is proposed as a novel method for the determination of eight 5-NDZs and three of their main metabolites in urine samples. As sample treatment, a molecular imprinted polymer solid phase extraction (MISPE) procedure was considered. Samples were hydrodynamically injected at 50 mbar for 40 s. 5-NDZ separation was performed in a fused silica capillary (110 cm x 50 µm) using formic acid 1.0 M (pH 1.8) as background electrolyte (BGE). A separation voltage of 28 kV and a temperature of 25°C were considered. Moreover, a pressure of 50 mbar was applied to the inlet vial during separation in order to improve migration time reproducibility. A sheath flow interface was employed for the CE-MS coupling, considering a sheath liquid composition of isopropanol/water/acetic acid (60:38.8:0.2, (v/v/v)) with a flow rate of 0.2 mL/min. Electrospray ionization (ESI) in positive mode was used under a nebulization pressure of 7.4 psi, a dry gas flow of 6 L/min and a dry gas temperature of 160°C. Matrix-matched calibration curves showed satisfactory linearity (R2 ≥ 0.994) and detection limits from 0.02 to 0.31 µg/mL were obtained. Precision studies resulted in

317www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
relative standard deviations (RSDs) lower than 16.1%. Recoveries over 79.2% were obtained for all studied compounds.References: [1] C. Mahugo-Santana, Z. Sosa-
Ferrera, M.E. Torres-Padrón, J.J. Santana-Rodríguez, Anal. Chim. Acta, 665:113-122 (2010).
Acknowledgements: The “Junta de Andalucía” has supported this work (Excellence Project Ref: P12-AGR-1647). MHM thanks to the Plan Propio of the University of Granada for a post-doctoral contract.Disclosure of Interest:None DeclaredKeywords: 5-nitroimidazoles, CE-MS/MS, urine
P03-003-051 - Towards a chromatographic similarity index to establish localized QSRR models for retention prediction: use of retention time ratio
E. Tyteca 1 2,*, M. Talebi 2, R. Amos 2, S. H. Park 2, M. Taraji 2, Y. Wen 2, R. Szucs 3, P. Haddad 2
1 Department of Chemical Engineering, Vrije Universiteit Brussel, Elsene, Belgium,
2 Department of Chemistry, University of Tasmania, Hobart, Australia,
3 Pfizer, Sandwich, United Kingdom
Content: Quantitative Structure Retention Relationships (QSRRs) have the potential to speed up the screening phase of the chromatographic method development process. The initial exploratory experiments are replaced by prediction of the retention solely based on the structure of the molecule, and allow the choice of chromatographic technique (RPLC, IC, HILIC, SFC), stationary phase, pH and solvent composition. Prediction errors up to 10% can be tolerated. The so-called localized QSRR modeling uses only these compounds in the database which are most
similar to the unknown compound to build the actual model. The prediction accuracy is dependent on the size of the database, the employed similarity index (which acts as a filter) and the number of selected similar compounds selected.
In this study, we investigated the use of tR as the most pure chromatographic similarity index, to deliver proof of the concept of federation of local models. Furthermore, the effectiveness of tR was compared with Tanimoto Similarity (TS), which is considered the ‘gold standard’ of structural similarity indices. Since there exists a trend towards small tR-ratios at high TS-values (TS > 0.8) constructing a high-TS training in fact clusters the compounds according to similar tR-values.
The use of logD as similarity index for RPLC was investigated as well, as there exists a clear relationship between the solute hydrophobicity (reflected by logD) and tR in RPLC. The effectiveness of these filters was compared with a reference case in which the whole database, except for the unknown compound, was used to generate the predictive QSRR model (global QSRR modeling).
The similarity ranking, descriptor selection and QSRR modeling were performed in an automated fashion using Matlab software. To enable this, the original GA-PLS Matlab routines from Leardi et al. [1] were modified.References: 1. R. Leardi, A. L. González, Chemom.
Intell. Lab. Syst. 41 (1998) 195–207Disclosure of Interest: None DeclaredKeywords: Automation, QSRR, Similarity searching

www.ISC2016.ie @isc2016 #isc2016 /isc2016318
POSTER ABSTRACTS Pharmaceutical Analysis
P03-003-052 - Development of Stability Indicating Analytical Methods for Simultaneous Estimation of Olanzapine & Fluoxetine
N. K. Rangra*, K. K. Pradhan 1
1 Department of Pharmaceutical Sciences & Technology, Birla Institute of Technology, Mesra, Ranchi, Jharkhand, India, Ranchi, India
Content: A very simple, accurate and precise stability indicating analytical method was developed for the simultaneous estimation of Olanzapine (OLZ) and Fluoxetine (FLX) in bulk and pharmaceutical dosage forms. Primarily the λmax of OLZ and FLX was determined as 224 and 254 nm respectively. The accuracy was determined by preparing various formulations like 80%, 100%, 120% using the concentrations of 24, 30 and 36 µg/ ml of the pure drug of OLZ and FLX. For RP-HPLC method, the mobile phase used is acetonitrile and 10 mM potassium dihydrogen phosphate (pH 4.5) in a ratio of 50:50 v/v, delivered at a flow rate of 1.0ml/min at wavelength 235 nm and injection volume was 20 μl. The retention time of OLZ and FLX were 2.654 min and 4.274 min respectively. The linearity was found to be in the range of 0.2-1.2 µg/ml and 0.8-4.8 µg/ml; and precision of the method was determined by using three different concentrations of the 0.60, 0.80, 1.0 µg/ml and 2.4, 3.2, 4.0µg/ml for OLZ and FLX, respectively. For HPTLC, the separation was carried out on Merck TLC aluminum sheets of silica gel 60 F254 using toluene: ethyl acetate: methanol in a ratio of 4:4:2, as mobile phase. The accuracy was determined by preparing various formulations like 80%, 100%, 120% using the concentrations of 2, 4 and 8 µg/ ml of the pure drug of OLZ and
FLX. The forced degradation studies were carried out in various stressed conditions and the drugs OLZ and FLX were found to degrade in alkaline, thermal and oxidative stress. The statistical tests indicate that the proposed method reduces the duration of analysis and appears to be equally suitable for the routine analysis of pharmaceutical formulation in quality control laboratories where the economy and time are the essential factors.References:1. S.Patel, N. J.Patel, Indian J. pharm. sci., 4(477-
80) (2009)2. A.Pathak,S.J.Rajput, J.chromatogr.sci.,7(605-11)
(2009)3. M.M.Eswarudu,M.Anitha,N.Gayathri,
T.Chaithanya, Int.res.J.pharm., 4(310-313)(2012)Disclosure of Interest:None Declared
Keywords: Fluoxetine, Olanzapine, RP-HPLC, HPTLC
P03-003-053 - Quality control of vitamin D3 by means of a fully validated UHPSFC method
B. Andri*, P. Lebrun 1 2, A. Dispas 1, R. Klinkenberg 3, B. Streel 3, R. Marini Djang’eing’a 1, P. Hubert 1
1 University of Liege - Laboratory of Analytical Chemistry, CIRM, 2Arlenda SA, Liege, 3Galephar Research Center M/F, Marche en Famenne, Belgium
Content: The quantitative performances of a modern UHPSFC method were challenged on a real-life case study: the quality control of vitamin D3.
Regarding vitamin D, a worldwide deficiency is currently described for the large extent of the population – children included - due to its small exogenous intakes and low endogenous synthesis.

319www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
Therefore, to take advantage of its benefits and obtain satisfying levels of vitamin D, supplementation with medicines or pharmaceuticals is often necessary1.
To ensure the quality of these products, it is mandatory to proceed to quality control of the raw material used, which is usually performed with NPLC. Though, in the context of green analytical chemistry, SFC is often suggested as an alternative to NPLC2 because it provides fast, efficient and green separations3.A rapid UHPSFC method was optimized thanks to the Design of Experiment–Design Space methodology. A high quality and robust separation was obtained using a gradient of ethanol as co-solvent of the carbon dioxide. The analytical method was then fully validated according to the total error approach, demonstrating the compliance of the method to the specifications of USP (97.0-103.0%) and EP (97.0-102.0%) for an interval of [50-150%] of the target concentration. In order to allow quantification of impurities with vitamin D3 as an external standard in SFC-UV, correction factors were determined and confirmed during method validation. Thus, accurate quantification of impurities was demonstrated at the specified levels (0.1 and 1.0% of the main) in a 70.0-130.0% dosing range.
This work demonstrates the feasibility and validity of the application of an UHPSFC-UV method for the QC of vitamin D3. It also supports the switch to a greener and faster alternative to NPLC in industrial frameworks.References: 1. M. Wacker, M.F. Holick, Nutrients. 5 (2013) 111–
148.2. L. Taylor, Past, LC GC North America. 31 (2013)
s44–s49.3. A. Tarafder, TrAC. (2016) in press.Disclosure of Interest:None DeclaredKeywords: Accuracy profile , DoE-DS methodology, Vitamin D3
P03-003-054 - Direct determination of oxidation-sensitive amino acids and their dipeptides by HPLC-UV method
Z. Temova 1, A. Kristl 1, R. Roskar 1,*
1 Chair of Biopharmacy and Pharmacokinetics, University of Ljubljana, Faculty of Pharmacy, Ljubljana, Slovenia
Content: Amino acids are, because of their polarity and lack of chromophores, most commonly determined by ion-exchange or reversed-phase chromatography with pre- or post-column derivatization1. Our aim was to establish simple HPLC-UV method without derivatization for analysis of several amino acids and dipeptides in stability studies.
Materials and methods: Amino acids, chosen according to oxidation susceptibility: methionine, histidine, tryptophan, tyrosine and cysteine, some of their dipeptides and glutathione were obtained from Sigma-Aldrich. Eight columns with different stationary phases (C18, C8, C4, NH2) were obtained from two manufacturers (Waters, Phenomenex). An Agilent 1100/1200 HPLC system, equipped with diode array detector was used.
Results: Method development was challenging due to highly hydrophilic nature of some analytes and lipophilic nature of aromatic amino acids and their dipeptides. The aforementioned columns

www.ISC2016.ie @isc2016 #isc2016 /isc2016320
POSTER ABSTRACTS Pharmaceutical Analysis
in temperature range 25-40ºC, various gradient elution runs using acetonitrile and phosphate buffer solutions (15-50 mM), with pH 3.0-8.5 and flow rates (0.5-1.5 mL/min) were tested in order to retain polar analytes and optimise separation of 12 selected analytes. Sufficient separation of all analytes and their degradation products was obtained on Luna C8 250×4.6 mm column (Phenomenex) with initial isocratic elution with phosphate buffer solution (25 mM, pH 8.3), followed by gradient elution (up to 40% acetonitrile) at 25ºC. Detection was carried out at 210, 250 and 275 nm. This method was successfully validated according to ICH guidelines Q2(R1).
Conclusion: The presented method is beneficial because of its simplicity, especially as derivatization is not required, suitable for evaluation of selected analytes in stability studies and also applicable for their quantitative analysis. References: 1. Bartolomeo P, Maisano F. Validation
of a reversed-phase HPLC method for quantitative amino acid analysis. J Biomol Tech 17, 131–7, 2006.
Disclosure of Interest: None DeclaredKeywords: amino acids , direct determination, HPLC-UV method
P03-003-055 - Multiresidue analysis of selected pharmaceuticals in water samples by stir-bar sorptive extractionA. Klančar*, J. Trontelj 1, R. Roškar 1
1 Chair of Biopharmacy and Pharmacokinetics, Faculty of pharmacy, Ljubljana, Slovenia
Content: Stir-bar sorptive extraction is a microextraction which enables the isolation
and concentration of pharmaceuticals in one step. Furthermore, it fully complies with green chemistry principles and provides high recoveries for target analytes with low consumption of organic solvents. Our aim was to select 20 pharmaceuticals and to develop and validate a stir-bar sorptive extraction coupled to LC-MS/MS and apply it to wastewater samples.
Materials: Standards amitriptyline, azithromycin, carbamazepine, desipramine, diazepam, donepezil, escitalopram, fluoxetine, haloperidol, loperamide, loratadine, metoprolol, propranolol, raloxifene, risperidone, selegiline, sertraline, venlafaxine, verapamil, tramadol, triclosan were purchased from Sigma-Aldrich.
Stir-bars (1×10 mm) were purchased from GERSTEL. Wastewater effluents were obtained from five sewage treatment plants in Slovenia as 24-hours composite samples.
Methods: Stir-bars were preconditioned in 2 mL of MeOH for 30 minutes. The extraction was performed in 20 mL of heated pre-filtered samples adjusted to pH 9, lasted for 150 minutes at 990 rpm. Liquid desorption was accomplished in 2 mL of acetonitrile and MeOH mixture (50/50, v/v) in 15 minutes. Finally, samples were analysed by LC-MS/MS (Agilent 6460 QQQ).
Results: The validated method provided good recoveries (55-101 %) and a wide linear range (1.25-1250 ng/L, R2 >0.99) with very low limit of detection (1.25 ng/L). Half of the monitored analytes were detected in all five samples. The found concentrations were in broad range; mostly in low ng/L while the carbamazepine, azithromycin and venlafaxine exceeded the limit of 100

321www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
ng/L. Tramadol was detected in the highest average concentration (18 µg/L). Our results show that such a method may present a simple and efficient alternative to the classic extraction methods for monitoring the aquatic contamination.References: He M. et al. (2014). Recent
developments in stir bar sorptive extraction. Anal Bioanal Chem, 406(8), 2001-26
Disclosure of Interest: None DeclaredKeywords: pharmaceuticals, stir-bar, water samples
P03-003-056 - Capillary Electrophoretic Method for Kinetic and Inhibition Studies of β-Secretase in Nanoliter-Scale Format
R. Řemínek*, J. Schejbal 1, Z. Glatz 1
1 Department of Biochemistry, Faculty of Science, Masaryk University, Brno, Czech Republic
Content: Alzheimer’s disease (AD) represents the most common cause of the dementia. Development of a cure for AD, however, is hindered by limited knowledge about its cause; currently available medications may only ease its symptoms. Since amyloid plaques found in a brain tissue of AD patients cause the inflammatory reaction leading to degradation of neurons, specific inhibition of β-secretase (BACE), enzyme responsible for formation of these plaques, appears to be a promising way for slowing down or even stopping the progression of the disease.
Capillary electrophoresis (CE) represents a suitable technique to assess the enzyme activity in vitro due to high separation efficiency and minuscule sample
consumption. Furthermore, a fused-silica capillary can serve as a nanoliter-scale reaction vessel. Such procedures are referred to as on-line CE methods that integrate incubation of an enzymatic reaction, separation of reaction products, and their detection and quantitation into a single fully automated analysis.
For these reasons, the main goal of this work was to develop an on-line CE method for studies of BACE activity. A principle of reported diffusion-based technique was adopted for mixing of the enzyme and model substrate, decapeptide SEVNLDAEFR, inside the capillary [1]. Conceptually, their solutions were injected by hydrodynamic pressure as a series of consecutive plugs with parabolic profiles allowing rapid mixing of the enzyme and substrate by transverse diffusion. After the incubation was finished, reaction products were separated in less than 6 min. The fragment DAEFR was detected and quantified with UV-Vis detector in this study, 5% acetic acid used as a BGE nonetheless enables utilization of MS detection as well. The optimized method was validated and used for kinetic and inhibition study of BACE. Results obtained were in good agreement with literature and conducted control off-line study.References: [1] V., Okhonin, et. al., Anal. Chem. 77 (5925-5929)
(2005).Acknowledgements: This work was supported by grant No. GA16-06106S from the Czech Science Foundation.Disclosure of Interest:None DeclaredKeywords: Alzheimer’s disease, Capillary electrophoresis, β-secretase

www.ISC2016.ie @isc2016 #isc2016 /isc2016322
POSTER ABSTRACTS Pharmaceutical Analysis
P03-003-057 - Analysis of selected antineoplastic drugs from wipe samples using micro-liquid chromatography tandem mass spectrometry
T. Hetzel 1 2,*, T. Teutenberg 1, T. C. Schmidt 2
1 Research analysis, Institut für Energie- und Umwelttechnik e. V., Duisburg,
2 Instrumental Analytical Chemistry, University Duisburg-Essen, Essen, Germany
Content: Micro-LC offers several advantages that can be exploited for routine analysis. Due to the flow rate reduction, the costs for organic solvents as well as their disposal can be significantly reduced [1]. In addition, the combination of flow rates in the range of several microliters per minute (µL min-1) and decreased column inner diameter (i.d.) between 0.3 to 0.5 mm leads to high linear velocities. In order to achieve similar linear velocities with a conventional column i.d. (e.g. 4.6 mm), higher flow rates up to the mL min-1 range must be applied. Moreover, the reduced gradient delay volume of dedicated micro-LC systems is a key feature to achieve fast separations and reduced analysis cycle times. As a consequence, the sample throughput can be significantly increased which represents a major advantage for routine analysis. Despite all these advantages, the applicability of micro-LC for routine analysis is often being questioned because of the supposed decreased system robustness and aggravated handling. As a consequence, it is doubted whether this technique can satisfy the requirements of method validation. Another important aspect is the sensitivity that has to be reached for a certain application, which is frequently assumed to be difficult with
micro-LC. Therefore, the aim of this study is the successful method development and validation of a micro-LC-MS/MS method for routine analysis of antineoplastic drugs. It was found that general requirements in interday and intraday stability as well as sensitivity can be fulfilled.References: [1] G. Desmet, S. Eeltink, Analytical
Chemistry 85 (2013) 543.Disclosure of Interest: None DeclaredKeywords: Large volume injection, Method development and validation, Peak distortion
P03-003-058 - Comparison of the Selectivity of C18, CD-Screen® and CD-Screen®-IEC Stationary Phases for HPLC Analysis of Single-isomer Cyclodextrin DerivativesJ. Szemán 1,*, K. Csabai 1, E. Varga 1, G. Benkovics 2, M. Malanga 1, K. Ludányi 3, L. Szente 1
1 CycloLab Ltd., Budapest, Hungary, 2 Department of Organic Chemistry, Charles University in
Prague, Prague, Czech Republic, 3 Department of Pharmaceutics, Semmelweis University,
Budapest, Hungary
Content: Cyclodextrins (CDs) and their derivatives are widely used in various fields of pharmaceutical industry, e.g. for drug delivery, stabilization of drugs, as additives in biotechnology. Moreover, CDs have the ability to discriminate between structurally related compounds, isomers and even enantiomers, therefore, they are employed as separating additives in several analytical techniques. Besides the statistically substituted CD derivatives single-isomer CD-derivatives were prepared for analytical purposes; they provide a better control over the molecular recognition processes and more predictable separations. The

323www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
first CD active pharmaceutical substance on the market called Sugammadex is also a single-isomer CD derivative. The above applications require substances of high purity, therefore, sophisticated analytical methods are needed for identification and control of their quality.
In this work three different stationary phases are used for characterization negatively charged single-isomer CD derivatives, such as carboxymethylated and sulfated β-CDs, as well as Sugammadex. Besides a C18 column the separation potency of two special stationary phases – which were developed for CD analysis - are examined. The principle of separation is inclusion complex interaction in case of CD-Screen®
column(1), while CD-Screen®-IEC HPLC stationary phase combines the ion exchange
and complex formation interactions.
During the work not only the pure end-products, but also crude samples are examined. The main impurities are identified with HPLC-MS technique. The different selectivity of the tested stationary phases will be discussed.References: 1. J. Szemán, K. Csbai, K. Kékesi, L.
Szente, G. Varga, J. Chromatogr. A 1116, (76-82) (2006)
Acknowledgements: The financial support from the Marie Curie Programme Initial Training Network, Project No 608407 (CyclonHit) is highly appreciated. Disclosure of Interest:None DeclaredKeywords: CD-Screen, HPLC-ELSD, Single-isomer cyclodextrin
P03-003-059 - cAMP and cGMP analysis in rat brain microdialysate
H. Darwinkel*
Content: Phosphodiesterases (PDE) are drug targets for various diseases (dementia, depression and schizophrenia). Inhibition of these enzymes can increase cyclic adenosine 3’-5’ monophosphate (cAMP) and cyclic guanosine 3’-5’ monophosphate (cGMP) levels. cAMP and cGMP are intracellular second messengers playing a key role in physiological processes. Previous work has demonstrated that cAMP and cGMP, which is formed intracellularly, can be transported into the extracellular space. With microdialysis it is possible to monitor these nucleotides in the extracellular space over time from the same subject. The microdialysis technique is based on diffusion of analytes over a membrane. Therefore, the recovery of the analyte of interest is low and a sensitive analytical method is required.
An RP-LC-MS/MS method was developed and validated to quantify endogenous c AMP and cGMP in rat brain microdialysate in freely moving animals using aCSF as a surrogate matrix. Chromatographic separation was achieved using a Atlantis C18 T3 column (Waters, Milford, USA), 150 mm x 2.1 mm, 3 µm particle size. Gradient elution with solvent A (0.1% FA) and solvent B (0.1% FA in 70% ACN) at a flow rate of 0.2 ml/min was applied in combination with a make up flow (2% FA in IPA/ACN (50:50)) at a flow rate of 0.1 ml/min to enhance sensitivity.
cAMP and cGMP and their stable labeled internal standards were detected in positive

www.ISC2016.ie @isc2016 #isc2016 /isc2016324
POSTER ABSTRACTS Pharmaceutical Analysis
mode electrospray ionization using multiple reaction monitoring (MRM). The validation and in-vivo results show that this method offers robustness, speed and sensitivity over a linear range of 0.02-8.0 nM for both analytes.References: .Disclosure of Interest: None DeclaredKeywords: Atlantis T3, cyclic nucleotides, microdialysis
P03-003-062 - LC/MS/MS Detection with Online Solid Phase Extraction of Thyroid Hormones in Biological Fluids
E. G. Jones 1,*, X. Lu 1, D. S. Bell 1, A. Fridström 2
1 MilliporeSigma, Bellefonte, United States, 2 Merck, St. Gallen, Switzerland
Content: Thyroid hormones play critical roles in the regulation of biological processes such as growth, metabolism, protein synthesis, and brain development. Specifically, thyroid hormones, 3,3’, 5,5’-tetraiodo-L-thyronine (thyroxine or T4) and 3,3’,5-triiodo-L-thyronine (T3), are essential for development and maintenance of normal physiological functions. For a clinical laboratory, measurements of total T4 and total T3, along with estimates of free T4 (FT4) and free T3 (FT3), are important for the diagnosis and monitoring of thyroid diseases. Immunoassay-based methods offer a relatively rapid, high patient sample throughput that lends itself to automation but are significantly compromised by problems with assay interference and are perturbed by changes in protein levels that alter the free hormone availability.1 Liquid chromatography mass
spectrometry (LC/MS) has been reported to offer superior specificity and speed over the immunoassays for determination of thyroid hormones in biological matrices such as serum and tissues.1-3 Nevertheless, the reported sample preparation procedures, typically by liquid-liquid extraction followed by solid phase extraction (SPE), involve multiple steps, being time consuming and less compatible with automation.1-2 The present work devises an online SPE-LC/MS/MS method for the determination of thyroid hormones in biological fluids. The method exploits reversed phases, including RP-Amide and C8, as the trapping columns and Phenyl phase as the separation column, respectively. The preliminary experiments demonstrated that, under the optimized conditions, both RP-Amide and C8 effectively trapped the thyroid hormones extracted from spiked rabbit plasma sample which had been protein precipitated.References:1. N. Kahric-Janicic, S. Soldin, O. Soldin, T. West, J.
Gu, J. Jonklaas. Thyroid. 2007;17(4): 303-11. 2. S. Taia, L. Sniegoski, M. Welch. Clinical Chemistry.
2002; 48(4): 637-642.3. D. Wang, H. Stapleton. Anal Bioanal Chem. 2010;
397(5): 1831–1839.Disclosure of Interest:None DeclaredKeywords: LC-MS/MS, SPE extraction
P03-003-063 - Aspirin HPLC: artefacts, impurities and modernisation
W. J. Lough 1, S. I. Yarima 1,*, L. Singh 1
1 Sunderland Pharmacy School, University of Sunderland, Sunderland, United Kingdom
Content: In carrying out pharmacopoeial methods for the determination of aspirin related substances, it proved necessary to

325www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
establish whether a previously unreported peak was attributable to a significant related substance, an over-estimated trace peak running close to the main aspirin peak or merely an artefact arising from the very high test concentrations used. In fact, all of these applied, depending on the conditions used. Subsequently it was demonstrated that, by using a lower test solution concentration and slightly modified conditions from the pharmacopoeial method to give greater retention, that for certain aspirin samples, there was indeed a trace impurity running close to the main aspirin peak. To investigate this further, it was decided to apply more modern LC technology. Having failed to observe impurities at the first attempt at uHPLC and having previously observed an artefact when using a linear gradient, a 10-min gradient method incorporating an initial isocratic stage for the resolution of aspirin and close-running impurities using fused core-shell technology was developed. The method, using ACE Ultracore SuperC18 (100 x 4.6 mm i.d.) column was faster, gave better resolution, in particular giving resolution of previously unresolved impurities, could be used for all pharmacopoeial named related substances and was suitable for use on conventional HPLC systems. Following validation studies, including work on the stability of sample solutions over long autosampler runs, the method was used to explore the extent to which the trace impurities occurred in a range of aspirin api and product samples.References: 1. The British Pharmacopoeia (BP): The British
Pharmacopoeia Volumes I and II Monographs: Medicinal and Pharmaceutical Substances (2015 [accessed online]
Acknowledgements: The authors wish to acknowledge the British Pharmacopoeia Laboratory for the supply of ‘impure aspirin’ and University of Sunderland MPharm and MSc Drug Discovery students for drawing attention to the initial issue and trialling the methodology in ‘research-led’ practical classes respectively.Disclosure of Interest:None DeclaredKeywords: aspirin, fused core-shell technology, related substances
P03-003-064 - Evaluation and Exploitation of Stationary Phase Selectivity
V. Tang 1,*, W. J. Lough1 Sunderland Pharmacy School, University of Sunderland,
Sunderland, United Kingdom
Content: With sets of drugs and their structurally-related impurities (confidential studies on Drug Development candidates), it had been found previously that only alkyl-bonded silicas containing residual silanol groups, Hypercarb and ion-exchange phases had exhibited LC selectivity significantly orthogonal to that for alkyl-bonded silicas operating almost exclusively through hydrophobic partition. Several stationary phases with claimed alternative selectivity had shown only limited promise due to the dominance of the hydrophobic partition mechanism in RP-HPLC. Nonetheless, because of the unmet need for orthogonal RP-HPLC stationary phase selectivity in several pharmaceutical LC applications, it was decided to continue to monitor phases to look for those that might operate to a lesser degree via hydrophobic partition and in particular to study the selectivity offered by a RaptorTM Biphenyl phase with claimed more aromatic selectivity than ordinary phenyl-hexyls1.

www.ISC2016.ie @isc2016 #isc2016 /isc2016326
POSTER ABSTRACTS Pharmaceutical Analysis
Retention on RaptorTM Biphenyl correlated well with that on ACE 3 C18, a typical alkyl-bonded silica phase, for chlorpheniramine and related substances (r2 = 0.9619), tryptophan analogues (r2 = 0.9135) and phenols (r2 = 0.9425). However there was some promise with respect to non-steroidal anti-inflammatory drugs r2 = 0.5094) and steroids (r2 = 0.4920) and there was poor correlation for a diverse set (r2 = 0.0365). From this, it was possible to identify drug types that interacted strongly with the biphenyl- phase and therefrom identify applications where the use of the biphenyl- phase might be beneficial. This led to the very effective use of the Raptor™ Biphenyl phase in RP-HPLC methods for paroxetine, flurbiprofen and dothiepin and their related substances and for steroid mixtures. So, overall, this phase exhibited encouraging and useful levels of alternative selectivity for certain compound classes.References: 1. Restek, “Raptor™ Biphenyl LC
Columns,” 2015. [Online]. http://www.restek.com/catalog/ view/40873.
Disclosure of Interest: None DeclaredKeywords: biphenyl-, drug related substances, stationary phase selectivity
P03-003-065 - Prospective study of substandard and counterfeit medicines in the North African regionA. M. Abushoffa*, A. A. Ashames 1, J. S. Mezogi 2
1 Pharmaceutical Chemistry, Faculty of Pharmacy, 2 Pharmacognosy, Faculty of Pharmacy, University of
Tripoli, Tripoli, Libya
Content: Thakling and combat of substandred or falsified medicines is one of the global chalengs, these sort of medicines
is a global socio-economic problem which requires an enforcement of an international, regional and national strong and tuff measures.
Laws and acts must be revised in order to enhance the way of combating the problem in a proper way, pharmaceutical quality control, frontier and ports customs staff should also be trained in order to be able to be more efficient in talking such phenomenon.
Awareness to the Community pharmacists, national quality control laboratories staff, hospital pharmacists, warehouses staff should also be considered as a sin part of the war against counterfeiting and trafficking of medicines which are imported in an illegal way.
This study has emphasised the importance of this task and to participate in the efforts leading to minmizing the disaster caused by such problem in the North African countries.References: SE. Nsemba, East Afr J Public Health, 5(3), (205-10)
(2008).D. Sato, Yakugoku Zasshi 134(2), (213-22)
(2014). Review JapaneseG. Onishchenko, Gig. Saint, , 2, (4-15) (2008).Reviw Russian Acknowledgements: Directorates of Pharmacy and Medical appliances in the North African countries, Ministry of Health, Libya are great fully acknowledged for providing data related to the study.Disclosure of Interest:None DeclaredKeywords: Substandard medicines; counterfeiting; Combat

327www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
P03-003-066 - Rationalizing Drug-Receptor Kinetics Using an Immobilised Artificial Membrane
S. M. Mutuku 1,*
1 School of Life Sciences, University of Nottingham, Nottingham, United Kingdom
Content: The accurate determination of kinetics of drug-receptor binding is vital in understanding the pharmacology of drugs. Several approaches described in literature to ascertain the molecular mechanism of sustained duration of action of long acting β2-adrenoceptor agonists have resulted in hypotheses that have varied consensus among pharmacologists. This research project examined the influence of lipophilicity on drug interaction with the phospholipid membrane and receptor affinity. Using an IAM-PC-DD2 column as a model of the phospholipid bilayer, marker compounds were eluted in the absence of an organic modifier and their log k’w
IAM values correlated well with measured log D7.4 (r2=0.80, P<0.0001). This inferred that the lipophilic properties of various compound classes can be reliably predicted by this approach. A rapid gradient-HPLC method using 10mM pH 7.4 phosphate buffer and acetonitrile mobile phase composition established the CHIIAM values of alkylphenone calibrants. These values offered a convenient lipophilicity scale to derive the log k’w
IAM values for a series of considerably lipophilic β2-adrenoceptor ligands which correlated significantly with experimental kon data (r2=0.71, P<0.0001). This can be explained by drug accumulation around the receptor microenvironment within the lipid bilayer - attributable to a drug’s lipophilic/basic properties. It is envisaged that these findings will offer a clearer perception of localised pharmocokinetics/pharmacodynamics
relationships for clinical drugs.Abbreviations: k’
w – chromatographic
retention factor
at 100% aqueous phase, kon
– kinetic association rate, IAM-PC-DD2 – immobilised artificial membrane – phosphatidylcholine - drug discovery type 2 column, CHI
IAM – chromatographic hydrophobicity
index values of IAM chromatography at pH 7.4.References: D. Sykes, British Journal of Pharmacology, 165:
2672-2683 (2012)K. Valkó, Journal of Chromatography A, 1037: 299-
310 (2004) Acknowledgements: Dr. Patrick Barton, Associate Professor in Drug Discovery, School of Life Sciences, The University of Nottingham Medical SchoolDr. David J. Tooth, Manager Bio-mass Spectrometry Facility, Faculty of Medicine & Health Sciences, The University of Nottingham Medical SchoolDisclosure of Interest:None DeclaredKeywords: chromatographic hydrophibicity index, drug-receptor kinetics, immobilised artificial membrane
P03-003-067 - Quantification of Small Molecule Impurities of a Safe Drug Carrier, Sulfobutyl ether-beta-cyclodextrin by HPLC-ELSDK. Temesvári*
Content: Cyclodextrins are cyclic oligosaccharides, differing structurally in the number of glucopyranose units. The parent α-, β- and γ-cyclodextrins (CDs) contain six, seven and eight glucopyranose units, respectively. Owing to their characteristic truncated cone-shaped structure with a hydrophobic internal cavity and hydrophilic exterior surface, CDs are applied in drug formulation as carriers. Hydrophobic drug molecules

www.ISC2016.ie @isc2016 #isc2016 /isc2016328
POSTER ABSTRACTS Pharmaceutical Analysis
penetrate into the cavity, forming inclusion complexes. The hydrophilic exterior surface improves the water solubility, while the inclusion may increase the stability of the drug molecule. Several β-cyclodextrin formulations for oral pharmaceutical products are on the market worldwide. The parenteral usage of CDs is limited due to their hemolytic and renal toxicity observed after systemic exposure.
Derivatization of the hydroxyl groups of native β-CD with sulfobutyl ether substituents (SBE) further improves its water solubility and affects favourably the safety profile. The anionic SBE groups help the kidney excrete these excipients rapidly, thus minimizing their contact time with the kidney cells.
The production of SBE-β-cyclodextrin involves the reaction of β-CD and 1,4-butane sultone, in which two small-molecule impurities, 4-hydroxybutane-1-sulfonic acid and bis(4-sulfobutyl) ether disodium can also be formed.
These impurities are removed by ultrafiltration. There exists a USP monograph for the impurity profiling of SBE-β-cyclodextrin, but the recommended method could not be applied in our laboratory. Therefore a novel HPLC method with light scattering detection has been developed for the determination and quantification of these molecules both in the filtrate samples and the final product SBE-β-CD. The poster presents the analytical challenges and advantages of this new method.References: 2016 USPC Official 5/1/16-7/31/16 NF
Monographs: Betadex Sulfobutyl Ether SodiumDisclosure of Interest: None DeclaredKeywords: cyclodextrin, drug carrier, HPLC-ELSD
P03-003-068 - Quantitative Structure-Retention Relationships for Dissociating Compounds Aided by Mass SpectrometryL. Kubik*, W. Struck-Lewicka 1, R. Kaliszan 1, P. Wiczling 1
1 Department of Biopharmacy and Pharmacodynamics, Medical University of Gda−sk, Gda−sk, Poland
Content: The Quantitative Structure-Retention Relationships (QSRR) are mathematical models developed for structure-based prediction of chromatography related parameters, such as hydrophobicity (logkw) and consequently retention times. QSRR equations can improve the search for optimal separation of chemical compounds in a mixture. It can also be applied for determination of physicochemical parameters of a drug candidate substance. Such parameters (like logkw, pKa) can be used for predicting pharmacokinetic properties of the analyte, its druglikeness and the likely behavior in the biological systems. In this work we have presented QSRR models developed for chromatographic parameters: logkw, pKa and S (slope factor from Snyder-Soczewinski equation).
Over 100 structures of compounds in their neutral and ionic forms were optimized using molecular modelling approach. Geometry optimization was performed in water solvent with the application of DFT B3LYP quantum chemistry method with 6-31G basis set. Optimization was done in the Gaussian 09 software. 4885 molecular descriptors were calculated using Dragon 6 program. Experimental values of logkw, pKa and S were measured with the application of double gradient (organic modifier and pH) reversed phase high performance

329www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
liquid chromatography (RP HPLC) with the time-of-flight mass spectrometry (TOF MS). Electrospray ionization (ESI) was used as the ion source. MS experiments were performed in positive and negative polarity. QSRR equations were developed using three different statistical techniques: Lasso Penalized Regression, Stepwise Regression and Partial Squares Regression.
The obtained models were characterized by simplicity and good predictive properties (average Q2>0.6), assessed by 10-fold cross validation and external validation.References: Ł. Kubik, W. Struck-Lewicka, R. Kaliszan, P.
Wiczling, J. Chromatogr. A 1416 (31-37) (2015)Ł. Kubik, P. Wiczling, J. Pharm.
Biomed. Anal. (2016) doi:10.1016/j.jpba.2016.02.050
Acknowledgements: Authors would like to thank the National Science Centre Poland (grant 2014/13/N/NZ7/04218) and MUG Faculty of Pharmacy (National Leading Scientific Centre) for funding and Academic Computer Centre in Gda−sk for access to computer cluster.Disclosure of Interest:None DeclaredKeywords: HPLC, mass spectrometry, QSRR
P03-003-069 - Batman peaks in action: rate constant determination from chromatograms
A. Sepsey*, D. Németh 1, G. Németh 2, A. Felinger 3
1 Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics,
2 Drug Substance Development Division, Egis Pharmaceuticals PLC, Budapest,
3 Department of Analytical and Environmental Chemistry and Szentágothai Research Center, University of Pécs, Pécs, Hungary
Content: Enantiomers are present everywhere in nature, including the human body. We must take into account that the synthetized drugs are in most cases also chiral molecules which can be present in either racemic or in pure form depending on the synthetization process. Once a drug is in racemic form, both enantiomers must be tested whether they are toxic or not. This is especially important because several enantiomer molecules can dynamically interconvert to each other depending on the environment (solvent, temperature, etc.).
“Batman peaks” appear as a plateau between two peaks because of the dynamic interconversion of the enantiomers to each other during the chromatographic run. The rate of the interconversion and thus the peak shape depend on the rate constant of the dynamic reaction.
The examination of the enantiomer separation is important to investigate the molecular processes. Thus, we extended the stochastic model of chromatography to the case of interconverting enantiomers for deeper understanding of the molecular processes behind the transformation. The characteristic function, the band profile and the main moments of the elution profiles were determined by mathematical formulas and we investigated the effects of the parameters affecting the separation. We used this novel model for an interconverting drug molecule to evaluate the rate constant based on the enantiomerization at various flow rates and obtained good agreement with the rate constant evaluated from optical rotation experiments.Disclosure of Interest: None DeclaredKeywords: Enantiomer separation, rate constant determination, stochastic model

www.ISC2016.ie @isc2016 #isc2016 /isc2016330
POSTER ABSTRACTS Pharmaceutical Analysis
P03-003-070 - Chromatographic methods for measuring lipophilicity and phospholipophilicity in predicting cellular accumulation of compounds
D. Sanchez Garcia, M. Sjödin, M. Hellstrandh*, U. Norinder, J. Lindberg, V. Munic Kos
Content: Lipophilicity gives a better understanding of the biological and physico chemical properties influencing bioaccumulation. In this study commercially available drugs were tested to investigate if there is a correlation between the degree of lipophilicity of the compounds and their accumulation and retention in cells. The distribution coefficient, logD, was calculated based on their specific retention on a C18 column. For comparison compounds were also investigated for binding to an immobilized artificial membrane column, IAM PC DD2. The separation using this column attempt to mimic the interaction of the xenobiotic with the phosphatidylcholine in the lipid bilayer, phospholipophilicity using the chromatographic hydrophobicity index IAM, CHIIAM.
The calibration curve calculating logD7.4 based on 6 compounds with known logD7.4 values (-2 to 5.5) was reproducible with r2=0.999. CHIIAM was calculated using 6 compounds and plotting the obtained retention times against their CHIIAM 7.4 literature values. This experiment was also reproducible with r2=0.997. LogD7.4 and CHIIAM 7.4 showed moderate correlation to each other (r2=0.51). In addition, it was observed that CHIIAM 7.4 values, in contrast to logD, highly correlates with cellular accumulation of drugs. The CHIIAM 7.4
value >40 turned out to be a good predictor for high accumulation of drugs in cells.
In conclusion, logD7.4 is not able to represent drug partition in membranes, whereas CHIIAM 7.4 also contains information about the interaction with charged lipids. This is of interest because many drugs are basic amines and they show higher partition into membranes and higher accumulation in the cells than the one predicted with logP/logD due to the amphiphilic nature of the lipid bilayer. CHIIAM 7.4 covers for this and is therefore a more suitable parameter to study and predict drug distribution into cells.References: 1. Jiang Z, Reilly J. J Pharm Biomed
Anal. 2012;61:184-190. Disclosure of Interest: None DeclaredKeywords: Bioaccumulation, Lipophilicity, Phospholipophilicity
P03-003-071 - A Stability-indicating Method for the Quantitation of Povidone in an Ophthalmic Solution by Size-exclusion HPLC
S. Stover 1, M. Selman 1,*, S. Hepburn 1
1 Analytical Development, The Mentholatum Co, Horsham, United States
Content: Poly(1-vinyl-2-pyrrolidinone), aka povidone, is a common ingredient in the pharmaceutical and food industries, and is typically used as a binder in tablets and as a thickening agent. Povidone is also an approved active pharmaceutical ingredient (API) with the function of a lubricant permitted at levels of 0.1 – 2.0% in over-the-counter ophthalmic eye drops.1 The subject povidone raw material used as the API has a molecular weight range of 1 – 1.5M Daltons [Kollidon 90F]. The development and validation

331www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSPharmaceutical Analysis
of a high-performance liquid chromatography (HPLC) stability-indicating method is presented, including specificity, linearity, accuracy, repeatability, intermediate precision, reproducibility and a filter study. The separation was achieved isocratically using a Tosoh Bioscience TSKgel Alpha-6000 size exclusion column (exclusion limit of 10M Daltons) and UV detection at 210 nm. Extinction coefficients of povidone solutions along with solutions of its reduced monomer and the proposed degradant model compound were determined to support the stability-indicating conclusion. Refractive index (RI) detection was also utilized in series to estimate the apparent molecular weight of the povidone raw material and other povidone materials against a calibration curve established with polyethylene oxide standards. References: 1. U.S. Code of Federal Regulations,
Title 21 §349.12.Disclosure of Interest: None DeclaredKeywords: povidone, size-exclusion, stability-indicating

www.ISC2016.ie @isc2016 #isc2016 /isc2016332
POSTER ABSTRACTS Process Chromatography and Process Analytical Technology (PAT)
Process Chromatography and Process Analytical Technology (PAT)
P01-001-026 - The effect of bed compression on the quality of column packing for protein separationD. Kong*
Content: Models to predict pressure drop across preparative chromatography beds has been studied for soft or partially rigid gel filtration media. However, different methods of compression applied to new and cycled media is not yet well understood enough to provide reliable information for protein separation.
In this study the relationship between column efficiency and protein separation with further compression by mechanical and liquid flow compression of Sepharose CL-6B and Q Sepharose Fast Flow was evaluated.
We determined the asymmetry and height equivalent of a theoretical plate (HETP) by using 2% v/v acetone, as well as the void volume and intraparticle porosity by loading and blue dextran. A protein mixture of ovalbumin (chicken), bovine serum albumin (BSA) and ɣ′- globulin (bovine) with molecular weights of: 44, 67 and 158 kDa was analysed using a wavelength of 280nm.
Results showed that step by step gradual mechanical compression was found to be highly correlated with better asymmetry, pore diffusion and product purity and yields. Further compression tended to reduce the ratio between accessible internal volume to void volume for this gel resin.
The results suggest that the presence of mechanical bed compression of Sepharose CL-6B and Q Sepharose Fast Flow can be used to improve column efficiency. Disclosure of Interest:None DeclaredKeywords: Anion exchange, Compression, Gel filtration
P03-003-072 - Similarity Searching in Quantitative Structure-Retention Relationship for Retention Prediction in Reversed-Phase Liquid Chromatography
Yabin Wen1, Paul R. Haddad1, Mohammad Talebi1, Ruth I.J. Amos1, Robert A. Shellie1, Roman Szucs2, John W. Dolan3 and Christopher A. Pohl4
1 Australian Centre for Research on Separation Science (ACROSS), School of Physical Sciences-Chemistry, University of Tasmania, Private Bag 75, Hobart 7001, Australia
2 Pfizer Global Research and Development, Sandwich, UK3 LC Resources Inc., Amity, OR, USA4 Thermo Fisher Scientific, Sunnyvale, CA, USA
Content: Quantitative structure-retention relationship (QSRR) analysis is a useful technique capable of relating chromatographic retention time to the chemical structure. In this work, three training datasets and QSRR were employed to predict the retention time for unknown compounds: for each unknown compound, a cluster of approximately the top 20 similar compounds was built. Then, compound descriptors (CDs) were calculated using Dragon. Finally, a genetic algorithm (GA) combined with partial least squares (PLS) regression was utilised to build the QSRR models to predict the retention time for unknown compound and the results were compared to the experimental retention

333www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSProcess Chromatography and Process Analytical Technology (PAT)
time. In addition, clustering methodologies based on chemical similarity (Tanimoto), chromatographic similarity (retention factor) and the physical-chemical property logD were compared. Current outcomes shows logD and Tanimoto similarity clustering do not compare well with the artificial chromatographic similarity found by retention factor. However, logD clustering was more accurate than Tanimoto structural similarity clustering. It can be concluded that the better the chromatographic similarity of the compounds in clusters to the unknown compound, the better the results in predicted retention values. Now a combination research is in progress and the hydrophobic subtraction model (HSM) will be applied to new solutes for retention prediction.References: 1. N. S. Wilson. et al. J. Chromatogr. A. 961.2 (2002)
171-193.2. L. R. Snyder, J. W. Dolan, P. W. Carr. J.
Chromatogr. A. 1060.1 (2004) 77-116.Acknowledgements: I would like to acknowledge my supervisors, Paul Haddad, Robert Shellie, Mohammad Talebi, Ruth Amos, and ACROSS (UTAS) for the support, and the Australian Research Council with industrial partners: Pfizer, Inc. (Roman Szucs), Thermo Fisher Scientific, Inc. (Chris Pohl), LC Resources, Inc. (John Dolan) for the grant.Disclosure of Interest:None DeclaredKeywords: Molecular Descriptors, Quantitative Structure-Retention Relationships , Retention Prediction
P03-003-073 - Carnosol purification from rosmarinus officinalis by centrifugal partition chromatography, from laboratory to industry
A. Berthod*, E. Bouju 1, M. Batteau 1, K. Faure 1
1 Institut of Analytical Sciences - CNRS, Villeurbanne, France
Content: Rosemary (Rosmarinus Officinalis) is an aromatic herbal plant belonging to the Lamiaceae family and known for its medicinal and taste properties. Recent studies have shown its pharmacologic activities for cancer chemoprevention and therapy due to phenolic compound presence such as carnosol, carnosic acid and rosmarinic acid. Carnosol was more specifically evaluated for anti-cancer properties in prostate, breast, skin, leukemia and colon cancer showing promising results. Its purification is required at lab-scale for toxicology studies and at industrial scale for production as an active ingredient. In this context, we present the centrifugal partition chromatography (CPC) method development and carnosol purification from a Rosemary leaves extract on a lab-scale instrument, highlighting the advantages of the CPC technique on natural products purification. After a rapid method development on a small-scale 35-mL CPC instrument that allowed for the determination of the solvent system and maximum sample concentration and volume, the purification was transferred on an industrial-scale 1-liter instrument using the “free space between peaks” method. The method takes into account the technical limitations of the larger instrument, such as pressure and/or maximum centrifugal field, and allows, by simply running an analytical-sized injection on the large scale rotor, to

www.ISC2016.ie @isc2016 #isc2016 /isc2016334
POSTER ABSTRACTS Process Chromatography and Process Analytical Technology (PAT)
give an accurate prediction of the maximum sample load and best throughput. The 0.27 g of rosemary extract maximum load on the 35-mL CPC was transferred as 9 g load (33 times bigger load) on the 1000-mL CPC (28 times larger column volume). If the scaling-up in CPC instruments is not directly homothetic, the described protocol makes it highly predictable through few simple experiments.References: 1. Elodie Bouju, Alain Berthod, Karine
Faure, J. Chromatogr. A, submitted (2016).Acknowledgements: The Kromaton Rousselet Robatel company (Annonay, France) and the CIFRE program of the French government are greatly acknowledged for the PhD grant supporting EB for three years.Disclosure of Interest:None DeclaredKeywords: Carnosol purification, countercurrent chromatography, gram-scale

335www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
Sample Preparation Techniques And Methods
P01-001-004 - Development of immunosorbents coupled to immobilized pepsin reactors for the quantitative analysis of human butyrylcholinesterase.
M. Bonichon 1,*, A. Combès 1, C. Desoubries 2, A. Bossée 2, V. Pichon 1
1 Department of Analytical, Bioanalytical Sciences and Miniaturization (LSABM), ESPCI Paris Tech, UMR CBI 8231, PSL Research University, Paris,
2 Analytical chemistry department, DGA CBRN Defence, Vert-le-Petit, France
Content: Human butyrylcholinesterase (HuBuChE) has been widely used as a biomarker of exposure to organophosphorus nerve agents (OPs). Indeed, the analysis of a specific HuBuChE peptide on which OPs covalently bind allows the detection of nerve agent intoxication for several weeks by LC-MS/MS analysis. Nevertheless, current HuBuChE analyses require tedious and time consuming digestion steps combined with a non-selective extraction procedure. Therefore, we aim to develop a fast and total on-line analysis of HuBuChE from plasma including immunoextraction (using HuBuChE antibodies), digestion on Immobilized Enzyme Reactors (IMER), and microLC-MS/MS analysis of the HuBuChE digest. This work focuses on the extraction and digestion steps, with the synthesis and the characterization of various immunosorbents (IS) and IMERs.
IS were made by the covalent grafting of three different anti-HuBuChE antibodies on CNBr-sepharose and polymetacrylate supports packed in pre-columns. Antibodies
grafting yields on these supports were evaluated for each antibody and sorbent. HuBuChE recoveries were assessed by LC/UV to determine the best antibodies/support combination for HuBuChE extraction. A control sorbent prepared with a nonspecific antibody was developed to confirm the selectivity brought by the anti-HuBuChE IS.
Several IMERs were prepared by varying the amount of immobilized pepsin on CNBr-sepharose supports. HuBuChE digestion yields on IMERs were quantified by microLC-MS/MS and IMERs were characterized in term of pepsin grafting yields. The digestion of HuBuChE on IMERs was optimized and performed in less than 20 minutes. Digestions on IMERs led to higher digestion yields than the digestion performed with free pepsin in solution. Finally, IMER synthesis was reproduced (n=3) and showed repeatable grafting process and HuBuChE digestion yields.
Therefore, the coupling of IS to this brief and repeatable IMER digestion will be discussed for a fast and total on-line analysis of BuChE-OP adducts.Disclosure of Interest:None DeclaredKeywords: Immobilized Enzyme Reactor, Immunosorbent, OP-HuBUCHE adducts
P01-001-037 - Development and validation of a quantitative method of carotenoids determination in organic extracts obtained from agro industrial waste materials
I. Rubashvili 1,*, M. Fillet 2, M. Tsitsagi 1

www.ISC2016.ie @isc2016 #isc2016 /isc2016336
POSTER ABSTRACTS Sample Preparation Techniques and Methods
1 Petre Melikishvili Institute of Physical and Organic Chemistry, Ivane Javakhishvili Tbilisi State University, Tbilisi, Georgia,
2 Laboratory for the Analysis of the Medicines, University of Liege, Liege, Belgium
Content: The present research concerns the development and validation of a new, rapid, modern, effective and selective method for carotenoids – beta-carotene and lycopene determination in organic extracts obtained from tomato, tangerine and orange agro industrial waste materials using supercritical fluid and sequential ultrasonication-assisted extraction procedures and high performance liquid chromatography.
The method was developed using RP-18 endcapped LiChroCART 4 x 250 mm, 5 µm column with gradient elution; The flow rate – 1.5 mL/min; The detector wavelength - 272 nm for lycopene and 455 nm for beta-carotene; The injection volume – 50 µL; Column temperature - 40 0C; The method was validated with respect to system suitability test, specificity, linearity-range, accuracy, precision, limit of detection (LOD) and quantitation (LOQ). The stability of solutions was studied as well. The calibration curve is linear over a concentration range 6.497 µg/mL – 0.0081 µg/mL for beta-carotene (the r2=0.99924) and 18.76 µg/mL – 0.034 µg/mL for lycopene (the r2=0.99990); The LOQ and the LOD are 0.0081 µg/mL and 0.0041 µg/mL for beta-carotene, 0.034 µg/mL and 0.0085 µg/mL for lycopene, respectively; No interference was observed; The main accuracy is 106.8 % for beta-carotene and 101.4 % for lycopene.
Hence, the proposed validated HPLC method with appropriate extraction procedure can be applicable for identification and quantitative determination of
carotenoids in organic extracts and is able to give the precise, reliable results in routine analysis.References: 1. ICH Harmonized tripartite guideline, validation of
analytical procedures, text and methodology Q2 (R1), 2005.
2. Eurachem Guide: The fitness for purpose of analytical methods – A laboratory guide to method validation and related topics, 2nd ed., 2014.
Acknowledgements: The research was supported financially By Shota Rustaveli National Science FoundationDisclosure of Interest:None DeclaredKeywords: Analytical method validation, High
performance liquid chromatography, Carotenoids
P03-003-074 - Identification and Detection of the new designer drug 4-Chloro-N-Ethylamphetamine using GC-MS, LC-Q(TOF)-MS, ATR-FTIR and NMR in the FSL-South Africa
Z. A. Mangombo 1,*, M. P. Mthembi 1
1 Chemistry, Forensic Science Laboratory-SAPS, Pretoria, South Africa
Content: The abuse of the designer Amphetamine-type stimulants (ATS) is increasing around the world. Their detection is the utmost important issue. The substitution of the hydrogen atoms from the group results in a large class of compounds that provide new designer drugs with the desired pharmacological properties, and/or avoid detection through normal testing protocol such as 4-Chloro-N-ethylamphetamine. This new designer drug, 4-Chloro-N-ethylamphetamine (4-CEA), is a homolog of the N-ethylamphetamine (EA) and/or neurotoxin 4-Chloroamphetamine.

337www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
Street samples consisting of capsules seized by the South African Police were submitted to the Forensic Science Laboratory (FSL) for further analysis. The following techniques such as Gas chromatography-mass spectrometry (GC-MS), Liquid chromatography quadrupole coupled with (time-of-flight) mass spectrometry (LC-Q(TOF)-MS), Attenuated total reflectance-Fourier transform infrared spectrometry (ATR-FTIR) and Nuclear Magnetic Resonance (NMR) spectroscopy were used to identify the samples. The study of the samples was tentatively identified as 4-Chloro-N-ethylamphetamine [1].References: [1] Lin TC, Lin DL., Lua AC., J.
Analytical Toxicology 35 (2011) 205-210Acknowledgements: The authors are thankful for the support of the Forensic Science Laboratory (FSL), the Quality Management Component, the Divisional Commissioner of Forensic Services as well as the South African Police Service (SAPS). Disclosure of Interest:None DeclaredKeywords: 4-Chloro-N-ethylamphetamine, Forensic Science Laboratory (FSL), Gas chromatography-mass spectrometry (GC-MS)
P03-003-075 - Design of strategies to study the metabolic profile of highly polar compounds in plasma by Liquid Chromatography-Mass Spectrometry
E. Sánchez-López 1,*, A. L. Crego 1, M. L. Marina 1
1 Departamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcala, Alcala de Henares, Spain
Content: Amino acids and their metabolites are important molecules which are involved in numerous metabolic pathways, serve
as building blocks for proteins, and act as biomarkers of several disorders [1]. When employing Liquid Chromatography (LC) with Reversed-Phase stationary phases most of these compounds are unable to be retained due to their high hydrophilic character. An interesting and easy strategy is to reduce their polarity through their derivatization with a labelling reagent, such as the commercially available 9-fluorenylmethyloxycarbonyl (FMOC) which offers many advantages such as forming stable complexes with primary and secondary amine moieties rapidly, improves the chromatographic separation, and also enhances the selectivity and sensitivity. Although some derivatization reagents have been employed in the study of metabolic profiles, FMOC has never been employed for this purpose.
In this work, it is demonstrated that the use of LC-MS employing a C18 column and using FMOC as labelling agent enables the determination of a larger number of hydrophilic compounds (proteinogenic amino acids, non-proteinogenic amino acids, and biogenic amines) when compared with the use of more polar stationary phases without using compound labelling. Different strategies for protein elimination were also carefully evaluated in plasma and results revealed that ultrafiltration gave rise to a larger number of compounds with a lower variability from sample to sample when compared with the protein precipitation method. Finally, the suitability of the proposed methodology in the metabolic profiling of plasma samples was also shown.References: [1] C. Nagata C, K. Wada, M. Tsuji, M.
Hayashi, N. Takeda, K. Yasuda, Cancer Cause Control 2 (143-149) (2014).

www.ISC2016.ie @isc2016 #isc2016 /isc2016338
POSTER ABSTRACTS Sample Preparation Techniques and Methods
Acknowledgements: Authors thank financial support from the Spanish Ministry of Economy and Competitiveness (project CTQ2013-48740-P) and the University of Alcalá (project CCG2014/EXP-059). Elena Sánchez-López thanks the University of Alcalá for her pre-doctoral contract.Disclosure of Interest:None DeclaredKeywords: Hydrophilic compounds, Liquid Chromatography Mass Spectrometry, Metabolic profile
P03-003-076 - Determination of Geosmin and 2-Methylisoborneol In Environmental Waters. Which is the Best Methodology?M. R. Boleda 1,*, L. Vegué 1
1 Aigües de Barcelona, Empresa Metropolitana de Gestió del Cicle Integral de l’Aigua, S.A., Barcelona, Spain
Content: Geosmin and 2-methylisoborneol are strong odour compounds produced mainly by cyannobacteria and actynomicets. These compounds are not associated to known adverse effects over human health, but their musty-earthy odour at low levels can disrupt the perception that consumers have about the safety of drinking water (odour thresholds around 10 ng/L for 2-MIB [1] and Geosmin [2]). This is the reason why their occurrence and behaviour in different water matrixes have been widely studied over the last years, and several analytical methods have been developed for their monitoring. The Grob Closed-Loop Stripping Analysis (CLSA) method [3] has been traditionally chosen for the analysis of these off-flavour compounds. This is a useful technique in case of odour and taste problems episodes, but its disadvantage arises from the long time required for the analysis of each sample. In order to overcome this problem, two
alternative methods have been optimized and tested: A) purge and trap (P&T) and Gas Chromatography (GC) coupled to Mass Spectrometry (MS), and B) Stir Bar Sorptive Extraction (SBSE) and GC coupled to Mass Spectrometry in tandem (MS/MS). P&T-GC/MS reduces at maximum the sample preparation and allows obtaining good recoveries with lower sample volume. Otherwise SBSE-GC-MS/MS allows an unequivocal identification of the analytes, together with a simple preparation process. This work discusses advantages and disadvantages of each methodology and summarizes the selected parameters for the extraction, detection and quantification in the analysis of geosmin and 2-MIB in water. The best methodology, in terms of sensitivity, selectivity, precision and robustness, is proposed and used in systematic analysis of these compounds.References: [1] BK Olsen, MF Chislock, AE Wilson - Water
research 92 (2016) 228-234.[2] F. Tan, H. Chen, D. Wu, N. Wang, Z. Gao, L. Wang
- Journal of Residuals Science&Technology, Vol 13,No 1 (2016).
[3] K. Grob, Journal of Cromatography 84 (1973) 255-273.
Disclosure of Interest: None DeclaredKeywords: Off-flavour, Purge and Trap, Stir Bar Sorptive Extraction

339www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
P03-003-077 - Rapid Determination of Drug Protein Binding Affinity using Solid Phase Micro Extraction
C. R. Aurand 1,*, D. S. Bell 1
1 Advanced Analytics, Merck, Bellefonte, United States
Content: Determination of free circulating drug is important in establishing the pharmacokinetic activity. In most cases, drug-protein complexes are formed thus affecting the active level of circulating drugs. Techniques used for determining drug protein binding levels consist of ultrafiltration, ultracentrifugation and micro dialysis. Automation can be used in the case of micro dialysis, but processing may be greater than 6 hours for equilibrium to be reached. In this study, a novel BioSPME micro extraction device is evaluated as a rapid means of determining drug protein binding affinities from plasma. Here, a model set of drugs with varied range of protein binding affinities were utilized to evaluate the utility of the BioSPME sampling technique. Initial studies demonstrate that drug binding affinities can be determined in less than 30 minutes using the micro extraction technique.
Protein binding affinities for compounds propranolol, warfarin and diclofenac are performed by comparative BioSPME extraction from plasma and phosphate buffered saline. Plasma and buffer samples are plated into 96well plates and extracted by immersion of the BioSPME fibers. The BioSPME fibers are then directly desorbed into solvent direct LC/MS quantitation. Correlation between BioSPME sampling technique and published micro dialysis data has demonstrated to be within 1%.
Preliminary data has demonstrated this as a rapid techinique for determining drug protein binding affinities. Additional studies to evaluate broader protein binding affinity drugs are being conducted.References: Journal of Laboratory
Automation February 2011 16: 56-67Disclosure of Interest: None DeclaredKeywords: drug protein binding, micro sampling, solid phase micro-extraction
P03-003-078 - Fast analysis of natural and artificial vanilla flavorings Computer assisted development of a robust, fast and sensitive UHPLC method
P. Jochems*
Content: Vanilla is one of the most important flavors worldwide and is widely used in foods, beverages and perfumes[1]. Natural vanilla extract contains up to several hundred substances with vanillin, vanillic acid, 4-hydroxybenzoic acid and 4-hydroxybenzaldehyd as the major components[2]. Due to the continuously increasing demand and the resulting high costs of natural vanilla extracts, artificial flavorings are often used instead. Vanillin can be obtained through various methods like chemical synthesis and biotransformation[3][4]. These artificial flavors can contain synthetic vanillin, ethyl vanillin, eugenol, guaiacol, vanillin mandelic acid and others.
As authenticity criteria for vanilla, the ratios of the major components vanillin, 4-hydroxybenzaldehyde, vanillic acid

www.ISC2016.ie @isc2016 #isc2016 /isc2016340
POSTER ABSTRACTS Sample Preparation Techniques and Methods
and 4-hydroxybenzoic acid are frequently used. In order to monitor the composition and therefore quality of vanilla flavors contained in food, an analytical method needs to enable individual quantification of any of these ingredients as well as possible ingredients like the precursors from the synthesis of vanillin.
This poster shows the development and optimization of a rapid UHPLC screening test for the separation and quantification of natural and artificial vanilla flavoring substances as well as some precursors for the quality control of vanilla products using an automated method scouting / method optimization workflowReferences: [1] A.S. Ranadive; in Charalambous G. 34 (pp. 517-
577) (1994)[2] J. Adeji, T.G. Hartmann, C.T. Ho; in Perf. Flav. 18
(pp. 25-33) (1993)[3] J.R. Desmurs et al.; in Perf. Flav. 29 (pp. 32-42)
(2004)[4] R. Berger; in Flavours and Fragrances 1 (2007)Disclosure of Interest:None DeclaredKeywords: computer assisted method development, flavourings, UHPLC
P03-003-079 - Application of Headspace-SPME Coupled with GC/MS for the Characterization of Polymeric MaterialsP. Kusch*
Content: Solid-Phase Microextraction (SPME) is a very simple and efficient, solventless sample preparation method, invented by Pawliszyn and co-workers in 1989 [1-3]. This method has been widely used
in different fields of analytical chemistry since its first applications to environmental and food analysis. SPME integrates sampling, extraction, concentration and sample introduction into a single solvent-free step. The method saves preparation time, disposal costs and can improve detection limits. It has been routinely used in combination with gas chromatography (GC) and gas chromatography/mass spectrometry (GC/MS) and successfully applied to a wide variety of compounds, especially for the extraction of volatile and semi-volatile organic compounds from environmental, biological and food samples [4].
Since the last twenty years, SPME in headspace (HS) mode is used as a valuable sample preparation technique for identifying degradation products in polymers and for determination of rest monomers and other light-boiling substances in polymeric materials [5-7]. For more than ten years, our laboratory has been involved in projects focused on the application of HS-SPME-GC/MS for the characterization of polymeric materials from many branches of manufacturing and building industries. This presentation describes the application examples of this technique for the characterization of polymer composites, rubber and packaging materials.References: [1] C. L. Arthur, J. Pawliszyn, Anal. Chem., 62(19),
1999, 2145–2148.[2] T. Górecki, J. Pawliszyn, Anal. Chem.,
67(18), 1995, 3265–3274.[3] J. Pawliszyn (Ed.), Application of Solid Phase
Microextraction, RSC, Cambridge, UK, 1999.[4] G. Vas, K. Vékey, J. Mass Spectrom., 39, 2004,
233-254.[5] M. Hakkarainen, S. Karlsson, A.-Ch. Albertsson, J.

341www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
Environ. Polym. Degrad., 5(2), 1997, 67-73.[6] P. Kusch, G. Knupp, J.Sep. Sci, 25(8), 2002, 539-
542.[7] P. Kusch, G. Knupp, J. Polym. Environ. 12(2),
2004, 83-87. Disclosure of Interest: None DeclaredKeywords: GC/MS, HS SPME, Polymeric materials
P03-003-080 - A simplified solid phase extraction procedure combined with liquid chromatography tandem mass spectrometry to assess pharmaceuticals in liquid samples
C. Afonso-Olivares 1,*, Z. Sosa-Ferrera 1, J. J. Santana-Rodríguez 1
1 Instituto Universitario de Estudios Ambientales y Recursos Naturales (i-UNAT), Universidad de Las Palmas de Gran Canaria, Las Palmas de Gran Canaria, Spain
Content: Direct analytical technique, to avoid sample treatment, is within twelve “Green Analytical Chemistry” (GAC) principles [1]. However, the existence of micropollutants, such as pharmaceuticals, which are compounds in the range from ng·L-1 to µg·L-1 [2], does not allow to follow this goal due to the necessity of a pre-concentration technique before to analysis by the modern analytical equipments.
Solid phase extraction (SPE) is the most commonly extraction technique and has become a reference standard method to compare with the novel extraction procedures. Standard SPE use a protocol with five steps that include conditioned, equilibrated, load, wash step and elution, therefore, a relative elevated consumption of time and quantity of solvent is required. However, thanks to the characteristic of
hydrophilic-lipophilic balanced copolymer sorbents, it could be used with a simplified system in three steps (without two first steps of standard SPE). As a result, we can obtain a reduction of consumed time and the quantity of solvent according to another GAC principle [3].
In this sense, we use N-vinylpyrrolidone-divinylbenzene copolymer (OASIS HLB) cartridges in a simplified SPE procedure, combined with Liquid Chromatography tandem Mass Spectrometry (LC-MS/MS) in order to determine twenty-three pharmaceuticals in liquid environmental samples. The optimal SPE conditions were 250 mL sample volume and pH 7, wash step of 5 mL of Milli-Q water and 5 mL of eluent (methanol). All analytical parameters were determined in order to validate the proposed analytical method and the applicability was evaluated in different wastewater samples.References: 1. A. Gałuszka, Z. Migaszewski, J. Namieśnik. TrAC-
Trend Anal. Chem. 50 (78–84) (2013).2. Y. Luo, W. Guo, H. Hao Ngo, L. Duc Nghiem, F.
Ibney Hai, J. Zhang, S. Liang, X. C. Wang. Sci. Total Environ. 473–474 (619–641) (2014).
3. X. Zhang, P. C. Iraneta, F. J. Marszalkowski, K. J. Fountain. Application Note. Waters. (2014)
Acknowledgements: Cristina Afonso-Olivares thanks to FPU Grant Program of the Spanish Ministry of Education, Culture and SportDisclosure of Interest: None DeclaredKeywords: LC-MS/MS, Pharmaceuticals, Simplified SPE

www.ISC2016.ie @isc2016 #isc2016 /isc2016342
POSTER ABSTRACTS Sample Preparation Techniques and Methods
P03-003-081 - Natural product purification by Centrifugal Partition Chromatography
Y. G. Hou 1,*, D. Rutterschmid 1, Z. Kovács 1, L. Lorántfy 1, L. Németh 1, Z. Misek 1
1 Rotachrom Technológiai kft., Dabas, Hungary
Content: Napalese yew (Taxus wallachiana) is used to produce paclitaxel (taxol), a chemotherapeutic agent.Natural taxol can be isolated from the bark of the tree in a small quantities, such work has put the yew on the IUCN red list of threatened species.
We have received dryed methanolic samples of different yew leaves containing baccatin-III, paclitaxel and 10-DAB-III as well. For the purification we have used Centrifugal Partition Chromatography (CPC),a liquid-liquid chromatography technique, where the liquid stationary phase is immobilized by a strong centrifugal force while the liquid mobile phase is pumped through the stationary phase.
We have evaulated different solvent systems for the possible purification of these mixtures. However due to some selectivity problems it was necessary to use two separation steps, to achieve high loadings and high yields, while mainting high output purity (>98%).
We decided to use a solvent system incorporating dichloromethane for the first step, where we experienced multy-gramm loadings of the dryed curde extract on the smallest CPC instruments. After the first step we achieved purities of >70%, while maintint complete (>98%) recovery of 10-DAB-III, and easy evaporation of the chlorinated solvent during the sample
concentration. For the second purification steps we had multiple choices for purification. Using a mixture of methyl-tert-buthyl-ether, acetone and water we were able to achieve 10-DAB-III purities of 98% purity and one-step recovery of 70%.
This combined purification process proved low production cost, a quick and efficient way to purification of 10-DAB-III,with the aprralel purification of side-products like natural paclitaxel.References: A.P. Foucault, Centrifugal Partition Chromatography
Chapter 1 (1994)Vanek T, Vesela D, Pospisilova R. Study of the
influence of year season on the taxanes content in Taxus baccata bark. Planta Med 1993; 59:A698.
Y. K. Park, K. H. Row, S. T. Chung, Separation and Purification Technology 27-37 p. (2000)
Disclosure of Interest:None DeclaredKeywords: anti-cancer drug, Centrifugal Partition
Chromatography, liquid-liquid chromatography
P03-003-082 - Analysis of Hormones in Sludge Samples using Microwave-Assisted Extraction and Ultra-High Performance Liquid Chromatography Tandem Mass Spectrometry
R. Guedes-Alonso 1,*, S. Santana-Viera 1, C. Afonso-Olivares 1, S. Montesdeoca-Esponda 1, Z. Sosa-Ferrera 1, J. J. Santana-Rodríguez 1
1 Instituto Universitario de Estudios Ambientales y Recursos Naturales (i-UNAT), Universidad de Las Palmas de Gran Canaria, Las Palmas de Gran Canaria, Spain
Content: Steroid hormones are a group of natural and synthetic compounds which are considered as endocrine disruptor

343www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
compounds due to their characteristics and possible toxic effects over aquatic biota like changes in fertility or hermaphroditism [1]. Wastewater treatment plants (WWTPs) are the major sources of these compounds into the environment and because of their non-polar nature, they can be adsorbed on solid matrices.
Nowadays, studies about determination of steroid hormones in solid samples are scarce in comparison with studies in liquid samples. However, as happen in liquid samples, concentrations of steroid hormones in solids are very low and it is necessary the development of methodologies of extraction and preconcentration which permits the determination of this kind of micropollutants at measurable concentrations [2].
Microwave-assisted extraction (MAE) is an easy technique which uses small solvent volumes, short times and it allows the extraction of analytes in complex matrices [3]. In this work a methodology based in MAE for the extraction of sex hormones and corticosteroids from sewage sludge is developed. All the parameters involved in the extraction as, irradiation power (100–500 W), time (2–10 minutes) or volume of extractant (5–10 mL) were evaluated using two experimental designs which were applied to assess the effects of each variable in the extraction procedure.
After the optimization of the variables involved in the procedure, all the analytical parameters were calculated in order to evaluate the quality of the extraction and determination method. Finally, it was applied to real samples of sewage sludge of a WWTP located in Gran Canaria (Spain).
References: 1. A. Pal, K.Y.-H. Gin, A.Y.-C. Lin, M. Reinhard, Sci.
Total Environ. 408 (6062–6069) (2010)2. R. Guedes-Alonso, S. Montesdeoca-Esponda, Z.
Sosa-Ferrera, J.J. Santana-Rodríguez, Trends Environ. Anal. Chem. 3–4 (14-27) (2014)
3. T. Vega-Morales, Z. Sosa-Ferrera, J.J. Santana-Rodríguez, Talanta 85 (1825-1834) (2011)
Acknowledgements: This work was supported by funds provided by the Spanish Ministry of Economy and Competitiveness, Research Project CTM2015-66095-C2-1-R. Rayco Guedes-Alonso thanks the University of Las Palmas de Gran Canaria (Spain) for his Ph.D. student grant.Disclosure of Interest:None DeclaredKeywords: Microwave-assisted extraction, Steroid hormones, UHPLC-MS/MS
P03-003-083 - Development of ion imprinted polymers for the selective extraction of lanthanide ions
M. Moussa 1, T. Pinta 1, V. Pichon 1 2,*, T. vercouter 3, N. Delaunay 1
1 LSABM-UMR CBI 8231- CNRS/ESPCI, 2 UPMC, Sorbonne universités, Paris, France, Paris, 3 DEN, DANS, DPC, SEARS, LANIE, CEA Saclay, Gif-sur-
Yvette, France
Content: The increased use of lanthanides (Ln) in industrial applications has induced their release into the environment. Since Ln are present at trace level in complex environmental samples, an extraction and preconcentration step is often necessary prior to their quantification [1]. Solid phase extraction (SPE) using ion imprinted polymers (IIPs) as sorbents is a selective method to extract Ln [2]. This project aims to develop IIP SPE sorbents to selectively extract Ln ions from natural water, before their analysis by Inductively Coupled Plasma Mass Spectrometry (ICP-MS).

www.ISC2016.ie @isc2016 #isc2016 /isc2016344
POSTER ABSTRACTS Sample Preparation Techniques and Methods
Binary complex Ln-IIP was synthesized in acetonitrile using Nd, methacrylic acid (MAA) and ethylene glycol dimethacrylate (EGDMA) as template, ligand and crosslinker, respectively. Other IIPs were synthetized by forming ternary complexes between either Nd3+ or Er3+, MAA and 2-vinylpyridine (2-VP) in acetonitrile, EGDMA was used as crosslinker. Non-imprinted polymers (NIPs) were synthetized under the same conditions as for IIPs but without the ion templates.
After the template ions elimination, all the synthesized polymers were packed in SPE cartridges and each SPE step was optimized, i.e. conditioning, loading, washing, and elution.
Recovery yields of more than 80% were obtained with the binary complex IIP for a mixture of light and heavy Ln (La, Nd, Sm, Gd, Dy, Er and Lu), against less than 25% in the case of the corresponding NIP. Ternary complex IIP using Nd3+ as template, offered similar results as for the binary complex IIP. However, IIPs synthetized with Er3+ and Nd3+ as templates showed a different behavior towards light Ln which seems to confirm the imprinting effect. Reproducibility of synthesis was also successively demonstrated. Finally, extraction of Ln from real water samples was realized to demonstrate the potential of these SPE sorbents. References: [1] K. Pyrzynska, A. Kubiak, I. Wysocka, Talanta. 154
(2016) 15–22.[2] T. Prasada Rao, S. Daniel, J. Mary Gladis, TrAC
Trends Anal. Chem. 23 (2004) 28–35.Disclosure of Interest:None Declared
P03-003-084 - High performance liquid chromatographic analysis of amino acids and biogenic amines in fermented fishZ. Joungmunkong 1, S. Boonchiangma 2, C. Yenjai 2, S. Srijaranai 2,*
1 Faculty of Pharmaceutical Sciences, Ubon Ratchathani University, Ubon Ratchathani,
2 Chemistry, Khon Kaen University, Khon Kaen, Thailand
Content: Biogenic amines (BAs) are the products of the decarboxylation of amino acids (AAs). BAs usually occur in fermented food such as sausages, fermented fish, beer etc. The important BAs in food are histamine, putrescine and tyramine, which are produced from the AAs; histidine, arginine and tyrosine, respectively. Analysis of AAs and BAs in foods is very interesting. Nutritional data on type and quantity of AAs in food are benefit for health, while the presence of BAs is toxic to consumers.
This study demonstrates the simultaneous analysis of AAs and BAs by HPLC. The studied analytes are nine AAs (i.e. arginine, asparagines, glutamic acid, histidine, leucine, lysine, phenylalanine, tryptophan and g-aminobutyric acid) and five BAs (i.e. cadaverine, histamine, putrescine, tryptamine and tyramine). AAs and BAs were pre-column derivatized with o-phthaladehyde-ethanethiol (OPA-ET) before the analysis by reversed phase HPLC. The optimum conditions for both derivatization and HPLC were studied. The optimum conditions for derivatization were the molar ratio of AAs and BAs to OPA-ET of 1:20 and reaction time 5 min. Chromatographic separation was carried out on a Waters Symmetry C8 column, using gradient elution of methanol and water at a flow rate of 0.5 mL/min. Fluorescent

345www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
detection was employed with the excitation wavelength at 278 nm and emission wavelength at 360 nm. The studied analytes were completely separate within 25 min. Linear ranges were 0.02-10.00 mg/mL with the correlation coefficients higher than 0.9990. Limit of detections based on 3 signal to noise ratio were 0.01 mg/mL (tyramine)-0.10 mg/mL (lysine). The proposed method was successfully applied for fresh and fermented fish samples.References: 1. Ezzat M.A., Zare D., Karim R., Ghazali H.M., Food
Chemistry 172 (893-899) (2015) .2. Dai Z., Wu Z., Jia S., Wu G., Journal of
Chromatography B 964 (116-127) (2014).Acknowledgements: Financial support from the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Commission on Higher Education, Ministry of Education, Thailand is gratefully acknowledged. Disclosure of Interest:None DeclaredKeywords: amino acid, biogenic amine, o-phthaladehyde-ethanethiol
P03-003-085 - Analysis of carboxylic acids in raw sugar and cane juice by ion exchange chromatography
S. Boonchiangma 1, W. Wannachai 2, S. Srijaranai 1, C. Ruangviriyachai 1,*
1 Chemistry, Khon Kaen University, Khon Kaen, 2 United Farmer & Industry co., Ltd., Chaiyaphum, Thailand
Content: Carboxylic acids play important roles in sugar production from sugarcane such as affecting clarification process and crystallization of sugar. In addition, content of carboxylic acids is an index for cane juice and raw sugar quality. Therefore, monitoring of carboxylic acids is an essential task in sugar industry.
A simple method for the determination of carboxylic acids using ion exchange chromatography equipped with conductometric detection was developed. Five carboxylic acids studied are lactic acid, succinic acid, oxalic acid, citric acid and aconitic acid. The separation was performed using an AS4A-Sc Dionex anionic exchange column and the gradient elution of sodium hydroxide ranged from 50 to 140 mmol/L at a flow rate of 0.7 mL/min. The complete separation was achieved within 14 min. The linearity of all the studied analytes was in the range of 0.10-100 mg/mL, except succinic acid, with the correlation coefficients higher than 0.997. Limit of detections, based on signal to noise ratio of 3, were in the range 0.05 mg/mL (oxalic acid and citric acid) – 0.50 mg/mL (lactic acid). As previous report, the highest content of carboxylic acids found in both cane juice and raw sugar samples is aconitic acid, which in the ranges of 0.17-0.34 % and 0.08-0.18% for cane juice and raw sugar samples, respectively. References: 1. Kemmei T., Kodama S., Yamamoto A., Inoue Y.,
Hayakawa K., Journal of Chromatography A 1375 (49-53) (2015).
2. Jaffé W.R., Journal of Food Composition and Analysis 43 (194-202) (2015).
Acknowledgements: Financial support from the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Commission on Higher Education, Ministry of Education, Thailand is gratefully acknowledged.Disclosure of Interest:None DeclaredKeywords: carboxylic acid, ion exchange chromatography, raw sugar

www.ISC2016.ie @isc2016 #isc2016 /isc2016346
POSTER ABSTRACTS Sample Preparation Techniques and Methods
P03-003-086 - A Novel and Quick QuEChERS Method To Determine Residues Of B-Lactam Antibiotics In Pork By UHPLC-MS/MS
D. Barron Bueno 1,*, L. Domínguez-Ruiz 1, A. Junza 1, J. F. López-Sánchez 2
1 Food and Nutrition Torribera Campus, 2 Analytical Chemistry, Universitat de Barcelona,
Barcelona, Spain
Content: Antibiotics are widely used in the veterinary field for the treatment of diseases in food-producing animals such as cows, pigs and chickens. The widespread use of such compounds represents a potential risk to human health. Residues of these compounds may be present in edible tissues destined for human consumption. To protect human health, the EU has established safe maximum residue limits (MRLs) for veterinary drugs in animal tissues destined to be consumed (EU Regulation 37/2010).
A method based on QuEChERS (Quick, Easy, Cheap, Effective, Rugged and Safe) methodology has been optimized and validated for the determination of residues of blactam antibiotics (penicillins and cephalosporins) in edible pork tissues (muscle, liver and kidney) by ultra-high performance liquid chromatography coupled to tandem mass spectrometry (UHPLC-MS/MS). The optimization of compositions of QuEChERS was determined applying a Box-Behnken design for 3 factors (C18, PSA and MgSO4). The optimum conditions were determined to be 400 mg PSA and 300 mg MgSO4 for muscle. However, 100 mg PSA and 850 mg MgSO4 resulted to be the best QuEChERS composition for liver and kidney analysis.
The method was validated according to the European guidelines (Decision 2002/657/EC) using matrix-matched standard calibration followed by a recovery assay with spiked samples. LOQs were lower than the established MRL for all antibiotics and matrixes. In general, the precision in samples prepared the same day (intra-day) and different days (inter-day) was lower than 15%, as required by the regulation. Finally, in order to evaluate the applicability of the method, samples of muscle, liver and kidney of medicated pigs with amoxicillin were analysed.References: R. Pérez-Burgos, E.M. Grzelak, G. Gokce, J.
Saurina, J. Barbosa, D. BarrónJ. Chromatogr. B, 899, 57-65 (2012) Acknowledgements: The authors are gratefull to the Ministerio de Economia y Competitividad (Project CTQ2013-44077-R) for finantial supportDisclosure of Interest:None DeclaredKeywords: b-lactamic antibiotics, QuEChERS method, UHPLC-MS/MS
P03-003-087 - Synthesis Of Molecular Imprinted Polymers (MIPs) For The Extraction Of Antibacterial QuinolonesB. Benedetti 1, A. Lascorz 1, A. Ruiz-Garrido 1, E. Megías 1, I. Gutiérrez-Mas 1, A. Junza 1, D. Barrón 1, C. Minguillón 1,*
1 Food and Nutrition Torribera Campus, Universitat de Barcelona, Barcelona, Spain
Content: The presence of residues of antibiotics in foodstuff of animal origin can involve a risk on consumer’s health. Quinolones are among the most used antibacterial agents in human and veterinary medicine. European regulations

347www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
establish MRL (maximum residue limits) for eight compounds in this series.
Classical sample treatment techniques such as solid phase extraction (SPE) and liquid-liquid extraction (LLE) are the most used methods in the determination of residues of antibiotics in food. However, these are procedures which involve a low selectivity. In this regard, it has been proved that molecularly imprinted polymers (MIPs) permit to increase selectivity for a diversity of compounds. In this study to improve extraction selectivity for antibacterial quinolones, the preparation and optimization of MIPs has been undertaken.
The synthesis of MIPs involves the polymerization of a functional monomer with a crosslinker, in the presence of a porogenic solvent and a template. Acetonitrile, methanol and chloroform were studied as porogen solvents. Diethylaminoethyl methacrylate (DAM), methyl metacrylate (AM) and 4vinylpyridine (VP) were tested individually as monomers, with ethylene dimethacrylate (EDMA) as a crosslinker. 2,2’Azobisisobutyronitrile (AIBN) was used as radical initiator. Levofloxacin was chosen as a template. The choice of this compound was determined by the fact of being levofloxacin a not regulated quinolone. The diverse polymers prepared were evaluated against enrofloxacin retention. Moreover, MIP polymers were characterized by SEM and BET. Also, NMR was used in the assessment of the cleaning quality (removal of template) and retention (enrofloxacin) of the considered MIPs. Finally, selectivity and competitiveness tests for diverse quinolones (sarafloxacin, norfloxacin, enrofloxacin and ciprofloxacin) were performed.
References: J.L. Urraca, M. Castellari, C.A. Barrios, M.C. Moreno-Bondi. J. Chromatogr. A, 1343, 1-9 (2014)
Acknowledgements: The authors are grateful to the Ministerio de Economia y Competitividad (Project CTQ2013-44077-R) for finantial supportDisclosure of Interest:None DeclaredKeywords: Molecular Imprinted Polymer (MIP), Optimisation, quinolone
P03-003-088 - Determination of 17 common organic acids found in milk-based beverages using ion exclusion chromatography.
M. D. Jones 1,*, M. Breen 1 2, D. Connolly 1 2
1 PMBRC, Waterford Institute of Technolog, 2 Science, PMBRC, Waterford, Ireland
Content: Milk-based beverages contain a number of essential bio-active compounds such as organic acids. Regulation of organic acid content is stringent and so their qualitative and quantitative analysis is of huge importance in the scientific community. Organic acids in milk occur naturally due to hydrolysis of fats, biochemical metabolic processes and bacterial growth [1]. They are also added during the manufacturing process to enhance specific flavours, regulate shelf life or to increase the quality of the end product. An ion exclusion chromatographic method has been developed for the separation of 17 organic acids found in milk, those being: oxalic, orotic, citric, tartaric, pyruvic, D-malic, succinic, uric, lactic, formic, acetic, propionic, iso-butyric, n-butyric, iso-valeric, n-valeric & hippuric acid. Separation was achieved using an HPLC system fitted with a strong cation exchange column (Aminex HPX-87H, 300 x 7.8 mm x 9 µm). The mobile

www.ISC2016.ie @isc2016 #isc2016 /isc2016348
POSTER ABSTRACTS Sample Preparation Techniques and Methods
phase consists of 2 mM sulphuric acid & 10% MeCN with a flow rate of 0.4 mL/min. UV detection was optimised using wavelength switching at 210 & 220 nm with an injection volume of 20 µL. To achieve resolution between all analytes, two methods were required at different temperatures: 45 oC & 65 oC with approximate run times of 15 & 35 minutes, respectively. A novel extraction technique known as cloud point extraction is undergoing preliminary investigations using the environmentally benign, non-ionic surfactant Triton X-100 to simultaneously pre-concentrate and extract target analytes.References: R.T. Marsili, J. Food Sci. 46 52–57
(1981)Acknowledgements: Waterford Institute of Technology Scholarship ProgramDisclosure of Interest:None DeclaredKeywords: cloud point extraction , ion exclusion chromatography, Organic acids in milk
P03-003-089 - The Study of Potential use of Nanofiber Polymers as Sorbents for the On-Line Extraction In HPLC
M. Parmová 1,*, L. Chocholoušová Havlíková 1, D. Šatínský 1, J. Fibigr 1, J. Chvojka 2
1 Department of Analytical Chemistry, Faculty of Pharmacy in Hradec Králové, Charles University in Prague, Hradec Králové,
2 Technical university of Liberec, Liberec, Czech Republic
Content: Nowadays, SPE (solid phase extraction) is the most popular sample pre-treatment technique. Sorbents for SPE are still innovated and one of these innovations is the using of nanofibers as an extraction phase. Electrospinning is one of the most used processes for forming the nanofibers.
Nanofibers are formed from solution or melt of polymer thanks to electrostatic field.1 Polymers which are most often used as sorbents for SPE are polyamide 6, polypyrrole and polystyren.
Recent trends in sample pre-treatment are also focused on the possibility of automation of all steps and the on-line connection of the sample pre-treatment with suitable analytical technique. Sample pre-treatment in on-line connection is realized in the HPLC system, and after extraction the analytes are directly injected to the analytical column.
This work was focused on the application of nanofiber polymers as sorbents in on-line SPE-HPLC. Tested analytes were chosen from the groups of pyrethroids and carbamates. Tested nanofibers were polyamide 6, polycaprolactone and polystyrene. C18 sorbent was chosen as a referent sorbent. Retention of the analytes using various SPE conditions (valve switching time, sample washing step) was observed. These conditions and HPLC mobile phase composition for SPE-HPLC were optimized. Linearity and repeatability were tested and evaluated for each polymer individually using the optimal HPLC conditions.References: 1 N., Sasithorn, L., Martinová, J.
Nanomater. (1-9) (2014).Acknowledgements: The study was supported by specific research, no. SVV 260 292.Disclosure of Interest:None DeclaredKeywords: HPLC, Nanofibers, on-line SPE

349www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
P03-003-090 - Evaluation of the potential of carbosilane dendrimers coated nanotubes in protein sample preparationE. González-García 1,*, C. E. Gutiérrez Ulloa 2, F. J. de la Mata 2, M. L. Marina 1, M. C. García 1
1 Departamento de Química Analítica, Química Física e Ingeniería Química,
2 Departamento de Química Orgánica y Química Inorgánica, CIBER-BBN, Universidad de Alcalá, Alcalá de Henares, Spain
Content: Carbosilane dendrimers are nanostructured hyperbranched macromolecules whose silicon-carbon skeleton confers them unique properties. Interactions between proteins and carbosilane dendrimers functionalized with sulphonate or carboxylate groups have been carried out. Moreover, carboxylate-terminated dendrimers have demonstrated a great potential in protein sample preparation. Nevertheless, a practical limitation appears in the separation of proteins from the dendrimer itself.
On the other hand, single-walled carbon nanotubes (SWCNTs) are allotropes of carbon with cylindrical structure of a graphene sheet. Due to the extraordinary combination of mechanical and optical properties and electrical and thermal conductivities, SWCNTs have been used in a wide range of bioapplications since they appeared 25 years ago. Many of these applications are based on interactions between proteins and SWCNTs [1]. Functionalization of SWCNTs with sulphonate dendrons could improve the interaction between proteins and SWCNTs and make easier their separation.
The aim of this work was to explore the nature and characteristics affecting the
interaction between functionalized SWCNTs and standard proteins and to evaluate their potential in protein purification/enrichment.
SWCNTs unfunctionalized and functionalized with three different generations of sulphonate-terminated carbosilane dendrons were used. Different pHs and concentrations were tried and three standard proteins covering a wide range of isoelectric points and molecular weights were employed. Variation of proteins fluorescence intensity and emission wavelength were used as probes to study the interaction between proteins and SWCNTs. Both parameters resulted affected when SWCNTs and proteins were in contact. Most significant changes were observed at neutral and basic pHs and when increasing dendron generations. A static proteins quenching effect of SWCNTs was found.References: [1] L. Li, et al., J. Lumines. 145 (2014)
125-131.Acknowledgements: Authors thank the Spanish Ministry of Economy and Competitiveness (AGL2012-36362 and CTQ2014-54004-P) and the Comunidad of Madrid (Spain) and FEDER program (S2013/ABI-3028). E.G.-G. thanks the University of Alcalá for her pre-doctoral contract.Disclosure of Interest:None Declared
P03-003-091 - Fatty acid profiling in bovine milk in selected breeds using dispersive liquid-liquid microextraction coupled with GC-FIDA. Quigley*, S. Walsh 1, W. Cummins 1, D. Connolly 1
1 Science, Waterford Institute of Technology, Waterford, Ireland

www.ISC2016.ie @isc2016 #isc2016 /isc2016350
POSTER ABSTRACTS Sample Preparation Techniques and Methods
Content: Bovine milk is an important source of energy, protein and essential vitamins and minerals. It lends itself to the manufacture of a range of dairy products. Fatty acid composition has a marked influence on the nutritional quality of milk and hence the properties of dairy products. Some of these fatty acids have health benefits (conjugated linoleic acid [1]), while others are detrimental to human health (saturated fatty acids [2]). Intrinsic (breed, animal genetics, stage of lactation, health status) and extrinsic (diet, management and season) factors as well as their interactions can alter fatty acid composition. Holstein-Friesians produce milk with the lowest fat content (~3.5%), while Jerseys the highest (~6%). However, there is limited research carried out in beef cow breeds. Seasonal production systems have a marked effect on milk fat, which varies from ~3% in early lactation to >4.5% in late lactation. Milk samples (n=3) were collected from Rotbunt and Holstein-Friesian cows in glass bottles at two different time-points during the lactation cycle and immediately frozen at -20 °C prior to analysis. This current work focuses on a novel extraction and pre-concentration protocol for fatty acids in bovine milk, followed by separation and identification using GC-FID (gas chromatography-flame ionisation detection). Dispersive liquid-liquid micro-extraction (DLLME) involves the rapid injection of an immiscible extraction solvent into an aqueous sample in the presence of a third dispersion solvent, producing a stable emulsion comprised of micro-droplets of extractant into which the analyte rapidly partitions. Centrifugation of the ternary mixture facilitates recovery of the sedimented extraction solvent prior to GC analysis. Pre-concentration factors (several hundred-fold) can be optimised
by fine-tuning of ternary solvent volume ratios, pH, aqueous phase ionic strength etc. results are presented herein.References: [1] K. Koba. 8 (1–8) (2013) .[2] Y.S. Diniz. 20 (230–234) (2004).Disclosure of Interest:None DeclaredKeywords: DLLME, gas chromatography, milk
P03-003-092 - On-line extraction on molecularly imprinted polymers coupled with chromatographic determination of patulinI. Lhotská 1,*, M. Parmová 1, A. Holznerová 1, D. Šatínský 1
1 Department of Analytical Chemistry, Faculty of Pharmacy in Hradec Králové, Charles University in Prague, Hradec Králové, Czech Republic
Content: Patulin, mycotoxin produced by moulds of genera Penicillium, Aspergillus or Byssochlamys, is important contaminant of apples. As patulin is intestine-irritating and cytotoxic and genotoxic, it is controlled in apple products by EU legislation.
The AOAC (The Association of Analytical Communities) Official Method for patulin determination uses liquid-liquid extraction with ethyl acetate and treatment with sodium carbonate solution1. However, patulin is unstable in alkaline solutions2. Several methods using solid-phase extraction were published. To achieve more selective extraction of patulin, molecularly imprinted polymers (MIPs) are also commercially available.
In our study, MIP sorbent was packed into holder and connected on-line in column-switching system. This enabled on-line extraction and preconcentration of patulin.

351www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
However, juice matrix is very complex and the interfering compounds were not removed completely which made reaching lower concentration of patulin impossible. Several approaches were tested to improve the elimination of interfering matrix.
This work deals with on-line coupling of patulin MIPs to HPLC for the first time. On-line extraction minimizes time of whole analysis, human errors by handling with sample and solvent consumption, in comparison with off-line procedure.References: 1 MacDonald et al.: Journal of AOAC International
83 (6) 20002 N. De Clercq et al., Food Analytical Methods (370-
377) (2016)Acknowledgements: This study was supported by the Charles University - project of specific research, SVV No. 260 292, and project of Charles University Grant Agency, GAUK No. 726316.Disclosure of Interest:None DeclaredKeywords: molecularly imprinted polymers (MIP), on-line SPE, patulin
P03-003-093 - Isolation of Human Milk Whey Proteins by Solid Phase Extraction with a Ground Monolith modified with Gold NanoparticlesJ. M. Herrero-Martínez*, I. Ten-Doménech 1, E. F. Simó-Alfonso 1
1 Analytical Chemistry, University of Valencia, Burjassot, Spain
Content: Human milk is considered the optimal way of providing young infants with the necessary nutrients for healthy growth and development. In particular, the protein fraction in milk plays an important role in achieving many of the benefits of
breast-feeding. In human milk, the whey fraction contains the majority of the protein, whereas casein has a smaller fraction. The isolation of human whey proteins has some difficulties, since some whey proteins may coprecipitate with caseins at pH 4.6. For these reason, a relative simple method to allow rapid analysis of the major whey proteins in human milk might be of interest.
In this work, a method for the isolation of human milk whey proteins by solid-phase extraction (SPE) with a ground monolith modified with gold nanoparticles (AuNPs) is described. Firstly, the effect of pH in the loading and washing step was studied. For this purpose, proteins with very different isoelectric point (pI), such as, human serum albumin (pI 4.7), alpha-lactalbumin (pI 4.9), lactoferrin (pI 8.7) and lysozyme (pI 11) were used as test solutes. Relatively high retentions were achieved for all four proteins regardless of the loading pH. The ionic strength of the elution solution was also optimized. Good recoveries were obtained with a phosphate buffer solution at pH 12 with 1 M NaCl. A human milk whey sample and a direct dilution of a milk sample were passed through the SPE cartridge at optimized pH with retention values higher than 90% of their protein content, which demonstrates the effectiveness of the proposed method. References: .Acknowledgements: Project CTQ2014-52765-R (MINECO of Spain and FEDER). I. T-D thanks the MINECO for an FPU grant for PhD studies.Disclosure of Interest:None DeclaredKeywords: gold nanoparticles, human milk proteins, SPE extraction

www.ISC2016.ie @isc2016 #isc2016 /isc2016352
POSTER ABSTRACTS Sample Preparation Techniques and Methods
P03-003-094 - A new proposal for the sustainable extraction of proteins based on carbosilane dendrimer coated gold nanoparticles
R. Vásquez-Villanueva 1,*, C. Peña-González 2, F. J. de la Mata 2, M. L. Marina 1, M. C. García 1
1 Departamento de Química Analítica, Química Física e Ingeniería Química,
2 Departamento de Química Orgánica y Química Inorgánica. CIBER-BBN, Universidad de Alcalá, Alcalá de Henares, Spain
Content: Dendrimers are synthetic nanostructures made up of a central core surrounded by branches called dendrons. Dendrimers can be customized with suitable terminated groups for their interaction with biological molecules [1]. On the other hand, gold nanoparticles (GNP) have become an attractive material widely employed in biochemistry, medicine and analytical chemistry due to their physical and chemical properties. GNPs have been functionalized with dendrimers to increase their affinity to proteins, stability in physiological media, and surface functionality [2]. Interactions between dendrimer coated GNPs and proteins have been used for the development of a biosensor and its application to the monitorization of proteins [3]. The aim of this work was to propose a new strategy for protein sample preparation based on carbosilane dendrimer coated GNPs as an alternative to usual methods that require the use of high amounts of organic solvents and reagents.
Carbosilane dendrimer coated GNPs of first, second, and third generation functionalized with sulfonate and carboxylate groups were employed. Their interaction with three standard proteins
(bovine serum albumin, myoglobin, and lysozyme) was monitored under different conditions (protein: GNP ratio, pH, polarity, and dendrimer generation). Intrinsic fluorescence of proteins due to tryptophan residues was quenched when adding different concentrations of dendrimer-GNPs. Gel electrophoresis was employed to further support the interactions between proteins and dendrimer-GNPs. Most suitable dendrimer-GNP and favorable conditions were used in the evaluation of the performance of these nanosystems in protein sample preparation.References: [1] C. E. Peña-González, et al. Chem.-Eur. J., 22
(2016) 2987-2999.[2] N. Bogdan, et al. RSC Advances, 2 (2012) 985-
991.[3] T. J. Choo, et al. Lagmuir, 30 (2014) 3883-3893. Acknowledgements: Authors thank the Spanish Ministry of Economy and Competitiveness (AGL2012-36362 and CTQ2014-54004-P) and the Comunidad of Madrid (Spain) and FEDER program (S2013/ABI-3028). R.V.V. thanks the University of Alcalá for her pre-doctoral contract.Disclosure of Interest:None Declared
P03-003-095 - Enantiomeric separation of non-protein amino acids by Electrokinetic Chromatography
R. Pérez Míguez 1,*, M. L. Marina 1, M. Castro-Puyana 1
1 Departamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcalá, Alcalá de Henares, Spain
Content: A high number of non-protein amino acids are chiral compounds that have demonstrated to be relevant in the pharmaceutical, clinical and food fields.

353www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
The study of their chiral behavior is a matter of interest since L-enantiomers are present in nature while D-enantiomers can occur through different metabolic pathways, by different processing conditions employed by the food industry, synthesized in enzymatic pathways by the action of microorganisms or they can even be present in supplemented foodstuffs due to the fraudulent addition of racemic mixtures [1, 2].
The aim of this work was to develop new analytical methodologies by Electrokinetic Chromatography (EKC) to achieve the enantiomeric separation of non protein amino acids of interest. Pyroglutamic acid, norvaline, norleucine, 3,4-dihydroxy-phenylalanine, selenomethionine, 2-aminoadipic acid, citrulline and pipecolic acid were derivatized with FMOC (9-fluorenylmethoxycarbonyl chloride) prior to their analysis by EKC using a formate buffer at acidic pH and anionic cyclodextrins as chiral selectors. The developed methodologies enabled the enantiomeric separation of the compounds studied showing changes in the enantiomeric migration order depending on the structure of the amino acids.References: 1] R. Pérez-Míguez, M. L. Marina, M. Castro-Puyana,
J. Chromatogr. A, 1428 (2015) 97-114.[2] R. Pérez-Míguez, M. Castro-Puyana, M. L.
Marina, Recent applications of chiral capillary electrophoresis in food analysis, in: A. Haynes (Ed.), Advances in Food Analysis Research, Nova Science publishers, New York, 2015, pp. 89-120
Acknowledgements: Authors thank the Spanish Ministry of Economy and Competitiveness (CTQ2013-48740-P) and the Comunidad of Madrid (Spain) and European funding from FEDER program (S2013/ABI-3028, AVANSECAL-CM). M.C.P. also
thanks the Ministry of Economy and Competitiveness (Spain) for her “Ramón y Cajal” research contract (RYC-2013-12688). R.P.M. thanks the University of Alcalá for her pre-doctoral contract.Disclosure of Interest:None Declared
P03-003-096 - Polymer Solid-Phase Extraction Monolith Modified with Gold and Silver Nanoparticles for Isolation of ProteinsM. Vergara-Barberán, M. J. Lerma-García, E. F. Simó-Alfonso, J. M. Herrero-Martínez*
Content: Protein isolation and purification are essential steps in protein analysis schemes and proteomic techniques. In this sense, solid-phase extraction (SPE) has been widely used as attractive alternative in sample preparation. However, the number of sorbents developed for proteins at present has been scarcely investigated. In this study, a new SPE sorbent has been developed to isolate proteins from complex samples. For this purpose, a polymeric bed was first synthesized and subsequently modified with different metallic nanoparticles (NPs). These sorbents were prepared from a ground poly(glycidyl-co-ethylene dimethacrylate) based polymer and then treated with different ligands (ammonia or cysteamine). The resulting stationary phases (containing amine or thiol groups, respectively) were modified with gold and silver NPs, on the basis of the strong interaction between these groups and both types of NPs. To evaluate the performance of these SPE supports, BSA was selected as test solute, being the extraction conditions and other parameters (loading capacity and regenerative ability of sorbent) established. The sorbent based on thiol-modified polymers containing AuNPs

www.ISC2016.ie @isc2016 #isc2016 /isc2016354
POSTER ABSTRACTS Sample Preparation Techniques and Methods
and AgNPs provided the highest recoveries (<90% for both of them) and loading capacities (29.3 and 17.6 µg/mg sorbent, respectively). The applicability of these sorbents was demonstrated by isolating thionins, which are small basic sulphur-rich proteins (≃5 kDa), in European mistletoe extracts. The isolated fractions were subjected to analysis by SDS-PAGE and MALDI-TOF to demonstrate the selectivity of the developed SPE sorbent. Acknowledgements: Project CTQ 2014-52765-R (MINECO of SPAIN and FEDER); M. V-B. thanks the MEC for an FPU grant for PhD studies.Disclosure of Interest:None DeclaredKeywords: Metallic nanoparticles, Proteins, Solid-phase extraction
P03-003-097 - Design of Experiments in QuEChERS method: Determination of persistent organic pollutants in soil by Gas Chromatography coupled to Ion Trap (GC/ITQ)
N. Castro*, M. I. Páez
Content: Soils are the main environmental reservoir of persistent organic pollutants (POPs) and this make a major contribution to the global inventory of POPs. These Pollutants are strongly retained for this matrix due to the high soil organic matter content (SOM) 1, therefore, this represent a big challenge in the sample preparation methods to improve and achieve the determination of them at trace level. Usually, in complex matrices like it, numerous stages have to be set up the best experimental conditions to obtain good results 2. In this
work, different versions of QuEChERS method was applied to select the best according to recoveries and repeatability. It was carried out with completely randomized designs at four levels: 1. Original unbuffered QuEChERS, 2. AOAC 2007.01, 3. AOAC 2007.01 + NaCl, 4. EN 15662, where recoveries were the response variables. Then, Full factorial design was applied to obtained the sample and water amount necessary for extraction phase, this design had 3 level for water: 6 mL, 9 mL and 12 mL, and 3 levels for sample amount: 8 g, 10 g and 12 g. The samples were spiked at 10 µg.Kg-1 and 20 µg.Kg-1 for organochlorine pesticides and polychlorinated biphenyls, respectively.
The results showed that the best QuEChERS method was the original method with recoveries between 75 y 109 % with CV below at 11 %. The sample and water amount necessary to the extraction phase were 8 g and 6 mL, respectively. This obtained recoveries between 90 and 109 % with CV below at 12 %. DOE allowed to know that at low level of water and high sample amount there wasn’t separation of water and organic phases, on the other hand, high water levels can produce pesticides losses due at high temperature in the extraction phase, when the salts are added.References: 1. A,Cabrerizo, Environ. Sci. Technol. 45, (4785–92 )
(2011)2. M,Callao,Trends Anal. Chem. 62, (86–92)(2014Disclosure of Interest:None DeclaredKeywords: Design of Experiments, QuEChERS
method, Soil

355www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
P03-003-098 - Two simple cleanup methods combined with LC-MS/MS for quantification of steroid hormones in in-vivo and in-vitro assays
J. J. Weisser 1,*, C. Cornett 1, B. Styrishave 1, C. H. Hansen 1, R. Poulsen 1, L. Larsen 1
1 Department of pharmacy, University of Copenhagen, Faculty of Health and Medical Sciences, København K, Denmark
Content: Measuring both progestagens, androgens, corticosteroids as well as estrogens with a single method makes it possible to investigate the effects of endocrine disrupting chemicals (EDC´s) on the main pathways in the mammalian steroidogenesis. This paper presents two simple methods for the determination of the major steroid hormones in biological matrixes using liquid chromatography tandem mass spectrometry (LC-MS2). A novel method was developed for the determination of 14 steroids in the H295R in-vitro assay without the need for solid phase extraction (SPE) purification. The in-vitro assay was validated by exposing H295R cells to Prochloraz (PRO) for inhibiting of steroid hormones secretion and by exposing cells to forskolin (FOR) for inducing steroid hormones secretion. The method fulfills the recommendations for the H295R assay suggested by the OECD. Furthermore, a simple offline C18 SPE methodology was developed for the necessary clean-up of in-vivo assays. Samples, such as gonad tissue, plasma and serum, are complex biological matrixes and the SPE methodology was optimized to remove salts and proteins prior to elution of target analytes. At the same time lipophilic compounds were retained on the SPE cartridge during elution. This,
combined with the multi steroid LC-MS2 method made it possible to determine 10 steroids in male Sprague Dawley rat gonad tissue. Furthermore, it was possible to quantify 6 steroids in the plasma. In general, the observed concentration of steroid hormones in plasma, testes and H295R cell medium corresponded well with previous studies. The off-line SPE method was validated using spiked charcoal-stripped serum. Method recovery, accuracy, precision and robustness were all good. Instrument sensitivity was in the range of 55 – 530 pg / mL (LLOQ).References: J J weisser, C H Hansen, R Poulsen, L
W Larsen, C Cornett and B Styrishave. J of Anal Bioanal Chem. Received: 14 January 2016 /Revised: 8 April 2016 /Accepted: 18 April 2016 (not available online yet)
Disclosure of Interest: None DeclaredKeywords: Online clean-up, plasma, testis, gonads, H295R cell assay, solid phase extraction
P03-003-099 - Preparation of methacrylate polymer-coated gold nanoparticles for solid-phase extraction of fungicides in water samples
M. Catalá-Icardo 1, S. Meseguer-Lloret 1, S. Torres-Cartas 1, C. Gómez-Benito 1, A. Calabuig-Belda 1, M. Vergara-Barberán 2, E. Simó-Alfonso 2, J. M. Herrero-Martínez*
1 Research Institute for Integrated Management of Coastal Areas, Universitat Politècnica de València, Valencia,
2 Analytical Chemistry, University of Valencia, Burjassot, Spain
Content: Solid-phase extraction (SPE) is the most popular sample preparation procedure, being widely employed in

www.ISC2016.ie @isc2016 #isc2016 /isc2016356
POSTER ABSTRACTS Sample Preparation Techniques and Methods
a great variety of sample matrices. This pretreatment technique has been extensively used in the determination of pollutants in environmental waters due to its advantages (high recoveries and enrichments factors, ease of operation, automation, etc). Within commercial sorbents, standard C18 material has been used popularly as SPE sorbent; however, it provides poor recoveries for polar pesticides, a narrow pH stability range and a limited reusability. Thus, the choice of appropriate SPE sorbent is a critical factor to achieve a high extraction efficiency and preconcentration level.
Polymeric monoliths have been used to develop stationary phases for both separation techniques and sample pretreatment. The performance of extraction efficiency of these monoliths could be changed by tailoring its pore structure and surface chemistry. In particular, glycidyl methacrylate (GMA)-based monolith can be considered as the most commonly support of choice to produce chromatographic beds with different functionalities. Also, its powder have been employed for holding gold nanoparticles (AuNPs) to enhance surface-enhanced Raman spectroscopy detection [1] or for the isolation of proteins in vegetal extracts [2].
In this work, a polymeric sorbent modified with AuNPs was used for extraction and preconcentration of residues of fungicides (azoxystrobin and chlorothalonil, which are the most-widely used for fungal pathogens control) in environmental water samples. These pollutants contain available cyano groups in their structure for interactions with AuNPs-based sorbent. The extraction conditions and efficiency of this sorbent were investigated. The optimized methodology
was applied to the determination of these fungicides in environmental water samples.References: 1. X. Wang, Food Control 46 (108-114) (2014)2. M. Vergara-Barberán, Anal. Chimica Acta 917 (37-
43) (2016)Acknowledgements: Project CTQ2014-52765-R (MINECO of Spain and FEDER).Disclosure of Interest:None DeclaredKeywords: fungicides, gold nanoparticles, solid-phase extraction
P03-003-100 - Preparation of magnetic polymethacrylate-based sorbent for selective analysis of organothiophosphorous pesticides in environmental waters
S. Meseguer-Lloret 1, S. Torres-Cartas 1, M. Català-Icardo 1, C. Gómez-Benito 1, B. Jacobs-Fantassi 1, E. J. Carrasco-Correa 2, E. F. Simó-Alfonso 2, J. M. Herrero-Martínez*, G. Ramis-Ramos 2
1 Research Institute for Integrated Management of Coastal Areas, Universitat Politècnica de València, Grao de Gandía,
2 Analytical Chemistry, University of Valencia, Burjassot, Spain
Content: Organothiophosphorous (OTP) pesticides have been employed to protect agricultural crops due to their insecticidal properties. However, the use of these compounds must be monitored due to their adverse effects in the environmental and human health. Then, the presence of their residues in foods and drinking waters must be evaluated. According to the EU Directive on water quality (98/83/EC), the maximum allowed concentration for pesticides in drinking water is 0.1 µg L-1, and 0.5 µg L-1 for total pesticides. Therefore, the analysis

357www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
of pesticides requires sensitive techniques, being the chromatography methods the most widely used. In addition, solid-phase extraction (SPE) procedures have been commonly adopted in environmental water samples. In particular, SPE with C18-based material is the current and efficient method to reach a good extraction efficiency and enrichment of these pesticides. However, this method has some disadvantages: long extraction time, column blocking, limited selectivity and poor reusability. In this sense, the development of new SPE sorbents with enhanced properties is highly desirable.
In this work, a SPE sorbent consisting of magnetite nanoparticles (MNPs) bonded to glycidyl methacrylate (GMA)-based polymer was developed, and its application for preconcentration of OTP pesticides, in combination with HPLC-DAD detection, was studied. Thus, the sorbent was prepared from a ground GMA-based polymer modified with a silanizing agent following by attachment of MNPs. The developed cartridges were applied to the extraction of several OTP pesticides from aqueous solutions. Extraction parameters as loading pH, flow rate, eluent type and volume and other variables (breakthrough volume, loading capacity and reusability) influencing on the analytical performance were established. Under optimized conditions, the detection limits of the proposed method were below than the legislated values. Good recoveries (higher than 70%) were also achieved.References: .Acknowledgements: Project CTQ2014-52765-R (MINECO of Spain and FEDER)Disclosure of Interest:None DeclaredKeywords: magnetic polymer-based material, pesticides, solid-phase extraction
P03-003-101 - Application of Experimental Design in Optimisation of Sample Pretreatment for Quantitative Determination of Tocochromanols in Human Adipose Tissue
E. Bartosińska 1,*, A. Borsuk 1, M. Walentynowicz 1, D. Siluk 1
1 Department of Biopharmacy and Pharmacokinetics, Medical University of Gdańsk, Gdańsk, Poland
Content: Tocopherols and tocotrienols belong to a group of fat-soluble substances generally considered as vitamin E. Recent studies indicate the anticancer potential of these compounds, in addition to their antioxidant properties [1].
The complexity of adipose tissue sample matrix causes a number of difficulties during its preparation. Therefore some additional steps should be established in order to obtain efficiently purified extracts, for instance the application of solid-phase extraction technique. The aim of this study was to optimize the homogenization and extraction steps of tocopherols and tocotrienols from human adipose tissue. In order to select the optimal solvent mixtures and conditions for adipose tissue homogenization, Plackett-Burman design with 8 experiments and 23 full-factorial design were used. Then, the SPE conditions were assessed with the application of Plackett-Burman design with 12 experiments, which was carried out with standard solutions of tocochromanols. The previously established RP-HPLC-FLD separation conditions were reevaluated by Central Composite Design (CCD).
The content of isopropanol-ethanol phase in extracting mixture appeared to be an

www.ISC2016.ie @isc2016 #isc2016 /isc2016358
POSTER ABSTRACTS Sample Preparation Techniques and Methods
important feature of homogenization step. In the case of SPE procedure, the percentage of organic solution (10-80%) used for sample clean-up was the most significant among all factors. The chromatographic separation conditions were proven to be properly determined previously. Satisfactory recovery results (at least 70%) confirmed the utility of the developed method. Relevant identified factors enabled the final development of the RP-HPLC-FLD method for quantitative determination of tocopherols and tocotrienols in human adipose tissue.References: [1] B. B. Aggarwal, C. Sundaram,
S. Prasad, R. Kannappan, Biochemical Pharmacology 80 (1613-1631) (2010).
Acknowledgements: The project is supported by National Science Centre (grant no 2013/11/N/NZ5/00164).Disclosure of Interest:None DeclaredKeywords: HPLC, Sample clean-up, SPE extraction
P03-003-102 - Characterization of Essential Oil and Extracts of Leaves and Fruits from Morinda citrifolia (L.) using GC×GC/qMS
E. B. Caramão 1,*, D. B. D. M. Lima 1, A. L. dos Santos
2, M. I. S. Melecchi 3, L. C. Krause 1
1 Biotechnology, UNIT-ITP, Aracaju, 2 Chemistry, UFRGS, 3 Chemistry, CMPA, Porto Alegre, Brazil
Content: Morinda citrifolia (Rubiaceae), commonly called Noni, is a plant typically found in northeastern Brazil. Extracts from bark, stem, root, leaf and fruit of Noni has been used for several years in folk medicine for many diseases (diabetes, hypertension, cancer), but few studies related to their chemical characterization
have been published. The goal of this work is to study the extracts of Noni, obtained by Hydro Distillation with Clevenger (HDC) and Ultrasonic Assisted Extraction (UAE) using hexane and ethyl acetate. The extracts (UAE) and essential oils (HDC) were analyzed by comprehensive two-dimensional gas chromatography with quadrupole mass spectrometry detection (GCxGC/qMS) with the aid of the retention indices. As various fatty acids were identified in the extracts, they were saponified and esterified generating two fractions: the ester fraction, rich in methyl esters of many fatty acids (C8, C16 and C18) and the unsaponifiable fraction, with phenols, alcohols and hydrocarbons. The extract of the fruits also presented vitamins (β-sitosterol, carotenoids, vitamin A and C), which should be new object of study using LC/MS-MS. The yield of essential oils was very low and only the essential oils of the leaves were analyzed, with similar composition to the unsaponifiable fraction of the hexane extract.References: PÉRES, V. F.; SAFFI, J.; INÊS, M.; MELECCHI, S.;
ABAD, F. C.; JACQUES, R. A.; MARTINEZ, M.; OLIVEIRA, E. C.; CARAMÃO, E. B. , Journal of Chromatography A, 1105, 115–118, 2006.
JACQUES, R.A.; DARIVA, C.; OLIVEIRA, J.V.; CARAMÃO, E.B. Analytica Chimica Acta., 625, 70- 76, 2008.
SINGH, D.R. J. Diabet. Endocrinol., 3 (6),77–91, 2012.Acknowledgements: CNPq, FAPITEC, FINEPDisclosure of Interest:None DeclaredKeywords: essential oils, GCxGC, sonication

359www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSample Preparation Techniques and Methods
P03-003-103 - Chemical Characterization of Bio-Oil from the Pyrolysis of Harvest Waste’s of Basic Germoplasm of Energy-CaneE. B. Caramão 1,*, J. A. S. Barros 1, M. E. Machado 1, M. C. Krause 2, A. L. do Amaral 3, L. C. Krause 1
1 Biotechnology, 2 Engenharia de Petróleo, UNIT-ITP, 3 Biotechnology, EMBRAPA, Aracaju, Brazil
Content: In Brazil, sugar cane is used to produce sugar and alcohol. The sugar-alcohol sector has relevance in the Brazilian economy, once Brazil is the world’s premier productor of ethanol, a renewable fuel. In this context, there is an evolution of the research of second generation energy, using lignocellulosic materials for producing biofuels. The Active Germplasm Bank of Saccharum located in the Brazilian Agricultural Research Corporation (Embrapa Tabuleiros Costeiros -SE / Brazil), has been developing research through the crossing between species Saccharum sp. (Commercial variety) and Saccharum Robustum for breeding of sugar cane with the purpose of better adaptation to adverse environmental conditions and improve the characteristic of fiber production by the entire plant for using in energy generation. Beside the cane production, many residues are disposed during the harvest, mainly straw and bagasse. These residues can be transformed by pyrolysis (thermodegradation) generating (bio-oil), non-condensable gases and biochar. The goal of this work is to investigate the chemical composition of the bio-oil from the pyrolysis of harvest residues germoplasms produced by crossing wild species of sugarcane, through comprehensive two-dimensional gas chromatography (GCxGC/qMS), aiming the identification and quantification
of its major constituents. The results showed high amount of hydrocarbons in the hybrid species, confirming the success of the improvement of the cane. Beside hydrocarbons, some phenols and aldehydes are also identified.References: [1] MORAES, M.S.A.; GEORGES, F.; ALMEIDA,
S.R.; DAMASCENO, F.C.; MACIEL, G.P.S.; ZINI C.A.; JACQUES, R.A.; CARAMÃO, E.B. Fuel Process Technology, 101, 35–43, 2012.
[2] KIM, M.; DAY, D. Journal of Industrial Microbiology & Biotechnology, 38, 803 – 807, 2011.
Acknowledgements: FAPITEC, CNPq, FINEPDisclosure of Interest:None DeclaredKeywords: GCxGC, pyrolysis, sugar cane
P03-003-104 - Novel downstream processing technique for Cyclosporine
Y. G. Hou 1, D. Rutterschmid 1, Z. Kovács 1, Z. Misek
1,*, L. Lorántfy 1, L. Németh 1
1Rotachrom Technológiai kft., Dabas, Hungary
Content: Cyclosporine is a cyclic oligo peptide, produced by a fungi called Tolypocladium inflatum. This generic active pharmaceutical ingredient (API) has an immunosuppressant effect, and is widely used in organ transplantations to prevent rejection.
The original downstream processing technique for cyclosporine involves multiple recrystallization and normal phase liquid chroamtographic steps. As downstream processing costs may take up to 80% of production costs, specially when using liquid chromatography, there is a high pressure on generic API manufacturers

www.ISC2016.ie @isc2016 #isc2016 /isc2016360
POSTER ABSTRACTS Sample Preparation Techniques and Methods
to develop more and more cost effective techniques.
RotaChrom iCPC platform is an industrial realization of Centrifugal Partition Chromatography (CPC), where mobile and stationary phase are liquid, and the liquid stationary phase is immobilized by a strong centrifugal forcewhile the liquid mobile phase is pumped through. [1]
The CPC techniques was applied after the first crystallization of cyclosporine, containing lot of impurities, with cyclopsorine assays of 80-85%. Some impurities, like dihydro-cyclosporine could not be removed from the crude with high yields.
Various solvent systems were tested, while the chosen biphasic solvent system contained hexane-aceton-water, providing proper range of partition coefficients, good selectivitiy, and system stability.
Some gram of crude sample was successfully purificated on an Armen lab scale instrument, before linear scale up to a RotaChrom rCPC instrument, with column volume of 2 l and flow rate of 200 ml/min, providing productivity over 150 g per workday. The impurity levels were controlled under desired limits.
Our developed method provides efficient way to purificate cyclosporine. With one iCPC instrument it is possible to process more than 5 kgs of crude material day, providing a cost-effective and scalable downstreamprocessing method for the API. References: [1] Alain P. Foucault, ”Centrifugal
Partition Chromatography,” Chromatographic Science Series vol. 68, New Jersey, 1994
Disclosure of Interest: None DeclaredKeywords: Centrifugal Partition Chromatography, cyclosporine A

361www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSupercritical Fluid Chromatography
Supercritical Fluid Chromatography
P01-001-003 - Analyze of pesticides in food product using SFE-SFC-MSMS
S. Baumgarten 1,*, M. Panov 2
1 Shimadzu Europa, Duisburg, Germany, 2 Chem Analytical Services Ltd, Sofia, Bulgaria
Content: Conventionally, analysis of residual pesticides in foods has involved pesticide extraction by QuEChERS method before analysis by LC/MS or GC/MS. However, it is time-consuming, and a large amount of organic solvent is used.
Supercritical fluid extraction (SFE) that uses supercritical carbon dioxide as the extraction solvent provides good extraction efficiency, where the solvent is similar to gas in terms of low viscosity and high diffusivity, and similar to fluid in terms of high solubility. This allows for extraction within a short period of time. Hydrophilic pesticides can be extracted from sample as well as hydrophobic ones by adding a modifier in the SFE process. We introduce an example GC/MS analysis of pesticides extracted from agricultural products using the Nexera UC off-line SFE system and the GCMS-TQ8040 (Shimadzu Corporation, Japan). Supercritical fluid chromatography (SFC) is one of the separation techniques and it can separate numerous pesticides with a wide range of different polarities simultaneously. We have established an on-line SFE-SFC-MS system that combines SFE and SFC in a seamless flow line and applied this to the analysis of residual pesticides. On-line SFE-SFC-MS analysis was carried out using Nexera UC system coupled with a LCMS-8050 triple quadruple mass spectrometer (Shimadzu Corporation, Japan).
A mixed standard solution of more than 500 pesticides was spiked in different food matrices (sunflower seeds, wheat, and animal fat) to different concentrations.
Two different analyses were performed: • After trapping the extracted material
on a trap column, eluate containing the target components was collected in a fraction collector, prior to analysis by GCMS-MS.
• During dynamic extraction, extracted substances were eluted from the extraction vessel into the C18 reversed phase column and analyzed by SFC-MS-MS.
As a result more than 500 pesticides were reliably analyzed and showed good linearity within a range of 10-300 ng/g for most of them. Disclosure of Interest:None DeclaredKeywords: mass spectrometry, Pesticides, SFC-SFE
P01-001-041 - Estimating the theoretical limits of temperature deviations in Supercritical Fluid Chromatography (SFC)A. Tarafder 1,*
1Waters Corporation, Milford, United States
Content: In SFC, due to mobile-phase (MP) compressibility, pressure (P) drop along the column leads to volumetric expansion, causing temperature (T) deviations, which may result in thermal-heterogeneity (TH) inside the column [1]. TH leads to efficiency loss and should be avoided in any chromatographic process [1]. The

www.ISC2016.ie @isc2016 #isc2016 /isc2016362
POSTER ABSTRACTS Supercritical Fluid Chromatography
word “deviation” is used here to refer to uncontrolled T variations in column, away from the set point.
The extent of T deviation, and thus the extent of TH, however, depends on the adiabatic compressibility of the mobile phase (β). Neat CO2 compressibility (βCO2) can widely vary with the P and T of the MP. E.g. βCO2 at 40C, 2000 psi is about 650% more than that at 25C, 5000 psi [2]. At high P and low T, CO2 is more liquid-like. Here βCO2 is much lower and should not lead to significant T deviations, even with high pressure drop. With high modifier percent, SFC MP may not experience any change in T at all, or may heat up like in UPLC [3]. This demonstrates the importance of having a clear idea on the effect of β on T deviations during SFC method development.
Although the above discussion sounds quite complex, a graphical representation of CO2 and CO2+modifier enthalpy (H), as function of P and T, can greatly clarify the situation [3]. From the constant H (isenthalpic) curves, plotted on (P-T) plane of the MP compound(s), we can clearly and easily estimate the theoretical limits of T deviations caused by a given pressure drop [3]. A comparison between the max extents of T deviations, between different methods, can guide the selection of competing SFC methods. In this presentation, working principle behind this graphical approach will be explained, and with the help of isenthalpic plots of CO2 and CO2+MeOH mixtures, a general guideline on method selection to avoid TH in SFC will be discussed.References: [1] D. P. Poe et al, J. Chrom A, 1216 (2009) 7915.[2] E. W. Lemmon et al, NIST, Std Ref Data Prog,
Gaithersburg 9.0.
[3] A. Tarafder et al, J. Chrom A, 1366 (2014) 126.Disclosure of Interest:None Declared
Keywords: Thermal-heterogeneity, efficiency-loss
P01-001-047 - Unified Chromatography, transforming the approach to complex separations
R. Wong*
Content: Recently UHPLC systems have been deployed to produce separations with peak capacities over one thousand. This has been achieved by exploiting sub-2 µm superficially porous particle technology. However, since the column backpressure is inversely proportional to the square of the particle diameter; the by-product of achieving such high efficiencies result in higher operating backpressures. This is further compounded by the fact the optimal linear velocity increases with smaller particle diameters.
Unified Chromatography (UC) takes advantage of the lower viscosity of supercritical fluid in order to improve both peak capacity and chromatographic resolution, without the same pressure limitations currently encountered by modern UHPLC technology. The release of the Nexera-UC chromatograph systems makes it possible to take advantage of this. In addition, for challenging separations, chromatographers can now use the Nexera-UC in order to improve the number of options with regards to both selectivity and retention; in order to resolve complex mixtures.
Permethrins are widely used as an insecticide and insect repellent. They are

363www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSupercritical Fluid Chromatography
also the first-line treatment for scabies. Chiral resolution of permethrins have often proven challenging since some possess 3 chiral centres with 8 stereoisomers. As part of a recent study the new Nexera-UC chromatograph system was challenged to resolve these stereoisomers using SFC.Disclosure of Interest:None DeclaredKeywords: Complex Separations, Permethrins, SFC-SFE
P01-001-050 - Separation of fatty acid positional and cis and trans isomers by SFC
J. Yang*
Content: Routine simultaneous identification and quantification of a variety of fatty acids is a common analysis in food testing labs. Although gas chromatography (GC) methods are traditionally used for fatty acid analyses (ref 1), they often have long runtime (about 60 minutes), peak shifting issue, and need derivatization. In order to overcome some of these shortcomings, UltraPerformance Convergence ChromatographyTM (UPC2) has been investigated for fast analysis of free fatty acids. UPC2 is a next-generation supercritical fluid chromatography. It leverages the unique properties of compressed CO2 at or near its supercritical state, such as low viscosity and high diffusivity, and sub-two micron particle packed columns to improve separation efficiency, speed, and selectivity.
This study will look into the separation of the critical pairs in common fatty acids by UPC2. The critical pairs are isomers that are either differ in the trans and cis configuration, such
as C16:1(9Z) and C16:1(9E), or the double bond position along the fatty acid chain, such as C18:1(Δ6), C18:1(Δ9), C18:1(Δ11), or combination of both, such as C18:1(6Z) and C18:1(9E). Various UPC2 separation conditions, including mobile phase co-solvent and column stationary phase will be investigated. References: 1) Determination of labeled fatty acids content in milk
products and infant formula, AOAC Official Method 2012.13 First Action 2012, AOAC International
Disclosure of Interest:None DeclaredKeywords: fatty acid, SFC, trans fatty acid
P03-003-105 - A Novel Unified Analytical Workflow with Online SFE/SFC
Y. Watabe 1,*, Y. Hayakawa 1, S.-I. Kawano 1, T. Hattori 1, H. Terada 1, T. Uchikata 1, Y. Funada 1
1 Shimadzu Corporation, Kyoto, Japan
Content: Current analytical methodology for quantitative determination of complex sample matrices typically requires off-line sample preparation technique that is followed by a chromatographic analysis. The sample preparation and the following manual transfer to the analytical instrument are generally tedious and time-consuming so that they interfere with the enhancement of efficiency of lab work and the increase of experimental throughput. Recently, an innovative new concept of SFE/SFC has been introduced that greatly reduces sample preparation time and the quantitative variability associated with manual procedures. This new technique provides fully automated sample preparation followed by chromatographic

www.ISC2016.ie @isc2016 #isc2016 /isc2016364
POSTER ABSTRACTS Supercritical Fluid Chromatography
separation of target compounds using supercritical fluid. When on-line SFE/SFC is employed, extracted compounds from the sample matrix are introduced into the analytical column for separation without any human intervention. A number of applications in various fields including food, environmental, and pharmaceutical are expected to prove the flexibility of this technique. Here we present automated online extraction and analysis for labile compounds and phospholipids with significant reliability. References: T. Uchikata, et al, J. Chromatogr.A 1250
(69-75) (2012)Disclosure of Interest: None DeclaredKeywords: online extraction, SFC-HPLC switching, SFE-SFC
P03-003-106 - A unified analytical tool for green extraction and determination of both targeted and untargeted analysisM. Zoccali 1,*, G. Purcaro 1, M. Nishimura 2, L. Mondello 1 3
1 Chromaleont srl, Messina, Italy, 2 Shimadzu Scientific Instruments, Inc., Columbia, MD,
United States, 3 Dipartimento di Scienze Chimiche, Biologiche,
Farmaceutiche ed Ambientali, University of Messina, Messina, Italy
Content: The on-line coupling between supercritical fluid extraction (SFE) and supercritical fluid chromatography (SFC), generate a powerful tool for the automatic extraction and detection of target compounds from food matrices. The on-line nature of the system, compared to off-line approaches, improves run-to-run precision, enables the setting of batch-type
applications, and reduces the risks of sample contamination. Supercritical carbon dioxide (CO2) presents unique characteristics, which make it an excellent solvent, it shows a relatively high density and consequently, a high solvation power. It presents low viscosity and high diffusion coefficient, which allow fast extraction. From the environmental point of view, the use of supercritical CO2 for both purposes, SFE and SFC, greatly reduces the use of organic solvents. The density of the supercritical fluid can be easily handled by changing its pressure and/or temperature, changing the solvent strength. The same considerations can be made regarding the SFC, obtaining both high speed and resolution analysis. Finally, the QqQ MS detector due to its high sensitivity and selectivity allows the identification and structural analysis of targeted and untargeted compounds. The analytical potential of the system described are shown in a proof of principle study involving the carotenoids study from food samples.Disclosure of Interest: None DeclaredKeywords: supercritical fluid chromatography, supercritical fluid extraction
P03-003-107 - Selectivity comparison and behavior evaluation of an amide-based stationary phase for supercritical fluid chromatographyA. C. Borges-Muñoz 1,*, L. A. Colón 1
1 Chemistry, University at Buffalo, State University of New York, Buffalo, United States
Content: A stationary phase containing a polar group embedded in a hydrophobic backbone (i.e., ACE®C18-amide) was evaluated for use in supercritical fluid

365www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSupercritical Fluid Chromatography
chromatography. The amide-based column was compared with columns packed with bare silica, C18 silica, and a terminal amide silica phase. The system was held at supercritical pressure and temperature with a mobile phase composition of carbon dioxide and methanol as co-solvent. The linear solvation energy relationship (LSER) model was used to evaluate the behavior of these stationary phases, relating the retention factor of selected probes to specific chromatographic interactions. A five-component test mixture, consisting of a group of drug-like molecules was separated isocratically. The results extracted from the LSER show that the C18-amide stationary phase provided a combination of interactions contributing to the retention of the probe compounds. Although hydrophobic interactions exist, the electron donating ability of the embedded amide group shows a large positive interaction between the solutes and the stationary phase. Under the chromatographic conditions used, the C18-amide column was able to provide baseline resolution of all the drug-like probe compounds in a text mixture, while the other columns tested did not. Details of the chromatographic evaluation will be discussed.References: 1. J. G. C., Dorsey; T., William. Analytical
Chemistry, 66 (857A-867A) (1994).2. U. D., Neue. HPLC Columns: Theory, Technology,
and Practice, Wiley-VCH. Inc. (1997).3. C., West; E. J., Lesellier. Chromatogr.
A, 1169 (205-219) (2007).4. C., West; E. J., Lesellier. Chromatogr.
A, 1191 (21-39) (2008).5. C. R., Mitchell; N. J., Benz; S. J., Zhang. Sep.
Sci., 33 (3060-3067) (2010).6. M., Vitha; P. W., Carr. J. Chromatogr. A, 1126 (143-
194) (2006).
Disclosure of Interest: None DeclaredKeywords: Linear solvation energy relationship, stationary phases, Supercritical fluid chromatography (SFC)
P03-003-108 - Supercritical fluid chromatography with mass spectrometry detection for the separation of novel chiral compounds
G. J. Schad 1,*, D. Van den Heuvel 2
1Analytical Business Unit, Shimadzu Europa GmbH, Duisburg, Germany, 2Shimadzu Benelux B.V., ‘s-Hertogenbosch, Netherlands
Content: During API (active pharmaceutical ingredient) development, drug stereoisomerism is recognized as an issue having clinical and regulatory implications. Enantiomers have essentially identical physical and chemical properties, while potentially showing large differences in toxicity. Therefore, the stereoisomeric composition of a drug with a chiral center should be well documented. To evaluate the pharmacokinetics of a single enantiomer or any mixture of enantiomers, manufacturers must develop quantitative assays for individual enantiomers early in drug development.
A fast and simple SFC – PDA/MS method for the screening of chiral compounds on different columns was developed within two days, using a column screening system with 6 columns and 2 different organic modifiers. The method was optimized with regards to separation and sensitivity. Simultaneous detection of PDA signal and MS scanning was used for confirmation of the isomer signals and resolution of > 1.5

www.ISC2016.ie @isc2016 #isc2016 /isc2016366
POSTER ABSTRACTS Supercritical Fluid Chromatography
was obtained for all compounds of interest with RSD < 2.0 % for retention time using PDA and MS detection (without splitting).
SFC-MS was shown to be a reliable, robust and simple alternative to routine LC analysis. Use of SFC in terms of complexity of the instrument and method development was very similar to the HPLC approach. However, SFC is known to be superior to HPLC for chiral separations.References: [1] L.A. Nguyen, et al.; Int J Biomed
Sci. 2006 Jun; 2(2): 85–100.Disclosure of Interest: None DeclaredKeywords: Chiral drug, Method development, supercritical fluid chromatography
P03-003-109- Extraction and Quantification of Mycotoxins in Peanut Butter Using SFE–SFC–MS
G. J. Schad 1,*, T. Ogura 2, K. Tanaka 2
1 Shimadzu Europa GmbH, Duisburg, Germany, 2Shimadzu Scientific Instruments Inc., Columbia, United States
Content: Aflatoxins are mycotoxins produced by fungal molds of the Aspergillus species, which prefer a warm and damp climate. Aflatoxins can be produced by fungal infestation during or after harvest of peanuts and can therefore end up in processed products such as peanut butter or satay sauce. In addition to being acutely toxic, they are also known to be carcinogenic [1]. To ensure food safety, manufacturers of food and beverages have to strictly manage risks from such contaminants, and sensitive methods to assay mycotoxins in complex matrices are essential.
A conventional, manual method for sample extraction of aflatoxins from peanut
butter is time consuming[2]. The use of “supercritical” CO2 for extraction as well as chromatographic separation can offer a more efficient extraction process while reducing solvent consumption and the risk of losses or error of measurement during sample preparation. Furthermore, extraction selectivity can be carefully controlled by adjusting the solvating power of the CO2 through changes in pressure and temperature, making it faster, more efficient, and more versatile than liquid and SPE [3].
The online SFE–SFC–MS approach was successfully used to extract and analyze aflatoxin B1 and B2 from spiked peanut butter samples in only a fraction of the time that a conventional liquid extraction followed by SPE clean-up sample preparation method would take. The procedure was shown to be linear in a concentration range of 0–10 ppm with good reproducibility.References: [1] European Food Safety Authority: http://www.efsa.
europa.eu/de/topics/topic/aflatoxins[2] Application Note on Extraction of Aflatoxins (B1,
B2, G1, and G2) from Corn, Peanuts and Peanut Butter 2012 Avantor Performance Materials, Inc.
[3] T. Letzel and S. Bieber, Analytik News 8 (2015)Disclosure of Interest: None DeclaredKeywords: mycotoxins, SFC-MS/MS, supercritical fluid extraction

367www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSupercritical Fluid Chromatography
P03-003-110 - Chiral Separation and Quantification of Metabolites using Super Critical Fluid Chromatography Coupled with Triple Quadrupole Mass Spectrometry
S. Joseph 1, S. Krieger 2,*, U. Huber 2
1 Agilent Technologies India Pvt.Ltd, Bangalore, India, 2 Agilent Technologies, Waldbronn, Germany
Content: Introduction: Risperidone, an antipsychotic drug, is metabolized by different cytochrome P-450 enzymes, to predominantly form the 9-hydroxy metabolite. The 9-hydroxylation results in the formation of a chiral carbon atom yielding two enantiomers, (+)- and (-)-9-hydroxyrisperidone. To a lesser extent N-dealkylation and 7-hydroxylation may also occur. To distinguish stereoisomers and identify trace level metabolites, baseline resolved chromatographic separation and sensitive mass spectrometric detection are required. Supercritical Fluid Chromatography (SFC) provides baseline resolution using chiral columns with several advantages such as rapid and green separations over conventional normal phase separations. Here, a chiral separation and sensitive quantitation of major hydroxylated metabolites of risperidone from an in-vitro study using an SFC-QQQ system will be presented.
Methods: An SFC/QQQ system with ESI source was used for the study(1). The electrospray source was equipped with thermal gradient focusing technology to enhance sensitivity and operated in positive mode. Protonated precursors of risperidone and its metabolites were selected for MRM based quantification
from rat in-vivo samples.
Results: SFC chiral analysis using AD-3 column resulted in baseline separation of metabolite enantiomers and the drug compound within 6 minutes. The method validation criteria include method selectivity, detection limits and linearity ranges of each metabolite. Precision and accuracy were within the acceptable levels. Using this validated SFC-MS/MS method, quantitative analysis of trace metabolites were performed from in-vitro samples.References: 1. Siji Joseph, Agilent Hybrid
SFC/UHPLC-TripleQuadrupole System for Achiral andChiral Metabolite Analysis, Agilent Publication 5991-5725EN, 2015.
Disclosure of Interest: None DeclaredKeywords: Chiral Separation, Quantification, Metabolites, Super Critical Fluid Chromatography, Triple Quadrupole
P03-003-111 - Feasibility of the modelling of the retention behaviour in Supercritical Fluid Chromatography with pure in silico descriptors.
B. Andri 1,*, A. Dispas 1, P. Hubert 1, R. Al Bakain 2, P. Sassiat 3, D. Thiebaut 3, J. Vial 3
1 University of Liege - Laboratory of Analytical Chemistry, CIRM, Liege, Belgium,
2 University of Jordan - Faculty of Science, Amman, Jordan, 3 ESPCI Paris - LSABM - CNRS UMR 8231, Paris, France
Content: The present work assesses the feasibility to establish a simple and predictive model of the retention in Supercritical Fluid Chromatography on the basis of molecular descriptors obtained only from calculations.

www.ISC2016.ie @isc2016 #isc2016 /isc2016368
POSTER ABSTRACTS Supercritical Fluid Chromatography
Different molecular descriptors were calculated with PaDEL-Descriptor for a selection of 15 compounds. These compounds, having pharmaceutical origins and sharing different physicochemical properties, were used as probes of the retention on a 2-ethylpyridine column.
The experimental conditions were defined with the Design of Experiment (DoE) methodology and a full factorial DoE was used to study the effects of the pressure, temperature and gradient slope on the retention. Prior to any attempts of the establishment of a global model, an individual multi-linear regression model was fitted to each compound to verify their response and confirm it could be modelled. A good agreement was found between the variation of the retention and the experimental conditions investigated, proving the relevance of the approach and the quality of the experiments.
Then, a global modelling of the retention was investigated with different input parameters (calculated molecular descriptors on the one hand and chromatographic conditions on the other) and various multivariate techniques. Using Molecular Linear Free Energy Relationship (MLFER) descriptors, Partial Least Square (PLS) regression showed really promising results, which permitted to predict the retention of the different model compounds in presence of two different co-solvents under standard experimental conditions.
Owing to the unavailability of truly predictive model of retention in SFC1, this work, presenting a rather universal approach to study the retention, should ultimately provide the SFC users with an efficient tool
to favour and fasten the development of green and efficient separations.References: 1. E. Lesellier et al., J.Chrom.A, 1382 (2-46) (2016) Disclosure of Interest: None DeclaredKeywords: Design of Experiments, Molecular Descriptors, Multivariate Analysis
P03-003-112 - Supercritical Co2
Extraction of Volatiles from Rose Petals
S. Surmová 1,*, A. Eisner 1, P. Kašparová 1, K. Ventura 1
1 University of Pardubice, Department of Analytical Chemistry, Pardubice, Czech Republic
Content: Among the different extraction techniques used at analytical and preparative scale, supercritical fluid extraction is one of the most used, especially for natural products (1-2). This paper describes extraction of volatiles from rose petals using supercritical fluid extraction (CO2) and following gas chromatography-mass spectrometry analysis. This work was focus on finding the optimized conditions of extraction of volatiles and identification with the largest number of compounds. Different parameters including extraction pressure (116–384 bar), extraction temperature (36.50–103.5 °C) and volume of carbon dioxide (990 mL–6010 mL) with CO2 flow rate 80-120 mL/min and polar modifier (methanol, acetonitrile and toluene), were investigated. Extraction solution, such as toluene, hexane, methanol, were also studied and results were compared.References: (1) M. Herrero, J. A. Mendiola, A. Cifuentes, E.
Ibáñez. Supercritical fluid extraction: Recent advances and applications. Journal of Chromatography A. 1217(16), 2495-2511, 2010.

369www.ISC2016.ie @isc2016 #isc2016 /isc2016
POSTER ABSTRACTSSupercritical Fluid Chromatography
(2) S. Maksimovic, Z. Kesic, I. Lukic, S. Milovanovic, M. Ristic, D. Skala. Supercritical fluid extraction of curry flowers, sage leaves, and their mixture: Recent advances and applications. The Journal of Supercritical Fluids. 84(16), 1-12, 2013.
Disclosure of Interest: None DeclaredKeywords: Gas chromatography-mass spectrometry (GC-MS), rose, supercritical fluid chromatography
P03-003-113 - On-Line SFE/SFC-MS/MS of Polycyclic Aromatic Hydrocarbons and Metabolites in Soils Related to Energy ExplorationD. D. Carlton 1 2,*, A. P. Wicker 1, K. E. Tanaka 3, E. A. McAllister 3, K. A. Schug 1 2
1Department of Chemistry and Biochemistry, 2Collaborative Laboratories for Environmental Analysis and Remediation, Univ. of Texas at Arlington, Arlington, 3Shimadzu Scientific Instruments, Columbia, United States
Content: Polycyclic aromatic hydrocarbons (PAHs) are a class of organic compounds that have been connected to a range of health and environmental implications. Relevant sources of PAHs for this work are crude oil and incomplete combustion products of organic matter. Soil in close vicinity to energy exploration can be at greater risk for these types of compounds due to the possibility of spills, leaks, and gas flaring. Supercritical fluid extraction of sixteen PAH and five metabolite (Oxy-PAH) compounds from certified reference material (CRM) soil and mixtures was performed with supercritical CO2 and methanol modifier. The extract was loaded on-line to a biphenyl column for supercritical fluid chromatography. Detection and quantitation was achieved through tandem mass spectrometry. Soil samples were collected in Texas from a drilling waste water disposal site and from a location of dense unconventional
drilling and natural gas flaring, previously identified to be contaminated with single-ring aromatic compounds.
The selected PAH and metabolite compounds, spanning 2-6 fused rings, were eluted with much less organic modifier than the comparable reversed-phase LC. An estimated LOD for these compounds by SFC-MS/MS was sub-picogram on column. Matrix effects for the extraction were observed for the compounds when using CRM sand, clay, and sediment reference materials. The preliminary extraction efficiency of the total PAHs and Oxy-PAHs was approximately 85% from a soil classified as sandy clay with a 55% sand, 7% sediment, and 28% clay composition. A number of compounds, including Pyrene, Benzo(a)pyrene, and Benzo(g,h,i)perylene were extracted and detected in the three different soils contaminated by waste water at the disposal well. The ability for these rapid online extractions and analyses fosters the ability to recommend locations of soil for remediation, especially since these instances will be unique based on each site’s energy extraction processes.References: 1. None Acknowledgements: The authors would like to acknowledge the Shimadzu Institute for Research Technologies at UTA for the use of the NexeraUC instrument throughout this project.Disclosure of Interest: None DeclaredKeywords: Polycyclic Aromatic Hydrocarbons, Supercritical fluid chromatography (SFC), supercritical fluid extraction

www.ISC2016.ie @isc2016 #isc2016 /isc2016370
POSTER ABSTRACTS Supercritical Fluid Chromatography
P03-003-114 - Evaluation of Critical Column Parameters for Use in Supercritical Fluid Chromatography
A. P. Wicker 1, D. D. Carlton 1,*, T. Kahler 2, K. A. Schug 1
1 Department of Chemistry and Biochemistry, Univ. of Texas at Arlington, Arlington,
2 Restek Corporation, Bellefonte, United States
Content: The advancements in instrumentation has brought SFC techniques out of niche applications and placed them amongst the ranks of GC and HPLC. To use this technique to the greatest of its ability, it is needed to establish an understanding of the effects of LC column parameters for these SFC separations. A mixture of compounds was separated across ten biphenyl columns to evaluate the effects of the column phase and dimensions towards the efficiency and quality of SFC separation. Column variables included particle type, particle diameter, column internal diameter, and column length.
Superficially porous particles (SPP) yielded less retention time drift and greater number of theoretical plates (N) compared to fully porous particles (FPP). FPP of diameters 5, 3, and 1.8 µm demonstrated 3 µm particles having the greatest N. The 1.8 µm particle generated the least N and the greatest amount of peak tailing. Between the 2.1, 3.0, and 4.6 mm ID FPP columns, the 4.6 mm column routinely possessed the greatest N. The 150 mm FPP column surpassed the 50 mm column with improved peak shape, reproducible retention times, and shortest HETP. Some of these conclusions are contrary to the popular reversed-phase
liquid chromatography and need to be established for SFC as chromatographers learn new platforms.References: 1. NoneDisclosure of Interest: None DeclaredKeywords: Column characterization and comparison, Supercritical fluid chromatography (SFC)


Late Breaking Posters

373www.ISC2016.ie @isc2016 #isc2016 /isc2016
LATE BREAKING POSTERS
LATE Breaking Posters
LBP-001 - Asymmetric Peroxidation of α,β-Unsaturated Aldehydes and Determination of Enantioselectivity by Chiral HPLC
Gandhi, H.a,b, and O’Sullivan T. P.a,b,c
a Department of Chemistry, University College Cork, Corkb Analytical and Biological Chemistry Research Facility,
University College Cork, Cork.c School of Pharmacy, University College Cork, Cork.
Content: Neglected Tropical diseases (NTDs) like Chagas disease, leishmaniasis, human African trypanosomiasis cause great suffering in many developing countries. Chagas disease is an infectious disease caused by the protozoan Trypanosoma cruzi. The current drugs used most extensively for treatment are benznidazole and nifurtimox. However, neither has been shown to eradicate infection during the chronic phase when most patients are diagnosed. Accordingly, the search for novel drug targets and new lead structures for the treatment of Chagas disease is an area of on-going research. One effective strategy is to develop and modify lead structures from the nature with proven bioactivity. Recently, the Plakortis species of marine sponges has proven to be rich source of bioactive cycloperoxides. Among these, Plakortide P was reported to be highly selective for Chagas disease and 15 times more potent than benznidazole (IC50 2.3µg/mL vs 36.3 µg/mL)1. The plaktorides typically contain a stereogenic carbon, to which a peroxide group is attached. Enantioselective peroxidation of α,β-unsaturated carbonyl compounds is one method for synthesising chiral cycloperoxides2. Herein we describe our work to date on the asymmetric synthesis
of chiral peroxides and the determination of the degree of enantioselectivity using chiral HPLC.1. A.J. Jones et al., Marine Drugs, 2013, 11, 4058. 2. L. Deng et al., J. Am. Chem. Soc, 2008, 130, 8134
LBP-002 - Dithiophosphonate Analogues of Acyclic Nucleoside Reverse Transcriptase Inhibitors Synthesis, Isolation and Analysis of Key Intermediates
Jones, D.a,b; O’Leary, E.M.a,b,c; O’Sullivan, T.P.a,b,c
a Department of Chemistry, University College Cork, Corkb Analytical and Biological Chemistry Research Facility,
University College Cork, Corkc School of Pharmacy, University College Cork, Cork
Content: Prodrug forms of the Acyclic Nucleoside Phosphonates (ANPs), Tenofovir (1) and Adefovir (2) are potent replication inhibitors of the Human Immunodeficiency Virus (HIV) (Figure 1). By strategic incorporation of a more labile phosphorous-sulfur bond into the phosphonate moiety, we hope to generate a series of second generation ANP analogues, dithiophosphonate ANPs (DTP-ANPs) DTP-1 and DTP-2. Introduction of the DTP moiety should enhance the rate at which these compounds inhibit the replication of HIV. This offers an attractive solution to suppress HIV replication and potentially combat newly emerging strains of HIV which are resistant to the currently existing first generation analogues.
As part of our on-going investigation into the synthesis of these compounds, we have investigated the formation of

www.ISC2016.ie @isc2016 #isc2016 /isc2016374
LATE BREAKING POSTERS
protected dithiophosphonates. This publication describes the application of 31P Nuclear Magnetic Resonance spectroscopy (31P-NMR) to facilitate monitoring of the incorporation and subsequent removal of this protecting group on a model substrate. Insights gained from this work will guide future synthetic exploration in this area and illustrates a potentially novel and exciting application of 31P-NMR which may be further applied to the monitoring of pharmaceutical processes.
Figure 1 – Proposed Antiviral Dithiophosphonate Analogues
LBP-003 - Synthesis and analysis of novel sulfur-containing antiviral analogues
O’Donovan, F.P.a,b; O’Leary, E.M.a,b,c; O’Sullivan, T.P.a,b,c
a Department of Chemistry, University College Cork, Cork b Analytical and Biological Research Facility, University
College Cork, Corkc School of Pharmacy, University College Cork, Cork
Content: There are currently 15.8 million people being treated with Antiretroviral Therapy (ART) for infection with Human Immunodeficiency Virus (HIV), according to the latest UNAIDS report [1]. HIV is a retrovirus that infiltrates the body’s immune system, hijacking its CD4+ T-cells’ replication system to reproduce its genome, destroying the T-cells in the process, leaving the host’s immune system susceptible to infection. The nucleoside reverse transcriptase inhibitor (NRTI) zidovudine (AZT) became the first FDA-approved treatment for HIV in 1987. Since then, a further six compounds in this class have received FDA-approval. The main problems attributed to NRTIs are associated toxicities and development of viral resistance, therefore there is a constant need for new improved therapy options.
4´-Thionucleosides have long been of interest to researchers for their ability to act as bioisosteres of cell-native nucleosides. Although none have made it to market, several have exhibited promising antiviral activity [2-4]. This work aims to synthesise and test several novel 4´-thionucleosides for biological activity. Using methodology published by Young et al we have adapted and optimised their strategy to suit our synthesis of sulfur analogues and derivatives of abacavir [5]. Analytical techniques have thus far played a huge part

375www.ISC2016.ie @isc2016 #isc2016 /isc2016
LATE BREAKING POSTERS
in the progression of this project. We have exploited the use of column chromatography to separate diastereoisomers and used 1H and 13C NMR spectrometry as key aids in compound characterisation. In this poster we will outline our synthetic approach and process optimisation strategies, and the analytical techniques which have guided us along the way. [1] UNAIDS, AIDS by the numbers 2015, UNAIDS,
Geneva, 2015[2] Kumamoto, H.; Nakai, T.; Haraguchi, K.;
Nakamura, K. T.; Tanaka, H.; Baba, M.; Cheng, Y.-C. J. Med. Chem. 2006, 49, 7861.
[3] Bobek, M.; Whistler, R. L.; Bloch, A. J. Med. Chem. 1972, 15, 168.
[4] Secrist, J. A.; Riggs, R. M.; Tiwari, K. N.; Montgomery, J. A. J. Med. Chem. 1992, 35, 533.
[5] Young, R. J.; Shaw-Ponter, S.; Thomson, J. B.; Miller, J. A.; Cumming, J. G.; Pugh, A. W.; Rider, P. Bioorg. Med. Chem. Lett. 1995, 5, 2599.
LBP-004 - Synthesis and Characterisation of Novel Analogues of Diffusible Signal FactorHorgan, C.a, Kumar, V. P.a,b and O’Sullivan T. P.a,b,c
a Department of Chemistry, University College Cork, Corkb Analytical and Biological Chemistry Research Facility,
University College Cork, Cork.
c School of Pharmacy, University College Cork, Cork.
Content: Cystic Fibrosis (CF) is a genetic, life-threatening disease that affects the lungs and digestive system of about 75,000 people worldwide. CF causes the body to produce abnormally thick and sticky mucus that builds up in the breathing passages of the lungs. The build-up of mucus predisposes the lungs of CF patients to colonisation by numerous bacterial species and this is the main cause of morbidity and mortality. These bacterial infections are difficult to treat due to the ability to resist frontline antibiotics and paucity of new antibiotics coming to market. This emphasises the need for alternative novel therapeutic strategies. Interactions between bacteria mediated by cell-to-cell signals can profoundly influence their behaviour to enhance resistance to antibiotics, promote bacterial persistence and the formation of bacterial biofilms. It has been proposed that interference with such signalling process may be an attractive target for interference. The signal molecule family, known as Diffusible Signal Factor family, has been found to play a role in biofilm formation and virulence in many bacteria1. Two such signals are cis-2-unsaturated fatty acids, Diffusible Signalling Factor and Burkholderia Diffusible Signalling Factor (BDSF). DSF family molecules have been detected in the airways of CF patients2. It has been found that interference with DSF signalling may inhibit biofilm formation in various species of bacteria increasing their susceptibility to antibiotic treatment. Herein, we describe the synthesis and characterisation of several, novel BDSF/DSF analogues.1. R. P. Ryan, et al., Trends Microbiol. 2011, 19, 145-
152.2. K. B. Twomey, et al., PLoS Pathogens 2012, 6,
939-950.

www.ISC2016.ie @isc2016 #isc2016 /isc2016376
LATE BREAKING POSTERS
LBP-005 - Uses of Chromatography and Spectroscopy in the Impurity Profiling Studies of Controlled Hallucinogenic Amphetamines
Roisin Jonesemail: [email protected]
Content: Amphetamine is a simple molecule to synthesise and alter, leading to a large number of readily available amphetamine-type stimulants (ATS). One such derivative ATS is BromoDragon-FLY (BDF) (1), a highly hallucinogenic amphetamine. The most common route to making amphetamines in clandestine labs is the Leuckart reaction, which converts a precursor ketone to the desired amine. The Leuckart reaction produces a number of characteristic route specific impurities that can be used to identify the synthetic pathway, most notably benzyl (2) and phenyl (3) pyrimidines and pyridines, with the first two being present in higher yields.
From a tetrahydrobenzodifuran (THBD) ketone precursor, there are three steps to BDF: bromination, aromatisation and the Leuckart reaction. Depending on the order that these steps are performed in, there are eight potential pyrimidine impurities from the formation of BDF.
The aims of this body of work are to: • Independently synthesise each of
these pyrimidines, purify them via chromatographic techniques and fully characterize them using spectroscopic techniques such as 1H and 13C NMR, FT-IR and mass spectrometry in order to use them as analytical standards which can later be used to identify them in reaction mixtures or illicit drug samples.
Thus far, six of the eight impurities have been successfully synthesised and characterised, and the work remains ongoing.
• Use chromatographic techniques such as HPLC and GC-MS to build profiles of these analytical standards so that the compounds can be identified in even in crude reaction mixtures, and in illicit drug samples. Thus far HPLC and GC-MS work has been carried out for the series of benzyl pyrimidines, with work still to be carried out on the series of phenyl pyrimidines.
• Run a series of Leuckart reactions which should produce the benzyl and phenyl pyrimidines described above ((2) and (3)), and analyse the crude reaction mixtures using optimized HPLC and GC-MS conditions, comparing the results to the chromatographic data gathered for the analytical standards to see if the standards can be used to identify route-specific impurities in Leuckart reactions. Thus far, a number of precursors to the required ketones have been synthesized, but work is still on-going in this section.
!

377www.ISC2016.ie @isc2016 #isc2016 /isc2016
LATE BREAKING POSTERS
LBP-006 - Development of an Integrated Sensing System for PAT Application in the Food and Beverage Industry Scanlon, S.1*, Hobbs, Elizabeth 2, Morris, Virginia 2 and Moore, E.J.11 Tyndall National Institute and the Department of
Chemistry, UCC, Cork2 PepsiCo International, Little Island, Corkemail: [email protected]
Abstract: Food and beverage quality and safety have become of significant importance over the past decade and assuring the highest standards of process control is a key priority. The FDA’s Process Analytical Technology (PAT) initiative emphasises that “quality cannot be tested into products; it should be built in or should be by design” [1].
This research proposes a new system for quality control in the food and beverage industry, through the development of a multi-parameter sensing device for PAT application. Following the miniaturization of various analytical techniques, used for quality control analysis, integration into a single device will create a multi-parameter sensing system for real-time process parameter analysis. To date, much of the research has been focused on the caffeine determination, which is currently carried out off-line, using High Performance Liquid Chromatography (HPLC), which can be time consuming, with expensive instrumentation. In this work, chemical sensors for the electrochemical determination of caffeine in real samples have been developed. Three electrodes were compared, using a Nafion® modification to increase electrode sensitivity. Figure 1 shows the comparison of the three modified electrodes for the detection of caffeine, with the gold electrode
proving the most efficient, even without prior pre-treatment. A low cost, re-useable sensor for the determination of caffeine in soft drink concentrate has been developed using a Nafion®-modified screen printed gold electrode. This is the first time that a sensor of its kind is used for such a purpose, with a high degree of recoverability. Work has begun on an electrochemical pH sensor, using iridium oxide modified electrodes, grown by potentiostatic cycling in an iridium complex solution, as shown in figure 2. This is the first time that an iridium oxide-modified screen printed electrode has been developed for pH determination. References:Guidance for Industry PAT-A Framework for Innovative Pharmaceutical Development, Manufacturing and Quality Assurance, United States Food and Drug Administration (FDA), September 2004
Figure 1: Cyclic voltammograms recorded at Nafion-modified glassy carbon (blue), screen printed carbon (red) and screen
printed gold (green) electrodes in 0.1 M Phosphoric Acid containing 300 uM caffeine.
Figure 2: Cyclic voltammograms for the growth of an Iridium Oxide pH film by potentiostatic cycling in an Iridium complex solution.

www.ISC2016.ie @isc2016 #isc2016 /isc2016378
LATE BREAKING POSTERS
LBP-007 - A ‘Smart’ Needle For Objective Nerve Localisation During Ultrasound Guided Peripheral Nerve BlockHelen, Lisa1*; O’Donnell, Brian2; and Moore, Eric1.1 Sensing and Separation Group, Chemistry Department
and Life Science Interface Group, Tyndall National Institute, University College Cork, Ireland
2 Department of Anaesthesia, Cork University Hospital, Cork, Ireland
email: [email protected]
Abstract: As the world of smart technology advances, insensate objects are being transformed to ‘smart’ devices by the application of sensors. Our research focuses on applying an impedance sensor to a hypodermic needle to create a ‘smart’ needle. This novel device will use bioimpedance to determine needle-to-nerve proximity for application in ultrasound guided peripheral nerve block (USgPNB) procedures. Bioimpedance data from the needle tip will allow for real-time tissue identification and thus provides the user with the exact needle tip location. For anaesthetists it will provide valuable information indicating needle contact with the nerve covering or dangerous needle position within the nerve. Introduction of this new technology to USgPNB will increase its safety, efficacy and training capacity thus increasing the use of the technique and reducing costs for the healthcare system, as it is a safer quicker alternative to general anaesthesia. The impedance sensor on the ‘smart’ needle is a miniaturised two electrode set-up. This first prototype ‘smart’ needle has been fabricated and characterisation is underway. Impedance values, using this prototype, for different substances including saline solutions, phantom gelatine models and different tissue types in meat will be
presented. Results have demonstrated that bioimpedance can be used to identify tissue type at the needle tip. The addition of ‘smart’ needle technology to USgPNB may provide a solution to a currently unmet clinical need.
LBP-008 - Sound Bending Chemical Reactions: Non-Titrimetric Determination of Acid-Base Reactions using BARDS.M. Rizwan Ahmed and Dara Fitzpatrickemail: [email protected]
Abstract: Is it possible to determine an acid-base reaction without a pH probe, indicator and titres of reagent from a burette?! Is it also possible to follow the same reaction with nothing more than a microphone or even by ear? We present data which demonstrates for the first time how acid-carbonate reactions can be monitored accurately by acoustic measurements.
The stoichiometric reactions yield specific quantities of carbon dioxide which cause reproducible changes to the compressibility of the reaction mixture. This in turn slows down the speed of sound in solution which is generated by allowing the reaction vessel to resonate, which is induced by the magnetic follower inside.
Broadband Acoustic Resonance Dissolution Spectroscopy (BARDS) harnesses this phenomenon for several applications.1 However, in this study, acid (HCl) and base (Carbonate) were reacted in stoichiometric equilibrium, to record a BARDS time –frequency acoustic spectra. The BARDS spectra obtained depend on the limiting

379www.ISC2016.ie @isc2016 #isc2016 /isc2016
LATE BREAKING POSTERS
reagent. Therefore an excess of one over the other will not produce a greater quantity of carbon dioxide. For this reason, one addition of a reagent in excess is enough to determine the end point without the need for titrimetry.
BARDS analysis also demonstrated a similar behaviour for reaction between HCl and carbonates irrespective of their counter ions (Na and K). The data also shows how half the amount of a diprotic acid is required to neutralise the same amount of carbonate as HCl to yield the same spectra for both. This is also irrespective of the counter ion of the carbonate. This method can be used to interrogate the acidity of any reaction solution in a rapid manner and may be important in situations where it is crucial that the acid is spent in a reaction mixture.References:[1] Fitzpatrick, D., Krüse, J., Vos, B., Foley, O.,
Gleeson, D., O’Gorman, E., O’Keefe, R., 2012. Principles and applications of broadband acoustic resonance dissolution spectroscopy (BARDS): A sound approach for the analysis of compounds. Anal. Chem. 84, 2202–2210.
LBP-009 - Application of Qualitative LC-UV-TOF/MS Techniques for the Profiling of Novel Ellipticine Compounds. Ryan Alam and Florence McCarthyDepartment of Chemistry and ABCRF, University College Cork, Western Road, Cork, Ireland.
Abstract: Liquid chromatography mass spectrometry (LC-MS) is a hyphenated analytical technique; effectively combining the versatile separation capabilities of LC with the high sensitivity and selectivity of MS detection. The ability to isolate the individual components of a sample in-line, prior to MS detection, permits the investigation of highly complex and varied sample matrices.1 These characteristics have facilitated the growing popularity of LC-MS, in areas such as pharmaceutical impurity profiling and structural analysis.2 Impurity profiling can be beneficial during the development and synthesis of potential drug substances, as it may provide guidance for the improvement of synthetic methods. Similarly, when investigating new chemical entity bioactivity, prior confirmation of structure is essential. The aim of this qualitative study was to develop a method for the separation and profiling of a series of novel ellipticines.3 To-date, no publication has documented the LC-MS analysis of this class of compounds. A reversed-phase LC method was developed that afforded the adequate separation of sample components and facilitated their identification. The isomeric separation of a novel bioisosteric ellipticine analogue was achieved, using analytical HILIC mode LC. All chromatographic separations were coupled with simultaneous UV/PDA and TOF/MS detection.

www.ISC2016.ie @isc2016 #isc2016 /isc2016380
LATE BREAKING POSTERS
References 1 Trufelli H., Palma P., Famiglini G., Cappiello
A. An overview of matrix effects in liquid chromatography-mass spectrometry. Mass Spectrom. Rev. 2011; 30(3): 491-509.
2 Gillespie T.A., Winger B.E. Mass spectrometry for small molecule pharmaceutical product development: A review. Mass Spectrom. Rev. 2011;30(3): 479 – 490.
Deane F.M., O’Sullivan E.C., Maguire A.R., Gilbert J., Sakoff J.A., McCluskey A., McCarthy F.O. Synthesis and evaluation of novel ellipticines as potential anti-cancer agents. Org, Biomol. Chem. 2013;11(8): 1334-1344.

381www.ISC2016.ie @isc2016 #isc2016 /isc2016
NOTES

www.ISC2016.ie @isc2016 #isc2016 /isc2016382
NOTES

383www.ISC2016.ie @isc2016 #isc2016 /isc2016
NOTES

www.ISC2016.ie @isc2016 #isc2016 /isc2016384
NOTES